Copy number variants selected during pig domestication inferred from whole genome resequencing
 Wei Zhang
Wei Zhang Chengliang Xu
Chengliang Xu Mei Zhou
Mei Zhou Linqing Liu1
Linqing Liu1  Chonglong Wang
Chonglong Wang- 1Institute of Animal Husbandry and Veterinary Medicine, Anhui Academy of Agricultural Sciences, Anhui Provincial Breeding Pig Genetic Evaluation Center, Key Laboratory of Pig Molecular Quantitative Genetics of Anhui Academy of Agricultural Sciences, Anhui Provincial Key Laboratory of Livestock and Poultry Product Safety Engineering, Hefei, China
- 2Anhui Provincial Livestock and Poultry Genetic Resources Conservation Center, Hefei, China
Over extended periods of natural and artificial selection, China has developed numerous exceptional pig breeds. Deciphering the germplasm characteristics of these breeds is crucial for their preservation and utilization. While many studies have employed single nucleotide polymorphism (SNP) analysis to investigate the local pig germplasm characteristics, copy number variation (CNV), another significant type of genetic variation, has been less explored in understanding pig resources. In this study, we examined the CNVs of 18 Wanbei pigs (WBP) using whole genome resequencing data with an average depth of 12.61. We identified a total of 8,783 CNVs (~30.07 Mb, 1.20% of the pig genome) in WBP, including 8,427 deletions and 356 duplications. Utilizing fixation index (Fst), we determined that 164 CNVs were within the top 1% of the Fst value and defined as under selection. Functional enrichment analyses of the genes associated with these selected CNVs revealed genes linked to reproduction (SPATA6, CFAP43, CFTR, BPTF), growth and development (NR6A1, SMYD3, VIPR2), and immunity (PARD3, FYB2). This study enhances our understanding of the genomic characteristics of the Wanbei pig and offers a theoretical foundation for the future breeding of this breed.
1 Introduction
The pig (Sus scrofa) was domesticated approximately 10,000 years ago, significantly influencing economic, social, and cultural aspects of human society (1–3). Pig domestication involves phenotypic and genomic alterations, including behavioral changes such as reduced aggression and watchfulness, morphological adaptations like brain size and skull shape alterations, and physiological improvements in growth and prolificacy. To adapt to varying environmental conditions and market demands over different periods, approximately 600 pig breeds have been established worldwide (4). Understanding the relationship between domestication and phenotypic changes is crucial not only for deciphering the genetic basis of complex economic traits but also for advancing future pig breeding practices.
In 2012, a significant milestone was reached in pig genome research with the assembly of the pig reference genome, providing a valuable resource for studying this important livestock species (5). Moreover, the continuous decrease in sequencing costs, driven by technological advancements, has enabled more scientists to explore germplasm characteristics extensively. To date, numerous functional genes and loci regulating important traits have been identified. For instance, the coat color trait, a prominent characteristic in pigs, has been the subject of several studies, identifying genes such as MITF, EDNRB, KIT, and MC1R associated with this trait (6–9). Additionally, genes associated with environmental adaptation traits like cold tolerance (TRPV5) (10), heat tolerance (VPS13A) (11, 12), and hypoxia (EPAS1) (13) have been discovered. However, these findings were predominantly based on single nucleotide polymorphism (SNP) detection. Copy number variations (CNVs), ranging from 50 bp to several Mb in size, represent another crucial type of genetic variation in the pig genome (14). The role of CNVs in domestication and their significant impact on phenotypic characteristics, gene function, evolutionary adaptation, and disease susceptibility (15) are less understood. CNVs have been shown to explain complex traits in humans and domesticated animals, such as starch-associated and high-altitude adaptation in humans (16, 17), pathogen and parasite resistance in cattle (18), fatty acid metabolism in dogs (19), and litter size in pigs (20).
The Wanbei pig (WBP), a valuable genetic resource in northern Anhui Province, China, exhibits high fertility, exceptional meat quality, and robust disease resistance. Understanding their germplasm characteristics is vital for their protection and utilization. Previous studies have investigated genomic SNPs in WBP, identifying selected genes based on SNP analysis (21). However, information on CNVs in WBP and selection based on CNVs remains unexplored. Given the significant role of CNVs in elucidating complex phenotypes, detecting CNVs in WBP and understanding their contribution to domestication is essential.
This study utilized resequencing data from 18 WBPs and 19 Asian wild boars (AWBs) to perform a CNV analysis. The study comprises three steps: (1) creating a comprehensive CNV landscape for the Wanbei pig, (2) conducting population structure analysis based on CNVs, and (3) identifying selected CNVs and important candidate genes. The findings of this research enhance our understanding of the impact of CNVs and provide new insights for the protection and utilization of the WBP population.
2 Materials and methods
2.1 Ethics statement
This study was conducted in accordance with and was approved by the Animal Care Committee of the Anhui Academy of Agricultural Sciences (Hefei, China; no. AAAS2020-04).
2.2 Sample collection and whole-genome resequencing
A total of 18 WBPs (Figure 1), approximately 2 years old, were analyzed through whole-genome resequencing with an average depth of 12.61. The WBPs were sourced from a conservation farm in Yingshang, China (longitude 116.26455E; latitude 32.62893 N). Genomic DNA was extracted from ear tissue samples employing the standard phenol–chloroform extraction method (22). The sequencing library was prepared through a series of steps including random fragmentation of the DNA, purification of the fragments to obtain the desired length, adapter ligation, and DNA clustering. Sequencing was performed on an Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, United States).

Figure 1. Pictures of Wanbei pig: (A) female; (B) male.
To compare population structures and identify selection signatures between WBPs and Asian wild boars (AWBs), we utilized a total of 19 resequenced AWB datasets. Six AWB were sequenced in a previous study, with the data accessible under accession number PRJNA699491 (23). Genomic data for the remaining 13 AWB were retrieved from the National Center for Biotechnology Information (NCBI) under accession numbers PRJNA213179, PRJNA186497, and PRJEB1683, respectively (5, 24, 25).
2.3 Read mapping and CNV detection
Prior to CNV detection, adapters and low-quality reads were removed using the NGSQC Toolkit (v.2.30) (26). The filtered reads were aligned to the pig reference genome (Sus scrofa 11.1) using the Burrows–Wheeler aligner (BWA) with default parameters. Manta (27) and Paragraph (28) were employed in combination to detect CNVs on autosomes. Specifically, Manta was used for CNV identification and Paragraph for genotyping the variants in each sample. The methods for obtaining high-quality CNVs were based on our previous study (29). PLINK software v.1.90 (30) was used to determine frequency, with visualization conducted using R (v4.2.0).
2.4 Principal component and phylogenetic analysis
Before analyzing population structure, VCF files were converted into map and ped formats using PLINK. Two analyses were conducted: principal component analysis (PCA) and phylogenetic trees. PCA was performed using EIGENSOFT (31), phylogenetic trees were created with MEGA v7.0 (32), and visualization was done with ITOL1 (33).
2.5 Identifying genomic signatures of selection
The fixation index (FST) was calculated to evaluate population differentiation. The formula for Fst calculation is Fst = (Ht − Hs)/Ht, where Ht is the expected heterozygosity of the population, and Hs is the expected heterozygosity of the subgroup. We selected the CNVs at the top 1% of the FST value as signatures of selection. For enrichment analysis of the selected genes within these regions, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were conducted using KOBAS.2
3 Results
3.1 Copy number variation identification
To detect genome-wide CNVs and compare the differentiation between WBP and AWB, whole-genome sequencing of 18 WBP and 19 AWB was conducted. The sequencing generated a total of 590.3 Gb for WBP with an average depth of 12.61, and 662.8 Gb for AWB with an average depth of 13.44. Detailed information on WBP and AWB is provided in Supplementary Table S1. Overall, 1253.1 Gb of data were used in this study. We identified 8,783 CNVs in WBP, covering ~30.07 Mb (1.20% of the pig genome), including 8,427 deletions (Del) and 356 duplications (Dup) (Table 1). The average and median lengths of CNVs in WBP are 3,423 bp and 282 bp, respectively (Supplementary Table S2). In AWB, 13,128 CNVs covering 33.32 Mb (1.33% of the pig genome) were detected, including 12,500 Del and 628 Dup (Table 1). The average length of CNVs in AWB is 2,538 bp, and the median length is 259 bp (Supplementary Table S2).
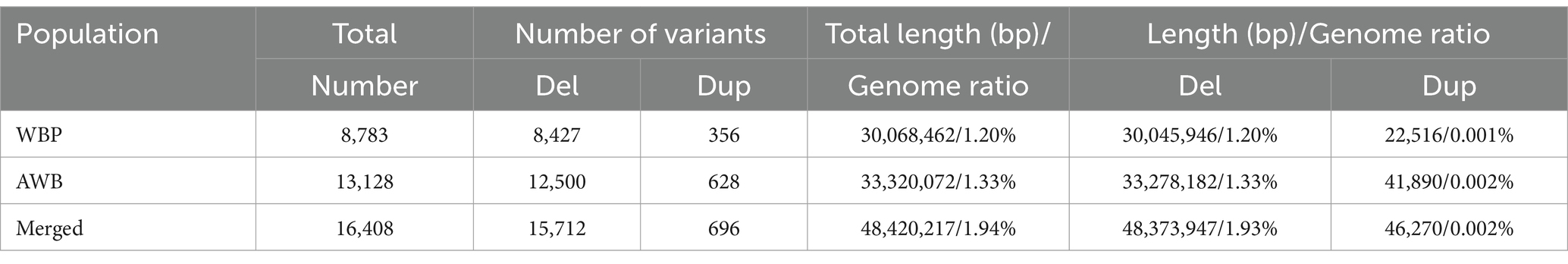
Table 1. The statistic of CNV in WBP and AWB.
After merging the CNVs, a total of 16,408 CNVs (15,712 Del and 696 Dup) were obtained, covering 48.42 Mb of the pig genome, corresponding to 1.94% of the pig genome (Table 1). The information on these CNVs is provided in Supplementary Table S3. The distribution of CNVs across the 18 autosomes varies (Figure 2A). A Venn diagram revealed that 5,506 CNVs (33.56% of the total 16,408) are common between the two populations, and 19.97% are unique to WBP (Figure 2B). Annotation analysis indicated that the CNVs were most abundant in intronic regions (45.75%), intergenic regions (43.44%), and exonic regions (1.23%, 202 CNVs) (Table 2). The frequency of CNVs was divided into ten groups (0–0.1 to 0.9–1), as shown in Supplementary Table S4 and Figures 2C,D. The 0–0.1 frequency group was the largest, covering 63.71% of WBP CNVs and 47.48% of AWB CNVs. The trend between frequency and the number of CNVs in Del and Dup is similar in both populations.
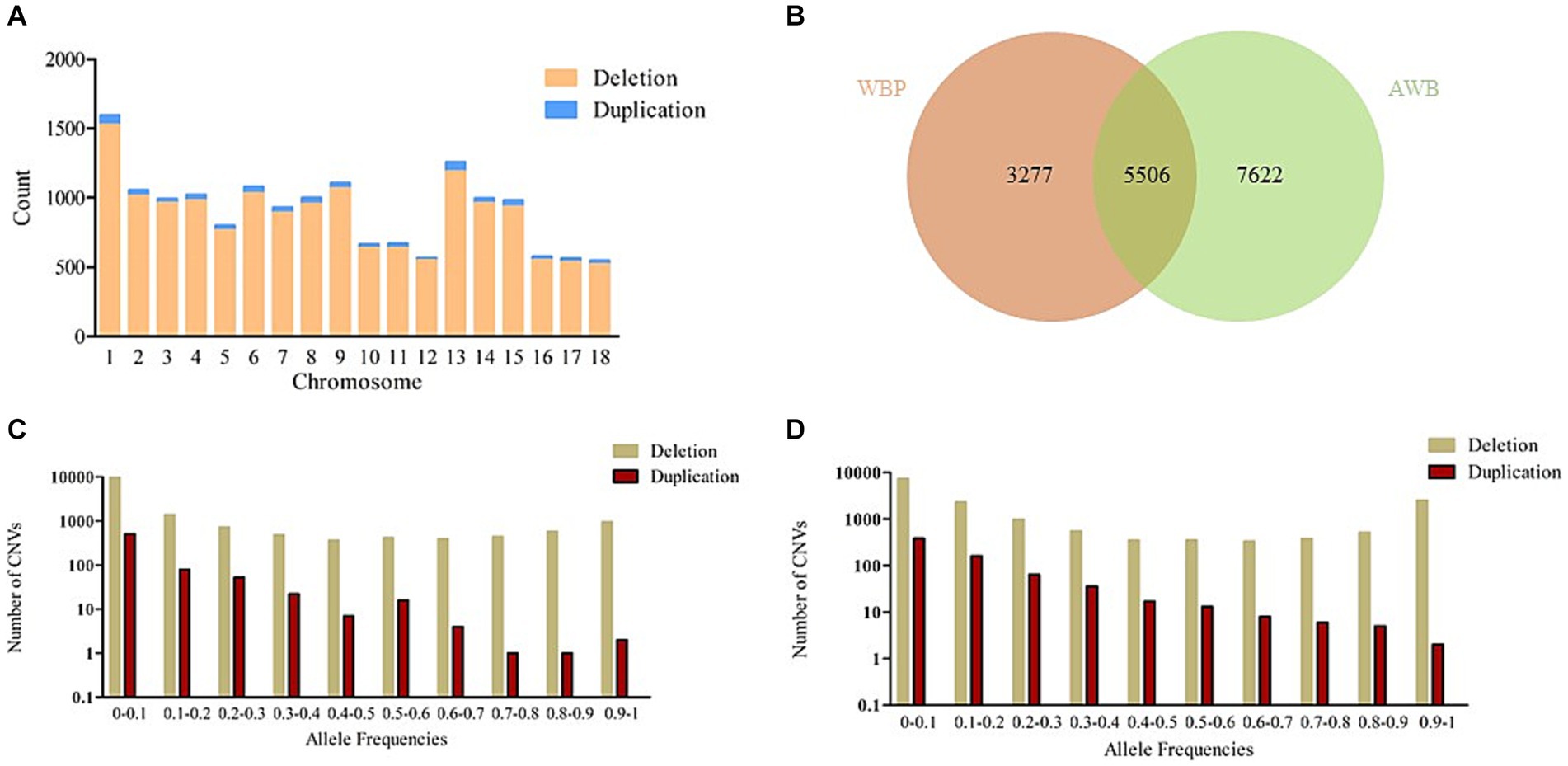
Figure 2. (A) The number of CNVs (deletion and duplication) in 18 autosomes. (B) The Venn diagram of CNVs between WBP and AWB. (C) The allele frequencies of variants in the WBP (n = 18). (D) The allele frequencies of variants in the AWB (n = 19).
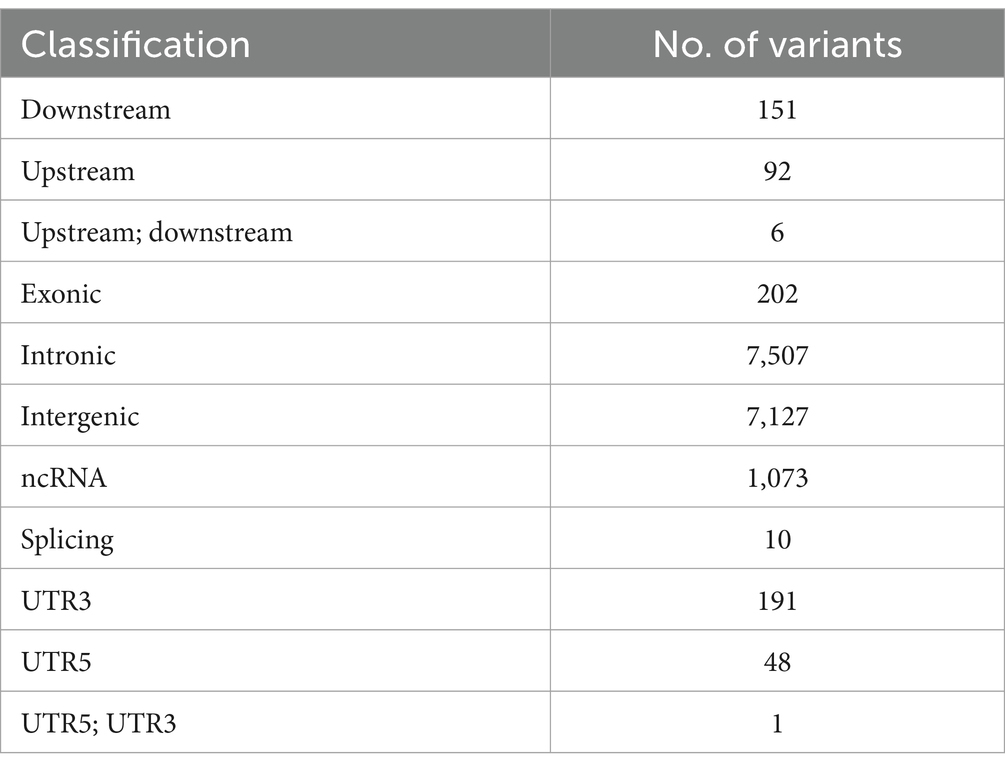
Table 2. Annotation of the merged CNVs.
3.2 PCA and phylogenetic analysis
PCA and Neighbor-Joining (NJ) tree analysis were conducted to elucidate the relationship between WBP and AWB populations. Figure 3A shows that PCA, performed with the first two principal components, clustered WBP and AWB separately. A similar pattern was observed in the NJ tree (Figure 3B).

Figure 3. (A) PCA plots for the first two PCs for all 37 individuals. (B) Neighbor-joining tree constructed from CNV data in study population.
3.3 Patterns of selection signatures
The Fst was used to identify CNVs under selection across autosomes. A total of 164 CNVs were selected within the top 1% of the Fst value (threshold: 1%, Fst = 0.7741, Supplementary Table S5). The Manhattan plot of the Fst statistic is shown in Figure 4. A total of 84 genes were detected (Supplementary Table S6). Gene ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were analyzed using KOBAS software. The GO term analysis enriched a total of 36 terms at level 2 GO enrichment (Supplementary Figure S1; Supplementary Table S7), with these genes associated with growth (GO:0040007, 2 genes), reproduction (GO:0000003, 5 genes), and immune system processes (GO:0002376, 4 genes). The KEGG analysis enriched 10 pathways (Supplementary Figure S2; Supplementary Table S8), including the Rap1 signaling pathway (ko04015, 5 genes), cAMP signaling pathway (ko04024, 4 genes), bile secretion (ko04976, 2 genes), and gastric acid secretion (ko04971, 2 genes).
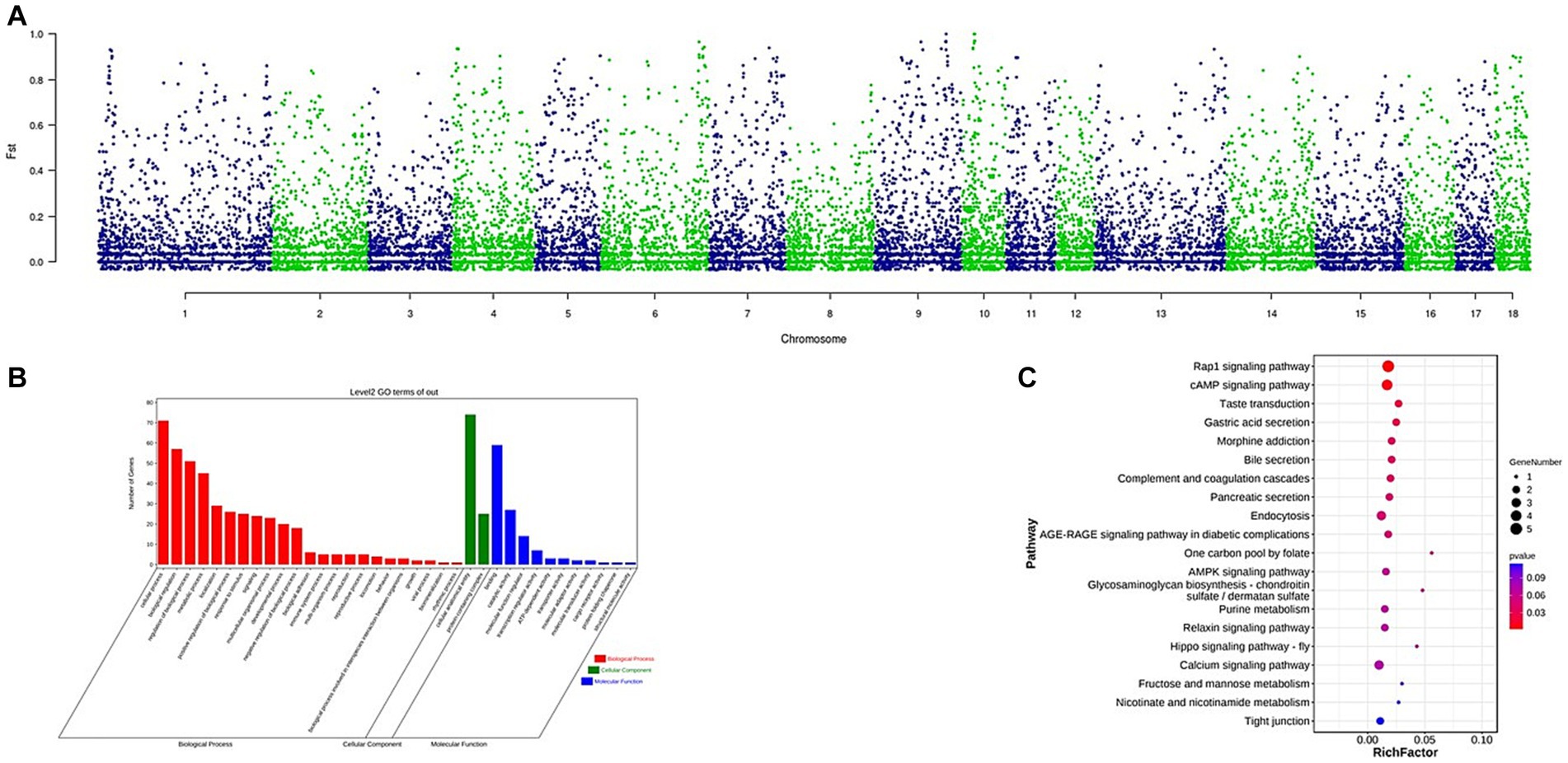
Figure 4. (A) Identification of regions with selection in Wanbei pig population compared to Asian wild boar, which are calculated with Fst. (B) GO analysis of the selected genes, red referring to biological process, green referring to cellular component and blue referring to molecular function. (C) KEGG analysis of the selected genes, the size of dot refers to the number genes related to pathway, and the red to blue indicate the significant value of p change.
4 Discussion
Pig germplasm resources are national strategic assets and form the foundation for the development of the modern pig industry. As the pig industry evolves towards intensification and large-scale operations, the interest in rearing indigenous pig breeds has diminished significantly. This shift has led to a reduction in the size of local pig populations, a gradual loss of genetic diversity, and the endangerment or even extinction of some local pig breeds. Protecting the genetic diversity of pig breeding resources is imperative, not only to support the future development and utilization of pig breeding but also as a cornerstone for the stable production of pig farming in the future. Additionally, pigs hold significant cultural value for the Chinese people. For instance, the Chinese character for “home” (家) is historically represented by an oracle bone inscription of a pig under a roof, symbolizing treasure and safety. Furthermore, numerous place names in China are associated with pigs, embedding an emotional connection to this animal in every corner of the land. In this study, we analyzed 18 unrelated WBP and 19 AWB to investigate population structure and selection signatures. We detected a total of 8,783 CNVs in WBP and 13,128 CNVs in AWB. Of these, 164 CNVs were under selection, and 84 genes were identified within these selected CNVs. Functional enrichment analysis revealed that the selected genes are associated with several vital traits.
Notably, some genes were linked to reproduction. For instance, the inactivation of Spermatogenesis-associated 6 (SPATA6) may lead to sterility (34), while its overexpression has been shown to induce the secretion of testosterone hormone (35). Cilia- and flagella-associated protein 43 (CFAP43) is linked with multiple morphological abnormalities of the sperm flagella (MMAF). CFAP43-null male mice were found to be infertile, exhibiting defects in sperm flagella (36–38), and significant associations between CFAP43 and litter size in goats were identified (39). Cystic fibrosis transmembrane regulator (CFTR) plays a role in regulating protein concentration in the reproductive tract (40), and its expression in spermatozoa correlates with sperm quality (41). Lastly, Bromodomain PHD finger transcription factor (BPTF) is crucial in establishing the anteroposterior axis of the embryo, with mutations in BPTF potentially leading to embryonic death (42). Recent research has indicated that knockdown of BPTF is associated with the proliferation and apoptosis of GCs cells (43).
Several genes associated with growth and development were identified. The nuclear receptor subfamily 6 group A member 1 (NR6A1) is known to influence the number of lumbar vertebrae (44) and regulate body size in pigs (45). Previous studies have linked NR6A1 with average daily gain (ADG) and ham weight (46). When comparing CNV positions with PigQTLdb,3 12 quantitative trait loci (QTLs) were identified, including those for ADG (ID = 659, 22,269), body weight (ID = 660, 661, 662), and ham weight (ID = 376). SET and MYND domain-containing protein 3 (SMYD3) is crucial in the development of skeletal muscle and myocardium and is involved in regulating myofibril assembly in both muscle types (47, 48). Analysis with PigQTLdb identified QTLs for percentage type I fibers (ID = 7,012, 7,026), and percentage type IIa fibers (ID = 7,034). The vasoactive intestinal peptide receptor 2 (VIPR2) gene is associated with insulin secretion (49) and cAMP production (50). Studies have shown that skeletal muscle mass positively correlates with increased cAMP levels upon the administration of a VIPR2-selective agonist (51). VIPR2 knockout resulted in inhibited growth, reduced fat mass, and increased lean mass (52).
Some genes were identified as immune-related. Porcine epidemic diarrhea virus (PEDV) poses a significant threat to the pig industry. Proteomic analysis comparing PEDV-infected and non-infected groups revealed that PARD3 is a significantly downregulated protein during virus infection (53). FYB2 encodes a T cell adaptor protein that can activate integrin and T cell adhesion (54).
Several genes associated with other important traits were also identified. Adenylate cyclase 8 (ADCY8), part of the adenylate cyclase family, regulates nutrient homeostasis in rodents (55–57) and has been found under selection in cattle (58) and pigs (21). Previous GWAS studies have linked ADCY8 positively with Mg and Fe levels (59), high-density cholesterol metabolism in humans (60), and total cholesterol and high-density lipoprotein cholesterol in pigs (61).
However, this study has limitations that should not be overlooked. Due to the limited number of sequenced samples, the detected CNVs may not represent the entire population’s variation. Additionally, the collection of samples to verify the identified CNVs was impeded by the impact of African swine fever. Future plans include collecting a larger sample set to validate the variations and conducting extensive sequencing to more comprehensively detect CNVs.
5 Conclusion
In this study, we initially identified the CNVs in the WBP. Subsequently, we analyzed the population structure, comparing WBP with AWB. Furthermore, we identified selection signatures in WBP and discovered several genes associated with reproduction, immunity, growth and development, and lipid metabolism. These insights broaden our understanding of the impact of CNVs in pigs and offer a valuable resource for future genetic breeding endeavors.
Data availability statement
The data presented in the study are deposited in the Genome Sequence Archive of “China National Center for Bioinformation” (https://ngdc.cncb.ac.cn/gsa/), accession number is CRA015150.
Ethics statement
The animal studies were approved by the Animal Care Committee of the Anhui Academy of Agricultural Sciences. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
WZ: Conceptualization, Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. CX: Resources, Writing – review & editing. MZ: Resources, Writing – review & editing. LL: Resources, Writing – review & editing. ZN: Resources, Writing – review & editing. SS: Resources, Writing – review & editing. CW: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the grants from Anhui Provincial Breeding Pig Genetic Evaluation Center, Anhui Provincial Financial Agricultural Germplasm Resources Protection and Utilization Fund Project; Anhui Province Academic and Technical Leader Candidate Project (no. 2022H300); the Special Fund for Anhui Agricultural Research System (AHCYJSTX-05-12, AHCYJSTX-05-23).
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing. We thank many people not listed as authors who provided help, encouragement, and feedback.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2024.1364267/full#supplementary-material
Footnotes
References
1. Giuffra, E, Kijas, JM, Amarger, V, Carlborg, O, Jeon, JT, and Andersson, L. The origin of the domestic pig: independent domestication and subsequent introgression. Genetics. (2000) 154:1785–91. doi: 10.1093/genetics/154.4.1785
2. Kijas, JMH, and Andersson, L. A phylogenetic study of the origin of the domestic pig estimated from the near-complete mtDNA genome. J Mol Evol. (2001) 52:302–8. doi: 10.1007/s002390010158
3. Larson, G. Worldwide phylogeography of wild boar reveals multiple centers of pig domestication. Science. (2005) 307:1618–21. doi: 10.1126/science.1106927
4. FAO. Status and Trends of Animal Genetic Resources 2020. Rome: Commission on Animal Genetic Resources (2021).
5. Groenen, MA, Archibald, AL, Uenishi, H, Tuggle, CK, Takeuchi, Y, Rothschild, MF, et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature. (2012) 491:393–8. doi: 10.1038/nature11622
6. Wilkinson, S, Lu, ZH, Megens, HJ, Archibald, AL, Haley, C, Jackson, IJ, et al. Signatures of diversifying selection in European pig breeds. PLoS Genet. (2013) 9:e1003453. doi: 10.1371/journal.pgen.1003453
7. Wang, C, Wang, H, Zhang, Y, Tang, Z, Li, K, and Liu, B. Genome-wide analysis reveals artificial selection on coat colour and reproductive traits in Chinese domestic pigs. Mol Ecol Resour. (2015) 15:414–24. doi: 10.1111/1755-0998.12311
8. Huang, M, Zhang, H, Wu, ZP, Wang, XP, Li, DS, Liu, SJ, et al. Whole-genome resequencing reveals genetic structure and introgression in Pudong white pigs. Animal. (2021) 15:100354. doi: 10.1016/j.animal.2021.100354
9. Zhang, W, Yang, M, Zhou, M, Wang, Y, Wu, X, Zhang, X, et al. Identification of signatures of selection by whole-genome resequencing of a Chinese native pig. Front Genet. (2020) 11:566255. doi: 10.3389/fgene.2020.566255
10. Zhang, DJ, He, XM, Wang, L, and Liu, D. Genomic sequence feature analysis of de novo assembly in min pig. J. Jilin Agric Univ. (2023). 600–608. doi: 10.13327/j.jjlau.2021.1324
11. Zhong, ZQ, Li, R, Wang, Z, Tian, SS, Xie, XF, Wang, ZY, et al. Genome-wide scans for selection signatures in indigenous pigs revealed candidate genes relating to heat tolerance. Animal. (2023) 17:100882. doi: 10.1016/j.animal.2023.100882
12. Zhang, L, Zhang, S, Zhan, F, Song, M, Shang, P, Zhu, F, et al. Population genetic analysis of six Chinese indigenous pig meta-populations based on geographically isolated regions. Animals (Basel). (2023) 13:1396. doi: 10.3390/ani13081396
13. Ma, YF, Han, XM, Huang, CP, Zhong, L, Adeola, AC, Irwin, DM, et al. Population genomics analysis revealed origin and high-altitude adaptation of Tibetan pigs. Sci Rep. (2019) 9:11463. doi: 10.1038/s41598-019-47711-6
14. Mills, RE, Walter, K, Stewart, C, Handsaker, RE, Chen, K, Alkan, C, et al. Mapping copy number variation by population-scale genome sequencing. Nature. (2011) 470:59–65. doi: 10.1038/nature09708
15. Marques-Bonet, T, Girirajan, S, and Eichler, EE. The origins and impact of primate segmental duplications. Trends Genet. (2009) 25:443–54. doi: 10.1016/j.tig.2009.08.002
16. Iskow, RC, Gokcumen, O, and Lee, C. Exploring the role of copy number variants in human adaptation. Trends Genet. (2012) 28:245–57. doi: 10.1016/j.tig.2012.03.002
17. OuzhuluobuHe, Y, Lou, H, Cui, C, Deng, L, Gao, Y, et al. De novo assembly of a Tibetan genome and identification of novel structural variants associated with high-altitude adaptation. Natl Sci Rev. (2020) 7:391–402. doi: 10.1093/nsr/nwz160
18. Bickhart, DM, Hou, Y, Schroeder, SG, Alkan, C, Cardone, MF, Matukumalli, LK, et al. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. (2012) 22:778–90. doi: 10.1101/gr.133967.111
19. Wang, GD, Shao, XJ, Bai, B, Wang, J, Wang, X, Cao, X, et al. Structural variation during dog domestication: insights from gray wolf and dhole genomes. Natl Sci Rev. (2019) 6:110–22. doi: 10.1093/nsr/nwy076
20. Ran, XQ, Pan, H, Huang, SH, Liu, C, Niu, X, Li, S, et al. Copy number variations of MTHFSD gene across pig breeds and its association with litter size traits in Chinese indigenous Xiang pig. J Anim Physiol Anim Nutr (Berl). (2018):24. doi: 10.1111/jpn.12922
21. Zhang, W, Liu, L, Zhou, M, Su, S, Dong, L, Meng, X, et al. Assessing population structure and signatures of selection in Wanbei pigs using whole genome resequencing data. Animals (Basel). (2022) 13:13. doi: 10.3390/ani13010013
22. Sambrook, J, and Russell, DW. Molecular Cloning: A Laboratory Manual. 3rd ed. New York, NY, USA: Cold Spring Harbor Laboratory Press (2001).
23. Wu, X, Zhou, R, Wang, Y, Zhang, W, Zheng, X, Zhao, G, et al. Genome-wide scan for runs of homozygosity in Asian wild boars and Anqing six-end-white pigs. Anim Genet. (2022) 53:867–71. doi: 10.1111/age.13250
24. Li, M, Tian, S, Jin, L, Zhou, G, Li, Y, Zhang, Y, et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat Genet. (2013) 45:1431–8. doi: 10.1038/ng.2811
25. Ai, H, Fang, X, Yang, B, Huang, Z, Chen, H, Mao, L, et al. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing. Nat Genet. (2015) 47:217–25. doi: 10.1038/ng.3199
26. Patel, RK, and Jain, M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. (2012) 7:e30619. doi: 10.1371/journal.pone.0030619
27. Chen, X, Schulz-Trieglaff, O, Shaw, R, Barnes, B, Schlesinger, F, Källberg, M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. (2016) 32:1220–2. doi: 10.1093/bioinformatics/btv710
28. Chen, S, Krusche, P, Dolzhenko, E, Sherman, RM, Petrovski, R, Schlesinger, F, et al. Paragraph: a graph-based structural variant genotyper for short-read sequence data. Genome Biol. (2019) 20:291. doi: 10.1186/s13059-019-1909-7
29. Zhang, W, Zhou, M, Liu, L, Su, S, Dong, L, Meng, X, et al. Population structure and selection signatures underlying domestication inferred from genome-wide copy number variations in Chinese indigenous pigs. Genes (Basel). (2022) 13:26. doi: 10.3390/genes13112026
30. Purcell, S, Neale, B, Todd-Brown, K, Thomas, L, Ferreira, MA, Bender, D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. (2007) 81:559–75. doi: 10.1086/519795
31. Price, AL, Patterson, NJ, Plenge, RM, Weinblatt, ME, Shadick, NA, and Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. (2006) 38:904–9. doi: 10.1038/ng1847
32. Kumar, S, Stecher, G, and Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. (2016) 33:1870–4. doi: 10.1093/molbev/msw054
33. Letunic, I, and Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. (2016) 44:W242–5. doi: 10.1093/nar/gkw290
34. Yuan, S, Stratton, CJ, Bao, J, Zheng, H, Bhetwal, BP, Yanagimachi, R, et al. Spata6 is required for normal assembly of the sperm connecting piece and tight head-tail conjunction. Proc Natl Acad Sci USA. (2015) 112:E430–9. doi: 10.1073/pnas.1424648112
35. Li, X, Yao, X, Xie, H, Deng, M, Gao, X, Deng, K, et al. Effects of SPATA6 on proliferation, apoptosis and steroidogenesis of Hu sheep Leydig cells in vitro. Theriogenology. (2021) 166:9–20. doi: 10.1016/j.theriogenology.2021.02.011
36. Tang, S, Wang, X, Li, W, Yang, X, Li, Z, Liu, W, et al. Biallelic mutations in CFAP43 and CFAP44 cause male infertility with multiple morphological abnormalities of the sperm flagella. Am J Hum Genet. (2017) 100:854–64. doi: 10.1016/j.ajhg.2017.04.012
37. Morimoto, Y, Yoshida, S, Kinoshita, A, Satoh, C, Mishima, H, Yamaguchi, N, et al. Nonsense mutation in CFAP43 causes normal-pressure hydrocephalus with ciliary abnormalities. Neurology. (2019) 92:e2364–74. doi: 10.1212/WNL.0000000000007505
38. Yu, Y, Wang, J, Zhou, L, Li, H, Zheng, B, and Yang, S. CFAP43-mediated intra-manchette transport is required for sperm head shaping and flagella formation. Zygote. (2021) 29:75–81. doi: 10.1017/S0967199420000556
39. Wang, Z, Pan, Y, He, L, Song, X, Chen, H, Pan, C, et al. Multiple morphological abnormalities of the sperm flagella (MMAF)-associated genes: the relationships between genetic variation and litter size in goats. Gene. (2020) 753:144778. doi: 10.1016/j.gene.2020.144778
40. Zondek, LH, and Zondek, T. Normal and abnormal development of the epididymis of the fetus and infant. Eur J Pediatr. (1980) 134:39–44. doi: 10.1007/BF00442401
41. Li, CY, Jiang, LY, Chen, WY, Li, K, Sheng, HQ, Ni, Y, et al. CFTR is essential for sperm fertilizing capacity and is correlated with sperm quality in humans. Hum Reprod. (2010) 25:317–27. doi: 10.1093/humrep/dep406
42. Landry, J, Sharov, AA, Piao, Y, Sharova, LV, Xiao, H, Southon, E, et al. Essential role of chromatin remodeling protein Bptf in early mouse embryos and embryonic stem cells. PLoS Genet. (2008) 4:e1000241. doi: 10.1371/journal.pgen.1000241
43. Wang, D, Lu, X, Jiang, Y, Pan, L, Zhu, F, Yu, A, et al. The chromatin remodeling protein BPTF mediates cell cycle, proliferation and apoptosis in porcine ovarian granulosa cells. Theriogenology. (2023) 211:172–81. doi: 10.1016/j.theriogenology.2023.08.017
44. Mikawa, S, Morozumi, T, Shimanuki, S, Hayashi, T, Uenishi, H, Domukai, M, et al. Fine mapping of a swine quantitative trait locus for number of vertebrae and analysis of an orphan nuclear receptor, germ cell nuclear factor (NR6A1). Genome Res. (2007) 17:586–93. doi: 10.1101/gr.6085507
45. Li, LY, Xiao, SJ, Tu, JM, Zhang, ZK, Zheng, H, Huang, LB, et al. A further survey of the quantitative trait loci affecting swine body size and carcass traits in five related pig populations. Anim Genet. (2021) 52:621–32. doi: 10.1111/age.13112
46. Palma-Granados, P, Muñoz, M, Delgado-Gutierrez, MA, Óvilo, C, Nuñez, Y, Fernández-Barroso, MA, et al. Candidate SNPs for meat quality and carcass composition in free-range Iberian pigs. Meat Sci. (2024) 207:109373. doi: 10.1016/j.meatsci.2023.109373
47. Du, SJ, Tan, X, and Zhang, J. SMYD proteins: key regulators in skeletal and cardiac muscle development and function. Anat Rec (Hoboken). (2014) 297:1650–62. doi: 10.1002/ar.22972
48. Fujii, T, Tsunesumi, S, Yamaguchi, K, Watanabe, S, and Furukawa, Y. Smyd3 is required for the development of cardiac and skeletal muscle in zebrafish. PLoS One. (2011) 6:e23491. doi: 10.1371/journal.pone.0023491
49. Tsutsumi, M, Claus, TH, Liang, Y, Li, Y, Yang, L, Zhu, J, et al. A potent and highly selective VPAC2 agonist enhances glucose-induced insulin release and glucose disposal: a potential therapy for type 2 diabetes. Diabetes. (2002) 51:1453–60. doi: 10.2337/diabetes.51.5.1453
50. Lutz, EM, Shen, S, Mackay, M, West, K, and Harmar, AJ. Structure of the human VIPR2 gene for vasoactive intestinal peptide receptor type 2. FEBS Lett. (1999) 458:197–203. doi: 10.1016/s0014-5793(99)01135-7
51. Hinkle, RT, Donnelly, E, Cody, DB, Sheldon, RJ, and Isfort, RJ. Activation of the vasoactive intestinal peptide 2 receptor modulates normal and atrophying skeletal muscle mass and force. J Appl Physiol (1985). (2005) 98:655–62. doi: 10.1152/japplphysiol.00736.2004
52. Asnicar, MA, Köster, A, Heiman, ML, Tinsley, F, Smith, DP, Galbreath, E, et al. Vasoactive intestinal polypeptide/pituitary adenylate cyclase-activating peptide receptor 2 deficiency in mice results in growth retardation and increased basal metabolic rate. Endocrinology. (2002) 143:3994–4006. doi: 10.1210/en.2002-220354
53. Huang, H, Li, Y, Wang, L, Song, Y, and Zhang, G. Membrane proteomic analysis identifies the polarity protein PARD3 as a novel antiviral protein against PEDV infection. J Proteome. (2022) 253:104462. doi: 10.1016/j.jprot.2021.104462
54. Jung, SH, Yoo, EH, Yu, MJ, Song, HM, Kang, HY, Cho, JY, et al. ARAP, a novel adaptor protein, is required for TCR Signaling and integrin-mediated adhesion. J Immunol. (2016) 197:942–52. doi: 10.4049/jimmunol.1501913
55. Takeshita, S, Moritani, M, Kunika, K, Inoue, H, and Itakura, M. Diabetic modifier QTLs identified in F2 intercrosses between Akita and a/J mice. Mamm Genome. (2006) 17:927–40. doi: 10.1007/s00335-005-0130-z
56. Kose, H, Bando, Y, Izumi, K, Yamada, T, and Matsumoto, K. Epistasis between hyperglycemic QTLs revealed in a double congenic of the OLETF rat. Mamm Genome. (2007) 18:609–15. doi: 10.1007/s00335-007-9031-7
57. Su, Z, Tsaih, SW, Szatkiewicz, J, Shen, Y, and Paigen, B. Candidate genes for plasma triglyceride, FFA, and glucose revealed from an intercross between inbred mouse strains NZB/B1NJ and NZW/LacJ. J Lipid Res. (2008) 49:1500–10. doi: 10.1194/jlr.M800053-JLR200
58. Moradian, H, Esmailizadeh Koshkoiyeh, A, Mohammadabadi, M, and Asadi, FM. Whole genome detection of recent selection signatures in Sarabi cattle: a unique Iranian taurine breed. Genes Genomics. (2020) 42:203–15. doi: 10.1007/s13258-019-00888-6
59. Guo, D, Zhou, Y, Wei, X, Zhang, S, Jin, T, Zhang, Y, et al. Preliminary study of genome-wide association identifies novel susceptibility genes for serum mineral elements in the Chinese Han population. Biol Trace Elem Res. (2022) 200:2549–55. doi: 10.1007/s12011-021-02854-4
60. Silver, M, Chen, P, Li, R, Cheng, CY, Wong, TY, Tai, ES, et al. Pathways-driven sparse regression identifies pathways and genes associated with high-density lipoprotein cholesterol in two Asian cohorts. PLoS Genet. (2013) 9:e1003939. doi: 10.1371/journal.pgen.1003939
61. Bovo, S, Mazzoni, G, Bertolini, F, Schiavo, G, Galimberti, G, Gallo, M, et al. Genome-wide association studies for 30 haematological and blood clinical-biochemical traits in large white pigs reveal genomic regions affecting intermediate phenotypes. Sci Rep. (2019) 9:7003. doi: 10.1038/s41598-019-43297-1
Keywords: Wanbei pig, Asian wild boar, copy number variation (CNV), selection signature, whole genome resequencing
Citation: Zhang W, Xu C, Zhou M, Liu L, Ni Z, Su S and Wang C (2024) Copy number variants selected during pig domestication inferred from whole genome resequencing. Front. Vet. Sci. 11:1364267. doi: 10.3389/fvets.2024.1364267
Edited by:
Ning Song, Anhui Agricultural University, ChinaReviewed by:
Zhong Xu, Hubei Academy of Agricultural Sciences, ChinaGuoqiang Yi, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, China
Copyright © 2024 Zhang, Xu, Zhou, Liu, Ni, Su and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chonglong Wang, ahwchl@163.com; Shiguang Su, ssg92@163.com
†These authors have contributed equally to this work