 Marta Monguió-Tortajada
Marta Monguió-Tortajada Ricardo Lauzurica-Valdemoros
Ricardo Lauzurica-Valdemoros Francesc E. Borràs
Francesc E. Borràs- 1Innovation in Vesicles and Cells for Application Therapy Group (IVECAT), Institut d’Investigació Germans Trias i Pujol, Badalona, Spain
- 2Nephrology Service, Hospital Universitari Germans Trias i Pujol, Badalona, Spain
Organ transplantation is often the unique solution for organ failure. However, rejection is still an unsolved problem. Although acute rejection is well controlled, the chronic use of immunosuppressive drugs for allograft acceptance causes numerous side effects in the recipient and do not prevent chronic allograft dysfunction. Different alternative therapies have been proposed to replace the classical treatment for allograft rejection. The alternative therapies are mainly based in pre-infusions of different types of regulatory cells, including DCs, MSCs, and Tregs. Nevertheless, these approaches lack full efficiency and have many problems related to availability and applicability. In this context, the use of extracellular vesicles, and in particular exosomes, may represent a cell-free alternative approach in inducing transplant tolerance and survival. Preliminary approaches in vitro and in vivo have demonstrated the efficient alloantigen presentation and immunomodulation abilities of exosomes, leading to alloantigen-specific tolerance and allograft acceptance in rodent models. Donor exosomes have been used alone, processed by recipient antigen-presenting cells, or administered together with suboptimal doses of immunosuppressive drugs, achieving specific allograft tolerance and infinite transplant survival. In this review, we gathered the latest exosome-based strategies for graft acceptance and discuss the tolerance mechanisms involved in organ tolerance mediated by the administration of exosomes. We will also deal with the feasibility and difficulties that arise from the application of this strategy into the clinic.
Introduction
Solid organ transplantation (SOT) is the unique solution for end-stage organ failure, and can be considered among the major accomplishments of the twentieth century in human health. Only in 2012, it is estimated that about 115,000 solid organ transplants were performed worldwide (1). Apart from saving lives, SOT is a cost-effective alternative to other medical options (when available). For instance, it is well established that kidney transplantation (by far the most transplanted organ worldwide, being 65% of total SOT), increases survival rates, guarantees a better quality-of-life and it is also less costly in the long term compared to hemodialysis.
The improved methodology in surgical techniques, technological advances, and research in biological and pharmaceutical products have profoundly improved the survival of transplanted patients. Remarkably, the maximum survival reported for a kidney transplanted patient is 46 years, and 39 years for a liver transplanted patient (2). However, the overall data indicates that the median graft survival of kidney transplants is 50% after 10 years. Most of these graft looses are due to chronic rejection episodes conducted by the recipient’s immune system against the graft. Therefore, one of the most important challenges in organ transplantation is achieving graft immunological tolerance, i.e., preventing the recipient’s immune system attack and destruction of the transplanted organ leading to graft rejection.
To prevent graft rejection, immunosuppresive drugs (ISd) have been successfully given to transplant recipients. Not in vain, these ISd are responsible for the increased survival of transplanted patients. Nonetheless, the chronic use of ISd leads to drug related-toxicity and to an un-specific and general suppression of the immune system, which may cause rising of opportunistic infections and malignancy. Therefore, alternative treatments to classical immunosuppression to induce donor-specific tolerance need to be found. In this review, we will briefly mention the mechanisms of graft rejection, the classical immunosuppression strategies, and how new extracellular vesicles (EVs)-based strategies may be an opportunity to induce organ tolerance. We will also discuss some critical points to be solved for the application of this strategy into the clinic.
Graft Rejection
Transplant rejection is a complex immune response directed against the alloantigens (antigenic alleles) specifically expressed by the graft, which are recognized as “non-self” by the host’s immune system. These alloantigens essentially include the major histocompatibility complex (MHC) molecules and also minor histocompatibility antigens (miHAs) expressed by graft cells. The final outcome of this immune response is the rejection of the organ, leading the recipient to a new transplantation or alternative replacement therapy, such as dialysis in the case of kidney failure.
According to the clinical and pathological course, graft rejection may be classified as (3) (i) Hyperacute rejection, which takes place after only few minutes to few hours of transplantation; (ii) Acute cellular rejection, mainly mediated by cells and occurring within a few weeks; (iii) Acute humoral rejection, arising at the first/second week after transplantation and mediated by antibodies directed against alloantigens; (iv) Chronic rejection, which may appear at any time (even years) after the acute phase and progressively deteriorates the graft function. This last type of rejection is responsible of most of rejected organs after 1 year from transplant.
In all graft rejection types, the effector mechanisms responsible for injury and destruction of the transplanted organ involve the participation of all (cellular and soluble) components of the immune system. These include not only T and B lymphocytes, the major effectors of the adaptive immune system (involved in antigen-specific rejection), but also cells of the innate immune system including endothelial cells, NK cells, macrophages, and/or polymorphonuclear cells. Also, antigen-specific (antibodies) and un-specific (complement) soluble molecules of the immune system are involved in the host’s attack to the graft cells. A recent review provides an excellent overview of the cellular and molecular mechanisms leading to graft rejection (4).
Alloantigens, the Inducers of Graft Rejection
As mentioned above, MHC molecules from donor origin are the main molecular targets triggering the immune attack suffered by the graft. MHC molecules are a high variable, codominantly expressed, and autosomically inherited genes expressed by most cell types (5). Physiologically, MHC molecules (HLA molecules in humans) are the essential bridge between innate immunity and adaptive (specific) antigenic responses. In short, antigen-presenting cells [APCs, such as dendritic cells (DCs) or Macrophages] capture pathogens and exhibit pathogen-derived processed antigens via MHC molecules to antigen-specific T cells. This recognition initiates both cellular and humoral adaptive immune responses, ideally leading to the eradication of the infective process.
In the transplant situation, graft cells expressing donor HLA molecules are recognized by the recipient T cells as “non-self” molecules, leading to a similar induction of the immune response (6). However, while in a “physiological” immune response against a pathogen, the number of activated T cell clones is rather low (approximately 1/100,000), in the transplant situation this number is increased to 1/100 or even more. Thus, the potency of inducing immune responses by HLA foreign alloantigens is much higher compared to a conventional immune response. This strong allorecognition is mainly based in two different aspects. First, the high level of polymorphism associated to HLA genes, the most polymorphic loci described in humans, that is continuously updated with the appearance of new allelic forms (7). Second, the wide repertoire of T cells able to respond to the allostimulation. This is due to the fact that priming and activation of T cells by alloantigens may occur through three different mechanisms, namely direct allorecognition, semi-direct allorecognition, and indirect allorecognition (Figure 1A) (8–11). In the direct allorecognition, recipient T cells “directly” recognize donor peptide-MHC complexes on donor APCs. This mechanism is responsible for the acute rejection and diminishes with time due to the progressive loss of donor APCs. Importantly, in acute rejection the inflammation caused in the organ by the surgical procedure or the period of ischemia-reperfusion may induce the expression of MHC and adhesion molecules (for instance in endothelial cells) and also the production of other inflammatory mediators that contribute to amplify the immune attack. The semi-direct allorecognition occurs when donor MHC molecules are recycled and presented as intact molecules on recipient APCs and presented to antigen-specific T cells (12). Finally, in the indirect allorecognition, recipient APCs capture and process donor alloantigens (as any exogenous antigen in a “physiological” immune response) and the derived peptides are exposed to T cell recognition via self (of the recipient) MHC molecules. Both, semi-direct and indirect allorecognition may be involved in chronic rejection.
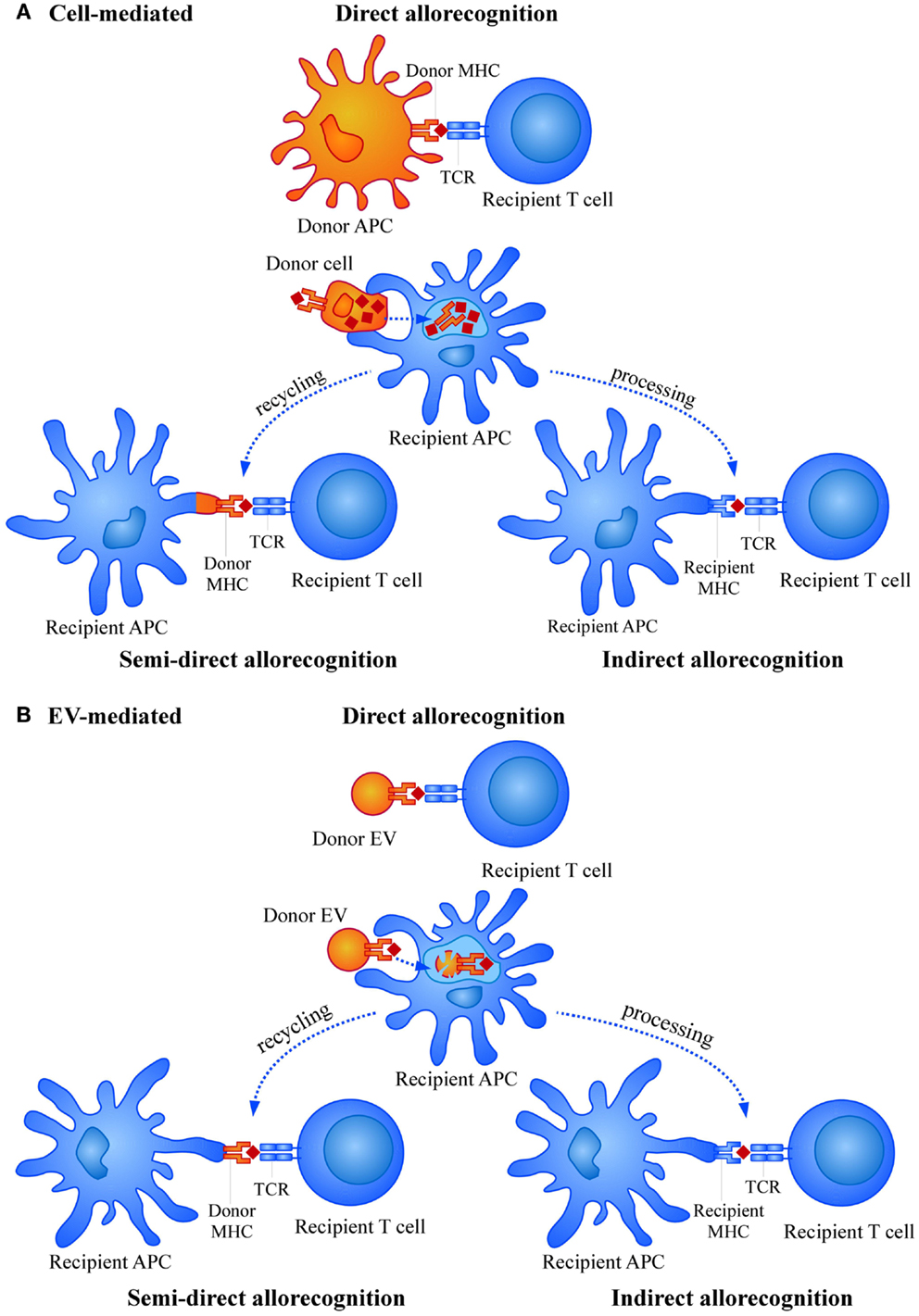
Figure 1. Mechanisms of allorecognition by (A) cells and (B) extracellular vesicles (EVs).
Of course, an ideal situation to avoid the immune response against alloantigens would be to find a complete HLA compatible donor, a possibility reduced to HLA-identical siblings. Considering the multiple combinations and possibilities derived from the number and diversity of MHC alleles and miHAs, it is extremely difficult to find a high level of HLA compatibility between a donor and a recipient. Thus, SOT has been routinely performed between ABO compatible donor–recipient pairs with no evidence of preformed anti-HLA antibodies (Cross-match negative). However, due to the shortage of organs from deceased donors, an important number of SOTs (mainly kidney transplantations) are performed over HLA or ABO antibody barriers. Desensitization strategies such as plasmapheresis, immune adsorption, anti-CD20 antibodies, and the use of complement inhibitors, may help those end-stage kidney disease patients (13). Yet, recipients with high titters of antibodies and HLA sensitized patients demonstrate a limiting long-term run.
The necessity to effectively block the immune response against the graft and therefore avoid transplant rejection has encouraged the study and generation of different immunosuppressive strategies.
Current Approaches to Immunosuppression
Clinically, immunosuppression (IS) in transplantation has the mission to prevent and treat acute rejection and to avoid chronic graft injury. These main objectives have to be in fine equilibrium with minimal adverse effects for the patient. Needless to say this balance is very difficult to achieve.
During the early years of organ transplantation, basic IS used corticosteroids and azathioprine. The appearance of Cyclosporine introduced a significant change in the field of transplantation, since the use of this drug dramatically reduced acute rejection episodes. Cyclosporine became the basic ISd until a new calcineurin inhibitor, Tacrolimus, was introduced in the 90s. Tacrolimus was more powerful compared to Cyclosporine, but shared a similar spectrum of adverse effects (basically worsening cardio-vascular profiles). Tacrolimus is still today the drug of reference for renal transplantation. When used in combination with antiproliferative drugs such as mycophenolate mophetil/mycophenolate sodium, the frequency of acute rejection episodes was set below 20%. The “Symphony” international study established the guidelines for IS in transplantation for the last 15 years. Following the study, more than 85% of current IS protocols are based on a combination of steroids, tacrolimus at low doses plus mycophenolate (14).
A new generation of ISd emerged with the synthesis of mTOR inhibitors, sirolimus, and everolimus. These drugs were initially applied in the so-called “no nephrotoxicity” protocols, which avoided using calcineurin inhibitors. However, a high frequency of adverse effects combined with increased rates of acute rejection episodes forced to stop treatments. Today mTOR inhibitors are combined with reduced doses of calcineurin inhibitors (15). Several independent studies support this strategy, although a large cohort study showing their efficacy and acceptable side effects is still missing.
Biological-derived drugs are the next generation of “conventional” IS. One of the most promising new drugs is Belatacept, a human fusion protein aimed to block the co-stimulation of T lymphocytes. Several studies suggest an efficacy comparable to calcineurin inhibitors, but preserving organ function (lower incidence of chronic kidney disease). Although Belatacept shows a good cardio-vascular profile (being this the main complication of Tacrolimus), an undesirable side effect is an increased incidence of post-transplant lymphomas, especially in Epstein-Barr virus seronegative patients. This treatment is therefore not used in this subgroup of patients (16, 17).
Many other agents are being evaluated in clinical trials to prevent acute rejection. Some examples are Sotrastaurine (potent and selective inhibitor isoform of protein kinase C), Tofacitinib (selective inhibitor of JAK 3 kinase), Alefacept (anti-CD2 humanized monoclonal antibody), and others (18–20). However, most of these studies are discontinued due to lower efficacy and safety profiles when compared to conventional ISd.
Most of the improvements and new drugs show efficacy in the short term after transplantation, thus controlling acute rejection (the first objective). Unfortunately, similar results have not been observed at long-term (21). The so-called chronic graft dysfunction is still a major cause of graft loss in kidney transplant [chronic kidney dysfunction (CKD)]. It is estimated that a 4% of transplanted kidneys are lost every year by this pathological process. In fact, CKD may be considered an epidemic itself, with similar prevalence to diabetes in the general population (22). Thus, the second mission of conventional IS aiming to control chronic graft injury is still not fully accomplished.
Another unsolved issue with ISd is the reduction of adverse side effects largely associated with these drugs. Remarkably, the main cause of loss of fully functional transplanted kidneys is due to death of the recipient patients affected by ISd side effects, including cardio-vascular diseases, opportunistic infections, and post-transplant neoplasia (23–25).
In summary, the use of ISd has markedly reduced the incidence of acute rejection and early graft loss. However, the numerous adverse side effects observed, and failure to effectively prevent chronic allograft dysfunction of conventional IS boosted the development of alternative strategies to avoid graft rejection.
New Approaches for Immune Regulation: Cell Therapy
Among the new approaches for the induction of allograft tolerance, the use of the regulatory properties of different cell types, such as regulatory T cells, DCs, and mesenchymal stem cells (MSCs) has been evaluated in animal models and also in some clinical trials using primates, with promising results (26, 27).
Regulatory T (Treg) cells have been widely studied for their capacities to modulate the immune response toward tolerance in different immunological contexts, being autoimmune diabetes the first to be considered for regulatory T cell therapy in a clinical trial (28). Their use in allograft acceptance has shown encouraging results (29–31), although no long term allograft tolerance has been attained so far. The mechanisms by which Treg cells manage to induce allograft tolerance are yet to be fully elucidated. Membrane-bound TGF-β and CTLA-4 expression is thought to mediate contact-dependent immunosuppression toward APCs and effector cells (32–35). Treg cells have been also reported to block the induction of IL-2 in T cells at the transcription level, leading to low proliferation and decreased activation of effector CD4+ and CD8+ T cells (36). Indirect recognition is thought to be the main allorecognition pathway suppressed by Treg cells as tolerance is deficient in animals lacking this mechanism of alloantigen presentation (37).
Different types of strategies have been tried to expand and activate Treg cells to enhance their immunosuppressive functions, thus several cytokine cocktails and immunosuppressants have been used to obtain more potent suppressor cells ex vivo, such as rapamycin (35). While some studies ensure IFN-γ induce Treg cells in vitro, other relate TGF-β and IL-10 as the most potent cytokines for Treg activation and survival (34, 38–42). On the other hand, there are reports showing the need for naïve T cells co-culture for obtaining alloantigen-specific Treg cells as the last depend on cytokines produced by ongoing Th1/Th2 immune response to develop (43). Anyway, the presence of CD4+ CD25+ T cells in the recipient is necessary to induce tolerance and achieve allograft acceptance (44).
Mesenchymal stem cells are an adult source of progenitor cells with the ability to self-replicate and differentiate to multiple lineages. MSCs have been proposed for their application in therapy of multiple diseases involving aberrant immune responses given their intrinsic immunoregulatory capabilities (35, 45–47) and ability to stimulate tissue repair and regeneration, as detailed further in the article by de Jong et al. published in this same issue. Treatment of injuries and diseases produced by unwanted inflammatory processes has been done with MSCs, and they have proven effective in kidney and cardiac injuries and in clinical trials for the treatment of Chron disease, graft versus host disease (GvHD), and diabetes (48, 49).
Mesenchymal stem cells are found to have different immunological policing according to the inflammatory milieu they are found in. For instance, under non-inflammatory conditions, MSCs promote regeneration and tissue repair, and have poor intrinsic immunogenicity due to the low expression of MHC or activation accessory molecules, which makes them suited to be used allogeneically in therapy and administered repeatedly (49, 50). On the other hand, MSCs become highly immunosuppressive when triggered by the inflammatory cytokines IFN-γ and TNF-α, and then importantly express MHC molecules (45, 48, 51), a combination that would be optimal for the purpose of achieving allograft tolerance.
In the transplantation set-up, there are some opposing studies (52–54), but the use of MSCs has been found also to help prolong heart and skin allograft survival and proves effective against refractory GvHD (45, 48). Relevantly, Ge et al. demonstrated the need of the right concomitant immunosuppressive for MSC engraftment and thus consecution of infinite allograft tolerance and also showed Ag-specific tolerance induction regardless of MSCs origin (55). A beneficial effect of MSC on experimental chronic graft nephropathy has been also reported in a rat kidney allograft model (56). Importantly, MSCs have been already used in clinical trials for allograft acceptance in the context of kidney transplantation, showing interesting results (57).
However, MSCs therapy has obvious concerns, which cannot be ignored, specifically their intrinsic tumorigenic potential given their self-replicating and differentiation capabilities. Moreover, their autologous use requires surgical intervention in compromised patients and exogenous expansion cultures, in which MSCs could modify their potency, efficiency, and safety (49).
Antigen-presenting cells have also been tested in the induction of tolerance. Among professional APCs, DCs actively participate in the physiological mechanisms of tolerance, through the induction of T cell anergy, depletion of antigen-specific T cells, and/or the promotion of regulatory T cells (Treg) (58–60). The tolerogenic potential of modified tolerogenic DCs (Tol-DCs) loaded with relevant antigens opened the possibility to treat certain autoimmune diseases in which antigenic proteins are at least partially characterized, such as multiple sclerosis or rheumatoid arthritis (61, 62). Similarly, the use of Tol-DCs has been tested in transplantation. Preliminary experiments in rodents have widely demonstrated that administration of different types of Tol-DCs prolong graft survival in combination with suboptimal doses of conventional immunosuppressant [reviewed in Ref. (63)]. As Tol-DCs may be obtained under GMP conditions (64), they are being used in clinical trials of tolerance induction in arthritis and allergy (www.clinicaltrials.gov) (65).
Some evidences support that unloaded Tol-DCs are sufficient for inducing allograft acceptance. In fact, it has been reported that recipient DCs are actually responsible for alloantigen presentation and tolerance attainment rather than injected DCs, from which they would capture alloantigens in a tolerogenic fashion (66). This would open the possibility to explore alternative cell-free alloantigen sources.
Alloantigens: Opportunities in Tolerance Induction in Transplantation
Several sources of alloantigenic material have been investigated for the induction of tolerance in transplantation. Immunodominant peptides were first explored for the induction of tolerance (67), which demonstrated the specificity of the response. In the case of SOT, due to the high variability associated to MHC antigens, it is virtually impossible to synthesize and cover all the antigenic polymorphisms of a given donor–receptor pair, thus pointing to the need of finding specific sources of these alloantigens in each transplant situation. Mimicking the experiments performed in DC-based tumor immunotherapy (68), cell-free lysates were initially chosen as a source of alloantigens, showing some encouraging results (69–73).
Apoptotic bodies from donor origin have been also proposed as another alloantigen source given their immunoregulatory capabilities and enhanced capture by APCs [reviewed by Ref. (74)]. Moreover, apoptotic lymphocytes would be a rich MHC source (75, 76), easy to prepare, and also would not require pre-loading DCs in vitro as their use per se has been proven sufficient. They have been used in transplant models in mice and rats, showing a prolonged allograft survival, promoting donor-specific tolerance, and proving to be safe by intravenous administration (75–79). Significantly, these studies highlighted the importance of the right timing of the therapy and demonstrated apoptosis’ intrinsic immunoregulatory capabilities, as necrosis did not show the same beneficial effects (76). Nevertheless, despite the promising results shown by several groups, infinite survival of the allograft has not been attained yet. Also, given the content in damaged DNA and high heterogeneity, other sources of alloantigens, such as EVs, are being considered. In contrast to cell lysates and apoptotic bodies, EVs represent a more stable and controlled source, can be cryopreserved and produced for clinical purposes (80, 81).
EVs as Alloantigens
Extracellular vesicles include a wide variety of lipid bilayered vesicles secreted by cells, ranging from nano to micrometric sizes and bearing distinct biochemical and physical properties. EVs mediate communication by transferring proteins and RNA between cells (82–84) not only at the paracrine level but also systemically. These vesicles are found in biological fluids like urine, blood, ascites fluid, cerebrospinal fluid, or semen [reviewed in Ref. (85)]. The term EVs refers to a broad spectrum of vesicles from different cell origin, biogenesis, function, and isolation method (86–88). Actually, most studies performed until now in this field refer to EVs as exosomes. While microvesicles are budded from the plasma membrane itself, exosomes are shed by many cell types upon the fusion of the multivesicular bodies (MVBs) with the plasma membrane and contain representative molecules from the cell they originate from, with functional proteins and RNA specifically sorted into them (89–91). Exosomes consistently express MHC antigens (92) and their composition is more homogeneous compared to apoptotic bodies (80) and less prone to inflammation compared to cell lysates. For this reason, exosomes, and extendedly EVs, have been proposed as a possible source for alloantigen presentation to the host.
Alloantigen presentation in vivo could be either directly mediated by the peptide-loaded MHC molecules found in the EVs or indirectly upon the capture and presentation by recipient APCs. Also, entire donor MHC molecules could be recycled by recipient’s APCs and presented to the recipient T cells (Figure 1B). There has been some controversy regarding the feasibility of direct presentation by EVs. Some studies proved the need of indirect presentation by DCs for exosomes to be able to stimulate T cells (93–95), while other groups did demonstrate direct functional presentation through exosomes themselves (96, 97).
Source of EVs for Therapeutic Use
Plasma EVs
Donor EVs containing MHC and miHAs may be obtained from multiple sources, each possessing intrinsic characteristics and advantages and being studied independently as strategies for allograft acceptance. The first biological fluid coming to mind given its ease of obtaining would be plasma. However, previous studies showed the little content of EVs and low MHC expression in healthy human plasma samples (98, 99), meaning plasma would not be the first choice in terms of alloantigen availability.
Cell-Derived EVs
Extracellular vesicles coming from cell-culture supernatants of different immunoregulatory cells would be the choice to modulate further the immune response triggered by alloantigen presentation. One of their main benefits would be to possess a stable phenotype that, contrary to cells, is not subject to further changes or alteration by the milieu. There are three main cell types being studied so far for the production of immunomodulatory EVs: regulatory T (Treg) cells, MSCs, and APCs, mainly DCs.
Regulatory T Cells EVs
As detailed information about Treg EVs and graft rejection can be found in the paper from Lesley Ann Smyth et al. published in this same issue, we will not discuss further this source.
Mesenchymal Stem Cells EVs
Mesenchymal stem cells were thought to mediate tissue repair and regeneration through replacement of injured cells by MSCs themselves. Lately, it has been found that rather than proliferating, MSCs promote the secretion of immunomodulatory cytokines and trophic factors in response to damage signaling, encouraging proliferation, and limiting apoptosis of the injured tissue (45, 48, 100). Recently, MSCs were found to secrete EVs, which would mediate MSCs’ signature effects (101–106), although there is some controversy regarding their full efficiency (107).
Mesenchymal stem cells-derived EVs have been used for the treatment of kidney, cardiac, and brain injuries showing regeneration and protective effect against injury, mainly at a paracrine level, and thought to be mediated through cytokines, growth factors and miRNAs delivered by EVs secreted by MSCs (47, 51, 108). In the context of transplantation, some studies have shown promising results, being the most outstanding the study reported by Kordelas et al. in which the infusion of MSC-derived EVs was able to treat a patient refractory to conventional IS therapy in GvHD (109). More information about MSC–EVs can be read on the study by Franquesa et al., in this same issue.
Dendritic Cells EVs
Since Raposo et al. demonstrated in 1996 the presence of MHC molecules in EVs secreted by B cells (110), APC-derived EVs as source of alloantigens has gained enormous interest. Later, studies focused on the description of the cargo molecules present in EVs coming from DCs indicated that DC-derived EVs were not only carrying class I and II MHC molecules, but also accessory molecules involved in T cell co-stimulation (81, 87, 111). As mentioned before, some groups demonstrated EVs were able to engage T cells through direct presentation (96, 112). Others suggested that exosomes required to be captured by DCs to induce an immune response by indirect presentation (94, 95). Moreover, content of MHC molecules in EVs was proven to be sufficient for effective and potent cross-presentation by host APCs. In this sense, it was reported that exosomes from tumor cells could trigger cross-priming of specific antitumor cytotoxic T lymphocytes (113, 114) and DC-derived EVs could induce tumor rejection in mice (68) and in human (115–117). Besides their use as cell-free vaccines in antitumoral therapies, DC-derived EVs have been studied as alternative therapies to induce tolerance in autoimmune diseases and in the transplantation setting.
On one hand, there are strategies focusing on strengthening the immunoregulatory properties of the EV-producing cells. For instance, several approximations engineered DC-derived-exosomes expressing Fas-L or IL-4-transduced BMDC-derived-exosomes, which were used as alternative treatments in models of delayed-type hypersensitivity (DTH) and collagen-induced arthritis (CIA) in mice. These approximations managed to delay the onset and severity of these immune-related diseases (118, 119). In a similar way, exosomes from IL-10-treated BMDCs or transduced with an adenovirus expressing IL-10 suppressed DTH responses (120).
Nevertheless, some effect was reported with mock exosomes, so immature DCs were suggested to secrete exosomes with regulatory properties. In fact, allogeneic exosomes from immature DCs can modulate the rejection of heart allografts (112). Therefore, it is important to consider that the activation state of the DCs producing EVs, may determine the immune response that these EVs will evoke in the host [(68, 112, 121), p. 200; (96)].
In the context of transplantation, donor EVs derived from immature Bone Marrow DC (BMDC) have been used as source of donor MHC antigens in animal models of heart and intestinal transplantation. A single iv administration of donor immature BMDC-derived EVs (imDex) prior to intestinal transplantation in a rat model reduced the host’s anti-donor cellular response, induced the generation of regulatory T cells, and temporally prolonged allograft survival (122). Interestingly, the double infusion of donor imDex before heterotopic heart transplantation prolonged allograft survival in a donor-specific manner (112). This effect was accompanied by a decrease in graft infiltrating leukocytes, a reduction of IFN-γ mRNA expression in the graft, and a decrease in the anti-donor cellular response post-transplantation.
Also, the combination of EV infusion along with non-specific immunosuppressive therapy to favor a tolerogenic microenvironment has also been tested in heterotopic models of heart transplantation (97, 123). Donor imDex administered post-transplantation in combination with suboptimal doses of the immunosuppressive drug LF15-0195 induced donor-specific tolerance, long term allograft survival, and delayed chronic rejection (97). Furthermore, the combination of rapamycin and donor imDex injected before and after transplantation promoted donor-specific tolerance, induced the generation of regulatory T cells and prolonged allograft survival, this time in a mouse model (123). In both cases, donor-specific tolerance was demonstrated to be transferable to naïve recipients, thus allograft survival would be mediated by cellular tolerance.
Critical Points
The use of EVs for tolerance induction share some of the critical points identified before in experimental cell therapies. As in these approaches, the route of administration, timing and frequency of administration, and the dose are some of the unsolved problems in EVs therapy.
Extracellular vesicles may be administered through several routes for tolerance induction, depending on the specific pathologies. For instance, intra-articular injection of anti-inflammatory exosomes has been used in rheumatoid arthritis patients [reviewed in Ref. (124)]. Also, intranasal administration has been tested in mice models of allergy (125). In the case of experimental transplantation, and similarly to cell therapeutic approaches, intravenous administration is the route of choice for injecting EVs in most of the experimental procedures.
The fate of (intravenously) injected EVs is still under discussion. It has been described that the expression of integrins, adhesion molecules, lipids, and other molecules on EVs contribute to the attachment and fusion of the injected vesicles to “acceptor” cells (126–130). In vitro experiments have shown that internalization of EVs is an active process (inhibited by cytochalasin D, EDTA, or low temperatures among others). In vivo imaging experiments revealed that intravenously administered EVs rapidly disappear from circulation and are firstly found in liver and lungs (131). Partially confirming these results, in vitro capture of EVs has been observed by liver and macrophage cell lines (98), and also described in splenic and peripheral blood DCs (127, 132). It is thus tempting to speculate that liver and spleen resident cells will be the main targets of intravenously injected EVs, and thus these cells will initially conduct the ongoing response (95).
A special mention needs to be paid to the different types of vesicles and their cellular origin. Obviously, different types of vesicles (such as apoptotic bodies and exosomes) from different cellular origins (MSCs, DCs, and others), or even the activation state of the EV-producing cells (for instance, immature versus mature DCs) will produce a specific response on target cells (and tissues) most probably through different mechanisms. In the past years, an outstanding effort has been made to elucidate the mechanisms of action of EVs. Recently, Robbins and Morelli reviewed the regulatory effect of EVs (from different cell origin) in the immune system (133). Yet, their regulatory effects in vivo are largely unknown, especially in humans. Probably, one of the first demonstrations of the in vivo regulation by EVs is the reported effect of MSC-derived EVs to treat a patient refractory to conventional IS therapy in graft-versus-host disease (109). Apparently, the mechanism of action was an impaired capability of the patient’s PBMCs to release pro-inflammatory cytokines in response to the EV treatment. Speculatively, this effect could be attributed to a higher IL10/IFNγ ratio in the infused EVs, although other mechanisms (generation of Tregs, miRNA regulation) may also contribute to the observed anti-inflammatory effect. The definition of a given type of vesicles to specifically apply for therapeutic purposes will undoubtedly depend on the pathology, mechanism(s) of action, and the feasibility to obtain sufficient amounts of EVs under GMP conditions to conduct the therapeutic approach.
Certainly, the definition of therapeutic doses is another important issue to be solved for the use of EVs in therapeutic applications. Interestingly, in SOT studies in mice and rats, a common observation seems to point to 10–25 μg of EV-associated protein per dose as the optimal quantity leading to increased survival of the graft (Table 1) (97, 121–123). Remarkably, exceeding or decreasing this quantity could only reproduce the results in part. It is noticeable though that this specific dose seems to work even when different concomitant IS regimes were used and also in spite of injecting different number of doses and at different days pre or post-transplantation. Given that the protocols used to enrich EVs do not preclude the presence of contaminant proteins in EV preparations, protein determination does not seem to be an accurate method to define the actual dose of EVs used. Beyond differences among several laboratories, this could also lead to variability among different preparations or batches in a given lab. Debates on the adoption of quantitative standards applicable to different laboratories, together with nomenclature of EVs, are underway in international forums.

Table 1. EV treatments in transplantation-related settings.
Scaling up this hypothetical working dose of 10 μg to a 60-kg human being would result in approximately 30 mg of EV-associated protein per dose. Data regarding this specific point is yet scarce. In the first phase I clinical trial using DC-derived EVs for melanoma vaccination, intradermal or subcutaneous EVs were injected at doses set up based on the concentration of MHC molecules in the EV preparations (116). More related to the induction of tolerance, Kordelas et al. defined one EV unit as the quantity of EVs recovered from 4 × 107 MSCs after 48 h in culture. The NTA analyses of this supernatant revealed a range of 1.5–3.5 × 1010 vesicles per unit and between 0.5 and 1.6 mg of protein (109). Further work is needed to define the optimal dose, number, and frequency of administrations for a given therapeutic situation. These may well benefit from the development of new methods aiming at an accurate quantification of EV preparations (134–136).
Thus, a number of additional questions need to be fully answered regarding EV therapy. Apart from the source of EVs, the optimal route, dose, and frequency of administration, other issues such as the standardization of EVs isolation/enrichment or quantitative issues will be necessarily solved in the incoming years. To date, most of the procedures for EVs isolation are based on differential ultracentrifugation. Other methods are based on the capacity of several precipitating agents to favor EV selective enrichment. Most of them though, do not preclude other proteins to be co-enriched with EVs. Further developments on size-exclusion chromatography and immune-based selection of EVs will contribute to reduce the presence of non-vesicular proteins and to improve EV preparations.
Despite these unsolved issues, EV therapy may have some advantages over cell therapy approaches. These include, among others, a non-tumorigenic potential (one of the main concerns on MSC therapy), and the possibility of sterilization by filtration, the capacity of EVs to cross tissue barriers (such as the blood–brain barrier) (137), or the fact that EVs cannot be influenced by the surrounding milieu (138). Some of these advantages may possibly favor the definition of less restrictive regulatory conditions, allowing an easier implementation of EV therapies.
Concluding Remarks
After a twentieth century, in which the development of IS has permitted outstanding advances in the field of transplantation, the next challenge of this discipline is a graft for life (139), that is, preventing chronic rejection of engrafted organs. Different approaches are exploring how to achieve this goal, including combination of organ and bone marrow transplantation (140). Hypothetically, discontinuation of IS after an initial acute phase would contribute to reduce side effects, thus importantly improving life expectancy after transplantation.
Given their particular characteristics, the contribution of EV therapy in organ transplantation for tolerance induction may be advantageous compared to other approaches in development, such as cell therapy. Together with their potential as drug-delivery carriers, cancer therapy, or in biomarker discovery, using EV strategies in tolerance induction will undoubtedly be one of the future areas of interest in biomedicine and biotechnology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by the Spanish “Ministerio de Ciencia e Innovación” through grant PI13/00050 from the “Fondo de Investigación Sanitaria” (FIS-ISCIII), also from Secretaria d’Universitats i Recerca del Departament d’Economia i Coneixement de la “Generalitat de Catalunya” (2014SGR804) and from REDinREN 2.0 (RD12/0021/0027). Francesc E. Borràs is sponsored by the “Researchers Stabilization Program” from the Spanish “Sistema Nacional de Salud” (SNS-ISCIII) and “Direcció d’Estratègia i Coordinació,” Catalan Health Department (CES07/015). Marta Monguió-Tortajada is sponsored by a grant (2014FI_B00649) from the “Agència de Gestió d’Ajuts Universitaris i de Recerca” (AGAUR) of the Catalan Government.
References
1. Spanish National Transplant Organization (ONT), World Health Organization (WHO). Global Observatory on Donation and Transplantation [Internet]. (2014). Available from: http://www.transplant-observatory.org/Pages/home.aspx
3. Rocha PN, Plumb TJ, Crowley SD, Coffman TM. Effector mechanisms in transplant rejection. Immunol Rev (2003) 196:51–64. doi:10.1046/j.1600-065X.2003.00090.x
4. Moreau A, Varey E, Anegon I, Cuturi M-C. Effector mechanisms of rejection. Cold Spring Harb Perspect Med (2013) 3. doi:10.1101/cshperspect.a015461 Available from: http://perspectivesinmedicine.cshlp.org/content/3/11/a015461.abstract,
5. Turner D. ES07.02 The human leucocyte antigen (HLA) system. Vox Sang (2004) 87:87–90. doi:10.1111/j.1741-6892.2004.00438.x
6. Burne MJ, Daniels F, El Ghandour A, Mauiyyedi S, Colvin RB, O’Donnell MP, et al. Identification of the CD4(+) T cell as a major pathogenic factor in ischemic acute renal failure. J Clin Invest (2001) 108:1283–90. doi:10.1172/JCI200112080
7. Marsh SGE. Nomenclature for factors of the HLA system, update March 2014. Int J Immunogenet (2014) 41:351–60. doi:10.1111/iji.12124
8. Rogers NJ, Lechler RI. Allorecognition. Am J Transplant (2001) 1:97–102. doi:10.1034/j.1600-6143.2001.10201.x
9. Lechler RI, Garden OA, Turka LA. The complementary roles of deletion and regulation in transplantation tolerance. Nat Rev Immunol (2003) 3:147–58. doi:10.1038/nri1002
10. Bolton EM, Bradley JA, Pettigrew GJ. Indirect allorecognition: not simple but effective. Transplantation (2008) 85(667–669). doi:10.1097/TP.0b013e3181664db3
11. Gökmen MR, Lombardi G, Lechler RI. The importance of the indirect pathway of allorecognition in clinical transplantation. Curr Opin Immunol (2008) 20:568–74. doi:10.1016/j.coi.2008.06.009
12. Herrera OB, Golshayan D, Tibbott R, Salcido Ochoa F, James MJ, Marelli-Berg FM, et al. A novel pathway of alloantigen presentation by dendritic cells. J Immunol (2004) 173:4828–37. doi:10.4049/jimmunol.173.8.4828
13. Maggiore U, Oberbauer R, Pascual J, Viklicky O, Dudley C, Budde K, et al. Strategies to increase the donor pool and access to kidney transplantation: an international perspective. Nephrol Dial Transplant (2014). doi:10.1093/ndt/gfu212
14. Ekberg H, Tedesco-Silva H, Demirbas A, Vítko Š, Nashan B, Gürkan A, et al. Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med (2007) 357:2562–75. doi:10.1056/NEJMoa067411
15. Cibrik D, Silva HTJ, Vathsala A, Lackova E, Cornu-Artis C, Walker RG, et al. Randomized trial of everolimus-facilitated calcineurin inhibitor minimization over 24 months in renal transplantation. Transplantation (2013) 95(933–942). doi:10.1097/TP.0b013e3182848e03
16. Vincenti F, Larsen C, Durrbach A, Wekerle T, Nashan B, Blancho G, et al. Costimulation blockade with belatacept in renal transplantation. N Engl J Med (2005) 353:770–81. doi:10.1056/NEJMoa050085
17. Rostaing L, Vincenti F, Grinyó J, Rice KM, Bresnahan B, Steinberg S, et al. Long-term belatacept exposure maintains efficacy and safety at 5 years: results from the long-term extension of the BENEFIT study. Am J Transplant (2013) 13:2875–83. doi:10.1111/ajt.12460
18. Ponticelli C. Present and future of immunosuppressive therapy in kidney transplantation. Transplant Proc (2011) 43:2439–40. doi:10.1016/j.transproceed.2011.06.025
19. Rostaing L, Charpentier B, Glyda M, Rigotti P, Hettich F, Franks B, et al. Alefacept combined with tacrolimus, mycophenolate mofetil and steroids in de novo kidney transplantation: a randomized controlled trial. Am J Transplant (2013) 13:1724–33. doi:10.1111/ajt.12303
20. Russ GR, Tedesco-Silva H, Kuypers DR, Cohney S, Langer RM, Witzke O, et al. Efficacy of sotrastaurin plus tacrolimus after de novo kidney transplantation: randomized, phase II trial results. Am J Transplant (2013) 13:1746–56. doi:10.1111/ajt.12251
21. Meier-Kriesche H-U, Schold JD, Srinivas TR, Kaplan B. Lack of improvement in renal allograft survival despite a marked decrease in acute rejection rates over the most recent era. Am J Transplant (2004) 4:378–83. doi:10.1111/j.1600-6143.2004.00332.x
22. Zoccali C, Kramer A, Jager KJ. Epidemiology of CKD in Europe: an uncertain scenario. Nephrol Dial Transplant (2010) 25:1731–3. doi:10.1093/ndt/gfq250
23. Marcén R. Immunosuppressive drugs in kidney transplantation. Drugs (2009) 69:2227–43. doi:10.2165/11319260-000000000-00000
24. Stewart JH, Vajdic CM, van Leeuwen MT, Amin J, Webster AC, Chapman JR, et al. The pattern of excess cancer in dialysis and transplantation. Nephrol Dial Transplant (2009) 24:3225–31. doi:10.1093/ndt/gfp331
25. Lim WH, Turner RM, Chapman JR, Ma MKM, Webster AC, Craig JC, et al. Acute rejection, T-cell-depleting antibodies, and cancer after transplantation. Transplantation (2014) 97(817–825). doi:10.1097/01.TP.0000442773.38510.32
26. Ezzelarab MB, Zahorchak AF, Lu L, Morelli AE, Chalasani G, Demetris AJ, et al. Regulatory dendritic cell infusion prolongs kidney allograft survival in nonhuman primates. Am J Transplant (2013) 13:1989–2005. doi:10.1111/ajt.12310
27. Geissler EK, Hutchinson JA. Cell therapy as a strategy to minimize maintenance immunosuppression in solid organ transplant recipients. Curr Opin Organ Transplant (2013) 18:408–15. doi:10.1097/MOT.0b013e328363319d
28. Leslie M. Immunology. Regulatory T cells get their chance to shine. Science (2011) 332:1020–1. doi:10.1126/science.332.6033.1020
29. Bradley JA. Transplant tolerance by Treg therapy. Am J Transplant (2004) 14:5–6. doi:10.1111/ajt.12510
30. Pu L-Y, Wang X-H, Zhang F, Li X-C, Yao A-H, Yu Y, et al. Adoptive transfusion of ex vivo donor alloantigen-stimulated CD4+CD25+ regulatory T cells ameliorates rejection of DA-to-Lewis rat liver transplantation. Surgery (2007) 142:67–73. doi:10.1016/j.surg.2007.02.014
31. Lee K, Nguyen V, Lee KM, Kang SM, Tang Q. Attenuation of donor-reactive T cells allows effective control of allograft rejection using regulatory T cell therapy. Am J Transplant (2014) 14:27–38. doi:10.1111/ajt.12509
32. Read S, Malmström V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med (2000) 192:295–302. doi:10.1084/jem.192.2.295
33. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med (2000) 192:303–10. doi:10.1084/jem.192.2.303
34. Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med (2001) 194:629–44. doi:10.1084/jem.194.5.629
35. Wood KJ, Bushell A, Hester J. Regulatory immune cells in transplantation. Nat Rev Immunol (2012) 12:417–30. doi:10.1038/nri3227
36. Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol (2000) 164:183–90. doi:10.4049/jimmunol.164.1.183
37. Yamada A, Chandraker A, Laufer TM, Gerth AJ, Sayegh MH, Auchincloss H. Recipient MHC class II expression is required to achieve long-term survival of murine cardiac allografts after costimulatory blockade. J Immunol (2001) 167:5522–6. doi:10.4049/jimmunol.167.10.5522
38. Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature (1997) 389:737–42. doi:10.1038/39614
39. Yamagiwa S, Gray JD, Hashimoto S, Horwitz DA. A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J Immunol (2001) 166:7282–9. doi:10.4049/jimmunol.166.12.7282
40. Sawitzki B, Kingsley CI, Oliveira V, Karim M, Herber M, Wood KJ. IFN-gamma production by alloantigen-reactive regulatory T cells is important for their regulatory function in vivo. J Exp Med (2005) 201:1925–35. doi:10.1084/jem.20050419
41. Feng G, Gao W, Strom TB, Oukka M, Francis RS, Wood KJ, et al. Exogenous IFN-gamma ex vivo shapes the alloreactive T-cell repertoire by inhibition of Th17 responses and generation of functional Foxp3+ regulatory T cells. Eur J Immunol (2008) 38:2512–27. doi:10.1002/eji.200838411
42. Feng G, Wood KJ, Bushell A. Interferon-gamma conditioning ex vivo generates CD25+CD62L+Foxp3+ regulatory T cells that prevent allograft rejection: potential avenues for cellular therapy. Transplantation (2008) 86:578–89. doi:10.1097/TP.0b013e3181806a60
43. Hall BM, Tran G, Hodgkinson SJ. Alloantigen specific T regulatory cells in transplant tolerance. Int Immunopharmacol (2009) 9:570–4. doi:10.1016/j.intimp.2009.01.016
44. Taylor PA, Lees CJ, Blazar BR. The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood (2002) 99:3493–9. doi:10.1182/blood.V99.10.3493
45. Hoogduijn MJ, Popp F, Verbeek R, Masoodi M, Nicolaou A, Baan C, et al. The immunomodulatory properties of mesenchymal stem cells and their use for immunotherapy. Int Immunopharmacol (2010) 10:1496–500. doi:10.1016/j.intimp.2010.06.019
46. Casiraghi F, Perico N, Remuzzi G. Mesenchymal stromal cells to promote solid organ transplantation tolerance. Curr Opin Organ Transplant (2013) 18:51–8. doi:10.1097/MOT.0b013e32835c5016
47. Lai RC, Yeo RWY, Tan KH, Lim SK. Mesenchymal stem cell exosome ameliorates reperfusion injury through proteomic complementation. Regen Med (2014) 8:197–209. doi:10.2217/rme.13.4
48. Kebriaei P, Robinson S. Mesenchymal stem cell therapy in the treatment of acute and chronic graft versus host disease. Front Oncol. (2011) 1:16. doi:10.3389/fonc.2011.00016
49. Patel AN, Genovese J. Potential clinical applications of adult human mesenchymal stem cell (Prochymal®) therapy. Stem Cells Cloning Adv Appl. (2011) 4:61–72. doi:10.2147/SCCAA.S11991
50. Sundin M, Barrett AJ, Ringdén O, Uzunel M, Lönnies H, Dackland A-L, et al. HSCT recipients have specific tolerance to MSC but not to the MSC donor. J Immunother (2009) 32:755–64. doi:10.1097/CJI.0b013e3181ab1807
51. Katsuda T, Kosaka N, Takeshita F, Ochiya T. The therapeutic potential of mesenchymal stem cell-derived extracellular vesicles. Proteomics (2013) 13:1637–53. doi:10.1002/pmic.201200373
52. Khakoo AY, Pati S, Anderson SA, Reid W, Elshal MF, Rovira II, et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi’s sarcoma. J Exp Med (2006) 203:1235–47. doi:10.1084/jem.20051921
53. Sudres M, Norol F, Trenado A, Grégoire S, Charlotte F, Levacher B, et al. Bone marrow mesenchymal stem cells suppress lymphocyte proliferation in vitro but fail to prevent graft-versus-host disease in mice. J Immunol (2006) 176:7761–7. doi:10.4049/jimmunol.176.12.7761
54. Nauta AJ, Westerhuis G, Kruisselbrink AB, Lurvink EGA, Willemze R, Fibbe WE. Donor-derived mesenchymal stem cells are immunogenic in an allogeneic host and stimulate donor graft rejection in a nonmyeloablative setting. Blood (2006) 108:2114–20. doi:10.1182/blood-2005-11-011650
55. Ge W, Jiang J, Baroja ML, Arp J, Zassoko R, Liu W, et al. Infusion of mesenchymal stem cells and rapamycin synergize to attenuate alloimmune responses and promote cardiac allograft tolerance. Am J Transplant (2009) 9:1760–72. doi:10.1111/j.1600-6143.2009.02721.x
56. Franquesa M, Herrero E, Torras J, Ripoll E, Flaquer M, Gomà M, et al. Mesenchymal stem cell therapy prevents interstitial fibrosis and tubular atrophy in a rat kidney allograft model. Stem Cells Dev (2012) 21:3125–35. doi:10.1089/scd.2012.0096
57. Pileggi A, Xu X, Tan J, Ricordi C. Mesenchymal stromal (stem) cells to improve solid organ transplant outcome: lessons from the initial clinical trials. Curr Opin Organ Transplant (2013) 18(672–681). doi:10.1097/MOT.0000000000000029
58. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392:245–52. doi:10.1038/32588
59. Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med (2001) 194:769–80. doi:10.1084/jem.194.6.769
60. Yamazaki S, Dudziak D, Heidkamp GF, Fiorese C, Bonito AJ, Inaba K, et al. CD8+CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T Cells. J Immunol (2008) 181:6923–33. doi:10.4049/jimmunol.181.10.6923
61. Stoop JN, Harry RA, von Delwig A, Isaacs JD, Robinson JH, Hilkens CMU. Therapeutic effect of tolerogenic dendritic cells in established collagen-induced arthritis is associated with a reduction in Th17 responses. Arthritis Rheum (2010) 62:3656–65. doi:10.1002/art.27756
62. Raϊch-Regué D, Grau-López L, Naranjo-Gómez M, Ramo-Tello C, Pujol-Borrell R, Martínez-Cáceres E, et al. Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients. Eur J Immunol (2012) 42:771–82. doi:10.1002/eji.201141835
63. Moreau A, Varey E, Bériou G, Bouchet-Delbos L, Segovia M, Cuturi MC. Tolerogenic dendritic cells and negative vaccination in transplantation: from rodents to clinical trials. Front Immunol (2012) 3:218. doi:10.3389/fimmu.2012.00218
64. Naranjo-Gomez M, Raich-Regue D, Onate C, Grau-Lopez L, Ramo-Tello C, Pujol-Borrell R, et al. Comparative study of clinical grade human tolerogenic dendritic cells. J Transl Med (2011) 9:89. doi:10.1186/1479-5876-9-89
65. Search of Dendritic Cells Tolerogenic – List Results. (2014). Available from: https://clinicaltrials.gov/ct2/results?term=dendritic+cells+tolerogenic
66. Divito SJ, Wang Z, Shufesky WJ, Liu Q, Tkacheva OA, Montecalvo A, et al. Endogenous dendritic cells mediate the effects of intravenously injected therapeutic immunosuppressive dendritic cells in transplantation. Blood (2010) 116:2694–705. doi:10.1182/blood-2009-10-251058
67. Garrovillo M, Ali A, Depaz HA, Gopinathan R, Oluwole OO, Hardy MA, et al. Induction of transplant tolerance with immunodominant allopeptide-pulsed host lymphoid and myeloid dendritic cells. Am J Transplant (2001) 1:129–37. doi:10.1034/j.1600-6143.2001.10206.x
68. Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell derived exosomes. Nat Med (1998) 4:594–600.
69. Taner T, Hackstein H, Wang Z, Morelli AE, Thomson AW. Rapamycin-treated, alloantigen-pulsed host dendritic cells induce Ag-specific T cell regulation and prolong graft survival. Am J Transplant (2005) 5:228–36. doi:10.1046/j.1600-6143.2004.00673.x
70. Turnquist HR, Raimondi G, Zahorchak AF, Fischer RT, Wang Z, Thomson AW. Rapamycin-conditioned dendritic cells are poor stimulators of allogeneic CD4+ T cells, but enrich for antigen-specific Foxp3+ T regulatory cells and promote organ transplant tolerance. J Immunol (2007) 178:7018–31. doi:10.4049/jimmunol.178.11.7018
71. Horibe EK, Sacks J, Unadkat J, Raimondi G, Wang Z, Ikeguchi R, et al. Rapamycin-conditioned, alloantigen-pulsed dendritic cells promote indefinite survival of vascularized skin allografts in association with T regulatory cell expansion. Transpl Immunol (2008) 18:307–18. doi:10.1016/j.trim.2007.10.007
72. Sacks JM, Kuo Y-R, Taieb A, Breitinger J, Nguyen VT, Thomson AW, et al. Prolongation of composite tissue allograft survival by immature recipient dendritic cells pulsed with donor antigen and transient low-dose immunosuppression. Plast Reconstr Surg (2008) 121(37–49). doi:10.1097/01.prs.0000293754.55706.7f
73. Ikeguchi R, Sacks JM, Unadkat JV, Solari M, Horibe EK, Thomson AW, et al. Long-term survival of limb allografts induced by pharmacologically conditioned, donor alloantigen-pulsed dendritic cells without maintenance immunosuppression. Transplantation (2008) 85:237–46. doi:10.1097/TP.0b013e31815e870e
74. Morelli A, Larregina A. Apoptotic cell-based therapies against transplant rejection: role of recipient’s dendritic cells. Apoptosis (2010) 15:1083–97. doi:10.1007/s10495-010-0469-9
75. Bittencourt M, Perruche S, Contassot E, Fresnay S, Baron MH, Angonin R, et al. Intravenous injection of apoptotic leukocytes enhances bone marrow engraftment across major histocompatibility barriers. Blood (2001) 98:224–30. doi:10.1182/blood.V98.1.224
76. Sun E, Gao Y, Chen J, Roberts AI, Wang X, Chen Z, et al. Allograft tolerance induced by donor apoptotic lymphocytes requires phagocytosis in the recipient. Cell Death Differ (2004) 11:1258–64. doi:10.1038/sj.cdd.4401500
77. Morelli AE, Wang Z, Shufesky WJ, Larregina AT, Zahorchak AF, Logar AJ. Use of donor apoptotic cells is a safe and effective means to prolong graft survival through interaction with recipient dendritic cells (Abstract). Am J Transplant (2003) 3:195.
78. Xu D-L, Liu Y, Tan J-M, Li B, Zhong C-P, Zhang X-H, et al. Marked prolongation of murine cardiac allograft survival using recipient immature dendritic cells loaded with donor-derived apoptotic cells. Scand J Immunol (2004) 59:536–44. doi:10.1111/j.1365-3083.2004.01427.x
79. Wang Z, Larregina AT, Shufesky WJ, Perone MJ, Montecalvo A, Zahorchak AF, et al. Use of the inhibitory effect of apoptotic cells on dendritic cells for graft survival via T-cell deletion and regulatory T cells. Am J Transplant (2006) 6:1297–311. doi:10.1111/j.1600-6143.2006.01308.x
80. Théry C, Boussac M, Véron P, Ricciardi-Castagnoli P, Raposo G, Garin J, et al. Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol (2001) 166:7309–18. doi:10.4049/jimmunol.166.12.7309
81. Lamparski HG, Metha-Damani A, Yao J-Y, Patel S, Hsu D-H, Ruegg C, et al. Production and characterization of clinical grade exosomes derived from dendritic cells. J Immunol Methods (2002) 270:211–26. doi:10.1016/S0022-1759(02)00330-7
82. Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia (2006) 20:1487–95. doi:10.1038/sj.leu.2404296
83. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJA. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol (2011) 29:341–5. doi:10.1038/nbt.1807
84. Gutiérrez-Vázquez C, Villarroya-Beltri C, Mittelbrunn M, Sánchez-Madrid F. Transfer of extracellular vesicles during immune cell-cell interactions. Immunol Rev (2012) 251:125–42. doi:10.1111/imr.12013
85. Witwer KW, Buzás EI, Bemis LT, Bora A, Lässer C, Lötvall J, et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles (2013) 2:20360. doi:10.3402/jev.v2i0.20360
86. Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol (2009) 19:43–51. doi:10.1016/j.tcb.2008.11.003
87. Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol (2009) 9:581–93. doi:10.1038/nri2567
88. Gould SJ, Raposo G. As we wait: coping with an imperfect nomenclature for extracellular vesicles. J Extracell Vesicles (2013) 2:20389. doi:10.3402/jev.v2i0.20389
89. Kalra H, Simpson RJ, Ji H, Aikawa E, Altevogt P, Askenase P, et al. Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol (2012) 10:e1001450. doi:10.1371/journal.pbio.1001450
90. Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan MLG, Karlsson JM, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood (2012) 119:756–66. doi:10.1182/blood-2011-02-338004
91. Villarroya-Beltri C, Gutiérrez-Vázquez C, Sánchez-Cabo F, Pérez-Hernández D, Vázquez J, Martin-Cofreces N, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun (2013) 4:2980. doi:10.1038/ncomms3980
92. Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol (2002) 2:569–79.
93. Vincent-Schneider H, Stumptner-Cuvelette P, Lankar D, Pain S, Raposo G, Benaroch P, et al. Exosomes bearing HLA-R1 molecules need dendritic cells to efficiently stimulate specific T cells. Int Immunol (2002) 14:713–22. doi:10.1093/intimm/dxf048
94. Théry C, Duban L, Segura E, Véron P, Lantz O, Amigorena S. Indirect activation of naïve CD4+ T cells by dendritic cell-derived exosomes. Nat Immunol (2002) 3:1156–62. doi:10.1038/ni854
95. Montecalvo A, Shufesky WJ, Beer Stolz D, Sullivan MG, Wang Z, Divito SJ, et al. Exosomes as a short-range mechanism to spread alloantigen between dendritic cells during T cell allorecognition. J Immunol (2008) 180:3081–90. doi:10.4049/jimmunol.180.5.3081
96. Segura E, Nicco C, Lombard B, Véron P, Raposo G, Batteux F, et al. ICAM-1 on exosomes from mature dendritic cells is critical for efficient naive T-cell priming. Blood (2005) 106:216–23. doi:10.1182/blood-2005-01-0220
97. Pêche H, Renaudin K, Beriou G, Merieau E, Amigorena S, Cuturi MC. Induction of tolerance by exosomes and short-term immunosuppression in a fully MHC-mismatched rat cardiac allograft model. Am J Transplant (2006) 6:1541–50. doi:10.1111/j.1600-6143.2006.01344.x
98. Bastos-Amador P, Royo F, Gonzalez E, Conde-Vancells J, Palomo-Diez L, Borras FE, et al. Proteomic analysis of microvesicles from plasma of healthy donors reveals high individual variability. J Proteomics (2012) 75:3574–84. doi:10.1016/j.jprot.2012.03.054
99. Muller L, Hong C-S, Stolz DB, Watkins SC, Whiteside TL. Isolation of biologically-active exosomes from human plasma. J Immunol Methods (2014). doi:10.1016/j.jim.2014.06.007
100. Koch M, Lehnhardt A, Hu X, Brunswig-Spickenheier B, Stolk M, Bröcker V, et al. Isogeneic MSC application in a rat model of acute renal allograft rejection modulates immune response but does not prolong allograft survival. Transpl Immunol (2013) 29:43–50. doi:10.1016/j.trim.2013.08.004
101. Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, et al. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia (2006) 20:847–56. doi:10.1038/sj.leu.2404132
102. Gatti S, Bruno S, Deregibus MC, Sordi A, Cantaluppi V, Tetta C, et al. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol Dial Transplant (2011) 26:1474–83. doi:10.1093/ndt/gfr015
103. Bruno S, Grange C, Collino F, Deregibus MC, Cantaluppi V, Biancone L, et al. Microvesicles derived from mesenchymal stem cells enhance survival in a lethal model of acute kidney injury. PLoS One (2012) 7:e33115. doi:10.1371/journal.pone.0033115
104. Cantaluppi V, Gatti S, Medica D, Figliolini F, Bruno S, Deregibus MC, et al. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int (2012) 82:412–27. doi:10.1038/ki.2012.105
105. Lai RC, Yeo RWY, Tan KH, Lim SK. Mesenchymal stem cell exosome ameliorates reperfusion injury through proteomic complementation. Regen Med (2013) 8:197–209. doi:10.2217/rme.13.4
106. Li T, Yan Y, Wang B, Qian H, Zhang X, Shen L, et al. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis. Stem Cells Dev (2013) 22:845–54. doi:10.1089/scd.2012.0395
107. Conforti A, Scarsella M, Starc N, Giorda E, Biagini S, Proia A, et al. Microvescicles derived from mesenchymal stromal cells are not as effective as their cellular counterpart in the ability to modulate immune responses in vitro. Stem Cells Dev (2014). doi:10.1089/scd.2014.0091
108. Dorronsoro A, Robbins PD. Regenerating the injured kidney with human umbilical cord mesenchymal stem cell-derived exosomes. Stem Cell Res Ther (2013) 4(2):39. doi:10.1186/scrt187
109. Kordelas L, Rebmann V, Ludwig AK, Radtke S, Ruesing J, Doeppner TR, et al. MSC-derived exosomes: a novel tool to treat therapy-refractory graft-versus-host disease. Leukemia (2014) 28(4):970–3. doi:10.1038/leu.2014.41
110. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med (1996) 183:1161–72. doi:10.1084/jem.183.3.1161
111. Denzer K, van Eijk M, Kleijmeer MJ, Jakobson E, de Groot CJ, Geuze H. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J Immunol (2000) 165:1259–65. doi:10.4049/jimmunol.165.3.1259
112. Pêche H, Heslan M, Usal C, Amigorena S, Cuturi MC. Presentation of donor major histocompatibility complex antigens by bone marrow dendritic cell-derived exosomes modulates allograft rejection. Transplantation (2003) 76:1503–10. doi:10.1097/01.TP.0000092494.75313.38
113. Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med (2001) 7:297–303. doi:10.1038/85438
114. André F, Chaput N, Schartz NEC, Flament C, Aubert N, Bernard J, et al. Exosomes as potent cell-free peptide-based vaccine. i. dendritic cell-derived exosomes transfer functional MHC class I/peptide complexes to dendritic cells. J Immunol (2004) 172:2126–36. doi:10.4049/jimmunol.172.4.2126
115. Chaput N, Schartz NEC, André F, Taïeb J, Novault S, Bonnaventure P, et al. Exosomes as potent cell-free peptide-based vaccine. II. Exosomes in CpG adjuvants efficiently prime naive Tc1 lymphocytes leading to tumor rejection. J Immunol (2004) 172:2137–46. doi:10.4049/jimmunol.172.4.2137
116. Escudier B, Dorval T, Chaput N, Andre F, Caby M-P, Novault S, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of the first phase I clinical trial. J Transl Med (2005) 3:10. doi:10.1186/1479-5876-3-10
117. Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM, et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med (2005) 3:9. doi:10.1186/1479-5876-3-9
118. Hee Kim S, Bianco N, Menon R, Lechman ER, Shufesky WJ, Morelli AE, et al. Exosomes derived from genetically modified dc expressing FasL are anti-inflammatory and immunosuppressive. Mol Ther (2006) 13:289–300. doi:10.1016/j.ymthe.2005.09.015
119. Kim SH, Bianco NR, Shufesky WJ, Morelli AE, Robbins PD. Effective treatment of inflammatory disease models with exosomes derived from dendritic cells genetically modified to express IL-4. J Immunol (2007) 179:2242–9. doi:10.4049/jimmunol.179.4.2242
120. Kim S-H, Lechman ER, Bianco N, Menon R, Keravala A, Nash J, et al. Exosomes derived from IL-10-treated dendritic cells can suppress inflammation and collagen-induced arthritis. J Immunol (2005) 174:6440–8. doi:10.4049/jimmunol.174.10.6440
121. Pêche H, Trinité B, Martinet B, Cuturi MC. Prolongation of heart allograft survival by immature dendritic cells generated from recipient type bone marrow progenitors. Am J Transplant (2005) 5:255–67. doi:10.1111/j.1600-6143.2004.00683.x
122. Yang X, Meng S, Jiang H, Zhu C, Wu W. Exosomes derived from immature bone marrow dendritic cells induce tolerogenicity of intestinal transplantation in rats. J Surg Res (2011) 171:826–32. doi:10.1016/j.jss.2010.05.021
123. Li X, Li J-J, Yang J-Y, Wang D-S, Zhao W, Song W-J, et al. Tolerance induction by exosomes from immature dendritic cells and rapamycin in a mouse cardiac allograft model. PLoS One (2012) 7:e44045. doi:10.1371/journal.pone.0044045
124. Yang C, Robbins PD. Immunosuppressive exosomes: a new approach for treating arthritis. Int J Rheumatol. (2012) 2012:573528. doi:10.1155/2012/573528
125. Prado N, Cañamero M, Villalba M, Rodríguez R, Batanero E. Bystander suppression to unrelated allergen sensitization through intranasal administration of tolerogenic exosomes in mouse. Mol Immunol (2010) 47:2148–51. doi:10.1016/j.molimm.2010.04.014
126. Clayton A, Turkes A, Dewitt S, Steadman R, Mason MD, Hallett MB. Adhesion and signaling by B cell-derived exosomes: the role of integrins. FASEB J (2004). doi:10.1096/fj.03-1094fje Available from: http://www.fasebj.org/content/early/2004/06/02/fj.03-1094fje.short,
127. Morelli AE, Larregina AT, Shufesky WJ, Sullivan MLG, Stolz DB, Papworth GD, et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood (2004) 104:3257–66. doi:10.1182/blood-2004-03-0824
128. Barrès CB, Blanc L, Bette-Bobillo P, André S, Mamoun R, Gabius HJ, et al. Galectin-5 is bound onto the surface of rat reticulocyte exosomes and modulates vesicle uptake by macrophages. Blood (2009) 115:696–705. doi:10.1182/blood-2009-07-231449
129. Izquierdo-Useros N, Lorizate M, Puertas MC, Rodriguez-Plata MT, Zangger N, Erikson E, et al. Siglec-1 is a novel dendritic cell receptor that mediates HIV-1 trans-infection through recognition of viral membrane gangliosides. PLoS Biol (2012) 10:e1001448. doi:10.1371/journal.pbio.1001448
130. Saunderson SC, Dunn AC, Crocker PR, McLellan AD. CD169 mediates the capture of exosomes in spleen and lymph node. Blood (2013) 123:208–16. doi:10.1182/blood-2013-03-489732
131. Takahashi Y, Nishikawa M, Shinotsuka H, Matsui Y, Ohara S, Imai T, et al. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J Biotechnol (2013) 165:77–84. doi:10.1016/j.jbiotec.2013.03.013
132. Bastos-Amador P, Pérez-Cabezas B, Izquierdo-Useros N, Puertas MC, Martinez-Picado J, Pujol-Borrell R, et al. Capture of cell-derived microvesicles (exosomes and apoptotic bodies) by human plasmacytoid dendritic cells. J Leukoc Biol (2012). doi:10.1189/jlb.0111054 Available from: http://www.ncbi.nlm.nih.gov/pubmed/22319103,
133. Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol (2014) 14:195–208. doi:10.1038/nri3622
134. Kanwar SS, Dunlay CJ, Simeone DM, Nagrath S. Microfluidic device (ExoChip) for on-chip isolation, quantification and characterization of circulating exosomes. Lab Chip (2014) 14:1891–900. doi:10.1039/c4lc00136b
135. Im H, Shao H, Park YI, Peterson VM, Castro CM, Weissleder R, et al. Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor. Nat Biotech. (2014) 32:490–5. doi:10.1038/nbt.2886
136. Rupert DLM, Lässer C, Eldh M, Block S, Zhdanov VP, Lotvall JO, et al. Determination of exosome concentration in solution using surface plasmon resonance spectroscopy. Anal Chem (2014) 86:5929–36. doi:10.1021/ac500931f
137. Xin H, Li Y, Cui Y, Yang JJ, Zhang ZG, Chopp M. Systemic administration of exosomes released from mesenchymal stromal cells promote functional recovery and neurovascular plasticity after stroke in rats. J Cereb Blood Flow Metab (2013) 33:1711–5. doi:10.1038/jcbfm.2013.152
138. Naslund TI, Gehrmann U, Qazi KR, Karlsson MCI, Gabrielsson S. Dendritic cell-derived exosomes need to activate both T and B cells to induce antitumor immunity. J Immunol (2013) 190:2712–9. doi:10.4049/jimmunol.1203082
Keywords: exosomes, extracellular vesicles, transplantation immunology, tolerance, graft rejection, graft survival, organ transplantation
Citation: Monguió-Tortajada M, Lauzurica-Valdemoros R and Borràs FE (2014) Tolerance in organ transplantation: from conventional immunosuppression to extracellular vesicles. Front. Immunol. 5:416. doi: 10.3389/fimmu.2014.00416
Received: 30 July 2014; Paper pending published: 08 August 2014;
Accepted: 18 August 2014; Published online: 17 September 2014.
Edited by:
Ana Maria Merino, Bellvitge Biomedical Research Institute, SpainReviewed by:
Marcella Franquesa, Erasmus University Medical Center, NetherlandsJuan Torras, Hospital Universitari Bellvitge, Spain
Copyright: © 2014 Monguió-Tortajada, Lauzurica-Valdemoros and Borràs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesc E. Borràs, IVECAT Group, Institut d’Investigació Germans Trias i Pujol, Ctra. Can Ruti, Camí de les Escoles s/n, Badalona 08916, Spain e-mail: feborras@igtp.cat