Comparison of whole genome sequences from human and non-human Escherichia coli O26 strains
 Keri N. Norman1
Keri N. Norman1  Michael L. Clawson1
Michael L. Clawson1  Nancy A. Strockbine2
Nancy A. Strockbine2  Robert E. Mandrell3
Robert E. Mandrell3  Roger Johnson4
Roger Johnson4  Kim Ziebell4
Kim Ziebell4  Shaohua Zhao5
Shaohua Zhao5  Pina M. Fratamico6 Robert Stones7
Pina M. Fratamico6 Robert Stones7  Marc W. Allard8
Marc W. Allard8  James L. Bono1*
James L. Bono1*- 1U.S. Meat Animal Research Center, United States Department of Agriculture, Agricultural Research Service, Clay Center, NE, USA
- 2Division of Foodborne, Waterborne and Environmental Diseases, National Center for Emerging and Zoonotic Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, GA, USA
- 3Western Regional Research Center, United States Department of Agriculture, Agricultural Research Service, Albany, CA, USA
- 4Laboratory for Foodborne Zoonoses, Public Health Agency of Canada, Guelph, ON, Canada
- 5Division of Animal and Food Microbiology, Center for Veterinary Medicine, Food and Drug Administration, Laurel, MD, USA
- 6Eastern Regional Research Center, United States Department of Agriculture, Agricultural Research Service, Wyndmoor, PA, USA
- 7Food and Environment Research Agency, Sand Hutton, York, UK
- 8Division of Microbiology, Center for Food Safety and Applied Nutrition, Office of Regulatory Science, Food and Drug Administration, College Park, MD, USA
Shiga toxin-producing Escherichia coli (STEC) O26 is the second leading E. coli serogroup responsible for human illness outbreaks behind E. coli O157:H7. Recent outbreaks have been linked to emerging pathogenic O26:H11 strains harboring stx2 only. Cattle have been recognized as an important reservoir of O26 strains harboring stx1; however the reservoir of these emerging stx2 strains is unknown. The objective of this study was to identify nucleotide polymorphisms in human and cattle-derived strains in order to compare differences in polymorphism derived genotypes and virulence gene profiles between the two host species. Whole genome sequencing was performed on 182 epidemiologically unrelated O26 strains, including 109 human-derived strains and 73 non-human-derived strains. A panel of 289 O26 strains (241 STEC and 48 non-STEC) was subsequently genotyped using a set of 283 polymorphisms identified by whole genome sequencing, resulting in 64 unique genotypes. Phylogenetic analyses identified seven clusters within the O26 strains. The seven clusters did not distinguish between isolates originating from humans or cattle; however, clusters did correspond with particular virulence gene profiles. Human and non-human-derived strains harboring stx1 clustered separately from strains harboring stx2, strains harboring eae, and non-STEC strains. Strains harboring stx2 were more closely related to non-STEC strains and strains harboring eae than to strains harboring stx1. The finding of human and cattle-derived strains with the same polymorphism derived genotypes and similar virulence gene profiles, provides evidence that similar strains are found in cattle and humans and transmission between the two species may occur.
Introduction
Shiga toxin-producing Escherichia coli (STEC) O26:H11/NM has emerged as an important human pathogen capable of causing severe cases of diarrhea and hemolytic uremic syndrome (HUS) (Misselwitz et al., 2003). Also, enteropathogenic E. coli (EPEC) O26 strains lacking Shiga toxins (stx) have been heavily implicated in cases of infantile gastroenteritis (Zimmerhackl et al., 2010). The true incidence of STEC O26 infections is largely underrepresented because most laboratories lack standardized methods for detecting non-O157 STEC strains. Current research has largely focused on developing reliable methods for detecting non-O157 strains in clinical samples, food animals, and animal products (Verstraete et al., 2012; Wang et al., 2012; Kalchayanand et al., 2013; Windham et al., 2013; Yonekita et al., 2013). Current data indicates that STEC O26:H11/NM are the second most common STEC serotypes responsible for infections following O157:H7 (Gould et al., 2013).
Outbreaks of STEC infection have been traced to the O26 serogroup in the United States (Brown et al., 2012), Europe (Werber et al., 2002; Ethelberg et al., 2009; Buvens et al., 2011), and Japan (Misselwitz et al., 2003; Sonoda et al., 2008; Tomita, 2008). The most common serotype implicated in outbreaks is O26:H11 (Kaspar et al., 2010). Several of the more recent O26:H11 outbreaks include one in a Colorado childcare center in 2011 (Brown et al., 2012), a 2007 ice cream implicated outbreak in Belgium (De Schrijver et al., 2008), and a 2007 outbreak in Denmark traced to beef sausage (Ethelberg et al., 2007). The 2011 outbreak in the childcare center in Colorado was the largest reported O26 outbreak in the United States with 18 confirmed cases and an additional 27 suspected cases (Brown et al., 2012). More recently in 2012, 29 individuals from 11 states were infected with an O26 STEC strain traced back to clover sprouts (Centers for Disease Control and Prevention, 2012).
E. coli O26 strains may contain a variety of virulence factors including Shiga toxin genes (stx1, stx2, or both stx1 and stx2), the enterohemolysin gene ehxA, and/or the eae gene that encodes for intimin involved in attachment. Human pathogenic O26 strains containing stx2 initially emerged in Germany in the 1990s (Zhang et al., 2000), but have become increasingly reported in the United States (Misselwitz et al., 2003; Brooks et al., 2005) and Europe (Allerberger et al., 2003; Käppeli et al., 2011). It has been hypothesized that O26 strains carrying stx2 have increased virulence and are associated with an increased risk of HUS, in comparison to strains that are predominant in cattle and only contain stx1 (Bielaszewska et al., 2013). The reservoirs for these emerging STEC O26:H11 stx2 strains are still largely unknown; however strains have been isolated from healthy domestic ruminants in Switzerland (Zweifel et al., 2013).
STEC O26 has been isolated from the feces of cattle, sheep, goats, and pigs and also has been detected in animal-derived meats (Hussein and Sakuma, 2005; Dambrosio et al., 2007) and dairy products (Steele et al., 1997). In addition to non-pathogenic E. coli O26 strains, cattle have been shown to harbor STEC and EPEC O26 strains that may cause serious disease in humans (Jeon et al., 2006; Sasaki et al., 2012; Paddock et al., 2013). In fact, STEC O26:H11 was among the top 10 most common STEC serotypes isolated from cattle and beef products in Canada (Johnson et al., 1996). The finding of STEC E. coli O26 in the feces of food animals and in animal products is a concern regarding potential foodborne transmission of this pathogen. STEC, EPEC, and enterohemorrhagic E. coli (EHEC) O26 strains from humans and food animals have been previously compared using pulsed-field gel electrophoresis (PFGE), multi locus sequence typing (MLST) and whole genome sequencing. Leomil et al. (2005) found that human and cattle strains were closely related and shared similar genotypic characteristics. Ju et al. (2012) showed that the genomes of all O26:H11 strains sequenced were similar and at least one human isolate clustered with several cattle and one swine strain using whole genome sequencing and MLST.
The objectives of this study were to identify unique nucleotide polymorphisms in a panel of human and non-human derived O26 E. coli strains in order to compare differences in polymorphism derived genotypes and virulence gene profiles between the two host species. The majority of the non-human strains were of bovine origin. The DNA from the human and non-human strains were pooled separately and sequenced for single nucleotide polymorphism (SNP) discovery. A set of SNPs was selected for MALDI-TOF genotyping and resulting polymorphism-derived genotypes were used to construct phylogenetic trees. Genotypes were compared to evaluate relationships between serotypes, virulence genes, and host both within and between phylogenetic clusters.
Materials and Methods
Strains
A total of 291 E. coli O26 strains were included in this study for sequencing, genotyping, or both; 182 were classified as human strains and 109 as non-human. Non-human strains included 84 bovine, seven strains of unknown origin, six avian, three fly, three water, two goat, two sheep, one antelope, and one feral hog. Serotypes represented among the strains included 259 O26:H11, 20 O26 strains with unknown H types, 7 O26:NM, 3 O26:H32, 1 O26:H36, and 1 O26:H46. The stx gene profiles consisted of 210 stx1, 15 stx2, 18 both stx1 and stx2, and 48 non-STEC. Other virulence genes included 233 that carried both ehxA and eae, 40 eae only, 1 ehxA only, and 17 neither ehxA nor eae (Supplemental Table 1). The strains came from various geographic locations worldwide, including 223 from the United States, 40 from Canada and one each from Mexico, Australia, France, and Switzerland. Strains were characterized by PCR for Shiga toxin (stx1 and stx2), intimin (eae), hemolysin (ehxA), O-antigen (rfbO26), and flagellar (fliCH11) genes (Paton and Paton, 1998; Durso et al., 2005).
DNA Extraction, O26 DNA Pools, and 454 GS FLX Sequencing
Two genomic DNA pools were created for sequencing. One pool contained DNA from 109 human-derived strains and the second pool contained DNA from 73 non-human-derived strains [59 bovine, three fly, three water, two sheep, two unknown origin, one antelope, one avian, one feral swine, and one goat (Supplemental Table 1)]. Among the sequenced strains there were 182 STEC (151 stx1 only, 15 stx2 only, and 14 both stx1 and stx2) and two non-STEC strains (Supplemental Table 1). Genomic DNA was extracted using the Qiagen Genomic-tip 100G (Qiagen, Valencia, CA) with modifications as previously described (Clawson et al., 2009). The DNA pools were constructed by adding 3 μg of DNA per strain into their respective pools, i.e., human or non-human. Genomic sequencing libraries were created from each pool using the Roche GS FLX Titanium Rapid Library Preparation Kit (Roche Diagnostics, Indianapolis, IN) followed by emulsion PCR according to the manufacturer's protocol and run using a Roche Genome Sequencer GS-FLX+. To prepare reads for mapping, sequencing reads were filtered using the Roche GS FLX Data Processing Software. For nucleotide polymorphism genotyping, Qiagen QIAamp DNA mini extraction kits were used to isolate genomic DNA according to the manufacturer's directions for an additional 109 strains [73 human, 25 cattle, five strains of unknown origin, four chicken, one goat, and one turkey (Supplemental Table 1)]. Among 109 strains that were genotyped but not part of the DNA pools for sequencing, there were 63 STEC (59 stx1 only and 4 both stx1 and stx2) and 46 non-STEC strains (Supplemental Table 1).
Nucleotide Polymorphism Discovery and Genotyping
Nucleotide polymorphisms were identified by mapping 454 GS FLX sequencing reads from the human and non-human pools onto the chromosome sequence of STEC O26:H11 strain 11368 (Ogura et al., 2009) using Geneious Mapper (Biomatters Ltd., Auckland, New Zealand). After the initial identification, a reduced number of nucleotide polymorphisms was selected from the two pools for subsequent genotyping of individual strains. These polymorphisms were selected based on their minor allele frequencies in the pools and their locations in the reference genome that were likely to be conserved amongst all strains of the O26 serogroup. The identified nucleotide polymorphisms were used by the MassARRAY® assay design software to design matrix-assisted laser desorption ionization-time-of-flight (MALDI-TOF) assays and multiplexing as recommended by the manufacturer (Sequenom, Inc., San Diego, CA). The software designed primers for multiplex PCR primer (Up to 36 polymorphisms into an individual assay) and the ensuing probes for genotyping. The assays were run with iPLEX Gold® chemistry (Sequenom, Inc.) on a MassARRAY® genotyping system per instructions of the manufacturer.
Identification of Tagging Nucleotide Polymorphisms
For 289 of 291 O26 strains used in this study, the alleles of 283 nucleotide polymorphisms were genotyped and concatenated to produce polymorphism-derived genotypes. A ClustalW alignment of the polymorphism-derived genotypes was produced in MacVector (Version 12.0.6). From that alignment, a minimal set of polymorphism-derived genotypes that contained a single representative of every unique polymorphism-derived genotype was identified in Tree-Puzzle (version 5.2). Putative tagging nucleotide polymorphisms were then identified from the minimal set of polymorphism-derived genotypes using the Tagger option of Haploview (version 4.2) with r2 = 1, and with the option to capture all pairwise alleles through pairwise tagging. The putative tagging nucleotide polymorphisms were then systematically subtracted; one SNP at a time, from Clustal W alignments, and Tree-puzzle was used to check for redundant sequences as previously described (Bono et al., 2012) to identify a minimal set of tagging polymorphisms.
Phylogenetic Analyses
Neighbor-Joining trees were produced from Clustal W alignments of full-length nucleotide polymorphism-derived genotypes, and those defined by the minimal set of tagging polymorphisms in PHYLIP (version 3.69). The trees were made using an F84 distance model of substitution with a transition/transversion ratio of 2, and were bootstrapped with 1000 pseudo-datasets. The trees were viewed using Treeview (version 1.6.6). Additionally, a Neighbor-joining network was constructed from a Clustal X alignment of a minimal set of tagging polymorphisms using SplitsTree4 (version 4.12.3). Maximum likelihood trees were constructed with the Tamura-Nei model, uniform rates among sites, and with gaps deleted. The search was performed with SPR level 5 and very strong branch swapping settings in MEGA (version 6) (Tamura et al., 2013). Parsimony analysis using TNT (version 1.1) included a new technology search with the sectorial search, ratchet, drift, and tree fusing options all enabled, with the minimal length tree found 10 times (Dereeper et al., 2008, 2010).
Pulsed-Field Gel Electrophoresis
PFGE was performed on 35 epidemiologically unrelated, human-derived O26 strains that were sequenced and genotyped. Thirty-one of the strains were O26:H11 and the remaining four were O26:NM. The strains had similar virulence gene profiles; 33 stx1, eae, and ehxA; 1 stx1 and eae; 1 stx1, stx2, eae, and ehxA. PFGE followed methods previously published for E. coli O157:H7 and utilized the restriction enzyme XbaI (Ribot et al., 2006; Bono et al., 2012). Data were analyzed using Bionumerics Software (Applied Maths, Austin, TX) and compared to gel images provided by the Centers for Disease Control and Prevention. The strains were categorized by PFGE banding patterns and these categories were compared to the polymorphism-derived genotypes.
Results
Sequencing Coverage of O26 Strains
A total of 1.233 GB of sequence was produced in the study, with 591 MB originating from the pool of 111 human strains, and 642 MB originating from the pool of 71 non-human strains. Given that the size of the STEC O26 strain 11386 chromosome (Ogura et al., 2009) is approximately 5.7 Mb, the mapped depth of coverage per sample for the human pool was 0.9X (103.7X total for the pool) and 1.5X (112.6X total for the pool) for the non-human pool.
Identification and Validation of Polymorphisms
Mapping sequencing reads onto STEC O26 strain 11386 chromosome (Ogura et al., 2009), using Geneious mapper, identified a total of 32,710 nucleotide/indel polymorphisms with a minor allele frequency of 0.05 or higher (data not shown). From these, 12,229 polymorphisms mapped to prophages, insertion sequences, transposable elements and other repetitive regions in the STEC O26 chromosome and were removed from consideration for SNP validation. Repetitive regions create problems for SNP validation because it is difficult to determine whether sequences that are virtually indistinguishable from one another result from a true variation at a single site or result from multiple copies of a gene in the genome. Also, plasmids were not used due to lack of conserved sequences between strains. Of the remaining 20,481 polymorphisms that placed within more conserved regions of the STEC O26 chromosome, 8864 were identified in the human pool and 11,617 in the non-human pool.
MALDI-TOF assays were developed for 342 polymorphisms that were chosen for validation according to their minor allele frequency in the DNA sequencing pools and genomic position. MALDI-TOF genotyping of the polymorphisms across 289 O26 strains yielded 29 polymorphisms with heterozygous genotypes or more than 10% no calls in addition to 30 monomorphic polymorphisms that were all removed from further analysis. A total of 283 polymorphisms were validated by MALDI-TOF genotyping of which 226 reside in open reading frames with 134 predicted as non-synonymous or premature stop codon allele variants (Supplemental Table 2). From the 283 polymorphisms, the minor alleles of 17 polymorphisms were observed exclusively in one of the DNA pools at a frequency of 15% or higher with eight in the human pool and seven in the non-human pool. For polymorphisms lower than 15% but no lower than 5%, 24 were in the human pool and 62 in the non-human pool. Additionally, 180 polymorphisms were observed in both pools where 84 had a minor allele frequency greater than 15% and 49 with a minor allele lower than 15%, but not lower than 5%. The remaining 47 polymorphisms had an allele frequency greater than 15% in only one of the pools.
Identification of Polymorphism Derived Genotypes in E. coli O26 Strains
The genotypes of 289 O26 strains (241 STEC and 48 non-STEC) were concatenated from 283 validated polymorphisms and yielded 64 unique polymorphism-derived genotypes (Supplemental Table 3). The 64 genotypes could be derived from a minimal set of 43 polymorphisms (Supplemental Table 4). From these, 24 genotypes were exclusively found in human-isolated strains, 19 exclusively from the non-human sources, and 21 were found in both human and non-human strains (Figure 1, Supplemental Table 1). Genotypes 14 and 25 had the highest frequency (21 strains) of STEC O26 human strains while genotype 32 had the highest frequency (nine strains) of STEC O26 non-human strains. The 15-stx2 only strains were placed in six genotypes (1, 34, 43, 46, 47, and 61) and were the exclusive stx profile in three of those genotypes (1, 46, and 47) (Supplemental Table 1). Of the strains that carried stx2 only, 66.7% fell within genotype 47 (Figure 2). The 210-stx1 only strains were placed in 39 different genotypes with the highest frequency (22 strains) in genotype 25 followed by genotypes 21 and 27 (21 strains each). The 18 strains with both stx1 and stx2 strains were placed in eight different genotypes with the highest frequency (six strains) in genotype 14. Genotypes 14 and 25 had the highest frequency (23 strains) of US strains while genotype 21 had the highest frequency (nine strains) of Canadian strains. The 48 non-STEC strains were placed in 24 different genotypes with the highest frequency (10 strains) in genotype 63 followed by genotype 57 (six strains).
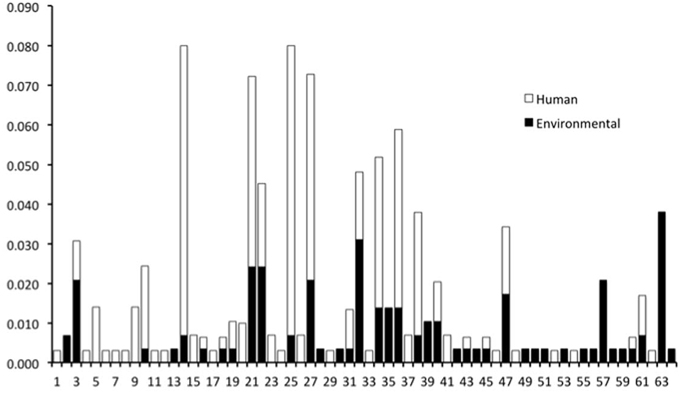
Figure 1. Frequency of human and non-human-derived strains for the 64 polymorphism-derived genotypes.
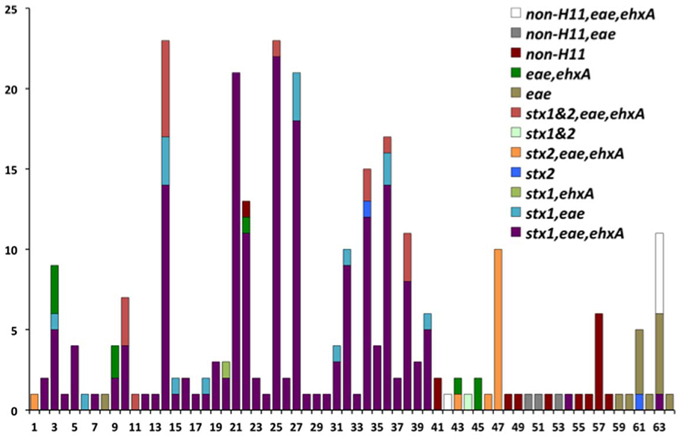
Figure 2. Virulence gene profiles within the 64 polymorphism-derived genotypes. Each bar or stacked bar represents the number of strains with the virulence gene profile per genotype.
Phylogenetic Analysis of O26 Polymorphism-Derived Genotypes
Neighbor-Joining trees were constructed from the 64 polymorphism-derived genotypes that were produced from (1) all 283-polymorphism alleles, and (2) a minimal set of 43 tagging polymorphisms (Figure 3, Supplemental Figure ). Of the two trees, the one representing 283 polymorphism alleles contained higher overall bootstrap support and was used for cluster identifications. Seven clusters were defined within the tree by the phylogenetic relatedness of the genotypes, and by the presence or absence of Shiga toxin genes (stx1 or stx2), eae, ehxA, and/or H11 serotype (Figure 3). The clusters were represented by as few as one genotype (Clusters 5, 6, and 7) and as many as 39 different genotypes (Cluster 1). Five of the seven clusters (1–2, 4–6) were well supported by bootstrap values, which indicated strong support of their placement in the tree. These results were supported by maximum parsimony and maximum likelihood analysis with the exception that Cluster 6 was collapsed into Cluster 3 (data not shown). To examine the support for Clusters 3 and 7 without using bootstrap values, a Neighbor-Joining network of the 64 polymorphism-derived genotypes based on all 283 polymorphism alleles was created (Figure 4). This network differed from the tree in that it allowed for recombination events. All seven clusters were defined within the network, and thus were reproduced between Neighbor-Joining trees and networks.
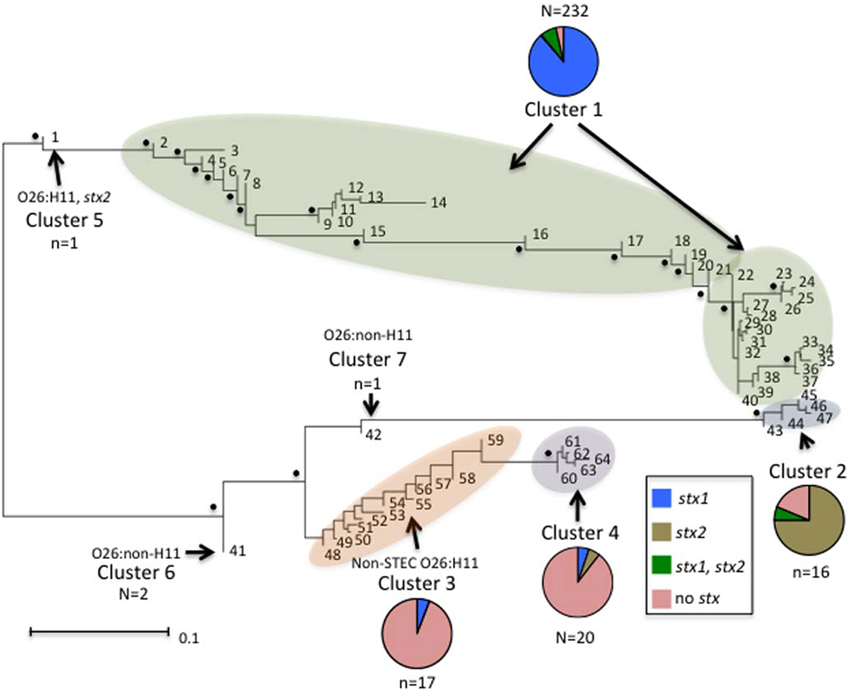
Figure 3. Neighbor-joining tree of the 64 full-length polymorphism-derived genotypes. Bootstrap values greater than 80 are indicated by a black dot. Outer taxonomic unit numbers correspond to the polymorphism-derived genotypes. The seven clusters are indicated and clusters containing multiple genotypes are highlighted. The frequencies of the different Shiga toxin profiles from each cluster are represented by pie charts with colors representing the different profiles. The scale bar represents substitutions per site.
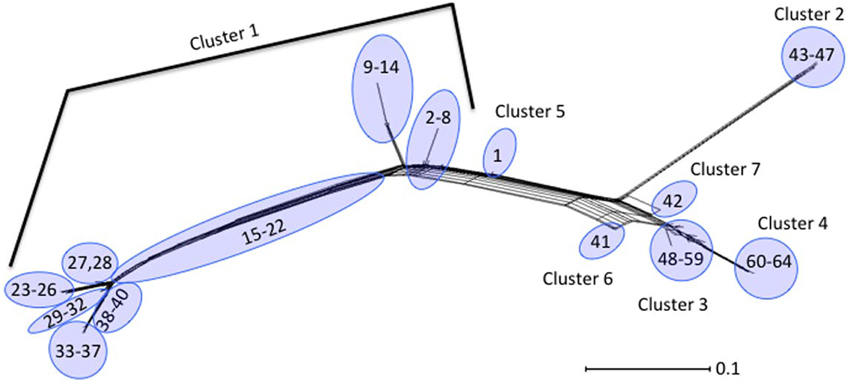
Figure 4. Neighbor-Joining network of the 64 polymorphism-derived genotypes based on all 283 polymorphism alleles. Outer taxonomic unit numbers correspond to the polymorphism-derived genotypes.
Distinct genetic determinants defined the polymorphism-derived genotypes within each of the seven clusters. Cluster 1 contained 39 genotypes that represented 232 O26 strains that were predominantly O26:H11 (231 strains), stx1 (223 strains), eae (230 strains), and ehxA (214 strains) positive and stx2 negative (214 strains) (Supplemental Table 1). The five genotypes of Cluster 2 represented 16 O26:H11 strains that were predominantly stx2 (13 strains), eae (15 strains), and ehxA (15 strains) positive and stx1 negative (15 strains). Cluster 3 contained 12 genotypes and 17 strains that were represented by 13 O26 non-STEC strains that were eae, ehxA, and H11 negative (12 had unknown H type and one was H46), three non-STEC strains of unknown H type that were positive for eae, and one STEC O26:H11 strain that was positive for stx1, eae, and ehxA. Cluster 4 contained five genotypes that were represented by 20 O26 strains, of which 13 were H11 and eae positive and five other strains that were H11 negative and eae positive. The remaining two strains in Cluster 4 were positive for ehxA and eae, with one carrying stx1 and the other stx2. Cluster 5 was represented by a single O26:H11 strain that was stx2, eae, and ehxA positive. Cluster 6 was represented by two O26 strains that had identical polymorphism-derived genotypes, and identical virulence gene profiles (negative for stx1, stx2, eae, ehxA, and H11). Lastly, Cluster 7 was represented by a single O26 isolate that was eae and ehxA positive, and negative for H11. As a whole, both human and non-human strains were represented in the clusters and virulence factor frequencies defined many of the clusters. In that regard, of the 17 strains with polymorphism-derived genotypes that placed in Cluster 3, only three were isolated from humans, which is notable as most of the strains in Cluster 3 were negative for many of the major virulence determinants.
Comparison of Polymorphism-Derived Genotypes and PFGE
Thirty-five epidemiologically unrelated, human STEC O26 strains were compared for genetic diversity based on PFGE and polymorphism-derived genotypes. PFGE profiles were determined using the restriction enzyme XbaI and banding patterns assigned using the CDC standard naming protocol. From the 35 strains, there were 26 PFGE patterns and 15 polymorphism-derived genotypes, with multiple PFGE patterns observed within six of the genotypes and seven manifested as singletons (Figure 5). Two genotypes contained the same PFGE profile, while three sets of strains with the same PFGE pattern were classified into different genotypes (data not shown).
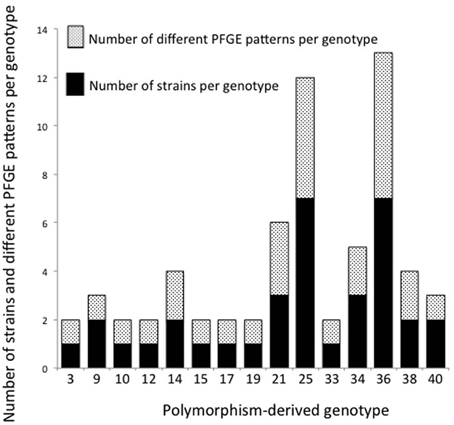
Figure 5. Number of strains and PFGE patterns in 15 polymorphism-derived genotypes. Stacked black bars or white speckled bars represent the number of strains and PFGE patterns per genotype, respectively.
Discussion
We sequenced and analyzed the genomes of 180 STEC and two non-STEC E. coli O26 strains from human and non-human sources. We identified 283 nucleotide polymorphisms in conserved regions of the chromosome that were utilized for MALDI-TOF genotyping of 180 sequenced strains and an additional 109 O26 E. coli strains. Genotyping data was used to compare polymorphism-derived genotypes and virulence gene profiles among strains from human and other non-human sources, particularly cattle. The concatenated polymorphism alleles identified 64 unique genotypes representing 289 strains that on phylogenetic analysis were grouped into seven clusters. Clusters were not distinguished by host species but did depend strongly on virulence gene profiles.
Similarities between human and cattle strains were found using whole genome sequencing and SNP genotyping. Among the 64 unique SNP genotypes, 21 were found in both human and non-human strains (79% of the non-human strains were of cattle origin) (Figure 1). Previous studies that have used PFGE, MLST, and whole genome sequencing to compare O26 strains from human and food animals have found similar results (Leomil et al., 2005; Ju et al., 2012). Using PFGE and MLST, Leomil et al. (2005) found two genetically distinct groups among 23 E. coli O26 strains. One group contained EPEC and EHEC strains from both humans and food animals and the second group contained EPEC strains from both humans and food animals that were phenotypically and genetically distinct from previously characterized O26 strains. Comparing seven O26:H11 strains using whole-genome sequencing, Ju et al. (2012) found that one human strain clustered among four cattle and one swine strain.
We found seven distinct clusters of O26 strains using whole genome sequencing, SNP analysis, and phylogenetics. The clusters were well supported by strong Neighbor-joining bootstrap values and reproducibility between phylogenetic algorithms. A similar whole genome sequencing study and SNP analysis was performed on a set of O26 EHEC strains from Europe (Bielaszewska et al., 2013; Bletz et al., 2013). The SNP genotyping of 120 human pathogenic strains that originated from Germany that harbored stx1, stx2 or both stx1 and stx2, resulted in 10 unique SNP profiles that phylogenetically grouped into four clonal complexes (clonal complexes shared ≥90% of the SNPs). When comparing our SNPs (Supplemental Table 2) with those of Bletz et al. (2013), we found that only 23 of the 283 SNPs and three of the 43 tagging polymorphisms matched. One reason for this low proportion of matched SNPs may be that O26 strains in Europe and the United States may have undergone phylogeographic stratification. Additionally, strains were isolated from different sources between the two studies; human and non-human STEC and non-STEC strains were sequenced in the current study; whereas, Bletz et al. (2013) focused mainly on virulent human STEC strains. The increased number of clonal complexes in our study may be the result of a larger and more diverse pool of O26 strains.
The phylogenetic clusters with multiple genotypes (1, 2, 3, 4) in our study were represented by both human- and cattle-isolated strains, suggesting that similar strains were found in humans and cattle. Human- and cattle-derived strains were also present in many of the genotypes; notably, 66.7% of our STEC O26 strains carrying only stx2 were in genotype 47, represented by similar numbers of human and cattle strains. The phylogenetic analysis by Bletz et al. (2013) suggests a source-sink evolutionary model, in that a stable population of O26 strains exists in a niche and only a few strains with evolutionary advantages are transferred to a new niche (Bletz et al., 2013). These new niches may account for the distinct clonal complexes and the emergence of the highly virulent strains of O26 in relevant reservoir and human hosts (Bletz et al., 2013). Our data support the hypothesis that a source population of non-pathogenic O26 strains exists in the environment, and that select strains have established themselves in cattle. These strains may have acquired virulence factors either before or after establishing in cattle as a result of selective pressures. These virulent strains may ultimately be transferred to humans where they are detected through disease.
Human and cattle strains in the current study also had similar virulence gene profiles. Among the 289 O26 strains genotyped, there were stx1 only, stx2 only, strains containing both stx1 and stx2, and non-STEC strains. There were six genotypes (1, 34, 43, 46, 47, and 61) with stx2 only strains; three of the genotypes had human and cattle strains (34, 43, and 47), two genotypes contained only human strains (1 and 46), and genotype 61 contained human strains and one strain from an unknown source (Figure 2; Supplemental Table 1). This is interesting because there has been a trend toward stx2 only O26 pathogenic strains (Zhang et al., 2000). STEC O26 strains from cattle have predominantly carried stx1 and there have been few reports of the prevalence of stx2 strains in cattle. Recently, two of 11 STEC O26:H11 strains from healthy cattle in Switzerland contained stx2 and eae (Zweifel et al., 2013). However, further investigation is required to determine if cattle are an important reservoir for the emerging virulent STEC O26 stx2 strains in humans. The finding of human and cattle strains with the same genotypes, and the similarity of virulence gene profiles between strains in both host species, suggests that transmission of E. coli O26 may occur between humans and cattle or both host species are infected by a common source.
Phylogenetic analysis of the genotypes revealed several interesting patterns regarding particular virulence gene profiles. In the neighbor-joining tree in Figure 3, seven clusters of O26 strains on two distinct branches are shown. The majority of O26 strains in Cluster 1 were stx1 only; however there were also both stx1 and stx2, stx2 only, and non-STEC strains. Cluster 5 was represented by a single O26:H11 stx2 only strain. Cluster 1 and Cluster 5 were on a distinct branch from Clusters 2, 3, 4, 6, and 7. In Cluster 2, 13 of 15 strains were stx2 only. All strains but one in Cluster 3 were non-STEC O26 strains. Cluster 4 contained O26 strains harboring eae with one stx1 only and one stx2 only strains. Cluster 6 contained two non-STEC O26:non-H11 strains, and Cluster 7 contained a single non-STEC O26:non-H11 strain harboring eae. This phylogenetic tree of the 64 unique genotypes provides evidence that O26 stx2 strains evolved separately from O26 stx1 strains and are more similar to non-STEC O26 strains. Interestingly, Bletz et al. (2013) also found stx to be an important virulence factor associated with phylogenetic clustering in O26 strains. Similar to our results, they found that the emerging highly virulent O26 strains harboring stx2 clustered separately from the other strains and suggested that a source-sink model is responsible for the distinct clonal complexes. However, there were few non-pathogenic strains included in this study so it is difficult to assess the phylogenetic relationship between the stx2 strains and non-STEC strains in comparison to the phylogenetic relationship between stx2 strains and stx1 strains. In regards to the source-sink model, our data would suggest that stx1 and stx2 strains have followed distinct evolutionary paths that may have been determined by differing selective pressures or fitness of the strains.
Several strains included in this study were among numerous diarrheagenic E. coli (DEC) strains previously genotyped using multilocus enzyme electrophoresis (Whittam et al., 1993). Across all the DEC strains, there were a total of 191 electrophoretic types (ETs) identified; however 15 ETs accounted for 70% of the strains in the study (Whittam et al., 1993). Twenty ETs were found among 93 O26 strains (Whittam et al., 1993). Four DEC9 and five DEC10 strains were included in our study. While most DEC9 strains lack stx and have eae and the H32 flagellar antigen (Whittam et al., 1993), those in our study were all O26:H11, but eae positive and stx and ehxA negative. On the other hand, the DEC10 strains typically were all O26:H11 and eae and ehxA positive, and three of the five were stx1 positive. Our results agreed with this classification scheme. The five DEC10 strains were classified in genotypes in Cluster 1, which were generally O26:H11 strains with eae and stx1 (Supplemental Table 1). The four DEC9 strains on the other hand, were classified in genotypes in Cluster 4, which were generally O26:H11 strains with eae but no stx. When looking at the 15 most common ETs associated with all the E. coli strains, Whittam et al. (1993) found DEC9 and DEC10 strains clustered separately, and although they are more closely related to each other than to other E. coli serogroups, they do have distinct evolutionary lineages. We also found that DEC9 and DEC10 strains had distinct evolutionary lineages. DEC10 strains were found in Cluster 1, which were on a separate branch of the phylogenetic tree from DEC9 strains in Cluster 4 (Figure 3).
A comparison of SNP derived genotypes with PFGE data for 35 epidemiologically unrelated, human-derived strains found a higher diversity among PFGE patterns (Figure 5). There were 26 PFGE patterns and 15 genotypes identified among the strains. Within the 15 genotypes, six contained multiple PFGE patterns. It was previously determined that SNP derived genotypes do not surpass the diversity provided by PFGE patterns in a study of 96 epidemiologically unrelated STEC O157 strains from ground beef (Bono et al., 2012). Bono et al. (2012) identified a total of 68 PFGE patterns using restriction enzymes XbaI and BlnI, and 41 polymorphism-derived genotypes were found using whole-genome sequencing. Within the 41 genotypes, 17 genotypes contained two or more PFGE profiles (Bono et al., 2012). Although these studies of STEC O26 and O157 provide evidence that PFGE provides better discriminatory resolution than polymorphism-derived genotypes for studying genetic diversity, PFGE does not provide data on the evolutionary relationships between strains.
In summary, whole genome sequencing and SNP-based phylogenetic analysis of E. coli O26 strains in this study revealed that human and cattle strains did not cluster in separate polymorphism-derived genotypes, and with some exceptions, they had similar virulence gene profiles. These findings suggest that similar strains are circulating between humans and cattle. In regards to virulence gene profiles, we found that strains harboring only stx1 clustered separately from strains harboring only stx2, strains harboring eae, and non-STEC strains. This would suggest that the emerging strains harboring only stx2 may have evolved separately from stx1 strains and are more closely related to non-STEC strains. The full extent of animal and non-human reservoirs for O26 with stx2 is unknown, however our findings suggest that cattle may be a reservoir for STEC O26 carrying stx2 that are emerging in humans. Further investigation of a larger collection of human and non-human STEC O26 strains with only stx2 beyond the 15 strains in this study, combined with other STEC O26 and stx variants is required to better identify their reservoirs and define their evolution.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Sandy Fryda-Bradley, Linda Flathman, and Chris Zimmerman for outstanding technical support. The.sff files for both human and non-human DNA pools have been deposited into the short read archives under BioProject accession PRJNA274335. Some strains used were collected as part of a USDA CSREES NRI grant awarded to Dr. James Keen. The use of product and company names is necessary to accurately report the methods and results; however, the USDA neither guarantees nor warrants the standard of the products, and the use of the names by the USDA implies no approval of the products to the exclusion of others that may also be suitable. USDA is an equal opportunity provider and employer.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fcimb.2015.00021/abstract
Supplemental Figure 1. Neighbor-joining tree of polymorphism-derived genotypes tagged with a minimal set of 43 polymorphisms. Bootstrap values greater than 80 are indicated by a black dot. The scale bar represents substitutions per site.
References
Allerberger, F., Friedrich, A. W., Grif, K., Dierich, M. P., Dornbusch, H. J., Mache, C. J., et al. (2003). Hemolytic-uremic syndrome associated with enterohemorrhagic Escherichia coli O26:H infection and consumption of unpasteurized cow's milk. Int. J. Infect. Dis. 7, 42–45. doi: 10.1016/S1201-9712(03)90041-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bielaszewska, M., Mellmann, A., Bletz, S., Zhang, W., Kock, R., Kossow, A., et al. (2013). Enterohemorrhagic Escherichia coli O26:H11/H-: a new virulent clone emerges in Europe. Clin. Infect. Dis. 56, 1373–1381. doi: 10.1093/cid/cit055
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bletz, S., Bielaszewska, M., Leopold, S. R., Kock, R., Witten, A., Schuldes, J., et al. (2013). Evolution of enterohemorrhagic Escherichia coli O26 based on single-nucleotide polymorphisms. Genome Biol. Evol. 5, 1807–1816. doi: 10.1093/gbe/evt136
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bono, J. L., Smith, T. P., Keen, J. E., Harhay, G. P., McDaneld, T. G., Mandrell, R. E., et al. (2012). Phylogeny of Shiga toxin-producing Escherichia coli O157 isolated from cattle and clinically ill humans. Mol. Biol. Evol. 29, 2047–2062. doi: 10.1093/molbev/mss072
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brooks, J. T., Sowers, E. G., Wells, J. G., Greene, K. D., Griffin, P. M., Hoekstra, R. M., et al. (2005). Non-O157 Shiga toxin–producing Escherichia coli infections in the United States, 1983–2002. J. Infect. Dis. 192, 1422–1429. doi: 10.1086/466536
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brown, J. A., Hite, D. S., Gillim-Ross, L. A., Maguire, H. F., Bennett, J. K., Patterson, J. J., et al. (2012). Outbreak of Shiga toxin-producing Escherichia coli serotype O26: H11infection at a child care center in Colorado. Pediatr. Infect. Dis. J. 31, 384–388. doi: 10.1097/INF.0b013e3182457122
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buvens, G., Posse, B., De Schrijver, K., De Zutter, L., Lauwers, S., and Pierard, D. (2011). Virulence profiling and quantification of verocytotoxin-producing Escherichia coli O145:H28 and O26:H11 isolated during an ice cream-related hemolytic uremic syndrome outbreak. Foodborne Pathog. Dis. 8, 421–426. doi: 10.1089/fpd.2010.0693
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Centers for Disease Control Prevention. (2012). Multistate Outbreak of Shiga Toxin-Producing Escherichia coli O26 Infections Linked to Raw Clover Sprouts at Jimmy John's Restaurants (Final Update). Available online at: http://www.cdc.gov/ecoli/2012/o26-02-12/index.html (Accessed March, 2014).
Clawson, M. L., Keen, J. E., Smith, T. P., Durso, L. M., McDaneld, T. G., Mandrell, R. E., et al. (2009). Phylogenetic classification of Escherichia coli O157:H7 strains of bovine and human origin using a novel set of nucleotide polymorphisms. Genome Biol. 10:R56. doi: 10.1186/gb-2009-10-5-r56
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dambrosio, A., Lorusso, V., Quaglia, N. C., Parisi, A., La Salandra, G., Virgilio, S., et al. (2007). Escherichia coli O26 in minced beef: prevalence, characterization and antimicrobial resistance pattern. Int. J. Food Microbiol. 118, 218–222. doi: 10.1016/j.ijfoodmicro.2007.07.041
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dereeper, A., Audic, S., Claverie, J. M., and Blanc, G. (2010). BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol. Biol. 10:8. doi: 10.1186/1471-2148-10-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dereeper, A., Guignon, V., Blanc, G., Audic, S., Buffet, S., Chevenet, F., et al. (2008). Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469. doi: 10.1093/nar/gkn180
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Schrijver, K., Buvens, G., Posse, B., Van den Branden, D., Oosterlynck, O., De Zutter, L., et al. (2008). Outbreak of verocytotoxin-producing E. coli O145 and O26 infections associated with the consumption of ice cream produced at a farm, Belgium, 2007. Euro Surveill. 131. Available online at: http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=8041
Durso, L. M., Bono, J. L., and Keen, J. E. (2005). Molecular serotyping of Escherichia coli O26:H11. Appl. Environ. Microbiol. 71, 4941–4944. doi: 10.1128/AEM.71.8.4941-4944.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ethelberg, S., Smith, B., Torpdahl, M., Lisby, M., Boel, J., Jensen, T., et al. (2007). An outbreak of Verocytotoxin-producing Escherichia coli O26:H11 caused by beef sausage, Denmark 2007. Euro Surveill. 12:E070531.4. Available online at: http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=3208
Ethelberg, S., Smith, B., Torpdahl, M., Lisby, M., Boel, J., Jensen, T., et al. (2009). Outbreak of non-O157 Shiga toxin-producing Escherichia coli infection from consumption of beef sausage. Clin. Infect. Dis. 48, e78–e81. doi: 10.1086/597502
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gould, L. H., Mody, R. K., Ong, K. L., Clogher, P., Cronquist, A. B., Garman, K. N., et al. (2013). Increased recognition of non-O157 Shiga toxin-producing Escherichia coli infections in the United States during 2000-2010: epidemiologic features and comparison with E. coli O157 infections. Foodborne Pathog. Dis. 10, 453–460. doi: 10.1089/fpd.2012.1401
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hussein, H. S., and Sakuma, T. (2005). Prevalence of shiga toxin-producing Escherichia coli in dairy cattle and their products. J. Dairy Sci. 88, 450–465. doi: 10.3168/jds.S0022-0302(05)72706-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jeon, B. W., Jeong, J. M., Won, G. Y., Park, H., Eo, S. K., Kang, H. Y., et al. (2006). Prevalence and characteristics of Escherichia coli O26 and O111 from cattle in Korea. Int. J. Food Microbiol. 110, 123–126. doi: 10.1016/j.ijfoodmicro.2006.01.035
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Johnson, R. P., Clarke, R. C., Wilson, J. B., Read, S. C., Rahn, K., Renwick, S. A., et al. (1996). Growing concerns and recent outbreaks involving Non-O157:H7 serotypes of Verotoxigenic Escherichia coli. J. Food Prot. 59, 1112–1122.
Ju, W., Cao, G., Rump, L., Strain, E., Luo, Y., Timme, R., et al. (2012). Phylogenetic analysis of Non-O157 Shiga toxin-producing Escherichia coli strains by whole-genome sequencing. J. Clin. Microbiol. 50, 4123–4127. doi: 10.1128/JCM.02262-12
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kalchayanand, N., Arthur, T. M., Bosilevac, J. M., Wells, J. E., and Wheeler, T. L. (2013). Chromogenic agar medium for detection and isolation of Escherichia coli serogroups O26, O45, O103, O111, O121, and O145 from fresh beef and cattle feces. J. Food Prot. 76, 192–199. doi: 10.4315/0362-028X.JFP-12-182
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Käppeli, U., Hächler, H., Giezendanner, N., Beutin, L., and Stephan, R. (2011). Human infections with non-O157 Shiga toxin-producing Escherichia coli, Switzerland, 2000–2009. Emerging Infect. Dis. 17, 180–185. doi: 10.3201/eid1702.100909
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kaspar, C., Doyle, M. E., and Archer, J. (2010). White Paper on Non-O157:H7 Shiga Toxin-Producing E. coli from Meat and Non-Meat Sources. FRI Food Saf. Rev. Available online at: http://fri.wisc.edu/files/Briefs_File/FRI_Brief_NonO157STEC_4_10.pdf (Accessed January 29, 2015).
Leomil, L., Pestana de Castro, A.F., Krause, G., Schmidt, H., and Beutin, L. (2005). Characterization of two major groups of diarrheagenic Escherichia coli O26 strains which are globally spread in human patients and domestic animals of different species. FEMS Microbiol. Lett. 249, 335–342. doi: 10.1016/j.femsle.2005.06.030
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Misselwitz, J., Karch, H., Bielazewska, M., John, U., Ringelmann, F., Ronnefarth, G., et al. (2003). Cluster of hemolytic-uremic syndrome caused by Shiga toxin-producing Escherichia coli O26:H11. Pediatr. Infect. Dis. J. 22, 349–354. doi: 10.1097/01.inf.0000059338.38673.ae
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ogura, Y., Ooka, T., Iguchi, A., Toh, H., Asadulghani, M., Oshima, K., et al. (2009). Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 106, 17939–17944. doi: 10.1073/pnas.0903585106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Paddock, Z. D., Renter, D. G., Cull, C. A., Shi, X., Bai, J., and Nagaraja, T. G. (2013). Escherichia coli O26 in feedlot cattle: fecal prevalence, isolation, characterization, and effects of an E. coli O157 vaccine and a direct-fed microbial. Foodborne Pathog. Dis. 11, 186–193. doi: 10.1089/fpd.2013.1659
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Paton, A. W., and Paton, J. C. (1998). Detection and characterization of Shiga toxigenic Escherichia coli by using multiplex PCR assays for stx1, stx2, eaeA, enterohemorrhagic E. coli hlyA, rfbO111, and rfbO157. J. Clin. Microbiol. 36, 598–602.
Ribot, E. M., Fair, M. A., Gautom, R., Cameron, D. N., Hunter, S. B., Swaminathan, B., et al. (2006). Standardization of Pulsed-field gel electrophoresis protocols for the subtyping of Escherichia coli O157:H7, Salmonella, and Shigella for PulseNet. Foodborne Pathog. Dis. 3, 59–67. doi: 10.1089/fpd.2006.3.59
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sasaki, Y., Usui, M., Murakami, M., Haruna, M., Kojima, A., Asai, T., et al. (2012). Antimicrobial resistance in Shiga toxin-producing Escherichia coli O157 and O26 isolates from beef cattle. Jpn. J. Infect. Dis. 65, 117–121.
Sonoda, C., Tagami, A., Nagatomo, D., Yamada, S., Fuchiwaki, R., Haruyama, M., et al. (2008). An enterohemorrhagic Escherichia coli O26 outbreak at a nursery school in Miyazaki, Japan. Jpn. J. Infect. Dis. 61, 92–93.
Steele, M. I., McNab, W. B., Poppe, C., Griffiths, M. W., Chen, S., Degrandis, S. A., et al. (1997). Survey of Ontario bulk tank raw milk for food-borne pathogens. J. Food Prot. 60, 1341–1346.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tomita, N. (2008). Enterohemorrhagic Escherichia coli O26 infection in City A in the eastern part of Ehime Prefecture in Japan. Nihon Koshu Eisei Zasshi 55, 163–169.
Verstraete, K., De Zutter, L., Robyn, J., Daube, G., Herman, L., Heyndrickx, M., et al. (2012). Validation of a method for simultaneous isolation of Shiga toxin-producing Escherichia coli O26, O103, O111, and O145 from minced beef by an international ring-trial. Foodborne Pathog. Dis. 9, 412–417. doi: 10.1089/fpd.2011.0998
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, F., Jiang, L., Yang, Q., Prinyawiwatkul, W., and Ge, B. (2012). Rapid and specific detection of Escherichia coli serogroups O26, O45, O103, O111, O121, O145, and O157 in ground beef, beef trim, and produce by loop-mediated isothermal amplification. Appl. Environ. Microbiol. 78, 2727–2736. doi: 10.1128/AEM.07975-11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Werber, D., Fruth, A., Liesegang, A., Littmann, M., Buchholz, U., Prager, R., et al. (2002). A multistate outbreak of Shiga toxin-producing Escherichia coli O26:H11 infections in Germany, detected by molecular subtyping surveillance. J. Infect. Dis. 186, 419–422. doi: 10.1086/341457
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Whittam, T. S., Wolfe, M. L., Wachsmuth, I. K., Orskov, F., Orskov, I., and Wilson, R. A. (1993). Clonal relationships among Escherichia coli strains that cause hemorrhagic colitis and infantile diarrhea. Infect. Immun. 61, 1619–1629.
Windham, W. R., Yoon, S. C., Ladely, S. R., Haley, J. A., Heitschmidt, J. W., Lawrence, K. C., et al. (2013). Detection by hyperspectral imaging of Shiga toxin-producing Escherichia coli serogroups O26, O45, O103, O111, O121, and O145 on rainbow agar. J. Food Prot. 76, 1129–1136. doi: 10.4315/0362-028X.JFP-12-497
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yonekita, T., Fujimura, T., Morishita, N., Matsumoto, T., and Morimatsu, F. (2013). Simple, rapid, and reliable detection of Escherichia coli O26 using immunochromatography. J. Food Prot. 76, 748–754. doi: 10.4315/0362-028X.JFP-12-423
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, W. L., Bielaszewska, M., Liesegang, A., Tschape, H., Schmidt, H., Bitzan, M., et al. (2000). Molecular characteristics and epidemiological significance of Shiga toxin-producing Escherichia coli O26 strains. J. Clin. Microbiol. 38, 2134–2140.
Zimmerhackl, L. B., Rosales, A., Hofer, J., Riedl, M., Jungraithmayr, T., Mellmann, A., et al. (2010). Enterohemorrhagic Escherichia coli O26:H11-associated Hemolytic Uremic Syndrome: bacteriology and clinical presentation. Semin. Thromb. Hemost. 36, 586–593. doi: 10.1055/s-0030-1262880
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zweifel, C., Cernela, N., and Stephan, R. (2013). Detection of the emerging Shiga toxin-producing Escherichia coli O26:H11/H- sequence type 29 (ST29) clone in human patients and healthy cattle in Switzerland. Appl. Environ. Microbiol. 79, 5411–5413. doi: 10.1128/AEM.01728-13
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Escherichia coli, O26, Shiga toxins, polymorphisms, phylogenetic
Citation: Norman KN, Clawson ML, Strockbine NA, Mandrell RE, Johnson R, Ziebell K, Zhao S, Fratamico PM, Stones R, Allard MW and Bono JL (2015) Comparison of whole genome sequences from human and non-human Escherichia coli O26 strains. Front. Cell. Infect. Microbiol. 5:21. doi: 10.3389/fcimb.2015.00021
Received: 22 December 2014; Accepted: 21 February 2015;
Published: 11 March 2015.
Edited by:
Eelco Franz, Centre for Infectious Disease Control, NetherlandsReviewed by:
Angela Van Hoek, Rijksinstituut voor Volksgezondheid en Milieu, NetherlandsNorval Strachan, University of Aberdeen, UK
Copyright © 2015 Norman, Clawson, Strockbine, Mandrell, Johnson, Ziebell, Zhao, Fratamico, Stones, Allard and Bono. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James L. Bono, U.S. Meat Animal Research Center, United States Department of Agriculture, Agricultural Research Service, State Spur 18D, PO Box 166, Clay Center, NE 68933-0166, USA jim.bono@ars.usda.gov