
- 1 Núcleo do Parque Tecnológico de São José dos Campos, Universidade Camilo Castelo Branco, São Paulo, Brazil
- 2 Gr. T. Popa Biomedical Research Center, University of Medicine and Pharmacy, Iasi, Romania
- 3 Max-Delbrück Center for Molecular Medicine, Berlin, Germany
Brain renin–angiotensin system (RAS) is significantly involved in the roles of the endocrine RAS in cardiovascular regulation. Our studies indicate that the brain RAS participates in the development of cardiac hypertrophy and fibrosis through sympathetic activation. Inhibition of sympathetic hyperactivity after myocardial infarction through suppression of the brain RAS appears beneficial. Furthermore, the brain RAS modulates the cardiovascular and fluid–electrolyte homeostasis not only by interacting with the autonomic nervous system but also by modulating hypothalamic–pituitary axis and vasopressin release. The brain RAS is also involved in the modulation of circadian rhythms of arterial pressure, contributing to non-dipping hypertension. We conclude that the brain RAS in pathophysiological states interacts synergistically with the chronically overactive RAS through a positive biofeedback in order to maintain a state of alert in diseased conditions, such as cardiac hypertrophy and failure. Therefore, targeting brain RAS with drugs such as renin or angiotensin converting enzyme inhibitors or receptor blockers having increased brain penetrability could be of advantage.
According to the WHO report Global atlas on cardiovascular disease prevention and control, cardiovascular diseases are the leading causes of death and disability in the world. Heart failure is a very common condition that is costly, disabling, and potentially deadly. In developed countries, around 2% of adults suffer from heart failure, but in those over the age of 65, this increases to 6–10% (Dickstein et al., 2010). Conditions that damage or overwork the heart muscle can cause heart failure. The most common causes of heart failure are coronary heart disease (CHD), high blood pressure, obesity, and diabetes (He et al., 2001). Treating these problems can prevent or improve heart failure. Drugs acting on the renin–angiotensin system (RAS), such as angiotensin converting enzyme (ACE) inhibitors or angiotensin receptor blockers are first-line therapy for all heart failure patients. Since the RAS has both endocrine and local tissue components, RAS drugs have been developed to attain increased tissue penetrability and volume of distribution and consequently an efficient inhibition/blockage of both RAS components. Of the tissue systems, the brain RAS is of particular interest for us. Accumulating evidence indicates that angiotensins produced locally in various brain nuclei involved in homeostasis control mainly in the hypothalamus and brain stem interact with several neurotransmitter systems to regulate cardiovascular and fluid–electrolyte homeostasis, their biology and mechanisms of action representing an active area of actual research interests (Baltatu et al., 2011; Diz et al., 2011). The brain RAS is physically separated from the endocrine one by the presence of the blood–brain barrier which hampers the penetration of angiotensin II (Ang II) from blood into the brain. Exceptions are some areas lacking the blood–brain barrier through which circulating Ang II can transmit its effects inside the brain. The brain RAS is involved in the modulation of cardiovascular and fluid–electrolyte homeostasis, complementing the classical roles of the endocrine RAS. Several lines of evidence demonstrate that chronic over activation of the brain RAS is responsible for the development and maintenance of hypertension in several animal models of disease (Baltatu et al., 2011; Diz et al., 2011).
To elucidate a contributory role of the brain RAS and its significance in diverse pathophysiological processes we developed a transgenic rat [TGR(ASrAOGEN)] to inhibit the production of angiotensinogen (AOGEN) specifically in the brain (Schinke et al., 1999). The brain levels of AOGEN in TGR(ASrAOGEN) rats are 90% low due to an antisense RNA expressed against AOGEN, induced by means of the astrocyte-specific glial fibrillary acidic protein promoter. As a consequence, these rats have low blood pressure and a diabetes insipidus-like syndrome with altered central vasopressinergic system (Campos et al., 2004).
The TGR(ASrAOGEN) rats were investigated in experimental conditions of hypertension and heart pathology. We demonstrated that the brain RAS significantly contributes to the development of hypertension in a transgenic model with overactive tissue RAS by crossbreeding the TGR(ASrAOGEN) rats with the hypertensive TGR(mREN2)27 strain (Schinke et al., 1999). We were further interested to investigate whether the brain RAS is participating in the development of hypertensive pathology in experimental reno-vascular hypertension. Ang II when infused at doses of 50–250 ng/kg/min subcutaneously, which do not produce direct vasoconstriction are described as “subpressor” or “slow-pressor” and can induce a gradual increase of blood pressure in days to weeks. It represents a model of reno-vascular hypertension with low plasma renin activity (PRA). The subpressor Ang II-induced hypertension at TGR(ASrAOGEN) rats was significantly attenuated supporting the importance of the brain RAS in this process (Baltatu et al., 2000). Furthermore, the Ang II-induced cardiac hypertrophy and fibrosis was considerably attenuated as well. This attenuation of the cardiac phenotype in this model of experimental hypertension might not only be due to the reduction of blood pressure but also because of a disturbed autonomic nervous system.
Several authors presented findings indicating that Ang II interacts with the autonomic nervous system at several levels, namely at postganglionic nerve terminals, sympathetic ganglia, and within the central nervous system (reviewed in Bader et al., 2001). Ang II signaling in the brain modulates reflex control of heart rate and sympathetic outflow (Dampney et al., 2007; Carlson and Wyss, 2008). Studies from our group provide evidence that a permanent inhibition of brain RAS may cause a decreased basal sympathetic outflow, as it has been observed in TGR(ASrAOGEN) rats (Campos et al., 2006a; Parrish et al., 2008). Power spectral analysis of heart rate and blood pressure indicates an altered autonomic nervous system activity with a dominance of vagal tone over sympathetic tone and exaggerated spontaneous baroreflex sensitivity (Baltatu et al., 2001). As a consequence to a diminished basal activity of sympathetic nervous system, the hearts of TGR(ASrAOGEN) exhibit an increased sensitivity to β-receptor agonists (Campos et al., 2006a; Parrish et al., 2008).
The brain RAS – sympathetic nervous system connection is important in the development of left ventricular dysfunction after myocardial infarction (Huang and Leenen, 2009). Sympathetic activity surges in parallel with the progress of damage in cardiac performance, contributing to the progression of the heart failure (Packer, 1992). TGR(ASrAOGEN) rats exhibit marked attenuation of both sympathetic hyperactivity and of left ventricular remodeling and dysfunction after myocardial infarction (Wang et al., 2004). As a consequence of a lower post-infarct sympathetic activation in the TGR(ASrAOGEN) rats, there was a blunted increase in tyrosine hydroxylase (TH; the rate-limiting enzyme in norepinephrine synthesis; Parrish et al., 2008). Lower cardiac TH levels may represent a mechanism contributing to the lower pathological elevation of norepinephrine.
Not only does peripheral Ang II interact with the brain RAS to activate the sympathetic nervous system in chronically overactive conditions, but also the brain RAS appear to influence renal renin release. The sympathetic nervous system, which is involved in the fight-or-flight reaction, is one of the main stimulators of renal renin release. During the stress reaction, there is an increase in blood levels of renin in addition to epinephrine. We provided evidence that brain RAS is an important player involved in the stress-induced renin release. The TGR(ASrAOGEN) rats with low brain AOGEN exposed to stress have elevations of only 50% on PRA levels (marker of both renal renin release and of the reactivity to different types of stressors) from those of the stressed control rats (Baltatu et al., 2004). Therefore, inhibition of the brain RAS may represent a protective strategy against chronic and generalized activation of both endocrine and local systems in disease.
Brain RAS modulates the cardiovascular and fluid–electrolyte homeostasis not only by interacting with the autonomic nervous system but also by modulating hypothalamic–pituitary axis and vasopressin release (Baltatu et al., 2004). Since the angiotensinogen-deficient TGR(ASrAOGEN) rats have lower plasma levels of vasopressin and an altered central vasopressinergic system (Schinke et al., 1999; Campos et al., 2004), we considered that this might be also one mechanism contributing to the attenuated Ang II-induced hypertension and cardiac hypertrophy. Therefore, we studied the Ang II-induced hypertension in vasopressin-deficient Brattleboro rats, as well. Basal systolic arterial pressure (SAP) monitored by telemetry was significantly lower in Brattleboro rats as in TGR(ASrAOGEN) in comparison with the respective parent strain Long–Evans (LE) rats (119.4 ± 1.1 vs. 129.4 ± 1.4 mmHg, p < 0.001, respectively) or Sprague-Dawley (SD; 122.5 ± 1.5 vs. 128.9 ± 1.9 mmHg, p < 0.05, respectively). The increase in SAP induced by 7 days of chronic Ang II infusion (100 ng/kg/min) in Brattleboro rats was similar to the LE rats (Figure 1; delta SAP: 25.1 ± 4.8 vs. 18.2 ± 7.7 mmHg, p > 0.05, respectively), in contrast to the significantly attenuated hypertension in TGR(ASrAOGEN; delta SAP: 29.8 ± 4.2 vs. 46.3 ± 2.5 mmHg, p < 0.005 vs. control SD rats). Heart hypertrophy measured as heart weight per body weight was evident neither in LE nor in Brattleboro rats, probably due to the mild hypertension levels. These results together with our previous results (Baltatu et al., 2000) indicate that the brain RAS but not vasopressin system plays an important role in mediating the hypertension induced by slow-pressor doses of Ang II.
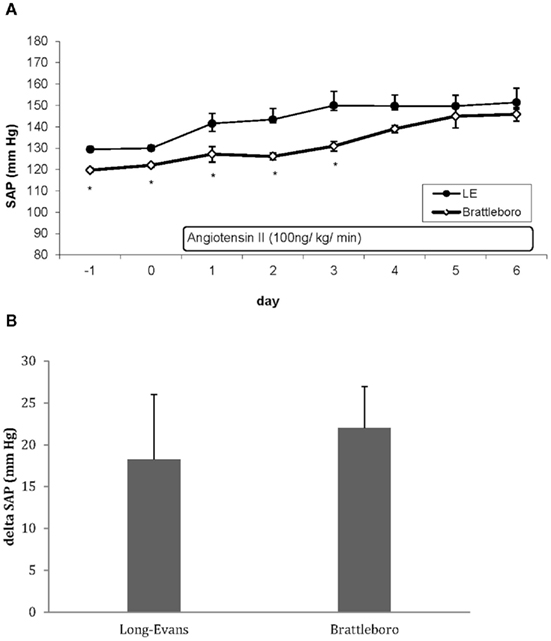
Figure 1. (A) Effect of Ang II infusion (100 ng/kg/min s.c.) on systolic arterial pressure (SAP). Data are extracted from telemetry recording and data acquisition as day mean. The start of Ang II infusion is at the end of day 0. LE indicates Long–Evans rats (n = 6); Brattleboro rats (n = 6). (B). Increase of systolic BP after Ang II infusion calculated by subtracting the 2-day mean basal values from the mean values of the last 2 days at the end of Ang II infusion [mean day (5, 6) – mean day (−1, 0)] (Long–Evans rats, n = 6; Brattleboro rats, n = 6). Data are expressed as mean ± SE.
Circadian variability of blood pressure may constitute another mechanism through which brain RAS may participate to the hypertensive target organ injury and cardiovascular events. According to the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC7), ambulatory blood pressure monitoring is warranted for evaluation of “white-coat” hypertension in the absence of target organ injury (Chobanian et al., 2003). It is also helpful to assess patients with apparent drug resistance, hypotensive symptoms with antihypertensive medications, episodic hypertension, and autonomic dysfunction. In most individuals, BP decreases by 10–20% during the night; those in whom such reductions are not present are at higher risk for cardiovascular events. A “non-dipping” blood pressure (reversed nighttime BP dipping pattern) was associated with a 2.2-fold higher incidence of heart failure (Ingelsson et al., 2006). Our group was the first to determine that slow-pressor Ang II infusion may induce a shift in BP circadian rhythm, making it a convenient and reproducible experimental model of Ang II-induced non-dipping hypertension (Baltatu et al., 2001). These alterations in BP circadian rhythm are not synchronized with alterations of heart rate or locomotor activity, contributing to the concept that the circadian variability of blood pressure and heart rate are differentially regulated (Baltatu et al., 2001; Campos et al., 2006b). The brain RAS appears to be also an important modulator of the circadian BP rhythm (Baltatu et al., 2001; Campos et al., 2006b). Ang II not only induces alterations in the circadian rhythm of BP but also its local production in the brain modulates the synthesis of both melatonin and vasopressin, which are important hormones regulating circadian rhythms (Baltatu et al., 2002; Campos et al., 2002, 2004). Furthermore, melatonin itself may alter the short-term variability of cardiovascular parameters, including the baroreflex (Campos-Santos et al., 2000). With these studies we demonstrated that the brain RAS is an important regulator of circadian rhythm of blood pressure. The experimental model of Ang II-induced hypertension might be useful to further dissect the pathophysiology and molecular biology of non-dipping hypertension.
In summary, several lines of evidence indicate that the brain RAS is involved in cardiovascular diseases including heart failure contributing to pathophysiological alterations (Figure 2). Chronically overactive RAS is thus recruiting tissue RASs through a positive biofeedback in order to maintain a state of alert diseased conditions. A circadian pattern becomes quite obvious in the occurrence of acute cardiovascular diseases, such as ischemia, infarction, stroke, and sudden death, and new chronotherapeutic approaches in antihypertensive therapy are trying to exploit the knowledge of circadian rhythms in order to reduce these events. Therefore, targeting brain RAS with drugs such as renin or ACE inhibitors or receptor blockers having increased brain penetrability could be of benefit. These RAS-targeting drugs are first-line therapy for all heart failure patients. Since the RAS has both endocrine and local tissue components, RAS drugs are being developed to attain increased tissue penetrability and volume of distribution and consequently an efficient inhibition of both RAS components.
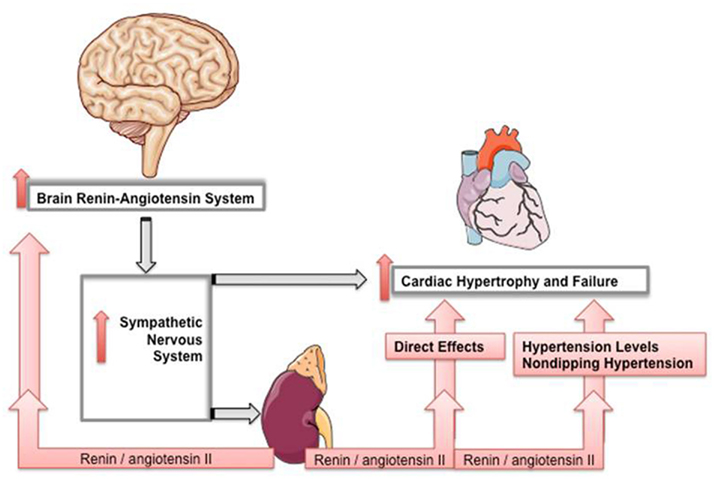
Figure 2. Brain RAS in pathophysiological states interacts synergistically with the chronically overactive RAS through a positive biofeedback in order to maintain a state of alert diseased conditions, such as cardiac hypertrophy and failure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work of the authors was supported by the Deutsche Forschun gsgemeinschaft (BA1374/13-2, BA1374/16-1, and BA1374/20-1).
References
Bader, M., Peters, J., Baltatu, O., Muller, D. N., Luft, F. C., and Ganten, D. (2001). Tissue renin-angiotensin systems: new insights from experimental animal models in hypertension research. J. Mol. Med. 79, 76–102.
Baltatu, O., Afeche, S. C., Jose dos Santos, S. H., Campos, L. A., Barbosa, R., Michelini, L. C., Bader, M., and Cipolla-Neto, J. (2002). Locally synthesized angiotensin modulates pineal melatonin generation. J. Neurochem. 80, 328–334.
Baltatu, O., Campos, L. A., and Bader, M. (2004). Genetic targeting of the brain renin-angiotensin system in transgenic rats: impact on stress-induced renin release. Acta Physiol. Scand. 181, 579–584.
Baltatu, O., Janssen, B. J., Bricca, G., Plehm, R., Monti, J., Ganten, D., and Bader, M. (2001). Alterations in blood pressure and heart rate variability in transgenic rats with low brain angiotensinogen. Hypertension 37, 408–413.
Baltatu, O., Silva, J. A. Jr., Ganten, D., and Bader, M. (2000). The brain renin-angiotensin system modulates angiotensin II-induced hypertension and cardiac hypertrophy. Hypertension 35, 409–412.
Baltatu, O. C., Campos, L. A., and Bader, M. (2011). Local renin-angiotensin system and the brain – a continuous quest for knowledge. Peptides 32, 1083–1086.
Campos, L. A., Afeche, S. C., Plehm, R., Michelini, L. C., Cipolla-Neto, J., Baltatu, O., and Bader, M. (2002). Altered circadian rhythm reentrainment and pineal indoles in rats with low brain angiotensinogen. J. Hypertens. 20, P1086.
Campos, L. A., Couto, A. S., Iliescu, R., Santos, J. A., Santos, R. A., Santos, R. A., Ganten, D., Campagnole-Santos, M. J., Bader, M., and Baltatu, O. (2004). Differential regulation of central vasopressin receptors in transgenic rats with low brain angiotensinogen. Regul. Pept. 119, 177–182.
Campos, L. A., Iliescu, R., Fontes, M. A., Schlegel, W. P., Bader, M., and Baltatu, O. C. (2006a). Enhanced isoproterenol-induced cardiac hypertrophy in transgenic rats with low brain angiotensinogen. Am. J. Physiol. Heart Circ. Physiol. 291, H2371–H2376.
Campos, L. A., Plehm, R., Cipolla-Neto, J., Bader, M., and Baltatu, O. C. (2006b). Altered circadian rhythm reentrainment to light phase shifts in rats with low levels of brain angiotensinogen. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R1122–R1127.
Campos-Santos, L., Cipolla-Neto, J., and Michelini, L. (2000). Beneficial effect of melatonin on baroreceptor reflex control of heart rate is blocked in spontaneously hypertensive rats. J. Hypertens. 18, S95–S95.
Carlson, S. H., and Wyss, J. M. (2008). Neurohormonal regulation of the sympathetic nervous system: new insights into central mechanisms of action. Curr. Hypertens. Rep. 10, 233–240.
Chobanian, A. V., Bakris, G. L., Black, H. R., Cushman, W. C., Green, L. A., Izzo, J. L. Jr., Jones, D. W., Materson, B. J., Oparil, S., Wright, J. T. Jr., Roccella, E. J., Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure, and National Heart, Lung, and Blood Institute, National High Blood Pressure Education Program Coordinating Committee. (2003). Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 42, 1206–1252.
Dampney, R. A., Tan, P. S., Sheriff, M. J., Fontes, M. A., and Horiuchi, J. (2007). Cardiovascular effects of angiotensin II in the rostral ventrolateral medulla: the push-pull hypothesis. Curr. Hypertens. Rep. 9, 222–227.
Dickstein, K., Vardas, P. E., Auricchio, A., Daubert, J. C., Linde, C., McMurray, J., Ponikowski, P., Priori, S. G., Sutton, R., van Veldhuisen, D. J., ESC Committee for Practice Guidelines (CPG), Vahanian, A., Auricchio, A., Bax, J., Ceconi, C., Dean, V., Filippatos, G., Funck-Brentano, C., Hobbs, R., Kearney, P., McDonagh, T., Popescu, B. A., Reiner, Z., Sechtem, U., Sirnes, P. A., Tendera, M., Vardas, P., Widimsky, P., Document Reviewers, Tendera, M., Anker, S. D., Blanc, J. J., Gasparini, M., Hoes, A. W., Israel, C. W., Kalarus, Z., Merkely, B., Swedberg, K., and Camm, A. J. (2010). 2010 Focused Update of ESC Guidelines on device therapy in heart failure: an update of the 2008 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure and the 2007 ESC guidelines for cardiac and resynchronization therapy. Developed with the special contribution of the Heart Failure Association and the European Heart Rhythm Association. Eur. Heart J. 31, 2677–2687.
Diz, D. I., Arnold, A. C., Nautiyal, M., Isa, K., Shaltout, H. A., and Tallant, E. A. (2011). Angiotensin peptides and central autonomic regulation. Curr. Opin. Pharmacol. 11, 131–137.
He, J., Ogden, L. G., Bazzano, L. A., Vupputuri, S., Loria, C., and Whelton, P. K. (2001). Risk factors for congestive heart failure in US men and women: NHANES I epidemiologic follow-up study. Arch. Intern. Med. 161, 996–1002.
Huang, B. S., and Leenen, F. H. (2009). The brain renin-angiotensin-aldosterone system: a major mechanism for sympathetic hyperactivity and left ventricular remodeling and dysfunction after myocardial infarction. Curr. Heart Fail Rep. 6, 81–88.
Ingelsson, E., Bjorklund-Bodegard, K., Lind, L., Arnlov, J., and Sundstrom, J. (2006). Diurnal blood pressure pattern and risk of congestive heart failure. JAMA 295, 2859–2866.
Packer, M. (1992). The neurohormonal hypothesis: a theory to explain the mechanism of disease progression in heart failure. J. Am. Coll. Cardiol. 20, 248–254.
Parrish, D. C., Gritman, K., Van Winkle, D. M., Woodward, W. R., Bader, M., and Habecker, B. A. (2008). Postinfarct sympathetic hyperactivity differentially stimulates expression of tyrosine hydroxylase and norepinephrine transporter. Am. J. Physiol. Heart Circ. Physiol. 294, H99–H106.
Schinke, M., Baltatu, O., Bohm, M., Peters, J., Rascher, W., Bricca, G., Lippoldt, A., Ganten, D., and Bader, M. (1999). Blood pressure reduction and diabetes insipidus in transgenic rats deficient in brain angiotensinogen. Proc. Natl. Acad. Sci. U.S.A. 96, 3975–3980.
Keywords: angiotensin, brain, sympathetic nervous system, heart failure, hypertrophy, fibrosis
Citation: Campos LA, Bader M and Baltatu OC (2012) Brain renin–angiotensin system in hypertension, cardiac hypertrophy, and heart failure. Front. Physio. 2:115. doi: 10.3389/fphys.2011.00115
Received: 06 October 2011; Accepted: 14 December 2011;
Published online: 03 January 2012.
Edited by:
Jacqueline Kathleen Phillips, Macquarie University, AustraliaReviewed by:
Robert A. Augustyniak, Oakland University William Beaumont School of Medicine, USAMichael McKinley, Florey Neuroscience Institutes, Australia
Copyright: © 2012 Campos, Bader and Baltatu. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Michael Bader, Max-Delbrück Center for Molecular Medicine, Robert-Rössle Str. 10, 13125 Berlin, Germany. e-mail: mbader@mdc-berlin.de