Questions, data and models underpinning metabolic engineering
 Rik P. van Rosmalen
Rik P. van Rosmalen Vitor A. P. Martins dos Santos
Vitor A. P. Martins dos Santos Maria Suarez-Diez
Maria Suarez-Diez- 1Laboratory of Systems and Synthetic Biology, Wageningen University & Research, Wageningen, Netherlands
- 2Bioprocess Engineering, Wageningen University & Research, Wageningen, Netherlands
- 3LifeGlimmer GmbH, Berlin, Germany
Model-driven design has shown great promise for shortening the development time of cell factories by complementing and guiding metabolic engineering efforts. Still, implementation of the prized cycle of model predictions followed by experimental validation remains elusive. The development of modelling frameworks that can lead to actionable knowledge and subsequent integration of experimental efforts requires a conscious effort. In this review, we will explore some of the pitfalls that might derail this process and the critical role of achieving alignment between the selected modelling framework, the available data, and the ultimate purpose of the research. Using recent examples of studies successfully using modelling or other methods of data integration, we will then review the various types of data that can support different modelling formalisms, and in which scenarios these different models are at their most useful.
1 Introduction
“All models are wrong”. This famous quote by George Box (Box, 1976) is often mentioned in discussions about mathematical models or their predictions, often followed by the qualifier “but some are useful”. This, naturally, turns this sentiment into practical advice: just find the model that is useful and off one goes. However, it is important to consider that on its own, a mathematical model is never wrong; it just fails to represent the intended phenomenon. You were wrong when building the model. A mathematical model is a way to formalize expert knowledge into an objective decision-making framework. “Errors” in a model, or the differences between predictions and experiments, thus expose incorrect assumptions, knowledge gaps or inconsistencies in the data, or as summarized by another famous idiom: “Garbage in, garbage out”.
In metabolic engineering, model-driven experiments have been long thought of as the future and many studies have shown that models can be useful tools to predict or validate biological mechanisms. For instance, Satanowski et al. (2020) use a constraint-based, genome-scale model of Escherichia coli to find possible carbon-fixating cycles that consume CO2 as a sole carbon source to produce pyruvate. A complementary model based on thermodynamic considerations was then used to assess the feasibility of successfully implementing these cycles, after which they show experimentally that CO2 fixation is feasible using one of these pathways. This study shows two important ways a metabolic model can be utilized, first by offering potential designs and next by evaluating these potential strategies. The model by Klipp et al. (2005) about the response of Saccharomyces cerevisiae to osmotic shock is a strong example of a model integrating different biological processes: metabolic, regulatory, and homeostatic. Integrating these parts into a single model allowed for the prediction of independent experiments, and led to an increased understanding of the system as a whole.
Still, expert knowledge is often preferred to guide experimental designs and most metabolic engineering studies published right now do not make use of a model. For example, a survey of recent metabolic engineering studies (Q4 2020) in the journals Metabolic Engineering and Microbial Cell Factories, show that only 32% (n = 54) and 17% (n = 21), respectively, make use of a metabolic model in their study.
Reasons for this could relate to the difficulties in constructing a quality model or the amount of data required. Alternatively, models often predict the obvious or the predictions of a model are not actionable and thus do not translate well to engineering strategies. Finally, the quality of the tools or a lack of training could be additional reasons for the lack of use of predictive models in metabolic engineering studies. Cvijovic et al. (2016) discusses possible strategies to address the last possibility, in particular on how to foster awareness of both experimental and modelling techniques and how they fit into a systems biology curriculum.
In this review, we will discuss how to avoid these pitfalls, how to design experiments with models in mind, and how to find the model that is not just wrong but also useful. In the first section, (Box 1) we will introduce some commonly used modelling terms and we will discuss the aspects that turn a model into something insightful and actionable. Next, we will give a high-level overview of the most common and relevant modelling techniques used for metabolic engineering, what kind of data they require and when they are the most useful. Finally, we will discuss what we believe to be the most important part of the model: the data. What kind of data is both useful and available for metabolic engineering studies? What are the limitations of different measurement techniques? And how does all this impact a potential model?
2 What is the right model?
To choose a model that is useful, start by considering the different aspects influencing the decision to choose for a specific model (Figure 1). First, figure out the question it needs to answer. What problems need to be solved? Or, more technically: What is the objective to be optimized? The more specific the problem can be formulated, the easier it becomes to solve the problem and to verify model predictions experimentally.
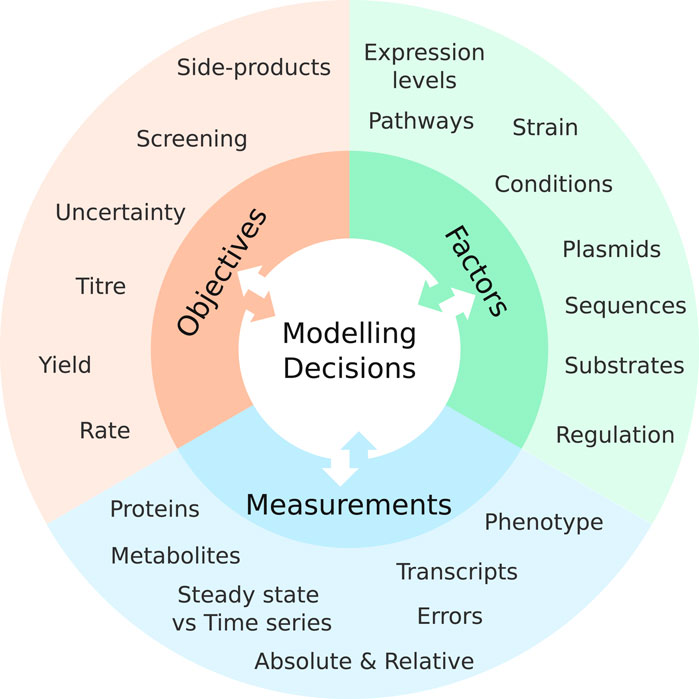
FIGURE 1. Research objectives, experimental factors, and the data that can be measured all have an impact on which type of model is the most suitable for their study. Conversely, the data the model can integrate and the predictions it can make also impact these three components in return. Balancing the requirements and advantages of each is essential for successfully utilizing a computational model for a study.
BOX 1
Different modelling fields often use their own specific jargon. Thus, we list here a number of common modelling terms for the context of this review.• Model: A mathematical representation of a (biological) system, describing, for example, flux through a metabolic pathway or a population of cells growing in a bioreactor.• Variable: A variable describes something that can be quantified in the system. In a model describing a metabolic pathway, this could be the concentration of a metabolite or enzyme. The complete set of variables describing the model is called the state. Note that a variable does not necessarily need to represent something that can be experimentally measured, but can also represent something abstract. A variable can thus also be binary, such as the presence or absence of a gene, or discrete, such as the choice of one of several plasmid backbones.• Parameter: A parameter is similar to a variable, but can be considered fixed during the simulation of the model, such as an enzyme rate constant or the relative strength of a ribosomal binding site in a genetic construct.• Input: A variable in the model that can be changed experimentally in order to achieve an objective, for example, the concentration of a medium component, or which enzyme variant to use in a pathway.• Output: A variable in the model that represents a property of interest resulting from the simulation, for example, a variable that can be measured experimentally and can thus be used to verify the model.• Objective function: A function quantifying the goal to achieve based on the state of the model. For example, the growth rate of an organism, the concentration of a product at a certain time, or the yield of a product relative to the consumption of the substrate. There can also be multiple objectives, possibly signifying a trade-off within the system. In constraint-based models specifically, the objective function is used to represent the biological objective of the organism, such as growth. A common objective is therefore the biomass reaction: a reaction that consumes the different metabolites the organism requires to grow.• Parametrization: The process of finding the parameter values best describing the system based on its agreement with experimental data, also known as fitting or optimization. During parametrization, the objective is to reproduce the measured data as closely as possible, thus an objective function is minimized that quantifies the difference between variables predicted by the model and experimental measurements of the same variables.• Constraint: A limitation on the range of one or more variables or parameters in the model, often due to practical or experimental reasons. A set of constraints specifically on the minimum and maximum value of a variable or parameter during optimization is often called bounds.
Different modelling fields often use their own specific jargon. Thus, we list here a number of common modelling terms for the context of this review.
• Model: A mathematical representation of a (biological) system, describing, for example, flux through a metabolic pathway or a population of cells growing in a bioreactor.
• Variable: A variable describes something that can be quantified in the system. In a model describing a metabolic pathway, this could be the concentration of a metabolite or enzyme. The complete set of variables describing the model is called the state. Note that a variable does not necessarily need to represent something that can be experimentally measured, but can also represent something abstract. A variable can thus also be binary, such as the presence or absence of a gene, or discrete, such as the choice of one of several plasmid backbones.
• Parameter: A parameter is similar to a variable, but can be considered fixed during the simulation of the model, such as an enzyme rate constant or the relative strength of a ribosomal binding site in a genetic construct.
• Input: A variable in the model that can be changed experimentally in order to achieve an objective, for example, the concentration of a medium component, or which enzyme variant to use in a pathway.
• Output: A variable in the model that represents a property of interest resulting from the simulation, for example, a variable that can be measured experimentally and can thus be used to verify the model.
• Objective function: A function quantifying the goal to achieve based on the state of the model. For example, the growth rate of an organism, the concentration of a product at a certain time, or the yield of a product relative to the consumption of the substrate. There can also be multiple objectives, possibly signifying a trade-off within the system. In constraint-based models specifically, the objective function is used to represent the biological objective of the organism, such as growth. A common objective is therefore the biomass reaction: a reaction that consumes the different metabolites the organism requires to grow.
• Parametrization: The process of finding the parameter values best describing the system based on its agreement with experimental data, also known as fitting or optimization. During parametrization, the objective is to reproduce the measured data as closely as possible, thus an objective function is minimized that quantifies the difference between variables predicted by the model and experimental measurements of the same variables.
• Constraint: A limitation on the range of one or more variables or parameters in the model, often due to practical or experimental reasons. A set of constraints specifically on the minimum and maximum value of a variable or parameter during optimization is often called bounds.
Next, consider what experimental factors, i.e., which properties of the system that can affect the research outcome, can be changed. Since these are the inputs the model can tune to predict different scenarios, it is essential that the model allows these factors to be represented. Vice versa, this also makes sure that the solutions and predictions of the model can be validated in practice.
Together with the research question and the inputs, consider the availability of data. What can be measured? Do not only consider here the different molecules or properties that can be measured, e.g. proteins, metabolites concentrations, or enzyme kinetics, but also other factors such as the throughput, the context in which it is measured (in vivo versus in vitro) or whether measurements can be done as a time-series, or in steady-state. Just as it is important to make sure that the inputs can be modelled by the model, the model should be able to integrate the data measured.
Finally, which other practical limitations exist? Think about the data that can or cannot easily be measured, the scale at which this can be done due to cost or time limitations and whether the assumptions that certain modelling techniques bring are valid. For example, how valid is the steady-state assumption that a constraint-based model would bring, or is it reasonable to assume that reactions inside the cell take place on a much faster time-scale then changes in the conditions of the medium, such as assumed by a dynamic Flux Balance Analysis (dFBA) model. Often, there is a trade-off to be made here. High-level models are built on more assumptions but do not require as much data to be useful. Low-level models make less assumptions about what happens inside the cell and describe processes in more detail, but do require more data to be useful.
As different models can offer distinct insights, there is no need to limit oneself to a single model for the whole study. Similar to how the objective or research question of a study might shift when new information becomes available, the most suitable model for the task might change as well. This might require a whole new modelling approach, or can be accomplished by adding new processes to the existing model. Imagine, for example, a study where potential targets are first identified using a data-driven approach based on comparative transcriptomics, after which a subset of these targets are evaluated and further studied with a constraint-based metabolic model. The design-build-test-learn (DBTL) cycle is a process that embraces this idea. In this iterative process, a genetic design is created, built and tested, after which the experimental data is used to improve a model (learn) to suggest new designs.
Reusing an existing model from a previous study can also be the right approach. However, be aware that it can be challenging to make use of a model if it is not clear exactly what data and assumptions the model is built on. Especially with kinetic models this can be problematic. Tiwari et al. (2021) report that around half of the systems biology models in the BioModels repository (Malik-Sheriff et al., 2020) were not reproducible. Porubsky et al. (2020) give an in-depth overview of the best practices to follow in order to do reproducible and FAIR research and how they apply to metabolic models. These best practices can also be used to evaluate the potential of a model for re-use.
2.1 What is the problem to be solved?
The problem to be solved, or the aim of the model is critical. Formulating a research question well is important in science in general, but especially essential when using a model, as the type of model that is useful critically depends on the question to be answered. For example, if the objective is to compare the co-factor usage of different pathways to produce a metabolite, flux balance analysis on a constraint-based model will easily perform this task. Ask the same model to predict the effect of metabolic regulation or feedback inhibition and the model will start to show its limitations.
In Gaspari et al. (2020) the authors aimed to find a defined medium for the growth of the fastidious pathogen Mycoplasma pneumoniae. Their modelling approach, a constraint-based model, was well suited for this as both the objective (a high growth rate) and the factors to change (medium components), were directly represented in the constraint-based model and could easily be measured or modified experimentally. Another example of using a model well suited to the objective is the study by Krambeck and Betenbaugh (2005), which investigated the glycosylation of glycoproteins in Chinese hamster ovary (CHO) cells. Since the glycosylation pathways involve many similar building blocks that can be assembled in different orders, enzyme kinetics play a large role in determining the exact products formed. By using a dynamic model describing the enzyme kinetics in detail, the authors were able to predict the effect of expression changes on the final distribution of glycoproteins and design strategies to increase the production of their preferred product. The problem or research question does not necessarily need to be something concrete, but can also be abstract. In de Groot et al. (2020) the authors aimed to unite different theories of why overflow metabolism takes place. By relating the works analysed back to a shared mathematical formulation, they showed that these different models share a common principle, i.e., that overflow metabolism is caused by two growth-limiting constraints.
In addition to the research question, the problem to be solved can also be regarded in a broader sense when considering aspects such as the number of solutions, tolerance for errors or other factors. In explorative studies, generating many leads for further research is the goal and missing good candidates might be a worse outcome than generating a few false-positive results. A modelling method that covers the entirety of metabolism such as a genome-scale constraint-based model, could thus be suitable (Figure 2). Due to its wide scope, different designs can be created for a single objective, even if some of them might not turn out to be feasible in practice. In contrast, when the objective is more constrained or the investment per experiment is higher it might become more important to minimize prediction errors rather than having the genome-scale scope of a constraint-based model. In this case, it would be preferred to use a method where it is possible to test the sensitivity of the prediction to different parameters or assumptions that were made in the design of the model such as a kinetic model. For example, in the previously mentioned study by Satanowski et al. (2020) they used both of these strategies in turn. First they predicted possible carbon fixation routes using a genome-scale constraint-based model purely based on reaction stoichiometry. Next, they performed a more detailed analysis of the predicted reaction thermodynamics for each potential pathway taking into account the thermodynamic potentials and possible concentration range of each intermediate metabolite. This allowed them to evaluate the potential design based on both the difficulty of implementing the required number of new enzymes, as well as the likelihood of the pathway being thermodynamically feasible.
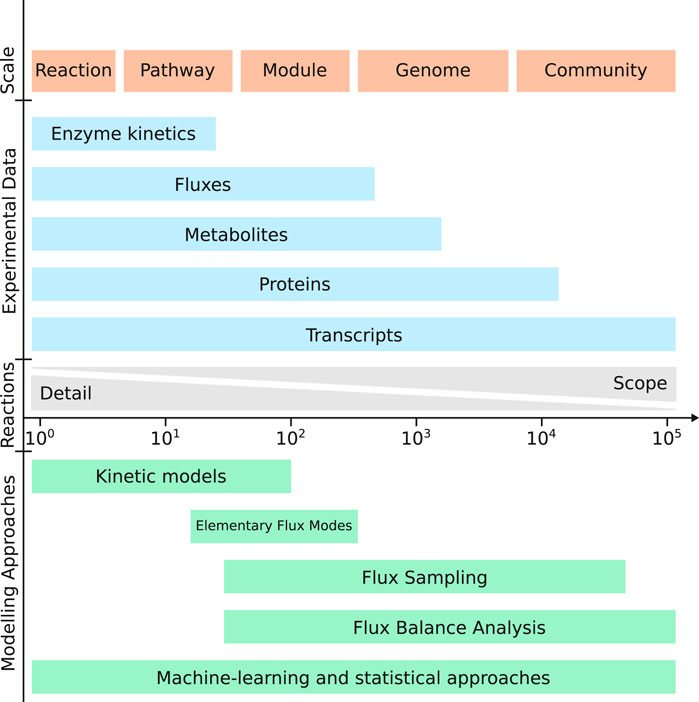
FIGURE 2. Different experimental and modelling techniques work best at certain scales. Often there is a trade-off between the methods scope and the amount of detail that is measured or simulated. Note that machine-learning and statistical approaches critically depend on the scale of the data collected and rather than on the scope of the model itself.
2.2 What are the factors that can be modified?
In order to figure out what the model should include and what type of model could be appropriate, consider the factors that can be changed experimentally to achieve their research goal. In metabolic engineering studies, for example, common factors are which strain or organism to use, or which genes to knock in or out. Also important are the growth conditions, such as the temperature, medium, and choice of substrate. To further optimize a strain, expression levels or the usage of enzyme variants with different kinetics or regulation become important targets. For dynamic processes, perturbations such as adding more substrate or an activating or inhibiting metabolite can also be a factor for optimization, as the strength and timing of the perturbation can be significant.
To optimize indigoidine production in Pseudomonas putida, Banerjee et al. (2020) tried to couple production to growth. Here they chose a method called minimal cut sets on a genome-scale constraint-based model to identify a minimal set of reactions to deactivate. Because of the mapping of reactions to genes supplied by the genome-scale constraint-based model, they could directly link the factors to be modified (i.e., the activity of specific genes) to their model. Alternatively, instead of modifying their organism, Li et al. (2019) controlled their medium by changing feed rates for different substrates of the algae Chlorella vulgaris using a constraint-based model to manage the trade-off between high growth, which requires nitrogen, and high production, which happens under nitrogen starvation. With the model, they devised feeding strategies that provided the required nitrogen for high growth but kept the concentration in the medium low. They tried this strategy both with cell density data from previous experiments to predict the consumption, as well as real-time data measured during the experiment itself. Using the constraint-based model as controller determining their feed rates, they managed to significantly increase their product concentration.
Expression levels are common targets for both screening and optimization. Lian et al. (2019), for instance, used CRISPR targeted activation, inhibition, and deletion for a whole-genome screening of furfural tolerance in S. cerevisiae, while Carbonell et al. (2018) used a combinatorial library of genetic constructs to vary gene order, promoters, and plasmid backbones in order to optimize a key precursor for the production of flavonoids in E. coli.
Usually, there are many factors that can be changed. However, some might be more practical to implement experimentally, while others might allow more granularity or a higher throughput of experiments. In addition, consider whether the effect of the change can be measured directly, or only in the end result. Having many steps between the changed factors and the measured readouts will make it harder to accurately model their effect as signals get mixed or diluted. A model can be a tool to help estimate the value of a proposed experimental factor, as the changes can be analysed computationally to estimate their effect at different levels. Using techniques such as sensitivity analysis and optimal experimental design, a model can thus assist by providing an expected return of investment versus the cost of doing an experiment.
2.3 What can be measured?
In order to create a quality model, data is usually required to fit the model. High-throughput “omics” data can often be an important source of data, such as transcriptomics, proteomics, metabolomics and fluxomics. In addition, many other types of data can be highly relevant and can be obtained from targeted experiments or assays, such as growth rates, kinetic parameters, toxicity of metabolites or localization of proteins. Furthermore, data on the “network” or interaction level, such as the presence of reactions, regulation or other interactions between species in the system are essential for an accurate model.
In addition to the type of data, the context of the measurements also has to be considered, as it impacts how samples relate to one another. For studying the effect of metabolic regulation, Link et al. (2015) opt to take metabolomics samples at very short intervals to capture the period of time when metabolic regulation is dynamically active while regulation on the gene level still has to come into effect. Contrast this with Gerosa et al. (2015), where they also looked at metabolic regulation and included metabolomics data from multiple steady-state conditions instead. Even though both studies aimed to study the impact of metabolic regulation and use metabolomics data to do so, they required an entirely different method of analysis. Similar to dynamics in time, a spatial component can also be considered. In Harcombe et al. (2014), the authors used a spatio-temporal model of competing microbial strains, including the relative positioning of the colonies as a factor to describe the interactions. A spatial component can also mean the presence of multiple compartments. For example, Alvarez-Vasquez et al. (2000) showed that transport is likely the limiting step for citrate production in Aspergillus niger using a dynamic model based on S-systems. Regardless of whether spatial or temporal interactions are considered, it is important to consider whether the time or space between samples is sufficiently small to match the expected temporal or spatial scale of the process in question, or, in the case of multiple compartments, whether metabolites in the different compartments can be measured independently.
As another example of how the context of a measurement can matter, in Shen et al. (2020) the authors used a cell-free system to optimize the Weimberg pathway to degrade xylose into α-ketoglutarate. Since they were able to control all species in the system and measure all changes of metabolite concentrations over time, they could fit a detailed kinetic model for the whole pathway. Alternatively, for Teusink et al. (2000), one of their goals was to fit a kinetic model of S. cerevisiae glycolysis using in vitro measured kinetic parameters. Even though this is a well-studied model system, they reported that only half of the in vitro measurements of enzyme kinetics described the in vivo enzyme activity accurately. While these two studies are not directly comparable, it goes to show that in this case the in vitro versus in vivo measurements, and also the cell-free versus in vivo systems, bring different constraints and thus impact the model.
The context can also mean the difference between single-cell data, where stochasticity plays a large role, or the data of an entire culture of cells in different metabolic states pooled together where these differences are averaged out. Co-cultures or microbial communities can add another layer of complexity, due to the extra level of interactions possible between different species. For microbiome studies, “meta-omics” pool together measurements for all species in the sample, where the exact composition of the species in the sample, or even the species themselves might not be known. This leads again to a new set of modelling challenges as the traditional compartmentalization of species in the model, such as in a co-culture model, might not be possible. In Delogu et al. (2020) the authors analyse the dynamics and metabolic capabilities of such a community, SEM1b, with the ability to degrade cellulose and produce methane using several “meta-omics” technologies.
Finally, care has to be taken whether the data measured is absolute or relative, as many of the current “omics” methods yield relative data. Evaluate whether integrating the relative data into the model is feasible, or switching to an alternative measurement method with absolute quantification is required. Similar to the difference between relative or absolute measurements, some methods generate data where multiple signals are combined into a single measurable output, imagine for example, an optical assay where the spectra of two species overlap. In this case, it can be considered whether integrating this relation directly into the model might allow for a more straightforward or robust interpretation than trying to isolate the signals beforehand, potentially losing the correlated structure of the two measurements.
Overall, it is important to assess what is feasible to measure, how it can be integrated into the model and how much information it contains about the process to be optimized. In Section 4, we will elaborate on the specific types of data and their suitability for modelling in depth.
2.4 What type of model is suitable?
Many different types and variants of models have seen use in metabolic engineering, both data-driven and knowledge-based approaches. Machine-learning and statistical models can be considered data-driven models, where the data is the main driving factor behind the model to find significant differences or correlations pointing out potential areas of interest to investigate. Mechanistic models such as constraint-based or kinetic models, on the other hand, are based on a description of the behaviour of the system according to the biological processes happening. They aim to use the experimental data to validate these descriptions and provide a deeper understanding of the underlying process and can thus be considered knowledge-based models. In metabolic engineering genome-scale, constraint-based metabolic models have been highly successful, but it is far from the only usable method and even within constraint-based models there are many variations.
All of these models come with their own set of assumptions. Genome-scale constraint-based models assume steady-state and optimality of a certain objective, while Michaelis-Menten kinetics assume that the concentration of the enzyme is significantly lower than that of the substrate. Statistical tests might assume normally distributed values or independence of two variables. Closely related to the assumptions made is the level of abstraction of a model. Statistical models work at a high level of abstraction, incorporating little about the details of the underlying process. Kinetic models, on the other hand, aim to describe the physical interactions between enzymes and metabolites. However, even within kinetic models there are degrees of abstraction, ranging from mass-action kinetics describing every interaction between metabolites and enzymes, to composite rate laws such as Michaelis-Menten, or lin-log kinetics, which trades physical accuracy for ease of modelling. Generally, lower-level models offer increased insights into the underlying processes, while higher-level models trade this extra detail for simplicity in both creating and using the model, also allowing for a wider scope of the model.
Apart from the use of a model to drive decision-making, another important aspect is the generation of “knowledge” from raw data, i.e., turning raw measurements into verifiable theories or quantifiable properties of the system. Depending on the research question, this might be a primary or secondary objective. However, even if it is not the primary objective, gaining more knowledge about the system can assist in further engineering efforts or serve to test and validate assumptions to simplify the system. Choosing a mechanistic model can help with achieving this knowledge-based objective, as system properties in these models often depend less on factors not included in the model and are thus easier to translate to a different system or model. Still, to make use of a lower-level model, more data is needed with a greater level of detail than for a higher-level model or the scope of the model has to be reduced. Relating to this is the concept of the design-build-test-learn (DBTL) cycle, where knowledge or data from each round of experiments is fed back into the model to get a better understanding of the system for subsequent rounds of experiments. While within a research project often a single type of model is utilized, it can also be considered to start the project with a high level and gradually move to a lower level of abstraction as the knowledge about the system increases.
Together with the considerations that we have discussed in the previous sections, these factors result in some models being more suitable for a research question than others. Summarizing, certain research questions align better with certain types of models and data. We explore three successful studies utilizing different types of models in Table 1 to highlight how they achieved this match between experiment and model. In Section 3, we will discuss the different types of models and how they have been successfully applied for metabolic engineering.

TABLE 1. Analysis of the objective, measured data, and model for three selected studies. It is highlighted how the experimental and modelling techniques have been aligned to create a successful study integrating models and experiments.
3 Metabolic engineering: Most relevant models
3.1 Knowledge-based models
3.1.1 Constraint-based models
Genome-scale constraint-based models are one of the more common types of models used to study metabolism, most often in combination with methods based on flux balance analysis (FBA). By assuming steady-state conditions, this method eliminates the need for describing reaction kinetics and can be simplified to a linear programming problem, which scales well to thousands of reactions and can thus span the full genome-scale reaction network. These models can be reconstructed from genomics data by matching genes to annotated enzymes catalysing known reactions, often starting from a known reaction network from a related organism. The metabolic network can then be further curated with studies of growth on specific substrates, metabolomics, knock-out or essentiality studies. Multiple tools exist to facilitate this task, several of which were compared by Mendoza et al. (2019).
Because these networks are usually built to study the full metabolic potential of the organism in question, it can be necessary to further tailor these networks to only contain reactions active in specific conditions. Especially for multi-cellular organisms, where regulation becomes more important, this can be relevant, although it proves challenging in practice. A comparison of multiple methods to integrate expression data for E. coli and S. cerevisiae by Machado and Herrgård (2014) showed large differences between methods but no clear advantage. Opdam et al. (2017) also applied several algorithms to generate context-specific networks for four human cancer cell lines using transcriptomic and exometabolomic data. They note that the choice of method and threshold settings to qualify a reaction as active have a great impact on the final model produced and the predictions. Despite this, Montero-Blay et al. (2020) showed that using proteomics and essentiality data from a transposon study active metabolic pathways can be accurately determined for the Mycoplasma agalactiae and M. pneuomiae, although for these two minimal organisms the number of possible alternative pathways is very limited.
Flux balance analysis (FBA) can be used to make predictions of growth and production rates using these constraint-based models by constraining metabolite uptake or secretion, and reaction directions in combination with an objective such as maximal growth or production. By changing medium conditions, objectives or by introducing or removing reactions, different scenarios can be simulated. For example, in Keller et al. (2020) E. coli was screened for strain designs where methanol assimilation is a strict requirement for growth, but an additional carbon source can be utilized to boost the growth rate. This extra requirement was set in order to find a strain that would be suitable to be further optimized through laboratory evolution. FBA can also be used for multi-species models, for example, to analyse a co-culture or microbial community. For example, Benito-Vaquerizo et al. (2020) used FBA to analyse a co-culture of two Clostridium species to convert syngas to medium-chain fatty acids. Using this model, the authors managed to predict strategies to increase the fatty-acid production rate, one of which was also previously found experimentally. There are also many alternative formulations of FBA, such as parsimonious FBA, where after optimization of the objective to a certain threshold the total sum of flux is minimized, based on the concept that each unit of flux carries some burden in enzyme cost. This method was used by Davidi et al. (2016) as a stand-in for fluxomics data to study enzyme kinetics, and by Zhang et al. (2020) to investigate which reactions became more or less active when over-producing tryptophan in S. cerevisiae.
Further extensions of FBA include factors such as kinetics, thermodynamics, or protein cost more explicitly. For example, the enzyme-constrained model where the protein cost for each reaction is added as a constraint scaled by the catalytic rate of the enzyme. This type of model was used by Ye et al. (2020) to increase the production of lysine in E. coli by optimizing high-demand proteins for lysine productions, and lowering the required protein expression for core metabolic proteins by comparing the required expression profiles on different nitrogen sources. Li et al. (2021) used a similar model formulation based on catalytic rates of the enzymes, but also included the effect of temperature on the enzyme stability and kinetics and use this to identify potential rate-limiting enzymes at higher temperatures. Other methods that include similar constraints are ME models, where apart from the metabolic (M) reactions, the metabolic costs for activating these reactions through expression (E) of the relevant RNA and proteins are explicitly included. Finally, methods such as OptKnock Burgard et al. (2003), offer the possibility of using a constraint-based model to directly design interventions to achieve improved production and growth coupling, where the production of a product of choice is a requirement for biomass generation. Many other computational strain-design methods have been created since; Machado and Herrgård (2015) and Maia et al. (2016) review the different methods and their successful applications.
An alternative usage of constraint-based models is the analysis through elementary flux modes (EFM). With this method, the metabolic network is decomposed into a set of minimal, unique pathways that flux can flow through. Any flux through the network can be represented as a linear combination of these EFMs. A variety of methods based on this principle exist, however, most do not scale well to genome-scale networks as the number of EFMs increases exponentially with the complexity of the network. However, this method can be useful for smaller networks, such as the core carbon metabolism, as once the enumeration of the EFMs is performed, strain design methods are easy to apply. For example, Poblete-Castro et al. (2012) analysed the potential of P. putida for the production of polyhydroxyalkanoates, while Jol et al. (2012) integrated metabolomics data to study the feasibility of specific EFMs under thermodynamic constraints.
Do note, however, that generally the solutions from FBA based approaches are not unique, as in large genome-scale metabolic models there can be multiple flux profiles that achieve the optimal objective value. An alternative method to utilize genome-scale constraint-based models that embraces this property is flux sampling. In contrast to FBA, where the fluxes are optimized according to some objective, flux sampling aims to explore the entire feasible flux space without imposing an objective. This can be considered a more robust approach, as assuming a single objective such as growth to be optimal is not always biologically valid, especially when dealing with engineered strains or multi-cellular organisms. Quantifying a large space of possible fluxes also offers the advantage of being able to add an error or uncertainty margin to predicted fluxes, offering additional guidance of for example, the feasibility of a pathway design that can be lacking when looking solely at a single optimal flux profile. In Herrmann et al. (2019), the authors used flux sampling to study cold acclimation of Arabidopsis thaliana and also compare several flux sampling implementations. By constraining the model with the measured input flux of CO2 and output fluxes of accumulated metabolites, they could compare the possible flux distributions in both conditions and predict which reactions are important for the adaption to low temperature conditions.
Genome-scale constraint-based metabolic models shine when exploring the potential routes through metabolism a metabolite can take, and investigating the impact of integrating new pathways or knocking out native genes. For most model organisms a high-quality model is readily available, and if not, the steps to build new models are well documented, albeit sometimes laborious due to the sheer scope of a genome-scale model. Methods for further integrating experimental data exist but can be inconsistent, as having sufficient coverage in the experimental data is often problematic due to the large scope of these models. The main downside of using constraint-based models is their steady-state assumption, making it problematic to integrate metabolite concentrations or to investigate dynamic processes such as metabolite regulation. In these cases, other models can be used to supplement the constraint-based model.
3.1.2 Kinetic models
Kinetic models describe a system through reaction rates based on the concentration of metabolites and the kinetic properties of the enzyme. Usually based on ordinary differential equations, they offer a flexible way to model metabolite dynamics and easily integrate with other dynamic processes. Since dynamic models are common to many fields, many tools are available for simulation, parameter fitting and analysis. Klipp et al. (2005) offers a good example of a kinetic metabolic model integrating with regulatory processes, such as enzyme phosphorylation and osmotic pressure. Another example is Maeda et al. (2019), where alternative modes of ammonia assimilation were modelled for E. coli. Here they integrated the uptake kinetics, related metabolic reactions and the phosphorylation of regulatory proteins to conclude that active transport of ammonia is more likely than the passive alternative given the available data.
One of the major downsides of kinetic models is that they contain many parameters, most of which are hard to measure directly. These parameters are thus fitted computationally, but the available experimental data often does not match well to what is optimal for parametrization, such as measurements of enzyme kinetics or high temporal resolution measurements of metabolite and enzyme concentrations. In addition, the computational effort to fit parameters scales exponentially with the number of parameters, limiting the size of kinetic metabolic models. However, this is more of an issue relating to the available data versus the scope of the model rather than an inherent problem with kinetic models. In Shen et al. (2020) the authors studied the Weimberg pathway of Caulobacter crescentus for xylose degradation to α-ketoglutarate. Here they used enzyme assays and quantified intermediates using NMR metabolomics every 5 min in a cell-free system, leading to a data set well suited to fitting a kinetic model. Improved methods for parametrization have been proposed to increase the scope of kinetic models by advanced methods to speed up parameter estimation such as by Fröhlich et al. (2018), or by Yuan et al. (2021), which takes advantage of recent advances in the infrastructure for training large-scale machine-learning models.
An alternative approach to analyse kinetic models is the use of sampling methods to characterize the model without fitting the parameters explicitly, also known as ensemble modelling, not to be confused with the sampling of constraint-based models generally referred to as flux sampling. By simulating the model with many scenarios of combinations of parameter values and statistically analysing the resulting simulations, system properties can be identified that hold true in a wide range of scenarios. In Rizk and Liao (2009) the authors utilized ensemble modelling to study the production of aromatic compounds in E. coli and showed that the model correctly predicts known phenotypes, while Murabito et al. (2014) used ensemble modelling to explore the metabolic regulation and control in the core metabolism of Lactococcus lactis. Finally, Chen et al. (2020) applied this ensemble method to assess the system robustness of an engineered E. coli methylotroph, obtaining targets for modulating enzyme expression that allowed for successful growth on methanol which was then further improved upon using laboratory evolution.
Kinetic models have a strong advantage over constraint-based methods when stoichiometric constraints are not the limiting factor in a system and additional detail is needed. Dynamic systems, as well as systems where metabolic regulation, toxicity, or non-linear kinetics play a role, are good targets where a kinetic model can be beneficial. However, due to their increased number of parameters and non-linear nature, they require not only more data to fit, but also more detailed data such as time-courses of metabolites and enzyme kinetic parameters. Furthermore, it often takes significant computational work to estimate the parameters even when this data is available. Still, kinetic models can be highly informative as they provide explanations for system behaviour supported directly by biochemical mechanisms. Foster et al. (2021) provides a review of tools for the creation and parametrization of kinetic models, as well as a number of examples of their application.
3.2 Data-driven models
In addition to constraint-based and kinetic models, statistical or data-driven models can be used to great effect, by identifying relevant areas of metabolism based on large-scale datasets which can then be further analysed in detail with other models. Alternatively, methods such as flux sampling of constraint-based models or ensemble models generate large results sets that require additional statistical analysis to process and explore.
Albeit not stated explicitly so far, a requirement for constructing a useful mechanistic is that there is enough known about the general metabolic structure of the organism of interest. However, if this is not the case, large scale data-driven analysis of “omics” data using statistical methods is often a good approach to start identifying important nodes in a system. Another case where this might be a better approach is if the data is too far off from the process that is supposed to be predicted. For example, trying to predict production rates of a metabolite by modifying the concentration of metal co-factors in the medium. While it is clear this could affect the production of a metabolite through changes in reaction fluxes via metal-dependent enzymes, there are multiple levels of interactions in-between. Since metal-protein interactions are often not well characterized, if known at all, building a mechanistic model describing all these interactions could prove challenging. A statistical or machine-learning model on the other hand can likely capture enough of the relevant relationships that the system can be optimized system without having to explicitly encode these interactions.
Machine-learning methods have found their way into metabolic engineering and can be a powerful complement or alternative to existing methods. Zhang et al. (2020) used constraint-based modelling to predict initial targets for modulating expression in order to optimize tyrosine production in S. cerevisiae. A library of genetic constructs with these targets was then tested, feeding a black-box machine-learning algorithm. Their method was then used to generate new designs both to maximize tyrosine production and to maximize the information gained to improve the model further. While machine-learning methods are often seen solely as black-box predictors, efforts have been made to ease the translation from predictive models to biological understanding. Although not directly modelling metabolism, Yuan et al. (2021) showed how “interpretable” machine learning can be applied to dynamic systems. In their case a model of drug responses of a melanoma cell line using a simplified dynamic description of the perturbation, decay, and interaction terms for measured proteins and phenotypes. Analysing these interaction terms showed that many known interactions could be recovered, highlighting the potential of combining mechanistic methods with machine-learning approaches. Zampieri G. et al. (2019) and Lawson et al. (2020) review the application of different machine-learning methods in the context of metabolic engineering.
3.3 Multi-scale modelling
Finally, multi-scale modelling is a method to combine modelling approaches where models are integrated together, each covering their own subject area. This method is often applied when there is a logical separation to be made in the system: spatial, temporal or otherwise. One of the most straightforward examples of this approach is dynamic FBA (dFBA). Normally, a constraint-based model would not be usable under dynamic conditions due to its steady-state assumptions. However, by assuming that the cells internal metabolic state quickly adapts to an optimal steady-state corresponding to the current medium conditions and by having another model simulate these medium conditions dynamically, we can integrate the two models into a hybrid multi-scale model. To simulate the model, the constraint-based model is re-optimized at every time step using the current medium conditions to determine growth and production rates which in turn are used to update the external conditions. Here, the separation of the models is based on the spatial separation of the external medium and internal metabolism of the cell, but also on the temporal level, with the assumption being that the steady-state conditions in the cell are adapted to the external medium every time step.
This technique can be useful to study dynamic systems such as co-cultures of different microbes or can be further extended to a spatial-dynamic model to include factors such as localized interactions between microbes or as metabolite gradients. Pacheco et al. (2019) applied both FBA and dFBA to study whether the exchange of “cost-less” metabolites can be a driving force of community interactions, while Harcombe et al. (2014) also integrated a spatial component to study interaction dynamics of a three strain consortium. In Øyås and Stelling (2018) additional applications of dFBA are reviewed, while Heinken et al. (2021) provides an in-depth review of dFBA in the context of microbial community modelling.
One of the more famous examples of multi-scale modelling is the work by Karr et al. (2012), who created a whole-cell model of Mycoplasma genitalium integrating 26 models into a single simulation framework. While multi-scale models can be informative, care has to be taken on the technical side as simulating and optimizing these models can be challenging. Existing tools such as ODE solvers are often unsuitable for the specific requirements of these models, thereby necessitating the parallel development of both the model and the tooling required to simulate and analyse the model.
4 Metabolic engineering: Most relevant types of data
4.1 Interactions
Although it can be easy to overlook because of its widespread availability in model organisms, possibly the most essential source of information about metabolism is the reaction network, connecting metabolites through enzyme catalysed reactions. These networks can be reconstructed from genomics and transcriptomics data by matching genes to annotated enzymes catalysing known reactions. Apart from the metabolite and reaction network, protein regulation is an important part of the interaction network as it determines which enzymes are active and in which amounts. Finally, subcellular location is important to consider as it limits which reactions or interactions can take place. Enzymes can be localized to a particular organelle, and metabolites can be transported between compartments with different mechanisms such as diffusion, passive or active transport. Knowing all these different interactions between the metabolites, enzymes or other species in a system of interest is essential, as this is the basis of structure of the model.
Many of these interactions have been previously characterized and can now be obtained from databases such as UniProt (The UniProt Consortium, 2021), which contains information on genes and proteins and provide cross-references to other databases with different areas of focus such as protein interactions or enzyme kinetics. In addition, there are databases focussing on metabolic networks in particular, such as KEGG (Kanehisa and Goto, 2000) and BioCyc (Karp et al., 2019), and even further specialized databases such as BRENDA (Chang et al., 2021) (enzyme properties), Rhea (Lombardot et al., 2019) (biological reactions) or BiGG (King et al., 2016) (reactions and metabolites from curated constraint-based models).
4.2 Metabolites
Knowing the presence and concentration of metabolites is key for studying metabolism. However, metabolomics is also one of the more finicky “omics” technologies, due to the inherent problem that, unlike DNA or proteins which are built from a limited set of similar subunits, metabolites are extremely chemically diverse. At the same time, due to the strong specificity of enzymes, structurally very similar metabolites can require entirely different pathways biologically, making it essential that they are differentiated correctly. Furthermore, metabolite concentrations span a wide range of magnitudes (Milo and Phillips, 2015), from micro- or even picomolar up to high millimolar concentrations, adding further difficulties to high-throughput analysis. Many metabolites are also reactive or have an extremely high turnover rate, requiring specialized procedures for sample preparation. Finally, when doing metabolomics experiments, care has to be taken to separate the metabolites inside and outside the cell, or even from different organelles.
Nonetheless, methods based on different technologies have been used to quantify metabolites. Mass-spectrometry (MS) based techniques have seen broad applicability and use, often in combination with chromatographic methods for separating metabolites such as liquid chromatography (LC) or gas chromatography (GC). For example, Zampieri M. et al. (2019) used untargeted MS to characterize the exometabolome of E. coli as well as LC-MS to measure a selection of intracellular metabolites. In combination with a constraint-based model they use this data to predict internal fluxes and to investigate the regulatory mechanisms underlying glucose and amino acid metabolism.
Nuclear magnetic resonance (NMR) (Nikolaev et al., 2019; Shen et al., 2020) and infrared (IR) spectroscopy (Sayqal et al., 2016) have also seen use, and thanks to their non-destructive nature, can be used to follow samples in real-time. However, when using these methods signals often cannot be assigned to specific metabolites. Although data analysis is not always straightforward, metabolic models can help in this case by providing a framework for separating and interpreting these signals with appropriate error margins.
In addition to these more high-throughput methods more specific assays exist, although they are usually limited to a single metabolite. These assays, especially in the case of optical assays, can be of great use due to their ease of measuring in common laboratory set-ups as well as being able to integrate well into large-scale screening efforts, such as shown for the optimization of tryptophan production by Zhang et al. (2020). Although not all metabolites are optically visible, often they can be linked to metabolites that are or can be made visible using different assays. For example, Yang et al. (2018) screened for knock-down targets that overproduce malonyl-CoA in an E. coli library made with small regulator RNAs. To measure malonyl-CoA overproduction, they introduced a gene to convert the malonyl-CoA to the detectable compound flaviolin.
Finally, there are many known regulatory proteins that interact with different metabolites that can drive the expression of a reporter protein such as GFP. Even if no known regulators are available, Hanko et al. (2020) showed that new regulatory proteins can be identified by screening for transcriptional regulators in close genetic proximity to enzymes interacting with a certain metabolite. Through this method, they identified 15 novel biosensors in Cupriavidus necator and showed that the majority of these also function in model strains such as E. coli and P. putida. Although the dynamic range of these biosensors can be low for the wild-type genes it can be improved (Chen et al., 2018; Meyer et al., 2019). Combining metabolic and regulatory pathways can further expand the space of detectable metabolites as shown by Voyvodic et al. (2019), who transformed an undetectable metabolite to a detectable signal by using metabolic transducers to arrive at a metabolite that can be detected using regulatory proteins.
4.3 Isotope tracing and fluxomics
Isotope labelling experiments in combination with metabolomics can be used to trace the flow of a labelled isotope, for example, 13C in a carbon source such as glucose, through the metabolic network by determining the ratio of labelled versus unlabelled metabolite. When engineering microbes to use alternative carbon sources, in particular, isotope labelling can be used to prove that a certain carbon source is successfully integrated into different components of the cells’ biomass. For example, in Keller et al. (2020) the authors aimed to engineer an alternative carbon assimilation cycle into E. coli starting from methanol and used isotope labelling experiments in combination with LC-MS to both prove the incorporation of carbon from methanol and to assess whether the flux profile as simulated by FBA was accurate. NMR metabolomics can also be used in combination with isotope labelling, as illustrated by Perrin et al. (2020), where the authors used NMR metabolomics to study co-utilization and diauxie of carbon sources in Pseudoalteromonas haloplanktis.
The isotope ratios are not only useful to determine which pathways connect to one another, but can also be applied at a larger scale to calculate relative flux rates through reactions, also known as fluxomics. Fluxes are not measured directly but approximated using metabolic flux analysis (MFA) of the isotope ratios and a model of the reaction network, similar to what is used for constraint-based metabolic modelling. This is a great example of how a good alignment of the data and model can be used to generate something more informative than the original data by itself but also serves to highlight that often a dataset comes with its own assumptions that have to be considered. In the case of fluxomics data, this is the structure of the reaction network, meaning that the flux rates can not be applied directly to a model using an updated network structure without reanalysing the original metabolomics data. As an example of the use of fluxomics, in Christodoulou et al. (2018) the authors used isotopically labelled glucose to differentiate flux going through glycolysis or the pentose phosphate pathway in E. coli by the ratio of labelled versus unlabelled fructose-6-phosphate. Gerosa et al. (2015) studied the regulation of central carbon metabolism of E. coli by comparing the fluxes when growing on eight isotopically labelled carbon sources, in conjunction with metabolomics and transcriptomics data.
4.4 Proteins and transcripts
Proteomics and transcriptomics are some of the most mature “omics” technologies, which shows in their widespread availability. However, due to the many layers of post-transcriptional and post-translational regulation between expression levels and reaction flux, transcriptomics in particular often do not contain as much information as expected. For one reaction a doubling of the transcript coding for the enzyme catalysing the reaction could correspond to an equivalent doubling of the reaction flux, while for another the effect is insignificant, due to regulation or other limitations. Nonetheless, transcriptomics and proteomics data sets often offer genome-wide coverage and can be integrated into many modelling approaches, often as a proxy for enzyme concentrations. In addition, they can be essential to place metabolic findings in a broader, whole-organism context and to relate changes in metabolism back to changes in microbial behaviour. For example, in Sastry et al. (2019) the authors analysed a large set of transcriptomics data from E. coli and used this to find independent transcriptomic modules, and link these to known regulators and changes in medium conditions. While not necessarily a predictive modelling approach this work shows how omics datasets can be used to connect different processes and can be used to put other experimental results in context.
In addition to “omics” technologies, measurements of individual transcripts can also be done using qPCR, which has the advantage of being able to provide an absolute quantification of the number of transcripts. Although absolute quantification of proteomics and transcriptomics is feasible (Schmidt et al., 2016; Delogu et al., 2020), often data is provided as relative levels between conditions which can make it harder to compare measurements between experiments. Finally, similar to the measurement of metabolites using biosensors, expression can also be monitored using reporter proteins such as GFP. Especially for small-scale models, such as kinetic models, these techniques can be invaluable as they can allow for improved time-resolution on the enzymes or regulators of interest.
4.5 Enzymes
In particular for kinetic models, enzyme kinetics are an important source of information. Unfortunately, the “omics” technologies have not caught up to the measurement of enzyme kinetics, as highlighted by Tummler and Klipp (2018). In addition to the lack of experimental measurements of enzyme kinetic parameters, there exists a gap between in vivo and in vitro measurements, as was shown by Davidi et al. (2016) in a large-scale study of catalytic rates in E. coli. Still, specialized enzyme databases such as BRENDA (Chang et al., 2021) aggregate large amounts of information about enzyme kinetics, co-factor utilization and regulation.
Apart from enzyme kinetics of the products and substrates, other interaction partners can be important such as which co-factors an enzyme can utilize as availability is different depending on the organism. Small molecule regulation of enzymes can be a significant factor for the regulation of (core) metabolism, as was shown in a study by Reznik et al. (2017) where they aimed to make an exhaustive inventory of these interactions in E. coli, and also compared the conservation of these interactions across domains. Recently, novel proteomics-based methods have shown to be effective to find interactions between enzymes and metabolites on a larger scale, by utilizing the binding of the regulating metabolites to the enzyme, which can affect the protein structure or stability. Piazza et al. (2018) showed this principle using a technique based on differential protein cleavage called LiP-SMap, while Mateus et al. (2020) applied a technique called thermal proteome profiling in E. coli to find enzyme interaction partners. While direct measurements are thus possible, inferring these regulatory interactions using metabolic modelling is also an option. Link et al. (2015) utilized short time-scale metabolomics (<30 s) to study the effect of metabolite regulation before expression based regulation has a chance to kick in. By sampling directly after switching from starvation to growth conditions, they can use a kinetic model to investigate regulation in amino acid and purine metabolism. Christodoulou et al. (2018) used an ensemble of kinetic models to find the most likely enzyme metabolite interactions in the pentose phosphate pathway of E. coli using a metabolomics data set taken before and after E. coli was challenged by oxidative stress.
4.6 Phenotypes
Apart from these data sources based on different classes of biologically active molecules and complexes, there is a multitude of other phenotypic traits that can be important to study or measure in relation to metabolism. Growth rates are essential to calibrate genome-scale constraint-based models, as well as the organisms’ ability to use different sources of carbon, nitrogen, or phosphate. Assays such as Biolog phenotype microarrays can be used to quickly screen the metabolic potential of a micro-organism, but can also be used for more in-depth analysis, such as done by Yang et al. (2019) to uncover the metabolic mechanisms of antibiotic resistance in E. coli by comparing the lethality of several antibiotics in multiple conditions. Transposon mutagenesis studies are useful to assess the essentiality of genes, and as such are often used to curate genome-scale constraint-based models. Other phenotypic markers, such as morphology or the tolerance to toxic compounds can also be an interesting source of high-level data on the state of an organism, such as shown in Caldera et al. (2019), where the authors analyse the effect of different drugs on morphological markers to compare the underlying mechanisms, coining the term “perturbome”.
5 Perspective
In this review, we have outlined the three factors we consider to be essential for the successful application of computational models to assist in metabolic engineering studies. We have also provided a selection of recent examples on the importance of aligning the modelling approach with these three factors: the research question and objectives, the experimental data, and the factors that can be experimentally modified. As with any tool, experimentally or computationally, a method can excel at certain uses and can be useless in others. The main idea, therefore, is that it is worthwhile to align the research objective not only with the model used but also with the experimental constraints, data, and vice versa. In larger projects, going through multiple design-build-test-learn cycles, the most appropriate model might shift. During the early stages, a data-driven approach could be the best but as more data and knowledge becomes available, mechanistic models such as constraint-based or kinetics models could become more suitable. This can require flexibility from both the experimental and the computational side of a project to adapt to shifted objectives and to judge whether the data being gathered and the model being used are still the most appropriate.
Automation is increasingly transforming the field of metabolic engineering and large-scale automated facilities, such as biofoundries, will likely make new experimental methods feasible to apply. As the scale of both data collection and experiments grows, and more detailed experiments become feasible, modelling methods will likewise have to adapt. Data-driven approaches and constraint-based methods offer the advantage of being able to predict possible experimental interventions on the whole-genome scale, however, more detailed models such as kinetic models could also become increasingly feasible with more data becoming available. Increased automation could also lead to shorter feedback cycles, which emphasizes the ability of a model for in-depth analysis of its own uncertainty and sensitivity in order to predict new experiments. These predictions can not only serve to optimize the objectives of the study but also to improve the accuracy of the model itself.
In conclusion, successful integration of experimental and modelling approaches is becoming more and more essential. In this paper, we have provided an overview of current approaches and outlined the factors we deem important to achieve this successful integration.
Author contributions
RvR drafted the initial manuscript under supervision of MS-D and VMdS. All authors contributed to manuscript conception and revision, and read and approved the final version.
Funding
This work was supported by EU Horizon 2020, grant numbers 634942 (Mycosynvac) and 635536 (Empowerputida).
Acknowledgments
This work has been previously published as part of a PhD thesis (van Rosmalen, 2022).
Conflict of interest
VMdS is employed by LifeGlimmer GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alvarez-Vasquez, F., González-Alcón, C., and Torres, N. V. (2000). Metabolism of citric acid production by Aspergillus Niger: Model definition, steady-state analysis and constrained optimization of citric acid production rate. Biotechnol. Bioeng. 70, 82–108. doi:10.1002/1097-0290(20001005)70:1¡82::AID-BIT10¿3.0.CO;2-V
Banerjee, D., Eng, T., Lau, A. K., Sasaki, Y., Wang, B., Chen, Y., et al. (2020). Genome-scale metabolic rewiring improves titers rates and yields of the non-native product indigoidine at scale. Nat. Commun. 11, 5385. doi:10.1038/s41467-020-19171-4
Benito-Vaquerizo, S., Diender, M., Parera Olm, I., Martins dos Santos, V. A. P., Schaap, P. J., Sousa, D. Z., et al. (2020). Modeling a co-culture of Clostridium autoethanogenum and Clostridium kluyveri to increase syngas conversion to medium-chain fatty-acids. Comput. Struct. Biotechnol. J. 18, 3255–3266. doi:10.1016/j.csbj.2020.10.003
Box, G. E. P. (1976). Science and statistics. J. Am. Stat. Assoc. 71, 791–799. doi:10.1080/01621459.1976.10480949
Burgard, A. P., Pharkya, P., and Maranas, C. D. (2003). Optknock: A bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 84, 647–657. doi:10.1002/bit.10803
Caldera, M., Müller, F., Kaltenbrunner, I., Licciardello, M. P., Lardeau, C.-H., Kubicek, S., et al. (2019). Mapping the perturbome network of cellular perturbations. Nat. Commun. 10, 5140–5214. doi:10.1038/s41467-019-13058-9
Carbonell, P., Jervis, A. J., Robinson, C. J., Yan, C., Dunstan, M., Swainston, N., et al. (2018). An automated Design-Build-Test-Learn pipeline for enhanced microbial production of fine chemicals. Commun. Biol. 1, 66–10. doi:10.1038/s42003-018-0076-9
Chang, A., Jeske, L., Ulbrich, S., Hofmann, J., Koblitz, J., Schomburg, I., et al. (2021). BRENDA, the ELIXIR core data resource in 2021: New developments and updates. Nucleic Acids Res. 49, D498–D508. doi:10.1093/nar/gkaa1025
Chen, F. Y. H., Jung, H.-W., Tsuei, C.-Y., and Liao, J. C. (2020). Converting Escherichia coli to a synthetic methylotroph growing solely on methanol. Cell 182, 933–946. e14. doi:10.1016/j.cell.2020.07.010
Chen, Y., Ho, J. M. L., Shis, D. L., Gupta, C., Long, J., Wagner, D. S., et al. (2018). Tuning the dynamic range of bacterial promoters regulated by ligand-inducible transcription factors. Nat. Commun. 9, 64. doi:10.1038/s41467-017-02473-5
Christodoulou, D., Link, H., Fuhrer, T., Kochanowski, K., Gerosa, L., and Sauer, U. (2018). Reserve flux capacity in the pentose phosphate pathway enables Escherichia coli’s rapid response to oxidative stress. Cell Syst. 6, 569–578. e7. doi:10.1016/j.cels.2018.04.009
Cvijovic, M., Höfer, T., Aćimović, J., Alberghina, L., Almaas, E., Besozzi, D., et al. (2016). Strategies for structuring interdisciplinary education in Systems Biology: An European perspective. NPJ Syst. Biol. Appl. 2, 16011–16017. doi:10.1038/npjsba.2016.11
Davidi, D., Noor, E., Liebermeister, W., Bar-Even, A., Flamholz, A., Tummler, K., et al. (2016). Global characterization of in vivo enzyme catalytic rates and their correspondence to in vitro k cat measurements. Proc. Natl. Acad. Sci. U. S. A. 113, 3401–3406. doi:10.1073/pnas.1514240113
de Groot, D. H., Lischke, J., Muolo, R., Planqué, R., Bruggeman, F. J., and Teusink, B. (2020). The common message of constraint-based optimization approaches: Overflow metabolism is caused by two growth-limiting constraints. Cell. Mol. Life Sci. 77, 441–453. doi:10.1007/s00018-019-03380-2
Delogu, F., Kunath, B. J., Evans, P. N., Arntzen, M. Ø., Hvidsten, T. R., and Pope, P. B. (2020). Integration of absolute multi-omics reveals dynamic protein-to-RNA ratios and metabolic interplay within mixed-domain microbiomes. Nat. Commun. 11, 4708. doi:10.1038/s41467-020-18543-0
Foster, C. J., Wang, L., Dinh, H. V., Suthers, P. F., and Maranas, C. D. (2021). Building kinetic models for metabolic engineering. Curr. Opin. Biotechnol. 67, 35–41. doi:10.1016/j.copbio.2020.11.010
Fröhlich, F., Kessler, T., Weindl, D., Shadrin, A., Schmiester, L., Hache, H., et al. (2018). Efficient parameter estimation enables the prediction of drug response using a mechanistic pan-cancer pathway model. Cell Syst. 7, 567–579. e6. doi:10.1016/j.cels.2018.10.013
Gaspari, E., Malachowski, A., Garcia-Morales, L., Burgos, R., Serrano, L., Martins dos Santos, V. A. P., et al. (2020). Model-driven design allows growth of Mycoplasma pneumoniae on serum-free media. NPJ Syst. Biol. Appl. 6, 33–11. doi:10.1038/s41540-020-00153-7
Gerosa, L., Haverkorn van Rijsewijk, B. R., Christodoulou, D., Kochanowski, K., Schmidt, T. S., Noor, E., et al. (2015). Pseudo-transition analysis identifies the key regulators of dynamic metabolic adaptations from steady-state data. Cell Syst. 1, 270–282. doi:10.1016/j.cels.2015.09.008
Hanko, E. K. R., Paiva, A. C., Jonczyk, M., Abbott, M., Minton, N. P., and Malys, N. (2020). A genome-wide approach for identification and characterisation of metabolite-inducible systems. Nat. Commun. 11, 1213–1214. doi:10.1038/s41467-020-14941-6
Harcombe, W. R., Riehl, W. J., Dukovski, I., Granger, B. R., Betts, A., Lang, A. H., et al. (2014). Metabolic resource allocation in individual microbes determines ecosystem interactions and spatial dynamics. Cell Rep. 7, 1104–1115. doi:10.1016/j.celrep.2014.03.070
Heinken, A., Basile, A., and Thiele, I. (2021). Advances in constraint-based modelling of microbial communities. Curr. Opin. Syst. Biol. 27, 100346. doi:10.1016/j.coisb.2021.05.007
Herrmann, H. A., Dyson, B. C., Vass, L., Johnson, G. N., and Schwartz, J.-M. (2019). Flux sampling is a powerful tool to study metabolism under changing environmental conditions. NPJ Syst. Biol. Appl. 5, 32–38. doi:10.1038/s41540-019-0109-0
Jol, S. J., Kümmel, A., Terzer, M., Stelling, J., and Heinemann, M. (2012). System-level insights into yeast metabolism by thermodynamic analysis of elementary flux modes. PLoS Comput. Biol. 8, e1002415. doi:10.1371/journal.pcbi.1002415
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi:10.1093/nar/28.1.27
Karp, P. D., Billington, R., Caspi, R., Fulcher, C. A., Latendresse, M., Kothari, A., et al. (2019). The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 20, 1085–1093. doi:10.1093/bib/bbx085
Karr, J. R., Sanghvi, J. C., Macklin, D. N., Gutschow, M. V., Jacobs, J. M., Bolival, B., et al. (2012). A whole-cell computational model predicts phenotype from genotype. Cell 150, 389–401. doi:10.1016/j.cell.2012.05.044
Keller, P., Noor, E., Meyer, F., Reiter, M. A., Anastassov, S., Kiefer, P., et al. (2020). Methanol-dependent Escherichia coli strains with a complete ribulose monophosphate cycle. Nat. Commun. 11, 5403. doi:10.1038/s41467-020-19235-5
King, Z. A., Lu, J., Dräger, A., Miller, P., Federowicz, S., Lerman, J. A., et al. (2016). BiGG models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 44, D515–D522. doi:10.1093/nar/gkv1049
Klipp, E., Nordlander, B., Krüger, R., Gennemark, P., and Hohmann, S. (2005). Integrative model of the response of yeast to osmotic shock. Nat. Biotechnol. 23, 975–982. doi:10.1038/nbt1114
Krambeck, F. J., and Betenbaugh, M. J. (2005). A mathematical model of N-linked glycosylation. Biotechnol. Bioeng. 92, 711–728. doi:10.1002/bit.20645
Lawson, C. E., Martí, J. M., Radivojevic, T., Jonnalagadda, S. V. R., Gentz, R., Hillson, N. J., et al. (2020). Machine learning for metabolic engineering: A review. Metab. Eng. 63, 34–60. doi:10.1016/j.ymben.2020.10.005
Li, C.-T., Yelsky, J., Chen, Y., Zuñiga, C., Eng, R., Jiang, L., et al. (2019). Utilizing genome-scale models to optimize nutrient supply for sustained algal growth and lipid productivity. NPJ Syst. Biol. Appl. 5, 33–11. doi:10.1038/s41540-019-0110-7
Li, G., Hu, Y., Jan, Z., Luo, H., Wang, H., Zelezniak, A., et al. (2021). Bayesian genome scale modelling identifies thermal determinants of yeast metabolism. Nat. Commun. 12, 190. doi:10.1038/s41467-020-20338-2
Lian, J., Schultz, C., Cao, M., HamediRad, M., and Zhao, H. (2019). Multi-functional genome-wide CRISPR system for high throughput genotype–phenotype mapping. Nat. Commun. 10, 5794. doi:10.1038/s41467-019-13621-4
Link, H., Fuhrer, T., Gerosa, L., Zamboni, N., and Sauer, U. (2015). Real-time metabolome profiling of the metabolic switch between starvation and growth. Nat. Methods 12, 1091–1097. doi:10.1038/nmeth.3584
Lombardot, T., Morgat, A., Axelsen, K. B., Aimo, L., Hyka-Nouspikel, N., Niknejad, A., et al. (2019). Updates in Rhea: SPARQLing biochemical reaction data. Nucleic Acids Res. 47, D596–D600. doi:10.1093/nar/gky876
Machado, D., and Herrgård, M. J. (2015). Co-evolution of strain design methods based on flux balance and elementary mode analysis. Metab. Eng. Commun. 2, 85–92. doi:10.1016/j.meteno.2015.04.001
Machado, D., and Herrgård, M. (2014). Systematic evaluation of methods for integration of transcriptomic data into constraint-based models of metabolism. PLoS Comput. Biol. 10, e1003580. doi:10.1371/journal.pcbi.1003580
Maeda, K., Westerhoff, H. V., Kurata, H., and Boogerd, F. C. (2019). Ranking network mechanisms by how they fit diverse experiments and deciding on E. coli ’s ammonium transport and assimilation network. NPJ Syst. Biol. Appl. 5, 14. doi:10.1038/s41540-019-0091-6
Maia, P., Rocha, M., and Rocha, I. (2016). In silico constraint-based strain optimization methods: The quest for optimal cell factories. Microbiol. Mol. Biol. Rev. 80, 45–67. doi:10.1128/MMBR.00014-15
Malik-Sheriff, R. S., Glont, M., Nguyen, T. V. N., Tiwari, K., Roberts, M. G., Xavier, A., et al. (2020). BioModels—15 years of sharing computational models in life science. Nucleic Acids Res. 48, D407–D415. doi:10.1093/nar/gkz1055
Mateus, A., Hevler, J., Bobonis, J., Kurzawa, N., Shah, M., Mitosch, K., et al. (2020). The functional proteome landscape of Escherichia coli. Nature 1, 473–478. doi:10.1038/s41586-020-3002-5
Mendoza, S. N., Olivier, B. G., Molenaar, D., and Teusink, B. (2019). A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol. 20, 158. doi:10.1186/s13059-019-1769-1
Meyer, A. J., Segall-Shapiro, T. H., Glassey, E., Zhang, J., and Voigt, C. A. (2019). Escherichia coli “Marionette” strains with 12 highly optimized small-molecule sensors. Nat. Chem. Biol. 15, 196–204. doi:10.1038/s41589-018-0168-3
Milo, R., and Phillips, R. (2015). Cell biology by the numbers. 1st edn. New York: Garland Science. doi:10.1201/9780429258770
Montero-Blay, A., Piñero-Lambea, C., Miravet-Verde, S., Lluch-Senar, M., and Serrano, L. (2020). Inferring active metabolic pathways from proteomics and essentiality data. Cell Rep. 31, 107722. doi:10.1016/j.celrep.2020.107722
Murabito, E., Verma, M., Bekker, M., Bellomo, D., Westerhoff, H. V., Teusink, B., et al. (2014). Monte-carlo modeling of the central carbon metabolism of Lactococcus lactis: Insights into metabolic regulation. PLOS ONE 9, e106453. doi:10.1371/journal.pone.0106453
Nikolaev, Y., Ripin, N., Soste, M., Picotti, P., Iber, D., and Allain, F. H.-T. (2019). Systems NMR: Single-sample quantification of RNA, proteins and metabolites for biomolecular network analysis. Nat. Methods 16, 743–749. doi:10.1038/s41592-019-0495-7
Opdam, S., Richelle, A., Kellman, B., Li, S., Zielinski, D. C., and Lewis, N. E. (2017). A systematic evaluation of methods for tailoring genome-scale metabolic models. Cell Syst. 4, 318–329. e6. doi:10.1016/j.cels.2017.01.010
Øyås, O., and Stelling, J. (2018). Genome-scale metabolic networks in time and space. Curr. Opin. Syst. Biol. 8, 51–58. doi:10.1016/j.coisb.2017.12.003
Pacheco, A. R., Moel, M., and Segrè, D. (2019). Costless metabolic secretions as drivers of interspecies interactions in microbial ecosystems. Nat. Commun. 10, 103. doi:10.1038/s41467-018-07946-9
Perrin, E., Ghini, V., Giovannini, M., Di Patti, F., Cardazzo, B., Carraro, L., et al. (2020). Diauxie and co-utilization of carbon sources can coexist during bacterial growth in nutritionally complex environments. Nat. Commun. 11, 3135. doi:10.1038/s41467-020-16872-8
Piazza, I., Kochanowski, K., Cappelletti, V., Fuhrer, T., Noor, E., Sauer, U., et al. (2018). A map of protein-metabolite interactions reveals principles of chemical communication. Cell 172, 358–372. e23. doi:10.1016/j.cell.2017.12.006
Poblete-Castro, I., Escapa, I. F., Jäger, C., Puchalka, J., Chi Lam, C. M., Schomburg, D., et al. (2012). The metabolic response of P. putida KT2442 producing high levels of polyhydroxyalkanoate under single- and multiple-nutrient-limited growth: Highlights from a multi-level omics approach. Microb. Cell Fact. 11, 34. doi:10.1186/1475-2859-11-34
Porubsky, V. L., Goldberg, A. P., Rampadarath, A. K., Nickerson, D. P., Karr, J. R., and Sauro, H. M. (2020). Best practices for making reproducible biochemical models. Cell Syst. 11, 109–120. doi:10.1016/j.cels.2020.06.012
Reznik, E., Christodoulou, D., Goldford, J. E., Briars, E., Sauer, U., Segrè, D., et al. (2017). Genome-scale Architecture of small molecule regulatory networks and the fundamental trade-off between regulation and enzymatic activity. Cell Rep. 20, 2666–2677. doi:10.1016/j.celrep.2017.08.066
Rizk, M. L., and Liao, J. C. (2009). Ensemble modeling for aromatic production in Escherichia coli. PLoS ONE 4, e6903. doi:10.1371/journal.pone.0006903
Sastry, A. V., Gao, Y., Szubin, R., Hefner, Y., Xu, S., Kim, D., et al. (2019). The Escherichia coli transcriptome mostly consists of independently regulated modules. Nat. Commun. 10, 5536–5614. doi:10.1038/s41467-019-13483-w
Satanowski, A., Dronsella, B., Noor, E., Vögeli, B., He, H., Wichmann, P., et al. (2020). Awakening a latent carbon fixation cycle in Escherichia coli. Nat. Commun. 11, 5812. doi:10.1038/s41467-020-19564-5
Sayqal, A., Xu, Y., Trivedi, D. K., AlMasoud, N., Ellis, D. I., Rattray, N. J. W., et al. (2016). Metabolomics analysis reveals the participation of efflux pumps and ornithine in the response of Pseudomonas putida DOT-t1e cells to challenge with propranolol. PLOS ONE 11, e0156509. doi:10.1371/journal.pone.0156509
Schmidt, A., Kochanowski, K., Vedelaar, S., Ahrné, E., Volkmer, B., Callipo, L., et al. (2016). The quantitative and condition-dependent Escherichia coli proteome. Nat. Biotechnol. 34, 104–110. doi:10.1038/nbt.3418
Shen, L., Kohlhaas, M., Enoki, J., Meier, R., Schönenberger, B., Wohlgemuth, R., et al. (2020). A combined experimental and modelling approach for the Weimberg pathway optimisation. Nat. Commun. 11, 1098–1113. doi:10.1038/s41467-020-14830-y
Teusink, B., Passarge, J., Reijenga, C. A., Esgalhado, E., Van Der Weijden, C. C., Schepper, M., et al. (2000). Can yeast glycolysis be understood terms of vitro kinetics of the constituent enzymes? Testing biochemistry. Eur. J. Biochem. 267, 5313–5329. doi:10.1046/j.1432-1327.2000.01527.x
The UniProt Consortium (2021). UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. doi:10.1093/nar/gkaa1100
Tiwari, K., Kananathan, S., Roberts, M. G., Meyer, J. P., Sharif Shohan, M. U., Xavier, A., et al. (2021). Reproducibility in systems biology modelling. Mol. Syst. Biol. 17, e9982. doi:10.15252/msb.20209982
Tummler, K., and Klipp, E. (2018). The discrepancy between data for and expectations on metabolic models: How to match experiments and computational efforts to arrive at quantitative predictions? Curr. Opin. Syst. Biol. 8, 1–6. doi:10.1016/j.coisb.2017.11.003
van Rosmalen, R. (2022). “Model-driven engineering of microbial metabolism,” (Wageningen: Wageningen University). Ph.D. thesis. doi:10.18174/559240
Voyvodic, P. L., Pandi, A., Koch, M., Conejero, I., Valjent, E., Courtet, P., et al. (2019). Plug-and-play metabolic transducers expand the chemical detection space of cell-free biosensors. Nat. Commun. 10, 1697. doi:10.1038/s41467-019-09722-9
Yang, D., Kim, W. J., Yoo, S. M., Choi, J. H., Ha, S. H., Lee, M. H., et al. (2018). Repurposing type III polyketide synthase as a malonyl-CoA biosensor for metabolic engineering in bacteria. Proc. Natl. Acad. Sci. U. S. A. 115, 9835–9844. doi:10.1073/pnas.1808567115
Yang, J. H., Wright, S. N., Hamblin, M., McCloskey, D., Alcantar, M. A., Schrübbers, L., et al. (2019). A white-box machine learning approach for revealing antibiotic mechanisms of action. Cell 177, 1649–1661. doi:10.1016/j.cell.2019.04.016
Ye, C., Luo, Q., Guo, L., Gao, C., Xu, N., Zhang, L., et al. (2020). Improving lysine production through construction of an Escherichia coli enzyme-constrained model. Biotechnol. Bioeng. 117, 3533–3544. doi:10.1002/bit.27485
Yuan, B., Shen, C., Luna, A., Korkut, A., Marks, D. S., Ingraham, J., et al. (2021). CellBox: Interpretable machine learning for perturbation biology with application to the design of cancer combination therapy. Cell Syst. 12, 128–140.e4. e4. doi:10.1016/j.cels.2020.11.013
Zampieri, G., Vijayakumar, S., Yaneske, E., and Angione, C. (2019a). Machine and deep learning meet genome-scale metabolic modeling. PLoS Comput. Biol. 15, e1007084. doi:10.1371/journal.pcbi.1007084
Zampieri, M., Hörl, M., Hotz, F., Müller, N. F., and Sauer, U. (2019b). Regulatory mechanisms underlying coordination of amino acid and glucose catabolism in Escherichia coli. Nat. Commun. 10, 3354. doi:10.1038/s41467-019-11331-5
Keywords: metabolic engineering, design-build-test-learn cycle, metabolic modelling, experimental design, model selection, data integration
Citation: van Rosmalen RP, Martins dos Santos VAP and Suarez-Diez M (2022) Questions, data and models underpinning metabolic engineering. Front. Syst. Biol. 2:998048. doi: 10.3389/fsysb.2022.998048
Received: 19 July 2022; Accepted: 09 September 2022;
Published: 10 October 2022.
Edited by:
Kristin Tøndel, Norwegian University of Life Sciences, NorwayReviewed by:
Birgitta Elisabeth Ebert, Australian Institute for Bioengineering and Nanotechnology, University of Queensland, AustraliaMiguel Rocha, University of Minho, Portugal
Copyright © 2022 van Rosmalen, Martins dos Santos and Suarez-Diez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Suarez-Diez, maria.suarezdiez@wur.nl