 Maria C. D'Adamo1,2†
Maria C. D'Adamo1,2† Constanze Gallenmüller3,4,5†
Constanze Gallenmüller3,4,5† Ilenio Servettini1
Ilenio Servettini1 Elisabeth Hartl3
Elisabeth Hartl3 Stephen J. Tucker6
Stephen J. Tucker6 Larissa Arning7Saskia Biskup8
Larissa Arning7Saskia Biskup8 Alessandro Grottesi9
Alessandro Grottesi9 Luca Guglielmi1
Luca Guglielmi1 Paola Imbrici10
Paola Imbrici10 Pia Bernasconi11
Pia Bernasconi11 Giuseppe Di Giovanni2,12
Giuseppe Di Giovanni2,12 Fabio Franciolini13
Fabio Franciolini13 Luigi Catacuzzeno13
Luigi Catacuzzeno13 Mauro Pessia1,2*‡
Mauro Pessia1,2*‡ Thomas Klopstock3,4,5,14*‡
Thomas Klopstock3,4,5,14*‡- 1Section of Physiology and Biochemistry, Department of Experimental Medicine, School of Medicine, University of Perugia, Perugia, Italy
- 2Section of Neurophysiology and Biophysics, Istituto Euro-Mediterraneo di Scienza e Tecnologia, Palermo, Italy
- 3Department of Neurology, Friedrich-Baur-Institute, Ludwig-Maximilians-University, Munich, Germany
- 4German Network for Mitochondrial Disorders (mitoNET), Ludwigshafen, Germany
- 5DZNE - German Center for Neurodegenerative Diseases, Munich, Germany
- 6Clarendon Laboratory, Department of Physics, University of Oxford, Oxford, UK
- 7Department of Human Genetics, Ruhr-University Bochum, Bochum, Germany
- 8Center for Genomics and Transcriptomics (CeGaT) GmbH Tübingen, Tübingen, Germany
- 9Department of Supercomputing Applications and Innovation, CINECA (Consorzio Inter-Universitario per il Calcolo Automatico), Rome, Italy
- 10Department of Pharmacy, University of Bari, Bari, Italy
- 11Neurology IV – Neuromuscular Diseases and Neuroimmunology Unit, Foundation IRCCS Neurological Institute “Carlo Besta”, Milan, Italy
- 12Department of Physiology and Biochemistry, Faculty of Medicine and Surgery, University of Malta, Msida, Malta
- 13Department of Chemistry, Biology and Biotechnology, University of Perugia, Perugia, Italy
- 14German Center for Vertigo and Balance Disorders, Munich, Germany
Episodic ataxia type 1 (EA1) is an autosomal dominant K+ channelopathy which manifests with short attacks of cerebellar ataxia and dysarthria, and may also show interictal myokymia. Episodes can be triggered by emotional or physical stress, startle response, sudden postural change or fever. Here we describe a 31-year-old man displaying markedly atypical symptoms, including long-lasting attacks of jerking muscle contractions associated with hyperthermia, severe migraine, and a relatively short-sleep phenotype. A single nucleotide change in KCNA1 (c.555C>G) was identified that changes a highly conserved residue (p.C185W) in the first transmembrane segment of the voltage-gated K+ channel Kv1.1. The patient is heterozygous and the mutation was inherited from his asymptomatic mother. Next generation sequencing revealed no variations in the CACNA1A, CACNB4, KCNC3, KCNJ10, PRRT2 or SCN8A genes of either the patient or mother, except for a benign variant in SLC1A3. Functional analysis of the p.C185W mutation in KCNA1 demonstrated a deleterious dominant-negative phenotype where the remaining current displayed slower activation kinetics, subtle changes in voltage-dependence and faster recovery from slow inactivation. Structural modeling also predicts the C185W mutation to be functionally deleterious. This description of novel clinical features, associated with a Kv1.1 mutation highlights a possibly unrecognized relationship between K+ channel dysfunction, hyperthermia and migraine in EA1, and suggests that thorough assessments for these symptoms should be carefully considered for all patients affected by EA1.
Introduction
Episodic ataxia type 1 (EA1) is an autosomal dominant K+ channelopathy which manifests as brief episodes of cerebellar dysfunction with paroxysmal ataxia, dysarthria, diplopia, vertigo and tremor often lasting for seconds or minutes. Some patients also show interictal myokymia. Additional ictal symptoms may comprise spastic contractions, stiffening of the body, visual disturbances, seizures and mild headache (VanDyke et al., 1975). The episodes can be triggered by emotional or physical stress, startle response, sudden postural change, and even caffeine. Fever or high temperatures occurring after a hot bath have also been reported to precipitate attacks (Eunson et al., 2000; Klein et al., 2004). The onset of symptoms is typically within the first or second decade of life and the frequency of attacks can vary from less than once per month to 30 times a day.
The disorder results from loss-of-function mutations in the KCNA1 gene which encodes the voltage-gated K+ channel, Kv1.1 (Browne et al., 1994; Adelman et al., 1995; D'Adamo et al., 1998; Imbrici et al., 2003, 2006, 2007, 2011; Cusimano et al., 2004; Pessia et al., 2012). Most individuals with EA1 have a pedigree suggestive of autosomal dominant inheritance with at least one affected family member. However, one de novo mutation has also been reported (Demos et al., 2009). Diagnosis is primarily based on clinical findings and molecular genetic testing of KCNA1, and these investigations in several affected families have broadened the clinical spectrum of EA1 to now include phenotypes with delay in motor development, choreoathetosis, cognitive dysfunction, transient postural abnormalities in infancy, shortening of the Achilles tendon in children and epileptic seizures (Zuberi et al., 1999; Demos et al., 2009; D'Adamo et al., 2012, 2013; Pessia et al., 2012). Electromyographic myokymia can be confirmed in nearly all individuals with EA1, but in some cases the myokymic activity only becomes apparent after application of regional ischemia. Mutations in KCNA1 are also known to be associated with isolated neuromyotonia (Eunson et al., 2000; Kinali et al., 2004). Moderate muscle hypertrophy, a generalized increase in muscle tone and bilateral calf hypertrophy have also been reported for EA1 patients (VanDyke et al., 1975; Kinali et al., 2004; Demos et al., 2009). Recently, direct evidence identified the motor nerve as an important generator of myokymic activity and showed that dysfunction of juxtaparanodal Kv1.1 channels alters Ca2+ homeostasis in motor axons (Brunetti et al., 2012; D'Adamo et al., 2014). In a large Brazilian family harboring a KCNA1 mutation, low blood levels of magnesium with recurrent muscle cramps, tetanic episodes, limb muscle weakness and electromyographic myokymia were also found (Glaudemans et al., 2009). Since Kv1.1 channels are widely expressed in the nervous system, particularly in the cerebellum, hippocampus and hypothalamus (Albrecht et al., 1995; Geiger and Jonas, 2000; Herson et al., 2003), it is therefore perhaps not so surprising that EA1 patients exhibit such a variable clinical phenotype (Graves et al., 2010).
In this study, we now describe a 31-year-old man carrying a dominant-negative, loss-of-function mutation in KCNA1 who displays a new phenotype characterized by episodes of hyperthermia, short-sleep duration, and severe migraine.
Materials and Methods
Genetic Analysis
Patients were studied after giving informed consent and investigations were conducted in accordance with protocols approved by the institutional review boards of the Friedrich-Baur-Institute of the Ludwig-Maximilians-University, Munich, Germany. DNA was isolated from the peripheral blood by standard methods. The coding regions and exon-intron boundaries of KCNA1 were PCR amplified using primer oligonucleotides designed with the Primer Express tools. PCR conditions are available upon request. Direct sequencing of KCNA1 was performed using an automated Sanger dideoxy method.
Electrophysiology
The human Kv1.1 cDNA was subcloned into the pBF oocyte expression vector. The p.C185W mutation was introduced by site-directed mutagenesis and verified by automated sequencing. The concentration of in vitro transcribed cRNA was quantified by electrophoresis and ethidium bromide staining, and spectrophotometric analysis. Expression of wild-type and mutant channels in Xenopus laevis oocytes and two-electrode voltage-clamp recordings (TEVC) and patch-clamp recordings were performed as previously described (D'Adamo et al., 1998). Briefly, TEVC recordings were performed from oocytes at ~22°C and 1–8 days after cRNA injection by using a GeneClamp 500 amplifier (Axon) interfaced to a PC computer with an ITC-16 interface (Instrutech Corp., USA). Microelectrodes were filled with 3M KCl and had resistances of 0.1–0.5 MΩ. The extracellular solution contained (mM): NaCl 96, KCl 2, MgCl2 1, CaCl2 1.8, HEPES 5, pH 7.4. Recordings were filtered at 2 kHz and acquired at 5 kHz with Pulse software and analyzed with either PulseFit (HEKA, Germany) or Origin 8 program. Leak and capacitative currents were subtracted using a P/4 protocol. Kinetic parameters were evaluated from current amplitudes <10 μA using low resistance electrodes.
Patch-clamp recordings were performed with an Axopatch 200B amplifier (Axon Instruments). The pipette solution contained (mM): NaCl 120, KCl 2, HEPES 5, pH 7.4 whereas the cytoplasmic solution contained (mM): KCl 120, EGTA 1, HEPES 5, pH 7.4. Recording electrodes were pulled from borosilicate glass, coated with Sylgard and had resistances of 5–15 MΩ. The excised inside-out patch recordings were low-pass filtered at 0.5–2 kHz with an 8 pole bessel filter (Frequency Devices, MA) and acquired with a Pulse + PulseFit program (HEKA elektronik GmbH, Germany). Channel activity was analyzed with a TAC-TACfit program (Bruxton Co. WA), by inspecting all transitions and their slope conductance was assessed at different potentials from all events histograms.
Xenopus laevis were deeply anesthetized with an aerated solution containing 3-aminobenzoic acid ethyl ester methanesulfonate salt (5 mM) and sodium bicarbonate (60 mM), pH 7.3. Stage V–VI Xenopus oocytes were isolated, injected with 50 nl RNA and stored at 16°C in fresh ND96 medium containing (mM): NaCl 96, KCl 2, MgCl2 1, CaCl2 1.8, HEPES 5, gentamicin 50 μg/ml. Xenopus laevis underwent no more than two surgeries, separated by at least 3 weeks. Animal handling and electrophysiological experiments were conducted in accordance with international standards on animal care, and the regulations of the Italian Animal Welfare Act, approved by the local Veterinary Service Authority, and with the NIH Guide for the Care and Use of Laboratory Animals.
Homology Modeling
A 3D structural model of Kv1.1 was built through comparative modeling using the software Modeler (Sali and Blundell, 1993) and the crystal structure of the Kv1.2/2.1 chimera (PDB id.: 2R9R) as a template (Nishida et al., 2007). Sequence alignment of the target sequence vs the template was generated using ClustalX, and further refined using Muscle (Edgar, 2004). Twenty homology models were generated and scored against the minimum number of constraint violations. Among them, the five lowest energy models were selected and analyzed using Procheck (Laskowski et al., 1996). The final model was chosen according to the highest percentage of residues in the allowed region of the Ramachandran plot (N90%). The model was then immersed in a pre-equilibrated palmitoyl-oleoyl-phosphatidylcholine (POPC) lipid bilayer, and all overlapping lipid molecules (within 3 Å from any protein atoms) removed. The final model was further minimized to reduce steric hindrance with neighboring atoms using GROMACS4 and the GROMOS96 forcefield (Hess et al., 2008).
Results
Case Report
The proband (II.2, Figure 1A) is a 31-year-old man who has been suffering from episodes of ataxia, dysarthria, diplopia and oscillopsias from childhood. The frequency of his attacks was mostly low (1–2 per week or less). Typical episodes lasted 2–3 min, were triggered by physical exercise and sudden movements, but not by alcohol or caffeine. Attacks were also precipitated by febrile illnesses. However, several episodes witnessed in-hospital were characterized by ataxia/dysarthria, lasting for hours and days, with concurrent hyperthermia up to 40.3°C that was not caused by an obvious focus of infection with the exception of streptococcal tonsillopharyngitis being detected in one episode.
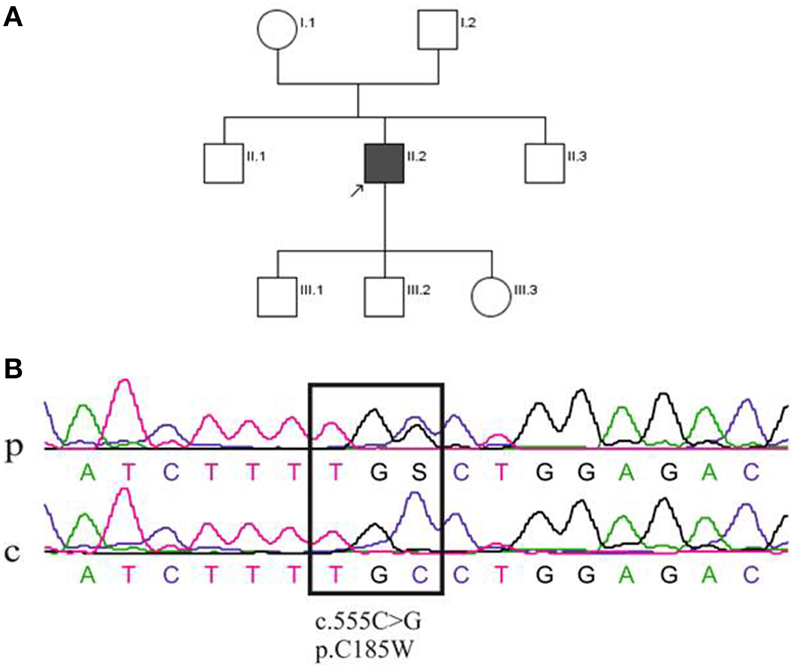
Figure 1. (A) Pedigree of the family of the patient (II.2). (B) Sequence analysis of the KCNA1 gene mutation. DNA sequence from the patient (p; II2) and a control individual (c). The upper sequencing profile shows the heterozygous single nucleotide change at position c.555C > G, which leads to a cysteine (C) to tryptophan (W) substitution (p.C185W) in the protein Kv1.1.
With regard to his sleeping habits, the patient reported that he has always been quite active at night and had chosen to work as a night watchman. He also reported that 5–6 h of sleep were typically sufficient. Polysomnography was unremarkable (sleep stages N1, N2, N3 and REM within normal ranges, sleep latency 42 min, REM latency 56 min, apnea-hypopnea index 3, mean SaO2 95.4%, min SaO2 90.5% with a total sleep time being 376.6 min. No periodic leg movements or apneas were detected. The observations prompted us to investigate the sleeping habits in another EA1 patient, whose clinical case had been described previously (Imbrici et al., 2008). Interestingly, this patient also reported that just 5–6 h of sleep was sufficient although polysomnography analysis was not performed.
The proband (II.2) has suffered from severe migraine since childhood and these attacks manifested with hemicrania, nausea, photo-, and phonophobia lasting for 4–5 h. Most of migraine attacks occurred independently from ataxia, although in some instances both symptoms manifested at the same time. The clinical interictal examination of the patient was completely normal; in particular there was no myokymia, ataxia, dysarthria or oculomotor abnormalities. Electromyography without application of regional ischemia of the right biceps brachii, right abductor digiti minimi and left orbicularis oris muscles revealed no myokymia or other abnormalities. Except for a mild increase of serum osmolality, laboratory tests were normal and no abnormalities of sodium, potassium, calcium, magnesium and phosphate in blood and urine were found. Brain MRI revealed no evidence of circumscribed atrophy, and the proband was treated with acetazolamide, but this therapy had to be stopped due to tiredness. Neither of the patient's parents, aged 48 (mother, I.1) and 54 (father, I.2) years, nor his two brothers (aged 30, II.1 and 16, II.3 years), or his two sons (aged 5, III.1 and 3, III.2 years) and daughter (aged 1, III.3 year) suffered from similar episodes of ataxia or dysarthria (Figure 1A).
Genetic Analysis
Despite a negative family history, direct sequencing was performed that revealed a single nucleotide change in KCNA1 at position c.555C > G (Figure 1B). The resulting p.C185W mutation, which changed a highly conserved residue located in the first transmembrane segment (S1) of Kv1.1, has been previously reported (Tomlinson et al., 2013). This heterozygous mutation was inherited from his mother, who nevertheless, appeared asymptomatic. Thus, to further associate this unusual phenotype with the c.555C > G mutation in KCNA1, next generation sequencing (NGS) of all known EA genes was performed. The panel comprised the following genes: CACNA1A, CACNB4, KCNA1, KCNC3, KCNJ10, PRRT2, SCN8A, and SLC1A3. This analysis confirmed the presence of the KCNA1 c.555C > G mutation in the index patient and his asymptomatic mother. The only other sequence change found among the EA genes mentioned above was a c.1559A > G, p.K520R change in the SLC1A3 gene of both the index patient and his asymptomatic mother. However, bioinformatics analysis suggested that p.K520R was a benign variant (PolyPhen-2: benign variant; SIFT: benign variant; Mutation Taster: pathogenic variant). These findings indicate that the c.555C > G, p.C185W mutation in KCNA1 is likely to be causative in this patient and suggest that the asymptomatic status of his mother may be due to low penetrance of the mutation.
Functional Analysis of Kv1.1 C185W Mutation
KCNA1 encodes the pore-forming subunit of the voltage-gated Kv1.1 potassium channel. The channel assembles as a tetramer and each subunit contains six transmembrane segments (S1–S6). Both wild-type and C185W mutant channels were expressed in Xenopus oocytes and whole-cell currents recorded by TEVC to assess possible differences in the expression levels caused by the mutation. Expression of the homomeric C185W mutant channel resulted in either no measurable functional channel activity, or current amplitudes barely above the background (Figures 2A3, B). By marked contrast, expression of wild-type Kv1.1 (WT) gave rise to typical delayed-rectifier K+ currents of large current amplitudes (Figures 2A1, B; cf. Figure 1 in D'Adamo et al., 1998).
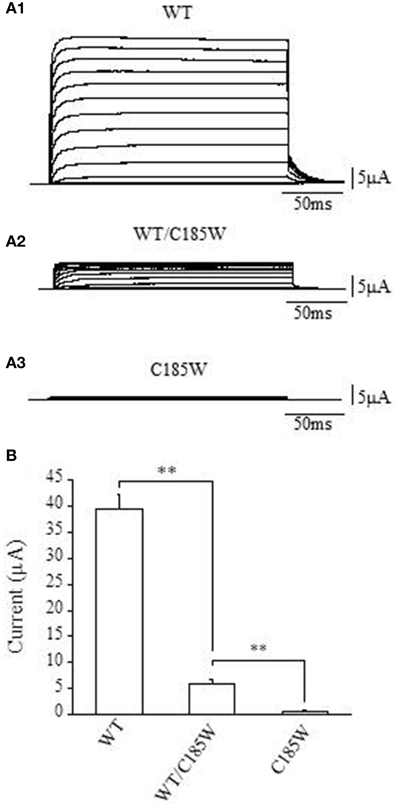
Figure 2. The C185W mutation results in non-functional homomeric channels and exerts a dominant negative effect on WT channels. (A1–3) Sample current families recorded from oocytes expressing the indicated channels. Outward currents were evoked by 200 ms depolarizing commands from a holding potential of −80 mV. (B) Representative bar graph showing the average whole-cell current recorded at +60 mV from oocytes injected with WT cRNA (2 ng, left column), co-injected with WT and C185W cRNAs (1 ng WT + 1 ng C185W, central column) or C185W cRNA (2 ng, right column). Similar results were obtained from several independent experiments carried out using different batches of oocytes (not shown). Data are mean ± SEM of 20–50 cells. The statistical significance was determined by using an unpaired Student's t-test (**P < 0.001).
The proband was heterozygous for the p.C185W mutation and thus both normal and mutant alleles can be expressed. We therefore examined whether channels composed of a mixture of WT and mutant subunits might be formed. Equal amounts of WT and mutant RNAs were co-injected into the same oocyte and the average current amplitudes then compared with those calculated from cells injected with an equivalent amount of WT RNA. The co-injection of WT and C185W RNAs (1:1 ratio) resulted in a markedly reduced average current amplitude, that was ~13% of the control WT current measured at +60 mV (Figures 2A2, B). Taken together these findings clearly demonstrate that in a homomeric channel the p.C185W mutation causes a near complete loss of function, and also exerts a dominant negative effect when combined with WT channels as would be observed in the heterozygous state.
Kv1.1 and Kv1.2 subunits are co-localized in several subcellular brain regions important for the control of movement (e.g., juxtaparanodal regions of myelinated axons and cerebellar Basket cell terminals). These subunits heteromultimerize to form channels whose function are also markedly altered by EA1 mutations (D'Adamo et al., 1999). Consistent with this, we also found that co-expression of Kv1.2 with C185W RNAs (1:1 ratio) resulted in whole-cell current that was ~20% of the control WT, at +60 mV (WT: 27.1 ± 3.2 μA; WT + C185W: 5.4 ± 0.6 μA; C185W: 0.8 ± 0.2 μA; n = 6). This demonstrates that the C185W mutation also exerts a dominant negative effect on Kv1.2 channels.
We next focused our investigations on the functional effects of the p.C185W mutation in heteromeric WT/C185W channels. cRNAs for both subunits were co-injected (1:1 ratio) into oocytes. This results in a mixed population of channels where the contribution of non-functional homomeric C185W channels will be negligible whilst functional homomeric WT channels will account for <7% of the channels present. This view is consistent with previous studies demonstrating that EA1 mutations, which do not alter surface expression of the protein (e.g., C185W; Tomlinson et al., 2013), predominantly form heteromeric channels (Imbrici et al., 2011).
Whole-cell currents were recorded using tailored voltage-clamp protocols (Figure 3) and the activating/deactivating current traces of either WT or WT/C185W channels were fitted with double- and single-exponential functions, respectively. The calculated time constants were then plotted as a function of membrane potential (Figures 3A,B). This analysis revealed that currents resulting from WT/C185W channels had slower kinetics of activation (Figures 3A,B), whereas the kinetics of deactivation were slightly faster (Figures 3A,B). To determine the voltage-dependence of WT/C185W current activation, tail current families were recorded at −50 mV after pre-pulse commands to several voltages (Figure 3C). The Boltzmann fit to either WT or WT/C185W tail current-voltage data points, revealed that the midpoint activation voltage (V1/2) for the WT/C185W current was unchanged (WT: −28.9 ± 0.5 mV; WT/C185W: −28.1 ± 0.5 mV; p > 0.05; Figure 3D). However, the slope factor k calculated from the Boltzmann fit of tail currents was slightly decreased by the mutation (WT: 6.9 ± 0.3 mV; WT/C185W: 5.1 ± 0.3 mV; p < 0.05; Figure 3D).
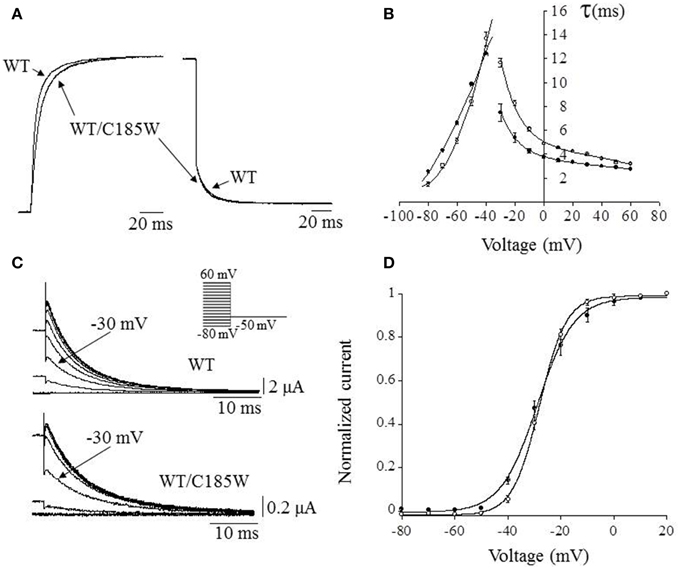
Figure 3. Effects of the mutation on activation/deactivation kinetics and voltage-dependence of heteromeric channels. (A) Representative current trace recorded at +60 mV for WT channels, overlain with that resulting from the co-injection of WT + C185W cRNAs (1:1). (B) The activating and deactivating current traces were fitted with a double and single exponential function, respectively, in order to assess the effects of the C185W mutation on the activation and deactivation kinetics of the channel. The relevant time constants for the WT (•), WT/C185W (°; 1:1 ratio) channels were calculated and plotted as a function of the test pulse in (B). This plot shows that the mutation affects channel activation (τfast) which can be directly observed also from (A). (C) Sample tail current families for the indicated channels recorded at −50 mV, after 200 ms pre-pulse commands from −80 mV to 60 mV (HP = −80 mV; ΔVincrement = 10 mV, inset). Note that although tail currents for WT/C185W channels are significantly smaller than the WT (see calibration bars) the relative current amplitudes, after a pre-pulse potential to −30 mV (arrows) are similar, denoting that both channel types have analogous V1/2 values. (D) The voltage-dependence of channel activation was determined by normalizing tail current-voltage data points for WT (•) or WT/C185W (°; 1:1 ratio) and by calculating the V1/2 and slope factor k from fits with the Boltzmann function I = 1/1 + exp{–(V – V1/2)/k} (solid curves). Notice that the mutation increases the steepness of the voltage-dependence of heterozygous channels.
WT channels are characterized by a slow process of inactivation named C-type inactivation that increases progressively during intense neuronal activity, modifying both the firing rate and the shape of the action potentials (Aldrich et al., 1979). Thus, possible modifications of this distinct channel property may be of pathogenic relevance for EA1. Slow inactivation was estimated by fitting the time course of current decay with double-exponential functions and calculating the fast (τfast) and slow (τslow) time constants and relevant amplitudes by using the equation: Afast(%) = Afast/(Afast+Aslow) × 100. This analysis shows that the τfast for heteromeric WT/C185W channels is slightly slower than the WT (Figures 4A,B). By contrast, the τslow is not statistically different (WT: τ = 34.8 ± 2.0 s, amplitude = 64%; WT/C185W: τ = 30.6 ± 1.2 s; amplitude = 73%; p > 0.05). Remarkably, WT/C185W channels display a much faster recovery from slow inactivation than the WT (Figures 4C–F). These results therefore suggest that this mutation destabilizes the slow inactivated state.
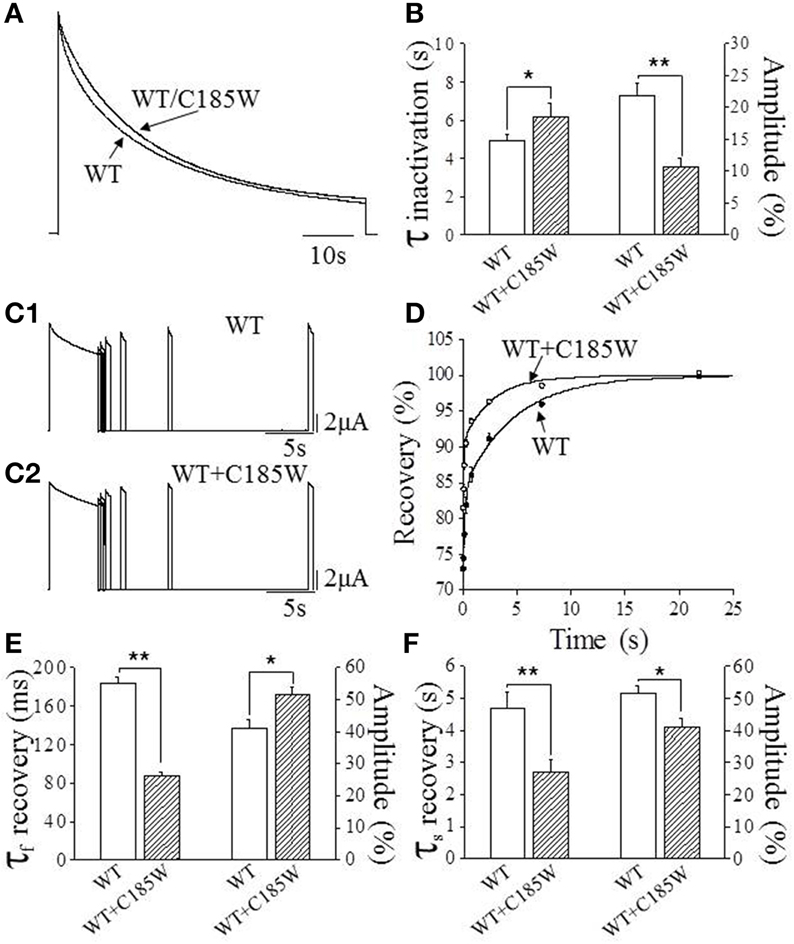
Figure 4. Effects of the C185W mutation on slow inactivation. (A) Normalized current traces resulting from either the expression of WT alone or from the co-expression of WT and C185W cRNA at 1:1 ratios. To determine slow inactivation kinetics, a test pulse to +60 mV for 3.5 min was delivered to oocytes expressing WT and WT/C185W channels and the decaying phase of the current fitted with a double-exponential function from which the time constants were calculated. (B) Bar graphs showing the fast time constants of the slow inactivation for the indicated channels (left y-axis) and the relevant amplitude of time constants (right y-axis). These results show that the mutation slightly slows-down the inactivation kinetics of WT/C185W channels. (C1,C2) Sample current traces evoked by the two-pulse protocol for the indicated channels. The recovery from slow inactivation was determined for either WT (C1) or WT/C185W (C2) channels by using a double-pulse protocol to +60 mV, separated by inter-pulse intervals of increasing duration (range: 0.010–21.87 s). The current amplitudes evoked by the second pulse (test) were divided by the first pulse (conditioning), normalized and plotted in (D) as a function of the interpulse interval. The solid curves in (D) indicate the fit of the data points with a double-exponential function from which the time constants were calculated for WT (•) or WT/C185W (°; 1:1). Bar graphs showing the fast (E) and slow (F) time constants for the recovery from slow inactivation for the indicated channels (left y-axis) and the relevant amplitude of time constants (right y-axis). Note that the mutation speeds-up the recovery from inactivation. A careful evaluation of the biophysical properties of homomeric C185W channels was not possible due to marked current reduction. The amplitudes of the time constants were calculated by using the equation: Afast (%) = Afast/(Afast+Aslow) × 100. Data are means ± SEM of 10–15 cells. Student's t-test: *, P < 0.05; **P < 0.001.
To examine the effects of the p.C185W mutation at the single-channel level, inside-out patch-clamp recordings were performed from oocytes expressing WT or WT/C185W subunits (Figure 5). The single channel slope conductance was unchanged by the mutation (WT: 10.8 ± 0.6 pS; WT/C185W: 11.2 ± 0.5 pS; n = 4). Moreover, the open-probability calculated at +20 mV was also unchanged (WT: 0.877 ± 0.022; WT/C185W: 0.882 ± 0.031; p = 0.9).
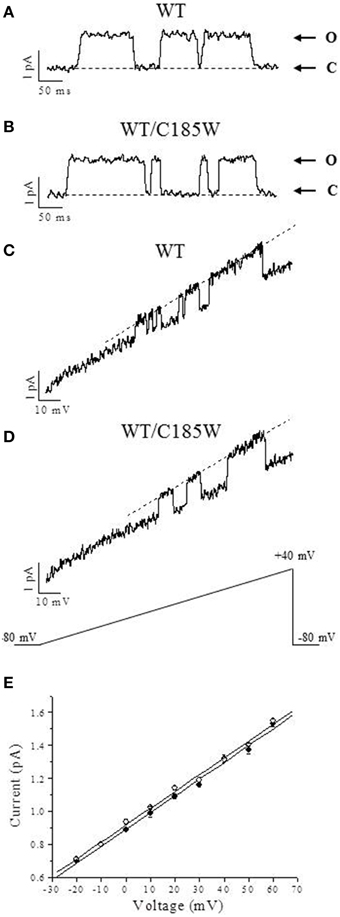
Figure 5. Analysis of single-channel currents. Representative single-channel currents recorded in the inside-out configuration of the patch-clamp from oocytes expressing WT (A) and WT/C185W (B) channels. The openings were evoked by 200 ms depolarizing voltage commands to +20 mV from a holding potential of −80 mV. Channel openings are up-ward deflections and arrows denote closed (C) and open (O) levels. Current traces were filtered at 0.5 kHz. Averaged sweeps with no openings were used to subtract leak and capacity currents. (C, D) Single-channel currents recorded from oocytes injected with WT (C) or WT/C185W (D) cRNAs and evoked by voltage-ramps of 1000 ms duration, from −80 mV to +40 mV. The single-channel current-voltage relationships were calculated from fits of the open state (dashed lines in C and D) which yielded 11.4 pS and 10.9 pS for WT and WT/C185W, respectively. (E) Single-channel current amplitudes calculated at several voltages from 4 different patches pulled from oocytes injected with WT (•) or WT/C185W (°; 1:1) cRNAs. Note that also these single-channel slope conductance calculations from all event histograms showed no differences between WT (10.8 pS) or WT/C185W (11.2 pS). Data are means ± SEM of 4 patches.
In summary, this detailed functional analysis shows that the C185W mutation: (i) causes a nearly complete loss-of-function in homomeric channels; (ii) exerts a severe dominant-negative effect on WT current amplitudes, and (iii) has significant effects on the gating parameters of WT/C185W currents.
Structural Consequences of the p.C185W Mutation
To investigate possible structural defects associated with this mutation, we used the crystal structure of the Kv1.2/2.1 channel (Long et al., 2007) to generate a homology model of Kv1.1. Residue C185 in Kv1.1 resides in the S1 segment of the voltage-sensor domain (Figure 6A). Also when mutated in silico to a tryptophan the side-chain sterically clashes with the pore-helix domain of the adjacent subunit (Figure 6) and is therefore likely to be highly deleterious to channel function.
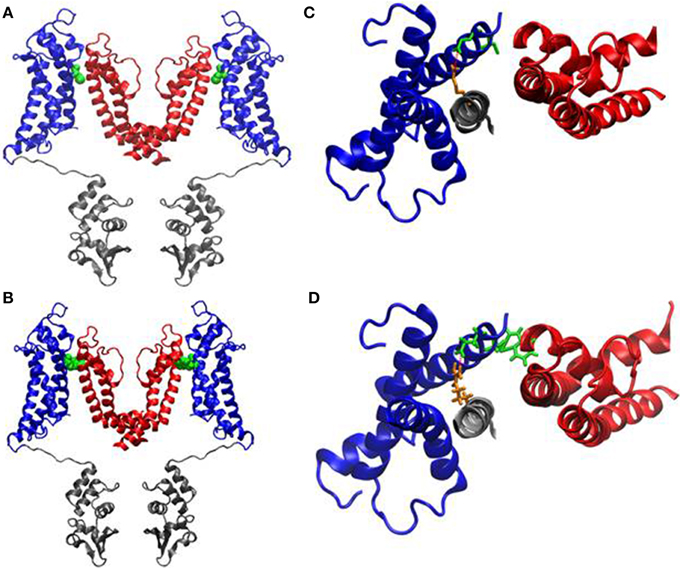
Figure 6. Homology modeling of the C185W channel. (A, B) Side stereoviews of a ribbon representation of the pore region (red), voltage-sensor (blue) and of the T1 domain (gray) of the channel. The pictures show only two monomers side by side for clarity. The location of the C185 residue (A) and of the C185W mutation (B) in the S1 helices are highlighted as Van der Waals spheres (green). Close-up views of the C185 residue (C, green sticks) and of the C185W mutation (D, green sticks) from top, showing their molecular interactions with the P α-helix of the adjacent subunit. The location of the salt-bridge between Glu187 (green) and Arg298 (orange) is also highlighted as a red dashed line.
Discussion
Here we describe a single nucleotide mutation that changes a highly conserved residue in the S1 segment (p.C185W) of the Kv1.1 channel subunit in a proband affected by EA1. The mutation produces marked effects on both the channel structure and function, which most likely underscore the several atypical symptoms displayed by the patient. First, we witnessed several episodes of hyperthermia in our proband without an apparent focus of infection. Although spontaneous myokymia was absent in our patient, the episodes of hyperthermia were associated with long lasting attacks of jerking muscle contractions. Thus, a possible explanation for the observed hyperthermia is that such prolonged muscle activity could be responsible for the abnormal rises in body temperature. Exposure to warm temperature has been reported to provoke attacks of ataxia (Eunson et al., 2000; Klein et al., 2004). There are also anecdotal reports of cold temperatures exacerbating symptoms in EA1 patients, which led to the suggestion of temperature extremes triggering EA1 symptoms (Demos et al., 2009). More recently, in Kv1.1 knock-in ataxic mice, spontaneous myokymic activity has been shown to be exacerbated by several types of stimuli including low temperature (Brunetti et al., 2012). Notably, the neuromuscular transmission of EA1 individuals and of animal models of EA1 is temperature sensitive, and attacks of ataxia may be brought on by fever or a hot bath (Tomlinson et al., 2013; D'Adamo et al., 2014). Altogether, this evidence poses the question as to whether loss-of-function mutations in Kv1.1 channels may be responsible for abnormal raises in body temperature by altering the activity of distinct CNS and PNS circuits. Indeed, possible sources of this thermoregulatory defect are hypothalamic neurons that mediate rapid core body temperature changes. Interestingly, these neurons normally express Kv1.1 channels which, if mutated, might produce such effects (Albrecht et al., 1995; Rhodes et al., 1996). In addition, the involvement of distinct subpopulation(s) of peripheral nerves that transmit temperature signals and which also express Kv1.1 channels cannot be excluded (Tsantoulas and McMahon, 2014).
Another atypical symptom, not previously described, concerns the sleeping habits of the patient who reported to need only 5–6 h per day. Interestingly, similar sleeping habits were found in another EA1 patient we have previously investigated (Imbrici et al., 2008). Most people sleep 7–8 h per night, although, a few perform apparently well with just 3–4 h of sleep. This phenotypic trait seems to run in families, although the underlying genes remain unclear. It is interesting to note that mutant mini-sleep Drosophila, which sleep ~70% less than WT flies, also carries a loss-of-function point mutation in the Shaker (Kv1.1) gene. This mutation also occurs in the S1 domain of the Drosophila Kv1.1 ortholog, just three residues beyond the p.C185W mutation (Cirelli et al., 2005). Moreover, mice lacking the Kcna2 gene, encoding for the Shaker-related Kv1.2 channel, also have reduced non-rapid eye movement (NREM) sleep and die early (Douglas et al., 2007). Both Kv1.1 and Kv1.2 can form heteromeric channels in several brain regions, including those involved in sleep-wake rhythm. Moreover, a number of studies have shown that heteromeric Kv1.1/Kv1.2 channels play an important role in the control of neuronal excitability, action potential propagation and synaptic transmission. Previously, we have shown that dominant-negative EA1 mutations can affect the function of heteromeric Kv1.1/Kv1.2 channels (D'Adamo et al., 1999). Here we also demonstrate that C185W markedly reduces the functional activity of Kv1.2 channels. Thus, although polysomnography analysis of the proband was borderline, a role for these channel types in the neuronal processes that control sleep in humans cannot be entirely excluded. Indeed, it is possible that other EA1 patients may display an unrecognized short-sleep duration trait which, if confirmed, could represent the first evidence showing a human K+ channel-dependent alteration in sleepbehavior.
Headache is also a symptom over-represented in EA1 individuals. Nevertheless, the presence of long-lasting episodes of severe migraine characterized by nausea, photo- and phonophobia, occurring independently of attacks of EA1, also demonstrate that our probands's symptoms do not fall within the spectrum previously reported for KCNA1 mutations.
Owing to a noticeable intrafamilial and interfamilial phenotypic variability, genotype-phenotype correlations for EA1 can often be difficult to establish reliably. Indeed, striking differences in severity and frequency of EA1 attacks have been reported even in identical twins (Graves et al., 2010). Thus, the asymptomatic status of the proband's mother and the lack of myokymia in the index patient, although unusual, could be due to low penetrance of the mutation or other epigenetic factors.
Structural and Functional Implications
Our structural model shows that the C185W mutation is located very close to a highly conserved salt-bridge (R298/E187 in Kv1.1), occurring between the S1 segment and the S4 helix, which represents the main voltage-sensor of the channel. This interaction, in the Kv1.2/2.1 chimeric channel, has been shown to be important for the control of voltage-dependent gating (Tao et al., 2010). Indeed, these conclusions are also consistent with the tryptophan-substitution mutagenesis of a Kv1.1 channel ortholog (Shaker) performed by Hong and Miller, who showed that the C245W mutation (equivalent to C185W) alters the voltage-dependence and stabilizes the closed-state of homomeric channels (Hong and Miller, 2000). On the other hand, our structural modeling suggest consistency between the C185W-induced perturbation of the pore-helix and the observed loss-of-function effect, possibly via steric-dependent stabilization of the closed-state in homomeric channel. Severe trafficking defects, underlying the almost complete loss-of-function of homomeric C185W channel, appear unlikely because the mutant protein traffic normally to plasmamembrane when expressed in the human HEK293 cell line (Tomlinson et al., 2013). The structural location of C185W is also coherent with its effects on slow inactivation, a process thought to involve conformational movements of the external pore and selectivity filter (Grissmer and Cahalan, 1989; Stuhmer et al., 1989; Hoshi et al., 1991; Pardo et al., 1992; Lopez-Barneo et al., 1993; Baukrowitz and Yellen, 1995; Molina et al., 1997; Pessia, 2004; González et al., 2012). In particular, the faster recovery from inactivation indicates that the C185W substitution destabilizes the slow inactivated state.
From a neurophysiological perspective, such faster kinetics of recovery from inactivation would render Kv1.1 expressing neurons more readily re-excitable by decreasing the refractory period of action potentials. This would further exacerbate neuronal hyper-excitability induced by the mutation in the heterozygous state.
In conclusion, we report a new EA1 phenotype characterized by hyperthermia associated with long lasting attacks of ataxia and severe migraine. Detailed genetic and functional analysis of this mutant allele are consistent with a pathogenic role for this KCNA1 mutation. These results therefore indicate that a thorough assessment for hyperthermia, short-sleep trait and migraine should be carefully considered for all patients affected by EA1.
Author Contributions
Mauro Pessia and Thomas Klopstock have been the principal investigators of this study. Thomas Klopstock has been the investigator on industry-sponsored trials funded by Santhera Pharmaceuticals Ltd. (idebenone in LHON, idebenone in Friedreich ataxia) and by H. Lundbeck A/S (carbamylated erythropoietin in Friedreich ataxia); has received research support from government entities (German Research Foundation; German Federal Ministry of Education and Research; European Commission 7th Framework Programme) and from commercial entities (Santhera Pharmaceuticals Ltd.; Actelion Pharmaceuticals Ltd.; H. Lundbeck A/S); has been serving on scientific advisory boards for commercial entities (Santhera Pharmaceuticals Ltd; Actelion Pharmaceuticals Ltd.) and for non-profit entities (Center for Rare Diseases Bonn, Germany; Hoffnungsbaum e.V., Germany); received speaker honoraria and travel costs from commercial entities (Dr. Willmar Schwabe GmbH & Co. KG; Eisai Co., Ltd.; Santhera Pharmaceuticals Ltd; Actelion Pharmaceuticals Ltd; Boehringer Ingelheim Pharma GmbH & Co. KG, GlaxoSmithKline GmbH & Co. KG); has been doing consultancies for Gerson Lehrman Group, USA, and FinTech Global Capital, Japan; has been serving as a Section Editor for BMC Medical Genetics from 2011.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was supported by funds from the German Federal Ministry of Education and Research (German Center for Vertigo and Balance Disorders, grant 01 EO 0901), Telethon (GGP11188), MIUR-PRIN (20108WT59Y_004), Ministero della Salute (GR-2009-1580433) and Fondazione Cassa di Risparmio di Perugia. We thank Cecilia Bove for her contributions to this manuscript.
References
Adelman, J. P., Bond, C. T., Pessia, M., and Maylie, J. (1995). Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron 15, 1449–1454. doi: 10.1016/0896-6273(95)90022-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Albrecht, B., Weber, K., and Pongs, O. (1995). Characterization of a voltage-activated K-channel gene cluster on human chromosome 12p13. Recept. Channels 3, 213–220.
Aldrich, R. W. Jr. Getting, P. A., and Thompson, S. H. (1979). Mechanism of frequency-dependent broadening of molluscan neurone soma spikes. J. Physiol. 291, 531–544. doi: 10.1113/jphysiol.1979.sp012829
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baukrowitz, T., and Yellen, G. (1995). Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron 15, 951–960. doi: 10.1016/0896-6273(95)90185-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Browne, D. L., Gancher, S. T., Nutt, J. G., Brunt, E. R., Smith, E. A., Kramer, P., et al. (1994). Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat. Genet. 8, 136–140. doi: 10.1038/ng1094-136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brunetti, O., Imbrici, P., Botti, F. M., Pettorossi, V. E., D'Adamo, M. C., Valentino, M., et al. (2012). Kv1.1 knock-in ataxic mice exhibit spontaneous myokymic activity exacerbated by fatigue, ischemia and low temperature. Neurobiol. Dis. 47, 310–321. doi: 10.1016/j.nbd.2012.05.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cirelli, C., Bushey, D., Hill, S., Huber, R., Kreber, R., Ganetzky, B., et al. (2005). Reduced sleep in drosophila shaker mutants. Nature 434, 1087–1092. doi: 10.1038/nature03486
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cusimano, A., D'Adamo, M. C., and Pessia, M. (2004). An episodic ataxia type-1 mutation in the S1 segment sensitises the hKv1.1 potassium channel to extracellular Zn2+. FEBS Lett. 576, 237–244. doi: 10.1016/j.febslet.2004.09.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D'Adamo, M. C., Catacuzzeno, L., Di Giovanni, G., Franciolini, F., and Pessia, M. (2013). K± Channelepsy: progress in the neurobiology of potassium channels and epilepsy. Front. Cell. Neurosci. 7:134. doi: 10.3389/fncel.2013.00134
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D'Adamo, M. C., Di Giovanni, G., and Pessia, M. (2014). “Animal models of Episodic Ataxia type 1 (EA1),” in Movement Disorders: Genetics and Models, 2nd Edn, ed M. S. LeDoux (San Diego, CA: Academic Press), 797–807.
D'Adamo, M. C., Hanna, M. G., Di Giovanni, G., and Pessia, M. (2012). “Episodic Ataxia type 1,” in GeneReviews, eds R. A. Pagon, T. C. Bird, C. R. Dolan, and K. Stephens (Seattle, WA: University of Washington).
D'Adamo, M. C., Imbrici, P., Sponcichetti, F., and Pessia, M. (1999). Mutations in the KCNA1 gene associated with episodic ataxia type-1 syndrome impair heteromeric voltage-gated K(+) channel function. FASEB J. 13, 1335–1345.
D'Adamo, M. C., Liu, Z., Adelman, J. P., Maylie, J., and Pessia, M. (1998). Episodic ataxia type-1 mutations in the hKv1.1 cytoplasmic pore region alter the gating properties of the channel. EMBO J. 17, 1200–1207. doi: 10.1093/emboj/17.5.1200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Demos, M. K., Macri, V., Farrell, K., Nelson, T. N., Chapman, K., Accili, E., et al. (2009). A novel KCNA1 mutation associated with global delay and persistent cerebellar dysfunction. Mov. Disord. 24, 778–782. doi: 10.1002/mds.22467
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Douglas, C. L., Vyazovskiy, V., Southard, T., Chiu, S. Y., Messing, A., Tononi, G., et al. (2007). Sleep in Kcna2 knockout mice. BMC Biol. 5:42. doi: 10.1186/1741-7007-5-42
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eunson, L. H., Rea, R., Zuberi, S. M., Youroukos, S., Panayiotopoulos, C. P., Liguori, R., et al. (2000). Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann. Neurol. 48, 647–656. doi: 10.1002/1531-8249(200010)48:4<647
Geiger, J. R., and Jonas, P. (2000). Dynamic control of presynaptic ca(2+) inflow by fast-inactivating K(+) channels in hippocampal mossy fiber boutons. Neuron 28, 927–939. doi: 10.1016/S0896-6273(00)00164-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glaudemans, B., van der Wijst, J., Scola, R. H., Lorenzoni, P. J., Heister, A., van der Kemp, A. W., et al. (2009). A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J. Clin. Invest. 119, 936–942. doi: 10.1172/JCI36948
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
González, C., Baez-Nieto, D., Valencia, I., Oyarzún, I., Rojas, P., Naranjo, D., et al. (2012). K(+) channels: function-structural overview. Compr. Physiol. 2, 2087–2149. doi: 10.1002/cphy.c110047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Graves, T. D., Rajakulendran, S., Zuberi, S. M., Morris, H. R., Schorge, S., Hanna, M. G., et al. (2010). Non genetic factors influence severity of episodic ataxia type 1 in monozygotic twins. Neurology 75, 367–372. doi: 10.1212/WNL.0b013e3181ea9ee3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grissmer, S., and Cahalan, M. D. (1989). Divalent ion trapping inside potassium channels of human T lymphocytes. J. Gen. Physiol. 93, 609–630. doi: 10.1085/jgp.93.4.609
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Herson, P. S., Virk, M., Rustay, N. R., Bond, C. T., Crabbe, J. C., Adelman, J. P., et al. (2003). A mouse model of episodic ataxia type-1. Nat. Neurosci. 6, 378–383. doi: 10.1038/nn1025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hess, B., Kutzner, C., van der Spoel, D., and Lindahl, E. (2008). GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447. doi: 10.1021/ct700301q
Hong, K. H., and Miller, C. (2000). The lipid-protein interface of a Shaker K+ channel. J. Gen. Physiol. 115, 51–58. doi: 10.1085/jgp.115.1.51
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoshi, T., Zagotta, W. N., and Aldrich, R. W. (1991). Two types of inactivation in shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron 7, 547–556. doi: 10.1016/0896-6273(91)90367-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Imbrici, P., Cusimano, A., D'Adamo, M. C., De Curtis, A., and Pessia, M. (2003). Functional characterization of an episodic ataxia type-1 mutation occurring in the S1 segment of hKv1.1 channels. Pflugers Arch. 446, 373–379. doi: 10.1007/s00424-002-0962-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Imbrici, P., D'Adamo, M. C., Cusimano, A., and Pessia, M. (2007). Episodic ataxia type 1 mutation F184C alters Zn2+-induced modulation of the human K+ channel Kv1.4-Kv1.1/Kvbeta1.1. Am. J. Physiol. Cell Physiol. 292, C778–C787. doi: 10.1152/ajpcell.00259.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Imbrici, P., D'Adamo, M. C., Grottesi, A., Biscarini, A., and Pessia, M. (2011). Episodic ataxia type 1 mutations affect fast inactivation of K+ channels by a reduction in either subunit surface expression or affinity for inactivation domain. Am. J. Physiol. Cell Physiol. 300, C1314–C1322. doi: 10.1152/ajpcell.00456.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Imbrici, P., D'Adamo, M. C., Kullmann, D. M., and Pessia, M. (2006). Episodic ataxia type 1 mutations in the KCNA1 gene impair the fast inactivation properties of the human potassium channels Kv1.4-1.1/Kvbeta1.1 and Kv1.4-1.1/Kvbeta1.2. Eur. J. Neurosci. 24, 3073–3083. doi: 10.1111/j.1460-9568.2006.05186.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Imbrici, P., Gualandi, F., D'Adamo, M. C., Masieri, M. T., Cudia, P., De Grandis, D., et al. (2008). A novel KCNA1 mutation identified in an italian family affected by episodic ataxia type 1. Neuroscience 157, 577–587. doi: 10.1016/j.neuroscience.2008.09.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kinali, M., Jungbluth, H., Eunson, L. H., Sewry, C. A., Manzur, A. Y., Mercuri, E., et al. (2004). Expanding the phenotype of potassium channelopathy: severe neuromyotonia and skeletal deformities without prominent episodic ataxia. Neuromuscul. Disord. 14, 689–693. doi: 10.1016/j.nmd.2004.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klein, A., Boltshauser, E., Jen, J., and Baloh, R. W. (2004). Episodic ataxia type 1 with distal weakness: a novel manifestation of a potassium channelopathy. Neuropediatrics 35, 147–149. doi: 10.1055/s-2004-817921
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laskowski, R. A., Rullmannn, J. A., MacArthur, M. W., Kaptein, R., and Thornton, J. M. (1996). AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486. doi: 10.1007/BF00228148
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Long, S. B., Tao, X., Campbell, E. B., and MacKinnon, R. (2007). Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382. doi: 10.1038/nature06265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lopez-Barneo, J., Hoshi, T., Heinemann, S. H., and Aldrich, R. W. (1993). Effects of external cations and mutations in the pore region on C-type inactivation of shaker potassium channels. Recept. Channels 1, 61–71.
Molina, A., Castellano, A. G., and Lopez-Barneo, J. (1997). Pore mutations in shaker K+ channels distinguish between the sites of tetraethylammonium blockade and C-type inactivation. J. Physiol. 499(Pt 2), 361–367. doi: 10.1113/jphysiol.1997.sp021933
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nishida, M., Cadene, M., Chait, B. T., and MacKinnon, R. (2007). Crystal structure of a Kir3.1-prokaryotic kir channel chimera. EMBO J. 26, 4005–4015. doi: 10.1038/sj.emboj.7601828
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pardo, L. A., Heinemann, S. H., Terlau, H., Ludewig, U., Lorra, C., Pongs, O., et al. (1992). Extracellular K+ specifically modulates a rat brain K+ channel. Proc. Natl. Acad. Sci. U.S.A. 89, 2466–2470. doi: 10.1073/pnas.89.6.2466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pessia, M. (2004). “Ion channels and electrical activity,” in Molecular Biology of the Neuron, 2nd Edn, eds R. W. Davies and B. J. Morris (Oxford: Oxford University Press), 103–137. doi: 10.1093/acprof:oso/9780198509981.003.0005
Pessia, M., D'Adamo, M. C., and Imbrici, P. (2012). “The neurobiology of episodic ataxia type 1: a Shaker-like K+ channel disorder,” in Ataxia: Causes, Symptoms and Treatment, ed S. H. Hong (New York, NY: NOVA SCIENCE), 47–75.
Rhodes, K. J., Monaghan, M. M., Barrezueta, N. X., Nawoschik, S., Bekele-Arcuri, Z., Matos, M. F., et al. (1996). Voltage-gated K+ channel beta subunits: expression and distribution of kv beta 1 and kv beta 2 in adult rat brain. J. Neurosci. 16, 4846–4860.
Sali, A., and Blundell, T. L. (1993). Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815. doi: 10.1006/jmbi.1993.1626
Stuhmer, W., Ruppersberg, J. P., Schroter, K. H., Sakmann, B., Stocker, M., Giese, K. P., et al. (1989). Molecular basis of functional diversity of voltage-gated potassium channels in mammalian brain. EMBO J. 8, 3235–3244.
Tao, X., Lee, A., Limapichat, W., Dougherty, D. A., and MacKinnon, R. (2010). A gating charge transfer center in voltage sensors. Science (New York, N.Y.) 328, 67–73. doi: 10.1126/science.1185954
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tomlinson, S. E., Rajakulendran, S., Tan, S. V., Graves, T. D., Bamiou, D. E., Labrum, R. W., et al. (2013). Clinical, genetic, neurophysiological and functional study of new mutations in episodic ataxia type 1. J. Neurol. Neurosurg. Psychiatr. 84, 1107–1112. doi: 10.1136/jnnp-2012-304131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsantoulas, C., and McMahon, S. B. (2014). Opening paths to novel analgesics: the role of potassium channels in chronic pain. Trends Neurosci. 37, 146–158. doi: 10.1016/j.tins.2013.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
VanDyke, D. H., Griggs, R. C., Murphy, M. J., and Goldstein, M. N. (1975). Hereditary myokymia and periodic ataxia. J. Neurol. Sci. 25, 109–118. doi: 10.1016/0022-510X(75)90191-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: episodic ataxia type 1, hyperthermia, sleep, migraine, Shaker potassium channels, C185W
Citation: D'Adamo MC, Gallenmüller C, Servettini I, Hartl E, Tucker SJ, Arning L, Biskup S, Grottesi A, Guglielmi L, Imbrici P, Bernasconi P, Di Giovanni G, Franciolini F, Catacuzzeno L, Pessia M and Klopstock T (2015) Novel phenotype associated with a mutation in the KCNA1(Kv1.1) gene. Front. Physiol. 5:525. doi: 10.3389/fphys.2014.00525
Received: 21 October 2014; Accepted: 20 December 2014;
Published online: 15 January 2015.
Edited by:
Christoph Fahlke, Forschungszentrum Jülich, GermanyReviewed by:
Carmen Valenzuela, Instituto de Investigaciones Biomédicas CSIC-UAM, SpainNico Melzer, University of Münster, Germany
David Naranjo, Universidad de Valparaiso, Chile
Copyright © 2015 D'Adamo, Gallenmüller, Servettini, Hartl, Tucker, Arning, Biskup, Grottesi, Guglielmi, Imbrici, Bernasconi, Di Giovanni, Franciolini, Catacuzzeno, Pessia and Klopstock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauro Pessia, Section of Physiology and Biochemistry, Department of Experimental Medicine, University of Perugia School of Medicine, P.le Gambuli, 06132 San Sisto, Perugia, Italy e-mail: mauro.pessia@unipg.it;
Thomas Klopstock, Department of Neurology, Friedrich-Baur-Institute, Ludwig-Maximilians-University, Ziemssenstr. 1a, 80336 Munich, Germany e-mail: tklopsto@med.lmu.de
†These authors have contributed equally to this work as first authors.
‡These authors have contributed equally to this work as last authors.