 Wilma Barcellini
Wilma Barcellini Bruno Fattizzo
Bruno Fattizzo- 1UO Ematologia, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
- 2Università degli Studi di Milano, Milan, Italy
Autoimmune hemolytic anemia (AIHA) is a greatly heterogeneous disease due to autoantibodies directed against erythrocytes, with or without complement activation. The clinical picture ranges from mild/compensated to life-threatening anemia, depending on the antibody's thermal amplitude, isotype and ability to fix complement, as well as on bone marrow compensation. Since few years ago, steroids, immunesuppressants and splenectomy have been the mainstay of treatment. More recently, several target therapies are increasingly used in the clinical practice or are under development in clinical trials. This has led to the accumulation of refractory/relapsed cases that often represent a clinical challenge. Moreover, the availability of several drugs acting on the different pathophysiologic mechanisms of the disease pinpoints the need to harness therapy. In particular, it is advisable to define the best choice, sequence and/or combination of drugs during the different phases of the disease. In particular relapsed/refractory cases may resemble pre-myelodysplastic or bone marrow failure syndromes, suggesting a careful use of immunosuppressants, and vice versa advising bone marrow immunomodulating/stimulating agents. A peculiar setting is AIHA after autologous and allogeneic hematopoietic stem cell transplantation, which is increasingly reported. These cases are generally severe and refractory to standard therapy, and have high mortality. AIHAs may be primary/idiopathic or secondary to infections, autoimmune diseases, malignancies, particularly lymphoproliferative disorders, and drugs, further complicating their clinical picture and management. Regarding new drugs, the false positivity of the Coombs test (direct antiglobulin test, DAT) following daratumumab adds to the list of difficult diagnosis, together with the passenger lymphocyte syndrome after solid organ transplants. Diagnosis of DAT-negative AIHAs and evaluation of disease-related risk factors for relapse and mortality, notwithstanding improvement in diagnostic approach, are still an unmet need. Finally, AIHA is increasingly described following therapy of solid cancers with inhibitors of immune checkpoint molecules. On the whole, the double-edged sword of new pathogenetic insights and therapies has changed the landscape of AIHA, both providing enthusiastic knowledge and complicating the clinical management of this disease.
Introduction
Autoimmune hemolytic anemia (AIHA) has always been considered the simplest and most scholastic example of antibody-mediated autoimmune disease. As a matter of fact, autoantibodies (Ab) directed against erythrocytes, with or without complement (C) activation, are the main pathogenic mechanism of the disease (1). Clinically, it has long been considered a trouble-free disease, easy to treat, and with low clinical impact, compared with malignant hematologic conditions. This approach is quite similar to that of immune thrombocytopenia, which has been defined the “hematology's Cosette from Les Misérables.” More recently, AIHA has been identified as a greatly heterogeneous disease, due to several immunological mechanisms involved beyond antibodies, complement and antibody-dependent cell-mediated cytotoxicity (ADCC). Accumulating evidence demonstrates reduced CD4+ T-regs, imbalance of T-helper 1/2 cytokines, increased activity of cytotoxic CD8+ T lymphocytes, natural killer cells, and activated macrophages. More importantly, attention has grown on the pivotal role of bone marrow compensation, and on bone marrow characteristics that may reveal dyserythropoiesis, fibrosis, and clonal lymphoproliferation (1–4). Previously, steroids, immunesuppressants, and splenectomy were the mainstay of AIHA treatment (5–8). More recently, several new targeted therapies are increasingly used in the clinical practice or under development in clinical trials (7, 9). Along with new therapeutic options for patients, this growing armamentarium has complicated the clinical management of AIHA and increased the number of relapsed/refractory cases. Therefore, harnessing treatment and defining a risk-adapted therapy is an emerging unmet need. A peculiar setting is AIHA after autologous and allogeneic hematopoietic stem cell transplantation (HSCT), as well as cases described during therapy with immune checkpoint inhibitors for solid cancers (10). Finally, AIHAs may be associated with several conditions (lymphoproliferative, autoimmune and infectious diseases, immunodeficiencies, solid tumors, transplants, and drugs) where the several immunologic mechanisms are unpredictably involved (7, 11). The recent availability of next generation sequencing has improved the diagnosis of the several associated conditions, but at the same time has extended the proportion of “secondary” vs. “primary” AIHAs (4, 12). All these new insights in the pathogenesis of the disease and treatment opportunities have undoubtedly changed the landscape of AIHA.
In this review we will describe new diagnostic tools, clinical characteristics and therapeutic options of AIHA, focusing on relapsed/refractory cases, secondary forms, and AIHAs associated with HSCT or therapy with immune checkpoint inhibitors (CPIs). Moreover, we will approach the identification of risk factors for the development, clinical severity, response to therapy, and outcome of AIHA in order to start the basis for a risk-adapted therapy.
Clinical Characteristics and Classification of AIHA
The gold standard for the diagnosis of AIHA is the Coombs test or direct antiglobulin test (DAT) that enables the classification of the disease according to the isotype and thermal characteristics of the autoantibody. Warm AIHA (wAIHA), the most common type (60–70% of cases) is typically DAT positive for anti-IgG, or IgG plus C, while cold forms (cold agglutinin disease, CAD, 20–25%), are due to IgM, and the DAT is positive for C3d. Among cold AIHAs it is worth considering paroxysmal cold hemoglobinuria (PCH), usually observed in children and; this very rare type of AIHA (1–5% of cases) is caused by the Donath-Landsteiner autoantibody, a bithermic hemolysin able to fix complement at cold temperatures and to determine RBCs lysis at 37°C. Mixed forms show both characteristics of wAIHA and CAD, with a DAT positive for both IgG and C and high titer cold agglutinins. Finally, there is a heterogeneous group of atypical AIHAs that include DAT negative, IgA driven, and warm IgM types (7, 8, 13). All these forms have a variable degree of anemia, hemolysis and bone marrow compensation, as shown in Table 1 for Hb and LDH levels, and reticulocyte counts.
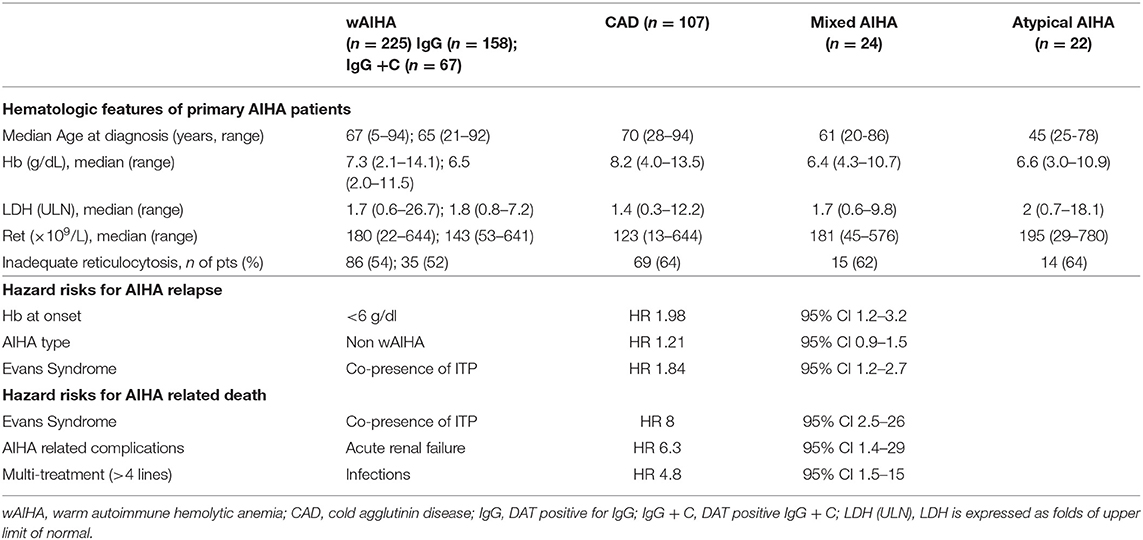
Table 1. Clinical and laboratory characteristics of patients at onset divided according to AIHA serological type.
Risk Factors for Relapse and Mortality
Given the great clinical heterogeneity of the various AIHA forms, an effort has been made to identify predictors of outcome, including complications, response to therapy and death. The severity of anemia at onset has been identified as the strongest predictor of relapse, with hazard ratios of 1.61, 1.74, and 1.98, for Hb levels of 8.1–10, 6.1–8, <6 g/dL, respectively (5, 8). Complement involvement and thermal characteristics of the autoantibody were also important, with warm IgG+C, mixed, CAD, and atypical forms more frequently needing second or further therapy lines. Moreover, the concomitant presence of immune thrombocytopenia (Evans syndrome) is associated with a higher risk of relapse and refractoriness to treatment. Overall, AIHAs other than warm forms, plus Evans syndrome and Hb <8 g/dL at onset had a 4-fold increased risk of multiple relapses (8). Moreover, bone marrow features impact on disease severity since the presence of reticular fibrosis, dyserithropoiesis, and hypercellularity correlated with shorter relapse-free survival and lower response rate to immunosuppressive therapies (3). Regarding fatal outcome, Hb <6 g/dL at onset, Evans' syndrome, multi-treatment, acute renal failure, and infections have been associated with 5-8 fold risk of increased mortality (8). A case series of 13 very severe relapsed/refractory primary AIHA reported a mortality of 57%, despite intensive treatment, including transfusions, steroid boli, intravenous immunoglobulins, rituximab, erythropoietin, and plasma-exchange (13). More recently, mortality was 30% in a series of 44 AIHA admitted to intensive care unit for severe anemia (14). It is worth remembering that about 15–20% of AIHAs display thrombotic events, including severe episodes (pulmonary embolism, stroke, cardiac infarction), which are generally proportional to active hemolysis (5, 7). Risk factors for these severe, although not fatal, complications are Hb levels <6 g/dL at onset, increased LDH levels, and previous splenectomy (8).
Secondary AIHAs
Several conditions represent a risk factor for the development of AIHA, including lymphoproliferative and autoimmune diseases, immunodeficiencies, infections, and solid tumors (Table 2). Concerning lymphoproliferative disorders, CLL patients show the highest risk with up to 5–10% developing AIHA, with an onset that may precede the diagnosis of lymphoproliferative disease (11, 15). The presence of unmutated IGHV status, sterotyped IGHV frames, and unfavorable cytogenetics (chromosome 17p and/or 11q deletions) represent a risk factor for the development of AIHA (4, 15–17). Other recently identified risk factors were several down-regulated miRNAs, some of them known to be involved in autoimmune phenomena (4). Of note, a positive DAT without hemolysis is frequent in CLL. AIHA prevalence in NHL is 2–3%, with higher frequencies in some subtypes (13–19% in angioimmunoblastic T-cell lymphoma and 50% in marginal zone lymphoma) (7, 11). A particular setting is CAD, which is associated with an indolent clonal lymphoid infiltrate distinct from other NHL (2, 6). In this disease recurrent mutations of KMT2D and CARD11 have been identified in 69 and 31% of cases, respectively (18). Similar mutations have also been reported in Kabuki syndrome, a congenital disorder characterized by malformations, immune-deficiency, and development of autoimmune diseases (4, 18).
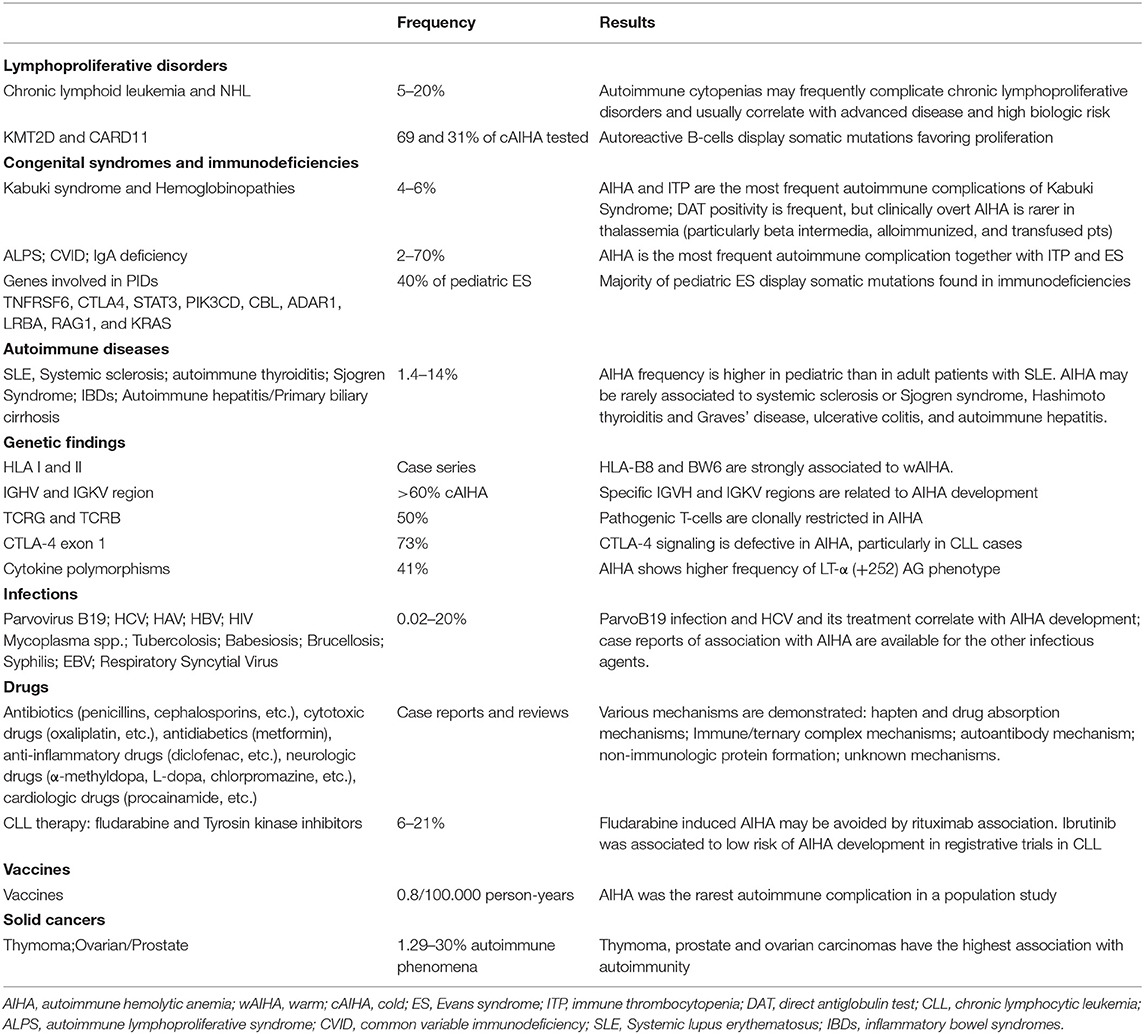
Table 2. Secondary conditions associated with autoimmune hemolytic anemia (AIHA).
Regarding AIHA in the context of immune dysregulation, patients with systemic lupus erythematosus develop AIHA in 14% of pediatric cases and 3% of adults (7, 11). A close association has also been reported with thyroid autoimmune disorders, such as Hashimoto thyroiditis and Graves' disease. Several case reports exist for AIHA association with systemic sclerosis, Sjögren syndrome (SS), autoimmune liver disorders, and inflammatory bowel diseases (7, 11). Moreover, various immunodeficiencies have been identified as predisposing conditions for AIHA, including common variable immunedeficiency (19), IgA deficiency, and autoimmune lymphoproliferative syndromes (ALPS) (20). Interestingly, mutations in genes implicated in primary immunodeficiencies (TNFRSF6, CTLA4, STAT3, PIK3CD, CBL, ADAR1, LRBA, RAG1, and KRAS) have been detected in about half of pediatric patients with AIHA and ITP (Evans Syndrome, ES); mutated patients showed more severe disease with higher treatment requirement and fatal outcome (12). These findings underline the close link between autoimmunity and immunodeficiency, i.e., a shared condition of dysregulated immune system.
Genetic Background and Exogenous Triggers for AIHA Development
Although not specifically involved in the changing landscape of AIHA, it is worth considering genetic factors and historically recognized exogenous triggers (4). Several old and recent studies demonstrated a strong association of AIHA with HLA-B locus, particularly HLA-B8 and BW6 (21), or a reduced frequency of the disease in subjects harboring the HLA-DQ6 locus (22) (Table 2). As regards humoral immune response, various variable regions of the immunoglobulin heavy and light chains (IGHV and IGKV) have been associated with AIHA, particularly IGHV4-34, IGHV3, and IGKV3-20 genes, responsible for I antigen binding, and mostly represented in CAD (6). Concerning cellular immunity, autoreactive clonal T- CD8+cells have been reported in about 50% of AIHA cases; moreover, polymorphism of the cytotoxic T-lymphocyte antigen-4 (CTLA-4) gene and of lymphotoxin-α (LT-α) may represent a risk factor for primary or secondary AIHA development (4).
Various infections have been associated with an increased incidence of AIHA, particularly Parvovirus B19 (associated with DAT positive hemolysis in up to 20% of cases and hepatotropic virus, mostly HCV and possibly related to interferon therapy (11). Moreover, cold agglutinin AIHA occurs in up to 3% of patients with infectious mononucleosis and Mycoplasma pneumoniae infection (7, 11). Finally, paroxysmal cold hemoglobinuria is almost invariably preceded by an infection, including syphilis and virus, particularly in children (7, 11). In addition there is a long list of drugs that have been proven or highly suspected to induce AIHA, including historical ones (α-methyldopa, procainamide, penicillins, cephalosporins, diclofenac, ibuprofen, thiazides, quinine, quinidine, metformin) and more recent molecules (cladribine, fludarabine, lenalidomide, oxaliplatin, teniposide, pentostatin) (7, 11). Concerning new small molecules (ibrutinib, venetoclax, and idelalisib) few case reports of treatment-emergent AIHAs have been published (4).
Comprehensive diagnostic Approach and New Diagnostic Tools
Given the several associated conditions, an accurate diagnostic approach to AIHA is fundamental for a comprehensive risk assessment and a proper therapy (Figure 1). Medical history and baseline evaluation is still fundamental to assess drug assumption, infections, signs of acute or chronic hemolysis, and bone marrow compensation (reticulocytes).

Figure 1. Diagnostic algorithm of autoimmune haemolytic anemia (AIHA). DAT, direct antiglobulin test or Coombs test; MS-DAT, mitogen-stimulated DAT; CAD, cold agglutinin disease; CT, computed tomography; G6PD, glucose-6-phosphate dehydrogenase deficiency; PKD, pyruvate kinase deficiency; EMA-binding, eosin-5′-maleimide-binding test.
The Standard DAT and More Sensitive Techniques
As mentioned earlier, the DAT with monospecific antisera (anti-IgG, anti- IgA, anti-IgM, anti C) is the cornerstone of diagnosis, and allows a proper distinction of the various AIHA forms, that have different responses to therapy and prognosis (23). A diagnostic challenge that may take advantage of new diagnostic tools is represented by DAT-negative AIHA, usually 5–10% of all forms. In these cases, excluding other common causes of hemolysis and pursuing the clinical suspicion of AIHA, it is recommended to ask for second-level tests in a reference center. The DAT negativity due to low-affinity antibodies may be overcome by low ionic strength solutions (LISS) or cold washings. The small amounts of RBC-bound antibodies (below the threshold of the test) may take advantage of more sensitive techniques, such as microcolumn and solid-phase antiglobulin tests. In fact, DAT tube effectively diagnoses AIHA when at least 500 molecules of autoantibodies are bound to RBCs, whereas microcolumn and solid phase require ~200–300 molecules per single RBC to yield a positive result. Consistently, DAT tube is the most specific but least sensitive test, whereas microcolumn and solid phase methods show reduced specificity but increased sensitivity (7, 24, 25). Smaller amounts of autoantibodies can be detected by new, even more sensitive techniques, such as flow cytometry (able to detect about 30–40 antibody molecules per RBC), the enzyme-linked and radiolabeled tests, or the mitogen-stimulated-DAT (able to amplify the autoimmune reaction in culture) (23, 24). Of particular importance is the identification of atypical AIHAs due to warm IgM that are potent activators of complement and often detach from the RBC during washing procedures, causing detrimental delay in diagnosis and therapy. In these cases the DDAT (Dual Direct Antiglobulin Test) may allow the diagnosis of these rare forms, usually severe and potentially lethal (26). In addition, as complement activation is recognized as negative prognostic factor, evaluation of baseline values of C3 and C4 fractions, would help completing the diagnostic workup. Notwithstanding extensive evaluation, a fraction of AIHA remains DAT-negative: in these cases the diagnosis is made after the exclusion of the many hemolytic disorders (congenital hemolytic anemias, paroxysmal nocturnal hemoglobinuria, thrombotic microangiopathies, mechanical and toxic noxae) and on the basis of an ex-adiuvantibus therapy with steroids.
Causes of Falsely Positive DAT
The DAT may be positive due to the presence of alloantibodies in recently transfused patients, in delayed hemolytic transfusion reactions, and in the hemolytic disease of the newborn (23). The coexistence of auto- and alloantibodies has been reported in about 30% of AIHA patients, and their presence is often masked by autoantibodies, possibly causing severe hemolytic reactions in case of RBC transfusion. In complex cases the distinction between allo- and autoantibody is advisable by immunoabsorbance techniques and by extended RBC genotyping (7, 24). It is worth reminding that daratumumab, the anti-CD38 antibody for the treatment of multiple myeloma, may give false DAT positivity. CD38 is also expressed on red-cell membranes, resulting in panreactive agglutination in the test used for antibody screening and cross-matching. Several methods have been proposed to overcome this interference, including pretreatment of red cells with dithiothreitol, use of antiidiotypic antibodies against daratumumab, supplementation of soluble CD38 to bind daratumumab in patient serum, use of red cells from newborns as test cells, and use of F(ab′)2 fragments of daratumumab by digestion with pepsin (27).
Bone Marrow Evaluation and Exclusion of Secondary AIHA Forms
Among “new” diagnostic approaches to AIHA there is the increasingly recommended (and performed) bone marrow evaluation (morphology, cytometry, cytogenetics and biopsy). Bone marrow evaluation may in fact give important information on adequate erythroid compensation, underlying lymphoproliferative disorder, and evidence of an early/subclinical or therapy-related myelodysplasia or bone marrow failure. These features may help in harnessing therapy, avoiding further detrimental immunesuppression, or selecting immunosupprors among the new targeted therapies, based on the type of bone marrow lymphocyte infiltrate (T or B). Moreover, the determination of endogenous EPO levels may indicate this treatment, which has recently shown effective particularly in relapsed/refractory and heavily treated subjects (7, 28). To properly identify secondary forms imaging and serologic investigation is fundamental (Figure 1). It is advised to test for anti-phospholipid antibodies (cardiolipin, beta-2, and lupus-like anticoagulant), given the known thrombotic diathesis of acute/severe AIHAs, and thus advising thromboprophylaxis. Finally, molecular analysis and next generation sequencing would help confirming associated conditions (primary immunodeficiencies, lymphoproliferative disorders, myelodysplastic syndromes, other coexisting congenital anemias) again harnessing therapy.
New Treatments for AIHA
The availability of several new treatments has undoubtedly boosted the therapeutic possibilities for patients, but at the same time has increased the number of heavily treated, relapsed/refractory cases. The immune-mediated pathogenic mechanisms in AIHA are different and may differently act at various degrees during the various phases of the disease. Therefore, the challenge for the future will be the selection and timing of administration of the several drugs available or under development. Firstly, distinction between wAIHA and CAD is pivotal, as therapy in quite different: the former usually respond to steroids, whereas the latter requires high and unacceptable doses. Splenectomy, although progressively abandoned and moved to third or further lines, is still a valid option for wAIHA; on the contrary it is ineffective and contraindicated in CAD, where RBC destruction occurs mainly in the liver and lymphoid organs. Likewise, it is poorly effective and discouraged in AIHA secondary to immunodeficiencies, autoimmune diseases, and lymphoproliferative disorders. Several target therapies are now in the clinical use or under development in AIHA (Table 3) (7, 9). Rituximab is becoming the preferred second-line for wAIHA and is recommended as first line in CAD. In the former, low-doses may be equally effective as standard ones, whereas in CAD the standard schedule is more effective. The drug is successfully administered in primary and secondary cases, alone or associated with chemotherapy (bendamustine, fludarabine, or other) in AIHAs secondary to lymphoproliferative diseases (6, 29, 30). The clinical challenges are wAIHAs relapsed after rituximab and unfit/refusing splenectomy, and CAD relapsed after rituximab monotherapy and unfit for rituximab-combined chemotherapy. In this setting, treatment selection would be ideally driven by disease-related risk factors, and/or associated conditions, as well as patient general comorbidities. Among monoclonal antibodies, ofatumumab (anti-CD20), alemtuzumab (anti-CD52), and daratumumab (anti-CD38) have shown promising results in case reports, mainly secondary AIHAs. Likewise, the orally administered B-cell receptor inhibitors ibrutinib and venetoclax seem particularly effective in secondary AIHAs, and the proteasome inhibitor bortezomib in 1/3 of refractory CADs (9). A further B-cell receptor target therapy is the PI3K inhibitor parsaclisib, which is under investigation in both wAIHA and CAD with very promising results. An interesting new approach for CAD is blocking complement activation, either at the C5 level (eculizumab) or more efficiently at the C3 (APL-2) or C1s level (sutimlimab), the latter with about 50% responses (31). Other drugs are directed at cellular immunity and cytokines, such as subcutaneous low-dose IL-2 and sirolimus (inhibitor of the serine treonine kinase mTOR). Targeting IgG driven extravascular hemolysis by inhibiting the spleen tyrosine kinase (Syk) is also an attractive approach in wAIHA (for example fostamatinib). Finally, inhibition of the neonatal crystallizable fragment receptor (FcRn) is a new interesting approach: these drugs avoid protection of circulating IgG, including pathogenic autoantibodies, from catabolism and thus regulate innate and adaptive responses initiated by IgG immune complexes (9).
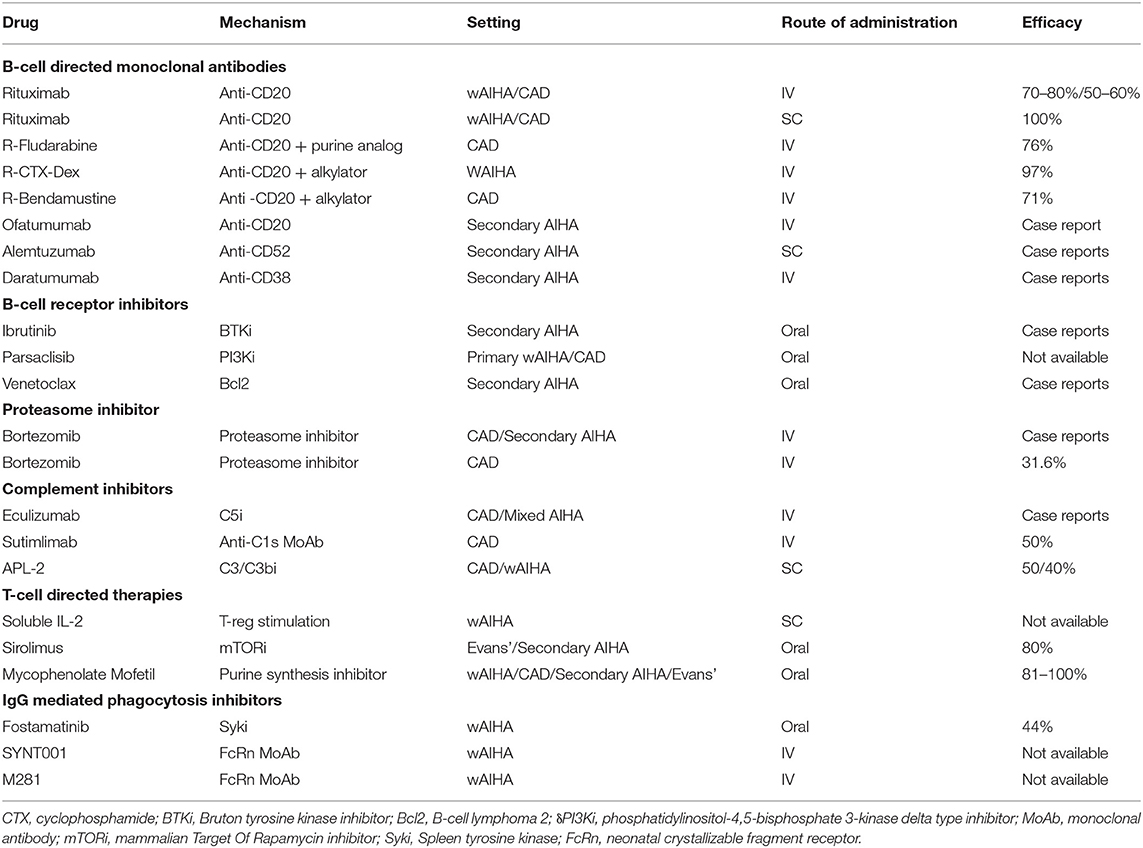
Table 3. Target therapies in autoimmune hemolytic anemia (AIHA).
It is worth commenting that several trials with these new drugs are ongoing or being planned. In order to achieve meaningful endpoints it will be essential to properly select patients, bringing into consideration the number of previous treatments and related complications, associated conditions, intrinsic AIHA-risk factors, and type and degree of the immunologic dysregulation. This would provide the basis of a risk-adapted therapy in AIHA, as it is now advised for malignant hematologic conditions.
The Transition From Chronic/Relapsing AIHA to Idiopathic Cytopenias/Dysplasias of Uncertain Significance (ICUS/IDUS)
Several lines of evidence support the existence of a relationship between MDS and autoimmunity, including their epidemiologic association, the existence of common immune-mediated physiopathologic mechanisms, and the response to similar immunosuppressive therapies. This relationship may be hypothesized also with the recently-identified conditions ICUS and IDUS, which are defined by unexplained cytopenia (hemoglobin <10 g/dL; platelet count <100 × 109/L; absolute neutrophil count <1.8 × 109/L) and/or dysplasia in <10% of bone marrow lineages (32, 33). More recently another category has been proposed, the clonal cytopenias of undetermined significance (CCUS), where both unexplained cytopenias and clonal mutation are found, without fulfilling WHO criteria for MDS (34). Of note, about 10% of the general population aged over 70 years carries mutations in genes associated with myeloid neoplasms, usually single mutations at a low variant allele frequency, whose pathophysiologic role is still unknown. These cytopenias may be considered milder MDS forms that may evolve, after a variable period, in overt MDS or other bone marrow failure syndromes. In this view, we described the presence of anti-erythroblast antibodies in a case of erythroblastic synartesis, a rare disease with an autoimmune pathogenesis against erythroid precursors (35). Moreover, we also described two cases of AIHA and Evan's syndrome with anti-erythroblast antibodies, which showed a clear-cut bone marrow erythrocyte precursor hyperplasia at diagnosis, but evolved into IDUS and AA after several years (33). In this setting it is tempting to speculate that refractory/relapsing AIHAs lose their predominant “peripheral” pattern over time, and shift toward a “central” autoimmunity (Figure 2), leading to a refractory anemia. Additional factors, like accumulating somatic mutations, increased apoptosis, overinflammatory response (inflammaging), unfavorable bone marrow cytokine microenvironment, and breakdown of DNA-repairing tools (telomere shortening) are likely to play a role and need to be addressed in large prospective studies. The increasing availability of NGS panels will also help in defining the genetic background of the immunologic dysregulation, both in terms of inability to clear pathogens/external triggers (chronic infection), or failure to tolerate autoantiges (chronic autoimmune stimulation). This would be of great importance to selectively modulate (potentiate, down-regulate, or re-direct) the innate and adaptive immune response and to avoid an excessively toxic approach to autoimmunity.
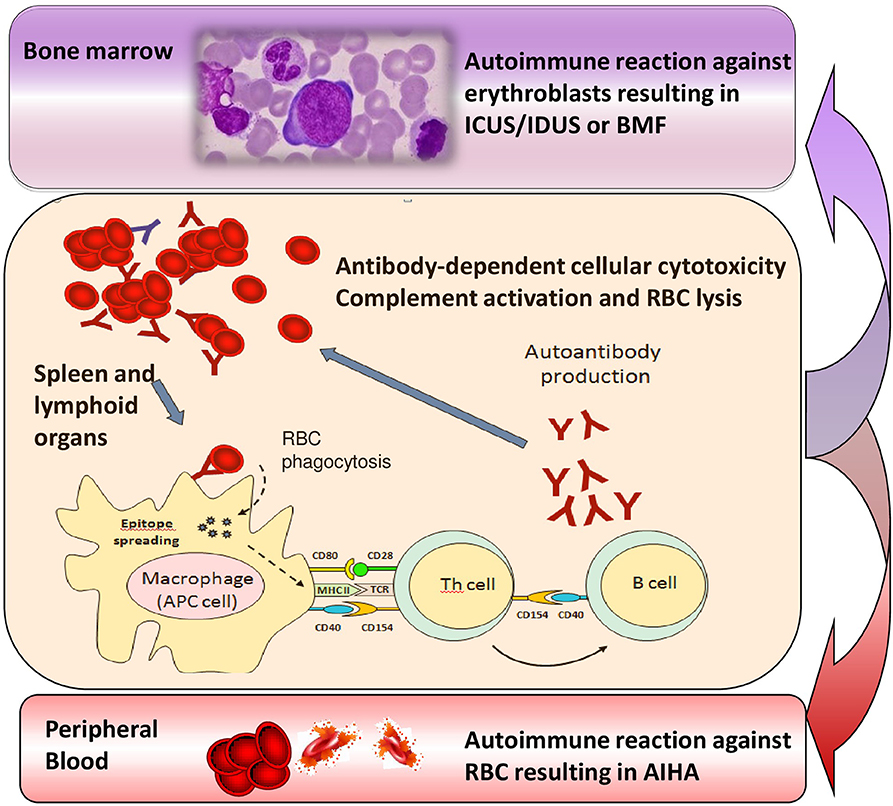
Figure 2. Transition from chronic/relapsing autoimmune hemolytic anemia (AIHA) to idiopathic cytopenias/dysplasias of uncertain significance (ICUS/IDUS) and bone marrow failure (BMF). Various immune effectors such as macrophages, antigen presenting cells (APC), T helper cells and B lymphocytes are involved into the immune attack, which is firstly directed against peripheral erythrocytes, but may persist and involve bone marrow precursors, possibly leading to ICUS/IDUS or BMF syndromes over time.
AIHAs Associated With Transplant
Organs and tissues transplants represent a challenging event for the recipient immune system and may evoke an “immunologic storm” resulting in either transplant rejection and/or devastating immune reactions. A particular mild picture is the passenger lymphocyte syndrome due to donor viable, immunocompetent lymphocytes present within the graft that can produce antibodies against donor RBCs. The syndrome involves mainly group O donors, though few cases have been described in AB recipients with non-AB donors. The risk of hemolysis is proportional to the burden of transplanted lymphocytes and ranges from 9 to 70% (kidney < liver < heart-lung transplants) (11, 36). Onset is between 3 and 24 days post-transplant and hemolysis is generally transient, since the lymphocytes transferred with the donor organ do not engraft. An emerging and more severe clinical entity is AIHA after hematopoietic stem cells transplant (HSCT). In this setting, autoantibodies are produced by the donor immune system against antigens on erythrocytes produced by the graft itself, and the clinical picture is generally severe (10). Several factors are implicated: the disease itself, the conditioning therapy preceding transplant, the subsequent immunosuppressive treatments, and the occurrence of HSCT complications such as viral infections reactivation. Moreover, the unfavorable immunologic microenvironment may lead to graft failure (as observed in transplanted patients with aplastic anemia), and graft immunocompetence may in turn induce graft vs. host disease (GVHD), further complicating the clinical course. Data from the literature report that immune hemolysis may complicate up to 2–4% of HSCTs after a median of 3–10 months. Both warm and cold forms are described, the former developing between 6 and 18 months, vs. 2–8 months for the latter. Risk factors for AIHA post HSCT are summarized in Table 4 and include use of unrelated donor and HLA-mismatch, occurrence of GVHD, use of cord blood, age < 15 years, CMV reactivation, alemtuzumab use, and non-malignant condition pre-HSCT (10). Mortality may be quite high and increases with infections (37).
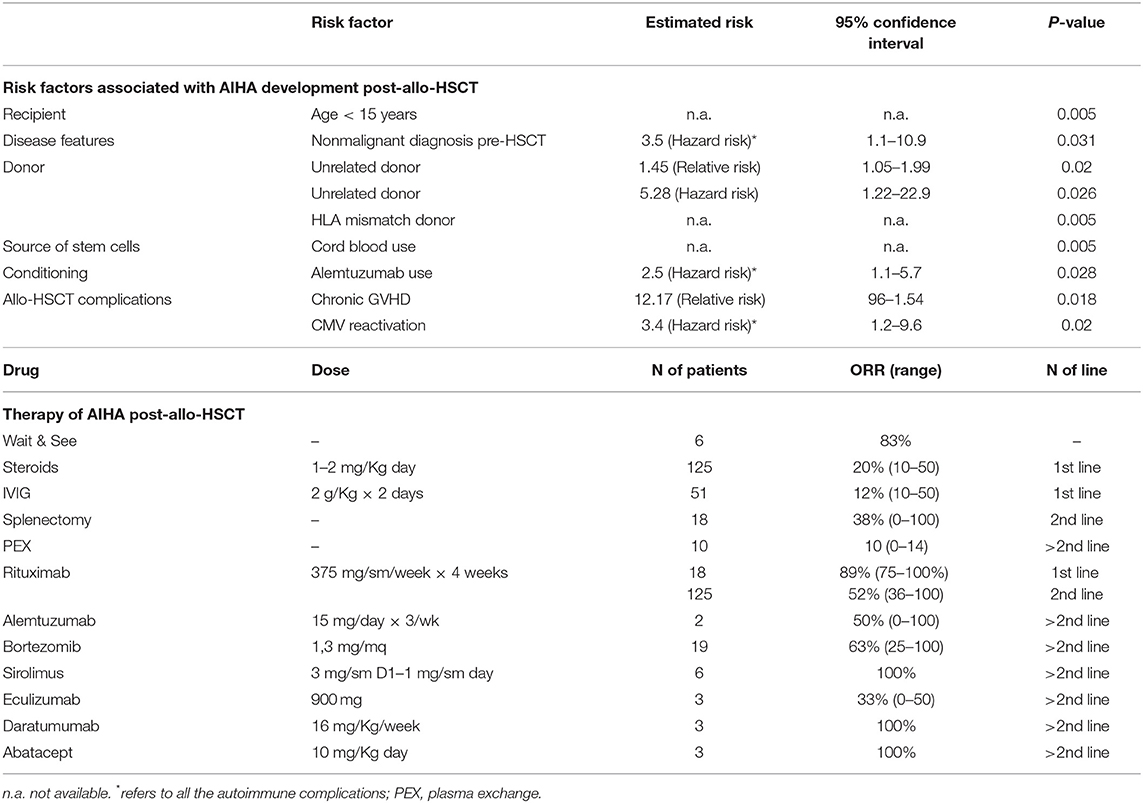
Table 4. Risk factors and therapies for post-allogenic hematopoietic stem cell transplant (allo-HSCT) AIHA.
Therapy of Post-Transplant AIHA
Table 4 recapitulates current and novel therapies that have been used in AIHA post-HSCT. It is evident that the total number of patients reported in the various studies is small, and case reports and series carry the bias of describing good outcomes only. However, first-line steroids seem to work less than in primary AIHA, being effective in about 20% of cases only. Moreover, frontline rituximab appears much more effective than in second line (89 vs. 52% responses), and most Authors suggest its early use, particularly in severe cases. Splenectomy is effective but its use is limited to selected cases given the high surgical, infectious, and thrombotic risk. Regarding novel targeted therapies, alemtuzumab, bortezomib, sirolimus, eculizumab, daratumumab, and abatacept have all been used in selected cases, as 3rd or further line, with heterogeneous outcomes. Finally, the passenger lymphocyte syndrome may occur also in this setting and is favored by: use of cyclosporine alone for GVHD prophylaxis, use of peripheral blood rather than bone marrow as source of the graft, use of reduced-intensity conditioning, use of a non-genotypically HLA-matched donor, and use of a female donor. Umbilical cord blood as source for stem cells appears protective. Careful transfusion procedures are warranted in transplanted patients, particularly in mismatched cases (10, 36, 38).
AIHAs Associated With New Biological Anti-Cancer Therapies
A fascinating field is that of anti-tumor immunotherapy, based on the understanding that tumor cells activate immune checkpoints such as molecular programmed death receptor-1 (PD-1) and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) signaling pathways to inhibit T lymphocyte activation and thus escape from immune surveillance, known as “immune brake.” Checkpoint inhibitors (CPIs) reactivate T lymphocytes to recognize cancer cells by blocking CTLA-4 or PD-1, and are therefore effective in numerous types of cancer. However, immune-related adverse effects have also been reported (39, 40) and hematologic ones are rare but potentially fatal. Most of them are monolineage cytopenia, or bilineage cytopenia, whilst acquired hemophilia A, eosinophilia, large granular lymphocytosis, and hemophagocytic lymphohistiocytosis are rare (41). A meta-analysis of 9,324 patients indicated that the incidence of anemia, neutropenia, and thrombocytopenia was 9.8, 0.94, and 2.8%, respectively (41). AIHA is the most commonly reported hematologic adverse event, with many case reports of fulminant course (40). A recent revision of the database of the Food and Drug Administration revealed a total of 68 cases: men to women ratio was similar, and the underlying diseases were mainly melanoma (41%), non-small cell lung cancer (NSCLC, 26%), and others including kidney cancer, Hodgkin's lymphoma or skin cancers. The reported cases were mostly from North America (49%) and Europe (34%), with a few from Asia (10%) and Australia (7%). Forty-three cases developed after nivolumab, 13 with pembrolizumab, 7 with ipilimumab, and 5 with atezolizumab, and 16% of cases had received two CPIs. The median time to AIHA onset was 50 days, four patients had concurrent thrombocytopenia, other four endocrine abnormalities (thyroiditis, adrenal insufficiency or hypophysitis), and three gastrointestinal adverse events (colitis or hepatitis). Most cases were IgG positive warm AIHA, whilst CADs were rarer. All episodes were severe, with 80% of cases developing grade 3–4 transfusion-dependent anemia, and the risk appeared higher with PD-1 or PD-L1 targeting agents (0.15–0.25%) than with CTLA-4 inhibitors (0.06%). Mortality was as high as 17%, mainly due to multi-organ failure and delayed diagnosis (42). In another recent analysis of 14 cases who developed AIHA after CPIs, median time to AIHA was 55 days (IQR 22–110 days). Compared to primary AIHA, these cases showed a higher proportion of DAT negativity (38%) and of severe anemia (median Hb 6.3 g/dL (IQR, 6.1–8.0 g/dL). Finally, 50% of cases relapsed after first line and 14% became chronic (43). Regarding therapy, prednisone 1.5 mg−2 mg/kg per day along with CPIs discontinuation is recommended, and evidences for the need of early rituximab or further immunosuppressive agents are lacking. The rechallenge of CPIs after AIHA has improved or is stable remains inconclusive. A patient with Hodgkin's lymphoma who developed nivolumab associated AIHA, that recovered after steroids and was later re-challenged with nivolumab without AIHA recurrence, has been described (44).
Conclusions
Nowadays the pathogenic and therapeutic landscape of AIHA is rapidly changing for several reasons. First, numerous AIHA-associated conditions have been identified, such as autoimmune diseases, immunodeficiencies, and tumors, which may have additional immune-mediated pathogenic mechanisms compared to primary disease, and deserve a specific therapeutic approach. In this view, the increasing use of molecular testing has disclosed several underlying conditions, questioning the distinction between primary and secondary forms. Second, the development of new drugs has offered additional therapeutic opportunities to “cure” the disease, but at the same time has increased the number of relapsed/refractory cases. Moreover, the future availability of even more target therapies will further puzzle the treatment algorithm of the disease. Third, there is increasing awareness of various pathogenic mechanisms that may differently act during the disease course, ranging from a predominant “peripheral” autoimmunity against erythrocytes to a “central” attack against erythroid precursors, possibly preceding a myelodysplastic or aplastic evolution. These findings have further therapeutic implications, suggesting to avoid heavy immunosuppression in favor of immunomodulating/stimulating agents. Finally, there is increasing emergence of complex and severe entities, particularly AIHA developing after HSCT and AIHA associated with novel anti-cancer drugs such as checkpoint inhibitors, which represent a clinical challenge for complications and fatal outcome. Diagnosis of DAT-negative AIHAs and evaluation of disease-related risk factors for relapse and mortality have improved, but are still an unmet need. The assessment of disease-related risk factor would be pivotal to design good clinical trials and to give hints for a risk-adapted therapy of AIHAs.
Author Contributions
WB designed the study, searched the literature, analyzed data, wrote the manuscript, and participated in the final revision. BF searched the literature, analyzed data, wrote the manuscript, and participated in the final revision.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
All authors participated in the design of the review, literature revision, manuscript writing, and final revision for important intellectual content.
References
1. Barcellini W. New insights in the pathogenesis of autoimmune hemolytic anemia. Transfus Med Hemother. (2015) 42:287–93. doi: 10.1159/000439002
2. Berentsen S, Bø K, Shammas FV, Myking AO, Ulvestad E. Chronic cold agglutinin disease of the “idiopathic” type is a premalignant or low-grade malignant lymphoproliferative disease. APMIS. (1997) 105:354–62. doi: 10.1111/j.1699-0463.1997.tb00581.x
3. Fattizzo B, Zaninoni A, Gianelli U, Zanella A, Cortelezzi A, Kulasekararaj AG, et al. Prognostic impact of bone marrow fibrosis and dyserythropoiesis in autoimmune hemolytic anemia. Am J Hematol. (2018) 93:E88–91. doi: 10.1002/ajh.25020
4. Fattizzo B, Barcellini W. Autoimmune cytopenias in chronic lymphocytic leukemia: focus on molecular aspects. Front Oncol. (2020) 9:1435. doi: 10.3389/fonc.2019.01435
5. Barcellini W, Fattizzo B, Zaninoni A, Radice T, Nichele I, Di Bona E, et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients. Blood. (2014) 124:2930–6. doi: 10.1182/blood-2014-06-583021
6. Berentsen S. How I manage patients with cold agglutinin disease. Br J Haematol. (2018) 181:320–30. doi: 10.1111/bjh.15109
7. Jäger U, Barcellini W, Broome CM, Gertz MA, Hill A, Hill QA, et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: recommendations from the First International Consensus Meeting. Blood Rev. (2019) 5:100648. doi: 10.1016/j.blre.2019.100648
8. Barcellini W, Zaninoni A, Fattizzo B, Giannotta JA, Lunghi M, Ferrari A, et al. Predictors of refractoriness to therapy and healthcare resource utilization in 378 patients with primary autoimmune hemolytic anemia from eight Italian reference centers. Am J Hematol. (2018) 93:E243–6. doi: 10.1002/ajh.25212
9. Barcellini W, Fattizzo B, Zaninoni A. Current and emerging treatment options for autoimmune hemolytic anemia. Expert Rev Clin Immunol. (2018) 14:857–72. doi: 10.1080/1744666X.2018.1521722
10. Barcellini W, Fattizzo B, Zaninoni A. Management of refractory autoimmune hemolytic anemia after allogeneic hematopoietic stem cell transplantation: current perspectives. J Blood Med. (2019) 10:265–78. doi: 10.2147/JBM.S190327
11. Hill QA, Stamps R, Massey E, Grainger JD, Provan D, Hill A. Guidelines on the management of drug-induced immune and secondary autoimmune, haemolytic anaemia. Br J Haematol. (2017) 177:208–20. doi: 10.1111/bjh.14654
12. Hadjadj J, Aladjidi N, Fernandes H, Leverger G, Magérus-Chatinet A, Mazerolles F. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. Blood. (2019) 134:9–21 doi: 10.1182/blood-2018-11-887141
13. Fattizzo B, Zaninoni A, Nesa F, Sciumbata VM, Zanella A, Cortelezzi A, et al. Lessons from very severe, refractory, and fatal primary autoimmune hemolytic anemias. Am J Hematol. (2015) 90:E149–51. doi: 10.1002/ajh.24047
14. Lafarge A, Bertinchamp R, Pichereau C, Valade S, Chermak A, Theodose I, et al. Prognosis of autoimmune hemolytic anemia in critically ill patients. Ann Hematol. (2019) 98:589–94. doi: 10.1007/s00277-018-3553-9
15. Visco C, Barcellini W, Maura F, Neri A, Cortelezzi A, Rodeghiero F. Autoimmune cytopenias in chronic lymphocytic leukemia. Am J Hematol. (2014) 89:1055–62. doi: 10.1002/ajh.23785
16. Maura F, Cutrona G, Fabris S, Colombo M, Tuana G, Agnelli L, et al. Relevance of stereotyped B-cell receptors in the context of the molecular, cytogenetic and clinical features of chronic lymphocytic leukemia. PLoS ONE. (2011) 6:e24313. doi: 10.1371/journal.pone.0024313
17. Maura F, Visco C, Falisi E, Reda G, Fabris S, Agnelli L, et al. B-cell receptor configuration and adverse cytogenetics are associated with autoimmune hemolytic anemia in chronic lymphocytic leukemia. Am J Hematol. (2013) 88:32–6. doi: 10.1002/ajh.23342
18. Malecka A, Troen G, Tierens A, Østlie I, Małecki J, Randen U, et al. Frequent somatic mutations of KMT2D (MLL2) and CARD11 genes in primary cold agglutinin disease. Br J Haematol. (2018) 183:838–42. doi: 10.1111/bjh.15063
19. Feuille EJ, Anooshiravani N, Sullivan KE, Fuleihan RL, Cunningham-Rundles C. Autoimmune cytopenias and associated conditions in CVID: a report from the USIDNET Registry. J Clin Immunol. (2018) 38:28–34. doi: 10.1007/s10875-017-0456-9
20. Bariş HE, Kiykim A, Nain E, Özen AO, Karakoç-Aydiner E, Bariş S. The plethora, clinical manifestations and treatment options of autoimmunity in patients with primary immunodeficiency. Turk Pediatri Ars. (2016) 51:186–92. doi: 10.5152/TurkPediatriArs.2016.3928
21. Abdel-Khalik A, Paton L, White AG, Urbaniak SJ. Human leucocyte antigens A, B, C, and DRW in idiopathic “warm” autoimmune haemolytic anaemia. Br Med J. (1980) 280:760–1. doi: 10.1136/bmj.280.6216.760
22. Wang-Rodriguez J, Rearden A. Reduced frequency of HLA-DQ6 in individuals with a positive direct antiglobulin test. Transfusion. (1996) 36:979–84. doi: 10.1046/j.1537-2995.1996.36111297091742.x
23. Petz LD, Garratty G. Immune Hemolytic Anemias. 2nd ed. Philadelphia: Churchill Livingstone (2004).
24. Barcellini W. Immune hemolysis: diagnosis and treatment recommendations. Semin Hematol. (2015) 52:304–12. doi: 10.1053/j.seminhematol.2015.05.001
25. Barcellini W, Fattizzo B. Clinical applications of hemolytic markers in the differential diagnosis and management of hemolytic anemia. Dis Markers. (2015) 2015:635670. doi: 10.1155/2015/635670
26. Bartolmäs T, Salama A. A dual antiglobulin test for the detection of weak or nonagglutinating immunoglobulin M warm autoantibodies. Transfusion. (2010) 50:1131–4. doi: 10.1111/j.1537-2995.2009.02533.x
27. Lancman G, Arinsburg S, Jhang J, Cho HJ, Jagannath S, Madduri D, et al. Blood transfusion management for patients treated with anti-CD38 monoclonal antibodies. Front Immunol. (2018) 9:2616. doi: 10.3389/fimmu.2018.02616
28. Fattizzo B, Michel M, Giannotta J, Guillet S, Frederiksen H, et al. Efficacy of recombinant erythropoietin in autoimmune hemolytic anemia: a multicenter international study. Haematologica. (2020). doi: 10.3324/haematol.2020.250522. [Epub ahead of print].
29. Berentsen S, Randen U, Vågan AM, Hjorth-Hansen H, Vik A, Dalgaard J, et al. High response rate and durable remissions following fludarabine and rituximab combination therapy for chronic cold agglutinin disease. Blood. (2010) 116:3180–4. doi: 10.1182/blood-2010-06-288647
30. Berentsen S, Randen U, Oksman M, Birgens H, Tvedt THA, Dalgaard J, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. (2017) 130:537–41. doi: 10.1182/blood-2017-04-778175
31. Röth A, Barcellini W, D'Sa S, Miyakawa Y, Broome CM, Michel M, et al. Inhibition of complement C1s with sutimlimab in patients with cold agglutinin disease (CAD): results from the Phase 3 Cardinal Study. Blood. (2019) 134 (Suppl_2). doi: 10.1182/blood-2019-132490
32. Barcellini W. The relationship between idiopathic cytopenias/dysplasias of uncertain significance(ICUS/IDUS) and autoimmunity. Expert Rev Hematol. (2017) 10:649–57. doi: 10.1080/17474086.2017.1339597
33. Barcellini W, Fattizzo B, Zaninoni A, Valli V, Ferri V, Gianelli U, et al. Clinical evolution of autoimmune cytopenias to idiopathic cytopenias/dysplasias of uncertain significance (ICUS/IDUS) and bone marrow failure syndromes. Am J Hematol. (2017) 92:E26–9. doi: 10.1002/ajh.24618
34. Bejar R. CHIP, ICUS, CCUS and other four-letter words. Leukemia. (2017) 31:1869–71. doi: 10.1038/leu.2017.181
35. Zaninoni A, Imperiali FG, Pomati M, Colombi M, Boschetti C, Barcellini W. Bone marrow mitogen-stimulated direct antiglobulin test in a case of erythroblastic synartesis. Clin Lab. (2010) 56:459–62.
36. Skeate R, Singh C, Cooley S, Geller M, Northouse J, Welbig J, et al. Hemolytic anemia due to passenger lymphocyte syndrome in solid malignancy patients treated with allogeneic natural killer cell products. Transfusion. (2013) 53:419–23. doi: 10.1111/j.1537-2995.2012.03942.x
37. González-Vicent M, Sanz J, Fuster JL, Cid J, de Heredia CD, Morillo D, et al. Autoimmune hemolytic anemia (AIHA) following allogeneic hematopoietic stem cell transplantation (HSCT): a retrospective analysis and a proposal of treatment on behalf of the Grupo Español De Trasplante de Medula Osea en Niños (GETMON) and the Grupo Español de Trasplante Hematopoyetico (GETH). Transfus Med Rev. (2018). doi: 10.1016/j.tmrv.2018.02.005. [Epub ahead of print].
38. Kruizinga MD, van Tol MJD, Bekker V, Netelenbos T, Smiers FJ, Bresters D, et al. Risk Factors, Treatment, and immune dysregulation in autoimmune cytopenia after allogeneic hematopoietic stem cell transplantation in pediatric patients. Biol Blood Marrow Transplant. (2018) 24:772–8. doi: 10.1016/j.bbmt.2017.12.782
39. Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. (2018) 378:158–68. doi: 10.1056/NEJMra1703481
40. Zhuang J, Du J, Guo X, Zhou J, Duan L, Qiu W, et al. Clinical diagnosis and treatment recommendations for immune checkpoint inhibitor-related hematological adverse events. Thorac Cancer. (2020) 11:799–804. doi: 10.1111/1759-7714.13405
41. Petrelli F, Ardito R, Borgonovo K, Lonati V, Cabiddu M, Ghilardi M, et al. Haematological toxicities with immunotherapy in patients with cancer: a systematic review and meta-analysis. Eur J Cancer. (2018) 103:7–16. doi: 10.1016/j.ejca.2018.07.129
42. Tanios GE, Doley PB, Munker R. Autoimmune hemolytic anemia associated with the use of immune checkpoint inhibitors for cancer: 68 cases from the Food and Drug Administration database and review. Eur J Haematol. (2019) 102:157–62. doi: 10.1111/ejh.13187
43. Leaf RK, Ferreri C, Rangachari D, Mier J, Witteles W, Ansstas G, et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am J Hematol. (2019) 94:563–74. doi: 10.1002/ajh.25448
Keywords: warm autoimmune hemolytic anemia, cold agglutinin disease, bone marrow transplant, checkpoint inhibitors, complement inhibitors, target therapy
Citation: Barcellini W and Fattizzo B (2020) The Changing Landscape of Autoimmune Hemolytic Anemia. Front. Immunol. 11:946. doi: 10.3389/fimmu.2020.00946
Received: 13 February 2020; Accepted: 22 April 2020;
Published: 03 June 2020.
Edited by:
Emira Ayroldi, University of Perugia, ItalyReviewed by:
Ursula Grohmann, University of Perugia, ItalyAnne Cooke, University of Cambridge, United Kingdom
Copyright © 2020 Barcellini and Fattizzo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wilma Barcellini, wilma.barcellini@policlinico.mi.it