CRISPR/Cas12a-Mediated Gene Editing in Geodia barretti Sponge Cell Culture
 Kylie Hesp1*
Kylie Hesp1*  John L. Flores Alvarez1
John L. Flores Alvarez1  Ana-Maria Alexandru1
Ana-Maria Alexandru1  Jip van der Linden1
Jip van der Linden1  Dirk E. Martens1
Dirk E. Martens1  Rene H. Wijffels1,2
Rene H. Wijffels1,2  Shirley A. Pomponi1,3
Shirley A. Pomponi1,3- 1Bioprocess Engineering, Wageningen University & Research, Wageningen, Netherlands
- 2Faculty of Biosciences and Aquaculture, Nord University, Bodø, Norway
- 3Sponge Biotechnology, Marine Biomedical & Biotechnology Research, Harbor Branch Oceanographic Institute, Florida Atlantic University, Fort Pierce, FL, United States
Sponges and their associated microorganisms are the most prolific source of marine natural products, and many attempts have been made at creating a marine sponge cell line to produce these products efficiently. However, limited knowledge on the nutrients sponge cells require to grow and poor genetic accessibility have hampered progress toward this goal. Recently, a new sponge-specific nutrient medium M1 has been shown to stimulate sponge cells in vitro to divide rapidly. In this study, we demonstrate for the first time that sponge cells growing in M1 can be genetically modified using a CRISPR/Cas12a gene editing system. A short sequence of scrambled DNA was inserted using a single-stranded oligodeoxynucleotide donor template to disrupt the 2′,5′-oligoadenylate synthetase gene of cells from the boreal deep-sea sponge Geodia barretti. A blue fluorescent marker gene appeared to be inserted in an intron of the same gene and expressed by a small number of G. barretti cells. Our results represent an important step toward developing an optimized continuous sponge cell line to produce bioactive compounds.
Introduction
Developing a marine sponge cell line to produce sponge-derived chemicals in vitro has been the holy grail of sponge biotechnology ever since it was discovered that marine sponges host a wide variety of bioactive secondary metabolites in marine sponges (Pomponi, 1999; Rinkevich, 2005; Mayer et al., 2010; Schippers et al., 2011; Newman and Cragg, 2016). Many of these compounds have the potential to be developed into new drugs to combat cancer (Nuijen et al., 2000; Schwartsmann et al., 2003; Jimenez et al., 2018; Khalifa et al., 2019), inflammatory disease (Alcaraz and Paya, 2006) and infections in humans (Laport et al., 2009; Abdelmohsen et al., 2017; Liu et al., 2019a). Others could be used in industrial and commercial applications, for example as anti-fouling agents (Qi and Ma, 2017), cosmetics and nutraceuticals (Balboa et al., 2015). Each of these applications potentially represents a multibillion-dollar market (Thoms and Schupp, 2005; Greco and Cinquegrani, 2016). However, lack of sufficient biomass to produce compounds at the scale required for clinical trials or industrial application has been a major bottleneck (Nuijen et al., 2000; Singh and Thakur, 2016). Harvesting of wild sponges is neither economically nor environmentally sustainable and chemical synthesis of many sponge-derived chemicals is time-consuming and expensive due to their complex nature (Nuijen et al., 2000; Thoms and Schupp, 2005; Montaser and Luesch, 2011; Singh and Thakur, 2016). Underlying metabolic pathways are often equally complex and poorly understood, making metabolically engineering yeasts or bacteria to produce compounds difficult (Thoms and Schupp, 2005; Montaser and Luesch, 2011). Although mariculture can be suitable in some cases (Brummer and Nickel, 2003; Duckworth and Battershill, 2003; Page et al., 2005), it does not allow to precisely control culture conditions or optimize productivity (Montaser and Luesch, 2011; Santhi et al., 2017). Culturing sponge cells in vitro has been proposed as a solution, with sponge cells producing the compound in a controlled environment that can be adapted and optimized. Furthermore, harvesting and downstream processing are simplified compared to mariculture.
Developing such sponge cell cultures has been challenging (Pomponi et al., 1997, Rinkevich, 2005; Schippers et al., 2011). Little was known about the nutrients sponge cells need to proliferate, resulting in short-lived primary cultures (Schippers et al., 2011). Cells in such cultures quickly became less viable and metabolically active, and were often overgrown by bacteria or fungi (Sipkema et al., 2005). Recently, it was shown that cells of the Caribbean sponge Dysidea etheria were more metabolically active in sponge-specific cell culture medium, named M1, which contains optimized amino acid concentrations (Munroe et al., 2019). Furthermore, cells of various other sponge species divided rapidly in M1. Cells of 3 species from the genus Geodia responded strongly to M1 with limited variation between individuals. Cells could be subcultured 3–5 times and reached a maximum of 7 population doublings (Conkling et al., 2019). One of these was Geodia barretti, one of the species that dominates the sponge grounds in the deep waters of the North Atlantic ocean (Klitgaard and Tendal, 2004; Murillo et al., 2012; Knudby et al., 2013). G. barretti has been found to produce multiple bioactive compounds, such as barretins, a group of 6-bromoindole derivatives with anti-inflammatory (Lind et al., 2013; Di et al., 2018) and anti-fouling (Sjogren et al., 2004; Hedner et al., 2008) activity, and more recently discovered anti-fouling peptides called barrettides (Carstens et al., 2015).
Having proliferating cells is an important step toward producing compounds in large-scale bioreactors. Moreover, it facilitates developing molecular tools and genetic engineering in particular, as dividing cells are not only more viable, but also more genetically accessible (Wang et al., 2014). Until now, the only molecular technique used successfully in sponge cell culture is transfecting cells with plasmids to transiently express the human telomerase reverse transcriptase oncogene (Pomponi et al., 2007) and green fluorescent protein (Schippers, 2013). No genetic engineering tools that can make permanent changes in sponge cells to create and optimize production strains have been reported. One of the most well-known genetic engineering tools is CRISPR/Cas gene editing. CRISPR (clustered regularly-interspaced palindromic repeat) arrays were discovered as adaptive immune systems in prokaryotes. Short sequences of foreign DNA incorporated into CRISPR arrays in the genome are transcribed and processed into CRISPR RNAs (crRNA), that guide endonucleases to cleave the complementary DNA sequence (Horvath and Barrangou, 2010). These systems use a protospacer-adjacent motif (PAM) to distinguish between foreign DNA and the host genome, since the PAM is present in the foreign DNA but not in the CRISPR array. By altering the crRNA sequence, CRISPR/Cas systems can be used to specifically target any DNA sequence that contains the PAM (Figure 1). The endonuclease induces a double-stranded break (DSB) that can be repaired in two ways: the first, non-homologous end joining (NHEJ), often leads to small insertions/deletions causing frameshifts, while the second, homology-directed repair (HDR), can be used to insert DNA sequences by providing a repair template with homology up- and downstream of the target site (Song and Stieger, 2017; Liu et al., 2019a; Figure 2A). CRISPR/Cas9 is the most commonly used CRISPR system for gene editing but requires 2 RNA molecules to induce a DSB: the crRNA that matches the target sequence and an additional trans-encoded small crRNA (tracrRNA). The crRNA and tracrRNA can be linked together to create a single guide RNA, but this requires an ~100 base pair (bp) long RNA molecule for each target sequence (Jinek et al., 2012). Another endonuclease, called Cas12a or Cpf1, was discovered more recently and is an attractive alternative to Cas9, because it does not require a tracrRNA, and instead uses 1 short (~42 bp) guide RNA (gRNA) molecule (Zetsche et al., 2015; Swarts and Jinek, 2018; Figure 1). CRISPR/Cas12a has been used to edit genomes in various cell cultures of plants, animals and humans (Safari et al., 2019).
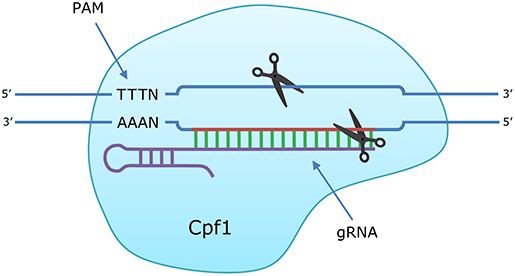
Figure 1. Introduction of a double stranded break (DSB) in DNA by CRISPR/Cas12a (Cpf1). The guide RNA (gRNA) directs the Cas12a endonuclease to the homologous target sequence. When the protospacer-adjacent motif (PAM) is recognized and the crRNA hybridizes with the target sequence, the nuclease domains of Cas12a introduce breaks in each strand: 19 nt (target strand) and 23 nt (complementary strand) downstream from the PAM sequence.
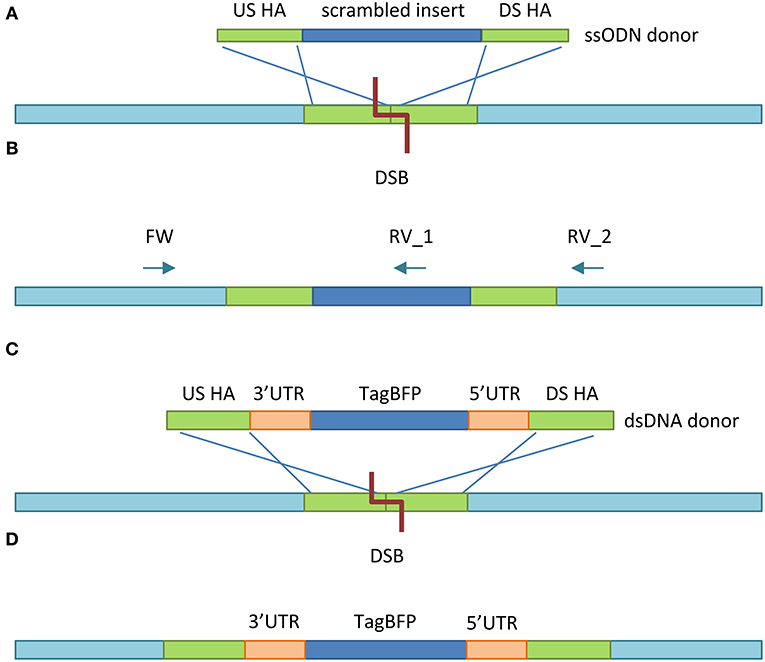
Figure 2. Experimental design. (A) After Cas12a introduces the DSB at the target site, HDR takes place between the US HA and DS HA of the ssODN donor template and the homologous regions US and DS from the DSB. (B) The insert is incorporated between the US and DS homologous region. Two PCR reactions determine whether mutation was successful: (1) reverse primer 1 (RV_1) anneals inside of the insert, yielding a product of 910 bp, providing HDR took place. (2) Reverse primer 2 (RV_2) anneals to the wild type gene sequence, yielding a 1,025 bp (wild type) or 1,127 (mutant) product. Both reactions use the same forward (FW) primer. (C) As with the ssODN donor, the US and DS HA recombine with their homologous regions US and DS of the DSB made by Cas12a. (D) The TagBFP gene with 5′ and 3′ UTRs of the S. domuncula actin locus is inserted into the genome in reverse orientation.
To test whether the highly versatile CRISRP/Cas12a system could also edit the genome of marine sponge cells in vitro, we designed a single-stranded oligodeoxynucleotide (ssODN) construct with short homology arms (HA) (30–40 bp). Donor templates provided as ssODN mediate HDR more efficiently than their double-stranded (dsDNA) counterparts (Yoshimi et al., 2016; Ferenczi et al., 2017) and shorter ssODN constructs are more efficient than longer ssODNs (Okamoto et al., 2019). The HAs matched regions upstream (US) and downstream (DS) of the target site in the 3rd OAS1Ab exon and flanked a 108 bp scrambled DNA insert (Figure 2A), containing stop codons in all 3 reading frames. To test whether the CRISPR/Cas12a system could be used to overexpress a gene, we designed a construct containing blue fluorescent marker gene TagBFP (Subach et al., 2008). Using the promoter and terminators sequences of a highly expressed gene in the host organism can ensure a heterologous gene can be efficiently recognized by the host cells' transcription machinery. However, no full genome is yet available for G. barretti, and no complete sequences of G. barretti 5′ and 3′ untranslated regions (UTRs), containing promoters, and terminators, respectively, have been reported. Therefore, we used the 5′ and 3′ UTRs of the Suberites domuncula actin locus, which have previously been used to transiently express a fluorescent marker in S. domuncula explants (Revilla-I-Domingo et al., 2018; Figure 2C). For the same reason, the 2′,5′-oligoadenylate synthetase 1 (OAS1Ab) gene (Accession Number HQ644329.1) was targeted, of which a partial genomic DNA sequence was reported (Vallmann et al., 2011). While the function of OAS1Ab in sponges has not been determined, OAS genes in humans, can induce apoptosis in tumor cells (Mullan et al., 2005). Therefore, OAS1Ab could also be an interesting future target gene to immortalize sponge cells. Heterologous genes would ideally be inserted in non-coding regions between 2 genes, to avoid interfering with the function of host genes. However, since no such intergenic sequences were available, the TagBFP gene would be inserted in the 2nd OAS1Ab intron in reverse orientation (Figure 2D), to reduce interfering of the marker gene with the function of the OAS1Ab gene. The ~2 kilobase (kb) dsDNA construct required longer HAs of 0.5–1 kb each (Zhang et al., 2017; Bier et al., 2018) and our design therefore featured a 533 bp US HA and 601 bp DS HA.
Expressing heterologous genes on plasmids in sponge cells has been a challenge in the past (Schippers, 2013). Therefore, pre-assembled ribonucleoprotein complexes (RNPs) of recombinant Cas12a protein and the guide RNA (gRNA) were used, rather than a plasmid encoding Cas12a and the gRNA. This method has been used to edit genes in mammalian cells (Hur et al., 2016; Liu et al., 2019a), zebrafish (Liu et al., 2019b), mouse embryos (Hur et al., 2016), and plants (Kim et al., 2017). Lipofection was used to deliver the Cas12a-gRNA complexes and donor construct, since this method has been used successfully to transfect sponge cells in vitro (Pomponi et al., 2007; Schippers, 2013). We confirmed that the scrambled DNA sequence was inserted by amplifying the region with polymerase chain reaction (PCR) (Figure 2B) and Sanger sequencing. Fluorescence microscopy was then used to verify that the TagBFP gene was inserted and expressed.
Materials and Methods
Sampling, Dissociation, and Cryopreservation
The G. barretti individual used in this study was collected with a triangular dredge at 500 meter depth in the Norwegian fjords (59°58.8″N 5°22.4′′E) and selected based on size (>15 cm3) to ensure sufficient cells could be obtained. The sponge was placed in a bucket filled with sea water as swiftly as possible to minimize exposure to air and ice packs were added to the bucket to keep the sponge at low temperature during transport to the laboratory. Sponge cells were dissociated by squeezing small pieces of tissue through a sterile gauze into artificial sea water (ASW) (23.30 g/L NaCl, 10.20 g/L MgCl2, 4.02 g/L Trizma HCl, 2.97 g/L Trizma Base, 1.1 g/L CaCl2, and 1 g/L KCl in dH2O, sterilized by filtering through a 0.22 μm mesh (Munroe et al., 2018, 2019). The cells were passed through a Falcon® 40 μm cell strainer (Corning, NY, USA), then centrifuged (300 × g, 5 min) and resuspended in ASW twice. The cells were diluted 100x in ASW and counted microscopically using a C-chipTM, Neubauer improved disposable hemocytometer (INCYTO, Cheonan, Republic of Korea). Cells were centrifuged once more (300 × g, 5 min) and resuspended in a cryoprotectant solution composed of 10% fetal bovine serum (FBS) and 10% dimethyl sulfoxide (DMSO) in ASW (Pomponi et al., 1997; Mussino et al., 2013; Munroe et al., 2018) at ~1.00E+08 cells/mL. Aliquots (1 mL) of this cell suspension in cryoprotectant, in FisherbrandTM 1 mL cryogenic vials (Thermo Fisher Scientific, MA, USA) were cooled at 1°C per min to −80°C in a Mr. Frosty freezing container (Nalgene®, NY, USA).
Medium Preparation
M1 medium was prepared following the original protocol (Munroe et al., 2019). Medium 199 powder (Sigma Aldrich, MO, USA) was dissolved in dH2O, then salts were added at concentrations like those in seawater, leading to a salinity of 33.5 ppt in M1, which is approximately the salinity of 35 ppt in seawater. The pH of the M1 medium (7.9) was also like that of seawater (8.1). Amino acids were added and the medium was sterilized by filtering through a 0.22 μm mesh. Finally, 30 μg/mL rifampicin (Sigma Aldrich) and 2.50 μg/mL amphotericin B (Sigma Aldrich) were added to control contamination by bacteria and fungi, respectively.
Cell Culture
Before cultures were inoculated, cryopreserved cells were thawed quickly in a water bath set at 50°C to prevent that cells would be damaged by ice crystals. The cells were washed twice with ASW by centrifuging at 300 × g for 5 min, removing the supernatant and resuspending them in 1 mL ASW. Next, cells were counted microscopically using disposable hemocytometers (C-chipTM, Neubauer improved) to determine their concentration. M1 medium was inoculated at 3.00E+06 cells/mL in 12-well plates with 1 mL cell suspension per well. To passage the cells, cell concentrations were determined, and the cell suspension was diluted back to 3.00E+06 cells/mL with fresh M1 medium.
gRNA Design and in vitro Assay
The online tool CRISPOR® (CRISPOR v4.97, RRID:SCR_015935) (Concordet and Haeussler, 2018) was used to design 3 gRNAs targeting the 3rd exon and 2 gRNAs targeting the 2nd intron (Supplementary Table 1) in the genomic G. barretti OAS1Ab sequence (Accession Number HQ644329.1) and ordered as synthetic Cas12a gRNA oligonucleotides at Integrated DNA Technologies (Leuven, Belgium). The recombinant Cas12a enzyme (EnGen® Lba Cas12a, derived from Lachnospiraceae bacterium ND2006, New England Biolabs, MA, USA) contains a poly-histidine tag and was overexpressed in Escherichia coli and purified using immobilized metal affinity chromatography (IMAC) (Loughran and Walls, 2011). All 3 crRNAs were reconstituted with nuclease free water (NFW) to a concentration of 300 nM and then tested in vitro for their efficiency in cleaving a 1,025 bp PCR product amplified from G. barretti genomic DNA (gDNA) [see “Genomic DNA extraction and PCR” section for DNA extraction, PCR conditions and primers (RV-WT)] in complex with the Cas12a enzyme following the manufacturer's instructions. For each crRNA, 3 μL of 300 nM stock solution (final concentration 30 nM) was mixed with 1 μL of 1 μM stock of Cas12a enzyme (final concentration 33.3 nM) to allow the ribonucleoprotein (RNP) complexes to form. The mixture was incubated for 10 min at 37°C in 3 μL 10x NEBuffer 2.1 Reaction Buffer (New England Biolabs) diluted with 20 μL NFW. Subsequently, 3 μL of 30 nM DNA fragment stock solution (final concentration 3 nM) was added to the reaction mixture and incubated with the RNPs for 10 min at 37°C. The final reaction volume after adding the PCR product was 30 μL. Samples were incubated for 10 min with 20 ng/μL proteinase K (Thermo Fisher Scientific) to stop the reaction. Finally, 10 μL of the reaction mixture was run on a 2% agarose gel at 135 V for 50 min.
ssODN and dsDNA Donor Template Design
Homology-directed repair of DSBs requires a donor template with homology arms up- and downstream of the target site. Synthetic single-stranded oligodeoxynucleotides (ssODNs) were ordered from Integrated DNA Technologies, designed featuring 30–40 bp homology arms flanking a 108 bp scrambled DNA insert containing stop codons in all 3 reading frames (Supplementary Table 1). A plasmid containing the dsDNA donor template, consisting of the TagBFP gene with S. domuncula actin 5′ and 3′ UTRs in reverse orientation, flanked by the US (533 bp) and DS (601 bp) HA, was ordered from Integrated DNA Technologies (Supplementary Figure 1).
Lipofection
Cells of 1 individual of G. barretti were cultured in 12-well plates with 1 mL cell suspension/well, starting at a density of 3.00E+06 cells/mL in M1 medium and a temperature of 4°C (Conkling et al., 2019). The ssODNs were transfected together with the RNPs using LipofectamineTM CRISPRMAXTM (Thermo Fisher Scientific). The TagBFP repair construct plasmid was introduced using LipofectamineTM 3000 (Thermo Fisher Scientific), 10 min before the RNPs were added using LipofectamineTM CRISPRMAXTM. For each sample, 0.5 μg of plasmid DNA and 5 μL of P3000TM reagent were added to 50 μL Opti-MEMTM I Reduced Serum Medium (GibcoTM, Thermo Fisher Scientific) in a 1.5 mL Eppendorf tube, and 3 μL LipofectamineTM 3000 in 50 μL Opti-MEMTM in a second tube. The contents of both tubes were mixed and incubated for 15 min at room temperature before the mixture was added dropwise to the sponge cells. RNPs were formed in vitro by preparing the following for each sample: 0.5 μg of gRNA E32 or I22, 2.5 μg of Cas12a, and 5 μL of Cas9 PlusTM Reagent in 50 μL Opti-MEMTM in 1 Eppendorf tube, and in another tube 3 μL of Lipofectamine® CRISPRMAXTM transfection reagent in 50 μL Opti-MEMTM. The contents of both tubes were combined and incubated for 10 min at room temperature. For transfection with the 108 bp scrambled DNA construct, 0.5 μg ssODN was mixed in at this point, and the solution was then added dropwise to the cells. For TagBFP plasmid transfection, no DNA was added at this stage.
Genomic DNA Extraction and PCR
After 4 days of incubation, gDNA was obtained from transfected and non-transfected cells using the High Pure PCR Template Preparation Kit (Roche Life Science, Penzberg, Germany). Successfully mutated cells were detected using 2 PCR reactions (Figure 2C). Both reactions used the same forward primer (FW 5′-ATGGCTAGCCCAGGACTTAGG), while the reverse primer bound either downstream of the target site (RV-wild-type 5′-AACAATGTGGTGCACTCGAA) or inside the scrambled DNA insert (RV-mutant 5′-CTATCCCCACCCCCACATTC). The 1st reaction should always yield a product, either of 1,025 bp in the wild-type, or 1,127 bp in case of mutated cells. The 2nd reaction would only amplify a 910 bp product if some cells in the culture were successfully mutated. The PCR mixture consisted of 25 μL Q5® High-Fidelity 2X Master Mix (New England Biolabs), 2.5 μL of working solution (10 μM) of both primers and ~20 ng of gDNA brought to a total reaction volume of 50 μL with MilliQ (MQ) water. PCR conditions were as follows: initial denaturation at 98°C for 15 s, followed by 35 cycles of denaturation at 98°C for 10 s, primer annealing at 59°C for 30 s, and extension at 72°C for 45 s, and final extension at 72°C for 10 min. Amplified products were purified using the DNA Clean and Concentrator-5 (capped) kit (Zymo Research, CA, USA) and eluted in a final volume of 10 μL MQ water. Product concentration in the purified samples was measured using NanoDropTM One (Thermo Fisher Scientific). Approximately 1 μg of purified PCR product per sample was run on a 1% agarose gel at 70 V for 35 min, with 500 ng GeneRuler 1 kb DNA Ladder (Thermo Fisher Scientific) as a marker on both sides.
Mutation Analysis
To verify that the 108 bp scrambled DNA sequence was successfully inserted into the 3rd exon of the OAS1Ab gene, 15 ng purified PCR product per 100 bp length (~140 ng for WT, ~155 ng for M products) for each sample was sent to BaseClear for Sanger sequencing as Long run (up to 1,100 nt) Quick Shot Premix samples with 25 pmol primer in a total volume of 20 μL. Sequences were aligned using SnapGene® 4.3 (SnapGene, RRID:SCR_015052) and to compare sequences obtained through Sanger sequencing to the genomic DNA sequence of the G. barretti OAS1Ab gene (Accession Number HQ644329.1). The 4′,6-diamidino-2-phenylindole (DAPI) filter on an EVOSTM FL Auto Cell Imaging System (Thermo Fisher Scientific), was used to determine whether TagBFP was successfully inserted into the 2nd intron of the OAS1Ab gene and expressed by the cells.
Results
gRNA Design and in vitro Assay
Synthetic gRNAs were designed for both target sites in the G. barretti OAS1Ab gene, 3 gRNAs targeted the 3rd exon and 2 gRNAs targeted the 2nd intron, as only 2 suitable target sequences for Cas12a were present in the intron that would leave space for >0.5 kb US and DS HAs (Figure 3A). An in vitro assay in which RNPs of Cas12a with each of the 5 gRNAs were added to a PCR product of the wild-type OAS1Ab gene was used to test how efficiently each gRNA could induce a DSB (Figure 2B, FW + RV primers). The same PCR product incubated with Cas12a but without a gRNA was used as a negative control (C). All 5 tested gRNAs induced cuts at their target sites (Figure 3B), but only exon 3 gRNA 2 (gRNA E32), and intron 2 gRNA 2 (gRNA I22) degraded all DNA present. We selected gRNA E32 and I22 to edit the G. barretti genome.
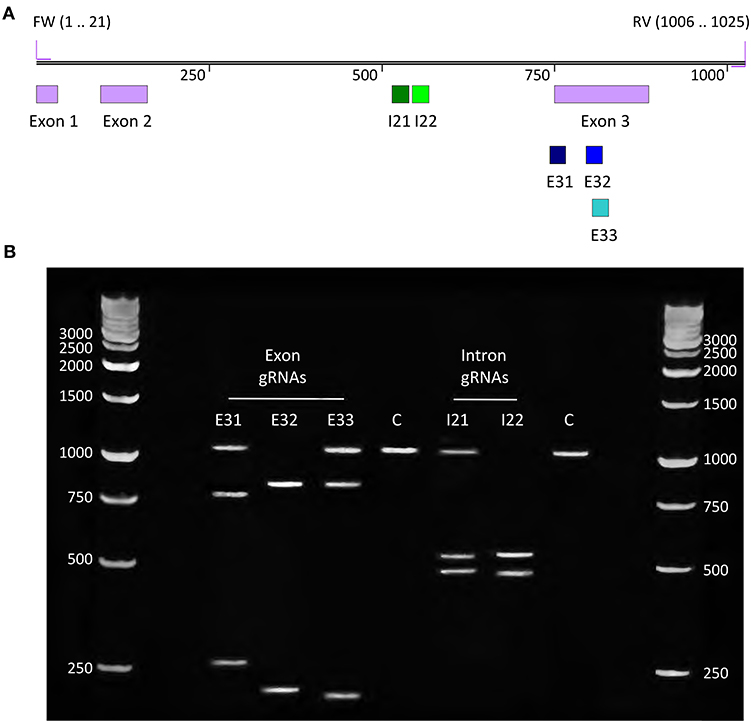
Figure 3. gRNA design & in vitro assay. (A) Sequence map showing locations of gRNAs on the OAS1Ab wild-type PCR product of 1,025 bp. (B) Gel electrophoresis results of the in vitro assay to determine how efficiently 5 gRNAs can direct Cas12a to cleave their respective target sites in the 1,025 bp wild-type PCR product. Most gRNAs partially degraded the target DNA, resulting in 3 visible bands (the full-length PCR product and the 2 cleavage products). gRNA E32 and I22 fully degraded the DNA, leaving only their 2 cleavage products as visible bands, and these gRNAs were selected to edit the G. barretti OAS1Ab gene.
Knock-Out OAS1Ab
Geodia barretti cells were transfected with RNPs of Cas12a and gRNA E32 accompanied by the ssODN donor template to insert the 108 bp scrambled DNA sequence and disrupt the OAS1Ab gene. Whether transfected cells were successfully mutated was checked using 2 PCR reactions (Figure 2B). The 1st reaction should always yield an either 1,025 bp (wild-type) or 1,127 bp (mutant) product or both, while the 2nd reaction only amplified a 910 bp product if mutated cells were present in the culture. Both the mutant and wild-type PCR reactions amplified their respective products in 2 out of 3 (T1 + T2) transfected cell populations (Figure 4A), suggesting the 108 bp scrambled DNA sequence was inserted into the OAS1Ab gene. Only the 1,025 bp wild-type product was detected in PCR 1 of T1 and T2, suggesting that mutated genes were present in small numbers and therefore not amplified due to the majority of wild-type genes in the extracted gDNA. The 3rd transfection (T3) yielded the same results as the negative control of cells transfected with Cas12a and ssODN donor, but without the gRNA (C), where only the wild-type PCR amplified its 1,025 bp product (Figure 4A). Sanger sequencing results of the mutant PCR product aligned with the expected sequence (Figure 4B), confirming the OAS1Ab gene was successfully edited.
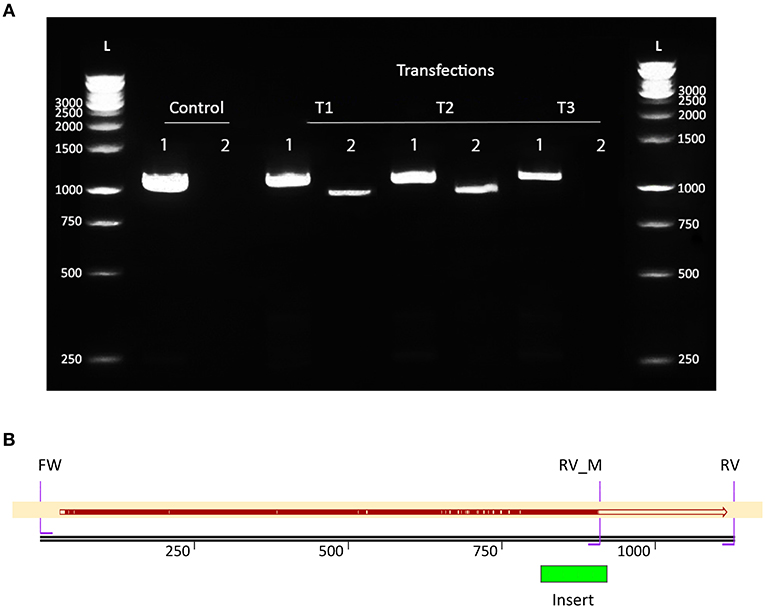
Figure 4. PCR and sequencing results confirm that the scrambled DNA sequence was inserted in populations of transfected cells. (A) Results of 2 PCR reactions amplifying part of the OAS1Ab gene in DNA extracted from 3 transfected G. barretti cell populations (T1, 2 and 3). Both reactions used the same FW primer. PCR 1 used the wildtype RV primer and should always amplify a 1,025 bp (W) or 1,127 bp (M) product. PCR 2 contained the mutant RV (RV_M) primer and only yields a 910 bp product (M) if the gene was successfully edited. The control (C) contained DNA extracted from cells that were treated the same as T1–3, except that no gRNA was provided in the transfection. T1 and T2 both yielded the M product, indicating the 108 bp scrambled DNA sequence was inserted at the target site in some cells. (B) Alignment of the sequencing results of the 910 bp mutant product with the sequence expected if the OAS1Ab gene was successfully edited. Red indicates matching sequences, while yellow indicates gaps. Since the sequenced PCR product was obtained using PCR 2, the sequence between RV_M and RV was not present in the sequence read.
Expression TagBFP Marker Gene
To insert and overexpress the blue fluorescent TagBFP gene, G. barretti cells were first transfected with the plasmid containing the TagBFP repair construct (Figure 2C), then after 10 min transfected again with the RNPs of Cas12a and gRNA I22. Whether cells were expressing TagBFP was determined by fluorescence microscopy. Sporadic blue fluorescence was observed in cells transfected with RNPs and the TagBFP repair construct (Figure 5). No fluorescent sponge cells were observed in controls transfected with the repair construct and Cas12a but without the gRNA. No clear PCR results were obtained, likely due to the low number of mutant compared to wild-type genes.
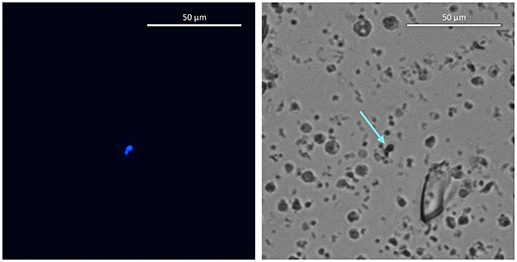
Figure 5. Blue fluorescing cells in images taken with DAPI (Left) and transmitted light (Right) filters at 80x magnification. Arrow indicates the cells that fluoresce blue in the DAPI filter image.
Discussion
We previously reported that cells from several species of marine sponges divided rapidly in M1 medium and could be subcultured for several weeks (Conkling et al., 2019). Considering that proliferating cells are currently available for multiple species, the next step is to improve various characteristics of the cells using molecular tools, to create efficient production strains for sponge-derived biopharmaceuticals. For example, by increasing how many population doublings cells can reach to produce more biomass, activating or optimizing pathways that synthesize potential drug candidates, or improving energy efficiency and other traits. CRISPR/Cas systems are currently the most prominent and promising tools, with new applications being reported in rapid succession (Zhang et al., 2018, 2020; Moon et al., 2019; Li et al., 2020). In this study, we report the first successful use of a CRISPR/Cas system in marine sponges.
We demonstrated that CRISPR/Cas12a (Cpf1) can be used to target and edit sites in the G. barretti genome, and that sequences can be inserted at these sites through HDR. We disrupted the OAS1Ab gene in G. barretti cells by inserting a 108 bp scrambled DNA sequence with stop codons in all 3 reading frames, ensuring no functional protein could be synthesized. As more genome sequence data from sponges become available, the method presented here could be used to disrupt other targets, like well-known tumor-suppressor genes, but also metabolic genes to direct energy and carbon sources toward producing compounds of interest, or unknown genes to dissect their functions. The role of OAS1Ab in sponges has not yet been determined (Vallmann et al., 2011), but examining how knocking out this gene affects sponge cells in culture would shed light on this question. If OAS1Ab acts as a tumor suppressor in sponges, as OAS1 does in humans, disrupting it could help immortalize sponge cells. In this study, we did not observe increased lifespan in transfected cells. However, cell lines of G. barretti in M1 medium have a limited lifespan of maximum 7 population doublings with little variation between individuals (Conkling et al., 2019). As it is unlikely that cells from different individuals are approximately the same number of doublings away from senescence, it seems that M1 medium is not able to support more than 7 doublings, regardless of whether the cells are immortalized. The number of doublings reached by G. barretti cells in the further optimized successor of M1 medium (OpM1) that was developed in our group far exceeded 7 doublings (Hesp et al., unpublished). Knocking out OAS1Ab or other potential tumor-suppressor genes in G. barretti cells cultured in OpM1 medium could therefore still result in immortalization.
We transfected cells with reporter constructs encoding TagBFP flanked by the promoter and terminator regions of the S. domuncula actin locus (Revilla-I-Domingo et al., 2018). TagBFP-transfected cells did sporadically display bright blue fluorescence that was not observed in controls that were transfected with Cas12a and the TagBFP repair construct, but without the gRNA. This excludes the possibility that cells were expressing TagBFP from the donor plasmid and suggests that edited cells could express TagBFP at high levels, but the editing efficiency was extremely low. We attempted to quantify the number of blue fluorescent cells with flow cytometry. However, the number of fluorescent cells was too low, and although the number was higher in transfected cells than controls, the difference was not large enough to draw any conclusions (data not shown). It is possible that other marker genes would be expressed more efficiently, such as green fluorescent protein (Chalfie et al., 1994) or the deep-red fluorescent mCherry (Merzlyak et al., 2007). Another well-documented method to increase heterologous gene expression is optimizing the use of codons in the gene to match the codon usage bias in the host organism's genome (Angov, 2011). Such bias has been observed in the sponges S. domuncula (Perina et al., 2009) and Geodia cydonium (Gamulin et al., 2001), and could affect TagBFP expression in G. barretti. More efficient gene editing in G. barretti could be achieved by optimizing the lipofection protocol or increasing how efficiently cells can perform HDR with the donor template. Varying the length of the homology arms can greatly influence HDR efficiency (Shin et al., 2014; Song and Stieger, 2017), and recently a new promising method was developed, where the dsDNA template is provided on a plasmid, flanked by target sites for the same gRNA as will be used to edit the genome (Zhang et al., 2017; Kanca et al., 2019). Another option would be to select for cells expressing a marker gene that protects them from an otherwise lethal chemical, such as geneticin (G418) or hygromycin B (Santerre et al., 1984). Once larger constructs can be inserted more efficiently, this technique could be used to permanently overexpress oncogenes or genes in pathways synthesizing potential biopharmaceuticals to increase productivity.
Our results represent the first venture into genetically modifying marine sponge cells. We used a CRISPR/Cas12a system to target and accurately disrupt a specific gene in cultured G. barretti cells. CRISPR/Cas systems are versatile and have been used in many types of organisms, now including G. barretti, which bodes well for using the methods developed in this study in other sponge species. However, because sponge species and individuals often respond differently to certain treatments (Conkling et al., 2019), it is likely that methods will need to be optimized on a case-by-case basis. More research is needed to improve HDR efficiency in sponge cells, in particular for inserting longer constructs needed to express heterologous genes. Nevertheless, we have shown that CRISPR/Cas12a holds promise as a powerful tool in sponge cell culture and that it should be pursued further. Once the method has been fully established and optimized in G. barretti and other sponges, it can be used to study gene functions to better understand fundamental aspects of sponge biology, as well as change or improve how genes work in sponge cells for biotechnological applications. for example, to develop promising sponge-derived drug candidates that are either very complex and therefore expensive to chemically synthesize, or present in sponges at concentrations too low for wild harvest or aquaculture (Duckworth and Battershill, 2003; Sipkema et al., 2005; Duckworth, 2009), such as anti-tumor compounds halichondrin B (Hirata and Uemura, 1986) and peloruside A (West et al., 2000). However, since in vitro cell cultures are optimizable and scalable, they are an attractive alternative production platform for any sponge-derived compound, even ones produced by symbiotic bacteria, which could be cultured together with sponge cell strains optimized using CRISPR-Cas12a. By demonstrating that CRISPR/Cas12a can be used to edit the genome of G. barretti cells in vitro, we have taken another important step toward creating and improving sponge cell strains to produce sponge-derived pharmaceuticals in quantities that allow clinical trials and ultimately, treat disease in patients with novel, sponge-derived drugs.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
KH conceived, designed, and conducted experiments, analyzed data, prepared figures for the paper, and wrote the paper. JF and A-MA conceived, designed, conducted experiments, and reviewed drafts of the paper. JvdL conceived and designed experiments, and reviewed drafts of the paper. DM conceived and designed experiments, analyzed data, contributed reagents, materials, analysis tools, and reviewed drafts of the paper. RW conceived and designed experiments, contributed reagents, materials, analysis tools, and reviewed drafts of the paper. SP conceived and designed experiments, collected the sponges, supervised the research, analyzed data, contributed reagents, materials, analysis tools, field expenses, and reviewed drafts of the paper. All authors contributed to the article and approved the submitted version.
Funding
DM, RW, and SP received funding from the European Union Horizon 2020 Project SponGES under Grant Agreement No. 679848.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully remember Dr. Hans Tore Rapp (University of Bergen) and his unrelenting positivity and willingness to help us and other colleagues, for example by collecting and transporting Geodia barretti samples used in this study. May he rest in peace. We thank Mihris Naduthodi (Microbiology Department, Wageningen University & Research) for providing the Cas12a enzyme used in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.599825/full#supplementary-material
References
Abdelmohsen, U. R., Balasubramanian, S., Oelschlaeger, T. A., Grkovic, T., Pham, N. B., Quinn, R. J., et al. (2017). Potential of marine natural products against drug-resistant fungal, viral, and parasitic infections. Lancet Infect. Dis. 17, 30–41. doi: 10.1016/S1473-3099(16)30323-1
Alcaraz, M. J., and Paya, M. (2006). Marine sponge metabolites for the control of inflammatory diseases. Curr. Opin. Invest. Drugs 7, 974–979.
Angov, E. (2011). Codon usage: nature's roadmap to expression and folding of proteins. Biotechnol. J. 6, 650–659. doi: 10.1002/biot.201000332
Balboa, E. M., Conde, E., Soto, M. L., Perez-Armada, L., and Dominguez, H. (2015). “Cosmetics from marine sources,” in Springer Handbook of Marine Biotechnology, ed S. Kim (Berlin: Springer), 1015–1042. doi: 10.1007/978-3-642-53971-8_44
Bier, E., Harrison, M. M., O'Connor-Giles, K. M., and Wildonger, J. (2018). Advances in engineering the fly genome with the CRISPR-cas system. Genetics 208, 1–18. doi: 10.1534/genetics.117.1113
Brummer, F., and Nickel, M. (2003). Sustainable use of marine resources: cultivation of sponges. Prog. Mol. Subcell. Biol. 37, 143–162. doi: 10.1007/978-3-642-55519-0_6
Carstens, B. B., Rosengren, K. J., Gunasekera, S., Schempp, S., Bohlin, L., Dahlstrom, M., et al. (2015). Isolation, characterization, and synthesis of the barrettides: disulfide-containing peptides from the marine sponge Geodia barretti. J. Nat. Prod. 78, 1886–1893. doi: 10.1021/acs.jnatprod.5b00210
Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., and Prasher, D. C. (1994). Green fluorescent protein as a marker for gene expression. Science 263, 802–805. doi: 10.1126/science.8303295
Concordet, J., and Haeussler, M. (2018). CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, 242–245. doi: 10.1093/nar/gky354
Conkling, M., Hesp, K., Munroe, S., Sandoval, K., Martens, D. E., Sipkema, D., et al. (2019). Breakthrough in marine invertebrate cell culture: sponge cells divide rapidly in improved nutrient medium. Sci. Rep. 9:17321. doi: 10.1038/s41598-019-53643-y
Di, X., Rouger, C., Hardardottir, I., Freysdottir, J., Molinski, T. F., Tasdemir, D., et al. (2018). 6-Bromoindole derivatives from the icelandic marine sponge Geodia barretti: isolation and anti-inflammatory activity. Mar. Drugs 16:437. doi: 10.3390/md16110437
Duckworth, A. (2009). Farming sponges to supply bioactive metabolites and bath sponges: a review. Mar. Biotechnol. 11, 669–679. doi: 10.1007/s10126-009-9213-2
Duckworth, A. R., and Battershill, C. (2003). Sponge aquaculture for the production of biologically active metabolites: the influence of farming protocols and environment. Aquaculture 221, 311–329. doi: 10.1016/S0044-8486(03)00070-X
Ferenczi, A., Pyott, D. E., Xipnitou, A., and Molnar, A. (2017). Efficient targeted DNA editing and replacement in Chlamydomonas reinhardtii using Cpf1 ribonucleoproteins and single-stranded DNA. Proc. Natl. Acad. Sci. U.S.A. 114, 13567–13572. doi: 10.1073/pnas.1710597114
Gamulin, V., Peden, J. F., Müller, I. M., and Müller, W. E. G. (2001). Codon usage in the siliceous sponge Geodia cydonium: highly expressed genes in the simplest multicellular animals prefer C- and G-ending codons. J. Zool. Syst. Evol. Res. 39, 97–102. doi: 10.1046/j.1439-0469.2001.00163.x
Greco, G. R., and Cinquegrani, M. (2016). Firms plunge into the sea. Marine biotechnology industry, a first investigation. Front. Mar. Sci. 2:124. doi: 10.3389/fmars.2015.00124
Hedner, E., Sjögren, M., Hodzic, S., Andersson, R., Göransson, U., Jonsson, P. R., et al. (2008). Antifouling activity of a dibrominated cyclopeptide from the marine sponge Geodia barretti. J. Nat. Prod. 71, 330–333. doi: 10.1021/np0705209
Hirata, Y., and Uemura, D. (1986). Halichondrins - antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 58, 701–710. doi: 10.1351/pac198658050701
Horvath, P., and Barrangou, R. (2010). CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170. doi: 10.1126/science.1179555
Hur, J. K., Kim, K., Been, K. W., Baek, G., Ye, S., Hur, J. W., et al. (2016). Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat. Biotechnol. 34, 807–808. doi: 10.1038/nbt.3596
Jimenez, P. C., Wilke, D. V., and Costa-Lotufo, L. V. (2018). Marine drugs for cancer: surfacing biotechnological innovations from the oceans. Clinics 73:e482s. doi: 10.6061/clinics/2018/e482s
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Kanca, O., Zirin, J., Garcia-Marques, J., Knight, S. M., Yang-Zhou, D., Amador, G., et al. (2019). An efficient CRISPR-based strategy to insert small and large fragments of DNA using short homology arms. Elife 8:e51539. doi: 10.7554/eLife.51539
Khalifa, S. A. M., Elias, N., Farag, M. A., Chen, L., Saeed, A., Hegazy, M. F., et al. (2019). Marine natural products: a source of novel anticancer drugs. Mar. Drugs 17:491. doi: 10.3390/md17090491
Kim, H., Kim, S. T., Ryu, J., Kang, B. C., Kim, J. S., and Kim, S. G. (2017). CRISPR/Cpf1-mediated DNA-free plant genome editing. Nat. Commun. 8:14406. doi: 10.1038/ncomms14406
Klitgaard, A. B., and Tendal, O. S. (2004). Distribution and species composition of mass occurrences of large-sized sponges in the northeast Atlantic. Prog. Oceanogr. 61, 57–98. doi: 10.1016/j.pocean.2004.06.002
Knudby, A., Kenchington, E., and Murillo, F. J. (2013). Modeling the distribution of Geodia sponges and sponge grounds in the Northwest Atlantic. PLoS ONE 8:e82306. doi: 10.1371/journal.pone.0082306
Laport, M. S., Santos, O. C., and Muricy, G. (2009). Marine sponges: potential sources of new antimicrobial drugs. Curr. Pharm. Biotechnol. 10, 86–105. doi: 10.2174/138920109787048625
Li, H., Yang, Y., Hong, W., Huang, M., Wu, M., and Zhao, X. (2020). Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Sig. Transduct. Targeted Ther. 5:1. doi: 10.1038/s41392-019-0089-y
Lind, F. K., Hansen, E., Østerud, B., Eilertsen, K., Bayer, A., Engqvist, M., et al. (2013). Antioxidant and anti-inflammatory activities of barettin. Mar. Drugs 11, 2655–2666. doi: 10.3390/md11072655
Liu, M., El-Hossary, E. M., Oelschlaeger, T. A., Donia, M. S., Quinn, R. J., and Abdelmohsen, U. R. (2019a). Potential of marine natural products against drug-resistant bacterial infections. Lancet Infect. Dis. 19, e237–e245. doi: 10.1016/S1473-3099(18)30711-4
Liu, P., Luk, K., Shin, M., Idrizi, F., Kwok, S., Roscoe, B., et al. (2019b). Enhanced Cas12a editing in mammalian cells and zebrafish. Nucleic Acids Res. 47, 4169–4180. doi: 10.1093/nar/gkz184
Loughran, S. T., and Walls, D. (2011). Purification of poly-histidine-tagged proteins. Methods Mol. Biol. 681, 311–335. doi: 10.1007/978-1-60761-913-0_17
Mayer, A. M., Glaser, K. B., Cuevas, C., Jacobs, R. S., Kem, W., Little, R. D., et al. (2010). The odyssey of marine pharmaceuticals: a current pipeline perspective. Trends Pharmacol. Sci. 31, 255–265. doi: 10.1016/j.tips.2010.02.005
Merzlyak, E. M., Goedhart, J., Shcherbo, D., Bulina, M. E., Shcheglov, A. S., Fradkov, A. F., et al. (2007). Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat. Methods 4, 555–557. doi: 10.1038/nmeth1062
Montaser, R., and Luesch, H. (2011). Marine natural products: a new wave of drugs? Future Med. Chem. 3, 1475–1489. doi: 10.4155/fmc.11.118
Moon, S. B., Kim, D. Y., Ko, J. H., and Kim, Y. S. (2019). Recent advances in the CRISPR genome editing tool set. Exp. Mol. Med. 51, 1–11. doi: 10.1038/s12276-019-0339-7
Mullan, P. B., Hosey, A. M., Buckley, N. E., Quinn, J. E., Kennedy, R. D., Johnston, P. G., et al. (2005). The 2,5 oligoadenylate synthetase/RNaseL pathway is a novel effector of BRCA1- and interferon-gamma-mediated apoptosis. Oncogene 24, 5492–5501. doi: 10.1038/sj.onc.1208698
Munroe, S., Sandoval, K., Martens, D. E., Sipkema, D., and Pomponi, S. A. (2019). Genetic algorithm as an optimization tool for the development of sponge cell culture media. In vitro cellular & developmental biology. Animal 55, 149–158. doi: 10.1007/s11626-018-00317-0
Munroe, S., Martens, D. E., Sipkema, D., and Pomponi, S. A. (2018). Comparison of cryopreservation techniques for cells of the marine sponge Dysidea etheria. Cryo Lett. 39, 269–278.
Murillo, F. J., Muñoz, P. D., Cristobo, J., Ríos, P., González, C., Kenchington, E., et al. (2012). Deep-sea sponge grounds of the flemish cap, flemish pass and the grand banks of newfoundland (Northwest Atlantic Ocean): distribution and species composition. Mar. Biol. Res. 8, 842–854. doi: 10.1080/17451000.2012.682583
Mussino, F., Pozzolini, M., Valisano, L., Cerrano, C., Benatti, U., and Giovine, M. (2013). Primmorphs cryopreservation: a new method for long-time storage of sponge cells. Mar. Biotechnol. 15, 357–367. doi: 10.1007/s10126-012-9490-z
Newman, D. J., and Cragg, G. M. (2016). Drugs and drug candidates from marine sources: an assessment of the current “State of Play”. Planta Med. 82, 775–789. doi: 10.1055/s-0042-101353
Nuijen, B., Bouma, M., Manada, C., Jimeno, J. M., Schellens, J. H., Bult, A., et al. (2000). Pharmaceutical development of anticancer agents derived from marine sources. Anticancer. Drugs 11, 793–811. doi: 10.1097/00001813-200011000-00003
Okamoto, S., Amaishi, Y., Maki, I., Enoki, T., and Mineno, J. (2019). Highly efficient genome editing for single-base substitutions using optimized ssODNs with Cas9-RNPs. Sci. Rep. 9:4811. doi: 10.1038/s41598-019-41121-4
Page, M. J., Northcote, P. T., Webb, V. L., Mackey, S., and Handley, S. J. (2005). Aquaculture trials for the production of biologically active metabolites in the New Zealand sponge Mycale hentscheli (Demospongiae: Poecilosclerida). Aquaculture 250, 256–269. doi: 10.1016/j.aquaculture.2005.04.069
Perina, D., Harcet, M., Mikoč, A., Vlahoviček, K., Müller, W. E. G., and Cetković, H. (2009). Highly expressed genes in marine sponge Suberites domuncula prefer C- and G-ending codons. Food Technol. Biotechnol. 47, 269–274.
Pomponi, S. A. (1999). The bioprocess-technological potential of the sea. J. Biotechnol. 70, 5–13. doi: 10.1016/S0079-6352(99)80092-7
Pomponi, S. A., Thompson, J., Bottesch, J., and Sennett, S. (2007). Methods for extending the life span of marine invertebrate cells. US Patent. Patent 2007/0254367 A1.
Pomponi, S. A., Willoughby, R., Kaighn, M. E., and Wright, A. E. (1997). “Development of techniques for in vitro production of bioactive natural products from marine sponges,” in Invertebrate Cell Culture: Novel Directions and Biotechnology Applications (Science Publishers), 231–237.
Qi, S. H., and Ma, X. (2017). Antifouling compounds from marine invertebrates. Mar. Drugs 15:263. doi: 10.3390/md15090263
Revilla-I-Domingo, R., Schmidt, C., Zifko, C., and Raible, F. (2018). Establishment of transgenesis in the demosponge Suberites domuncula. Genetics 210, 435–443. doi: 10.1534/genetics.118.301121
Rinkevich, B. (2005). Marine invertebrate cell cultures: new millennium trends. Mar. Biotechnol. 7, 429–439. doi: 10.1007/s10126-004-0108-y
Safari, F., Zare, K., Negahdaripour, M., Barekati-Mowahed, M., and Ghasemi, Y. (2019). CRISPR Cpf1 proteins: structure, function and implications for genome editing. Cell Biosci. 9:36. doi: 10.1186/s13578-019-0298-7
Santerre, R. F., Allen, N. E., Hobbs, J. N. Jr., Rao, R. N., and Schmidt, R. J. (1984). Expression of prokaryotic genes for hygromycin B and G418 resistance as dominant-selection markers in mouse L cells. Gene 30, 147–156. doi: 10.1016/0378-1119(84)90115-X
Santhi, L. S., Talluri, V. S. S. L. P, Nagendra, S. Y., and Krishna, R.E. (2017). Bioactive compounds from marine sponge associates: antibiotics from bacillus sp. Nat. Products Chem. Res. 5, 1–5. doi: 10.4172/2329-6836.1000266
Schippers, K. J., Martens, D. E., Pomponi, S. A., and Wijffels, R. H. (2011). Cell cycle analysis of primary sponge cell cultures. In vitro cellular and developmental biology. Animal 47, 302–311. doi: 10.1007/s11626-011-9391-x
Schippers, K. J. (2013). Sponge cell culture (Ph.D. dissertation), Wageningen: Wageningen University & Research.
Schwartsmann, G., Da Rocha, A. B., Mattei, J., and Lopes, R. (2003). Marine-derived anticancer agents in clinical trials. Expert Opin. Investig. Drugs 12, 1367–1383. doi: 10.1517/13543784.12.8.1367
Shin, J., Chen, J., and Solnica-Krezel, L. (2014). Efficient homologous recombination-mediated genome engineering in zebrafish using TALE nucleases. Development 141, 3807–3818. doi: 10.1242/dev.108019
Singh, A., and Thakur, N. L. (2016). Significance of investigating allelopathic interactions of marine organisms in the discovery and development of cytotoxic compounds. Chem. Biol. Interact. 243, 135–147. doi: 10.1016/j.cbi.2015.09.009
Sipkema, D., Osinga, R., Schatton, W., Mendola, D., Tramper, J., and Wijffels, R. H. (2005). Large-scale production of pharmaceuticals by marine sponges: sea, cell, or synthesis? Biotechnol. Bioeng. 90, 201–222. doi: 10.1002/bit.20404
Sjogren, M., Goransson, U., Johnson, A. L., Dahlstrom, M., Andersson, R., Bergman, J., et al. (2004). Antifouling activity of brominated cyclopeptides from the marine sponge Geodia barretti. J. Nat. Prod. 67, 368–372. doi: 10.1021/np0302403
Song, F., and Stieger, K. (2017). Optimizing the DNA donor template for homology-directed repair of double-strand breaks. Mol. Ther. Nucleic Acids 7, 53–60. doi: 10.1016/j.omtn.2017.02.006
Subach, O. M., Gundorov, I. S., Yoshimura, M., Subach, F. V., Zhang, J., Gruenwald, D., et al. (2008). Conversion of red fluorescent protein into a bright blue probe. Chem. Biol. 15, 1116–1124. doi: 10.1016/j.chembiol.2008.08.006
Swarts, D. C., and Jinek, M. (2018). Cas9 versus Cas12a/Cpf1: structure-function comparisons and implications for genome editing. Wiley Interdiscip. Rev. RNA 9:e1481. doi: 10.1002/wrna.1481
Thoms, C., and Schupp, P. J. (2005). Biotechnological potential of marine sponges and their associated bacteria as producers of new pharmaceuticals (Part II). J. Int. Biotechnol. Law 86, 1506–1512. doi: 10.1515/jibl.2005.2.6.257
Vallmann, K., Aas, N., Reintamm, T., Lopp, A., Kuusksalu, A., and Kelve, M. (2011). Expressed 2-5A synthetase genes and pseudogenes in the marine sponge Geodia barretti. Gene 478, 42–49. doi: 10.1016/j.gene.2011.01.014
Wang, B., Pfeiffer, M. J., Schwarzer, C., Arauzo-Bravo, M. J., and Boiani, M. (2014). DNA replication is an integral part of the mouse oocyte's reprogramming machinery. PLoS ONE 9:e97199. doi: 10.1371/journal.pone.0097199
West, L. M., Northcote, P. T., and Battershill, C. N. (2000). Peloruside A: a potent cytotoxic macrolide isolated from the new zealand marine sponge Mycale sp. J. Org. Chem. 65, 445–449. doi: 10.1021/jo991296y
Yoshimi, K., Kunihiro, Y., Kaneko, T., Nagahora, H., Voigt, B., and Mashimo, T. (2016). ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat. Commun. 7:10431. doi: 10.1038/ncomms10431
Zetsche, B., Gootenberg, J. S., Abudayyeh, O. O., Slaymaker, I. M., Makarova, K. S., Essletzbichler, P., et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-cas system. Cell 163, 759–771. doi: 10.1016/j.cell.2015.09.038
Zhang, C., Quan, R., and Wang, J. (2018). Development and application of CRISPR/Cas9 technologies in genomic editing. Hum. Mol. Genet. 27, R79–R88. doi: 10.1093/hmg/ddy120
Zhang, J. P., Li, X. L., Li, G. H., Chen, W., Arakaki, C., Botimer, G. D., et al. (2017). Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 18:35. doi: 10.1186/s13059-017-1164-8
Keywords: CRISPR/Cas, marine sponge cell culture, genome editing, CRISPRMAX, homologous recombination
Citation: Hesp K, Flores Alvarez JL, Alexandru A-M, van der Linden J, Martens DE, Wijffels RH and Pomponi SA (2020) CRISPR/Cas12a-Mediated Gene Editing in Geodia barretti Sponge Cell Culture. Front. Mar. Sci. 7:599825. doi: 10.3389/fmars.2020.599825
Received: 28 August 2020; Accepted: 01 December 2020;
Published: 23 December 2020.
Edited by:
Ellen Kenchington, Bedford Institute of Oceanography (BIO), CanadaReviewed by:
Bruno Francesco Rodrigues de Oliveira, Federal University of Rio de Janeiro, BrazilApril Hill, Bates College, United States
Copyright © 2020 Hesp, Flores Alvarez, Alexandru, van der Linden, Martens, Wijffels and Pomponi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kylie Hesp, kylie.hesp@wur.nl