 Magali Ranchou-Peyruse1,2,3
Magali Ranchou-Peyruse1,2,3 Marion Guignard1Franck Casteran2Maïder Abadie1Clémence Defois4
Marion Guignard1Franck Casteran2Maïder Abadie1Clémence Defois4 Pierre Peyret4David Dequidt5Guilhem Caumette3,6Pierre Chiquet3,6Pierre Cézac2,3
Pierre Peyret4David Dequidt5Guilhem Caumette3,6Pierre Chiquet3,6Pierre Cézac2,3 Anthony Ranchou-Peyruse1,3*
Anthony Ranchou-Peyruse1,3*- 1IPREM, Institut des Sciences Analytiques et de Physico-Chimie pour l’Environnement et les Matériaux, Université de Pau & Pays Adour/E2S-UPPA, Pau, France
- 2Laboratoire de thermique, énergétique et procédés IPRA, EA1932, Université de Pau & Pays Adour/E2S-UPPA, Pau, France
- 3Joint Laboratory SEnGA, UPPA-E2S-Teréga, Pau, France
- 4Université Clermont Auvergne, INRAE, UMR 0454 MEDIS, Clermont-Ferrand, France
- 5STORENGY – Geosciences Department, Bois-Colombes, France
- 6Teréga, Pau, France
Deep aquifers (up to 2km deep) contain massive volumes of water harboring large and diverse microbial communities at high pressure. Aquifers are home to microbial ecosystems that participate in physicochemical balances. These microorganisms can positively or negatively interfere with subsurface (i) energy storage (CH4 and H2), (ii) CO2 sequestration; and (iii) resource (water, rare metals) exploitation. The aquifer studied here (720m deep, 37°C, 88bar) is naturally oligotrophic, with a total organic carbon content of <1mg.L−1 and a phosphate content of 0.02mg.L−1. The influence of natural gas storage locally generates different pressures and formation water displacements, but it also releases organic molecules such as monoaromatic hydrocarbons at the gas/water interface. The hydrocarbon biodegradation ability of the indigenous microbial community was evaluated in this work. The in situ microbial community was dominated by sulfate-reducing (e.g., Sva0485 lineage, Thermodesulfovibriona, Desulfotomaculum, Desulfomonile, and Desulfovibrio), fermentative (e.g., Peptococcaceae SCADC1_2_3, Anaerolineae lineage and Pelotomaculum), and homoacetogenic bacteria (“Candidatus Acetothermia”) with a few archaeal representatives (e.g., Methanomassiliicoccaceae, Methanobacteriaceae, and members of the Bathyarcheia class), suggesting a role of H2 in microenvironment functioning. Monoaromatic hydrocarbon biodegradation is carried out by sulfate reducers and favored by concentrated biomass and slightly acidic conditions, which suggests that biodegradation should preferably occur in biofilms present on the surfaces of aquifer rock, rather than by planktonic bacteria. A simplified bacterial community, which was able to degrade monoaromatic hydrocarbons at atmospheric pressure over several months, was selected for incubation experiments at in situ pressure (i.e., 90bar). These showed that the abundance of various bacterial genera was altered, while taxonomic diversity was mostly unchanged. The candidate phylum Acetothermia was characteristic of the community incubated at 90bar. This work suggests that even if pressures on the order of 90bar do not seem to select for obligate piezophilic organisms, modifications of the thermodynamic equilibria could favor different microbial assemblages from those observed at atmospheric pressure.
Introduction
Among the different deep continental environments containing high total biomass levels, estimated at 2 to 6·1029 cells (Magnobosco et al., 2018), aquifers located in the uppermost two kilometers of the Earth’s crust are characterized by a diverse microbial biosphere (Wu et al., 2016; Hershey et al., 2018; Kadnikov et al., 2020). Oligotrophic conditions result in low cell concentrations; however, these aquifers have a considerable volume, estimated to be at 22.6 million km3 (Gleeson et al., 2015). Deep continental aquifers used as geological gas storage facilities are relevant ecosystems for studying the deep continental biosphere and the effect of human activity on it. These storage aquifers can be found all around the world, but are primarily located in North America, although some also occur in Europe and the Commonwealth of Independent States (Russia, Armenia, Azerbaijan, and Belarus; Cornot-Gandolphe, 2017). An understanding of these ecosystems makes it possible to predict the effects of future developments on these deep aquifers depending on their use, which include (i) the sequestration of CO2 or carbon molecules in the broad sense to address climate change; (ii) the temporary energy storage of CH4, H2 or air from renewable energy sources; (iii) rare metal mining; and (iv) exploitation of their water resources (De Silva et al., 2015; Flexer et al., 2018; Ranchou-Peyruse et al., 2019; Lemieux et al., 2020). Deep porous geological structures may present advantages such as their size, relative ubiquity and safety for gas storage, along with the proven know-how regarding their operation. Because of the high pressures deep underground, the volume of gas stored is less than that at atmospheric pressure, allowing the storage of large amounts of gas.
While the influence of high pressure on microbes has been widely studied in the context of marine environments (Jebbar et al., 2015; Zhang et al., 2015), very few such studies have been conducted on their continental counterparts (Escudero et al., 2018; Zhang et al., 2018). Nevertheless, international projects have aimed to fill the knowledge gaps in this theme, most notably including the following programs: the U.S Department of Energy’s Subsurface Program, Deep Underground Science and Engineering (DUSEL), the international Continental Scientific Drilling Program, the Deep Carbon Observatory’s Census of Deep Life, and the Center for Dark Energy Biosphere Investigations (C-DEBI). To our knowledge, no obligate piezophiles have yet been isolated from a deep continental environment, whereas they have been observed in marine subsurface environments. The temperature gradient of approximately 30°C km−1 implies that temperatures reached in deep continental environments prevent microbial life at depths of 3–4km. Multiple studies focusing on these environments were able to isolate and grow microorganisms at atmospheric pressure (Haveman and Pedersen, 2002; Baker et al., 2003; Basso et al., 2009; Leandro et al., 2018; Karnachuk et al., 2019). However, although the pressure encountered in these environments does not seem to select obligate piezophiles, pressure can influence the structure of the microbial community (Nyyssönen et al., 2014). In particular, pressure induces increased solubilization of gases and volatile carbon elements and could modify transfer rates through the cell membrane and accessibility to essential elements, including H2, CO2, and organic molecules (Follonier et al., 2012).
In the case of the geological storage of natural gas, deep aquifers are oligotrophic environments. The proximity of the stored gas has an impact on the microbial composition via the transfer of hydrocarbon molecules and other elements from the gas to water (Amigáň et al., 1990; Ranchou-Peyruse et al., 2019). Many of these environments contain sulfate, and heterotrophic sulfate-reducing microorganisms take advantage of this influx of organic molecules to prosper. In 2016, a study of the Pb_J_7 site in the Paris sedimentary basin closest to gas storage demonstrated the prevalence of sulfate-reducing microorganisms, particularly Desulfotomaculum spp., on the degradation of so-called BTEX monoaromatic hydrocarbons (Aüllo et al., 2016). These hydrocarbons are among the most soluble and thus the most likely ones to migrate in the aquifer. The study presented here concerns the Pb_J_6 site, which is part of the same Jurassic stratum as Pb_J_7 but is located at a distance of 1.3km. The main reason for this work was to test whether the monoaromatic hydrocarbon biodegradation potential was similar and involved the same microorganisms despite the aquifer being located farther away from the gas reservoir than the Pb_J_7 well. Moreover, with regard to deep aquifers used for natural gas storage, all biodegradation assays conducted to date have been carried out at atmospheric pressure, without taking into account the possible effects of in situ pressure on biodegradation capacities and microbial diversity. In this study, the Pb_J_6 monitoring well was cleaned using the same protocol used for sampling Pb_J_7 to prevent possible contamination by the biofilms developed in the pipes (Basso et al., 2005). The experiments carried out during this study combine classical microbiological approaches for assessing the BTEX biodegradation potential and molecular biology approaches for following changes of microbial diversity and understanding the behavior of these communities at atmospheric and in situ pressures.
Materials and Methods
Sample Site
The well is located approximately 20km west of Paris, and because it is a site of natural gas storage, it is classified as SEVESO-3.1 Its position with respect to other wells, particularly Pb_J_7, can be found in a publication by Ranchou-Peyruse et al. (2019). The well reaches a depth of 720 meters and has a temperature of 37°C. Depending on storage activity, the pressure can vary from 67 to 90bar. Formation water is contained in porous Jurassic rock primarily composed of sandstone, with some carbonates. Geochemical analyses of the waters were carried out by the CARSO Laboratory (France). The obtained data are presented in Table 1. The pH measured at the wellhead (i.e., surface) was 7.8. Samples were taken from the Pb_J_6 well located in the Paris sedimentary basin in September 2015 using the same aseptic and anoxic procedure described previously (Basso et al., 2005). Briefly, after brushing the entire well and circulating sodium hypochlorite (4.2L, 14% active chlorine) three times, the well was flushed (10 times the volume of the well, which is 2.4m3). A total of 99 Sterivex™-GP 0.22μm filter units (EMD Millipore, Molsheim, France) were used to filter the formation water and to concentrate the microbial biomass at a flow rate of 1.5m3 h−1 for 9h. The continuous flow of water made it possible to maintain anoxia inside the filters. At the end of the sampling campaign, the filters were sealed and immediately transported to the laboratory at anoxic conditions (GasPak™ EZ, BD) at room temperature. Several liters of additional formation water were also taken from the wellhead and stored in glass flasks at room temperature, respecting the conditions of anoxia during transport back to the laboratory. In the laboratory, the concentrated microbial biomass was stored at appropriate anoxic and sterile conditions in a glovebox at room temperature until use on the following day. Glass flasks with formation water were stored at 4°C until use, particularly for culture on the next day.
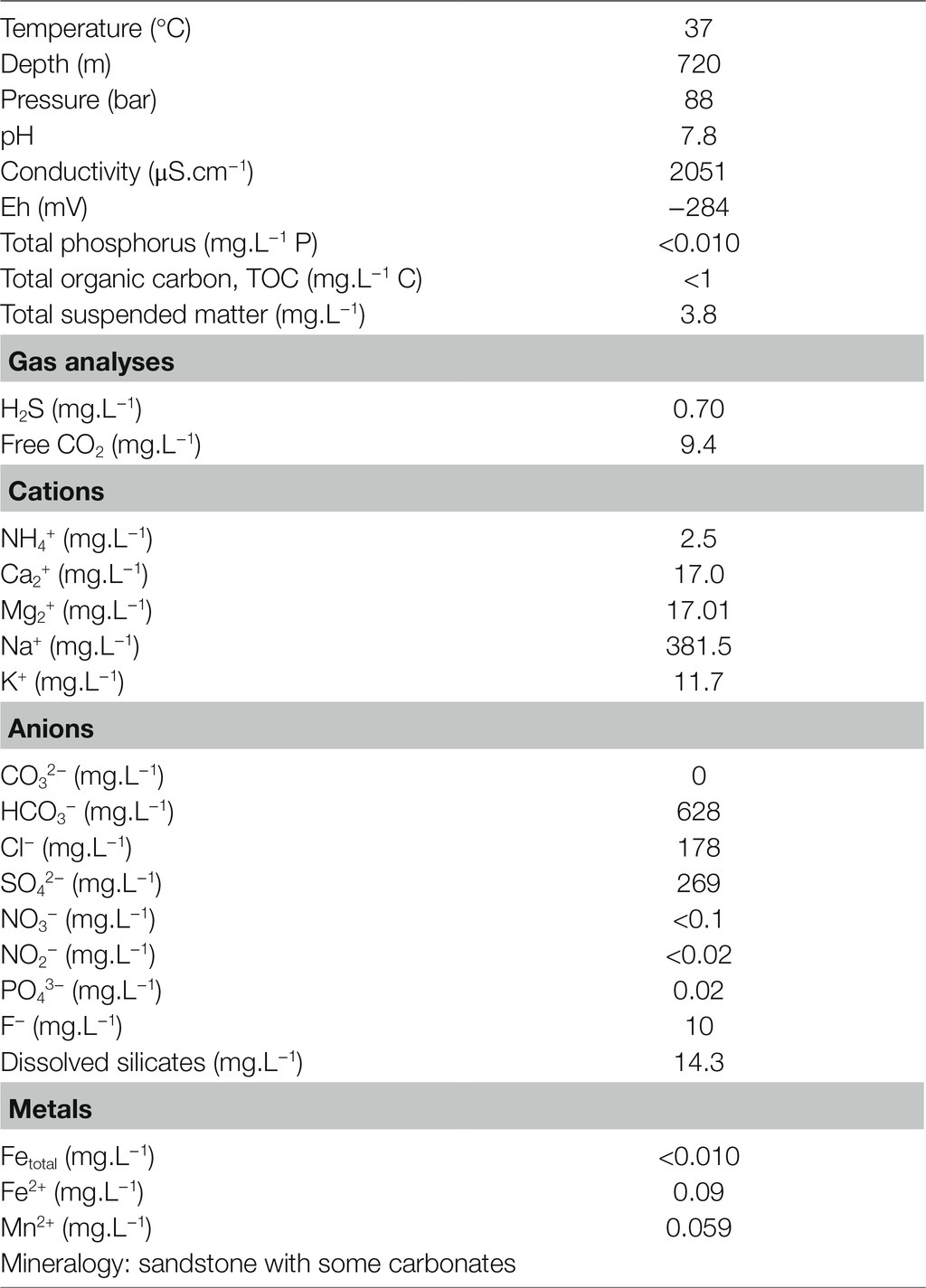
Table 1. Physicochemical parameters and components of the formation water from a deep aquifer analyzed at atmospheric pressure.
Culture Methods
Most Probable Number
In the laboratory, the formation water was used for cell counting via the most probable number (MPN) technique. One liter of the mineral base used to simulate formation water contained 0.0594g CaCl2·2H2O, 0.135g MgCl2·6H2O, 0.035g NaCl, 0.0152g SiO2, 0.02g KCl, 0.2g NH4Cl, 0.3g KH2PO4, 0.3g K2HPO4, 0.882g NaHCO3, 0.0013g FeCl2·4H2O, 0.5g cysteine as a reducing agent, 1mg resazurin as a redox indicator, and 10ml SL10 trace elements from a stock solution (Pfennig et al., 1981). Depending on the targeted metabolism (Table 2), the headspace of the basal medium was spiked with 1bar of H2/CO2 (80/20) for hydrogenotrophic methanogens; 5g of casein peptone, 1g of yeast extract, 6.8g of sodium acetate and 1.7g of NaNO3 for nitrate-reducing bacteria; or 15g of casein peptone, 10g of yeast extract and 2.5g of glucose for fermentative bacteria without the addition of a terminal electron acceptor (TEA). Following autoclaving (120°C, 20min), the various media were supplemented with 5ml of vitamins from a filtered and anoxic V10 stock solution (Pfennig et al., 1981). Finally, the pH was adjusted to 7.8. The media were then anoxically transferred into Hungate tubes. For MPN, 1ml of formation water was added to 9ml of medium. A series of successive dilutions were then performed until the final dilution of 10−6 (triplicate). All the cultures were incubated at 37°C. Growth was monitored daily by measuring turbidity (OD 600nm; spectrophotometer, Spectronic 401, Spectronic Instruments) and was finally confirmed by observation under a microscope (BX60, Olympus). The culture medium used to select sulfate reducers was a commercial medium (SRB Labège® Test kit, CFG, France). To count the sulfate-reducing spores present in the formation water, the water was heated to 80°C for 10min directly at the sampling site before being inoculated in the appropriate medium in the laboratory in accordance with the procedure described here.
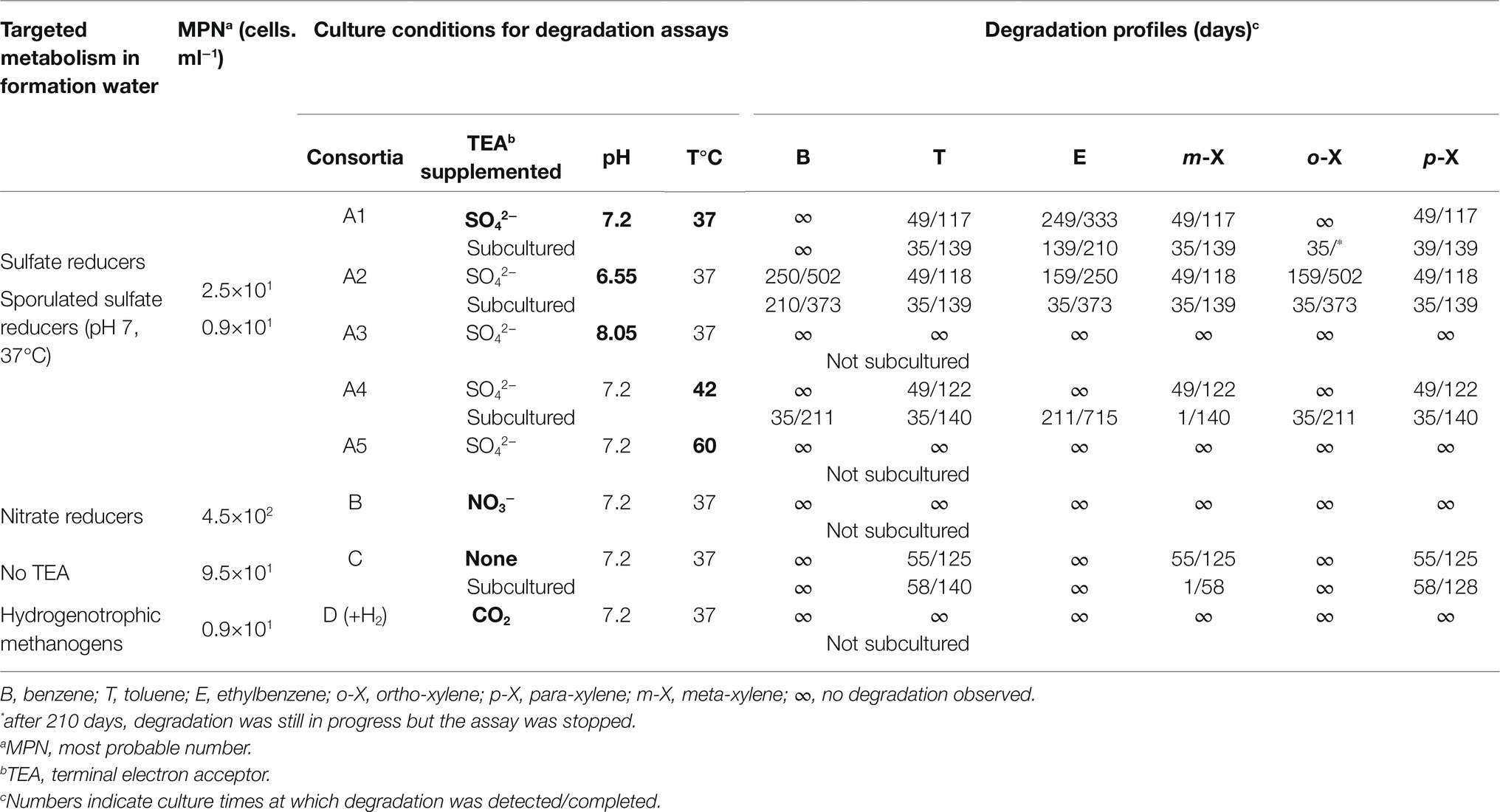
Table 2. Results of metabolic group counts by MPN and BTEX degradation according to targeted metabolism.
Biodegradation Assays
Biodegradation assays of BTEXs were performed with site water supplemented with concentrated biomass, as described by Berlendis et al. (2010). A sample of this water enriched with microorganisms was stored (80ml) at −20°C for molecular biology analyses. Between the sample being taken and the initiation of the biodegradation assays (less than 24h), the water continued to change, primarily under the effect of degassing, resulting in a pH of 7.2. The biodegradation assays were performed with 4 different media, A, B, C and D, to reveal the metabolic groups implicated in BTEX biodegradation (Table 2). Different conditions were tested in microcosms A, favoring sulfate-reducing microorganisms. The preparation of the different microcosms was carried out as follows: the formation water of microcosms A1 (pH 7.2; 37°C), A2 (pH 6.55; 37°C), A3 (pH 8.05; 37°C), A4 (pH 7.2; 42°C), and A5 (pH 7.2; 60°C) was supplemented with 10mM Na2SO4; microcosm B was supplemented with 10mM NaNO3; no TEA was added to microcosm C (beyond the TEAs potentially present in the formation water); and an H2/CO2 (80/20; 1bar) gas phase was added to microcosm D. The biomass that was concentrated on the 99 filters (Sterivex, EMD, Millipore, Molsheim, France) during sampling was resuspended in 200ml of formation water in an anaerobic glovebox (Getinge La Calhene, France) before being used to inoculate the biodegradation assays (1:10, v/v). Finally, a mixture of benzene, toluene, ethylbenzene, and m-, o-, and p-xylenes (Sigma–Aldrich) was added to a final concentration of 10ppm (v/v). Each condition was tested in duplicate, and a third vial was used as an abiotic control following the addition of 0.5ml of 10N HCl. Subcultures were then prepared in formation water sterilized by filtration (0.2μm porosity, cellulose nitrate filters, Sartorius Stedim) and appropriately supplemented to adapt them to the target metabolism. BTEX was monitored using a 7890A gas chromatograph (Agilent Technologies) equipped with a flame ionization detector, employing the same equipment and the same procedure described by Aüllo et al. (2016).
Culture at 90Bar Pressure
At the end of the biodegradation experiment conducted at atmospheric pressure, the two A2 replicates (sulfate-reducing conditions) were pooled and used as a 10% (v/v) inoculum in the formation water previously sterilized by filtration at 0.2μm porosity. As above, this water was supplemented with SL10 trace elements, vitamins, dithionite (0.2% w/v) as a reducing agent, 9mM NH4Cl, 2mM K2HPO4, 2mM KH2PO4, 10mM Na2SO4, and BTEX (10ppm each). When the initiation of degradation was observed for the p- and m-xylenes, 360ml of the culture was transferred under sterile and anoxic conditions to a flask at atmospheric pressure and to a reactor in which the pressure was gradually increased up to 90bar, both at a temperature of 37°C. The experiment was performed with an apparatus consisting of a one-liter stainless steel reactor in which it was possible to implement reactions at up to 300bar and 200°C. This reactor had three potential feeders and one outlet line equipped with a gas–liquid separator (cyclonic to limit the drive of the liquid phase). A constant reaction temperature was maintained by the circulation of oil in an external jacket connected to a thermostat. A variable-speed stirrer agitated the mixture. Safety measures such as a rupture disc and high-temperature cutoff were also implemented. A complete description of the device is presented by Kriaa et al. (2009). The reactor was sterilized with distilled water at 120°C for 4h. Inside the reactor, BTEX levels were monitored using the same approach described above in the “Biodegradation assays” section. Sampling was conducted using a PEEK tube with a diameter of 0.22mm and length of 4m (calculated internal volume of 125μl) and heat sterilized (100°C, overnight). A slight decrease in pressure was recorded throughout the experiment due to the uncompensated sample volumes (bacterial cell count and BTEX quantification).
Cell Counts
Twenty-four hours after sampling, the proportion of live cells was estimated by epifluorescence microscopy using a LIVE/DEAD BacLight Bacterial viability kit (Thermo Fisher Scientific) from the formation water sampled to determine the possible impact of depressurization during ascent through the control well. For this purpose, 10ml of formation water was spiked with 25μl of the SYTO9/PI (v/v) mix. After 15min in darkness, the sample was filtered through 0.2μm pore-size black polycarbonate (25mm, Millipore) under vacuum. Moreover, the total cell concentrations in the formation water and concentrated biomass were estimated using epifluorescence microscopy with DAPI staining (4',6'-diamidino-2-phenylindole, Sigma-Aldrich). Eighteen milliliters of the water was fixed with 2ml of 10% borax-buffered formaldehyde (37%, Sigma-Aldrich) before being stored at 4°C until observation. Next, 10ml of the fixed sample was spiked with 900μl of DAPI stock solution (200μg.ml−1) before being filtered through 0.2μm pore-size black polycarbonate (25mm, Millipore) under vacuum. In the experiment at 90bar pressure, cell counting was conducted using 500μl of culture fixed with 100μl of a 10% borax-buffered formaldehyde stock solution and then spiked with 25μl of DAPI before being filtered through 0.2μm pore-size black polycarbonate (25mm, Millipore) under vacuum. In each of the measurements, 10 randomly selected fields were used for cell counting which represented 202 to 507 microbial cells counted. The Zeiss Observer Z1c epifluorescence microscope was equipped with a mercury light source.
Molecular Biology Methods
Diversity of the Concentrated Biomass
Eighty milliliters of the water enriched with microorganisms used for the biodegradation tests was filtered (0.2μm porosity, cellulose nitrate filters, Sartorius Stedim). The filters were then ground in liquid nitrogen to a fine powder, and DNA was extracted using the Mobio-Power Soil DNA isolation kit (Mobio Laboratories, Carlsbad, CA) in line with the supplier’s recommendations. The genomic DNA was sent to a commercial company (MR DNA, Shallowater, Texas, United States). Following the amplification of the V4 hypervariable region of the 16S rRNA gene (PCR primers 515/806), the amplicons were sequenced via MiSeq 2 × 300bp sequencing (Illumina, California, United States) in line with the supplier’s recommendations. The sequences were converted and demultiplexed with QIIME1. The sequences were analyzed using the software package DADA2 (Callahan et al., 2016) in the QIIME2 pipeline. With this package, the sequences were treated by quality filtering, merging, dereplicating, and removing chimeras to determine amplicon sequence variants (ASVs). Taxonomy was assigned using VSearch (Rognes et al., 2016) against the SILVA SSU 138 NR database (Quast et al., 2013) without uncultured/environmental sequences. The raw data were deposited in the NCBI SRA under bioproject ID PRJNA739070.
Sequence Capture by Hybridization Experiment
The bssA gene encodes benzylsuccinate synthase, which initiates the anaerobic degradation of toluene and xylenes. A set of thirty-four 54- to 57-mer degenerate probes covering the bssA gene (Supplementary Table S1) was designed based on 17 bssA nucleic sequences from strains belonging to the phylum Desulfobacterota, which was recently defined from delta-Proteobacteria (EF123662, EF123663, EF123667, and EU780921), Firmicutes (EF123665, KJ398022, and GU357866), Geobacteraceae (AF123664, EF123666, and AF441130), and environmental sequences obtained with the 7772F/8546R primers set (KF995713, KF995719, KF995720, and KF995723). Three bssA sequences (KX576576, KX576577, and KX576575) were obtained from a previous study of deep aquifers (Ranchou-Peyruse et al., 2017). All of these sequences were processed using KASpOD software (Parisot et al., 2012), and we preferentially selected probes targeting the sequence encoding the Cys-loop (Funk et al., 2015). Adaptor sequences were added at each end of the probe to enable PCR amplification, resulting in the sequences “ATCGCACCAGCGTGT-N(31–38)-CACTGCGGCTCCTCA,” where N(31–38) represents the bssA-specific capture probe. Biotinylated RNA capture probes were then synthesized as described by Ribière et al. (2016).
A next-generation sequencing library was prepared from the DNA extracted from microcosm B using the Nextera XT Kit (Illumina) following the manufacturer’s instructions. Solution hybrid selection (SHS) was conducted according to the protocol described by Ribière et al. (2016). Briefly, 500ng of the heat-denatured libraries was hybridized to the set of biotinylated RNA probes for 24h at 65°C. Probe/target heterodimers were trapped by streptavidin-coated paramagnetic beads (Dynabeads M-280 Steptavidin, Invitrogen, Carlsbag, CA, United States). After several washing steps, the captured targets were eluted from the beads using NaOH and purified using AMPure beads (Beckman Coulter Genomics, Takeley, Essex, United Kingdom). The enriched products were PCR amplified using primers complementary to the library adapters and purified again using AMPure beads (Beckman Coulter Genomics). To increase enrichment, a second round of hybridization and amplification was performed using the obtained captured products.
Captured DNA products were sequenced using a MiSeq 2×300bp run (Illumina) according to the manufacturer’s specifications (Genoscreen). All raw reads were scanned for library adaptors and quality filtered using PRINSEQ-lite PERL script (Schmieder and Edwards, 2011) prior to assembly and analysis. The clean reads were assembled de novo using IDBA-UD (v1.1.1; Peng et al., 2012). The contigs generated were combined for a second round of assembly using CAP3 to generate longer contigs (Huang and Madan, 1999). The amino acid (AA) sequences were deduced from the final assembled nucleotide contigs and then aligned with reference open-reading frames sourced from public databases using MEGA version 6 (Tamura et al., 2013). The two obtained bssA sequences were deposited in the NCBI database (MW762607 and MW762608).
Diversity of the Communities Cultivated at 90Bar Versus Atmospheric Pressure
After 120days of incubation, samples were taken from each of the two cultures kept at high pressure (220ml) and atmospheric pressure (335ml) and filtered through a 0.22μm pore-size filter. The filters were immediately cut into three equal parts under sterile conditions and stored at −80°C for RNA conservation until use. Each piece of the filter was then ground in liquid nitrogen, and the RNA was extracted using the FastRNA® Pro Soil-Indirect kit (MP Biomedicals, Inc.) and separated from the DNA using the AllPrep DNA/RNA FFPE kit (Qiagen) in line with the manufacturer’s recommendations. Although only 16S rRNA gene data were employed in this study, the whole meta-transcriptomic experiment was performed on 2 × 3 replicates by a commercial company (Sequentia Biotech, Barcelona, Spain). Total RNA was quantified on a QUBIT fluorometer. The libraries were produced using retrotranscribed cDNA previously amplified with an Ovation Ultralow Library System V2 (NuGEN Technologies, Inc.). Library size and integrity were assessed using an Agilent Bioanalyzer (Santa Clara, CA). Sequencing was performed on the Illumina HiSeq 2,500 (Illumina, San Diego, CA), and 30M paired-end reads (2×125) per sample were generated. Sequences that were too small (<35bp) or had low-quality scores (<25) were eliminated with BBDuk software (version 12/2015) to improve the analysis quality. The raw data were processed via SortmeRNA (Kopylova et al., 2012) through the ASaiM framework with Galaxy2 to select the 16S rRNA gene sequences from the R1 and R2 files of each triplicate set of samples (Batut et al., 2018). Next, the R1 and R2 sequences from each file were interlaced into a single file using FASTQ interlacer. The paired R1/R2 sequences were processed with DADA2 in the QIIME pipeline, as described earlier. The raw data were deposited in the NCBI SRA under bioproject ID PRJNA715357.
Results
Physical Chemistry of the Aquifer Water
The formation water taken from a depth of 720m (Table 1) evolved at a pressure of 88bar in a rock formation composed of sandstone and carbonates dates back to the Jurassic period (Malm Kimmeridgian-Oxfordian). The water is mesothermal (37°C) with a pH of 7.8. It is brackish water with a conductivity of 2051 μS.cm−1. This salinity is primarily controlled by four predominant ions: Na+ and HCO3− (381.5 and 628mg.L−1, respectively), followed by SO42− (269mg.L−1) and Cl− (178mg.L−1). The sum of these ions (1.456g.L−1) represents 4% of marine salinity. According to the Langelier–Ludwig diagram, HCO3−/Cl−>1, Na+/Cl−>1, and Na+/K+=32.6 ratios are characteristic of formation water under the influence of the rock composition. Free CO2 could be detected (9.4mg.L−1). With total organic carbon (TOC)<1mg.L−1 and Eh=−284mV, the conditions are oligotrophic and highly reducing. The presence of oxygen, nitrate or nitrite could not be demonstrated, but an H2S content of 0.70mg.L−1 was recorded. The ammonium concentration was relatively high, at 2.5mg.L−1, whereas the phosphate concentration was low, at 0.02mg.L−1.
Composition of the Microbial Community From the Formation Water
Depending on the flow rate and the volume of the well, the rise of a microbial cell from the aquifer to the surface is estimated to last for approximately 100min. The impact of decompression from 88bar to atmospheric pressure during water wellhead sampling appears to be relatively low given the microorganism survival rate of 80% estimated using the LIVE/DEAD microscopy technique 24h after obtaining the sample. The cell number present in the water was 2.43·104 cells.ml−1. This biomass was concentrated approximately 25 times by the addition of biomass from Sterivex filters to reach a concentration of 6.10·105 cells.ml−1. Taxonomic diversity was then assessed based on the sequencing and analysis of 16S rRNA genes from the microbial community, and 10,741 amplicon sequence variants (ASVs) were generated (Figure 1). In total, 23 bacterial and 5 archaeal phyla were revealed in the concentrated biomass from the formation water. The microbial community was largely dominated by bacteria, which showed a relative abundance of 99.4% of ASVs. Excluding unassigned bacteria (12.4%), the microbial community was mainly composed of 10 different phyla: Firmicutes (19.7%), Desulfobacterota (17.4%), “Candidatus Acetothermia” (14.7%), Proteobacteria (10.4%), Nitrospirota (8.8%), Chloroflexi (3.3%), Sv0485 (3.0%), Actinobacteriota (1.6%), Bacteroidota (1.5%), and Elusimicrobiota (1.1%). The microbial community was driven by fermenters and sulfate reducers such as “Candidatus Acetothermia,” Peptococcaceae SCADC1_2_3, Anaerolineae, Desulfobacterota, Thermodesulfovibriona, and the candidate Sva0485 clade of the Deltaproteobacteria. Among the Proteobacteria, the microorganisms were mainly Nitrosococcaceae (6.7%), Acidovorax (1.6%), and Pseudomonas (1.0%).
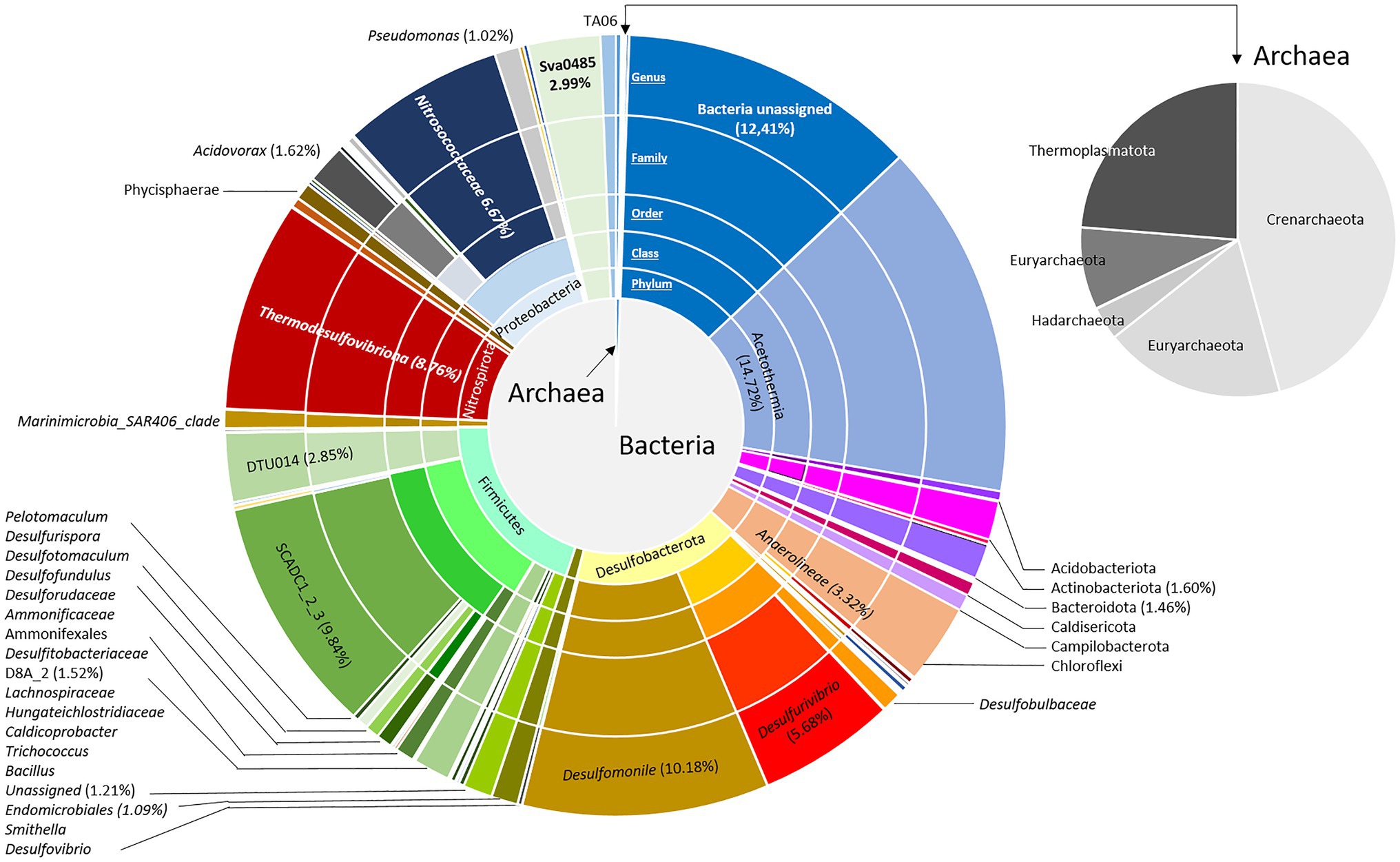
Figure 1. Bacterial and archaeal community composition in the formation water from a deep aquifer used for natural gas storage (Pb_J_6, Parisian sedimentary basin). Based on the V4 region of the 16S SSU rRNA, the pie charts represent the relative abundance as the percentage of each taxon with the interior toward the periphery: phylum, class, family, and genus.
The Archaean community was divided among Crenarchaeota (45.7%; represented by the Class Bathyarchaeia), Thermoplasmatota (23.7%; represented by the family Methanomassiliicoccaceae), Euryarchaeota (18.6%; represented by the family Methanobacteriaceae), Halobacterota (5%; represented by the family Methanosarcinaceae) and Hadarchaeota (3.4%; represented by the order Methanobacteriales).
When natural gas is stored in a deep aquifer, some of the BTEX contained in the gas can dissolve in the formation water and be degraded by microorganisms if these organisms have the potential to do so. Thus, the enrichment of this microbial community under several different conditions (different TEAs, temperatures or pH levels) was subsequently carried out.
BTEX Enrichment Cultures at Atmospheric Pressure
The results of metabolic group enumeration in the on-site samples and the BTEX degradation profiles under various culture conditions are shown in Table 2. Counting using the MPN technique revealed a sulfate reducer concentration of 2.5·101 cells.ml−1. On site, the samples were boiled for 10min to inactivate cells and evaluate the relative proportion of spores of sulfate-reducing bacteria, which showed a concentration of 0.9·101 cells.ml−1. Microorganisms capable of fermentation and those capable of lithoautotrophic growth based on hydrogen showed concentrations of 9.5·101 and 0.9·101 cells.ml−1, respectively. Even though no nitrate was detected in the formation water, the proportion of nitrate-reducing microorganisms was relatively high, with a concentration of 4.5·102 cells.ml−1. There was no growth in the presence of FeIII or oxygen in media targeting Pseudomonas, oligotrophs, methylotrophs and heterotrophs.
When selective enrichment conditions for the four metabolic groups for which growth had been demonstrated using the MPN technique were maintained, only sulfate-reducing (Conditions A) and fermentation (Condition C) conditions showed the capacity to degrade monoaromatic hydrocarbons. Without any TEA (fermentation, Condition C), only toluene, m- and p-xylene could be degraded. These compounds disappeared completely after approximately 4months. No degradation of benzene, ethylbenzene or o-xylene was observed.
Under sulfate-reducing conditions, several pH values and temperatures were tested. Degradation took place at 37 and 42°C but not at 60°C. While degradation was demonstrated at pH 7.2 and 6.55, no degradation was observed at pH 8.05. It should be noted that the aromatic hydrocarbons that were degraded first (toluene, m-xylene, and p-xylene) were the same than those that degraded under fermentation conditions. In addition to these three molecules, ethyl-benzene, o-xylene, and benzene were biodegraded under sulfate-reducing conditions over longer periods exceeding 1year.
While the anaerobic biodegradation capacity of BTEX has been the subject of numerous studies, the difficulty of accessing deep aquifers has caused these environments to be much less studied in general, particularly with regard to this type of metabolism. It is therefore important to detect the genes involved in this biodegradation process to reveal their diversity to better understand the phenomena in situ.
bssA and bssA-Like Sequence Captured by Hybridization
The enzyme responsible for the anaerobic biodegradation of hydrocarbons such as toluene and xylene isomers is benzylsuccinate synthase, which is encoded by the bssA gene. This gene was targeted in a biodegradation assay selecting for sulfate-reducing conditions (A1: pH 7.2, 37°C, Table 2), in which all the TEXs were degraded after 210days of incubation. Two amino acid sequences (contig456 with 522 AA and contig1660 with 807 AA) were related to bssA sequences. Figure 2 presents a phylogenetic tree constructed from fumarate-adding enzyme (FAE) amino acid sequences similar to enzymes reported to degrade hydrocarbons, particularly aromatics (2-methylnaphthalene, p-cresol, and toluene). Contig456 and contig1660 were in the same cluster as Desulfobacterium sp. (SPD74684), with 58% identity. This cluster was located between the bssA clades of p-cresol and toluene. While the Cys-loop, which plays a role in fumarate attachment, was clearly observed, the gly-loop contained the tyrosine (Y) amino acid characteristic of the p-cresol clade but the arginine (R) amino acid characteristic of the toluene clade.

Figure 2. Phylogenetic tree based on the partial bssA-like amino acid sequences from a BTEX biodegradation assay (in bold; this study) compared with the fumarate-adding enzyme (FAE) sequences available in international databases. The evolutionary history was inferred using the neighbor-joining method (Saitou and Nei, 1987). The optimal tree with the sum of branch length=6.67348502 is shown. The tree is drawn to scale, with branch lengths in the same units as the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Poisson correction method (Zuckerkandl and Pauling, 1965) and are presented in units of the number of amino acid substitutions per site. This analysis involved 34 amino acid sequences. All ambiguous positions were removed from each sequence pair (pairwise deletion option). There were a total of 914 positions in the final dataset. Evolutionary analyses were conducted in MEGA X (Kumar et al., 2018).
Culture at 90Bar vs. Culturing at Atmospheric Pressure
Using the same biodegradation assay employed for the gene capture technique previously described, incubation at 90bar pressure and in the presence of the 6 aromatic hydrocarbon molecules was performed for a period of 120days (Figure 3). Throughout this period, slow cell growth was observed from 1.15·106 to 5.11·106 cells.ml−1 after 105days of incubation, in parallel with the disappearance of monoaromatic hydrocarbons (between 20 and 40%) with the exception of benzene. The taxonomic diversity of the active microbial community was analyzed at the two different pressure conditions at the end of incubation following RNA extraction. This community capable of degrading BTEX was largely dominated by bacteria (99.97% of ASVs), and the archaeal component of the initial community (Figure 1) was considerably reduced during the two successive incubations of consortium A1 and its subculture under sulfate-reducing conditions (Table 2) preceding incubation at 90bar pressure. At atmospheric pressure, 10.8% of the ASVs could not be assigned taxonomically when they represented only 3.4% of the at high pressure. Among the 49 bacterial phyla identified in the community, the 6 phyla highlighted in Figure 4 represented 83.6 and 90.9% of the taxonomic diversity at Patm and P90bar, respectively. The phylum “Candidatus Acetothermia,” which was almost absent at atmospheric pressure, was highly overrepresented at high pressure (Patm: 0.3% vs. P90bar: 27.8%). The phylum Acidobacteriota was predominantly represented by the class Aminicenantia (Patm: 2.8% vs. P90bar: 0.8%). The Chloroflexi phylum was dominated by bacteria belonging to the Anaerolineae lineage in the Anaerolinaceae family, which represented the major microorganisms at atmospheric pressure, accounting for 44.1% of ASVs versus 18.7% at 90bar, as well as the SBR1031 lineage. The latter lineage was stable between the two culture conditions (2.9 vs. 2.5%). In the presence of 10mM sulfate, sulfate reducers were highlighted by the results as expected, particularly the phylum Desulfobacterota, which represented 14.9 and 24.9% of ASVs at Patm and P90bar, respectively. The Desulfobaccaceae (0.6 vs. 10.2%) and Desulfosarcinaceae (0.3 vs. 4.2%) families were favored by pressure, while to a lesser extent, the members of the Desulfomonilaceae family seemed to benefit from conditions involving atmospheric pressure (10.9 vs. 6.6%). Regarding the phylum Firmicutes, a not negligible proportion of the ASVs remained unassigned (4.6 vs. 1.9%). This phylum was dominated by the Peptococcaceae family, comprising the lineage SCADC1_2_3 (2.1 vs. 0.1%), but also the genera Desulfofarcimen (formerly Desulfotomaculum; 0.0 vs. 5.6%) and Cryptanaerobacter (2.7 vs. 0.2%). The last major phylum was the candidate phylum Marinimicrobia_SAR406 (1.5 vs. 0.5%). At least 13.2% at atmospheric pressure and 21.5% at 90 bar of the ASVs were unassigned or assigned to candidate lineages.
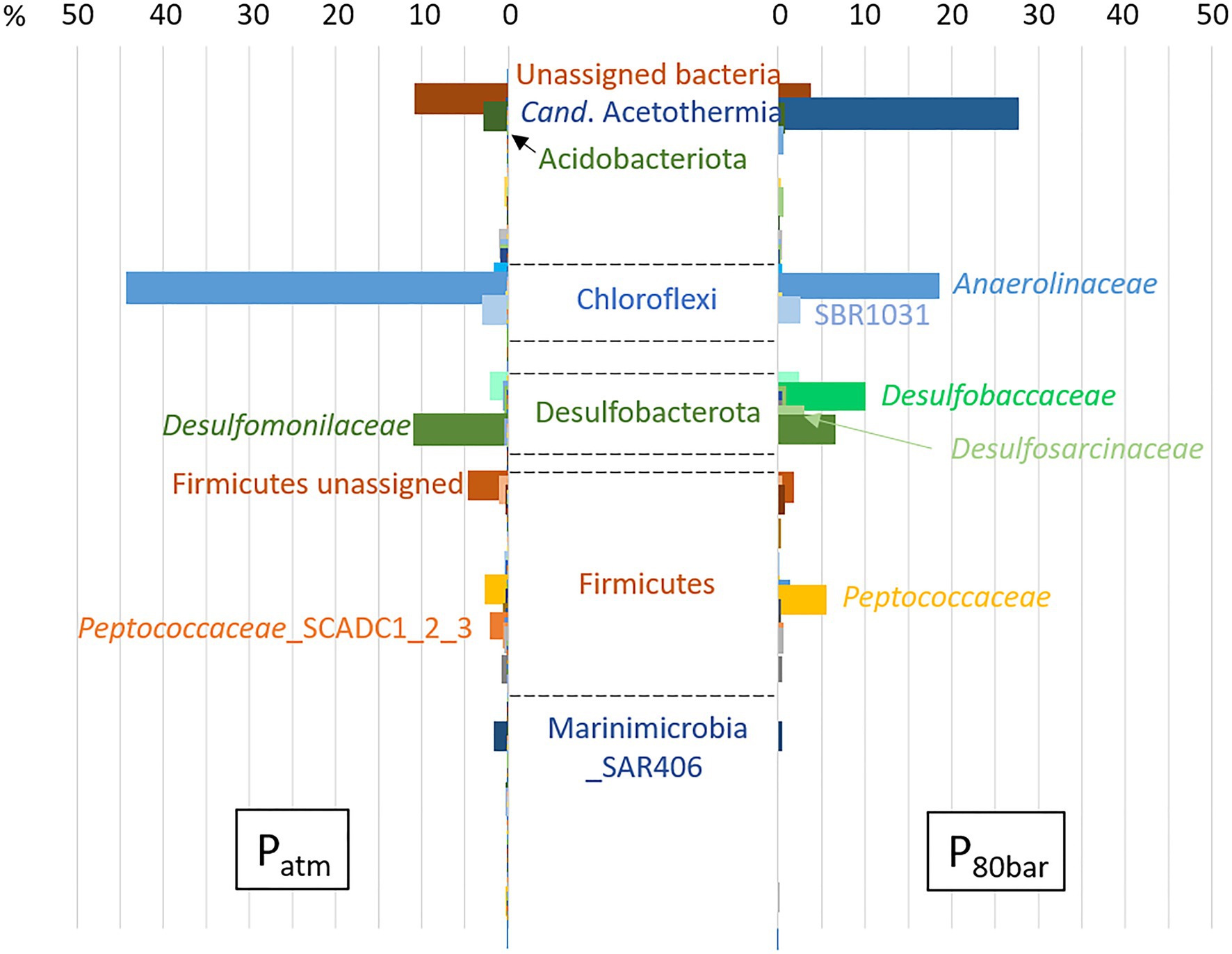
Figure 3. Degradation of BTEX by the bacterial community at 90 bar with monitoring of the cell concentration by microscopic counting.
Discussion
Approximately 85 natural gas storage sites in aquifers can be found around the world (Cornot-Gandolphe, 2017). The current investigation focused on water from the Pb_J_6 site, which forms part of a group of sites studied to examine the diversity of sulfate reducers and methanogens in 2019 by Ranchou-Peyruse and collaborators. This previous work showed the importance of the genus Desulfotomaculum within the family Peptococcaceae in the phylum Firmicutes as well as archaeal members of the family Methanosarcinaceae. The Pb_J_6 site is located 1.3km from another site (Pb_J_7; Ranchou-Peyruse et al., 2019) and has also been studied to evaluate the BTEX biodegradation capacity of its microbial community (Aüllo et al., 2016). Although the two sites belong to the same geological gas storage structure, their distance is relatively significant given the estimated natural displacement of the water mass of 1m/year, or possibly less (Jost et al., 2007). As shown in Table 1, the only terminal electron acceptors (TEAs) present in these ecosystems in sufficient quantities to have a significant influence driving the functioning of these communities were sulfate (269mg.L−1) and CO2 (9.4mg.L−1)/bicarbonate (628mg.L−1). A large part of the community is represented by microorganisms related to sulfate reducers: Desulfobulbaceae, Desulfomonile, Desulfovibrio, Desulforudaceae, Desulfofundulus, Desulfotomaculum, and Thermodesulfovibrio. Moreover, the candidate Sva0485 clade is assumed to be a potential sulfate reducer (Tan et al., 2019).
Taxonomic Diversity of the Microbial Community Found in Formation Water
During sampling and the 720-meter ascent to the surface, the dissolved gases escaped rapidly from the water and microorganisms. This degassing phenomenon was accentuated near stored natural gas and may lead to the deterioration of many cell membranes. By carrying out the ascent in approximately 100min, the objective was to limit microbial lysis, and the result was satisfactory, since 80% of the cells presented an intact membrane 24h after sampling. Almost half of the cultivable sulfate reducers (0.9·101 cells.ml−1) could be related to Peptococcaceae because of their capacity to sporulate and develop in sulfate-reducing conditions (Table 2). The presence of the microorganisms identified during this study is consistent with other studies relating to various deep continental environments, in particular deep aquifers (Basso et al., 2009; Mu et al., 2014; Frank et al., 2016; Kadnikov et al., 2017; Gulliver et al., 2019; Karnachuk et al., 2019; Soares et al., 2019; Stemple et al., 2021). However, the discovery of several taxa of bacteria associated with aerobic or nitrate-reducing metabolism identified here does not seem to be compatible with what we currently know about the physicochemical conditions of deep aquifers. While numerous studies have alluded to microorganisms described as being strictly aerobic or as nitrate-reducers in deep continental environments, their presence remains controversial (Pedersen et al., 2008; Puente-Sánchez et al., 2018; Kadnikov et al., 2020). Several examples of oxygen detection are described in the literature, even though this kind of environment is expected to be deprived of it (Frank et al., 2016; Kadnikov et al., 2018). In this study, no growth was revealed under oxic conditions. Conversely, nitrate-supplemented MPN presented higher concentrations of microorganisms (4.5·102 cells.ml−1) than MPN supplemented with other TEAs or without TEAs (fermentation), which were all below or equal to 9.5·101 cells.ml−1. The dissolution of nitrate salts in some rocks, such as siltstone and sandstone, is theoretically possible and was suggested as a possible explanation for the presence of nitrate in some groundwater from eastern Utah and other aquifers (Stewart and Peterson, 1914; Holloway and Dahlgren, 2002). In natural gas storage aquifers, the successive injection and withdrawal of natural gas could (i) increase the movement of the water mass; (ii) produce pressure variations inducing the displacement of mineral particles (i.e., geomechanics); and (iii) induce the dissolution of trapped nitrate salts consumed by nitrate reducers (Pseudomonas, Acidovorax, and Desulfurivibrio). On the other hand, microorganisms that are assumed to use oxygen, nitrate or nitrite as TEAs may be maintained in these environments via the use of other TEAs yet to be defined or by fermenting the rare organic molecules present. For example, the genus Pseudomonas has been suggested to carry out dissimilatory sulfate reduction (Kliushnikova et al., 1992), and some members, such as P. aeruginosa, have shown the capacity to ferment amino acids (arginine) or pyruvate (Vander Wauven et al., 1984; Schreiber et al., 2006).
The studied environment is naturally oligotrophic (TOC<1mg.L−1 C), and the only sources of organic carbon available for microorganisms are probably recalcitrant organic matter sequestered in the rock pore space following sedimentation as well as hydrocarbons from the stored gas (Parkes et al., 2000; Ranchou-Peyruse et al., 2019). Several of the microorganisms identified in the community (Figures 1, 3) have representatives that have been found in hydrocarbon-rich deep subsurface environments. For example, the lineage Peptococcaceae SCADC1_2_3, the genus Thermodesulfovibrio and the phylum “Ca. Acetothermia” have been identified in oil fields (Nunn, 2015; Hu et al., 2016; Liang et al., 2016). The genera Pelotomaculum, Desulfotomaculum, Desulfovibrio, Desulfomonile, Acidovorax, and Methanobacterium have been detected in contaminated aquifers and/or coal-bearing sediments (Ulrich and Edwards, 2003; Penner et al., 2010; Gründger et al., 2015; Aüllo et al., 2016; Ranchou-Peyruse et al., 2019). The sulfate-reducing bacterial group Sva0485 and the genus Thermodesulfovibrio have been shown to be responsible for the degradation of monoaromatic hydrocarbons such as benzene and alkanes, which is consistent with our observations (Ulrich and Edwards, 2003; Liang et al., 2016).
Deep subsurface environments have been shown to favor the sporulating sulfate-reducing and fermentative microorganisms of the Firmicutes phylum (nearly 20% of the observed community here), which were represented at the Pb_J_6 site by Peptococcaceae SCADC1_2_3 and the genera Pelotomaculum and Desulfotomaculum in particular (Aüllo et al., 2013). Beyond the phylum Firmicutes, sulfate reducers and fermenters dominated the community, as the taxa “Ca. Acetothermia,” Desulfobacterota, and Nitrospirota and the Sva0485 lineage were abundant. In addition, fermentative microorganisms may also have the capability to produce hydrogen. In such energy-poor environments, hydrogen is assumed to be rapidly consumed by other community members, such as hydrogenotrophic methanogens (with less than 0.55% ASVs), syntrophic acetate-oxidizing bacteria (Firmicutes DTU014; Dyksma et al., 2020), homoacetogens (“Ca. Acetothermia”; Youssef et al., 2019), or hydrogenotrophic sulfate-reducing bacteria (Desulfomonile, Desulfovibrio, and Desulfotomaculum; DeWeerd et al., 1991; Voordouw, 1995; Aüllo et al., 2013). Although the Pb_J_6 site is located farther from the stored gas than the Pb_J_7 site, the presence of stored gas may still be a key factor in the structuring of the community, particularly since it is a source of organic molecules. Sulfate reducers and fermenters may lead to the degradation of organic compounds ranging from short-chain alkanes to more complex polymers via monoaromatic hydrocarbons (Table 2) and other polyaromatic compounds.
Monoaromatic Hydrocarbon Degradation
Sulfate reducers (e.g., Desulfotomaculum and Desulfovibrio) and fermenters (e.g., Pelotomaculum) are known to belong to the key metabolic groups involved in the degradation of BTEX in deep aquifers (Berlendis et al., 2010; Kleinsteuber et al., 2012; Aüllo et al., 2016; Ranchou-Peyruse et al., 2019). In this study, BTEX compounds were used to model the transfer of organic molecules from the geologically stored gas phase to the aquifer. BTEX compounds were selected because these compounds, particularly benzene, are the most soluble hydrocarbons. In addition to fermentative microorganisms (degrading toluene, m-xylene, and p-xylene; Table 2), sulfate-reducing microorganisms may also degrade benzene, ethylbenzene and o-xylene if environmental factors are favorable. The Sva085 taxon consists of sulfate reducers, which have been shown to be capable of degrading benzene (Ulrich and Edwards, 2003). This capacity is consistent with the existence of these microorganisms in an oligotrophic environment near stored natural gas, in which hydrocarbons, particularly benzene, may be present and serve as nutrient resources. When BTEX compounds represent the only sources of carbon and energy accessible to this community, the microbial community undergoes simplification, with the disappearance of almost the entire archaeal fraction in particular (<0.03% of ASVs; Figure 4). The simplified microbial community remained capable of degrading all of the BTEX compounds but did so sequentially. The most rapidly degraded molecules were toluene, m- and p-xylene, with very similar degradation times. Once these three molecules disappeared, ethylbenzene and o-xylene were degraded. In the past, it has been demonstrated that while the presence of toluene does not interfere with the degradation of m-xylene (and inversely), the presence of one of these molecules delays the degradation of o- and p-xylene (Meckenstock et al., 2004; Morasch et al., 2004). Under certain pH and temperature conditions, benzene may finally be degraded by the microbial community. The positive effect of a pH of 6.5 on benzene degradation has already been demonstrated in a microbial community from another deep aquifer (Berlendis et al., 2010). Previous work suggested that biofilm growth should be largely dominant in deep subsurface environments, thus allowing microorganisms to benefit from more minerals by being in close contact to them (Wu et al., 2017; Casar et al., 2021). Based on these suggestions, we deduce that the degradation of BTEX compounds, particularly benzene, in deep aquifers should preferentially take place in biofilms at the rock–water interface, which less involvement of planktonic microorganisms in formation water at higher pH levels. It has been established that microorganisms can influence the pH in their immediate environment (Liermann et al., 2000). For example, research conducted on Desulfovibrio fructosovorans has shown that in medium with a pH of 8.5, this microorganism releases acid, thereby maintaining a pH of 6.7 in the close vicinity of its cells (Daumas et al., 1993).
The enzyme involved in anaerobic TEX degradation is benzylsuccinate synthase. Interestingly, previous studies of isolated bacterial strains capable of TEX degradation suggest that this enzyme can degrade toluene (TRM1 and TOL2 strains) and sometimes other alkylbenzenes (e.g., o- and p-xylene for the PRTOL1 strain; Rabus and Widdel, 1995; Heider et al., 2016). The metagenomic method applied in this study to evaluate the diversity of bssA-carrying organisms was shown to be very stringent and even made it possible to reveal rare diversity (Ranchou-Peyruse et al., 2017). Although the frequent degradation of methyl-substituted aromatic compounds was observed in this study, only two different, phylogenetically similar, benzylsuccinate synthase genes were identified within the low-diversity bacterial community. The two identified bssA genes were found to group closely with a bssA gene (SPD74684) affiliated with an uncultured Desulfobacterium species from a natural deposit of asphalt. They clustered between the BssA clade related to the degradation of toluene or other alkylbenzenes and the BssA clade related to p-cresol degradation. In the latter group, the model microorganisms Desulfobacula toluolica and D. phenolica are both capable of degrading toluene and p-cresol, while only D. phenolica can degrade phenol (Rabus and Widdel, 1995; Kuever et al., 2001). D. toluolica has also been shown to be capable of degrading o- and p-xylene but incapable of degrading m-xylene (Beller et al., 1996). It has been further demonstrated that this microorganism possesses an operon enabling the degradation of p-cresol and another operon enabling the degradation of toluene (Wöhlbrand et al., 2012). Like all glycyl radical enzymes, the two new BssA sequences described here possess a well-conserved Cys-loop. It should be noted that thse amino acid “methionine” directly following the “cysteine” active site is replaced by a “valine” in the case of contig456. The Gly-loop presents a “tyrosine” immediately after the “glycine” site, as do the BssA sequences of the clades of p-cresol, methylnaphthalene and alkanes. The ability to degrade aromatic compounds may represent a competitive advantage. Under pristine conditions, microbial degraders could use recalcitrant organic molecules trapped in rocks. Under natural gas storage conditions, a few ppm or even ppb of monoaromatic hydrocarbons available from the gas could feed these microorganisms.
Cultivation at High Pressure
In the context of BTEX degradation in deep subsurface environments, no study simulating in situ pressure conditions has ever been conducted to our knowledge. Here, the selected bacterial community capable of BTEX degradation was cultivated at a pressure of 90bar (Figure 3). Throughout the 120-day incubation period, alkylbenzene degradation was demonstrated, along with slow bacterial cell growth characteristic of deep oligotrophic ecosystems (Chapelle and Lovley, 1990; Hoehler and Jørgensen, 2013). The incubation time may have been insufficient to observe benzene degradation at the applied pressure conditions, since the degradation of benzene in the biodegradation assays began after 200days. Samples of the liquid phase necessary to monitor BTEX and microorganism concentrations led to a slow decrease in pressure, and the incubation stopped when the pressure was 80bar. At atmospheric and high-pressure conditions, the relative abundances of the active microbial populations presented in Figure 4 highlight the dominance of Bacteria. Independent of the pressure condition, the community was dominated by fermenters and sulfate reducers. These fermenters, which produce acetate and hydrogen, belonged to the phylum Chloroflexi, specifically to the Anaerolinae lineage, Anaerolinaceae family and SBR101 group, and to the phylum Firmicutes and family Peptococcaceae, comprising specific members of the genus Cryptanaerobacter and the group SCADC1_2_3 (Kunapuli et al., 2007; Tan et al., 2014; Liang et al., 2015; Xia et al., 2016). Sulfate reducers were mainly grouped together in the phylum Desulfobacterota, with the families Desulfobaccaceae, Desulfomonilaceae, and Desulfosarcinaceae, but also in the phylum Firmicutes, with the genus Desulfofarcimen (family Peptococcaceae), and the phylum Acidobacteriota (Hausmann et al., 2018). Members of Desulfobaccaceae and Desulfosarcinaceae have been reported to use acetate as the sole source of carbon and electron donors (Galushko and Kuever, 2019, Watanabe et al., 2020). Likewise, the genus Desulfofarcimen, formerly Desulfotomaculum, includes the type species D. acetoxidans and D. intricatum, both of which are capable of oxidizing acetate (Spring et al., 2009; Watanabe et al., 2013, 2018). Most of these sulfate-reducers or fermenting microorganisms have already been shown to be involved directly or indirectly in the degradation of aliphatic and/or aromatic hydrocarbons (Kunapuli et al., 2007; Tischer et al., 2013; Tan et al., 2014; Liang et al., 2015; Watanabe et al., 2017). In particular, the genus Cryptanaerobacter, formerly Desulfotomaculum subcluster Ih, has already been shown to be involved in the degradation of monoaromatic hydrocarbons via isotopic approaches (Kunapuli et al., 2007; Kleinsteuber et al., 2008; Herrmann et al., 2010; Taubert et al., 2012). The degradation of these compounds via fermentation pathways leads to the production of acetate, CO2 and H2 in particular. It is therefore probable that monoaromatic hydrocarbons could have fueled acetotrophic and/or hydrogenotrophic bacteria. Here, the candidate phylum Acetothermia, described as a producer of acetate from CO2 and H2 or via fermentation (Hao et al., 2018; Youssef et al., 2019), was characteristic of the community incubated at 90bar. While this phylum was not existent at atmospheric pressure (0.3%), its relative representativeness rose to 27.8% at high pressure. This lineage has been described in several oligotrophic thermal environments, such as hot springs, deep-sea sediments or deep aquifers (Takami et al., 2012; Badhai et al., 2015; Kadnikov et al., 2017; Zaitseva et al., 2017; Varliero et al., 2019). Although it is not possible to determine how incubation at 90bar could have favored the growth of these microorganisms, it is obvious that this condition favored its activity and is a parameter that must be considered to isolate bacteria of this candidate group. The majority of studies that have detected the presence of this lineage focused on taxonomic diversity analyses carried out directly on environmental samples. Nevertheless, a study carried out on anaerobic digesters, which contain a complex community, also identified these microorganisms (Hao et al., 2018). We show that these microorganisms can be maintained and active in a simplified community cultivated at oligotrophic conditions and high pressure. These elements should be taken into account in future procedures to isolate the first representative(s) from this candidate group.
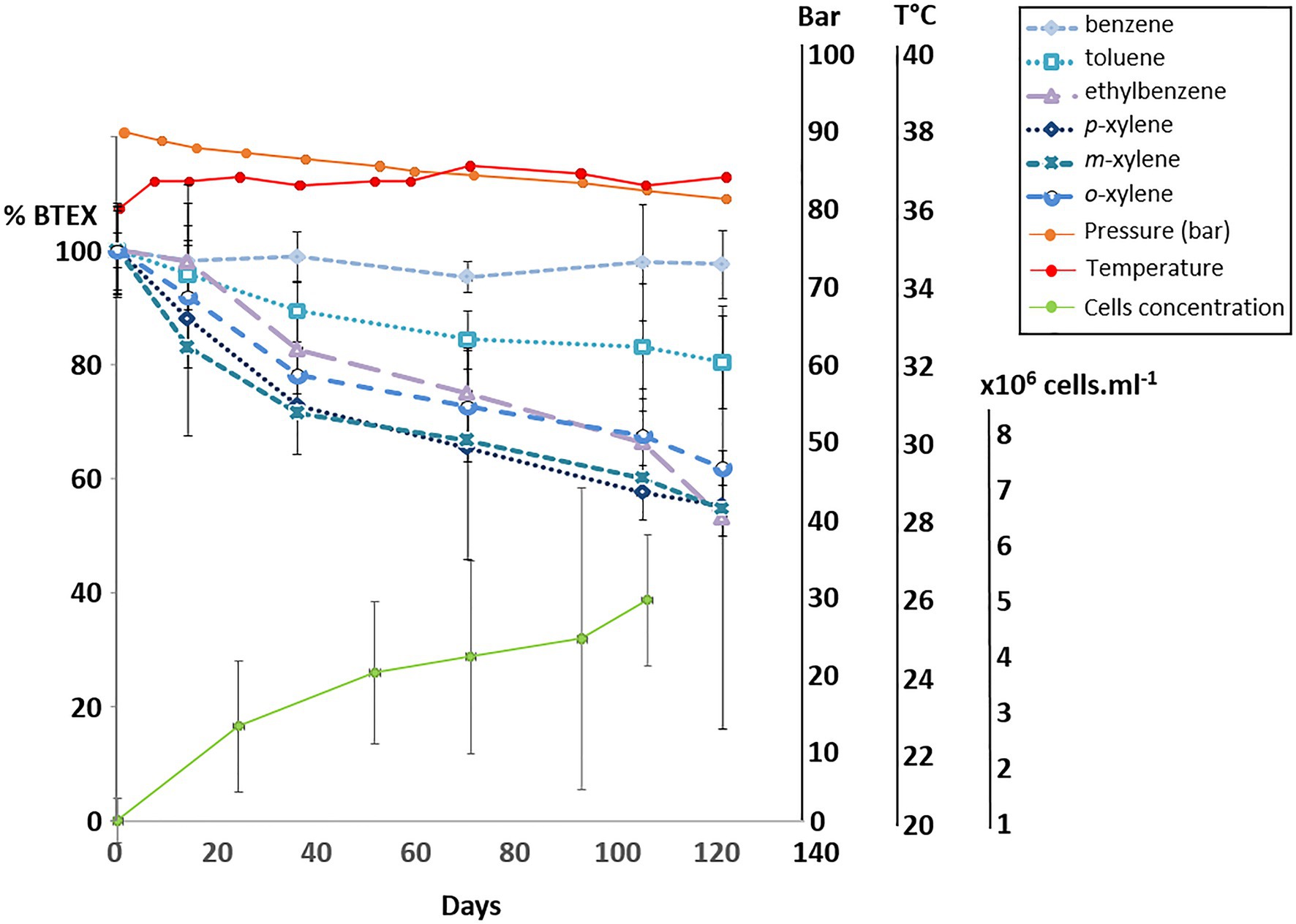
Figure 4. Relative abundance of active BTEX-degrading bacterial populations from a deep aquifer with or without pressure (90 bar). The percentage is shown on the X-axis. Total RNA was extracted from both communities, and taxonomic diversity analysis was performed based on 16S rRNA transcripts. The archaeal fraction accounted for less than 0.03% of ASVs.
Conclusion
The results showed that even at sites located away from stored gas, the microbial communities in deep aquifers retained the ability to degrade the 6 monoaromatic compounds at sulfate and/or fermentation conditions.
The experiment simulating the in situ pressure confirmed the maintenance of the biodegradation capacity when BTEX was the only source of carbon and electrons. On the other hand, the results showed that the pressure could influence the structure of the microbial community. Even at pressures below 100bar, the application of pressure seems to allow the culture of microorganisms that are not necessarily obligate piezophiles but have thus far been identified only through the observation of candidate lineages.
Finally, all the indicators led to the hypothesis that the driving force behind BTEX degradation in these environments should be biofilms. It is therefore necessary to study these processes in biofilm conditions and, hence, take into consideration a mineral phase colonized by microorganisms.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, from MW787045 to MW788027; MW762607, MW762608; PRJNA715357.
Author Contributions
MR-P, MG, FC, PCe, and AR-P co-conceived the study. MR-P, MG, MA, and AR-P performed the culture and molecular biology experiments. CD and PP carried out the DNA hybridization experiments. All authors contributed to the interpretation of the results and paper writing. All authors contributed to the article and approved the submitted version.
Funding
Storengy and Teréga funded this research project. MR-P’s salary was supported by E2S-UPPA.
Conflict of Interest
DD is employed by Storengy. GC and PCh are employed by Teréga.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors acknowledge the support of the Freiburg Galaxy Team funded by the Collaborative Research Centre 992 Medical Epigenetics (DFG grant SFB 992/1 2012) and the German Federal Ministry of Education and Research BMBF grant 031 A538A de.NBI-RBC. The authors thank Cyrielle Gasc for participating in probe design. The authors are grateful to the reviewers and the associate editor for their constructive comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.688929/full#supplementary-material
Supplementary Table S1 | Gene capture probes used in this study.
Footnotes
References
Amigáň, P., Greksák, M., Kozánková, J., Buezk, F., Onderka, V., and Wolf, I. (1990). Methanogenic bacteria as a key factor involved in changes of town gas stored in an underground reservoir. FEMS Microbiol. Ecol. 73, 221–224.
Aüllo, T., Berlendis, S., Lascourrèges, J. F., Dessort, D., Duclerc, D., Saint-Laurent, S., et al. (2016). New bio-indicators for long term natural attenuation of monoaromatic compounds in deep terrestrial aquifers. Front. Microbiol. 7:122. doi: 10.3389/fmicb.2016.00122
Aüllo, T., Ranchou-Peyruse, A., Ollivier, B., and Magot, M. (2013). Desulfotomaculum spp. and related gram-positive sulfate-reducing bacteria in deep subsurface environments. Front. Microbiol. 4:362. doi: 10.3389/fmicb.2013.00362
Badhai, J., Ghosh, T. S., and Das, S. K. (2015). Taxonomic and functional characteristics of microbial communities and their correlation with physicochemical properties of four geothermal springs in Odisha, India. Front. Microbiol. 6:1166. doi: 10.3389/fmicb.2015.01166
Baker, B. J., Moser, D. P., MacGregor, B. J., Fishbain, S., Wagner, M., Fry, N. K., et al. (2003). Related assemblages of sulphate-reducing bacteria associated with ultradeep gold mines of South Africa and deep basalt aquifers of Washington state. Environ. Microbiol. 5, 267–277. doi: 10.1046/j.1462-2920.2003.00408x
Basso, O., Lascourrèges, J. F., Jarry, M., and Magot, M. (2005). The effect of cleaning and disinfecting the sampling well on the microbial communities of deep subsurface water samples. Environ. Microbiol. 7, 13–21. doi: 10.1111/j.1462-2920.2004.00660.x
Basso, O., Lascourreges, J. F., Le Borgne, F., Le Goff, C., and Magot, M. (2009). Characterization by culture and molecular analysis of the microbial diversity of a deep subsurface gas storage aquifer. Res. Microbiol. 160, 107–116. doi: 10.1016/j.resmic.2008.10.010
Batut, B., Gravouil, K., Defois, C., Hiltemann, S., Brugère, J. F., Peyretaillade, E., et al. (2018). ASaiM: a galaxy-based framework to analyze microbiota data. Gigascience 7:giy057. doi: 10.1093/gigascience/giy057
Beller, H. R., Spormann, A. M., Sharma, P. K., Cole, J. R., and Reinhard, M. (1996). Isolation and characterization of a novel toluene-degrading, sulfate-reducing bacterium. Appl. Environ. Microbiol. 62, 1188–1196. doi: 10.1128/aem.62.4.1188-1196.1996
Berlendis, S., Lascourreges, J.-F., Schraauwers, B., Sivadon, P., and Magot, M. (2010). Anaerobic biodegradation of BTEX by original bacterial communities from an underground gas storage aquifer. Environ. Sci. Technol. 44, 3621–3628. doi: 10.1021/es100123b
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Casar, C. P., Kruger, B. R., and Osburn, M. R. (2021). Rock-hosted subsurface biofilms: mineral selectivity drives hotspots for intraterrestrial life. Front. Microbiol. 12:658988. doi: 10.3389/fmicb.2021.658988
Chapelle, F. H., and Lovley, D. R. (1990). Rates of microbial metabolism in deep coastal plain aquifers. Appl. Environ. Microbiol. 56, 1865–1874. doi: 10.1128/aem.56.6.1865-1874.1990
Cornot-Gandolphe, S. (2017). Underground gas storage in the world – 2017 Status. Cedigaz Insights No. 22.
Daumas, S., Magot, M., and Crolet, J. L. (1993). Measurement of the net production of acidity by a sulphate-reducing bacterium: experimental checking of theoretical models of microbially influenced corrosion. Res. Microbial. 144, 327–332. doi: 10.1016/0923-2508(93)90017-v
De Silva, G. P. D., Ranjith, P. G., and Perera, M. S. A. (2015). Geochemical aspects of CO2 sequestration in deep saline aquifers: A review. Fuel 155, 128–143. doi: 10.1016/j.fuel.2015.03.045
DeWeerd, K. A., Concannon, F., and Suflita, J. M. (1991). Relationship between hydrogen consumption, dehalogenation, and the reduction of sulfur oxyanions by Desulfomonile tiedjei. Appl. Environ. Microbiol. 57, 1929–1934. doi: 10.1128/aem.57.7.1929-1934.1991
Dyksma, S., Jansen, L., and Gallert, C. (2020). Syntrophic acetate oxidation replaces acetoclastic methanogenesis during thermophilic digestion of biowaste. Microbiome 8:105. doi: 10.1186/s40168-020-00862-5
Escudero, C., Oggerin, M., and Amils, R. (2018). The deep continental subsurface: The dark biosphere. Int. Microbiol. 21, 3–14. doi: 10.1007/s10123-018-0009-y
Flexer, V., Baspineiro, C. F., and Galli, C. I. (2018). Lithium recovery from brines: A vital raw material for green energies with a potential environmental impact in its mining and processing. Sci. Total Environ. 639, 1188–1204. doi: 10.1016/j.scitotenv.2018.05.223
Follonier, S., Panke, S., and Zinn, M. (2012). Pressure to kill or pressure to boost: a review on the various effects and applications of hydrostatic pressure in bacterial biotechnology. Appl. Microbiol. Biotechnol. 93, 1805–1815. doi: 10.1007/s00253-011-3854-6
Frank, Y. A., Kadnikov, V. V., Gavrilov, S. N., Banks, D., Gerasimchuk, A. L., Podosokorskaya, O. A., et al. (2016). Stable and variable parts of microbial community in siberian deep subsurface thermal aquifer system revealed in a long-term monitoring study. Front. Microbiol. 7:2101. doi: 10.3389/fmicb.2016.02101
Funk, M. A., Marsh, E. N., and Drennan, C. L. (2015). Substrate-bound structures of benzylsuccinate synthase reveal how toluene is activated in anaerobic hydrocarbon degradation. J. Biol. Chem. 290, 22398–22408. doi: 10.1074/jbc.M115.670737
Galushko, A., and Kuever, J. (2019). “Desulfobaccaceae,” in Bergey’s Manual of Systematics of Archaea and Bacteria. ed. W. B. Whitman, F. A. Rainey, P. Kämpfer, M. E. Trujillo, P. DeVos, and B. Hedlund (John Wiley & Sons.) doi: 10.1002/9781118960608.fbm00324s
Gleeson, T., Befus, K. M., Jasechko, S., Luijendijk, E., and Bayani Cardenas, M. (2015). The global volume and distribution of modern groundwater. Nat. Geosci. 9, 161–167. doi: 10.1038/ngeo2590
Gründger, F., Jiménez, N., Thielemann, T., Straaten, N., Lüders, T., Richnow, H.-H., et al. (2015). Microbial methane formation in deep aquifers of a coal-bearing sedimentary basin, Germany. Front. Microbiol. 6:200. doi: 10.3389/fmicb.2015.00200
Gulliver, D., Lipus, D., Ross, D., and Bibby, K. (2019). Insights into microbial community structure and function from a shallow, simulated CO2-leakage aquifer demonstrate microbial selection and adaptation. Environ. Microbiol. Rep. 11, 338–351. doi: 10.1111/1758-2229.12675
Hao, L., McIlroy, S. J., Kirkegaard, R. H., Karst, S. M., Fernando, W. E. Y., Aslan, H., et al. (2018). Novel prosthecate bacteria from the candidate phylum Acetothermia. ISME J. 12, 2225–2237. doi: 10.1038/s41396-018-0187-9
Hausmann, B., Pelikan, C., Herbold, C. W., Köstlbacher, S., Albertsen, M., Eichorst, S. A., et al. (2018). Peatland Acidobacteria with a dissimilatory sulfur metabolism. ISME J. 12, 1729–1742. doi: 10.1038/s41396-018-0077-1
Haveman, S. A., and Pedersen, K. (2002). Distribution of culturable microorganisms in Fennoscandian shield groundwater. FEMS Microbiol. Ecol. 39, 129–137. doi: 10.1111/j.1574-6941.2002.tb00914.x
Heider, J., Szaleniec, M., Martins, B. M., Seyhan, D., Buckel, W., and Golding, B. T. (2016). Structure and function of benzylsuccinate synthase and related fumarate-adding glycyl radical enzymes. J. Mol. Microbiol. Biotechnol. 26, 29–44. doi: 10.1159/000441656
Herrmann, S., Kleinsteuber, S., Chatzinotas, A., Kuppardt, S., Lueders, T., Richnow, H. H., et al. (2010). Functional characterization of an anaerobic benzene-degrading enrichment culture by DNA stable isotope probing. Environ. Microbiol. 12, 401–411. doi: 10.1111/j.1462-2920.2009.02077.x
Hershey, O. S., Kallmeyer, J., Wallace, A., Barton, M. D., and Barton, H. A. (2018). High microbial diversity despite extremely low biomass in a deep karst aquifer. Front. Microbiol. 9:2823. doi: 10.3389/fmicb.2018.02823
Hoehler, T. M., and Jørgensen, B. B. (2013). Microbial life under extreme energy limitation. Nat. Rev. Microbiol. 11, 83–94. doi: 10.1038/nrmicro2939
Holloway, J., and Dahlgren, R. A. (2002). Nitrogen in rock: occurrences and biogeochemical implications. Global Biogeochem. Cy. 16, 65–61. doi: 10.1029/2002GB001862
Hu, P., Tom, L., Singh, A., Thomas, B. C., Baker, B. J., Piceno, Y. M., et al. (2016). Genome-resolved metagenomic analysis reveals roles for candidate phyla and other microbial community members in biogeochemical transformations in oil reservoirs. mBio. 7, e1669–15. doi: 10.1128/mBio.01669-15
Huang, X., and Madan, A. (1999). CAP3: a DNA sequence assembly program. Genome Res. 9, 868–877. doi: 10.1101/gr.9.9.868
Jebbar, M., Franzetti, B., Girard, E., and Oger, P. (2015). Microbial diversity and adaptation to high hydrostatic pressure in deep-sea hydrothermal vents prokaryotes. Extremophiles 19, 721–740. doi: 10.1007/s00792-015-0760-3
Jost, A., Violette, S., Gonçalvès, J., Ledoux, E., Guyomard, Y., Guillocheau, F., et al. (2007). Long-term hydrodynamic response induced by past climatic and geomorphologic forcing: the case of the Paris basin. France. Phys. Chem. Earth 32, 368–378. doi: 10.1016/j.pce.2006.02.053
Kadnikov, V. V., Frank, Y. A., Mardanov, A. V., Beletsky, A. V., Ivasenko, D. A., and Pimenov, N. V. (2017). Variability of the composition of the microbial community of the deep subsurface thermal aquifer in western Siberia. Microbiology 86, 765–772. doi: 10.1134/S002626171706008X
Kadnikov, V. V., Mardanov, A. V., Beletsky, A. V., Banks, D., Pimenov, N. V., Frank, Y. A., et al. (2018). A metagenomic window into the 2-km-deep terrestrial subsurface aquifer revealed multiple pathways of organic matter decomposition. FEMS Microbiol. Ecol. 94:10 doi: 10.1093/femsec/fiy152
Kadnikov, V. V., Mardanov, A. V., Beletsky, A. V., Karnachuk, O. V., and Ravin, N. V. (2020). Microbial life in the deep subsurface aquifer illuminated by metagenomics. Front. Microbiol. 11:572252. doi: 10.3389/fmicb.2020.572252
Karnachuk, O. V., Frank, Y. A., Lukina, A. P., Kadnikov, V. V., Beletsky, A. V., Mardanov, A. V., et al. (2019). Domestication of previously uncultivated Candidatus Desulforudis audaxviator from a deep aquifer in Siberia sheds light on its physiology and evolution. ISME J. 13, 1947–1959. doi: 10.1038/s41396-019-0402-3
Kleinsteuber, S., Schleinitz, K. M., Breitfeld, J., Harms, H., Richnow, H. H., and Vogt, C. (2008). Molecular characterization of bacterial communities mineralizing benzene under sulfate-reducing conditions. FEMS Microbiol. Ecol. 66, 143–157. doi: 10.1111/j.1574-6941.2008.00536.x
Kleinsteuber, S., Schleinitz, K., and Vogt, C. (2012). Key players and team play: anaerobic microbial communities in hydrocarbon-contaminated aquifers. Appl. Microbiol. Biotechnol. 94, 851–873. doi: 10.1007/s00253-012-4025-0
Kliushnikova, T. M., Chernyshenko, D. V., and Kasatkina, T. P. (1992). Sul'fatvosstanavlivaiushchaia sposobnost’ bakteriĭ roda pseudomonas [The sulfate-reducing capacity of bacteria in the genus pseudomonas]. Mikrobiol. Zh. 54, 49–54.
Kopylova, E., Noé, L., and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217. doi: 10.1093/bioinformatics/bts611
Kriaa, K., Serin, J.-P., Contamine, F., Cézac, P., and Mercadier, J. (2009). 2-Butyne-1,4-diol hydrogenation in supercritical CO2: effect of hydrogen concentration. J. Supercrit. Fluids 49, 227–232. doi: 10.1016/j.supflu.2009.01.004
Kuever, J., Könneke, M., Galushko, A., and Drzyzga, O. (2001). Reclassification of Desulfobacterium phenolicum as Desulfobacula phenolica comb. nov. and description of strain SaxT as Desulfotignum balticum gen. Nov., sp. nov. Int. J. Syst. Evol. Microbiol. 51, 171–177. doi: 10.1099/00207713-51-1-171
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kunapuli, U., Lueders, T., and Meckenstock, R. U. (2007). The use of stable isotope probing to identify key iron-reducing microorganisms involved in anaerobic benzene degradation. ISME J. 1, 643–653. doi: 10.1038/ismej.2007.73
Leandro, T., Rodriguez, N., Rojas, P., Sanz, J. L., da Costa, M. S., and Amils, R. (2018). Study of methanogenic enrichment cultures of rock cores from the deep subsurface of the Iberian Pyritic Belt. Heliyon 16:e00605. doi: 10.1016/j.heliyon.2018.e00605
Lemieux, A., Shkarupin, A., and Sharp, K. (2020). Geologic feasibility of underground hydrogen storage in Canada. Int. J. Hydrog. Energy 45, 32243–32259. doi: 10.1016/j.ijhydene.2020.08.244
Liang, B., Wang, L. Y., Mbadinga, S. M., Liu, J. F., Yang, S. Z., Gu, J. D., et al. (2015). Anaerolineaceae and Methanosaeta turned to be the dominant microorganisms in alkanes-dependent methanogenic culture after long-term of incubation. AMB Express 5:117. doi: 10.1186/s13568-015-0117-4
Liang, B., Wang, L. Y., Zhou, Z., Mbadinga, S. M., Zhou, L., Liu, J. F., et al. (2016). High frequency of Thermodesulfovibrio spp. and Anaerolineaceae in association with Methanoculleus spp. in a long-term incubation of n-alkanes-degrading methanogenic enrichment culture. Front. Microbiol. 7:1431. doi: 10.3389/fmicb.2016.01431
Liermann, L., Barnes, A. S., Kalinowski, B. E., Zhou, X., and Brantley, S. (2000). Microenvironments of pH in biofilms grown on dissolving silicate surfaces. Chem. Geol. 171, 1–16. doi: 10.1016/S0009-2541(00)00202-3
Magnobosco, C., Lin, L.-H., Dong, H., Bomberg, M., Ghiorse, W., Stan-Lotter, H., et al. (2018). The biomass and biodiversity of the continental subsurface. Nat. Geosci. 11, 707–717. doi: 10.1038/s41561-018-0221-6
Meckenstock, R. U., Warthmann, R. I., and Schafer, W. (2004). Inhibition of anaerobic microbial o-xylene degradation by toluene in sulfidogenic sediment columns and pure cultures. FEMS Microbiol. Ecol. 47, 381–386. doi: 10.1016/S0168-6496(03)00303-9
Morasch, B., Schink, B., Tebbe, C. C., and Meckenstock, R. U. (2004). Degradation of o-xylene and m-xylene by a novel sulfate reducer belonging to the genus Desulfotomaculum. Arch. Microbiol. 181, 407–417. doi: 10.1007/s00203-004-0672-6
Mu, A., Boreham, C., Leong, H. X., Haese, R. R., and Moreau, J. W. (2014). Changes in the deep subsurface microbial biosphere resulting from a field-scale CO2 geosequestration experiment. Front. Microbiol. 5:209. doi: 10.3389/fmicb.2014.00209
Nunn, H. S. (2015). An Assessment of Microbial Communities and Their Potential Activities Associated with Oil Producing Environments. dissertation. Norman, OK: University of Oklahoma.
Nyyssönen, M., Hultman, J., Ahonen, L., Kukkonen, I., Paulin, L., Laine, P., et al. (2014). Taxonomically and functionally diverse microbial communities in deep crystalline rocks of the Fennoscandian shield. ISME J. 8, 126–138. doi: 10.1038/ismej.2013.125
Parisot, N., Denonfoux, J., Dugat-Bony, E., Peyret, P., and Peyretaillade, E. (2012). KASpOD - a web service for highly specific and explorative oligonucleotide design. Bioinformatics 28, 3161–3162. doi: 10.1093/bioinformatics/bts597
Parkes, R. J., Cragg, B. A., and Wellsbury, P. (2000). Recent studies on bacterial populations and processes in subseafloor sediments: a review. Hydrogeol. J. 8, 11–28. doi: 10.1007/PL00010971
Pedersen, K., Arlinger, J., Eriksson, S., Hallbeck, A., Hallbeck, L., and Johansson, J. (2008). Numbers, biomass and cultivable diversity of microbial populations relate to depth and borehole-specific conditions in groundwater from depths of 4-450 m in Olkiluoto, Finland. ISME J. 2, 760–775. doi: 10.1038/ismej.2008.43
Peng, Y., Leung, H. C. M., Yiu, S. M., and Chin, F. Y. L. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. doi: 10.1093/bioinformatics/bts174
Penner, T. J., Foght, J. M., and Budwill, K. (2010). Microbial diversity of western Canadian subsurface coal beds and methanogenic coal enrichment cultures. Int. J. Coal Geol. 82, 81–93. doi: 10.1016/j.coal.2010.02.002
Pfennig, N., Widdel, F., and Trüper, H. (1981). “The dissmilatory sulfate reducing bacteria,” in The Prokaryotes. eds. M. P. Starr, H. Stolp, H. G. Trüpper, A. Balows, and H. G. Schlegel (Berlin: Springer Verlag), 926–940.
Puente-Sánchez, F., Arce-Rodríguez, A., Oggerin, M., García-Villadangos, M., Moreno-Paz, M., Blanco, Y., et al. (2018). Viable cyanobacteria in the deep continental subsurface. Proc. Natl. Acad. Sci. U. S. A. 115, 10702–10707. doi: 10.1073/pnas.1808176115
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Rabus, R., and Widdel, F. (1995). Conversion studies with substrate analogues of toluene in a sulfate-reducing bacterium, strain Tol2. Arch. Microbiol. 164, 448–451. doi: 10.1007/BF02529744
Ranchou-Peyruse, M., Auguet, J. C., Mazière, C., Restrepo-Ortiz, C. X., Guignard, M., Dequidt, D., et al. (2019). Geological gas-storage shapes deep life. Environ. Microbiol. 21, 3953–3964. doi: 10.1111/1462-2920.14745
Ranchou-Peyruse, M., Gasc, C., Guignard, M., Aüllo, T., Dequidt, D., Peyret, P., et al. (2017). The sequence capture by hybridization: a new approach for revealing the potential of mono-aromatic hydrocarbons bioattenuation in a deep oligotrophic aquifer. Microb. Biotechnol. 10, 469–479. doi: 10.1111/1751-7915.12426
Ribière, C., Beugnot, R., Parisot, N., Gasc, C., Defois, C., Denonfoux, J., et al. (2016). Targeted gene capture by hybridization to illuminate ecosystem functioning. Microb. Environ. Gen. 1399, 167–182. doi: 10.1007/978-1-4939-3369-3_10
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Saitou, N., and Nei, M. (1987). The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Schreiber, K., Boes, N., Eschbach, M., Jaensch, L., Wehland, J., Bjarnsholt, T., et al. (2006). Anaerobic survival of Pseudomonas aeruginosa by pyruvate fermentation requires an Usp-type stress protein. J. Bacteriol. 188, 659–668. doi: 10.1128/JB.188.2.659-668.2006
Soares, A., Edwards, A., An, D., Bagnoud, A., Bomberg, M., Budwill, K., et al. (2019). A global perspective on microbial diversity in the terrestrial deep subsurface bioRxiv [Preprint]. doi: 10.1101/602672
Spring, S., Lapidus, A., Schröder, M., Gleim, D., Sims, D., Meincke, L., et al. (2009). Complete genome sequence of Desulfotomaculum acetoxidans type strain (5575). Stand. Genomic Sci. 1, 242–253. doi: 10.4056/sigs.39508
Stemple, B., Tinker, K., Sarkar, P., Miller, J., Gulliver, D., and Bibby, K. (2021). Biogeochemistry of the Antrim shale natural gas reservoir. ACS Earth Space Chem. 5, 1752–1761. doi: 10.1021/acsearthspacechem.1c00087
Stewart, R., and Peterson, W. (1914). The nitric nitrogen content in the country rock. Utah. Agric. Coll. Exp. Stn. Bull. 134, 421–465.
Takami, H., Noguchi, H., Takaki, Y., Uchiyama, I., Toyoda, A., Nishi, S., et al. (2012). A deeply branching thermophilic bacterium with an ancient acetyl-CoA pathway dominates a subsurface ecosystem. PLoS One 7:e30559. doi: 10.1371/journal.pone.0030559
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tan, B., Charchuk, R., Li, C., Nesbø, C., Abu Laban, N., and Foght, J. (2014). Draft genome sequence of uncultivated Firmicutes (Peptococcaceae SCADC) single cells sorted from methanogenic alkane-degrading cultures. Genome Announc. 2, e00909–e00914. doi: 10.1128/genomeA.00909-14
Tan, S., Liu, J., Fang, Y., Hedlund, B. P., Lian, Z. H., Huang, L. Y., et al. (2019). Insights into ecological role of a new deltaproteobacterial order Candidatus Acidulodesulfobacterales by metagenomics and metatranscriptomics. ISME J. 13, 2044–2057. doi: 10.1038/s41396-019-0415-y
Taubert, M., Vogt, C., Wubet, T., Kleinsteuber, S., Tarkka, M. T., Harms, H., et al. (2012). Protein-SIP enables time-resolved analysis of the carbon flux in a sulfate-reducing, benzene-degrading microbial consortium. ISME J. 6, 2291–2301. doi: 10.1038/ismej.2012.68
Tischer, K., Kleinsteuber, S., Schleinitz, K. M., Fetzer, I., Spott, O., Stange, F., et al. (2013). Microbial communities along biogeochemical gradients in a hydrocarbon-contaminated aquifer. Environ. Microbiol. 15, 2603–2615. doi: 10.1111/1462-2920.12168
Ulrich, A. C., and Edwards, E. A. (2003). Physiological and molecular characterization of anaerobic benzene-degrading mixed cultures. Environ. Microbiol. 5, 92–102. doi: 10.1046/j.1462-2920.2003.00390.x
Vander Wauven, C., Piérard, A., Kley-Raymann, M., and Haas, D. (1984). Pseudomonas aeruginosa mutants affected in anaerobic growth on arginine: evidence for a four-gene cluster encoding the arginine deiminase pathway. J. Bacteriol. 160, 928–934. doi: 10.1128/jb.160.3.928-934.1984
Varliero, G., Bienhold, C., Schmid, F., Boetius, A., and Molari, M. (2019). Microbial diversity and connectivity in deep-sea sediments of the South Atlantic polar front. Front. Microbiol. 10:665. doi: 10.3389/fmicb.2019.00665
Voordouw, G. (1995). The genus Desulfovibrio: the centennial. Appl. Environ. Microbiol. 61, 2813–2819. doi: 10.1128/aem.61.8.2813-2819.1995
Watanabe, M., Fukui, M., Galushko, A., and Kuever, J. (2020). “Desulfosarcina,” in Bergey’s Manual of Systematics of Archaea and Bacteria. ed. W. B. Whitman, F. A. Rainey, P. Kämpfer, M. E. Trujillo, P. DeVos, and B. Hedlund, et al. (John Wiley & Sons). doi: 10.1002/9781118960608.gbm01020.pub2
Watanabe, M., Higashioka, Y., Kojima, H., and Fukui, M. (2017). Desulfosarcina widdelii sp. nov. and Desulfosarcina alkanivorans sp. nov., hydrocarbon-degrading sulfate-reducing bacteria isolated from marine sediment and emended description of the genus Desulfosarcina. Int. J. Syst. Evol. Microbiol. 67, 2994–2997. doi: 10.1099/ijsem.0.002062
Watanabe, M., Kojima, H., and Fukui, M. (2013). Desulfotomaculum intricatum sp. nov., a sulfate reducer isolated from freshwater lake sediment. Int. J. Syst. Evol. Microbiol. 63, 3574–3578. doi: 10.1099/ijs.0.051854-0
Watanabe, M., Kojima, H., and Fukui, M. (2018). Review of Desulfotomaculum species and proposal of the genera Desulfallas gen. Nov., Desulfofundulus gen. Nov., Desulfofarcimen gen. Nov. and Desulfohalotomaculum gen. Nov. Int. J. Syst. Evol. Microbiol. 68, 2891–2899. doi: 10.1099/ijsem.0.002915
Wöhlbrand, L., Jacob, J. H., Kube, M., Mussmann, M., Jarling, R., Beck, A., et al. (2012). Complete genome, catabolic sub-proteomes and key-metabolites of Desulfobacula toluolica Tol2, a marine, aromatic compound-degrading, sulfate-reducing bacterium. Environ. Microbiol. 15, 1334–1355. doi: 10.1111/j.1462-2920.2012.02885.x
Wu, X., Holmfeldt, K., Hubalek, V., Lundin, D., Åström, M., Bertilsson, S., et al. (2016). Microbial metagenomes from three aquifers in the Fennoscandian shield terrestrial deep biosphere reveal metabolic partitioning among populations. ISME J. 10, 1192–1203. doi: 10.1038/ismej.2015.185
Wu, X., Pedersen, K., Edlund, J., Eriksson, L., Åström, M., Andersson, A. F., et al. (2017). Potential for hydrogen-oxidizing chemolithoautotrophic and diazotrophic populations to initiate biofilm formation in oligotrophic, deep terrestrial subsurface waters. Microbiome 5:37. doi: 10.1186/s40168-017-0253-y
Xia, Y., Wang, Y., Wang, Y., Chin, F. Y., and Zhang, T. (2016). Cellular adhesiveness and cellulolytic capacity in Anaerolineae revealed by omics-based genome interpretation. Biotechnol. Biofuels 9:111. doi: 10.1186/s13068-016-0524-z
Youssef, N. H., Farag, I. F., Rudy, S., Mulliner, A., Walker, K., Caldwell, F., et al. (2019). The wood-Ljungdahl pathway as a key component of metabolic versatility in candidate phylum Bipolaricaulota (Acetothermia, OP1). Environ. Microbiol. Rep. 11, 538–547. doi: 10.1111/1758-2229.12753
Zaitseva, S. V., Lavrentieva, E. V., Radnagurueva, A. A., Baturina, O. A., Kabilov, M. R., and Barkhutova, D. D. (2017). Distribution of acetothermia-dominated microbial communities in alkaline hot springs of baikal rift zone. PeerJ 5:e3492v1. doi: 10.7287/peerj.preprints.3492v1
Zhang, R., Hedrich, S., Ostertag-Henning, C., and Schippers, A. (2018). Effect of elevated pressure on ferric iron reduction coupled to sulfur oxidation by biomining microorganisms. Hydrometallurgy 178, 215–223. doi: 10.1016/j.hydromet.2018.05.003
Zhang, Y., Li, X., Bartlett, D. H., and Xiao, X. (2015). Current developments in marine microbiology: high-pressure biotechnology and the genetic engineering of piezophiles. Curr. Opin. Biotechnol. 33, 157–164. doi: 10.1016/j.copbio.2015.02.013
Keywords: deep aquifer, BTEX, biodegradation, deep subsurface biosphere, high pressure, benzyl-succinate synthase, gas storage
Citation: Ranchou-Peyruse M, Guignard M, Casteran F, Abadie M, Defois C, Peyret P, Dequidt D, Caumette G, Chiquet P, Cézac P and Ranchou-Peyruse A (2021) Microbial Diversity Under the Influence of Natural Gas Storage in a Deep Aquifer. Front. Microbiol. 12:688929. doi: 10.3389/fmicb.2021.688929
Edited by:
Axel Schippers, Federal Institute for Geosciences and Natural Resources, GermanyReviewed by:
Daniel Lipus, Helmholtz Centre Potsdam, GermanyBert Engelen, University of Oldenburg, Germany
Copyright © 2021 Ranchou-Peyruse, Guignard, Casteran, Abadie, Defois, Peyret, Dequidt, Caumette, Chiquet, Cézac and Ranchou-Peyruse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony Ranchou-Peyruse, anthony.ranchou-peyruse@univ-pau.fr