 Kathy L. Ryan1* Caroline A. Rickards2 Carmen Hinojosa-Laborde1 William H. Cooke2 Victor A. Convertino1
Kathy L. Ryan1* Caroline A. Rickards2 Carmen Hinojosa-Laborde1 William H. Cooke2 Victor A. Convertino1
- 1 U.S. Army Institute of Surgical Research, Fort Sam Houston, TX, USA11
- 2 Department of Health and Kinesiology, The University of Texas at San Antonio, San Antonio, TX, USA
Hemorrhage remains a major cause of mortality following traumatic injury in both military and civilian settings. Lower body negative pressure (LBNP) has been used as an experimental model to study the compensatory phase of hemorrhage in conscious humans, as it elicits central hypovolemia like that induced by hemorrhage. One physiological compensatory mechanism that changes during the course of central hypovolemia induced by both LBNP and hemorrhage is a baroreflex-mediated increase in muscle sympathetic nerve activity (MSNA), as assessed with microneurography. The purpose of this review is to describe recent results obtained using microneurography in our laboratory as well as those of others that have revealed new insights into mechanisms underlying compensatory increases in MSNA during progressive reductions in central blood volume and how MSNA is altered at the point of hemodynamic decompensation. We will also review recent work that has compared direct MSNA recordings with non-invasive surrogates of MSNA to determine the appropriateness of using such surrogates in assessing the clinical status of hemorrhaging patients.
Hemorrhage remains a major cause of mortality following traumatic injury in both military and civilian settings. Although many combat-related deaths are not survivable due to the severity of injury, approximately 51% of casualties who died of wounds (i.e., expired after reaching a hospital) in Operations Iraqi Freedom (OIF) and Enduring Freedom (OEF) have been classified as potentially survivable; of these, 80% of the deaths were due to the inability to control bleeding and effectively resuscitate hemorrhaging patients (Eastridge et al., 2011). In the civilian setting, estimates of mortality due to hemorrhage following trauma range from 21 to 39% of all trauma deaths, with exsanguination being the most common cause of death among those found dead upon arrival of emergency medical services (EMS) personnel (Sauaia et al., 1995; Stewart et al., 2003). Consistently, hemorrhage is the second leading cause of death after trauma, with only central nervous system (CNS) injury accounting for more mortality (Kauvar et al., 2006). Severe CNS injury, however, is often irreparable, while hemorrhage is more amenable to development of advanced interventions to reduce morbidity and mortality (Kauvar et al., 2006). Hence, research efforts have been concentrated on improving techniques used to diagnose the severity of blood loss, control bleeding, and return volume to the hemorrhaging patient.
To our knowledge, the first direct measurement of sympathetic nerve activity during hemorrhage was performed in chloralose-anesthetized cats by Gernandt et al. (1946). From this direct measurement as well as indirect measurements, primarily made in anesthetized animal models, it was clear by 1967 that the sympathetic nervous system was activated during the early phases of hemorrhage as a compensatory response to maintain arterial blood pressure (Chien, 1967). Subsequently, sympathoexcitation during the early stages of hemorrhage was demonstrated via direct measurement of activity in nerves supplying renal, hepatic, adrenal, splenic, and cardiac vascular beds in a variety of animal species (Ninomiya et al., 1971; Skoog et al., 1985; Koyama et al., 1988, 1992; Malpas et al., 1998). Schadt and Ludbrook (1991) later proposed that the physiological response to hemorrhage occurs in two phases. Phase I, evident in animals until 25–35% of blood volume is lost, consists of sympathoexcitation, which contributes to the maintenance of baseline levels of blood pressure. Phase II then occurs when compensatory mechanisms are exhausted, sympathoinhibition occurs, and blood pressure decreases precipitously (Schadt and Ludbrook, 1991).
Lower body negative pressure (LBNP) was introduced in the 1960s as an experimental perturbation to study the physiological responses produced by central hypovolemia in healthy humans; from the earliest studies, LBNP was seen as a model to study the acute responses to hemorrhage (Wolthuis et al., 1974). In 2001, the notion of using LBNP to develop clinical assessment tools for determination of the severity of hemorrhage and accurate definition of resuscitative strategies was advanced (Convertino, 2001). In 2004, we extended this concept by summarizing literature that suggested that LBNP could be used to investigate both the compensatory phase of hemorrhage and subsequent decompensation (defined herein as the loss of compensatory physiological responses that maintain blood pressure) in conscious humans (Cooke et al., 2004). Application of negative pressure to the lower body redistributes blood from the central thoracic and splanchnic circulations into the legs, thereby reducing venous return and cardiac filling, producing central hypovolemia without actual blood loss. Indeed, the regional vascular changes elicited by LBNP have been demonstrated to be similar to those induced by hemorrhage (Taneja et al., 2007). Furthermore, LBNP produces peripheral tissue dysoxia that is not sustained long enough to induce alterations in systemic lactate, pH, and base excess (Ward et al., 2010). While LBNP is a useful model to study the compensatory responses to loss of central blood volume and the subsequent loss of these responses, LBNP does not model prolonged hemorrhage eventuating in circulatory shock with accompanying metabolic acidosis. Additionally, physiological responses induced by LBNP are not accompanied by tissue trauma or pain as in hemorrhage induced by traumatic injury. Table 1 shows ranges of effective central blood loss (or fluid redistribution) produced by different levels of LBNP; we initially proposed these ranges of central hypovolemia based on comparison of cardiovascular responses to LBNP and hemorrhage (Cooke et al., 2004), but these equivalencies have more recently been confirmed via computer modeling (Summers et al., 2009).
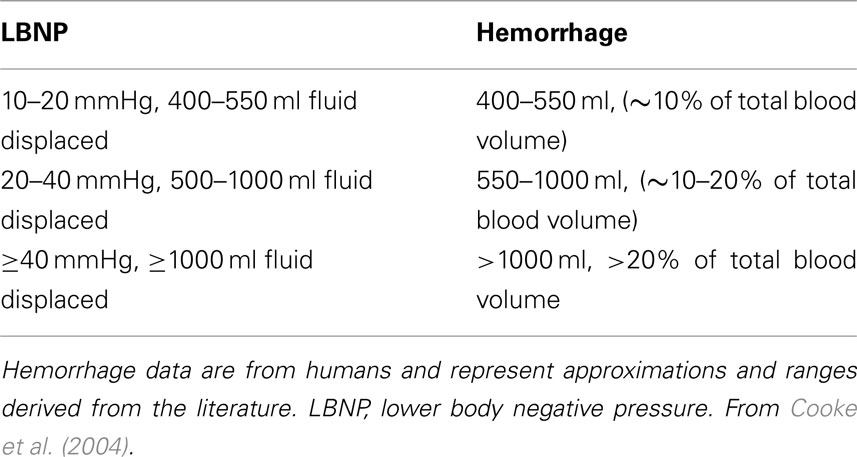
Table 1. Ranges of fluid displacement induced by lower body negative pressure and equivalent amounts of blood loss during hemorrhage.
Since 2004, we have used LBNP both to increase our understanding of physiological responses to hemorrhage and to develop new means of assessing the physiological status of bleeding patients (Convertino et al., 2008; Ward et al., 2010). Just as in animal models, one physiological compensatory mechanism that changes early in humans during the course of both LBNP and hemorrhage is a baroreflex-mediated increase in muscle sympathetic nerve activity (MSNA), usually accessed via placement of an electrode in the peroneal nerve (Rea et al., 1991). The purpose of this review is to describe recent results obtained using microneurography that have revealed new insights into how compensatory increases in MSNA occur during central hypovolemia, how MSNA is altered at the point of hemodynamic decompensation, and whether non-invasive surrogates of MSNA are appropriate for use in assessing the physiological status of hemorrhaging patients.
MSNA during the Compensatory Phase of Central Hypovolemia
At low levels of central hypovolemia elicited by mild LBNP in humans, MSNA increases in the absence of alterations in heart rate (HR) or blood pressure (Sundlof and Wallin, 1978a; Victor and Leimbach, 1987). This initial increase in MSNA occurs in a similar fashion whether measured from the radial nerve in the arm or the peroneal nerve in the leg, and is therefore a reflection of generalized sympathetic activation to muscle vascular beds (Rea and Wallin, 1989). Importantly, the response in MSNA to relatively low levels of LBNP (i.e., −10 mmHg) is identical to the MSNA response to blood loss of 450 ml, demonstrating the effectiveness of LBNP as an experimental model of hemorrhage (Figure 1; Rea et al., 1991).
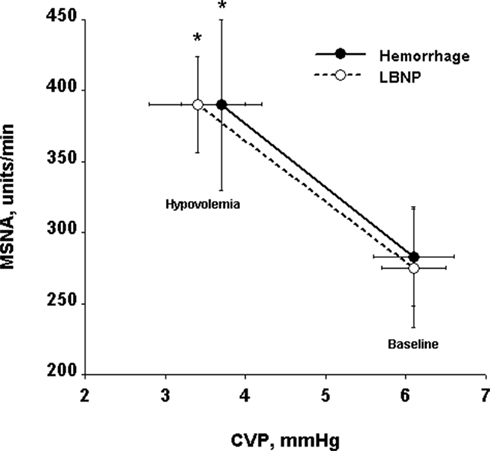
Figure 1. Comparison of relationships between central venous pressure (CVP) and muscle sympathetic nerve activity (MSNA) during −10 mmHg LBNP and 450 ml hemorrhage in nine human subjects. Circles and lines represent mean ± SE values. *P < 0.05 compared with baseline. From Cooke et al. (2004) using data modified from Rea et al. (1991).
Because arterial pressure was unchanged, the increase in MSNA elicited by low levels of LBNP or hemorrhage was originally ascribed to deactivation of cardiopulmonary baroreceptors alone, without contribution from arterial baroreceptors (Rea et al., 1991). In fact, so many investigators have used low levels of LBNP (≤20 mmHg) to “selectively” unload cardiopulmonary baroreceptors that this has recently been referred to as “dogma” (Fu et al., 2009). Subsequent work, however, has conclusively demonstrated that deactivation of arterial baroreceptors also contributes to sympathoexcitation induced by non-hypotensive hypovolemia. First, non-hypotensive hypovolemia reduces the diameter of both the ascending thoracic aorta (Taylor et al., 1995) and the carotid artery (Lacolley et al., 1992), sites of the stretch-sensitive aortic and carotid arterial baroreceptors. Second, Floras et al. (2001) demonstrated that, during LBNP of only −5 mmHg, MSNA increased while parasympathetic modulation of HR (assessed via power spectral analysis) decreased, which is a manifestation of arterial baroreceptor unloading. Third, MSNA increases during mild hypovolemia in both intact control subjects and in cardiac transplant patients, indicating a greater contribution to sympathoexcitation of sinoaortic baroreflexes than ventricular receptors (Jacobsen et al., 1993). Finally, Fu et al. (2009) recently observed that transient reductions in stroke volume and blood pressure [both systolic (SAP) and diastolic (DAP) pressures] occur at the onset of even mild levels of LBNP. Taken together, it has become clear that the sympathoexcitation produced by even low levels of central hypovolemia is a result of reflex-mediated deactivation of both cardiopulmonary and arterial baroreceptors. Additionally, recent evidence suggests that reflexes from muscle afferents stimulated during venous distension may also directly evoke increases in MSNA (Cui et al., 2009, 2011a). While venous distension should not occur during hemorrhage, the possibility that reflexes arising from distension of leg veins contribute to the sympathoexcitation observed during LBNP cannot be discounted.
Once initiated, MSNA has been proposed to linearly increase during progressive central hypovolemia, although much of the data supporting this suggestion until recently has been accrued using lower levels of LBNP (≤30 mmHg; Convertino and Cooke, 2002; Cooke et al., 2004). This is because of the inherent difficulty in maintaining nerve recordings during higher levels of negative pressure, as the microelectrode is often displaced during the LBNP procedure. To mitigate this issue, Khan et al. (2002) applied negative pressure to only one leg while performing microneurography in the other. Using this protocol, these investigators demonstrated that progressive reductions in LBNP (up to −50 mmHg) produce concomitant graded increases in MSNA, but they did not measure the degree of central hypovolemia induced by LBNP in order to quantify the relationship between volume loss and sympathetic activation (Khan et al., 2002).
Over the past 6 years, we have created a large database of human experiments (>200 subjects) in which progressive central hypovolemia has been used to produce hemodynamic decompensation (i.e., presyncope). In each subject, chamber decompression was applied for 5 min at −15, −30, −45, and −60 mmHg, and then additional increments of −10 mmHg were used until the onset of decompensation. In the course of this work, we have obtained microneurography recordings in a subset of subjects in which stroke volume, a measure of central hypovolemia, has also been assessed. We were able to maintain MSNA recordings throughout LBNP to either the point of presyncope or to a high (−80 mmHg) LBNP level in 20 subjects. This unique data set has provided the opportunity to develop new understandings of the MSNA response to intense levels of central hypovolemia. For example, Figure 2 shows MSNA recordings from a representative subject exposed to progressively higher levels of LBNP. Average values across subjects demonstrate that increases in MSNA burst frequency are inversely related to stroke volume levels during central hypovolemia in humans (Figure 3), thereby confirming the linearity of the relationship between central hypovolemia and the induced MSNA response. This same relationship between central hypovolemia and direct measurement of renal sympathetic nerve activity (RSNA) is also observed in anesthetized sheep during graded hemorrhage (Batchinsky et al., 2007b). It should be noted that progressive increases in sympathetic activation during LBNP are evident whether MSNA is expressed as frequency (bursts/min), incidence (bursts/100 heartbeats), or total activity (which takes into account burst amplitude; Cooke et al., 2009; Ryan et al., 2011).
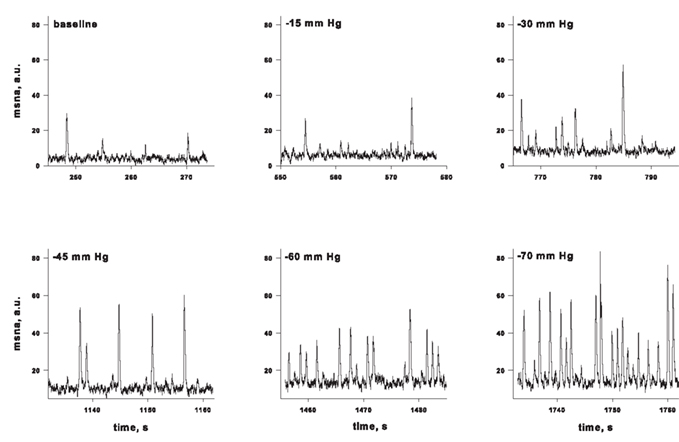
Figure 2. Muscle sympathetic nerve activity (MSNA) in a representative subject during progressive lower body negative pressure. From Cooke et al. (2008).
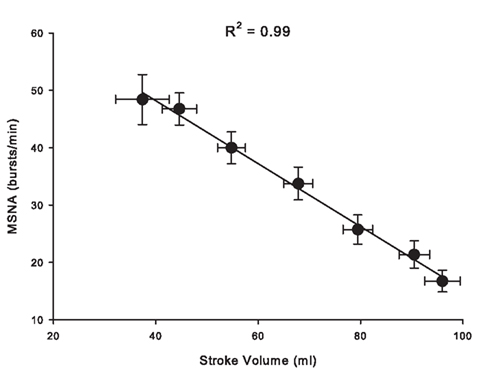
Figure 3. Relationship between stroke volume (SV) and muscle sympathetic nerve activity (MSNA) during LBNP in 20 human subjects. Data modified from Ryan et al. (2011).
In addition to increases in absolute MSNA, progressive central hypovolemia also changes the pattern of bursting activity. Even at rest, sympathetic nerve bursting is characterized by a rhythmic pattern that exists in the low frequency (LF) range (0.04–0.15 Hz); this oscillatory pattern in sympathetic nerve activity contributes to the 10-s arterial pressure Mayer waves (Preiss and Polosa, 1974). While it is beyond the scope of this review, there is a continued controversy regarding whether the oscillatory pattern of sympathetic nerve activity and Mayer waves is produced via a central oscillator mechanism or through the action of the baroreflex (for a review of this issue, see Julien, 2006). In animal models of hemorrhage, the amplitude of LF oscillations in blood pressure and sympathetic nerve activity increase (Guyton and Harris, 1951; Malpas et al., 1998). In humans, the amplitude of LF oscillations in MSNA (MSNALF) increase concomitantly with burst frequency and total activity during stresses that reduce venous return such as head-up tilt (Cooke et al., 1999; Furlan et al., 2000; Kamiya et al., 2005) and LBNP (Ryan et al., 2011). In addition to increases in the frequency and amplitude of sympathetic nerve firing, it is therefore possible that alterations in the oscillatory patterns of that firing may contribute to the ability of an individual to adequately compensate for progressive central hypovolemia (see below).
Sympathetic nerves normally fire in response to reductions of arterial pressure (particularly, DAP) and are silenced with the consequent SAP upstroke, resulting in almost constant latencies from preceding R-waves (Sundlof and Wallin, 1978b; Fagius and Wallin, 1980). At high levels of LBNP, however, an interesting phenomenon occurs in some subjects. Figure 4 depicts MSNA from a subject at baseline, during −60 and −90 mmHg of LBNP (Cooke et al., 2009). At −60 mmHg, coupling of bursts occurred followed by fusing of multiple bursts with greater central hypovolemia (i.e., at −90 mmHg). In our study, burst fusion occurred in 10 of the 17 subjects available for study at the time of publication (Cooke et al., 2009). Burst fusion did not appear to be artifactual, although we cannot completely discount the possibility that the 0.1-s time constant used to integrate MSNA may have contributed to their appearance (Cooke et al., 2009). Similar alterations in the typical pulse synchronous burst pattern of MSNA had previously been noted in subjects susceptible to fainting during head-up tilting; during the presyncopal (i.e., decompensation) phase, burst reflex latencies were shortened and burst durations were lengthened, effects which were reversed on return to the fully conscious state (Iwase et al., 2002). Our data extend this observation to the earlier compensatory phase of central hypovolemia, when blood pressure is still well-maintained. Importantly, fusion of RSNA has also been observed in anesthetized sheep made hypotensive by severe hemorrhage (Batchinsky et al., 2007b). We have suggested that this phenomenon may represent sympathetic baroreflex deafferentation (Cooke et al., 2009), as the fused bursts observed during intense LBNP are similar in both their pulse asynchrony and their structure to those observed after bilateral blocks of glossopharyngeal and vagus nerves (Fagius et al., 1985). It is also possible that loss of pulse-synchrony and continuous burst firing may occur in some individuals as a strategy to continue to maintain blood pressure during severe reductions in venous return. In support of this concept, Salmanpour et al. (2011) have recently shown that extreme baroreceptor unloading requiring high sympathetic outflow (induced by −80 mmHg LBNP) recruits a subpopulation of large postganglionic axons in some but not all individuals.
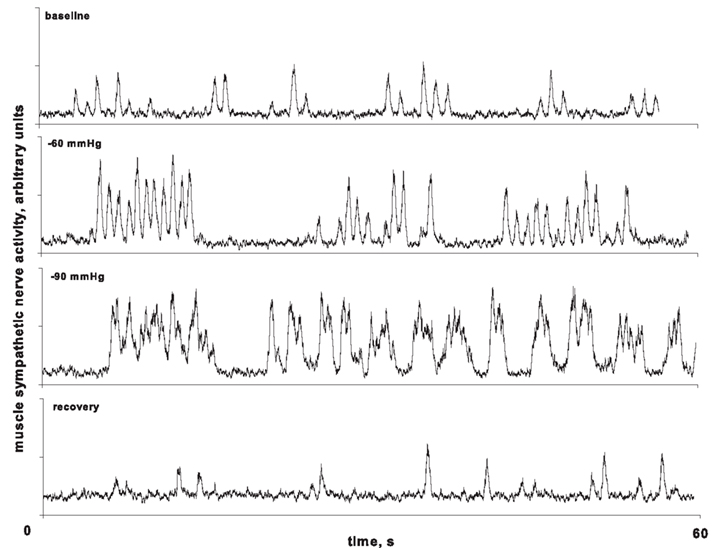
Figure 4. Coupling (−60 mmHg) and then fusing (−90 mmHg) of muscle sympathetic nerve activity (MSNA) in one subject during lower body negative pressure. From Cooke et al. (2009).
MSNA at Hemodynamic Decompensation
In a classic paper, Barcroft et al. (1944) observed that the onset of fainting produced by experimental hemorrhage in man was preceded by a profound vasodilation in the forearm and a decrease in systemic vascular resistance, suggesting inhibition of the compensatory sympathetic activation that had occurred before this point. As it became possible to directly measure sympathetic nerve activity in humans, a variety of case reports and experimental studies appeared showing an inhibition of sympathetic activity associated with the development of presyncope in both healthy humans (Burke et al., 1977; Sanders and Ferguson, 1989; Scherrer et al., 1990; Hayoz et al., 1996; Iwase et al., 2000; Cooke and Convertino, 2002) and in patient populations (Wallin and Sundlof, 1982; Yatomi et al., 1989; Converse et al., 1992; Jardine et al., 1996, 1998, 2002; Morillo et al., 1997). On the basis of these data, it was concluded that hemodynamic decompensation (i.e., presyncope) was associated with and possibly caused by sympathetic neural withdrawal (Convertino and Cooke, 2002; Cooke et al., 2004). Recently, however, we have observed that sympathetic withdrawal does not occur in all subjects at the point of hemodynamic decompensation (Figure 5); in fact, 41% of our subjects did not demonstrate any diminution of MSNA despite the onset of hypotension and presyncopal symptoms (Cooke et al., 2009). Subsequently, Vaddadi et al. (2010) showed that increases in MSNA induced by orthostatic stress were preserved through the point of syncope in 90% of patients previously diagnosed with vagovagal syncope. It is therefore apparent that sympathetic withdrawal is not an absolute requirement for the onset of hemodynamic decompensation in both healthy humans and in patients with a history of syncope. Indeed, careful perusal of some of the literature frequently cited to support the proposition that sympathetic withdrawal precipitates cardiovascular collapse reveals that blood pressure begins to decrease while MSNA remains elevated during the compensatory phase of hypovolemia (Wallin and Sundlof, 1982; Sanders and Ferguson, 1989; Converse et al., 1992; Jardine et al., 1996, 1998; Mosqueda-Garcia et al., 1997; Iwase et al., 2000), an observation confirmed more recently (Kamiya et al., 2005). Taken together, these data refute the concept that cessation of sympathetic nerve activity precedes and causes hypotension, the subsequent reduction in cerebral perfusion pressure, and syncope in all subjects.
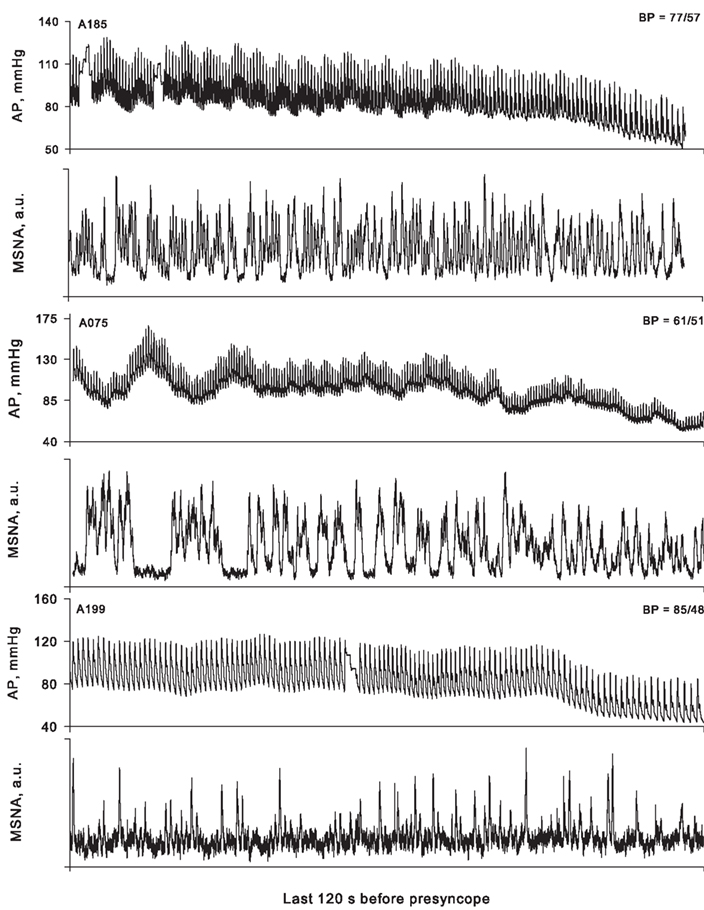
Figure 5. Arterial pressure (AP) and muscle sympathetic nerve activity (MSNA) for three representative subjects 2 min before the onset of presyncope. The lowest arterial pressure (BP) recorded for each subject is shown in the upper right corner of each AP panel. In subject A185, MSNA decreased in the last 2 min of LBNP (−120 to −60 s, 82 bursts/min; −60 s to presyncope, 64 bursts/min), but was still maintained at high levels relative to the pre-LBNP control (36 bursts/min). Subject A075 displayed burst fusion, elevated MSNA over control (25 bursts/min), and no withdrawal of MSNA at presyncope (−120 to −60 s, 53 bursts/min; −60 s to presyncope, 54 bursts/min). Subject A199 (a low tolerant subject) did not display large increases in MSNA from the pre-LBNP control value (17 bursts/min) and also did not demonstrate MSNA withdrawal at presyncope (−120 to −60 s, 22 bursts/min; −60 s to presyncope, 21 bursts/min). From Cooke et al. (2009).
Another possible mechanism that may contribute to the onset of cardiovascular collapse is resetting of baroreflexes, resulting in a loss of synchrony between arterial blood pressure and compensatory responses such as sympathetic activation. During mild to moderate central hypovolemia, the operating point of baroreflex-mediated control of sympathetic nerve activity is shifted upward without a change in gain, such that sympathetic nerve activity is increased at any given DAP (Ichinose et al., 2004a,b). Additionally, there is a tighter coupling between oscillations in arterial blood pressure and MSNA, as quantitated using cross-spectral analysis by an increase in the coherence function between these variables (Furlan et al., 2000; Kamiya et al., 2005; Cooke et al., 2009). This increase in coherence reflects greater baroreflex modulation of sympathetic activity compared with the baseline state and both the increase in coherence and operating point may be beneficial in mounting an appropriate compensatory response to hypovolemia. Evidence exists, however, to suggest that this tight coupling of blood pressure to sympathetic nerve activity may be lost at the point of hemodynamic decompensation. Based on their observation of sympathoinhibition (as measured by plasma catecholamines) despite hypotension, Jacobs et al. (1995) first suggested that the vasodepressor response at presyncope was due to a sudden central resetting of baroreflexes. Recently, this question was more fully investigated by Ichinose et al. (2006) using data from subjects who either exhibited presyncope or did not during central hypovolemia induced by LBNP. Approximately 1–2 min prior to the onset of hemodynamic decompensation, the gain of the baroreflex function relating DAP to MSNA was substantially reduced in those subjects exhibiting presyncope but was unchanged in non-presyncopal subjects (Ichinose et al., 2006). Likewise, we have also demonstrated a loss of linearity between the change in DAP and the change in MSNA just before the onset of presyncope, suggesting disruption in the normal baroreflex-mediated coordination between arterial pressure and sympathetic activation (Cooke et al., 2009; Convertino et al., 2010). Taken together, these results suggest that impairment of arterial baroreflex control over sympathetic vasomotor activity may contribute to the onset of hemodynamic decompensation. Such a sudden attenuation of baroreflex function before hemodynamic decompensation has also been noted for cardiovagal reflexes (Ogoh et al., 2004; Ocon et al., 2011).
As mentioned above, there is also evidence to suggest that the pattern of MSNA firing may be as important in determining the ability to withstand central hypovolemia as the absolute level of MSNA. In an elegant study, Kamiya et al. (2005) determined the temporal occurrence of events immediately preceding presyncope. During central hypovolemia induced by head-up tilt, both the absolute levels of MSNA (expressed as burst frequency and total activity) and the amplitude of LF oscillations in MSNA (MSNALF) increased markedly; increases in MSNALF were reflected in increases in LF oscillations of mean arterial pressure (MAPLF). As subjects moved toward presyncope, MSNALF and MAPLF decreased and were associated with a decrease in MAP, despite the maintenance of MSNA burst frequency and total activity at their elevated level. Interestingly, MSNA only decreased after MAP had begun to fall. MSNA, MSNALF, and MAPLF were maintained and did not decrease in those subjects who did not exhibit hypotension and presyncope (Kamiya et al., 2005). We have observed a similar phenomenon in a subject whose tolerance to central hypovolemia was improved through the use of inspiratory resistance breathing (Figure 6). In this experiment, subjects were exposed to LBNP to the point of presyncope in separate experiments performed at least 2 weeks apart. In one experiment, subjects breathed through a device that did not provide resistance to inspiration (sham), while in the other experiment, subjects breathed through a device that provided resistance to inspiration (active). Because resistance breathing improved LBNP tolerance, data were analyzed at the end of LBNP during the sham experiment and at this same absolute time point during the active experiment (i.e., well before the onset of presyncope during inspiratory resistance breathing). During breathing with the sham device, MSNA increased from 12 bursts/min at baseline to 44 bursts/min at the end of LBNP, while MSNA increased from 10 to 54 bursts/min at this same absolute time point (i.e., prior to presyncope) during resistance breathing. Importantly, MSNALF increased from 1.15 to 22.4 au2 at presyncope under the sham resistance breathing condition; with resistance breathing, MSNALF increased from 2.23 to 40.1 au2 at this same absolute time point. Moreover, the coherence between DAP and MSNA, a measure of the strength of the relationship between these two variables, was increased from 0.80 in the sham condition to 0.94 with resistance breathing at this same time point. Thus, in this one subject, it is apparent that inspiratory resistance breathing increased the amplitude of MSNA oscillations and coherence between MSNA and arterial blood pressure, and that this alteration in oscillatory pattern was associated with improved tolerance to central hypovolemia. These data are consistent with the concept that the maintenance of an increase in LF oscillations of both MSNA and MAP may be associated with the defense of blood pressure during severe central hypovolemia.
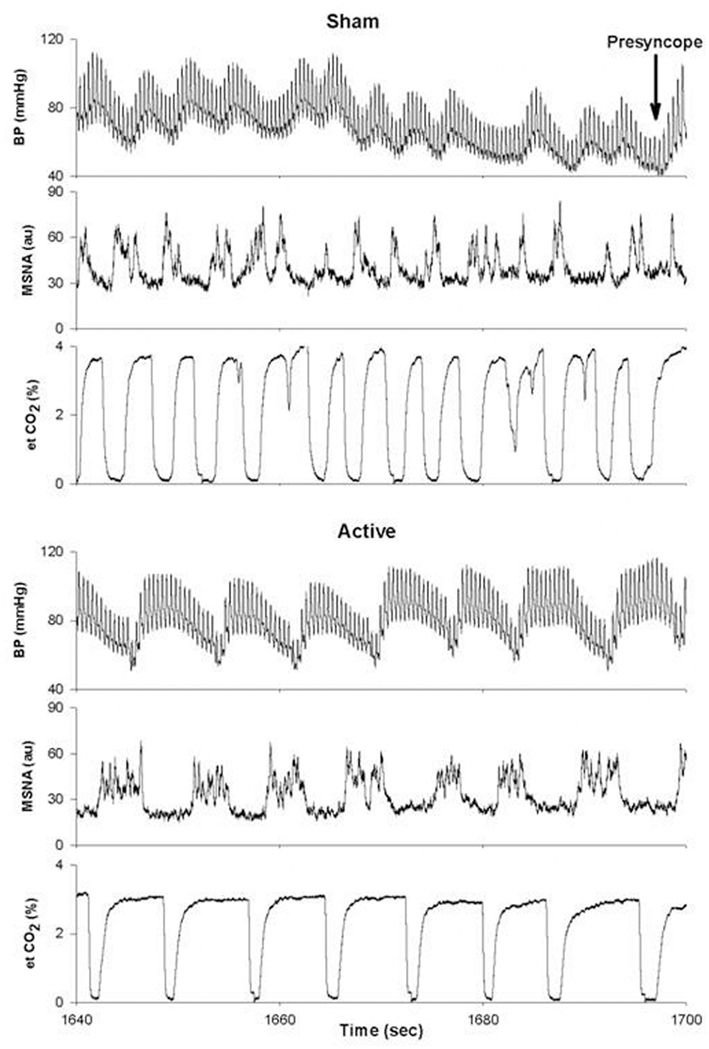
Figure 6. Blood pressure (BP), muscle sympathetic nerve activity (MSNA) and end-tidal CO2 (et CO2) tracings for a single subject under conditions of breathing with a sham (top) or active (bottom) device that provided resistance to inspiration. Au, arbitrary units. From Ryan et al. (2008).
Thus, there are several possible mechanisms involving alterations in activation of sympathetic nerve activity to explain hemodynamic decompensation during severe hypovolemia. Since sympathetic withdrawal occurs in some individuals before presyncope but not in others, it is no longer thought to be a prerequisite for the ensuing hypotension (Cooke et al., 2009). It is also possible that there is a central resetting of arterial baroreflex function that alters sympathetic outflow (Ichinose et al., 2006); this scenario may be especially prominent in those subjects in whom sympathetic inhibition occurs despite progressive hypotension. Finally, there is evidence to suggest that an increase in LF oscillations in both MSNA and MAP may be protective during central hypovolemia and that loss of these oscillations might precipitate hypotension (Kamiya et al., 2005). It is important to note that these mechanisms are not mutually exclusive and the contributions of each to the process have yet to be fully revealed. Furthermore, it is also possible and even probable that different mechanisms may predominate in different individuals. Because of the scope of this review, we have chosen to focus on those mechanisms preceding presyncope that involve loss of compensatory alterations in MSNA, but it is likely that loss of other compensatory responses may also contribute to the inability to maintain blood pressure during severe hypovolemia. One intriguing hypothesis put forward by Dietz et al. (1997) is that marked peripheral vasodilation is a major contributor to the fall in arterial pressure preceding vasovagal syncope. In this regard, it is of interest that, during prolonged LBNP at a low level (−15 mmHg), vasodilation of the forearm musculature was observed despite the continued presence of a sustained compensatory increase in MSNA, suggesting that “sympathetic escape” occurs (Joyner et al., 1990). This observation is currently being investigated.
Implicit in the preceding discussion is the notion that there are individual differences in the ability of healthy humans to tolerate central hypovolemia before reaching the point of cardiovascular collapse. Indeed, it has been known for many years that there is a great deal of variability in the ability of patients (Davis, 1949) and animals (Chien, 1967; Kim and Shoemaker, 1970) to survive traumatic hemorrhage; we have recently learned that there is a genetic basis underlying this variability (Klemcke et al., 2008, 2011). Likewise, individual differences in tolerance to central hypovolemia induced by LBNP have also been described (Sather et al., 1986) and attributed to differences in the release of vasoactive hormones (Convertino and Sather, 2000b; Greenleaf et al., 2000), compensatory tachycardia and vasoconstriction (Convertino and Sather, 2000a,b;Greenleaf et al., 2000), cardiac baroreflex gain (Convertino and Sather, 2000a; Convertino et al., in press), baroreflex gain of sympathetic nerve activation (Wijeysundera et al., 2001), and central blood volume and cerebral blood velocity (Levine et al., 1994). In this regard, we have recently shown that subjects demonstrating high tolerance (HT) to LBNP have a greater ability to increase MSNA than subjects with low tolerance (LT; Convertino et al., in press). Additionally, the ability to sustain the compensatory mechanisms described above involving both the baroreflex modulation of sympathetic activation and the oscillatory component of that activation may also act to determine tolerance to central hypovolemia. For example, Ichinose et al. (2006) described a sudden resetting of the DAP–MSNA baroreflex in subjects who became presyncopal (i.e., those demonstrating LT) during central hypovolemia but did not observe this resetting in those who did not (HT). Furthermore, Kamiya et al. (2005) observed a loss of MSNALF and MAPLF power in LT subjects before presyncope elicited by head-up tilt, but no such loss in HT subjects. As in previous reports (Sather et al., 1986; Convertino and Sather, 2000a,b), we recently confirmed and expanded on these results using our large database of subjects in which LBNP was applied to the point of presyncope in all individuals. We classified subjects as HT if they completed at least the −60-mmHg level of LBNP, and LT if they did not complete this level (Rickards et al., 2011). As in the previous study using head-up tilt (Kamiya et al., 2005), MAPLF increased in both LT and HT groups during early stages of LBNP, but MAPLF decreased to baseline levels in the LT group at −60 mmHg while it continued to increase in the HT group (Rickards et al., 2011). A similar relationship between the ability to increase orthostatic tolerance and the ability to increase blood pressure oscillations has previously been shown (Gulli et al., 2001). In our study, measurement of middle cerebral artery velocity (MCAv) by transcranial Doppler revealed a similar oscillatory pattern to that observed with MAP, indicating that the continued increase in amplitude of LF arterial pressure oscillations observed in HT subjects was transferred to the cerebral vasculature (Rickards et al., 2011). Although we did not report MSNA in our paper, Figure 7 shows these responses during LBNP in a HT and a LT subject. In the HT subject, MSNALF increased from baseline (4.4 au2) to a maximum of 85.8 au2 just prior to presyncope. In contrast, MSNALF did not increase in the LT subject at all (2.4 to 2.3 au2). Thus, we propose that the ability to increase LF oscillations in MSNA, MAP, and MCAv is an inherent characteristic associated with improved tolerance to central hypovolemia and therefore protects against the onset of hemodynamic decompensation during severe hypovolemia.
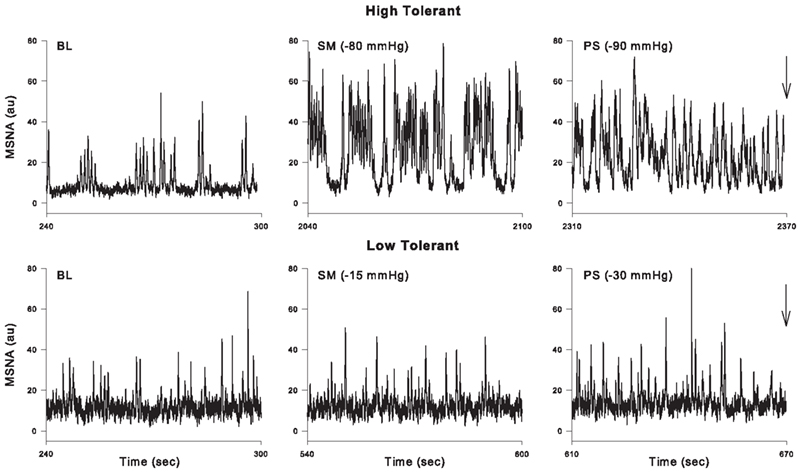
Figure 7. Representative tracings from a high tolerant and low tolerant subject at baseline (BL), sub-maximal LBNP (i.e., the LBNP level before the level at which presyncope was reached; SM) and immediately before presyncope (indicated by arrow; PS).
Non-Invasive Surrogate for MSNA for Assessing the Severity of Hemorrhage?
Because the autonomic nervous system serves to maintain cardiovascular homeostasis under a variety of physiological and pathological stresses, a non-invasive surrogate for sympathetic and/or vagal activation has long been sought for diagnostic use (Goldstein et al., 2011). Measures of HR variability (HRV) have received a great deal of attention, as some of the time and frequency domain measures have been associated with autonomic function. Specifically, power spectral analysis of intervals between R-waves (RRI) of the ECG yields LF (0.04–0.15 Hz) and HF (0.15–0.4 Hz) powers; LF power was originally thought to contain components of both cardiac sympathetic and vagal (parasympathetic) function, while HF power predominantly reflected parasympathetic function (Akselrod et al., 1981). Many investigators have also suggested that the ratio of LF/HF represents sympathovagal balance (Malliani et al., 1991; Goldstein et al., 2011) and that these or other HRV metrics might be useful in disclosing dysautonomia in such clinical conditions as myocardial infarction, cardiac arrhythmias, diabetes, and renal failure (Acharya et al., 2006; Montano et al., 2009). Indeed, a PubMed search on the term “heart rate variability” yields more than 14,300 references at the time of this writing (February 2012), yet we are unaware of any pathophysiological condition in which HRV is currently used as a standard of care for diagnosis and/or treatment. The use of HRV is appealing because calculation requires only non-invasive collection of a standard ECG.
Because increases in sympathetic activation occur in a linear fashion with decreases in stroke volume (Figure 3), we proposed that HRV metrics might be useful non-invasive surrogates of MSNA for assessing the degree and progression of hemorrhage in trauma victims (Cooke and Convertino, 2005). In 2008, we demonstrated using our LBNP model of simulated hemorrhage that, on average, RRIHF power and some time domain metrics associated with parasympathetic function are inversely related to direct measurement of MSNA. RRILF power, on the other hand, was not associated with MSNA (Cooke et al., 2008); it has been made clear more recently that RRILF is not a measure of cardiac sympathetic activity (Billman, 2011; Goldstein et al., 2011). From these data, we concluded that HRV might be of clinical utility in assessing autonomic function during hemorrhage, although we and others noted that correlations between MSNA and HRV in individual subjects were not as strong as those derived from group means (Floras et al., 2001; Cooke et al., 2008). Application of power spectral analysis to ECG recordings collected from actual trauma patients during air transport to the hospital further suggested that the use of these metrics might be appropriate for clinical assessment of the severity of hemorrhage, as group means of some HRV metrics differed between patients who lived and those who died up to 24 h later (Cooke et al., 2006a,b). Subsequent studies extended these observations to the use of non-linear HRV metrics to predict mortality (Batchinsky et al., 2007a) and to discriminate between trauma patients who required a life-saving intervention and those who did not (Cancio et al., 2008).
Importantly, all of the conclusions delineated above were developed based on standard analyses of group mean data. As our understanding evolved, however, we began to take the “next step” to assess whether HRV metrics could be useful in determining the physiological status of individuals rather than a group as a whole, a requirement that must be met for successful application of any metric for diagnosing individual patients. In doing so, we became aware of a number of challenges with all of the HRV metrics that we examined (time domain, frequency domain, and non-linear metrics). First, there are issues of large inter-individual variability and poor reproducibility within subjects during the same recording session (Rickards et al., 2010a) as well as across days (Tan et al., 2009). Second, accurate determination of HRV requires long (in some cases, up to 800 beats) segments of ECG recordings that do not contain electromagnetic noise or ectopic beats, but these occur more frequently in trauma patients than in healthy individuals (Sethuraman et al., 2010). Third, increases in HR will always be associated with decreases in HRV, purely as a mathematical function of the curvilinear nature of the relationship between HR and RRI (Sacha and Pluta, 2008). Decreases in HRV are therefore not specific to central hypovolemia but may be elicited by any physiological stressor that induces tachycardia such as physical movement (Rickards et al., 2008), pain, or anxiety, which are common in conscious trauma patients. Fourth, HRV metrics are not able to discriminate between HT and LT subjects early in the progression of hypovolemia; an effective triage tool should be able to alert medical personnel to those patients that will progress to hemodynamic decompensation more quickly (Hinojosa-Laborde et al., 2011). Finally, although group means of HRV metrics are highly correlated with decreases in stroke volume during central hypovolemia, analysis of the individual trajectories for these metrics demonstrated poor and inconsistent correlations with stroke volume at the individual subject level, even under controlled laboratory conditions (Ryan et al., 2010). Armed with this new understanding, re-analysis of ECG data collected from trauma patients in the prehospital phase demonstrated that, while means of some of these HRV metrics differed between groups that received a life-saving intervention and those that did not, there was such high overlap of individual patient HRV values between groups that it would have been impossible to accurately classify patients on the basis of HRV alone (Figure 8; Rickards et al., 2010b). It should be noted that, unlike in earlier studies (Cooke et al., 2006a,b; Batchinsky et al., 2007a; Cancio et al., 2008) in which differences existed between groups even in standard vital signs, only those trauma patients with normal vital signs were chosen for analysis in order to ascertain whether the use of HRV metrics provided added discriminatory value over standard vital signs. Based on these data, HRV metrics on their own fail to be useful for assessing the severity of hemorrhaging trauma patients in the prehospital and emergency department phases of diagnosis and treatment.
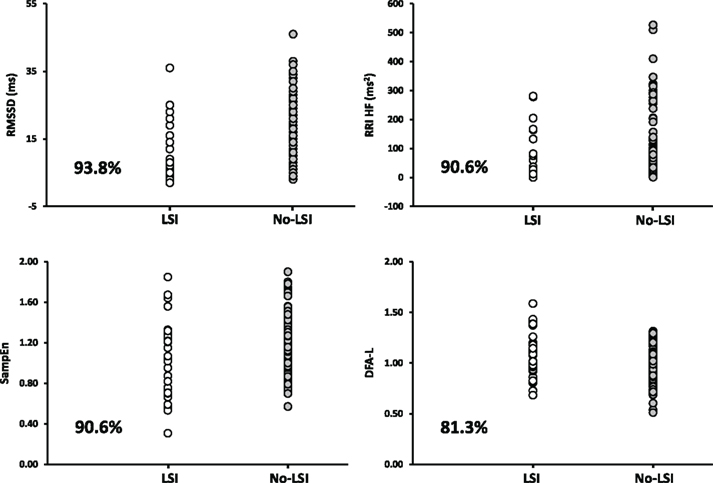
Figure 8. Individual values of four representative heart rate variability metrics for life-saving intervention (LSI; n = 32) and No-LSI (n = 127) patient groups (each point represents a single patient). For each HRV metric, group means statistically differ between LSI and No-LSI groups (P ≤ 0.03). The percentage of individual LSI patient values that fall within the range of the No-LSI group are presented in each plot. RMSSD, R–R intervals root mean squared SD (a time domain metric); RRIHF, R–R intervals high frequency (a frequency domain metric); SampEn, sample entropy (a non-linear metric); DFA-L, detrended fluctuations analysis – long range correlations (a non-linear metric). From Rickards et al. (2010b).
The amplitude of the LF oscillations in SAP has also been suggested as a potential non-invasive surrogate for sympathetic vasomotor tone and nerve activation (Pagani et al., 1986). Linear relationships between SAPLF and direct measurement of MSNA were later described in human subjects during sympathetic activation induced by sodium nitroprusside infusion (Pagani et al., 1997) and head-up tilt (Furlan et al., 2000). On the basis of such observations, SAPLF has been used as a non-invasive metric for the determination of dysautonomia in hypertension (Lucini et al., 2002) and Type 1 diabetes (Lucini et al., 2009). However, this viewpoint has been the subject of continued controversy (Parati et al., 2006; Taylor and Studinger, 2006). Data that argue against the use of SAPLF as a non-invasive metric of MSNA include the inability of SAPLF to adequately correlate with levels of MSNA in humans at rest (Taylor et al., 1998) or during various physiological or pathophysiological conditions (Radaelli et al., 1999; Cui et al., 2004b). We have recently investigated this question using our database of MSNA recordings obtained during central hypovolemia (Ryan et al., 2011). When group mean data were considered, there was a strong direct relationship (r2 = 0.98) between the increase in MSNA and SAPLF, but it was curvilinear rather than linear as had been previously described (Ryan et al., 2011). When we took the “next step” to determine correlations between SAPLF and MSNA within individual subjects, however, we found poor correlations in a significant portion of our subjects (range of r2: 0.09–0.96; Ryan et al., 2011). Because a strong relationship between these variables is not universally applicable to all healthy human subjects, SAPLF should not be used as a non-invasive surrogate of MSNA. Therefore, no reliable and accurate metric has been identified that is based on non-invasive recordings and that may be reliably used in the place of direct recordings of MSNA to assess levels of sympathetic activation during physiological stressors or disease states.
Conclusion
Hemorrhage continues to be a major cause of morbidity and mortality in both military and civilian settings, and research efforts continue to provide better means of both diagnosing the severity of blood loss and guiding resuscitation efforts. While important work continues in animal models, human experimental models such as LBNP have provided valuable insight as to the mechanisms of compensatory physiological responses to central hypovolemia without the confounding factors of anesthesia, species differences, and tissue injury. During even mild LBNP, both arterial and cardiopulmonary baroreceptors are unloaded, resulting in activation of the sympathetic nervous system to increase HR and vasomotor tone. Importantly, we now know that sympathetic activation entails not only an increase in sympathetic nerve firing but also an alteration in the pattern of firing, such that the amplitude of LF oscillations in MSNA and, consequently, blood pressure is also increased; both of these responses seem to be protective in that they are associated with improved tolerance to central hypovolemia. At some point, which occurs at different levels of central hypovolemia for individual subjects, this compensation fails and hypotension ensues. Although it was once thought that sympathetic withdrawal always precipitated hemodynamic decompensation and hypotension, it is now clear that a diminution of absolute levels of MSNA firing is not required in all individuals. Instead, it is possible that loss of the compensatory increase in MSNALF may also be involved, particularly in those subjects demonstrating HT to central hypovolemia. Additionally, there may be an acute resetting of the baroreflex at the level of the CNS such that coherence between arterial blood pressure and sympathetic nerve activity is lost. Determination of the mechanisms underlying the development of presyncope in both healthy human subjects and patients with diseases characterized by episodes of fainting continues to be an ongoing area of research.
The search for a non-invasive surrogate of MSNA for both research and clinical purposes continues. Certainly, an easily obtainable non-invasive metric of sympathetic activation could be of great importance for assessment of the severity of hemorrhage. However, metrics based on determination of variability in both RRIs and arterial blood pressure do not fulfill the necessary criteria to perform effectively in this role. Before implementation of any such metric, it is essential that investigators determine whether the metric will be able to reliably track MSNA within individual subjects rather than simply rely on analyses based on group mean data. Importantly, gender differences in the MSNA response to central hypovolemia must also be taken into account (Fu et al., 2005; Carter et al., 2009). For use in trauma victims suspected of hemorrhage, it will also be necessary for laboratory determination of the efficacy of the metric in the face of blood loss combined with other physiological stressors which can impact sympathetic activation, such as heat stress (Cui et al., 2004a, 2011b), dehydration (Kimmerly and Shoemaker, 2002, 2003; Fu et al., 2005), mental or emotional stress (Carter et al., 2008), and ingestion of alcohol (Carter et al., 2011), nicotine, or other drugs. All of these stressors are commonly observed in conjunction with traumatic hemorrhage in civilian and/or military settings. Because of the complexity of this problem, it is possible that clinical assessment of the severity of hemorrhage during the acute phase may be better realized using artificial intelligence technologies that reflect the integration of the sympathetic nervous response to hemorrhage with changes in circulatory pressure and volume (Convertino et al., 2011).
Disclaimer
The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Department of the Army or the Department of Defense.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank our research subjects, who cheerfully cooperated with the experiments described herein, and Mr. Gary Muniz for his superb technical assistance. All studies performed at the U.S. Army Institute of Surgical Research were conducted under protocols reviewed and approved by the US Army Medical Research and Materiel Command Institutional Review Board and in accordance with the approved protocols. This work was supported by the Combat Casualty Care Research Area Directorate of the US Army Medical Research and Materiel Command.
References
Acharya, U. R., Joseph, K. P., Kannathal, N., Lim, C. M., and Suri, J. S. (2006). Heart rate variability: a review. Med. Biol. Eng. Comput. 44, 1031–1051.
Akselrod, S., Gordon, D., Ubel, F. A., Shannon, D. C., Berger, A. C., and Cohen, R. J. (1981). Power spectrum analysis of heart rate fluctuation: a quantitative probe of beat-to-beat cardiovascular control. Science 213, 220–222.
Barcroft, H., Edholm, O. G., McMichael, J., and Sharpey-Schafer, E. P. (1944). Posthaemorrhagic fainting: study by cardiac output and forearm flow. Lancet 1, 489–490.
Batchinsky, A. I., Cancio, L. C., Salinas, J., Kuusela, T., Cooke, W. H., Wang, J. J., Boehme, M., Convertino, V. A., and Holcomb, J. B. (2007a). Prehospital loss of R-to-R interval complexity is associated with mortality in trauma patients. J. Trauma 63, 512–518.
Batchinsky, A. I., Cooke, W. H., Kuusela, T. A., Jordan, B. S., Wang, J. J., and Cancio, L. C. (2007b). Sympathetic nerve activity and heart rate variability during severe hemorrhagic shock in sheep. Auton. Neurosci. 136, 43–51.
Billman, G. E. (2011). Heart rate variability – a historical perspective. Front. Physiol. 2:86. doi:10.3389/fphys.2011.00086
Burke, D., Sundlof, G., and Wallin, G. (1977). Postural effects on muscle nerve sympathetic activity in man. J. Physiol. (Lond.) 272, 399–414.
Cancio, L. C., Batchinsky, A. I., Salinas, J., Kuusela, T., Convertino, V. A., Wade, C. E., and Holcomb, J. B. (2008). Heart-rate complexity for prediction of prehospital lifesaving interventions in trauma patients. J. Trauma 65, 813–819.
Carter, J. R., Durocher, J. J., and Kern, R. P. (2008). Neural and cardiovascular responses to emotional stress in humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R1898–R1903.
Carter, J. R., Lawrence, J. E., and Klein, J. C. (2009). Menstrual cycle alters sympathetic neural responses to orthostatic stress in young, eumenorrheic women. Am. J. Physiol. Endocrinol. Metab. 297, E85–E91.
Carter, J. R., Stream, S. F., Durocher, J. J., and Larson, R. A. (2011). Influence of acute alcohol ingestion on sympathetic neural responses to orthostatic stress in humans. Am. J. Physiol. Endocrinol. Metab. 300, E771–E778.
Converse, R. L. Jr., Jacobsen, T. N., Jost, C. M., Toto, R. D., Grayburn, P. A., Obregon, T. M., Fouad-Tarazi, F., and Victor, R. G. (1992). Paradoxical withdrawal of reflex vasoconstriction as a cause of hemodialysis-induced hypotension. J. Clin. Invest. 90, 1657–1665.
Convertino, V. A. (2001). Lower body negative pressure as a tool for research in aerospace physiology and military medicine. J. Gravit. Physiol. 8, 1–14.
Convertino, V. A., and Cooke, W. H. (2002). Relationship between stroke volume and sympathetic nerve activity: new insights about autonomic mechanisms of syncope. J. Gravit. Physiol. 9, P63–P66.
Convertino, V. A., Moulton, S. L., Grudic, G. Z., Rickards, C. A., Hinojosa-Laborde, C., Gerhardt, R. T., Blackbourne, L. H., and Ryan, K. L. (2011). Use of advanced machine-learning techniques for noninvasive monitoring of hemorrhage. J. Trauma 71, S25–S32.
Convertino, V. A., Ryan, K. L., Rickards, C. A., Salinas, J., McManus, J. G., Cooke, W. H., and Holcomb, J. B. (2008). Physiological and medical monitoring for en route care of combat casualties. J. Trauma 64, S342–S353.
Convertino, V. A., and Sather, T. M. (2000a). Effects of cholinergic and beta-adrenergic blockade on orthostatic tolerance in healthy subjects. Clin. Auton. Res. 10, 327–336.
Convertino, V. A., and Sather, T. M. (2000b). Vasoactive neuroendocrine responses associated with tolerance to lower body negative pressure in humans. Clin. Physiol. 20, 177–184.
Convertino, V. A., Rickards, C. A., and Ryan, K. L. (2010). Responses of sympathetic nerve activity to presyncope: new insights about mechanisms of fainting. J. Gravit. Physiol. 17, P27–P30.
Convertino, V. A., Rickards, C. A., and Ryan, K. L. (in press). Tolerance to central hypovolemia: autonomic mechanisms associated with heart rate and vasoconstrictor reserves in humans. Clin. Auton. Res.
Cooke, W. H., and Convertino, V. A. (2002). Association between vasovagal hypotension and low sympathetic neural activity during presyncope. Clin. Auton. Res. 12, 483–486.
Cooke, W. H., and Convertino, V. A. (2005). Heart rate variability and spontaneous baroreflex sequences: implications for autonomic monitoring during hemorrhage. J. Trauma 58, 798–805.
Cooke, W. H., Hoag, J. B., Crossman, A. A., Kuusela, T. A., Tahvanainen, K. U., and Eckberg, D. L. (1999). Human responses to upright tilt: a window on central autonomic integration. J. Physiol. (Lond.) 517, 617–628.
Cooke, W. H., Rickards, C. A., Ryan, K. L., and Convertino, V. A. (2008). Autonomic compensation to simulated hemorrhage monitored with heart period variability. Crit. Care Med. 36, 1892–1899.
Cooke, W. H., Rickards, C. A., Ryan, K. L., Kuusela, T. A., and Convertino, V. A. (2009). Muscle sympathetic nerve activity during intense lower body negative pressure to presyncope in humans. J. Physiol. (Lond.) 587, 4987–4999.
Cooke, W. H., Ryan, K. L., and Convertino, V. A. (2004). Lower body negative pressure as a model to study progression to acute hemorrhagic shock in humans. J. Appl. Physiol. 96, 1249–1261.
Cooke, W. H., Salinas, J., Convertino, V. A., Ludwig, D. A., Hinds, D., Duke, J. H., Moore, F. A., and Holcomb, J. B. (2006a). Heart rate variability and its association with mortality in prehospital trauma patients. J. Trauma 60, 363–370; discussion 370.
Cooke, W. H., Salinas, J., McManus, J. G., Ryan, K. L., Rickards, C. A., Holcomb, J. B., and Convertino, V. A. (2006b). Heart period variability in trauma patients may predict mortality and allow remote triage. Aviat. Space Environ. Med. 77, 1107–1112.
Cui, J., Leuenberger, U. A., Gao, Z., and Sinoway, L. I. (2011a). Sympathetic and cardiovascular responses to venous distension in an occluded limb. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R1831–R1837.
Cui, J., Shibasaki, M., Low, D. A., Keller, D. M., Davis, S. L., and Crandall, C. G. (2011b). Muscle sympathetic responses during orthostasis in heat-stressed individuals. Clin. Auton. Res. 21, 381–387.
Cui, J., McQuillan, P., Moradkhan, R., Pagana, C., and Sinoway, L. I. (2009). Sympathetic responses during saline infusion into the veins of an occluded limb. J. Physiol. (Lond.) 587, 3619–3628.
Cui, J., Wilson, T. E., and Crandall, C. G. (2004a). Muscle sympathetic nerve activity during lower body negative pressure is accentuated in heat-stressed humans. J. Appl. Physiol. 96, 2103–2108.
Cui, J., Zhang, R., Wilson, T. E., and Crandall, C. G. (2004b). Spectral analysis of muscle sympathetic nerve activity in heat-stressed humans. Am. J. Physiol. Heart Circ. Physiol. 286, H1101–H1106.
Davis, H. A. (1949). Shock and Allied Forms of Failure of the Circulation. New York: Grune & Stratton.
Dietz, N. M., Joyner, M. J., and Shepherd, J. T. (1997). Vasovagal syncope and skeletal muscle vasodilatation: the continuing conundrum. Pacing Clin. Electrophysiol. 20, 775–780.
Eastridge, B. J., Hardin, M., Cantrell, J., Oetjen-Gerdes, L., Zubko, T., Mallak, C., Wade, C. E., Simmons, J., Mace, J., Mabry, R., Bolenbaucher, R., and Blackbourne, L. H. (2011). Died of wounds on the battlefield: causation and implications for improving combat casualty care. J. Trauma 71, S4–S8.
Fagius, J., and Wallin, B. G. (1980). Sympathetic reflex latencies and conduction velocities in normal man. J. Neurol. Sci. 47, 433–448.
Fagius, J., Wallin, B. G., Sundlof, G., Nerhed, C., and Englesson, S. (1985). Sympathetic outflow in man after anaesthesia of the glossopharyngeal and vagus nerves. Brain 108, 423–438.
Floras, J. S., Butler, G. C., Ando, S. I., Brooks, S. C., Pollard, M. J., and Picton, P. (2001). Differential sympathetic nerve and heart rate spectral effects of nonhypotensive lower body negative pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281, R468–R475.
Fu, Q., Shibata, S., Hastings, J. L., Prasad, A., Palmer, M. D., and Levine, B. D. (2009). Evidence for unloading arterial baroreceptors during low levels of lower body negative pressure in humans. Am. J. Physiol. Heart Circ. Physiol. 296, H480–H488.
Fu, Q., Witkowski, S., Okazaki, K., and Levine, B. D. (2005). Effects of gender and hypovolemia on sympathetic neural responses to orthostatic stress. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R109–R116.
Furlan, R., Porta, A., Costa, F., Tank, J., Baker, L., Schiavi, R., Robertson, D., Malliani, A., and Mosqueda-Garcia, R. (2000). Oscillatory patterns in sympathetic neural discharge and cardiovascular variables during orthostatic stimulus. Circulation 101, 886–892.
Gernandt, B., Liljestrand, G., and Zotterman, Y. (1946). Efferent impulses in the splanchnic nerve. Acta Physiol. Scand. 11, 231–247.
Goldstein, D. S., Bentho, O., Park, M. Y., and Sharabi, Y. (2011). Low-frequency power of heart rate variability is not a measure of cardiac sympathetic tone but may be a measure of modulation of cardiac autonomic outflows by baroreflexes. Exp. Physiol. 96, 1255–1261.
Greenleaf, J. E., Petersen, T. W., Gabrielsen, A., Pump, B., Bie, P., Christensen, N. J., Warberg, J., Videbaek, R., Simonson, S. R., and Norsk, P. (2000). Low LBNP tolerance in men is associated with attenuated activation of the renin-angiotensin system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R822–R829.
Gulli, G., Wight, V. L., Hainsworth, R., and Cevese, A. (2001). Spectral and cross-spectral autoregressive analysis of cardiovascular variables in subjects with different degrees of orthostatic tolerance. Clin. Auton. Res. 11, 19–27.
Guyton, A. C., and Harris, J. W. (1951). Pressoreceptor-autonomic oscillation: a probable cause of vasomotor waves. Am. J. Physiol. 165, 158–166.
Hayoz, D., Noll, G., Passino, C., Weber, R., Wenzel, R., and Bernardi, L. (1996). Progressive withdrawal of muscle nerve sympathetic activity preceding vaso-vagal syncope during lower-body negative pressure. Clin. Sci. 91(Suppl.), 50–51.
Hinojosa-Laborde, C., Rickards, C. A., Ryan, K. L., and Convertino, V. A. (2011). Heart rate variability during simulated hemorrhage with lower body negative pressure in high and low tolerant subjects. Front. Physiol. 2:85. doi:10.3389/fphys.2011.00085
Ichinose, M., Saito, M., Fujii, N., Kondo, N., and Nishiyasu, T. (2006). Modulation of the control of muscle sympathetic nerve activity during severe orthostatic stress. J. Physiol. (Lond.) 576, 947–958.
Ichinose, M., Saito, M., Kitano, A., Hayashi, K., Kondo, N., and Nishiyasu, T. (2004a). Modulation of arterial baroreflex dynamic response during mild orthostatic stress in humans. J. Physiol. (Lond.) 557, 321–330.
Ichinose, M., Saito, M., Ogawa, T., Hayashi, K., Kondo, N., and Nishiyasu, T. (2004b). Modulation of control of muscle sympathetic nerve activity during orthostatic stress in humans. Am. J. Physiol. Heart Circ. Physiol. 287, H2147–H2153.
Iwase, S., Mano, T., Kamiya, A., Niimi, Y., Fu, Q., and Suzumura, A. (2002). Syncopal attack alters the burst properties of muscle sympathetic nerve activity in humans. Auton. Neurosci. 95, 141–145.
Iwase, S., Sugiyama, Y., Miwa, C., Kamiya, A., Mano, T., Ohira, Y., Shenkman, B., Egorov, A. I., and Kozlovskaya, I. B. (2000). Effects of three days of dry immersion on muscle sympathetic nerve activity and arterial blood pressure in humans. J. Auton. Nerv. Syst. 79, 156–164.
Jacobs, M. C., Goldstein, D. S., Willemsen, J. J., Smits, P., Thien, T., Dionne, R. A., and Lenders, J. W. (1995). Neurohumoral antecedents of vasodepressor reactions. Eur. J. Clin. Invest. 25, 754–761.
Jacobsen, T. N., Morgan, B. J., Scherrer, U., Vissing, S. F., Lange, R. A., Johnson, N., Ring, W. S., Rahko, P. S., Hanson, P., and Victor, R. G. (1993). Relative contributions of cardiopulmonary and sinoaortic baroreflexes in causing sympathetic activation in the human skeletal muscle circulation during orthostatic stress. Circ. Res. 73, 367–378.
Jardine, D. L., Ikram, H., and Crozier, I. G. (1996). Autonomic control of asystolic vasovagal syncope. Heart 75, 528–530.
Jardine, D. L., Ikram, H., Frampton, C. M., Frethey, R., Bennett, S. I., and Crozier, I. G. (1998). Autonomic control of vasovagal syncope. Am. J. Physiol. 274, H2110–H2115.
Jardine, D. L., Melton, I. C., Crozier, I. G., English, S., Bennett, S. I., Frampton, C. M., and Ikram, H. (2002). Decrease in cardiac output and muscle sympathetic activity during vasovagal syncope. Am. J. Physiol. Heart Circ. Physiol. 282, H1804–H1809.
Joyner, M. J., Shepherd, J. T., and Seals, D. R. (1990). Sustained increases in sympathetic outflow during prolonged lower body negative pressure in humans. J. Appl. Physiol. 68, 1004–1009.
Kamiya, A., Hayano, J., Kawada, T., Michikami, D., Yamamoto, K., Ariumi, H., Shimizu, S., Uemura, K., Miyamoto, T., Aiba, T., Sunagawa, K., and Sugimachi, M. (2005). Low-frequency oscillation of sympathetic nerve activity decreases during development of tilt-induced syncope preceding sympathetic withdrawal and bradycardia. Am. J. Physiol. Heart Circ. Physiol. 289, H1758–H1769.
Kauvar, D. S., Lefering, R., and Wade, C. E. (2006). Impact of hemorrhage on trauma outcome: an overview of epidemiology, clinical presentations, and therapeutic considerations. J. Trauma 60, S3–S11.
Khan, M. H., Sinoway, L. I., and MacLean, D. A. (2002). Effects of graded LBNP on MSNA and interstitial norepinephrine. Am. J. Physiol. Heart Circ. Physiol. 283, H2038–H2044.
Kim, S. I., and Shoemaker, W. C. (1970). Comparison of cardiorespiratory changes in surviving and nonsurviving shock dogs. Arch. Surg. 100, 275–279.
Kimmerly, D. S., and Shoemaker, J. K. (2002). Hypovolemia and neurovascular control during orthostatic stress. Am. J. Physiol. Heart Circ. Physiol. 282, H645–H655.
Kimmerly, D. S., and Shoemaker, J. K. (2003). Hypovolemia and MSNA discharge patterns: assessing and interpreting sympathetic responses. Am. J. Physiol. Heart Circ. Physiol. 284, H1198–H1204.
Klemcke, H. G., Baer, D. G., Pankratz, V. S., Cox, A., Cortez, D. S., Garrett, M. R., Joe, B., and Ryan, K. L. (2008). Is survival time after hemorrhage a heritable, quantitative trait? An initial assessment. Shock 29, 748–753.
Klemcke, H. G., Joe, B., Calderon, M. L., Rose, R., Oh, T., Aden, J., and Ryan, K. L. (2011). Genetic influences on survival time after severe hemorrhage in inbred rat strains. Physiol. Genomics 43, 758–765.
Koyama, S., Aibiki, M., Kanai, K., Fujita, T., and Miyakawa, K. (1988). Role of central nervous system in renal nerve activity during prolonged hemorrhagic shock in dogs. Am. J. Physiol. 254, R761–R769.
Koyama, S., Sawano, F., Matsuda, Y., Saeki, Y., Shibamoto, T., Hayashi, T. Jr., Matsubayashi, Y., and Kawamoto, M. (1992). Spatial and temporal differing control of sympathetic activities during hemorrhage. Am. J. Physiol. 262, R579–R585.
Lacolley, P. J., Pannier, B. M., Slama, M. A., Cuche, J. L., Hoeks, A. P., Laurent, S., London, G. M., and Safar, M. E. (1992). Carotid arterial haemodynamics after mild degrees of lower-body negative pressure in man. Clin. Sci. 83, 535–540.
Levine, B. D., Giller, C. A., Lane, L. D., Buckey, J. C., and Blomqvist, C. G. (1994). Cerebral versus systemic hemodynamics during graded orthostatic stress in humans. Circulation 90, 298–306.
Lucini, D., Mela, G. S., Malliani, A., and Pagani, M. (2002). Impairment in cardiac autonomic regulation preceding arterial hypertension in humans: insights from spectral analysis of beat-by-beat cardiovascular variability. Circulation 106, 2673–2679.
Lucini, D., Zuccotti, G., Malacarne, M., Scaramuzza, A., Riboni, S., Palombo, C., and Pagani, M. (2009). Early progression of the autonomic dysfunction observed in pediatric type 1 diabetes mellitus. Hypertension 54, 987–994.
Malliani, A., Pagani, M., Lombardi, F., and Cerutti, S. (1991). Cardiovascular neural regulation explored in the frequency domain. Circulation 84, 482–492.
Malpas, S. C., Evans, R. G., Head, G. A., and Lukoshkova, E. V. (1998). Contribution of renal nerves to renal blood flow variability during hemorrhage. Am. J. Physiol. 274, R1283–R1294.
Montano, N., Porta, A., Cogliati, C., Costantino, G., Tobaldini, E., Casali, K. R., and Iellamo, F. (2009). Heart rate variability explored in the frequency domain: a tool to investigate the link between heart and behavior. Neurosci. Biobehav. Rev. 33, 71–80.
Morillo, C. A., Eckberg, D. L., Ellenbogen, K. A., Beightol, L. A., Hoag, J. B., Tahvanainen, K. U., Kuusela, T. A., and Diedrich, A. M. (1997). Vagal and sympathetic mechanisms in patients with orthostatic vasovagal syncope. Circulation 96, 2509–2513.
Mosqueda-Garcia, R., Furlan, R., Fernandez-Violante, R., Desai, T., Snell, M., Jarai, Z., Ananthram, V., Robertson, R. M., and Robertson, D. (1997). Sympathetic and baroreceptor reflex function in neurally mediated syncope evoked by tilt. J. Clin. Invest. 99, 2736–2744.
Ninomiya, I., Nisimaru, N., and Irisawa, H. (1971). Sympathetic nerve activity to the spleen, kidney, and heart in response to baroceptor input. Am. J. Physiol. 221, 1346–1351.
Ocon, A. J., Medow, M. S., Taneja, I., and Stewart, J. M. (2011). Respiration drives phase synchronization between blood pressure and RR interval following loss of cardiovagal baroreflex during vasovagal syncope. Am. J. Physiol. Heart Circ. Physiol. 300, H527–H540.
Ogoh, S., Volianitis, S., Raven, P. B., and Secher, N. H. (2004). Carotid baroreflex function ceases during vasovagal syncope. Clin. Auton. Res. 14, 30–33.
Pagani, M., Lombardi, F., Guzzetti, S., Rimoldi, O., Furlan, R., Pizzinelli, P., Sandrone, G., Malfatto, G., Dell’orto, S., Piccaluga, E., Turiel, M., Baselli, G., Cerutti, S., and Malliani, A. (1986). Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagal interaction in man and conscious dog. Circ. Res. 59, 178–193.
Pagani, M., Montano, N., Porta, A., Malliani, A., Abboud, F. M., Birkett, C., and Somers, V. K. (1997). Relationship between spectral components of cardiovascular variabilities and direct measures of muscle sympathetic nerve activity in humans. Circulation 95, 1441–1448.
Parati, G., Mancia, G., Di Rienzo, M., and Castiglioni, P. (2006). Point: cardiovascular variability is/is not an index of autonomic control of circulation. J. Appl. Physiol. 101, 676–678; discussion 678–682.
Preiss, G., and Polosa, C. (1974). Patterns of sympathetic neuron activity associated with Mayer waves. Am. J. Physiol. 226, 724–730.
Radaelli, A., Perlangeli, S., Cerutti, M. C., Mircoli, L., Mori, I., Boselli, L., Bonaita, M., Terzoli, L., Candotti, G., Signorini, G., and Ferrari, A. U. (1999). Altered blood pressure variability in patients with congestive heart failure. J. Hypertens. 17, 1905–1910.
Rea, R. F., Hamdan, M., Clary, M. P., Randels, M. J., Dayton, P. J., and Strauss, R. G. (1991). Comparison of muscle sympathetic responses to hemorrhage and lower body negative pressure in humans. J. Appl. Physiol. 70, 1401–1405.
Rea, R. F., and Wallin, B. G. (1989). Sympathetic nerve activity in arm and leg muscles during lower body negative pressure in humans. J. Appl. Physiol. 66, 2778–2781.
Rickards, C. A., Ryan, K. L., and Convertino, V. A. (2010a). Characterization of common measures of heart period variability in healthy human subjects: implications for patient monitoring. J. Clin. Monit. Comput. 24, 61–70.
Rickards, C. A., Ryan, K. L., Ludwig, D. A., and Convertino, V. A. (2010b). Is heart period variability associated with the administration of lifesaving interventions in individual prehospital trauma patients with normal standard vital signs? Crit. Care Med. 38, 1666–1673.
Rickards, C. A., Ryan, K. L., Cooke, W. H., and Convertino, V. A. (2011). Tolerance to central hypovolemia: the influence of oscillations in arterial pressure and cerebral blood velocity. J. Appl. Physiol. 111, 1048–1058.
Rickards, C. A., Ryan, K. L., Cooke, W. H., Romero, S. A., and Convertino, V. A. (2008). Combat stress or hemorrhage? Evidence for a decision-assist algorithm for remote triage. Aviat. Space Environ. Med. 79, 670–676.
Ryan, K. L., Cooke, W. H., Rickards, C. A., Lurie, K. G., and Convertino, V. A. (2008). Breathing through an inspiratory threshold device improves stroke volume during central hypovolemia in humans. J. Appl. Physiol. 104, 1402–1409.
Ryan, K. L., Rickards, C. A., Hinojosa-Laborde, C., Cooke, W. H., and Convertino, V. A. (2011). Arterial pressure oscillations are not associated with muscle sympathetic nerve activity in individuals exposed to central hypovolaemia. J. Physiol. (Lond.) 589, 5311–5322.
Ryan, K. L., Rickards, C. A., Ludwig, D. A., and Convertino, V. A. (2010). Tracking central hypovolemia with ECG in humans: cautions for the use of heart period variability in patient monitoring. Shock 33, 583–589.
Sacha, J., and Pluta, W. (2008). Alterations of an average heart rate change heart rate variability due to mathematical reasons. Int. J. Cardiol. 128, 444–447.
Salmanpour, A., Brown, L. J., Steinback, C. D., Usselman, C. W., Goswami, R., and Shoemaker, J. K. (2011). Relationship between size and latency of action potentials in human muscle sympathetic nerve activity. J. Neurophysiol. 105, 2830–2842.
Sanders, J. S., and Ferguson, D. W. (1989). Profound sympathoinhibition complicating hypovolemia in humans. Ann. Intern. Med. 111, 439–441.
Sather, T. M., Goldwater, D. J., Montgomery, L. D., and Convertino, V. A. (1986). Cardiovascular dynamics associated with tolerance to lower body negative pressure. Aviat. Space Environ. Med. 57, 413–419.
Sauaia, A., Moore, F. A., Moore, E. E., Moser, K. S., Brennan, R., Read, R. A., and Pons, P. T. (1995). Epidemiology of trauma deaths: a reassessment. J. Trauma 38, 185–193.
Schadt, J. C., and Ludbrook, J. (1991). Hemodynamic and neurohumoral responses to acute hypovolemia in conscious mammals. Am. J. Physiol. 260, H305–H318.
Scherrer, U., Vissing, S., Morgan, B. J., Hanson, P., and Victor, R. G. (1990). Vasovagal syncope after infusion of a vasodilator in a heart-transplant recipient. N. Engl. J. Med. 322, 602–604.
Sethuraman, G., Ryan, K. L., Rickards, C. A., and Convertino, V. A. (2010). Ectopy in trauma patients: cautions for use of heart period variability in medical monitoring. Aviat. Space Environ. Med. 81, 125–129.
Skoog, P., Mansson, J., and Thoren, P. (1985). Changes in renal sympathetic outflow during hypotensive haemorrhage in rats. Acta Physiol. Scand. 125, 655–660.
Stewart, R. M., Myers, J. G., Dent, D. L., Ermis, P., Gray, G. A., Villarreal, R., Blow, O., Woods, B., McFarland, M., Garavaglia, J., Root, H. D., and Pruitt, B. A. Jr. (2003). Seven hundred fifty-three consecutive deaths in a level I trauma center: the argument for injury prevention. J. Trauma 54, 66–70; discussion 70–71.
Summers, R. L., Ward, K. R., Witten, T., Convertino, V. A., Ryan, K. L., Coleman, T. G., and Hester, R. L. (2009). Validation of a computational platform for the analysis of the physiologic mechanisms of a human experimental model of hemorrhage. Resuscitation 80, 1405–1410.
Sundlof, G., and Wallin, B. G. (1978a). Effect of lower body negative pressure on human muscle nerve sympathetic activity. J. Physiol. (Lond.) 278, 525–532.
Sundlof, G., and Wallin, B. G. (1978b). Human muscle nerve sympathetic activity at rest. Relationship to blood pressure and age. J. Physiol. (Lond.) 274, 621–637.
Tan, C. O., Cohen, M. A., Eckberg, D. L., and Taylor, J. A. (2009). Fractal properties of human heart period variability: physiological and methodological implications. J. Physiol. (Lond.) 587, 3929–3941.
Taneja, I., Moran, C., Medow, M. S., Glover, J. L., Montgomery, L. D., and Stewart, J. M. (2007). Differential effects of lower body negative pressure and upright tilt on splanchnic blood volume. Am. J. Physiol. Heart Circ. Physiol. 292, H1420–H1426.
Taylor, J. A., Halliwill, J. R., Brown, T. E., Hayano, J., and Eckberg, D. L. (1995). ‘Non-hypotensive’ hypovolaemia reduces ascending aortic dimensions in humans. J. Physiol. (Lond.) 483, 289–298.
Taylor, J. A., and Studinger, P. (2006). Counterpoint: cardiovascular variability is not an index of autonomic control of the circulation. J. Appl. Physiol. 101, 678–681; discussion 681.
Taylor, J. A., Williams, T. D., Seals, D. R., and Davy, K. P. (1998). Low-frequency arterial pressure fluctuations do not reflect sympathetic outflow: gender and age differences. Am. J. Physiol. 274, H1194–H1201.
Vaddadi, G., Esler, M. D., Dawood, T., and Lambert, E. (2010). Persistence of muscle sympathetic nerve activity during vasovagal syncope. Eur. Heart J. 31, 2027–2033.
Victor, R. G., and Leimbach, W. N. Jr. (1987). Effects of lower body negative pressure on sympathetic discharge to leg muscles in humans. J. Appl. Physiol. 63, 2558–2562.
Wallin, B. G., and Sundlof, G. (1982). Sympathetic outflow to muscles during vasovagal syncope. J. Auton. Nerv. Syst. 6, 287–291.
Ward, K. R., Tiba, M. H., Ryan, K. L., Filho, I. P., Rickards, C. A., Witten, T., Soller, B. R., Ludwig, D. A., and Convertino, V. A. (2010). Oxygen transport characterization of a human model of progressive hemorrhage. Resuscitation 81, 987–993.
Wijeysundera, D. N., Butler, G. C., Ando, S., Pollard, M., Picton, P., and Floras, J. S. (2001). Attenuated cardiac baroreflex in men with presyncope evoked by lower body negative pressure. Clin. Sci. 100, 303–309.
Wolthuis, R. A., Bergman, S. A., and Nicogossian, A. E. (1974). Physiological effects of locally applied reduced pressure in man. Physiol. Rev. 54, 566–595.
Keywords: hemorrhage, sympathetic activity, MSNA, baroreflex function, central hypovolemia, LBNP
Citation: Ryan KL, Rickards CA, Hinojosa-Laborde C, Cooke WH and Convertino VA (2012) Sympathetic responses to central hypovolemia: new insights from microneurographic recordings. Front. Physio. 3:110. doi: 10.3389/fphys.2012.00110
Received: 16 February 2012; Paper pending published: 15 March 2012;
Accepted: 03 April 2012; Published online: 26 April 2012.
Edited by:
Elisabeth Lambert, Baker IDI Heart and Diabetes Institute, AustraliaReviewed by:
Roland Veelken, University of Erlangen Nuremberg, GermanyDeborah A. Scheuer, University of Florida, USA
Copyright: © 2012 Ryan, Rickards, Hinojosa-Laborde, Cooke and Convertino. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Kathy L. Ryan, Research Division, U.S. Army Institute of Surgical Research, 3698 Chambers Pass, Fort Sam Houston, TX 78234, USA. e-mail: kathy.ryan@amedd.army.mil