In silico structural homology modeling and functional characterization of Mycoplasma gallisepticum variable lipoprotein hemagglutin proteins
 Susithra Priyadarshni Mugunthan
Susithra Priyadarshni Mugunthan Mani Chandra Harish
Mani Chandra Harish- Department of Biotechnology, Thiruvalluvar University, Vellore, India
Mycoplasma gallisepticum variable lipoprotein hemagglutin (vlhA) proteins are crucial for immune evasion from the host cells, permitting the persistence and survival of the pathogen. However, the exact molecular mechanism behind the immune evasion function is still not clear. In silico physiochemical analysis, domain analysis, subcellular localization, and homology modeling studies have been carried out to predict the structural and functional properties of these proteins. The outcomes of this study provide significant preliminary data for understanding the immune evasion by vlhA proteins. In this study, we have reported the primary, secondary, and tertiary structural characteristics and subcellular localization, presence of the transmembrane helix and signal peptide, and functional characteristics of vlhA proteins from M. gallisepticum strain R low. The results show variation between the structural and functional components of the proteins, signifying the role and diverse molecular mechanisms in functioning of vlhA proteins in host immune evasion. Moreover the 3D structure predicted in this study will pave a way for understanding vlhA protein function and its interaction with other molecules to undergo immune evasion. This study forms the basis for future experimental studies improving our understanding in the molecular mechanisms used by vlhA proteins.
Introduction
The bacteria of class Mollicutes are described as simplest self–replicating life forms due to their small cell size and complete lack of cell wall, limited metabolic pathway and reduced genome size (1). The Mycoplasmataceae family in Mollicutes includes majority of disease causing pathogens in medical and veterinary fields. A great number of Mycoplasma species are pathogenic to humans and animals which cause chronic infections consequential in infectious diseases. To adapt and survive the challenging and complex host environment, the mycoplasmas use combinational genetic machinery for phase and size variation of major surface components. Due to the lack of cell wall, the outer surface of the mycoplasma membrane plays a crucial role in the infection process, transport of nutrients, interaction with host cells, and host immune defense. Thus, gaining knowledge in the process of how and when the antigenic variation occurs can offer important insights to the tactics used by mycoplasmas to cause infection in host cells.
Mycoplasma gallisepticum is one of the most important avian pathogens which causes chronic respiratory disease (CRD) in chickens with the symptoms of cough, nasal discharge, low appetite, reduced hatchability and chick viability, loss of weight, and decreased egg production (1, 2). The responsible pathogenic events are due to genes that encode cytoadhesion and surface components with antigenic variation which involves the immune evasion of the host (3). M. gallisepticum infection results in infectious sinusitis in turkeys (swollen infraorbital sinuses) and conjunctivitis in finches.
The immune evasion of M. gallisepticum is regulated by the vlhA gene family. This family consists of 43 vlhA genes located in five loci (Table 1). The major function of this gene family is to engender antigenic diversity which assists in immune evasion during infection. The vlhA gene family shows phase variation during acute phase and immune evasion during the chronic phase of infection (4, 5). The phase variation may occur impulsively or by an immune attack and is crucial for survival of M. gallisepticum in host cells (6–8). Various mechanisms for phase variation like gene conversion, site specific recombination, DNA slippage, and reciprocal recombination were utilized by different species of Mycoplasma (9). The vlhA gene products are speculated to be engaged in the attachment of host apolipoprotein A1 (10, 11) and red blood cells (12). The phase variation of M. gallisepticum is exclusive and has not been studied yet. Among the other vlhA genes, vlhA 3.03, 2.02 and 4.01 genes are primarily expressed in the initial phase of infection, whereas vlhA 1.07 and 5.13 are expressed in the later stages of infection. The prototype followed by M. gallisepticum to express the dominant vlhA gene during the course of infection is stochastic and the mechanism is unknown and yet to be explored (4). This study employed computational tools to understand the evolutionary relationship of the vlhA proteins; structural studies which include its primary sequence analysis, and secondary and tertiary structural analysis, functional studies like the cellular localization, presence of the transmembrane helix and signal peptide in vlhA proteins, and finally identification of functional domain were performed. To date, no in silico structural and functional studies have been reported for M. gallisepticum vlhA proteins. The diagrammatic representation of the work flow is presented in Figure 1. The list of bioinformatics tools and servers employed in this study is given in Table 2.
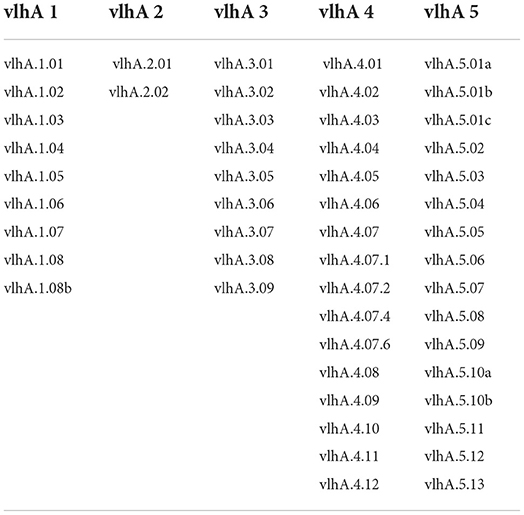
Table 1. List of vlhA genes based on their group analyzed in this study.
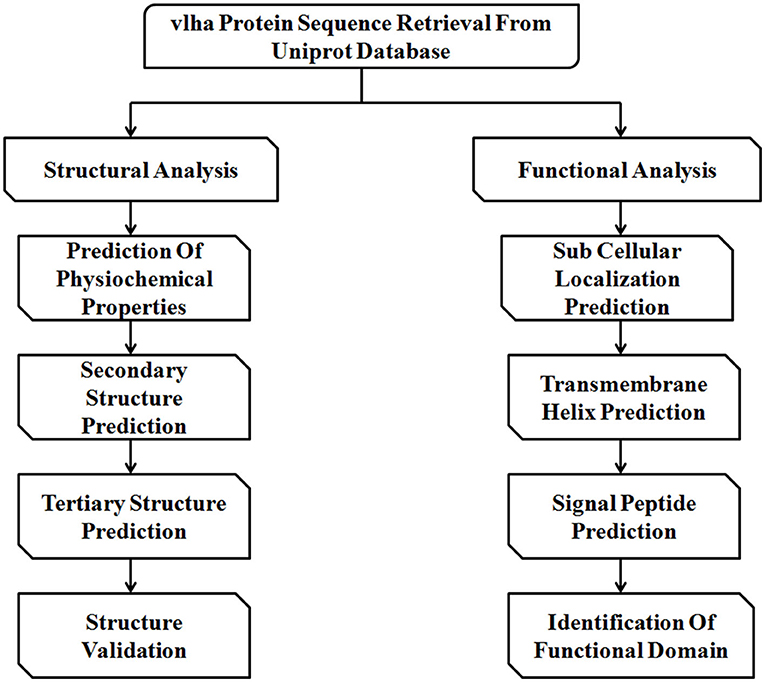
Figure 1. Schematic representation of the workflow followed in this study.
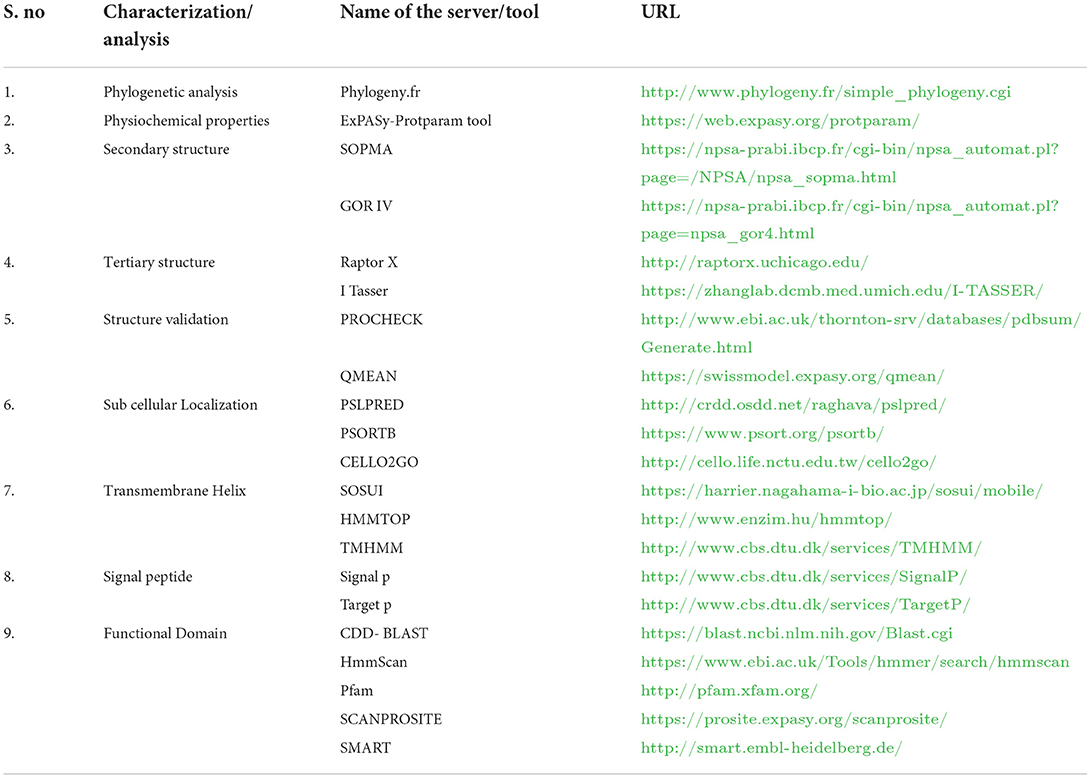
Table 2. List of bioinformatics tools and servers employed in the structural and functional analyses of vlhA proteins.
Understanding the structural and functional properties of vlhA proteins of M. gallisepticum will provide the first step/lead in the direction of understanding of underlying molecular mechanisms involved. In this study, we used in silico methods to determine the physical, structural, and functional characteristics of vlhA proteins.
Materials and methods
Sequence retrieval
The amino acid sequences of vlhA proteins from Mycoplasma gallisepticum strain R low used in this study were retrieved from UniProt in the FASTA format. The protein names and their unique UniProt IDs are shown in Supplementary Table 1.
Phylogenetic analysis
To understand the evolutionary relationships between the vlhA proteins, a phylogenetic tree was constructed using Phylogeny.fr, online software for phylogenetic analysis (13). The “One Click” option was used where the alignment was performed by MUSCLE, curation was performed by Gblocks, phylogeny was performed by PhyML, and Tree Rendering was performed by TreeDyn.
Structural analysis
Physiochemical properties
The ExPASyProtparam tool was used to analyze the physiochemical properties such as molecular weight (Mwt), amino acid composition (AA), theoretical isoelectric point (pI), number of negative residues (–R), number of positive residues (+R), extinction coefficient (EC), half-life (h), instability index (II),aliphatic index (AI), and grand average of hydropathy (GRAVY) of the protein sequence (37).
Secondary structure prediction
The secondary structure of protein was predicted by using SOPMA and GOR IV. The self-optimized prediction method (SOPMA) describes the three states of the protein structure (helices, turns, and coils). SOPMA predicts 90% of secondary structural information of proteins and it works under the homologous method and predicts 69.5% of amino acids for three states of the secondary structure. SOPMA is mainly classified into four steps. Step one involves the retrieval of homologous protein from UniProt. In step two, alignments of sequence compose the set of homologous proteins. Step three executes the SOPMA method with each and every aligned sequence. In the final step, the conformational state yielding the highest score is attributed to the given amino acid with the averaged conformational score (14).
GOR IV (Garnier-Osguthorpe–Robson) is another method to predict the secondary structure. In version I, GOR has information from the hydrophobic triplet. Hydrophobic triplet information does not significantly improve the predictive power (15). The method GOR IV is formed on information theory; GOR has a mean accuracy of 64.4% for a three state prediction when compared to another version. Version IV is more accurate. The GOR IV method analyzes the secondary structure of the protein and correlates it with net values of each amino acid position and three states (helices, turns, coils) (16).
Tertiary structure prediction
The tertiary structure of vlhA genes was constructed using the homology modeling server RaptorX (http://raptorx.uchicago.edu/) and I-TASSER server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) (17). Raptor X distinguishes itself from other servers by the quality of the alignment between a target sequence and one or multiple distantly related template proteins and by a novel nonlinear scoring function and a probabilistic-consistency algorithm. The predicted tertiary models can be used for binding site and epitope prediction; another application is found to be determining the binding topology of small ligand molecules to putative binding sites on the domain structure generated (54). The I-TASSER server employs ab initio modeling to predict 3D structures. The tertiary structures modeled by I-TASSER were subjected to refinement by the GalaxyRefine server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) (18). This server replaces amino acids with high-probability rotamers and applies molecular dynamic simulation for overall structural relaxation.
Structure validation
The refined structure was validated by PROCHECK (https://servicesn.mbi.ucla.edu/PROCHECK/), which analyzes the stereochemical quality of a protein structure by analyzing residue-by–Residue geometry and overall structure geometry (19).
QMEAN is used to analyze the quality of computationally predicted proteins. It is based on two distance-dependent interaction potentials of mean force, C-β atoms and is used to assess long–Range interactions (secondary structure dependent and torsion angle potential dependent). The QMEAN4 score is a linear combination of four statistical potential terms. It is trained to predict the IDDT (The Local Distance Difference Test) score in the range [0, 1]. To calculate the QMEAN Z-score, the normalized raw scores of a given model are compared with scores obtained for a representative set of high resolution X–Ray structures of similar size against the PDB reference set (20–22).
Functional analysis
Subcellular localization prediction
(A) PSLPRED
PSLpred is used for predicting the subcellular localization of prokaryotic proteins with an overall accuracy of 91.2%. It is a hybrid approach-based method. The prediction accuracies of 90.7, 86.8, 90.3, 95.2, and 90.6% were attained for cytoplasmic, extracellular, inner membrane, outer membrane, and periplasmic proteins, respectively (23).
(B) PSORTB
PSORTB is the most precise bacterial SCL (subcellular localization) prediction software that was introduced in 2005 and has been widely used. It provides quick and inexpensive means for gaining insight into the protein function, verifying experimental results, annotating newly sequenced bacterial genomes, detecting cell surface/drug targets, and identifying biomarkers for microbes. As a result, only ~50% of proteins encoded in gram-negative bacterial genomes and ~75% of proteins encoded in gram-positive bacterial genomes receive a prediction from PSORTb (24).
(C) CELLO2GO
CELLO2GO is a publicly available, web-based system for screening various properties of a targeted protein and its subcellular localization. It shows the exact location of the protein. CELLO2GO should be a useful tool for research involving complex subcellular systems because it combines CELLO and BLAST into one form (25).
Transmembrane helix prediction
(A) SOSUI
SOSUI is used for the discrimination of membrane proteins and soluble proteins and the prediction of the transmembrane helix, the accuracy of prediction was 99%, and the corresponding value for the transmembrane helix prediction was 97% (26).
(B) HMMTOP
A hidden Markov model with special architecture was developed to search transmembrane topology corresponding to the maximum likelihood among all the possible topologies of a given protein. The method is based on the hypothesis that the transmembrane segments and the topology are determined by the difference in the amino acid distributions in various structural parts of these proteins (27).
(C) TMHMM
TMHMM is a widely used bioinformatics tool, based on the hidden Markov model, which is used to predict transmembrane helices of integral membrane proteins. It is used to predict the number of transmembrane helices and discriminate between soluble and membrane proteins with a high degree of accuracy (28).
Signal peptide prediction
(A) Signal p
Signal p was the first publicly available method to predict signal peptide and its cleavage sites. It is based on deep neural network-based method combined with conditional random field classification and optimized transfer learning for improved signal peptide prediction. The input is given in FASTA format. The server predicts the presence of signal peptides, TAT signal peptides, and lipoprotein signal peptides from proteins present in Archaea, gram-positive bacteria, gram-negative bacteria, and eukaryotes (29).
(B) Target p
The target p server is used to predict the presence of signal peptides, and mitochondrial transit peptides and others were predicted using the FASTA sequence of the protein (30).
Identification of functional domain
The functional domain analysis was carried out using five publicly available tools (CDD-BLAST, HmmScan, Pfam, SCANPROSITE, and SMART). CDD-BLAST annotates the vlhA proteins by generating alignment models of the representative sequence fragment which were in agreement with domain boundaries as observed protein models in NCBI's Conserved Domain Database (31). HmmScan and SMART took a query sequence and searched it against the Pfam profile HMM library as a target database (32–34). Pfam was used to classify vlhA proteins functional families based on similarity (34). To predict the protein function, SCANPROSITE detects homologs and matches against signature from the PROSITE database (35).
Results
Phylogenetic analysis
Phylogenetic analysis was performed to examine the differences and relatedness among the vlhA proteins. A phylogenetic tree was constructed by using Phylogeny.fr. The computed data indicated that the expression of vlhA proteins during the course of infection varies greatly and vlhA from the five loci here clustered into different groups. The bootstrap values in the phylogenetic tree created for M. gallisepticum vlhA proteins showed that the proteins had less evolutionary similarity (Figure 2), and the divergence in sequence during evolution may have developed to evade host immune response and to adapt to each host. As a consequence, each protein has evolved due to strain during the course of infection, thus leading to antigenic variation (36).
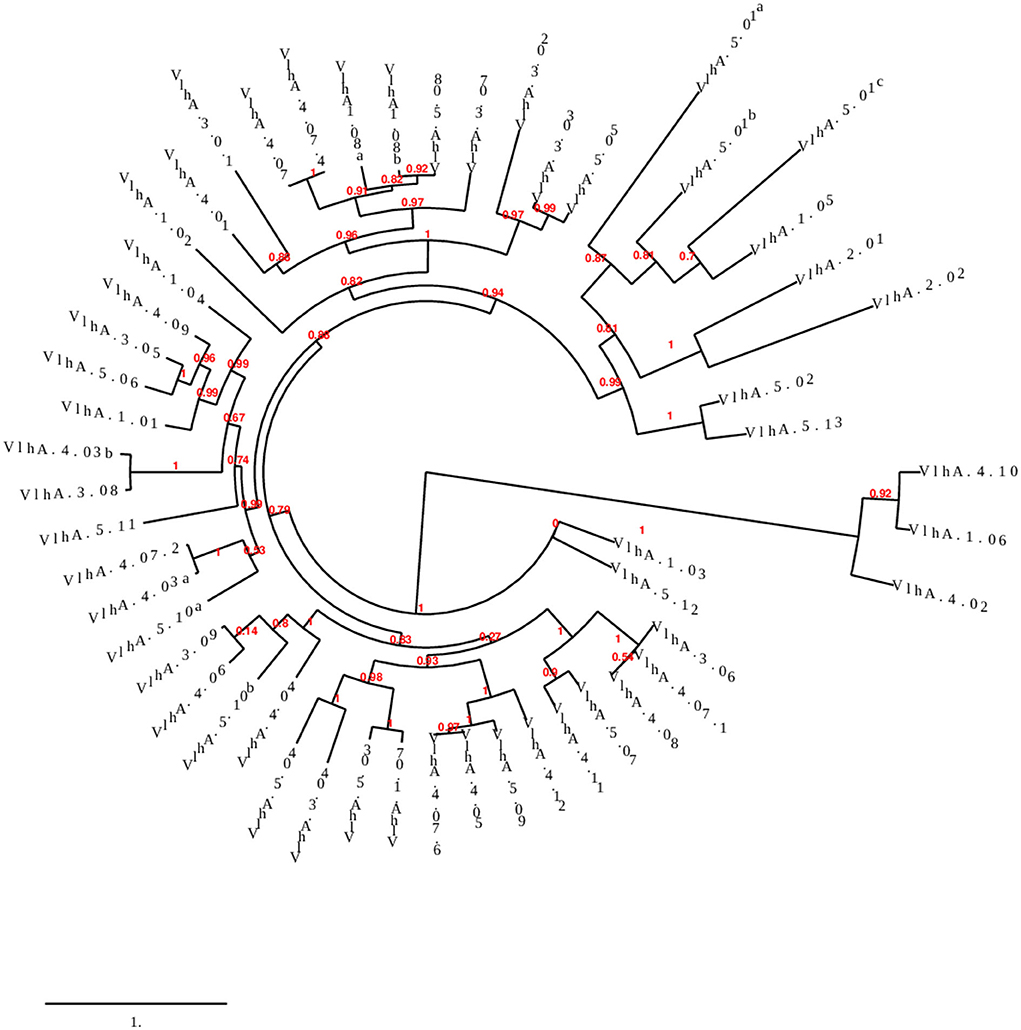
Figure 2. Phylogenetic tree showing the evolutionary relationship of different M. gallisepticum vlhA proteins. The numbers indicate bootstrap percentages and the scale indicates the divergence time.
Structural analysis
Physiochemical characterization
The ExPASy ProtParam was employed to analyze the protein primary structures and compute different parameters for their physiochemical properties. The number of amino acid residues in vlhA proteins varied from 77 to 795 amino acids. The composition of amino acid residues in each vlhA protein is presented in Figure 3. The molecular weight of these proteins varied from 8.12 to 85.3 kDa. The pI values of these proteins range from acidic pI 4.63 to alkaline pI 9.21. If the instability index (II) is above 40, the protein was considered to be unstable. As shown in Table 3, except a few vlhA (vlhA.1.08, vlhA.2.01, vlhA.5.01c, and vlhA.5.10b) proteins, other proteins were considerably stable. The aliphatic index (AI) of these vlhA proteins varied from 27.86 to 95.75. The high AI values indicated the thermal stability and hydrophobic nature of the proteins. When a protein was found to have a greater negative grand average of hydropathy (GRAVY) values, it indicated the hydrophilic nature of the protein and the possibility of better interactions between the protein and water (37). The complete physicochemical analysis of all the vlhA proteins is listed in Table 3.
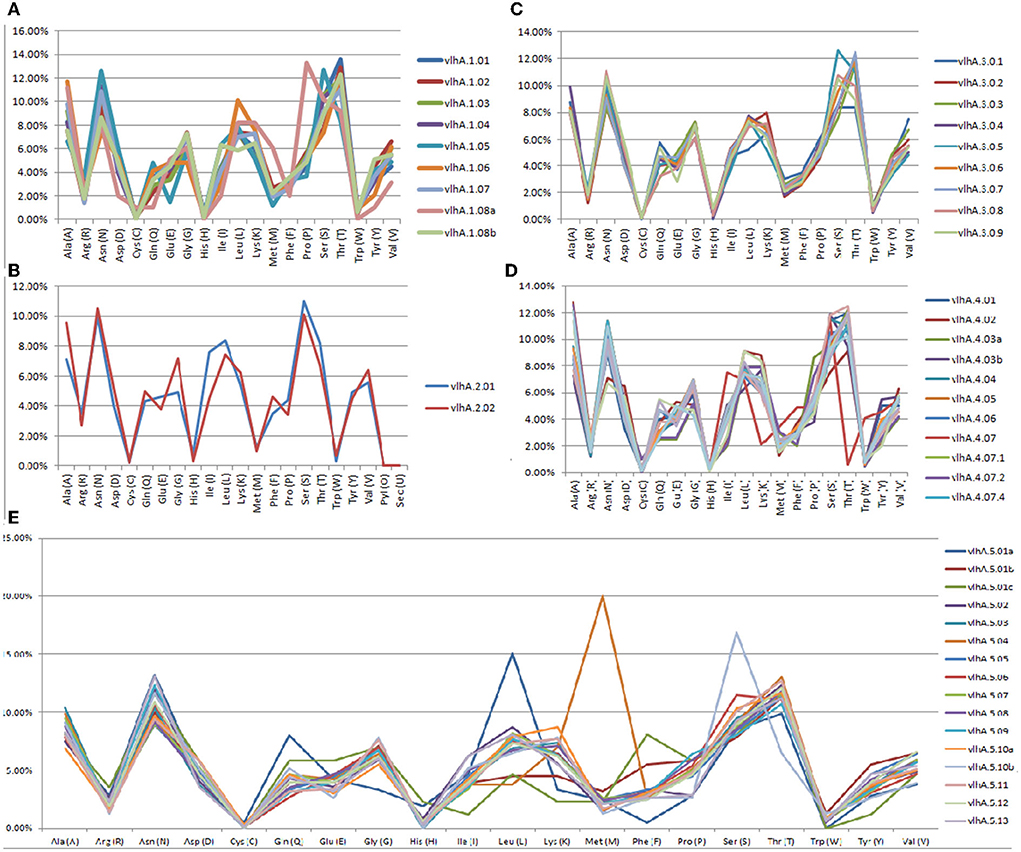
Figure 3. Graphical representation of amino acid composition of M. gallisepticum vlhA proteins. (A) vlhA 1 group, (B) vlhA 2 group, (C) vlhA 3 group, (D) vlhA 4 group, and (E) vlhA group 5.
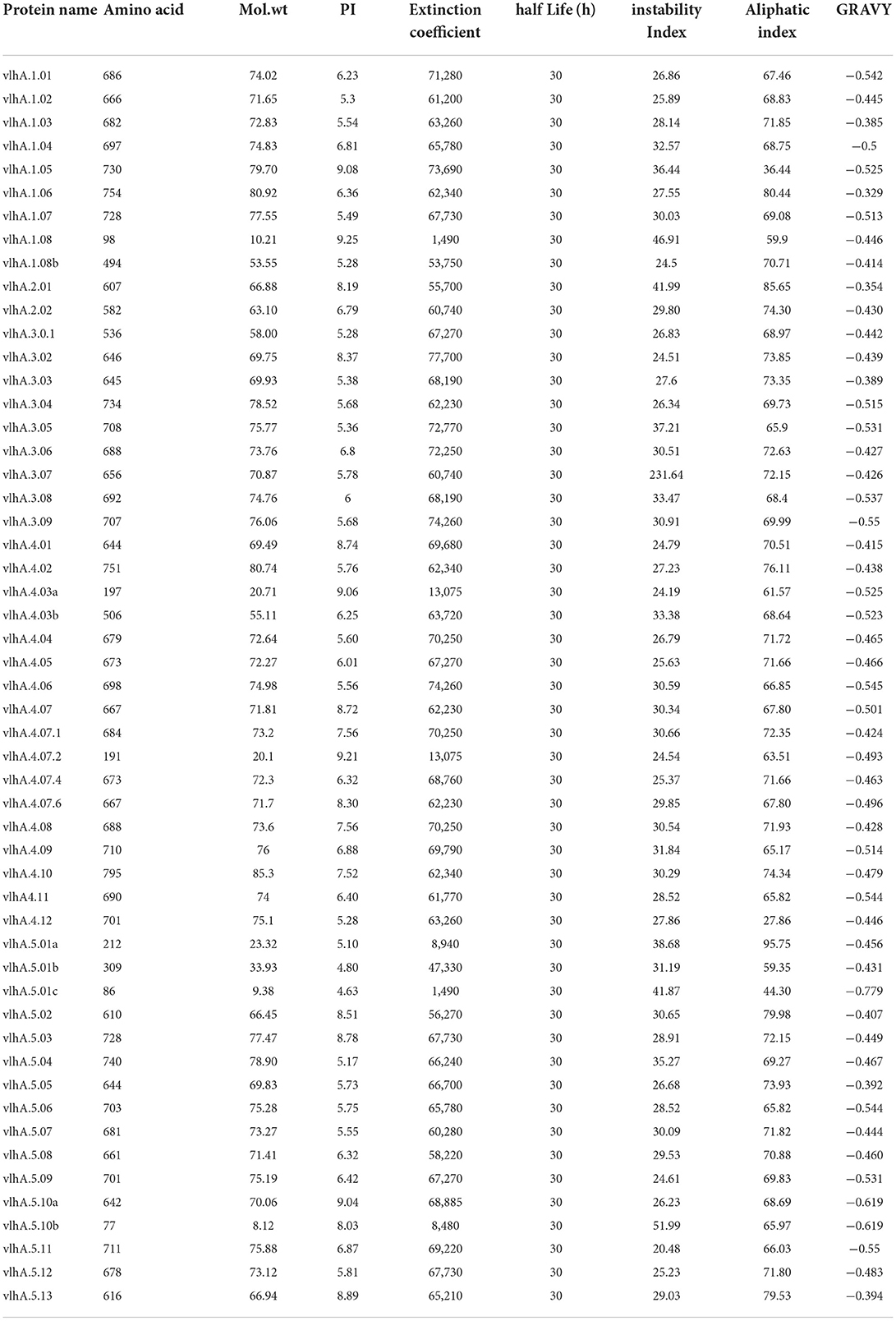
Table 3. Physiochemical properties like number of amino acids, molecular weight, isoelectric point, extinction coeffcient, half-life (h), instability index, aliphatic index, and GRAVY of M. gallisepticum vlhA proteins.
Secondary structure prediction
The secondary structure of vlhA proteins was predicted using SOPMA and GOR IV servers that showed similar results where the percentage of random coils was higher when compared with alpha helices and extended turns (Supplementary Table 2). Previous studies reported that the presence of a higher percentage of random coil structures in bacterial proteins facilitated the dimerization and/or colocalization process and also act as adaptor proteins (38–43).
Three-dimensional structure modeling and validation
The tertiary models of vlhA proteins were constructed using the server called RaptorX and I Tasser. In tertiary models predicted by Raptor X, the number of amino acids was less compared to the input sequence, and thus the model predicted by I-TASSER was used for further analysis. The results from I-Tasser are consistent with the secondary structure prediction where these proteins were predicted to have a high percentage of random coil structures (Figure 4).
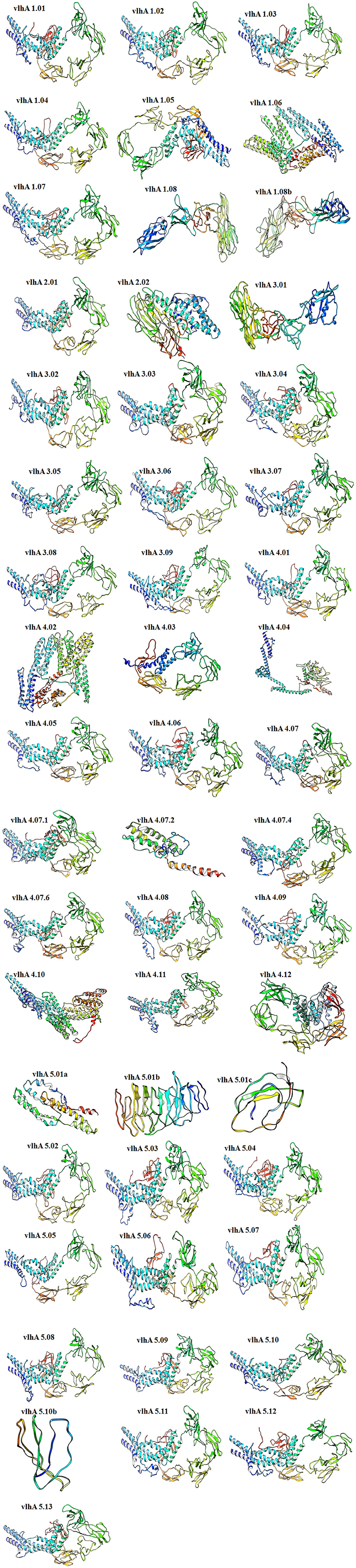
Figure 4. Three-dimensional ab initio models of vlhA proteins. Visualizations of model structures were performed by UCSF Chimera.
The PDBsum-PROCHECK program was used to validate the constructed three-dimensional models of these proteins. The Ramachandran Plot was used in the PROCHECK program to present the backbone conformation of proteins. The predicted models of vlhA proteins were analyzed and majority of the amino acid residues fall in the favored and allowed regions of the Ramachandran plot which indicates the good quality of the predicted models (Table 4).
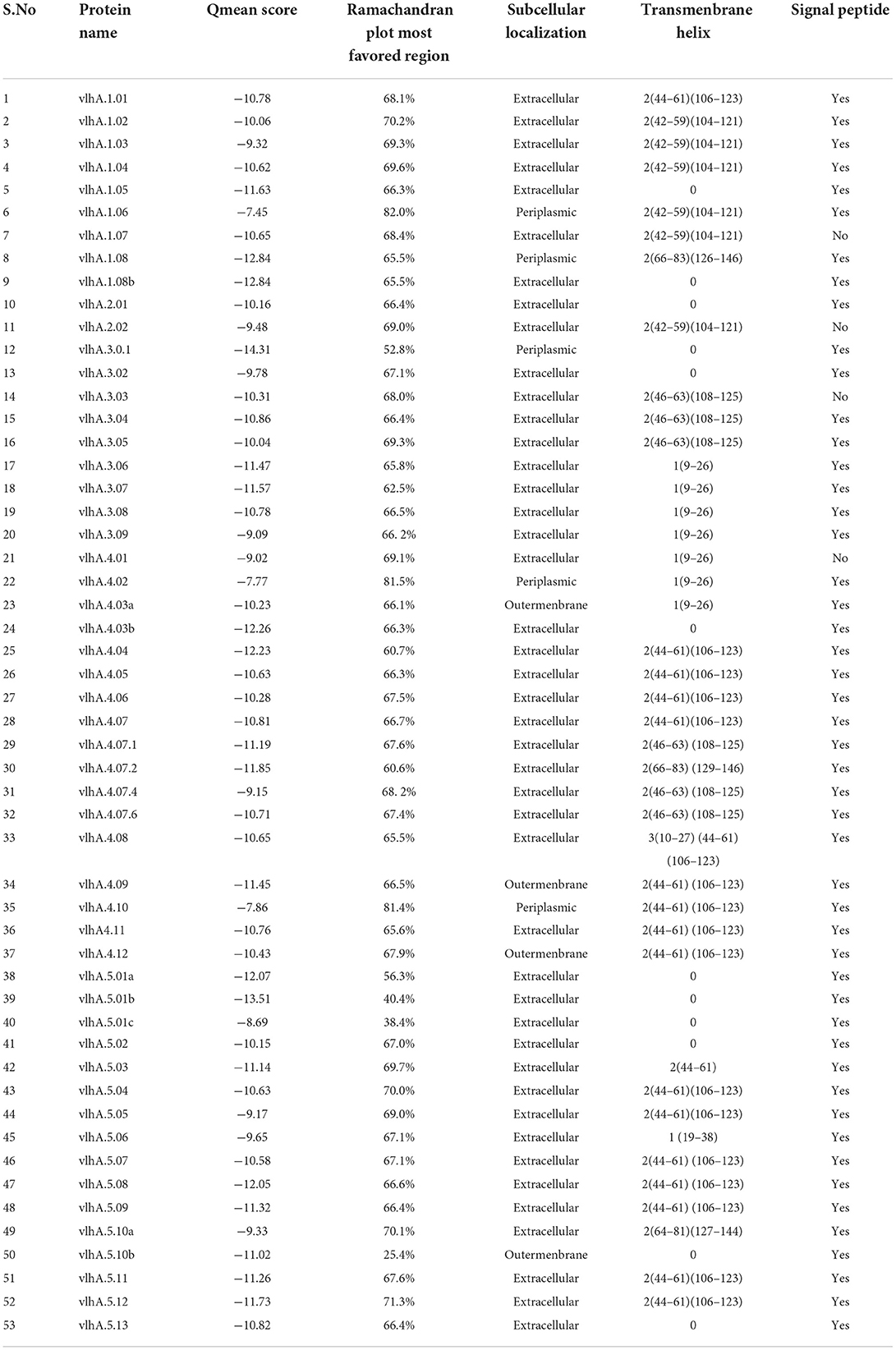
Table 4. Tertiary structural validation- Qmean Score, Ramachandran plot most favored region and functional analysis-Subcellular Localization, Transmembrane helix, Signal peptide of vlhA proteins.
QMEAN z-score was used to validate the good quality of these predicted tertiary models. This QMEAN software determined the closeness and similarity of the computationally predicted model with the existing PDB reference set. The normalized QMEAN score is provided in Table 4.
Functional analysis
Localization of vlhA proteins
In this study, 3 different servers (CELLO2GO, PSORTB, and PSLPRED) were used to predict the cellular location of vlhA proteins. As provided in Table 4, the vlhA proteins were predicted to be extracellular proteins which help in the host interactions and immune evasion. The results were similar for all the three servers. TMMHMM, HMMTOP, and SOSUI servers were used to predict the presence of transmembrane helices in these proteins. Except vlhA-−1.08b, 2.01, 2.02, 3.01, 3.02, 3.08, 4.01, 4.03b, 5.01a, 5.01b, 5.01c, 5.02, 5.08, 5.10b, and 5.13, other proteins were predicted to have transmembrane helices (Table 4). The prediction results are consistent among the servers. Based on the prediction using SignalP and TargetP servers, several vlhA proteins having lower values indicated the absence of signal peptides in them. In contrast, the vlhA proteins with higher values indicated the presence of signal peptides in their sequence (Table 4).
Identification of the functional domain
There are a large number of proteins that have no assigned function. For those proteins, the annotation generally depends on the sequence homology techniques (21). Functional domains were identified using CDD- BLAST, HmmScan, Pfam, SCANPROSITE, and SMART publicly available tools. After screening the vlhA proteins in the above mentioned servers, all the proteins were grouped under the mycoplasma hemagglutinin family by all the servers. Based on the similarity of the sequences of these proteins with mycoplasma hemagglutinin, these proteins were predicted to play a role in the hemagglutination process. The mycoplasma hemagglutinin family consists of several hemagglutinin sequences from mycoplasma species. The major plasma membrane proteins, vlhAs, of M. gallisepticum are cell adhesions or hemagglutinin molecules. The hemagglutination process of mycoplasma plays a crucial role in host immune evasion; the exact mechanism through which the hemagglutination mediated immune evasion occurs is yet to be explored (44, 45).
Discussion
Variable lipoprotein hemagglutinin A gene encodes immunodominant proteins that are believed to be responsible for M. gallisepticum's host cell interaction, pathogenesis, and immune evasion; however, their exact mechanism is unknown (46). The sound knowledge about the mechanism of immune evasion by this protein family will be valuable in the development of drugs and vaccines against M. gallisepticum infection in chickens. Protein structure and function identification is an essential step for understanding its cellular and molecular processes. In silico homology modeling studies provide an opportunity to establish a route for the structural modeling and analysis of vlhA proteins. With rapid advances in bioinformatics and computational biology, the prediction and validation of the structure and function of proteins have become easily accessible. The importance of functional analysis of proteins includes deeper knowledge in molecular mechanisms of disease progression, exploration of effective prophylactic targets, relationship, and interaction with other proteins in the same microorganism.
This study has analyzed the vlhA proteins from M. gallisepticum strain R low for its structural and functional characteristics. The amino acid sequences of vlhA proteins were retrieved in FASTA format from the UniProt database and used for further structural and functional analyses. The physiochemical characteristics such as amino acid composition, isoelectric point (pI), number of negative and positive residues, extinction coefficient, half-life, instability index (II), aliphatic index (AI), and grand average of hydropathy (GRAVY) of these proteins were predicted. According to the results obtained, a higher number of amino acids such as threonine, asparagine, serine, and alanine were observed whereas the amino acids such as cysteine, histidine, and tryptophan were low in amount. Cysteines are important for the formation of disulfide bonds in the protein structure which cannot be easily substituted or replaced and often acts together with histidines which are commonly present in the active or binding sites of the proteins (38). These vlhA proteins have the average molecular weight of 59.28 kDa, and are hydrophilic in nature and stable. The secondary structure of these proteins contains a higher percentage of random coils which are believed to facilitate in the dimerization and/or colocalization process and may also act as adaptor proteins (39–43, 53). The tertiary structures of vlhA proteins were predicted and validated for the good quality of the computationally predicted protein structure. These proteins have been predicted to be stable with the higher percentage of amino acids present in the most favored regions (>80%). The obtained QMEAN score indicated the good quality of these proteins with higher QMEAN values (20). As for the functional prediction of vlhA proteins, all of these proteins were predicted to be extracellular which may subsequently help in the immune evasion of the M. gallisepticum from the host immune system. The identification of the functional domain was performed by the sequence homology techniques. The result obtained showed that the domains of these proteins were similar to the mycoplasma hemagglutinin family as they consist of hemagglutinin sequences from the mycoplasma family and predicted to be involved in the hemagglutination process. It has been reported that the genetic determinants that code for the hemagglutinins are organized into a large family of genes and that only one of these genes is predominately expressed during the course of infection at a given time (44, 47–49). Antigenic variation or phenotypic switching occurs due to high frequency genetic mutations. Due to the lack of a rigid cell wall, the lipoproteins in the mycoplasma cell membrane function as the major elements that come into contact with the host environment (45, 46, 50). These proteins undergo antigenic variation through on/off switching, domain shuffling, and size variation to modify the antigenic components on their cell surface to produce heterotypes that allow mycoplasma to evade recognition and clearance by host immune cells that largely eliminate homo-types. Numerous human and animal mycoplasma species have the ability to go through antigenic variation so that these bacteria can evade recognition by the host humoral immune system (51, 52). In M. gallisepticum, the hemagglutination process may play a role in triggering the antigenic variation cascade leading to immune evasion. Since the exact function and machinery of these vlhA proteins are not determined at present, the in silico structural and functional prediction of these proteins may help in the determination of its cellular and molecular processes. To the best of our knowledge, this is the first study to explore the structural and functional properties of vlhA proteins. These findings may aid in understanding the mechanism of immune evasion by vlhA proteins.
Conclusion
Identifying the molecular processes by which the vlhA protein evades the host immune response is critical in understanding the pathogenicity of M. gallisepticum and will aid in the development of efficient infection control measures. In silico homology modeling studies allow researchers to build a pipeline for structural modeling and functional analysis of any protein as part of discovering the molecular mechanism of the protein's function and therapeutic targets. The physicochemical features of selected vlhA that are important for immune evasion were given in this work. The study also included secondary structure and tertiary model characteristics for the vlhA proteins. Furthermore, the functional analysis revealed that the vlhA proteins are clustered under the mycoplasma hemagglutinin family. For functional analysis of vlhA proteins, multiple servers like CDD- BLAST, HmmScan, Pfam, SCANPROSITE, and SMART were used and all the servers grouped the vlhA proteins under the mycoplasma hemagglutinin family; the results obtained were consistent, thus validating the uniqueness of our findings. The significance of this study is the analysis and exploration of unknown structural and functional characteristics of vlhA proteins through the application of latest bioinformatics software like Protparam, I Tasser, PSORTB, TMMHMM, SignalP, and Pfam, thus bridging the gap in knowledge in the role of vlhA proteins in M. gallisepticum pathogenesis. This research will serve as a foundation for future experimental studies aimed at clarifying the functional molecular mechanism of immune response.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
Author contributions
SM and MH designed and performed the experimental studies. SM carried out the in silico experiments. The manuscript was written by SM and MH. Both authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2022.943831/full#supplementary-material
References
1. Ley DH. Mycoplasma gallisepticum infection. In:. Saif YM, Fadly AM, Glisson JR, McDougald LR, Nolan LK, Swayne DE, eds Diseases of poultry, 12 ed. Ames, IA: Blackwell Publishing Professional (2008). 807–45.
2. Winner F, Rosengarten R, Citti C. In vitro cell invasion of Mycoplasma gallisepticum. Infect Immun. (2000) 68:4238–44. doi: 10.1128/IAI.68.7.4238-4244.2000
3. Tulman ER, Liao X, Szczepanek SM, Ley DH, Kutish GF, Geary SJ, et al. Extensive variation in surface lipoprotein gene content and genomic changes associated with virulence during evolution of a novel North American house finch epizootic strain of Mycoplasma gallisepticum. Microbiol. (2012) 158:2073–88. doi: 10.1099/mic.0.058560-0
4. Pflaum K, Tulman ER, Beaudet J, Liao X, Geary SJ. Global changes in Mycoplasma gallisepticum phase-variable lipoprotein gene vlha expression during in vivo infection of the natural chicken host. Infect Immun. (2015) 84:351–5. doi: 10.1128/IAI.01092-15
5. Noormohammadi AH. Role of phenotypic diversity in pathogenesis of avian mycoplasmosis. Avian Pathol. (2007) 36:439–44. doi: 10.1080/03079450701687078
6. Glew MD, Browning GF, Markham PF, Walker ID. pMGA phenotypic variation in Mycoplasma gallisepticum occurs in vivo and is mediated by trinucleotide repeat length variation. Infect Immun. (2000) 68:6027–33. doi: 10.1128/IAI.68.10.6027-6033.2000
7. Chopra-Dewasthaly R, Spergser J, Zimmermann M, Citti C, Jechlinger W, Rosengarten R, et al. Vpma phase variation is important for survival and persistence of Mycoplasma agalactiae in the immunocompetent host. PLoS Pathog. (2017) 13:e1006656. doi: 10.1371/journal.ppat.1006656
8. Czurda S, Hegde SM, Rosengarten R, Chopra-Dewasthaly R. Xer1-independent mechanisms of Vpma phase variation in Mycoplasma agalactiae are triggered by Vpma-specific antibodies. Int J Med Microbiol. (2017) 307:443–51. doi: 10.1016/j.ijmm.2017.10.005
9. Ma L, Jensen JS, Mancuso M, Myers L, Martin DH. Kinetics of Genetic Variation of the Mycoplasma genitalium MG192 gene in experimentally infected chimpanzees. Infect Immun. (2015) 84:747–53. doi: 10.1128/IAI.01162-15
10. Citti C, Nouvel LX, Baranowski E. Phase and antigenic variation in mycoplasmas. Future Microbiol. (2010) 5:1073–85. doi: 10.2217/fmb.10.71
11. Hu F, Zhao C, Bi D, Tian W, Chen J, Sun J, et al. Mycoplasma gallisepticum (HS strain) surface lipoprotein pMGA interacts with host apolipoprotein A-I during infection in chicken. Appl Microbial Biotechnol. (2016) 100:1343–54. doi: 10.1007/s00253-015-7117-9
12. Vogl G, Plaickner A, Szathmary S, Stipkovits L, Rosengarten R, Szostak MP, et al. Mycoplasma gallisepticum invades chicken erythrocytes during infection. Infect Immun. (2008) 76:71–7. doi: 10.1128/IAI.00871-07
13. Dereeper A, GuignonV, Blanc G, Audic S, Buffet S, Chevenet F, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. (2008) 36:W465–9. doi: 10.1093/nar/gkn180
14. Geourjon C, Deléage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci. (1995) 11:681–4. doi: 10.1093/bioinformatics/11.6.681
15. Garnier J, Osguthorpe DJ, Robson B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J Mol Biol. (1978) 120:97–120. doi: 10.1016/0022-2836(78)90297-8
16. Garnier J, Gibrat JF, Robson B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. (1996) 266:540–53. doi: 10.1016/S0076-6879(96)66034-0
17. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y, et al. The I-TASSER Suite: protein structure and function prediction. Nat Methods. (2015) 12:7–8. doi: 10.1038/nmeth.3213
18. Heo L, Park H, Seok C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. (2013) 41:W384–8. doi: 10.1093/nar/gkt458
19. Laskowski RA, MacArthur MW, Moss DS, Thornton J. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. (1993) 26:283–91. doi: 10.1107/S0021889892009944
20. Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinform. (2011) 27:343–50. doi: 10.1093/bioinformatics/btq662
21. Pearson WR. An introduction to sequence similarity (“homology”) searching. Curr Protoc Bioinform. (2013) Chapter 3:Unit3.1. doi: 10.1002/0471250953.bi0301s42
22. Mariani V, Biasini M, Barbato A, Schwede T. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinform. (2013) 29:2722–8. doi: 10.1093/bioinformatics/btt473
23. Bhasin M, Garg A, Raghava GP. PSLpred: prediction of subcellular localization of bacterial proteins. Bioinform. (2005) 21:2522–4. doi: 10.1093/bioinformatics/bti309
24. Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinform. (2010) 26:1608–1615. doi: 10.1093/bioinformatics/btq249
25. Yu CS, Cheng CW, Su WC, Chang KC, Huang SW, Hwang JK, et al. CELLO2GO: a web server for protein subCELlular LOcalization prediction with functional gene ontology annotation. PLoS ONE. (2014) 9:e99368. doi: 10.1371/journal.pone.0099368
26. Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinform. (1998) 14:378–9. doi: 10.1093/bioinformatics/14.4.378
27. Tusnády GE, Simon I. Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J Mol Biol. (1998) 283:489–506. doi: 10.1006/jmbi.1998.2107
28. Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. (2001) 305:567–80. doi: 10.1006/jmbi.2000.4315
29. Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. (2019) 37:420–23. doi: 10.1038/s41587-019-0036-z
30. Almagro Armenteros JJ, Salvatore M, Emanuelsson O, Winther O, von Heijne G, Elofsson A, et al. Detecting sequence signals in targeting peptides using deep learning. Life Sci Alliance. (2019) 2:e201900429. doi: 10.26508/lsa.201900429
31. Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, et al. CDD: NCBI's conserved domain database. Nucleic Acids Res. (2015) 43:D222–6. doi: 10.1093/nar/gku1221
32. Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. (2011) 39:W29–37. doi: 10.1093/nar/gkr367
33. Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. (2000) 28:231–4. doi: 10.1093/nar/28.1.231
34. Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. (2014) 42:D222–30. doi: 10.1093/nar/gkt1223
35. de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, et al. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. (2006) 34:W362–5. doi: 10.1093/nar/gkl124
36. Bencina D, Drobnic-Valic M, Horvat S, Narat M, Kleven SH, Dovc P, et al. Molecular basis of the length variation in the N-terminal part of Mycoplasma synoviae hemagglutinin. FEMS Microbiol Lett. (2001) 203:115–23. doi: 10.1111/j.1574-6968.2001.tb10829.x
37. Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A, et al. Protein identification and analysis tools on the ExPASy server. The Proteomics Protocols Handbook. Berlin/Heidelberg, Germany: Springer. (2005). p. 571–607.
38. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P, et al. Molecular Biology of the Cell. 4th edition. New York: Garland Science (2002). Available from: https://www.ncbi.nlm.nih.gov/books/NBK26830/
39. Alaidarous M, Ve T, Casey LW, Valkov E, Ericsson DJ, Ullah MO, et al. Mechanism of bacterial interference with TLR4 signaling by Brucella Toll/interleukin-1 receptor domain-containing protein TcpB. J Biol Chem. (2014) 289:654–68. doi: 10.1074/jbc.M113.523274
40. Xiong D, Song L, Geng S, Jiao Y, Zhou X, Song H, et al. Salmonella coiled-coil- and TIR-containing TcpS evades the innate immune system and subdues inflammation. Cell Rep. (2019) 28, 804–818.e7. doi: 10.1016/j.celrep.2019.06.048
41. Radhakrishnan GK, Yu Q, Harms JS, Splitter GA. Brucella TIR domain-containing protein mimics properties of the toll-like receptor adaptor protein TIRAP. J Biol Chem. (2009) 284:9892–8. doi: 10.1074/jbc.M805458200
42. Rana RR, Zhang M, Spear AM, Atkins HS, Byrne B. Bacterial TIR-containing proteins and host innate immune system evasion. Med Microbiol Immunol. (2013) 202:1–10. doi: 10.1007/s00430-012-0253-2
43. Ve T, Gay NJ, Mansell A, Kobe B, Kellie S. Adaptors in toll-like receptor signaling and their potential as therapeutic targets. Curr Drug Targets. (2012) 13:1360–74. doi: 10.2174/138945012803530260
44. Rosengarten R, Citti C, Glew M, Lischewski A, Droesse M, Much P, et al. Host-pathogen interactions in mycoplasma pathogenesis: virulence and survival strategies of minimalist prokaryotes. Int J Med Microbiol. (2000) 290:15–25. doi: 10.1016/S1438-4221(00)80099-5
45. Christodoulides A, Gupta N, Yacoubian V, Maithel N, Parker J, Kelesidis T, et al. The role of lipoproteins in mycoplasma-mediated immunomodulation. Front Microbiol. (2018) 9:1682. doi: 10.3389/fmicb.2018.01682
46. Pflaum K, Tulman ER, Beaudet J, Canter J, Geary SJ. Variable Lipoprotein Hemagglutinin A Gene (vlhA) expression in variant Mycoplasma gallisepticum strains in vivo. Infect Immun. (2018) 86:e00524–18. doi: 10.1128/IAI.00524-18
47. Liu L, Payne DM, van Santen VL, Dybvig K, Panangala VS. A protein (M9) associated with monoclonal antibody-mediated agglutination of Mycoplasma gallisepticum is a member of the pMGA family. Infect Immun. (1998) 66:5570–5. doi: 10.1128/IAI.66.11.5570-5575.1998
48. Markham PF, Glew MD, Sykes JE, Bowden TR, Pollocks TD, Browning GF, et al. The organisation of the multigene family which encodes the major cell surface protein, pMGA, of Mycoplasma gallisepticum. FEBS Lett. (1994) 352:347–52. doi: 10.1016/0014-5793(94)00991-0
49. Noormohammadi AH, Markham PF, Kanci A, Whithear KG, Browning GF. A novel mechanism for control of antigenic variation in the haemagglutinin gene family of mycoplasma synoviae. Mol Microbial. (2000) 35:911–23. doi: 10.1046/j.1365-2958.2000.01766.x
50. Barbosa MS, Spergser J, Marques LM, Timenetsky J, Rosengarten R, Chopra-Dewasthaly R, et al. Predominant single stable VpmaV expression in strain GM139 and major differences with Mycoplasma agalactiae type strain PG2. Animals. (2022) 12:265. doi: 10.3390/ani12030265
51. Pflaum K, Tulman ER, Canter J, Dhondt KV, Reinoso-Perez MT, Dhondt AA, et al. The influence of host tissue on M. gallisepticum vlhA gene expression. Vet Microbiol. (2020) 251:108891. doi: 10.1016/j.vetmic.2020.108891
52. Galyamina MA, Zubov AI, Ladygina VG, Li AV, Matyushkina DS, Pobeguts OV, et al. Comparative proteomic analysis of the Mycoplasma gallisepticum nucleoid fraction before and after infection. Bull Exp Biol Med. (2022) 172:336–40. doi: 10.1007/s10517-022-05388-4
53. Alaidarous M. In silico structural homology modeling and characterization of multiple N-terminal domains of selected bacterial Tcps. PeerJ. (2020) 8.e10143. doi: 10.7717/peerj.10143
Keywords: variable lipoprotein hemagglutin, immune evasion, bioinformatics, avian mycoplasmosis, M. gallisepticum
Citation: Mugunthan SP and Harish MC (2022) In silico structural homology modeling and functional characterization of Mycoplasma gallisepticum variable lipoprotein hemagglutin proteins. Front. Vet. Sci. 9:943831. doi: 10.3389/fvets.2022.943831
Received: 14 May 2022; Accepted: 01 July 2022;
Published: 04 August 2022.
Edited by:
Rajesh Kumar Pathak, Chung-Ang University, South KoreaReviewed by:
Dev Bukhsh Singh, Siddharth University Kapilvastu, IndiaNeetesh Pandey, Indraprastha Institute of Information Technology Delhi, India
Pallavi Shah, Indian Veterinary Research Institute (IVRI), India
Copyright © 2022 Mugunthan and Harish. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mani Chandra Harish, mc.harishin@tvu.edu.in