HIV- Bidirectional Encoder Representations From Transformers: A Set of Pretrained Transformers for Accelerating HIV Deep Learning Tasks
 Will Dampier1,2,3,4*
Will Dampier1,2,3,4*  Robert W. Link1,2,3
Robert W. Link1,2,3  Joshua P. Earl1,5 Mackenzie Collins1,2,3
Joshua P. Earl1,5 Mackenzie Collins1,2,3  Diehl R. De Souza1,2,3
Diehl R. De Souza1,2,3  Kelvin Koser1,2,3,6
Kelvin Koser1,2,3,6  Michael R. Nonnemacher1,2,3,4,7
Michael R. Nonnemacher1,2,3,4,7  Brian Wigdahl1,2,3,4,7*
Brian Wigdahl1,2,3,4,7*- 1 Department of Microbiology and Immunology, Drexel University College of Medicine, Philadelphia, PA, United States
- 2Center for Neurovirology and Translational Neuroscience, Institute for Molecular Medicine and Infectious Disease, Drexel University College of Medicine, Philadelphia, PA, United States
- 3Center for Pathogenic Emergence and Bioinformatics, Institute for Molecular Medicine and Infectious Disease, Drexel University College of Medicine, Philadelphia, PA, United States
- 4Center for Clinical and Translational Research, Institute for Molecular Medicine and Infectious Disease, Drexel University College of Medicine, Philadelphia, PA, United States
- 5Center for Genomic Sciences, Institute for Molecular Medicine and Infectious Disease, Drexel University College of Medicine, Philadelphia, PA, United States
- 6Department of Biochemistry and Molecular Biology, Drexel University College of Medicine, Philadelphia, PA, United States
- 7Sidney Kimmel Cancer Center, Thomas Jefferson University, Philadelphia, PA, United States
The human immunodeficiency virus type 1 (HIV-1) is a global health threat that is characterized by extensive genetic diversity both within and between patients, rapid mutation to evade immune controls and antiretroviral therapies, and latent cellular and tissue reservoirs that stymie cure efforts. Viral genomic sequencing has proven effective at surveilling these phenotypes. However, rapid, accurate, and explainable prediction techniques lag our sequencing ability. Modern natural language processing libraries, like the Hugging Face transformers library, have both advanced the technical field and brought much-needed standardization of prediction tasks. Herein, the application of this toolset to an array of classification tasks useful to HIV-1 biology was explored: protease inhibitor resistance, coreceptor utilization, and body-site identification. HIV-Bidirectional Encoder Representations from Transformers (BERT), a protein-based transformer model fine-tuned on HIV-1 genomic sequences, was able to achieve accuracies of 88%, 92%, and 89% on the respective tasks, making it competitive with leading models capable of only one of these tasks. This model was also evaluated using a data augmentation strategy when mutations of known function were introduced. The HIV-BERT model produced results that agreed in directionality 10- to 1000-fold better than traditional machine learning models, indicating an improved ability to generalize biological knowledge to unseen sequences. The HIV-BERT model, trained task-specific models, and the datasets used to construct them have been released to the Hugging Face repository to accelerate research in this field.
1 Introduction
The human immunodeficiency virus type 1 (HIV-1) is a global health threat that impacts millions of people worldwide. This lentivirus primarily infects the immune system by attaching to the CD4 receptor, entering the cell, integrating into the genome, and then producing viral copies to infect new cells, a process reviewed in greater detail by Mailler et al. (1). Antiretroviral therapy has proven to be an effective treatment for individuals; however, the error-prone replication process leads to rapid diversification of the virus and eventual resistance through the accumulation of mutations which impact the efficacy of the treatment (2, 3). This rapid evolution also allows the virus to evade host antibodies and invade new niches through the changes to the viral envelope and accessory proteins (4–7).
These phenotypes have drastic impacts on patient morbidity and mortality (5, 8, 9). Given their selective advantage, strains containing drug resistant mutations (DRMs) with low fitness requirements tend to proliferate within infected individuals and have been shown to drastically increase the potential for virologic failure (10, 11). At present, there are five classes of HIV-1 antiretroviral therapies: nucleoside reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), integrase inhibitors (INIs), and entry inhibitors (12, 13). Indeed, DRMs have been identified corresponding to each class of inhibitors, albeit at different proportions (12).
Since these DRMs and other genotypes/phenotypes are transmitted within the proviral genome, there has been a long history of techniques for predicting these traits from genomic sequence. This manuscript has chosen three diverse tasks from the HIV-1 field across the viral lifecycle as described in Figure 1 and below:
1. Protease inhibitor resistance – The protease gene is a 99 amino acid protein responsible for cleaving viral proteins that are translated as a single polyprotein into their active form; thus making it an ideal target for antiretroviral therapy. The Stanford HIV database maintains a prediction server for annotating HIV-1 sequences with known resistance mutations (14). Investigators have also developed predictive tools such as SHIVA, a decision tree classifier (15), generative machine learning algorithms (16), and the ANRS (Agence Nationale de Recherches sur le SIDA) algorithm, a well-regarded program for predicting protease receptor and reverse transcriptase resistance (17).
2. Coreceptor utilization – HIV-1 enters human cells by first binding the CD4 receptor and then recruiting one of two possible coreceptors: CCR5 or CXCR4. This allows for the entry into two different cellular reservoirs with CCR5 primarily responsible for T-cell infection and CXCR4 for macrophages (4, 18). This phenotype is primarily due to the 3rd variable loop of the envelope gene, the V3 loop. This 35 amino acid loop mediates the interaction between the envelope protein and coreceptor primarily at positions 13-21 (19). The earliest methods of coreceptor prediction used alignment-based methods to create a position-specific scoring matrix (PSSM) (20). Newer methodologies like decision trees (21) and XGboost (22) have been applied with great success.
3. Bodysite identification – Due to the ability of HIV-1 to infect immune cell populations, it spreads across the body rapidly and adapts to new compartments (23–26). When exploring HIV-1 cure strategies such as “shock & kill” and anti-HIV CRISPR-Cas9 gene editing, it is important to surveil these compartments (27–29). This task is traditionally tackled using phylogenetic methods in which the evolutionary relationship between sequences isolated from different bodysites is explored (30–32). In our review of the relevant literature this prediction task has not been previously attempted.
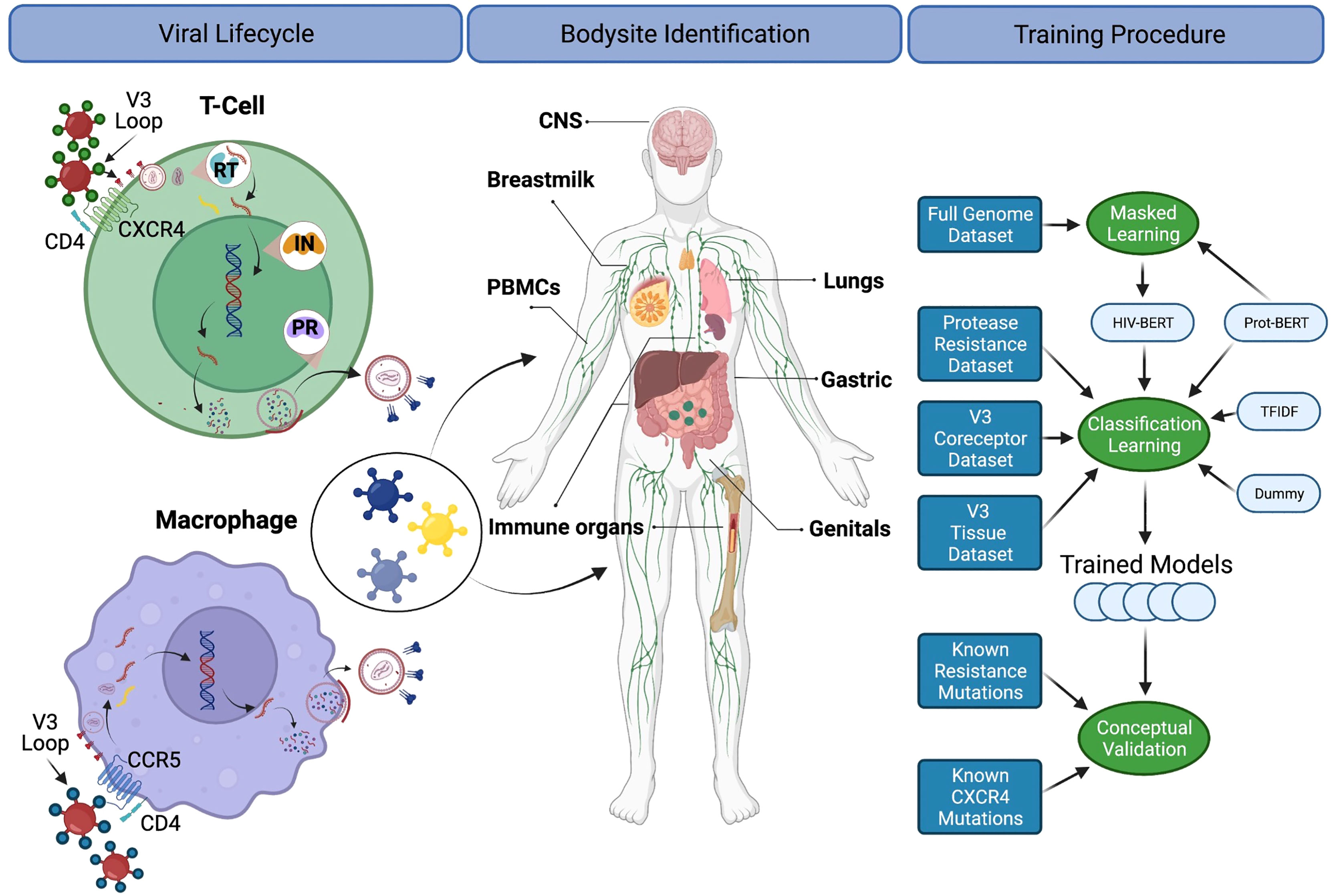
Figure 1 A diagram for the biological processes explored. The left column depicts the basic viral lifecycle starting with entry through the binding of CD4 and cell-specific coreceptors, followed by unpackaging of the capsid, reverse transcription of viral RNA, integration into the host genome, subsequent translation of viral proteins and repackaging. Steps where antiretroviral medications interfere with the lifecycle are also noted. The middle panel indicates bodysites that are known to be anatomic reservoirs of latent viral replication. The right panel shows a flow diagram of the experiments and training procedures described in the text. Figure created with BioRender.com.
Using sequence data for machine learning (ML) techniques poses multiple challenges. First, most ML techniques require numeric inputs. This requires a conversion of the amino acid sequence into a numeric form that can be consumed by downstream algorithms. Techniques for doing this can be grouped into two categories: alignment-based and alignment-free.
Alignment-based methods require using alignment tools like MUSCLE (33), T-coffee (34), or minimap2 (35) to associate each position in the query with a consistent column. This is critical when using machine learning methods like SVM, decision trees, or other classic techniques as they assume that each column consistently represents the same physical property. Once aligned, a feature matrix can be constructed by converting each column of the alignment into a number, or set of numbers, either through one-hot encoding or encoding physiochemical properties of each amino acid at each position. This matrix can then be used like any other ML dataset.
However, the rapid mutation of HIV-1 leads to difficulties with alignment-based methods as the insertion and deletion rate of the virus leads to alignments with copious gaps. Alignment-free methods sidestep this problem by summarizing a protein of any length into a fixed length vector. Tools like MathFeature (36) and ProtDCal (37), perform this through calculation of physiochemical properties of the protein. However, this presupposes that these properties are relevant to the prediction task. K-mer based approaches count occurrences of fixed-length subsequences of the protein. This generates a fixed-length vector for each protein that compares the presence, absence, or quantity of short amino acid sequences. However, this requires one to balance the length of the k-mer with the sparsity of the downstream dataset. As each k-mer is a distinct feature in the matrix, it also does not account for the similarities between amino-acids.
Many of these problems are also present in related fields like natural language processing (NLP): not all sentences are the same length, words have different meanings in different contexts, and similar words can be used interchangeably. This has spurred a great deal of crosstalk between the NLP field and the genomic language processing (GLP) field (38, 39). Of particular interest to this manuscript are advances in artificial intelligence (AI) methods, particularly the Transformer models (40, 41). While the initial investment is large (42), once trained, models can be reused on multiple tasks (43–45).
In 2021 a high-performance computing group led by Burkhard Rost set out to leverage Summit, the world’s second fastest computer, to accelerate GLP research. They leveraged 1096 GPU containing nodes to train a Bidirectional Encoder Representations from Transformers (BERT) model on a dataset of 393 million amino acids from Uniprot50, Uniref100, and the Big Fantastic Database (BFD) of human isolated metagenomic proteomes (46). This model architecture considers the entire protein at once (Bidirectional) and encodes proteins into fixed length vectors (Encoder Representation) for downstream predictions. Like all transformer style language models, it can be trained on unlabeled data for many tasks through a technique called Masked Language Modeling (MaskedLM). In this process, a subset of amino acids are ‘masked’ from the model during training and it is tasked with predicting them and its weights are updated through gradient back-propagation. Accomplishing this task pretrains the model for future tasks. The Rost-Lab showed that this model could be further refined in a process called “transfer learning” to predict new tasks such as subcellular localization, secondary structure, and enzyme activity (46).
This group released their prediction models into the Hugging Face Model Repository for open-source use (https://huggingface.co/models). The Hugging Face transformer library is a Python-based library for implementing state of the art AI models in a consistent, reproducible, and extensible fashion (47). Pretrained models can be downloaded with a single command and applied to new data with another Hugging Face transformer pipeline (47). The library also provides an interface for refining the pretrained models on new data (48). These studies reported herein applies these tools to the three HIV-1 prediction tasks described above. It explores the effects of pre-training, class weighting, and dataset size on the task. It also releases the models and datasets to the Hugging Face dataset and model hub for the community at large to accelerate future work. These studies also provide advice and expectations on adapting this to other applicable tasks.
2 Materials and Methods
The entire analysis pipeline was implemented as a Snakemake pipeline and is available on the public Github repository (49). This allows any researcher to download, reproduce, transform, or extend this analysis. The deep learning aspect of this pipeline requires access to sufficient computational resources. While the script auto-scales to the user’s available memory the training of the transformer models require at least 6GB of GPU RAM. Once trained, the models can be used on generally available hardware.
2.1 Explanation of Public Datasets
2.1.1 HIV-1 Full Genome Dataset
The Los Alamos National Laboratory HIV sequence (LANL) database maintains a standard dataset of 1,690 high-quality full-length genomes. The most recent version was downloaded (2016-Full-genome) on 12/21/2021 (https://www.hiv.lanl.gov). This was then processed using the LANL GeneCutter tool to extract and splice the DNA sequence of each gene (https://www.hiv.lanl.gov/content/sequence/GENE_CUTTER/cutter.html). The GeneCutter tool aligns the query to a gold-standard codon aligned multiple-sequence alignment using HMMER v 2.32. This ensures that any gene within the region is properly extracted in the proper reading-frame. These extracted DNA sequences were then translated into the appropriate protein sequence to the first stop-codon using the BioPython library Seq.translate function with a human codon table (50). The database was processed using the script workflow/scripts/process_lanl_flt.py.ipynb and deposited as a Hugging Face Dataset damlab/HIV_FLT.
2.1.2 Protease Drug Resistance
The high-quality interactions from the Stanford HIV Genotype-Phenotype database (51) were downloaded on 12/21/2021 and contained 1959 lines at the time of download. The file stores the amino-acid differences from the provided reference sequence, for example V30I|D46N, and an array of drug-susceptibility scores for each isolate. This was converted into a Hugging Face dataset by inferring the full protease sequence by exchanging the indicated amino acids from the pre-translated reference sequence and labeling any drug with a >4-fold increase in resistance as “True” conforming with the methodology described by Rhee et al. After filtering the dataset to only include the drugs FPV, IDV, NFV, and SQV, and dropping all missing items, there were 1733 total PR sequences for analysis with roughly half being resistant to at least one drug. The database was processed using the script workflow/scripts/process_stanford_pr.py.ipynb and deposited as a Hugging Face Dataset damlab/HIV_PI.
2.1.3 Coreceptor Tropism
V3-loop sequences were downloaded from the LANL database through the Search Interface (https://www.hiv.lanl.gov/components/sequence/HIV/search/search.html) on 12/20/21. The query was generated by limiting the Subtypes to A, B, C, and D; selecting “list in field output” for the Coreceptor and Sample Tissue fields; and selecting V3 in the Genomic Region Selection box. This generated approximately 220,000 results at the time of search. The LANL search tools were used to align, trim, and return the selected sequences with the associated background info. The background information was parsed to create an independent binary variable each for CCR5 and CXCR4 binding status. This search generated 2935 sequences with 91% being CCR5 tropic and 23% labeled as CXCR4 tropic. The database was processed using the script workflow/scripts/process_lanl_v3.py.ipynb and deposited as a Hugging Face Dataset damlab/HIV_V3_coreceptor.
2.1.4 Bodysite Identification
Using the same V3 dataset mentioned above, the Sample Tissue field was aggregated so similar bodysites were grouped together. The grouping was performed as follows:
● Periphery-tcell: plasma, PBMC, T cells, CD4+ T cells, resting CD4+ T cells, effector memory CD4+ T cells, transitional memory T cells, central memory T cells, serum, blood, lymph node, CD4+ T cell supernatant, lymph node CD4+ T cells, CD14+ monocytes, activated CD4+ T cells, naive CD4+ T cells, effector memory T cells, T-cell, CD8+ T cells, PMBC, PBMC supernatant, stem memory T cells, terminally differentiated T cells
● Periphery-monocyte: lamina propria mononuclear cells, CD14+ monocytes, monocyte, CD16+ monocytes
● CNS: brain, CSF, spinal cord, dendrites
● Lung: lung, BAL, sputum, diaphragm
● Breast-milk: breast milk
● Gastric: colon, rectum, jejunum, ileum, GALT, rectal fluid, intestine, feces, stomach, choroid plexus, sigmoideum, gastric aspirate, esophagus
● Male-genitals: semen, seminal plasma, foreskin, seminal cells, urethra, prostate, testis, prostatic secretion
● Female-genitals: vaginal fluid, cervix, vagina, vaginal cells, cervicovaginal secretions
● Umbilical-cord: umbilical cord plasma, placenta
● Organ: liver, kidney, epidermis, thymus, pancreas, adrenal gland, spleen, bone marrow
● Dropped sequence: supernatant, saliva, urine, meninges, skin tumor, qVOA, urine cells, breast milk supernatant, aorta, glioma
Then, sequences were grouped such that each unique sequence was annotated with all bodysites it was associated with. This allows a sequence to be annotated with multiple bodysites. Due to the over-representation of periphery-tcell tags, a random 95% were discarded. After processing, there were 5510 unique V3 sequences annotated with about half from the periphery-tcell tag and the rest ranging from 5-20% composition. The database was processed using the script workflow/scripts/process_lanl_v3.py.ipynb and deposited as a Hugging Face Dataset damlab/HIV_V3_bodysite.
2.2 Models
Four models were constructed to evaluate the real-world performance on these prediction tasks. The datasets above were split using 5-fold cross-validation with the folds preserved across the different model trainings. This allows for an honest comparison of each fold across each model.
2.2.1 Naive Models
First, a Dummy model was created using the sklearn.dummy.DummyClassifier class with stratified strategy. This model represents randomly guessing utilizing the known class distribution of the training data. For example, 91% of V3 sequences are CCR5 tropic, therefore this model will guess True 91% of the time. This naïve model represents the lowest reasonable bar to set for prediction tasks.
Next, a basic Random Forest Classifier was used as a biology naive model machine learning model. The variable length sequences were encoded into fixed length vectors using term frequency-inverse document frequency (TF-IDF). This technique creates a feature for each N-gram present in the database and encodes proteins by the sum of the number of times the N-gram occurs in the sequence divided by the number of times it occurs in the database. Sequences, V3 or PR, were transformed with the sklearn.feature_extraction.text.TfidfVectorizer class using a ngram_range = (1,3) and analyzer=‘char’. This creates a vector of each k-mer sized 1-3 with the count normalized by the inverse of its prevalence in the rest of the training dataset. This naturally highlights k-mers that are over-represented in the sequence. After a basic variance threshold to remove invariant columns, a classifier was built with the sklearn.ensemble.RandomForestClassifier class using the default parameters. This model represents a purely mathematical approach to the prediction problem. These models were trained using the workflow/scripts/sklearn_train.py script using scikit-learn version 1.0.2.
2.2.2 Transformer Models
Biologically informed Transformer models were imported using the Hugging Face AutoModelForSequenceClassification tool. The Hugging Face library provides a Trainer module for refining pretrained models on new datasets. However, the default Trainer cannot accommodate multi-label prediction problems like those posed here. As such, a CustomTrainer class was developed per recommendations in a related forum post (52) and documentation (53). In brief, there were three changes implemented. First, the CategoricalCrossEntropy loss function was replaced with a BinaryCrossEntropy function; this allows the model to predict multiple True values in each field (i.e. a protease sequence can be resistant to zero, one, or potentially all drugs). Second, as the class labels are not equally distributed for each field, 91% of V3 sequences are CCR5 tropic weighting was used to balance the imbalanced classes as described in the BCELoss documentation. Finally, the loss from each field of the prediction was added together in equal weights. This CustomTrainer is implemented in the module workflow/scripts/common.py. Training was performed using the same parameters across all models: learning rate of 1E-5, 50K warm-up steps, and a cosine_with_restarts learning rate schedule and continued until 3 consecutive epochs did not improve the validation loss metric.
For these experiments, the Prot-Bert-BFD model from the RostLab was used as the basis for pretraining (46), commit-tag 6c5c8a5. It has been trained across a wide array of proteins and is easily available in the Hugging Face library. This pre-trained model was used as the initial weights for training each of the three models described above. This is implemented in the script workflow/scripts/huggingface_train.py.
Previous research has shown that refining language models on domain-specific sequence can improve downstream performance (43–45). Using the whole genome sequence data described above, the Prot-BERT-BFD model was refined using MaskedLM training. In this task, a random set of amino acids are masked from the model, which is then asked to predict them. A training script was adapted from the HuggingFace transformers run_mlm.py script (54). Pre-training was performed by concatenating all protein sequences from the Full HIV Genome Dataset described above and chunked into 256 amino acid segments. 80% of the sequences were used for training and 20% were reserved for validation. This is implemented in the script workflow/scripts/huggingface_lm.py using transformers v4.15.0.
2.3 Conceptual Error
Conceptual error was calculated using a set of reference mutations known from other sources of information to induce a known effect. It relies on our knowledge of the structure-to-function relationships involved in these processes and quantifies the biological “unexpectedness” of a model’s output.
Protease resistance conceptual error was measured by generating a dataset of known protease inhibitor resistance mutations. Utilizing the Stanford HIV database mutation explorer, 10 mutations were identified as inducing a >4-fold increase in drug resistance when acting alone: D30N, V32I, M46I, M46L, G48V, I54V, V82F, I84C, N88S and L90M. For each sequence in the testing dataset of each fold, they were examined for these mutations. If absent, they were introduced; if present they were removed. Then, the original and altered sequences were inferred by each trained model. The logit output of each pair was subtracted and squared. Then, the squared-error for pairs which introduced resistance mutations that increased resistance were set to zero; as were pairs which removed resistance mutations that decreased resistance. The resulting masked vector was then averaged. This error represents the mean-squared error change in the unexpected direction.
V3 coreceptor prediction can also be interrogated in this way. Structural alignments by Fouchier et al. (55) have demonstrated an 11/24/25 rule: if there is a positive amino acid at positions 11, 24, or 25 of the V3 loop then it can bind to CXCR4, otherwise it binds to CCR5. Utilizing the same switching and error calculation strategy described above, pairs of sequences were made with S11H, S11R, S11K, G24H, G24R, G24K, E25H, E25R, and E25K.
As there are no known structure-function relationships that can be used as a gold-standard for bodysite identification, it was excluded from the conceptual error calculations.
2.4 Statistical Comparisons
As each prediction task used the same 5-fold cross-validation scheme, it is possible to compare different models trained on the same 80% and evaluated on the last 20%. For this reason, a paired t-test provides the most power to evaluate differences. For each comparison below, a paired t-test was implemented with scipy.stats.ttest_rel (v1.7.3). As these pairwise comparisons result in many tests per experiment, a Bonferroni correction was used to correct for false positives. A corrected p<0.05 was used as a threshold for declaring statistical significance.
3 Results
3.1 Dataset Release
This manuscript publicly releases four HIV-focused datasets. These have been prepared for other researchers by conforming to the Hugging Face Dataset Hub style. This allows the datasets to be downloaded using a simple command like:
from datasets import load_dataset
hiv_dataset = load_dataset(‘damlab/HIV_FLT’)
which are then available as high-speed data objects for downstream use.
As shown in Figure 2A, the whole genome dataset contains a mixture of genes of the correct length as well as those with premature stop codons, with 33.3% of genomes contained at least one gene with a premature stop-codon. When concatenated, this dataset contains 3.9 million characters, approximately 1% of the size of the original BFD training dataset of 393 million characters (46). The classification datasets are independent from the genome dataset as these full-genomes lack drug-resistance, coreceptor binding type, or tissue isolation information. As such, three datasets have been prepared for the three classification tasks relevant to HIV biology. Figure 2B shows the prevalence of drug resistance in Protease sequences across four drugs from the Stanford HIV Database. Out of the 1733 Protease sequences with known drug resistance, 55.8% of sequences have resistance to at least one drug, while 28.0% have resistance to all four. Figure 2C describes the profile of body-sites where 5510 unique V3 sequences have been isolated with 28.3% isolated from multiple locations. A partially overlapping set of 2935 V3 sequences contained coreceptor information with the majority being CCR5 binding 92.1%, 23.9% CXCR4 binding, and 16.0% dual tropic as shown in Figure 2D. Over 200,000 V3 sequences were discarded as having neither body-site nor coreceptor information.
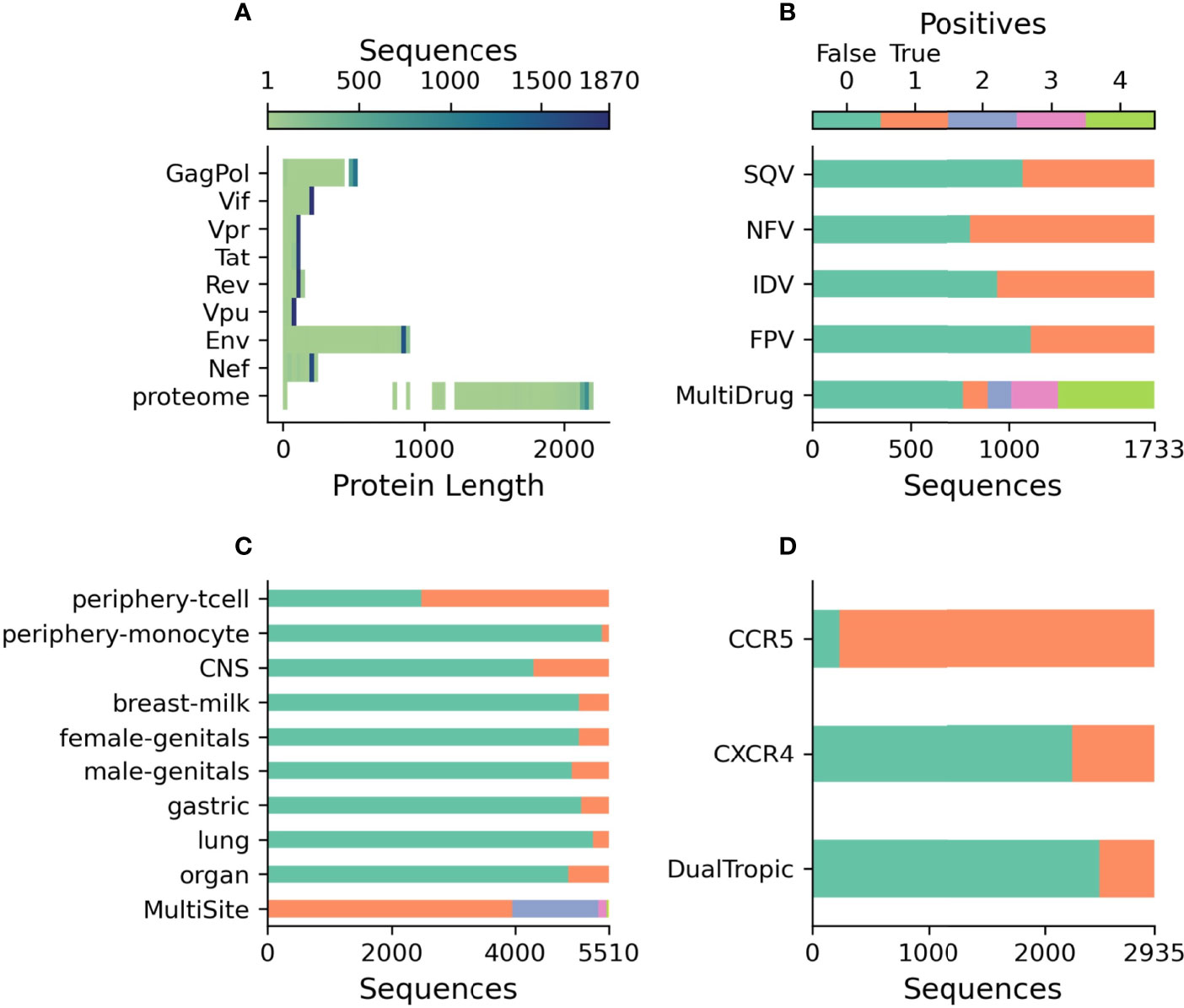
Figure 2 Description of publicly released datasets. (A) The length of each translated sequence is shown as a heatmap with darker regions indicating a greater concentration of sequences at that length. The proteome represents the total length of all translated sequences. (B) The number of protease sequences with observed resistance (orange) and no resistance (green) to each of four drugs. MultiDrug resistance represents the sum of individual drug resistances and is indicated by the key above. (C) The number of V3 sequences observed at each body-site (orange) and not observed (green) to each of the grouped sites. MultiSite represents the total number of sites that a unique V3 sequence was observed in. (D) The number of V3 sequences annotated with known coreceptor usage with those able to bind the coreceptor in orange and those not able to bind the coreceptor in green. DualTropic represents sequences that were determined to utilize both coreceptors in orange and those only able to bind to one are shown in green.
3.2 Classification Tasks
Table 1 and Figure 3 show the precision, recall, and accuracy of each of the trained models when a standard 50% cutoff was used to binarize the predictions. When considering accuracy, the percentage of correctly called sequences from the validation fold, the TF-IDF model best predicted protease resistance mutations with a 91.3% accuracy, while the HIV-BERT model performed the best at coreceptor prediction and bodysite identification with a 92.5% and 89.1% accuracy respectively. However, the TF-IDF and HIV-BERT models performed within 3% mean accuracy across all tasks, which is less than 1 standard deviation when considering models across folds.
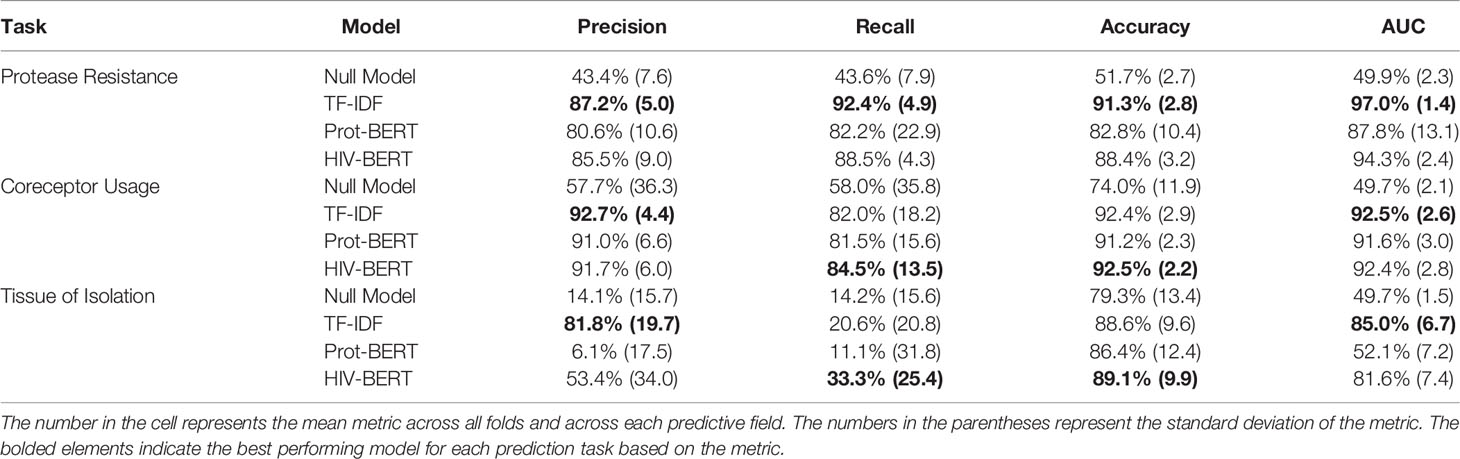
Table 1 Average model performance metrics across 5-fold cross-validation.
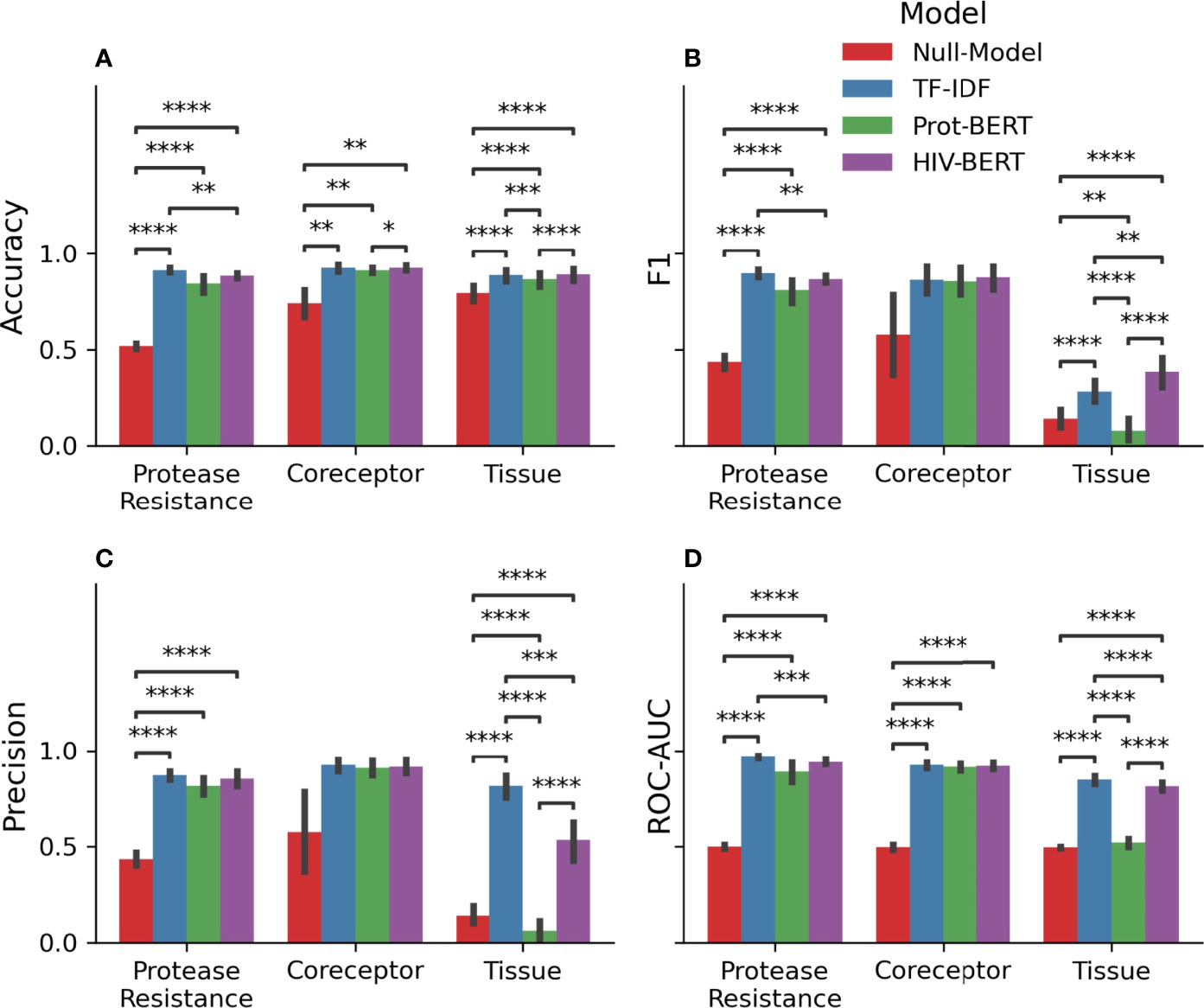
Figure 3 Pretraining improves prediction metrics across all tasks. The accuracy (A), F1-score (B), precision (C), and AUC (D) are shown for each model and each prediction task. The bar indicates the mean value and the error bars represent the 95% confidence interval of 5-fold cross validation. The Null model is shown in red, the TF-IDF model is show in blue, the Prot-BERT model in green, and the HIV-BERT model is in purple. The test-comparison bars represent the results of a paired t-test between each group; undrawn comparisons p<0.05, *(0.05<p<=0.01), **(0.01<p<=0.001), ***(0.001<p<=0.0001), ****(p<=0.00001). A Bonferroni correction based on all possible tests in the figure.
While commonly measured, accuracy may not be the ideal metric for comparing model performance in a clinical context. Depending on the situation, a researcher may wish to trade precision for recall by altering the cutoff between a positive and negative prediction. To account for this, the area under the receiver-operator characteristic curve (AUC) was calculated and shown in Table 1 and Figure 3D. Here the TF-IDF model performed the best across all tasks but again this was statistically significant in the protease resistance and tissue tasks but not coreceptor prediction.
As these tasks are multi-class predictions, it is important to examine the ability of the model to perform on each class. Figure 4 shows the accuracy of each model across each of the classes of each prediction task. When considering each model individually, there is a consistent level of prediction ability across classes with AUC scores within 5%. In the protease resistance task, TF-IDF and HIV-BERT have an increased ability to predict IDV relative to SQV (p<0.05). When examining the tissue prediction task, the TF-IDF and HIV-BERT did show differences across tasks with breast-milk having the highest accuracy and periphery-tcell having the lowest.
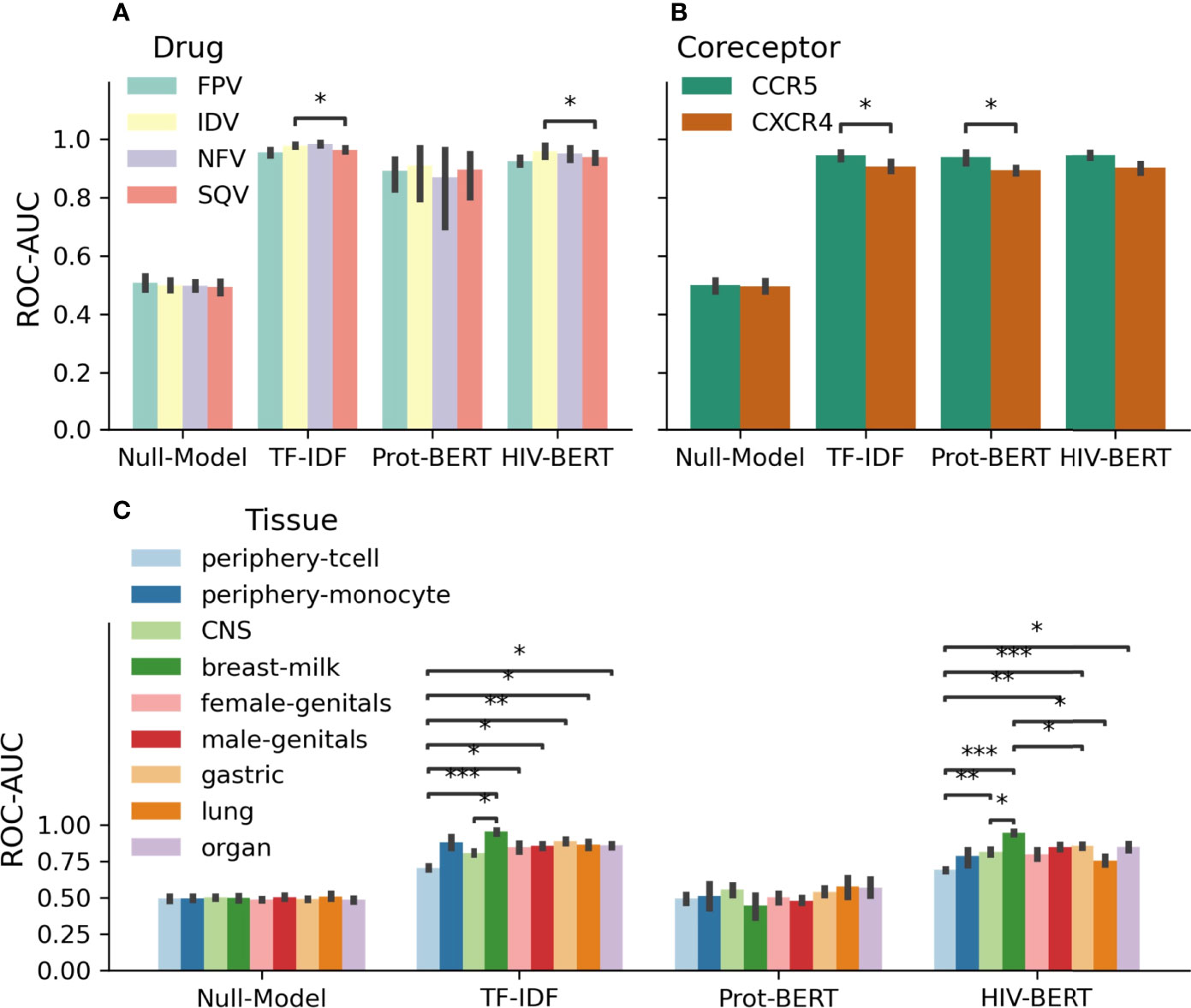
Figure 4 Area under the curve (AUC) scores for individual fields of drug resistance and coreceptor prediction are consistent, but tissue identification is not. The model AUC scores were disambiguated for each field of each prediction task. Each task is shown in (A) protease drug resistance, (B) coreceptor prediction, and (C) tissue isolation with colors indicating the prediction field. The bar indicates the mean value and the error bars represent the 95% confidence interval of 5-fold cross validation. The test-comparison bars represent the results of a paired t-test between each group; undrawn comparisons p<0.05, *(0.05<p<=0.01), **(0.01<p<=0.001), ***(0.001<p<=0.0001), ****(p<=0.00001). A Bonferroni correction based on all possible tests in the figure.
3.3 HIV-BERT Pretraining
Utilizing the full genome dataset described above, the RostLab/prot_bert_bfd model was refined for HIV specific tasks. This pretraining reduced the masked token cross-entropy loss from 1.85 nats for the unrefined model to 0.36 nats. This indicates that the average prediction for the correct amino acid improved from approximately 15% to 70%. This was visualized by subjecting the consensus subtype B V3 loop CTRPNNNTRKSIHIGPGRAFYTTGEIIGDIRQAHC (19) to single amino acid masking across all 35 positions. Figure 5 shows the difference between the unrefined model and the HIV-refined model across this masking task. The HIV-refined model has a greater predicted probability for the consensus amino acid at most positions (31/35) compared to the unrefined model.
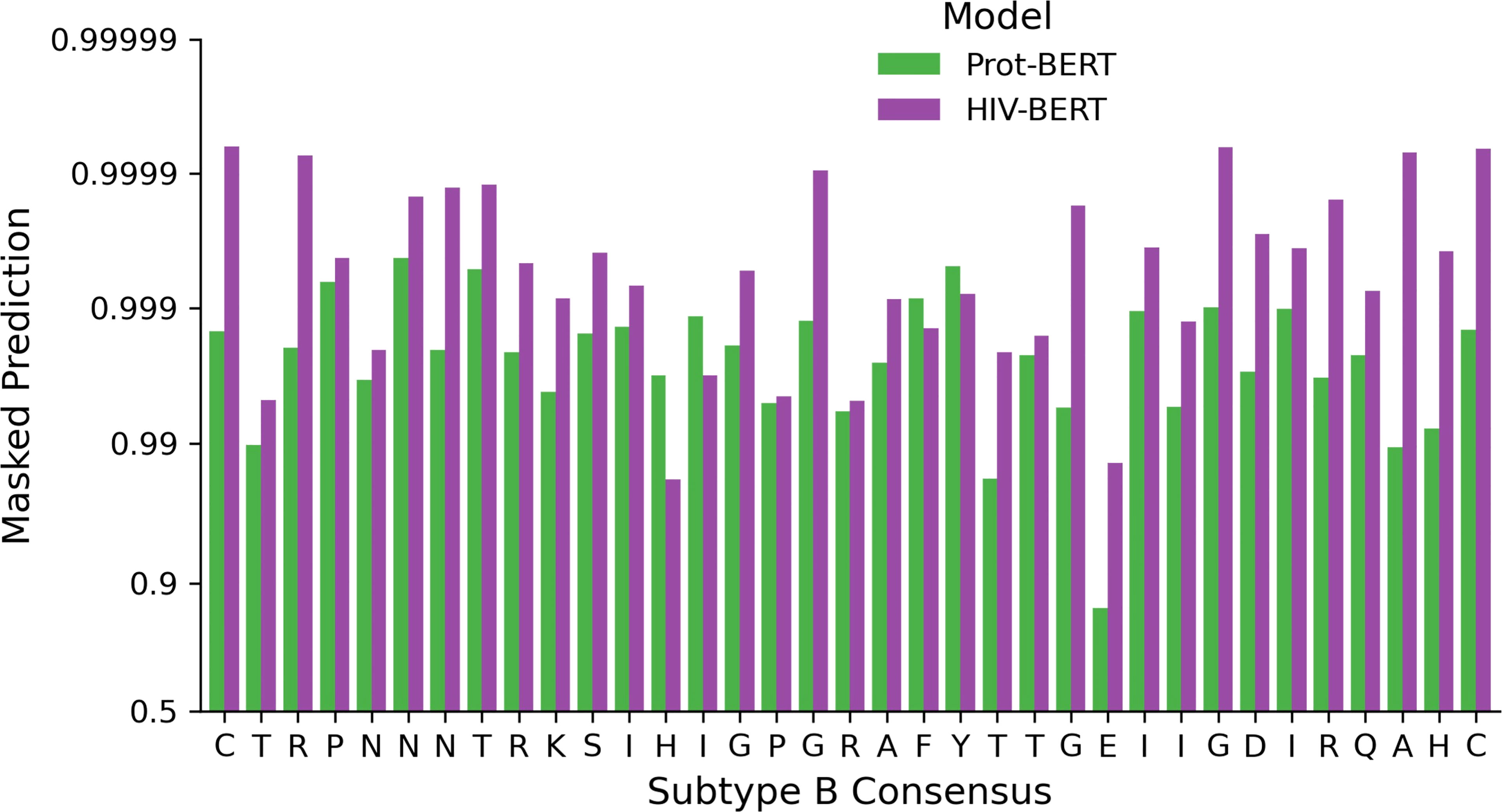
Figure 5 Full genome pretraining of the Prot-BERT model increases HIV-1 sequence awareness. The probability of each consensus amino acid of the V3 loop when performing masked prediction task. Green bars represent the prediction from the Prot-BERT model and red bars represent the HIV-BERT model.
3.4 Conceptual Error
Each sequence in the protease dataset was subjected to single amino acid mutations to either add or remove a known DRM, as listed in the methods above. Figure 6 shows the effect of those substitutions on the model’s prediction of the likelihood of drug resistance with red dots indicating a gain of a DRM and gray dots indicating a loss. A well performing model would show that an addition of a DRM (red dots) increases the resistance likelihood and would therefore be above the x=y line. The converse would be true when removing a known DRM (gray dots). When examining Figure 6, there are many instances in the TF-IDF and Prot-BERT models in which mutations mislead the prediction of the models; red dots that are below the line or grey dots that are above the line. This is noticeably less prevalent in the HIV-BERT model.
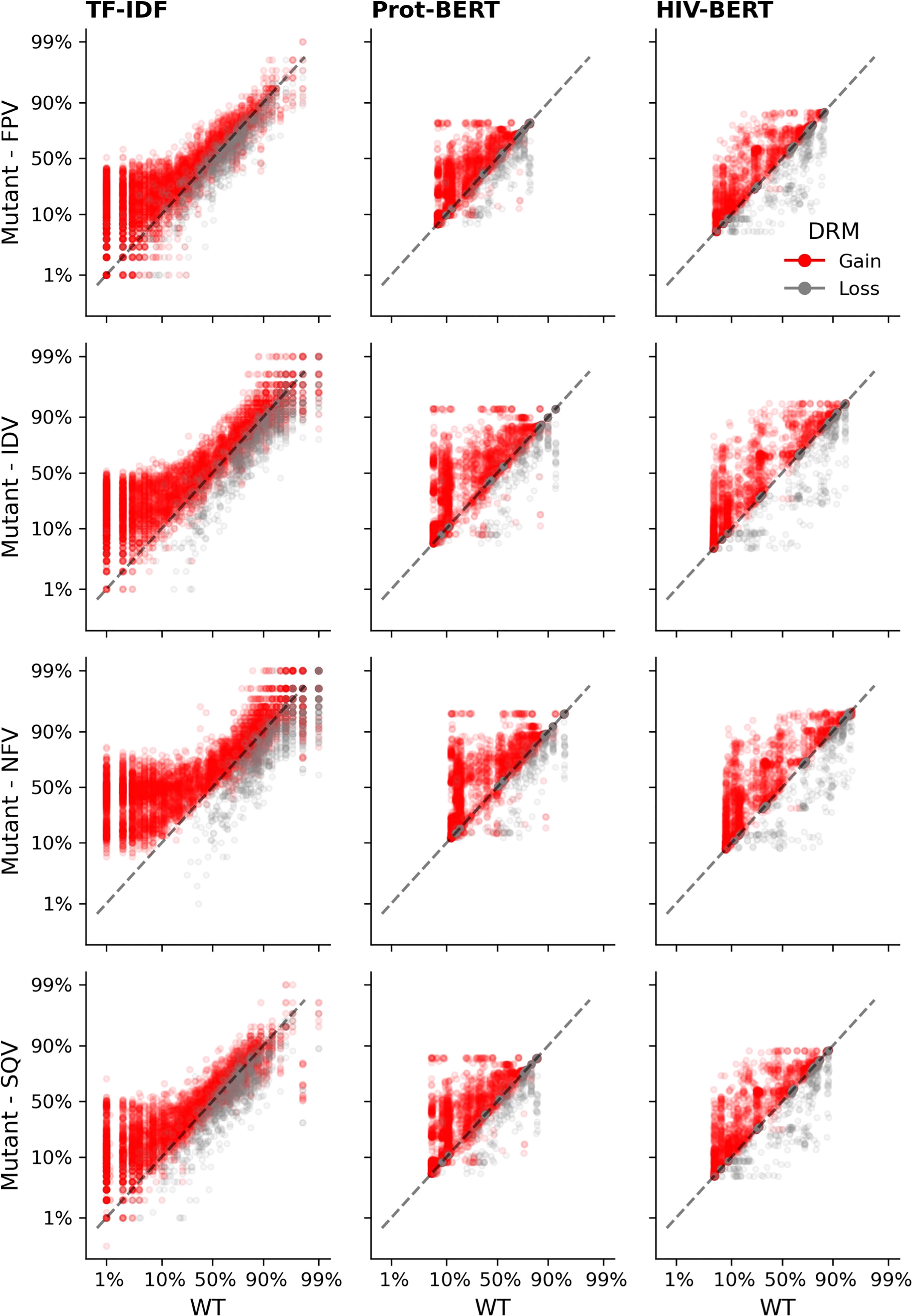
Figure 6 Transformer models accurately predict the outcome of drug resistance mutations (DRMs). Each sequence in the resistance dataset was mutated by each of the ten DRMs as described in the methods and each mutated sequence is shown as a single point; there are ten points per sequence. Sequences where a DRM was added are shown in red which should increase the probability of resistance. Points in grey indicate sequences where a DRM was removed and should decrease in probability of resistance. The dashed black line shows the x=y line and the assumption of no change due to the mutation. Each column of axes shows the predictions using each of the three models. With the first column indicating the TF-IDF model; the second indicating the Prot-BERT model; and the third showing the HIV-BERT results. Each row represents the prediction on a different drug in the order FPV, IDV, NFV, and SQV.
A similar pattern can be observed in Figure 7 when adding mutations known to increase CXCR4 binding (orange dots) and removing mutations known to indicate CXCR4 binding (green dots). The TF-IDF model also has poor confidence in classifying CXCR4 values after the addition of promoting mutations with few mutated sequences increasing past a 50% threshold. Prot-BERT and HIV-BERT do not suffer from this limitation.
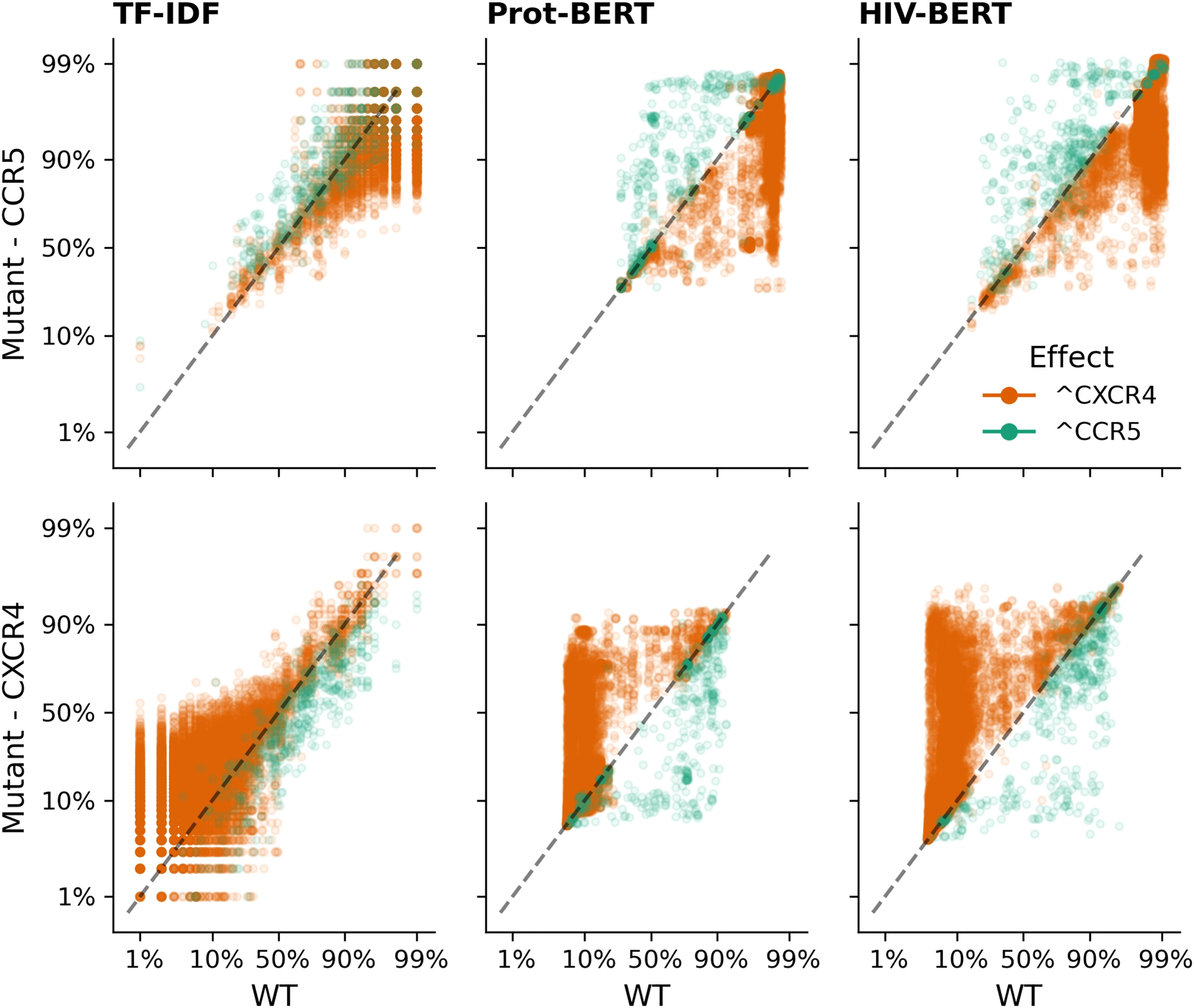
Figure 7 Transformer models accurately predict the outcome of CXCR4 enhancing mutations. Each sequence in the V3 coreceptor dataset was mutated by each of the nine CXCR4 promoting mutations as described in the methods and each mutated sequence is shown as a single point; there are nine points per sequence. Sequences where a CXCR4 promoting mutation was added is shown in orange which should increase the probability of CXCR4 binding and decrease the probability of CCR5 binding. Points in green indicate sequences where a CXCR4 promoting mutation was removed and should decrease CXCR4 binding and increase CCR5 binding. The dashed black line shows the x=y line and the assumption of no change due to the mutation. Each column of axes shows the predictions using each of the three models with the first column indicating the TF-IDF model; the second indicating the Prot-BERT model; and the third showing the HIV-BERT. Each row represents the prediction on a different coreceptor with CCR5 on the top and CXCR4 on the bottom.
In order to quantify this, a mean-squared error was calculated as described above such that only predictions in the unexpected direction were penalizing. Figure 8A shows the results of this analysis grouped by model. In the coreceptor prediction task, all models improve upon the naïve predictions but are not statistically significantly different from each other. However, for the protease resistance prediction task the Prot-BERT and HIV-BERT models are more statistically significant than the TF-IDF model but not from each other. Across all tasks, Figures 8B, C, the HIV-BERT model outperforms all other models but not to a statistically significant level.
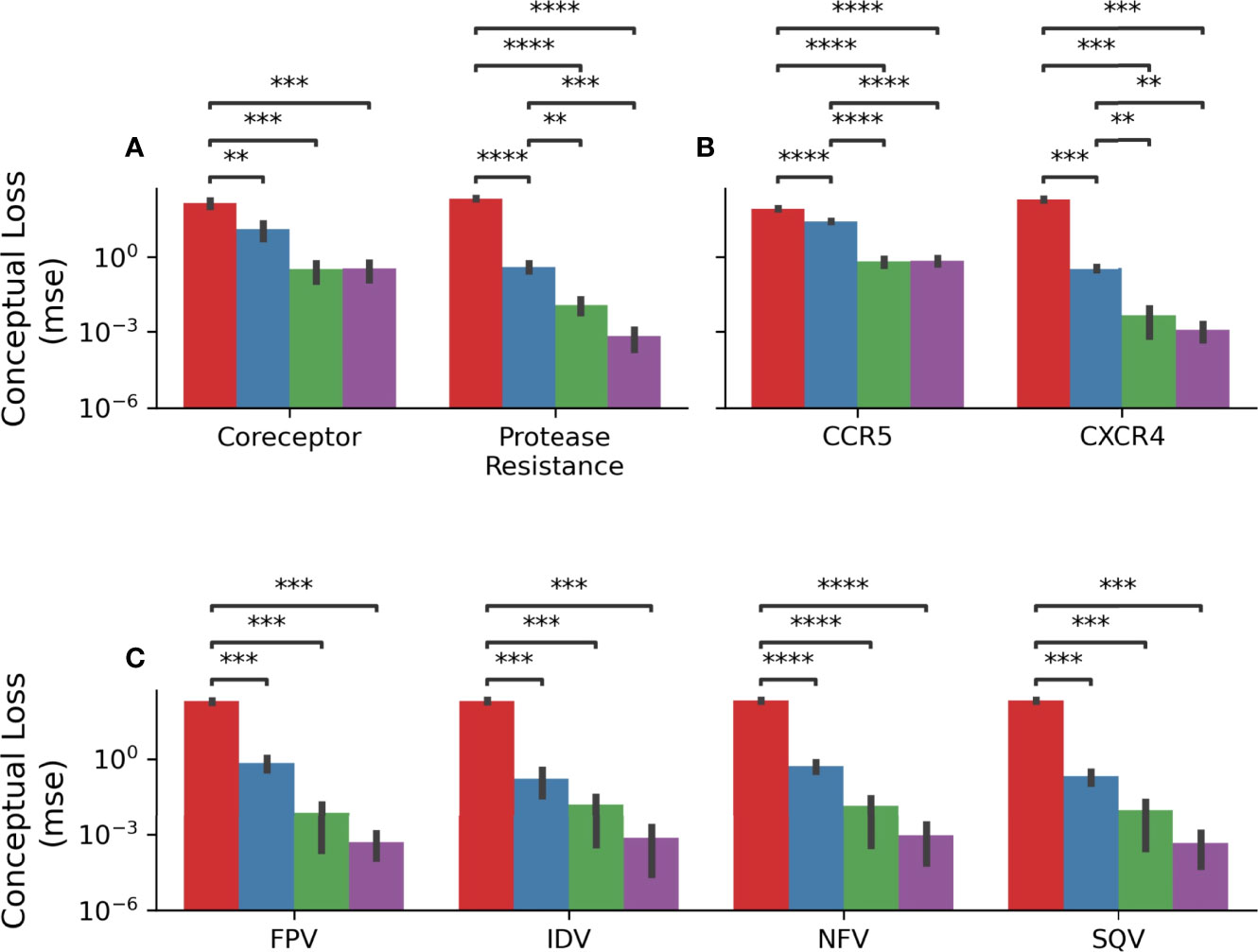
Figure 8 HIV-1 pretraining decreases conceptual error. (A) The average conceptual error of each model is shown across the coreceptor and protease resistance tasks for each model. (B) The conceptual error was disambiguated across each field of the coreceptor model. (C) The conceptual error was disambiguated across each field of the protease resistance model. Across all axes the Null model is shown in red, the TF-IDF model is shown in blue, the Prot-BERT model shown in green and the HIV-BERT model shown in purple. The test-comparison bars represent the results of a paired t-test between each group; undrawn comparisons p<0.05, *(0.05<p<=0.01), **(0.01<p<=0.001), ***(0.001<p<=0.0001), ****(p<=0.00001). A Bonferroni correction based on all possible tests in the figure.
4 Discussion
Over the past decade there has been an explosion in open-source natural language processing tools, especially in the AI field. However, biological datasets are rarely in a form amenable to easy implementation. This is particularly true of specialized datasets like those discussed in this analysis. Creating publicly accessible datasets in an easily retrieved form will help bridge the gap between AI and biological researchers. The study reported herein releases four HIV specific datasets to the public for new researchers to iterate upon. This is coupled with the release of a generically-trained HIV-BERT model as well as the three task-specific refinements discussed above. It is our hope that depositing these in the open-source Hugging Face transformers library will allow for a democratization of research and prevent issues like link-death, a common problem when attempting to build upon the work of others.
Examining previous machine learning attempts at these tasks reveals that our technique is competitive with existing methods. Early work in DRM mutation by Beerenwinkel et al. achieved sensitivities ranging from 58-92% and specificities ranging from 62-92% using decision trees across different inhibitors (56). Later work by Heider et al. was able to achieve AUCs ranging from 0.79-0.89 using chains of classifiers (57). Recent work published in 2020 by Steiner et al. tested older AI techniques like multi-layered perceptrons and recurrent networks achieving AUCs from 0.8 to 0.97 (58). The HIV-BERT model released on the Hugging Face repository has an average AUC of 0.94, making it competitive with current state of the art techniques. When examining our coreceptor prediction model we perform competitively with recent work by Chen et al. in 2019 using the XGBoost method (22).
It is important to note that none of the previous methods used the same dataset for training and validation, making direct comparison difficult. This is likely because HIV-1 sequence data is sequestered within databases that require domain specific knowledge to access and process for modern machine learning and AI tools. Our release of a standard-formatted dataset and processing scripts will help to alleviate these difficulties.
To our knowledge, the tissue identification task has never been posed as a classification problem in this manner. Previous research has used phylogenetic methods to identify the level of compartmentalization by finding mutations unique to a single tissue type when examining isolates from diverse tissues (59, 60). However, when performing routine surveillance sequencing of patients, invasive methods like lumbar punctures for CSF or bronchiolar lavage for lung sampling are impractical. Framing the problem as a classification task allows for sequencing of virus from peripheral blood to detect recent reactivation events from these latent reservoirs. This will be useful when evaluating the success of HIV cure strategies such as anti-HIV-1 CRISPR/Cas9 gene editing or latency reactivation (27).
In our analysis, a simple k-mer and tree-based prediction, TF-IDF, was able to outperform advanced AI models in many of the tasks when considering classic metrics such as accuracy and AUC. However, when it was subjected to biological investigation by introducing mutations with known function, it performed the worst. This reflects poor generalization and indicates that the TF-IDF model may be “memorizing” patterns that are correlated with the prediction task, but not causative. BERT-style models, pre-trained on millions of sequences, can distinguish between functional changes in the sequence and random mutations. This result indicates a need for detailed evaluation of prediction models to ensure that they reflect biological reality. The introduction of conceptual error described above is an initial attempt at performing this at-scale for HIV-1 tasks. Future work should expand this by incorporating deep mutational scanning datasets (61).
When examining the BERT style models across related tasks, there is a notable level of consistency across predicted fields. The AUC for each drug and coreceptor fall within 5% of each other. This may indicate that information is being shared within the model across these related fields, a common strength of transformer models. This pattern was not seen in the tissue classification task, this may indicate more sequences are needed or that other areas of the HIV-1 genome should be examined. Other published research has shown that regions across the genome have a role to play in tissue specificity and cellular tropism. It may be that the V3 loop is important for cellular tropism but the accessory proteins Vpr, Tat, and Nef may play a greater role in tissue specificity (62, 63). Future work should explore other proteins using similar prediction tasks.
The trained models have multiple biological applications. The PI- and Tropism-trained models are immediately applicable to predicting the most useful antiretroviral drug for a given patient. They can also be utilized for exploring the structure-function relationship between inhibitors and viral sequences. The bodysite prediction task allows for the tracking of leakages of strains from viral reservoirs. This may lead to new avenues of research around the exchange of viral quasispecies between compartments. Finally, the HIV-BERT model functions as a base that future HIV predictive models can be built upon.
Taken together, this work shows that AI models, particularly transformers, are well suited to biological prediction tasks. Refining the model on unlabeled sequences improves prediction accuracy with minimal upstream cost; the HIV-BERT model discussed above can be downloaded and finetuned for new prediction tasks with minimal additional effort and has been successful with fewer than 2000 labeled sequences. This puts many biologically relevant HIV-1 prediction tasks within reach and can accelerate any additional protein-to-function prediction task and will be useful to the HIV community as a whole.
Data Availability Statement
All trained models and datasets are available at https://huggingface.co/damlab. The training scripts and the code used to generate figures is available at https://github.com/DamLabResources/hiv-transformers.
Author Contributions
Conceptualization, WD; methodology, WD; software, WD; validation, WD; formal analysis, WD; investigation, WD; resources MN, BW, and WD; data curation, WD; writing—original draft preparation, WD; writing—review and editing, WD, RL, JE, MC, DS, KK, MN, and BW; visualization, WD; supervision, WD; project administration, WD; funding acquisition, MN, BW, and WD. All authors have read and agreed to the published version of the manuscript.
Funding
The authors were funded in part by the Public Health Service, National Institutes of Health, through grants from the National Institute of Mental Health (NIMH) R01 MH110360 (Contact PI, BW), the NIMH Comprehensive NeuroAIDS Center (CNAC) P30 MH092177 (KK, PI; BW, PI of the Drexel subcontract involving the Clinical and Translational Research Support Core), the Ruth L. Kirschstein National Research Service Award T32 MH079785 (PI, Tricia Burdo; BW, Principal Investigator of the Drexel University College of Medicine component) and from the National Institute of Neurological Disorders and Stroke (NINDS) R01 NS089435 (PI, MN).
Author Disclaimer
The contents of the paper are solely the responsibility of the authors and do not necessarily reflect the official views of the NIH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mailler E, Bernacchi S, Marquet R, Paillart JC, Vivet-Boudou V, Smyth RP. The Life-Cycle of the HIV-1 Gag-RNA Complex. Viruses (2016) 8(9). doi: 10.3390/v8090248
2. Mourad R, Chevennet F, Dunn DT, Fearnhill E, Delpech V, Asboe D, et al. A Phylotype-Based Analysis Highlights the Role of Drug-Naive HIV-Positive Individuals in the Transmission of Antiretroviral Resistance in the UK. AIDS (2015) 29(15):1917–255. doi: 10.1097/QAD.0000000000000768
3. Arias A, Ruiz-Jarabo CM, Escarmis C, Domingo E. Fitness Increase of Memory Genomes in a Viral Quasispecies. J Mol Biol (2004) 339(2):405–12. doi: 10.1016/j.jmb.2004.03.061
4. Aiamkitsumrit B, Dampier W, Antell G, Rivera N, Martin-Garcia J, Pirrone V, et al. Bioinformatic Analysis of HIV-1 Entry and Pathogenesis. Curr HIV Res (2014) 12(2):132–61. doi: 10.2174/1570162X12666140526121746
5. Peters PJ, Duenas-Decamp MJ, Sullivan WM, Clapham PR. Variation of Macrophage Tropism Among HIV-1 R5 Envelopes in Brain and Other Tissues. J Neuroimmune Pharmacol (2007) 2(1):32–41. doi: 10.1007/s11481-006-9042-2
6. Marino J, Maubert ME, Mele AR, Spector C, Wigdahl B, Nonnemacher MR. Functional Impact of HIV-1 Tat on Cells of the CNS and its Role in HAND. Cell Mol Life Sci (2020) 77(24):5079–99. doi: 10.1007/s00018-020-03561-4
7. Dampier W, Antell GC, Aiamkitsumrit B, Nonnemacher MR, Jacobson JM, Pirrone V, et al. Specific Amino Acids in HIV-1 Vpr are Significantly Associated With Differences in Patient Neurocognitive Status. J Neurovirol (2017) 23(1):113–24. doi: 10.1007/s13365-016-0462-3
8. Nonnemacher MR, Pirrone V, Feng R, Moldover B, Passic S, Aiamkitsumrit B, et al. HIV-1 Promoter Single Nucleotide Polymorphisms Are Associated With Clinical Disease Severity. PloS One (2016) 11(4):e0150835. doi: 10.1371/journal.pone.0150835
9. Gorry PR, Churchill M, Crowe SM, Cunningham AL, Gabuzda D. Pathogenesis of Macrophage Tropic HIV-1. Curr HIV Res (2005) 3(1):53–60. doi: 10.2174/1570162052772951
10. Wagner BG, Garcia-Lerma JG, Blower S. Factors Limiting the Transmission of HIV Mutations Conferring Drug Resistance: Fitness Costs and Genetic Bottlenecks. Sci Rep (2012) 2:320. doi: 10.1038/srep00320
11. Briones C, Domingo E. Minority Report: Hidden Memory Genomes in HIV-1 Quasispecies and Possible Clinical Implications. AIDS Rev (2008) 10(2):93–109.
12. Blassel L, Zhukova A, Villabona-Arenas CJ, Atkins KE, Hue S, Gascuel O. Drug Resistance Mutations in HIV: New Bioinformatics Approaches and Challenges. Curr Opin Virol (2021) 51:56–64. doi: 10.1016/j.coviro.2021.09.009
13. Ross L, Lim ML, Liao Q, Wine B, Rodriguez AE, Weinberg W, et al. Prevalence of Antiretroviral Drug Resistance and Resistance-Associated Mutations in Antiretroviral Therapy-Naive HIV-Infected Individuals From 40 United States Cities. HIV Clin Trials (2007) 8(1):1–8. doi: 10.1310/hct0801-1
14. Liu TF, Shafer RW. Web Resources for HIV Type 1 Genotypic-Resistance Test Interpretation. Clin Infect Dis (2006) 42(11):1608–18. doi: 10.1086/503914
15. Riemenschneider M, Hummel T, Heider D. SHIVA - A Web Application for Drug Resistance and Tropism Testing in HIV. BMC Bioinform (2016) 17(1):314. doi: 10.1186/s12859-016-1179-2
16. Pawar SD, Freas C, Weber IT, Harrison RW. Analysis of Drug Resistance in HIV Protease. BMC Bioinform (2018) 19(Suppl 11):362. doi: 10.1186/s12859-018-2331-y
17. Singh Y. Machine Learning to Improve the Effectiveness of ANRS in Predicting HIV Drug Resistance. Healthc Inform Res (2017) 23(4):271–6. doi: 10.4258/hir.2017.23.4.271
18. Gorry PR, Sterjovski J, Churchill M, Witlox K, Gray L, Cunningham A, et al. The Role of Viral Coreceptors and Enhanced Macrophage Tropism in Human Immunodeficiency Virus Type 1 Disease Progression. Sex Health (2004) 1(1):23–34. doi: 10.1071/sh03006
19. Tamamis P, Floudas CA. Molecular Recognition of CCR5 by an HIV-1 Gp120 V3 Loop. PloS One (2014) 9(4):e95767. doi: 10.1371/journal.pone.0095767
20. Jensen MA, Coetzer M, van 't Wout AB, Morris L, Mullins JI. A Reliable Phenotype Predictor for Human Immunodeficiency Virus Type 1 Subtype C Based on Envelope V3 Sequences. J Virol (2006) 80(10):4698–704. doi: 10.1128/JVI.80.10.4698-4704.2006
21. Lengauer T, Sander O, Sierra S, Thielen A, Kaiser R. Bioinformatics Prediction of HIV Coreceptor Usage. Nat Biotechnol (2007) 25(12):1407–10. doi: 10.1038/nbt1371
22. Chen X, Wang ZX, Pan XM. HIV-1 Tropism Prediction by the XGboost and HMM Methods. Sci Rep (2019) 9(1):9997. doi: 10.1038/s41598-019-46420-4
23. Borrajo A, Svicher V, Salpini R, Pellegrino M, Aquaro S. Crucial Role of Central Nervous System as a Viral Anatomical Compartment for HIV-1 Infection. Microorganisms (2021) 9(12):2537. doi: 10.3390/microorganisms9122537
24. Khan S, Telwatte S, Trapecar M, Yukl S, Sanjabi S. Differentiating Immune Cell Targets in Gut-Associated Lymphoid Tissue for HIV Cure. AIDS Res Hum Retroviruses (2017) 33(S1):S40–58. doi: 10.1089/AID.2017.0153
25. Salemi M, Rife B. Phylogenetics and Phyloanatomy of HIV/SIV Intra-Host Compartments and Reservoirs: The Key Role of the Central Nervous System. Curr HIV Res (2016) 14(2):110–20. doi: 10.2174/1570162x13666151029102413
26. Banga R, Munoz O, Perreau M. HIV Persistence in Lymph Nodes. Curr Opin HIV AIDS (2021) 16(4):209–14. doi: 10.1097/COH.0000000000000686
27. Atkins AJ, Allen AG, Dampier W, Haddad EK, Nonnemacher MR, Wigdahl B. HIV-1 Cure Strategies: Why CRISPR? Expert Opin Biol Ther (2021) 21(6):781–93. doi: 10.1080/14712598.2021.1865302
28. Stein J, Storcksdieck Genannt Bonsmann M, Streeck H. Barriers to HIV Cure. HLA (2016) 88(4):155–63. doi: 10.1111/tan.12867
29. Gantner P, Ghosn J. Genital Reservoir: A Barrier to Functional Cure? Curr Opin HIV AIDS (2018) 13(5):395–401. doi: 10.1097/COH.0000000000000486
30. Stam AJ, Nijhuis M, van den Bergh WM, Wensing AM. Differential Genotypic Evolution of HIV-1 Quasispecies in Cerebrospinal Fluid and Plasma: A Systematic Review. AIDS Rev (2013) 15(3):152–61.
31. Smit TK, Brew BJ, Tourtellotte W, Morgello S, Gelman BB, Saksena NK. Independent Evolution of Human Immunodeficiency Virus (HIV) Drug Resistance Mutations in Diverse Areas of the Brain in HIV-Infected Patients, With and Without Dementia, on Antiretroviral Treatment. J Virol (2004) 78(18):10133–48. doi: 10.1128/JVI.78.18.10133-10148.2004
32. Giatsou E, Abdi B, Plu I, Desire N, Palich R, Calvez V, et al. Ultradeep Sequencing Reveals HIV-1 Diversity and Resistance Compartmentalization During HIV-Encephalopathy. AIDS (2020) 34(11):1609–14. doi: 10.1097/QAD.0000000000002616
33. Edgar RC. MUSCLE: Multiple Sequence Alignment With High Accuracy and High Throughput. Nucleic Acids Res (2004) 32(5):1792–7. doi: 10.1093/nar/gkh340
34. Notredame C, Higgins DG, Heringa J. T-Coffee: A Novel Method for Fast and Accurate Multiple Sequence Alignment. J Mol Biol (2000) 302(1):205–17. doi: 10.1006/jmbi.2000.4042
35. Li H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics (2018) 34(18):3094–100. doi: 10.1093/bioinformatics/bty191
36. Bonidia RP, Domingues DS, Sanches DS, de Carvalho A. MathFeature: Feature Extraction Package for DNA, RNA and Protein Sequences Based on Mathematical Descriptors. Brief Bioinform (2022) 23(1). doi: 10.1093/bib/bbab434
37. Ruiz-Blanco YB, Paz W, Green J, Marrero-Ponce Y. ProtDCal: A Program to Compute General-Purpose-Numerical Descriptors for Sequences and 3D-Structures of Proteins. BMC Bioinform (2015) 16:162. doi: 10.1186/s12859-015-0586-0
38. Yandell MD, Majoros WH. Genomics and Natural Language Processing. Nat Rev Genet (2002) 3(8):601–10. doi: 10.1038/nrg861
39. Yue T, Wang H. Deep Learning for Genomics: A Concise Overview (2018) (Accessed February 01, 2018).
40. Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, et al. Attention Is All You Need (2017) (Accessed June 01, 2017). arXiv:1706.03762.
41. Zeng W, Wu M, Jiang R. Prediction of Enhancer-Promoter Interactions via Natural Language Processing. BMC Genomics (2018) 19(Suppl 2):84. doi: 10.1186/s12864-018-4459-6
42. Patterson D, Gonzalez J, Le Q, Liang C, Munguia L-M, Rothchild D, et al. Carbon Emissions and Large Neural Network Training (2021) (Accessed April 01, 2021).
43. Howard J, Ruder S. Universal Language Model Fine-Tuning for Text Classification, In: Arxiv (2018) (Accessed January 01, 2018).
44. Cohn D, Zuk O, Kaplan T. Enhancer Identification Using Transfer and Adversarial Deep Learning of DNA Sequences. bioRxiv (2018), 264200. doi: 10.1101/264200
45. Plekhanova E, Nuzhdin SV, Utkin LV, Samsonova MG. Prediction of Deleterious Mutations in Coding Regions of Mammals With Transfer Learning. Evol Appl (2019) 12(1):18–28. doi: 10.1111/eva.12607
46. Elnaggar A, Heinzinger M, Dallago C, Rehawi G, Wang Y, Jones L, et al. ProtTrans: Towards Cracking the Language of Lifes Code Through Self-Supervised Deep Learning and High Performance Computing. IEEE Trans Pattern Anal Mach Intell (2021). doi: 10.1109/TPAMI.2021.3095381
47. Wolf T, Debut L, Sanh V, Chaumond J, Delangue C, Moi A, et al. Transformers: State-of-the-Art Natural Language Processing. (2020).
48. Lhoest Q, del Moral AV, Jernite Y, Thakur A, von Platen P, Patil S, et al. Datasets: A Community Library for Natural Language Processing (2021) (Accessed September 01, 2021).
49. Koster J, Rahmann S. Snakemake-A Scalable Bioinformatics Workflow Engine. Bioinformatics (2018) 34(20):2520-2. doi: 10.1093/bioinformatics/bty350
50. Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics (2009) 25(11):1422–3. doi: 10.1093/bioinformatics/btp163
51. Rhee SY, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW. Human Immunodeficiency Virus Reverse Transcriptase and Protease Sequence Database. Nucleic Acids Res (2003) 31(1):298–303. doi: 10.1093/nar/gkg100
52. Tunstall L. Fine-Tune for MultiClass or MultiLabel-MultiClass (2021). Available at: https://discuss.huggingface.co/t/fine-tune-for-multiclass-or-multilabel-multiclass/4035/9 (Accessed 12/25/2021).
53. Paszke A, Gross S, Massa F, Lerer A, Bradbury J, Chanan G, et al. PyTorch BCELoss Documentation. Available at: https://pytorch.org/docs/stable/generated/torch.nn.BCELoss.html (Accessed 12/25/2021).
54. Thomas Wolf LD, Sanh V, Chaumond J, Delangue C, Moi A, Cistac P, et al. HuggingFace's Transformers: State-Of-the-Art Natural Language Processing (2019). Available at: https://github.com/huggingface/transformers/blob/master/examples/pytorch/language-modeling/run_mlm.py (Accessed 12/25/2021).
55. Fouchier RA, Groenink M, Kootstra NA, Tersmette M, Huisman HG, Miedema F, et al. Phenotype-Associated Sequence Variation in the Third Variable Domain of the Human Immunodeficiency Virus Type 1 Gp120 Molecule. J Virol (1992) 66(5):3183–7. doi: 10.1128/JVI.66.5.3183-3187.1992
56. Beerenwinkel N, Schmidt B, Walter H, Kaiser R, Lengauer T, Hoffmann D, et al. Diversity and Complexity of HIV-1 Drug Resistance: A Bioinformatics Approach to Predicting Phenotype From Genotype. Proc Natl Acad Sci USA (2002) 99(12):8271–6. doi: 10.1073/pnas.112177799
57. Heider D, Senge R, Cheng W, Hullermeier E. Multilabel Classification for Exploiting Cross-Resistance Information in HIV-1 Drug Resistance Prediction. Bioinformatics (2013) 29(16):1946–52. doi: 10.1093/bioinformatics/btt331
58. Steiner MC, Gibson KM, Crandall KA. Drug Resistance Prediction Using Deep Learning Techniques on HIV-1 Sequence Data. Viruses (2020) 12(5). doi: 10.3390/v12050560
59. van Marle G, Gill MJ, Kolodka D, McManus L, Grant T, Church DL. Compartmentalization of the Gut Viral Reservoir in HIV-1 Infected Patients. Retrovirology (2007) 4:87. doi: 10.1186/1742-4690-4-87
60. Sturdevant CB, Dow A, Jabara CB, Joseph SB, Schnell G, Takamune N, et al. Central Nervous System Compartmentalization of HIV-1 Subtype C Variants Early and Late in Infection in Young Children. PloS Pathog (2012) 8(12):e1003094. doi: 10.1371/journal.ppat.1003094
61. Fernandes JD, Faust TB, Strauli NB, Smith C, Crosby DC, Nakamura RL, et al. Functional Segregation of Overlapping Genes in HIV. Cell (2016) 167(7):1762–73.e12. doi: 10.1016/j.cell.2016.11.031
62. Antell GC, Dampier W, Aiamkitsumrit B, Nonnemacher MR, Pirrone V, Zhong W, et al. Evidence of Divergent Amino Acid Usage in Comparative Analyses of R5- and X4-Associated HIV-1 Vpr Sequences. Int J Genomics (2017) 2017:4081585. doi: 10.1155/2017/4081585
Keywords: HIV-1, deep learning, transformers, genetic variation, natural language processing, coreceptor tropism, compartmentalization, drug resistance
Citation: Dampier W, Link RW, Earl JP, Collins M, De Souza DR, Koser K, Nonnemacher MR and Wigdahl B (2022) HIV- Bidirectional Encoder Representations From Transformers: A Set of Pretrained Transformers for Accelerating HIV Deep Learning Tasks. Front.Virol. 2:880618. doi: 10.3389/fviro.2022.880618
Received: 21 February 2022; Accepted: 19 April 2022;
Published: 18 May 2022.
Edited by:
Manuela Sironi, Eugenio Medea (IRCCS), ItalyReviewed by:
Slim Fourati, Emory University, United StatesMoses Ekpenyong, University of Uyo, Nigeria
Copyright © 2022 Dampier, Link, Earl, Collins, De Souza, Koser, Nonnemacher and Wigdahl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Will Dampier, wnd22@drexel.edu; Brian Wigdahl, bw45@drexel.edu