 Eleonora Grandi
Eleonora Grandi Anthony W. Herren
Anthony W. Herren- Department of Pharmacology, University of California at Davis, Davis, CA, USA
Na+ homeostasis is a key regulator of cardiac excitation and contraction. The cardiac voltage-gated Na+ channel, NaV1.5, critically controls cell excitability, and altered channel gating has been implicated in both inherited and acquired arrhythmias. Ca2+/calmodulin-dependent protein kinase II (CaMKII), a serine/threonine kinase important in cardiac physiology and disease, phosphorylates NaV1.5 at multiple sites within the first intracellular linker loop to regulate channel gating. Although CaMKII sites on the channel have been identified (S516, T594, S571), the relative role of each of these phospho-sites in channel gating properties remains unclear, whereby both loss-of-function (reduced availability) and gain-of-function (late Na+ current, INaL) effects have been reported. Our review highlights investigating the complex multi-site phospho-regulation of NaV1.5 gating is crucial to understanding the genesis of acquired arrhythmias in heart failure (HF) and CaMKII activated conditions. In addition, the increased Na+ influx accompanying INaL may also indirectly contribute to arrhythmia by promoting Ca2+ overload. While the precise mechanisms of Na+ loading during HF remain unclear, and quantitative analyses of the contribution of INaL are lacking, disrupted Na+ homeostasis is a consistent feature of HF. Computational and experimental observations suggest that both increased diastolic Na+ influx and action potential prolongation due to systolic INaL contribute to disruption of Ca2+ handling in failing hearts. Furthermore, simulations reveal a synergistic interaction between perturbed Na+ fluxes and CaMKII, and confirm recent experimental findings of an arrhythmogenic feedback loop, whereby CaMKII activation is at once a cause and a consequence of Na+ loading.
Cardiac Na+ Handling
In cardiac myocytes, intracellular Na+ concentration ([Na+]i) is a key modulator of Ca2+ cycling, contractility and metabolism, and is controlled by the balance between Na+ influx and extrusion (Figure 1). The major contributors to Na+ entry during the cardiac cycle are the Na+/Ca2+ exchanger (NCX), the voltage-dependent Na+ channel (NaV), and the Na+/H+ exchanger (NHE) with relative contributions of NCX > NaV > NHE (see Figure 1). Smaller amounts of Na+ also enter the cell via the Na+/HCO3- and the Na+/K+/2Cl- cotransporters, and the Na+/Mg2+ exchanger. Na+ efflux is controlled primarily through the Na+/K+ ATPase (NKA) that keeps [Na+]i constant at steady-state [reviewed in (Despa and Bers, 2013)].
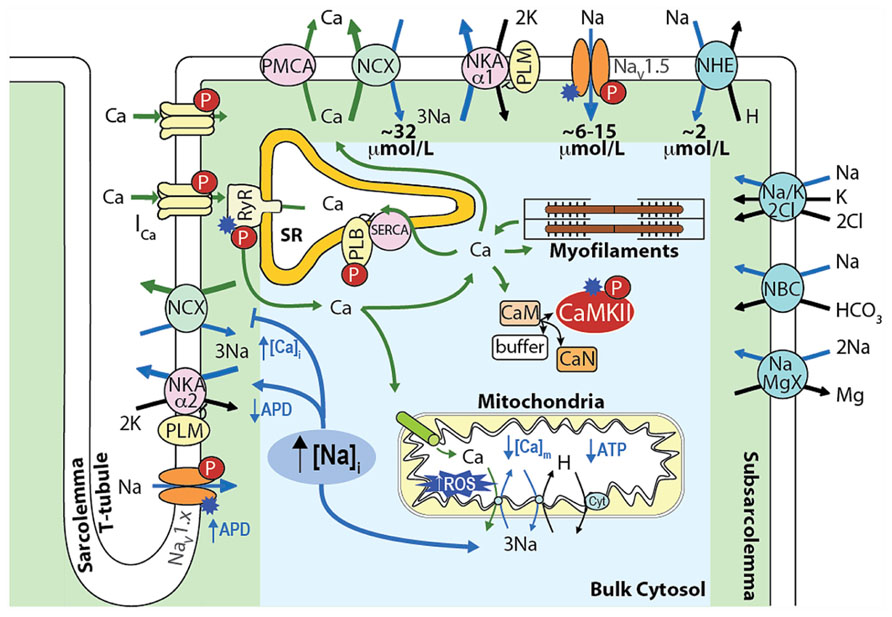
FIGURE 1. Schematic showing the main processes regulating [Na+]i and [Ca2+]i homeostasis in cardiac myocytes and the mechanisms by which an increase in [Na+]i affects [Ca2+]i, contractility, and metabolism. Upon cardiac myocyte electrical excitation, NaV opening initiates the AP upstroke and allows Na+ entry (limited to 6–15 μmol/L by rapid inactivation). Em depolarization causes LTCC openings and consequent Ca2+-induced SR Ca2+ release, which triggers contraction. During relaxation, Ca2+ is reuptaken into the SR by SERCA, and extruded out of the cytosol through NCX, which extrudes the ~10 μmol/L Ca2+ that enters via LTCCs and leads to an increase in [Na+]i by ~32 μmol/L during each AP. NHE brings in ~2 μmol/L Na+ at physiological intracellular pH (~16 μmol/L during intracellular acidosis). The Na+/HCO3- and the Na+/K+/2Cl- cotransporters, and the Na+/Mg2+ exchanger contribute minimally to the total Na+ entry (~40–45 μmol/L), which is then extruded by NKA [reviewed in (Despa and Bers, 2013)].
An integrative approach to cellular Na+ handling is critical to understand how these pathways interact spatially and temporally to affect cardiac cell function. Indeed, compelling evidence has accumulated that local [Ca2+]i and [Na+]i gradients exist close to the cell membrane (Carmeliet, 1992) that depend on the spatial localization of specific Na+ and Ca2+ handling proteins and their isoforms (Figure 1). For example, NKA-α2 is more concentrated at the t-tubules (whereas NKA-α1 is homogenously distributed), and this could be important in regulating local cleft [Na+]i and [Ca2+]i (Berry et al., 2007; Despa and Bers, 2007). NCX is concentrated at the t-tubules, but only a small NCX fraction colocalizes with proteins specific to the dyadic cleft [i.e., L-type Ca2+ channels (LTCCs) and ryanodine receptors (RyRs; Jayasinghe et al., 2009)]. Nevertheless, functional data indicate that NCX senses an early and high rise in local vs. bulk [Ca2+]i (Weber et al., 2003a) and Ca2+ entry via reverse mode NCX can even trigger sarcoplasmic reticulum (SR) Ca2+ release (Litwin et al., 1998). Moreover, a loss-of-function mutation in ankyrin-B results in reduced NKA and NCX protein levels and impaired targeting to the t-tubules, which causes altered Ca2+ signaling and after contractions (Mohler et al., 2003) and can affect local [Na+], cellular and SR Ca2+ cycling (Camors et al., 2012), and kinase/phosphatase balance (DeGrande et al., 2012).
Cardiac Na+ channel (NaV1.5) activity also requires proper sarcolemmal localization. For example, defective membrane targeting by impaired interaction of NaV1.5 and ankyrin-G causes Brugada syndrome (BrS; Mohler et al., 2004). NaV1.5 are found roughly homogenously distributed in the t-tubules and external sarcolemma, whereas non-cardiac NaV isoforms (whose role is still poorly understood) are more concentrated at the t-tubules (Brette and Orchard, 2006). However, immunofluorescence studies show NaV1.5 mostly at the intercalated disks, and it has been proposed that different pools of Na+ channels are located in the external sarcolemma vs. intercalated disks, where they interact within different macromolecular complexes and are regulated differently (Lin et al., 2011; Petitprez et al., 2011; Sato et al., 2011).
Altered Na+ homeostasis through the mechanisms described above (and in Figure 1) contributes to action potential (AP) and [Ca2+] cycling dysregulation, leading to arrhythmia, metabolic imbalance, remodeling and cell death, and is a hallmark of various cardiac diseases. The search for a common denominator has pointed to the Ca2+/calmodulin-dependent protein kinase II (CaMKII). CaMKII is a basophilic serine/threonine kinase that plays critical roles in cardiac physiology and disease (where it is often found hyperactive) through phosphorylation of several Ca2+ handling proteins and ion channels (Figure 1; Bers and Grandi, 2009; Anderson et al., 2011). NaV1.5, in particular, has emerged as a principle CaMKII target. Herein, we summarize growing evidence indicating an important role for CaMKII in regulating NaV1.5 gating and cardiomyocyte Na+ homeostasis, with an emphasis on the important interconnection between Na+ and Ca2+ handling, and consequences for arrhythmia.
Cardiac Na+ Channel Structure and Function
The cardiac Na+ channel is responsible for the upstroke of the cardiac AP and is a critical determinant of cardiac electrical excitability. The pore forming α-subunit, NaV1.5, is composed of four core domains (DI–DIV), each containing six transmembrane segments (S1–S6; ~230 kD; Figure 2). The positively charged S4 segments serve as the channel voltage sensors and the S5–S6 segments comprise the ion-conducting pore. Upon depolarization, the channel rapidly activates (<1 ms) producing a large (tens of nA), transient inward Na+-current, INa, and then undergoes fast inactivation (within a few ms) through interactions of the well-described DIII–IV linker IFM motif with the pore (similar to N-type inactivation in K+ channels). This is followed by poorly understood slower modes of inactivation (hundreds of ms to a few s) that likely involve rearrangements of the pore structure (similar to C-type inactivation in K+ channels). Channels that fail to completely inactivate, or close and then reopen, give rise to a persistent, or late Na+ current (INaL). NaV1.5α forms macromolecular complexes with β-subunits (β1 to β4) and many other accessory and regulatory proteins that modify channel gating [extensively reviewed by Abriel (2010); Wilde and Brugada (2011)]. The intracellular N- and C-termini and linker loops connecting DI–IV are also all involved in channel gating. In particular, the NaV1.5 I–II cytoplasmic linker loop is the subject of extensive post-translational regulation, and this region has received much attention as a hot spot for phosphorylation by CaMKII [reviewed in (Herren et al., 2013)].
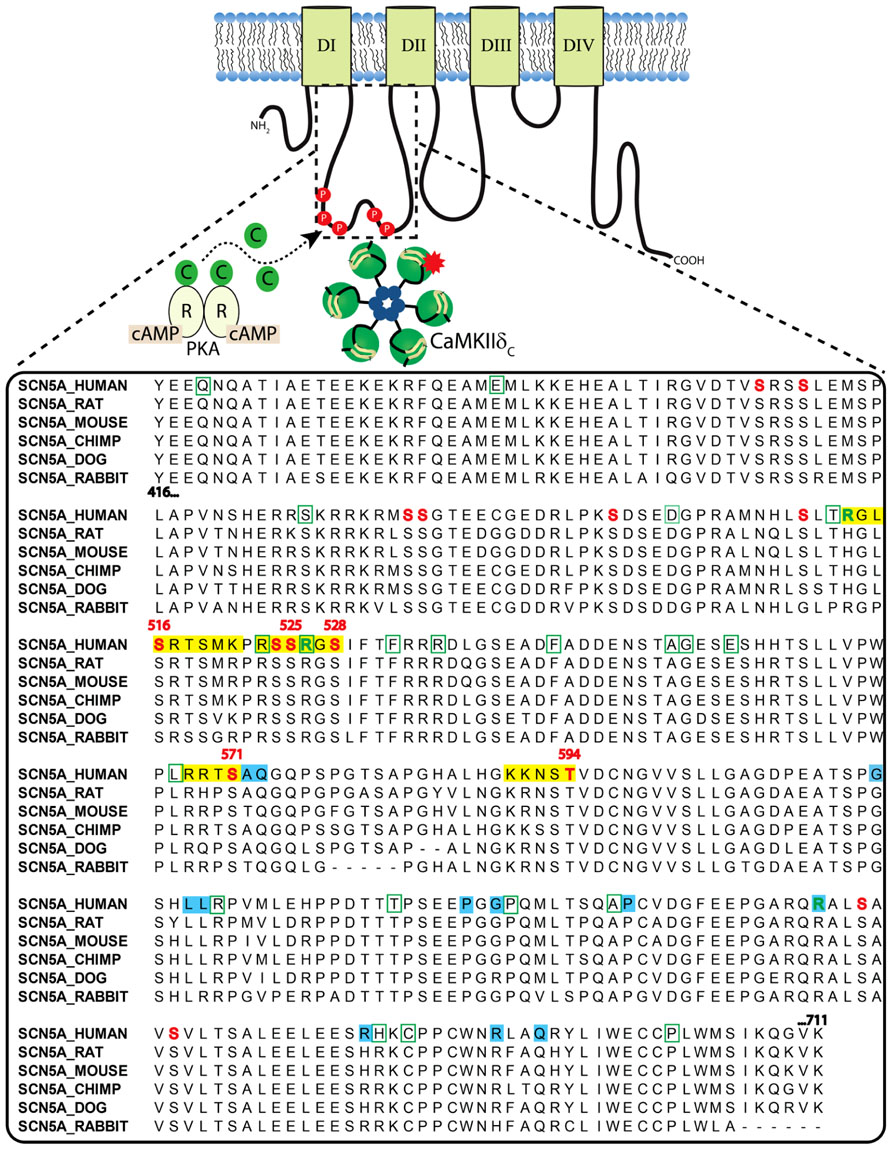
FIGURE 2. Clustal sequence alignment of NaV1.5 I–II loop across indicated species. Sites are color coded as follows: red font = phosphorylation; green font = methylation; yellow block = basophilic kinase consensus region; green bounding box = missense BrS mutation; blue block = LQTS mutation.
Phosphorylation of NaV1.5 by CaMKII
CaMKII activation has been shown to alter the gating properties of the Na+ channel, as summarized in Table 1. Specifically, CaMKII phosphorylation of NaV1.5 shifts the voltage dependence of inactivation to negative potentials without affecting channel activation, slows recovery from and enhances entry into slower forms of inactivation, and increases INaL (Wagner et al., 2006).
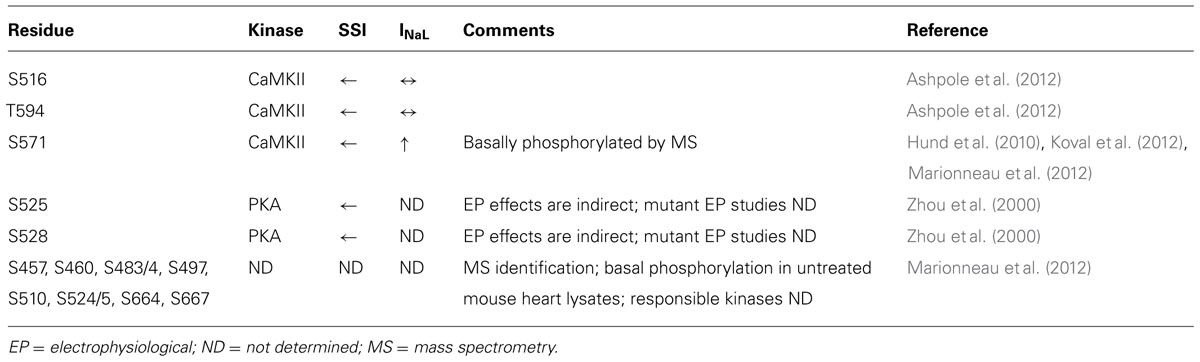
TABLE 1. NaV1.5 phosphorylation sites, their associated kinases, and biophysical effects.
The NaV1.5 I–II linker loop interacts with CaMKII (Ashpole et al., 2012) and contains multiple basophilic kinase consensus sequences. Mass spectrometry analysis of wild type NaV1.5 (purified from mouse heart lysates) revealed several sites within this loop that are basally phosphorylated (Marionneau et al., 2012; Figure 2, red color, and Table 1). Hund et al. (2010) mutated putative CaMKII consensus sites to non-phosphorylatable alanine residues and determined that CaMKII specifically phosphorylates S571. When expressed in a heterologous expression system, the S571A non-phosphorylatable mutant abolished constitutively active co-expressed CaMKII enhancement of channel inactivation and INaL. On the other hand, phosphomimetic S571E recapitulated CaMKII effects (but still in the presence of constitutively active CaMKII). S571 phosphorylation was increased in murine (Toischer et al., 2013) and human heart failure (HF) and canine ischemic cardiomyopathy (Koval et al., 2012).
Because CaMKII (and other kinases) can phosphorylate non-canonical sequences, we screened the entire NaV1.5 I–II loop for CaMKII phosphorylation with an in vitro kinase assay system (Ashpole et al., 2012). This assay consisted of a tiled peptide array spanning the entire loop region followed by in vitro phosphorylation with recombinant CaMKII and revealed that site S516 was phosphorylated most efficiently by CaMKII (Ashpole et al., 2012). Immobilized peptides containing S483/S484 and S593/T594 were also phosphorylated in this assay at much lower efficiency. It is important to consider peptide conformation may change when a peptide is immobilized or soluble, which can affect kinase access to phospho-acceptor sites (Bayer et al., 2006). In fact, subsequent studies using soluble peptides showed that T594 but not S483/4 could be phosphorylated, although only at low efficiencies. Furthermore, peptide conformation and kinase binding may be different for the full-length channel, thus affecting its ability to be phosphorylated. Therefore, additional phosphorylation studies using full-length NaV1.5 are needed.
Wild type or non-phosphorylatable mutant channels (S516A, T594A, S571A) were coexpressed with CaMKIIδC in HEK293 cells and voltage-clamped under pipette conditions to acutely activate CaMKII (with Ca2+ and calmodulin), with or without CaMKII inhibition [by autocamtide-2 inhibitory peptide (AIP); Ashpole et al., 2012]. CaMKII shifted steady-state inactivation (SSI) to hyperpolarizing potentials and increased entry into inactivation, and this was blocked with AIP or by mutating these sites to non-phosphorylatable alanine. Phosphomimetic S516E and T594E mutants recapitulated the hyperpolarizing shift in SSI even in the absence of CaMKII and presence of AIP. In our hands, however, S571E phosphomimetic mutants showed no statistically significant negative shift in SSI. Moreover, we observed no enhancement of late INaL in any of the mutants tested, but we did not coexpress β-subunits, which some have indicated to be important for INaL (Maltsev et al., 2009). Importantly, the phosphorylation status of S516 and T594 in native myocytes is yet to be determined, and more studies are needed to determine the relative contribution of these sites to specific channel gating properties.
Functional Consequences of I–II Loop Phosphorylation: Insight from Inherited Mutations
Structure-function studies of SCN5A channelopathies/inherited mutations may further an understanding of the functional consequences of NaV1.5 phosphorylation. CaMKII-dependent alterations of NaV1.5 gating are remarkably similar to those caused by the NaV1.5 mutation 1795insD (Bezzina et al., 1999), which is associated to patients with mixed long QT syndrome (LQTS) and BrS phenotypes. DelK1500 is another overlap mutation that causes both loss and gain of function channel effects and is functionally similar to the effects of CaMKII phosphorylation on NaV1.5 (Grant et al., 2002). Experiments and simulations have shown how the heart rate acts as a switch imparting LQTS or BrS phenotypes to the same genotype (Veldkamp et al., 2000; Clancy and Rudy, 2002), and we have hypothesized a similar scenario for CaMKII effects (Grandi et al., 2007). However, 1795insD and delK1500 are present on the NaV1.5 C-terminus and III–IV loop respectively, making structural correlation with CaMKII phosphorylation on the I–II loop difficult. To date, no overlap mutations have been identified anywhere within the I–II loop (Remme et al., 2008).
While no overlap mutations are present, several mutations or polymorphisms have been identified within the I–II loop phosphorylation hot spot through studies of LQTS and BrS patient cohorts. Examination of these mutations may be useful in dissecting out the structure-function relationship of kinase phosphorylation within this region. More than 30 putative BrS mutations have been identified in the I–II loop from residues 416–711 (Kapplinger et al., 2010; Figure 2). Unfortunately, not all of these have been followed up with functional studies. One mutation, L567Q, was identified in a family exhibiting BrS and sudden infant death syndrome (Priori et al., 2000). When expressed heterologously, the mutant NaV1.5 channel displayed a negative shift in inactivation and enhanced entry into inactivation that was not dependent on coexpression of β-subunits (Wan et al., 2001). Another mutation, T512I, was identified in a patient exhibiting cardiac conduction disease, and resulted in hyperpolarizing shifts in SSI and activation and enhanced slow inactivation (Viswanathan et al., 2003). Not only is this mutation juxtaposed to one of the known CaMKII phosphorylation consensus regions identified at R513 and phosphorylated at S516 [see Figure 2 and (Ashpole et al., 2012)], it also functionally mirrors the enhanced inactivation conferred by CaMKII phosphorylation at S516. Furthermore, PKA phosphorylation of nearby S525 and S528 (Murphy et al., 1996) has been previously described to similarly shift the voltage dependence of inactivation to negative potentials (Zhou et al., 2000). Thus, phosphorylation by either PKA (S525, S528) or CaMKII (S516) within a short ~10 amino acid stretch results in similar channel biophysics compared with the loss of function BrS mutation identified at T512. Moreover, a recent proteomics study demonstrated that residues R513 and R526 within this same region can be methylated, but the functional effect of this post-translational modification is unknown (Beltran-Alvarez et al., 2011). Another mutation, G514C, was identified in a family with cardiac conduction disease. Under voltage clamp, this mutation displayed a mixed biophysical phenotype of destabilized inactivation and decreased activation (Tan et al., 2001). These observations cement the importance of this short region in voltage-dependent inactivation of the channel.
CaMKII-dependent phosphorylation at S571 has been shown to result in both loss and gain of channel function (Hund et al., 2010). The functional effects of a negative charge at this site from CaMKII phosphorylation were suggested to phenocopy the adjacent inherited LQTS charge mutations at A572D and Q573E (Koval et al., 2012). Although A572D was initially identified as a causative LQTS mutation (Tester et al., 2005), a subsequent study showed that this mutation is actually a benign variant and does not cause LQTS in and of itself (Tester et al., 2010). Similarly, a mutation identified in a LQTS patient at 619 was also attributed to an observed LQTS phenotype in this patient. This L619F mutation was found to induce INaL when expressed in a heterologous expression system (Wehrens et al., 2003). However, a LQTS screening study from the Roden group failed to detect any late INaL (or gating changes) from L618F mutants expressed in heterologous cells and concluded this mutation is also a benign variant (Yang et al., 2002). Indeed, mutations within the NaV1.5 I–II loop have poor disease predictive value stemming from the high incidence of benign variants within this region (Kapa et al., 2009).
Thus, a clear and consistent role for the NaV1.5 I–II loop in loss of function inactivation gating emerges from studies of kinase phosphorylation (both CaMKII and PKA) and inherited mutations within this hotspot. There is less evidence for this loop region mediating INaL effects, with different groups providing contradictory results. It remains to be determined whether other unidentified CaMKII phospho-sites exist on other intracellular loops of NaV1.5 that may mediate (contribute to) enhancement of INaL (e.g., the C-terminus (Coyan et al., 2014) or III–IV loop where overlap mutations have been identified). It is also conceivable that INaL enhancement in pathological conditions could be due to an increase in neuronal Na+ channel isoforms with higher fractional INaL (Xi et al., 2009; Biet et al., 2012; Yang et al., 2012; Toischer et al., 2013). Alternatively, the effect of CaMKII to augment INaL may require accessory proteins not present in heterologous cell systems [such as regulatory β subunits (Maltsev et al., 2009)]. Indeed, this emphasizes the need for studies of these phosphorylation sites in native adult cardiomyocytes. Human induced pluripotent stem cells may also be used as suitable models, as has been done for some inherited mutations, such as 1795insD (Davis et al., 2012).
Enhanced inactivation and consequent reduction in channel availability due to CaMKII phosphorylation is expected to contribute to re-entrant arrhythmias from slowed conduction and enhanced dispersion of repolarization [as we have extensively reviewed previously (Herren et al., 2013)]. On the other hand, CaMKII enhancement of INaL, while not as well understood mechanistically, can also lead to arrhythmias arising from prolonged AP duration (APD) that makes the cell more vulnerable to triggered activity [via early after-depolarizations (EADs)]. EADs are favored by conditions leading to prolongation of the AP plateau within a voltage range permitting recovery from inactivation and reactivation of LTCC, or can be the consequence of SR Ca2+ release and consequent augmentation of NCX current. A novel and unique mechanism underlying phase 3 EADs in ventricular myocytes from CaMKIIδC overexpressing mice has been recently described (Edwards et al., 2012), which involves isoproterenol-induced exaggerated Ca2+ release, increased inward NCX current, and non-equilibrium reactivation of fast INa.
INaL and Cellular Na+ Loading in HF
In addition to directly affecting myocytes electrical stability, INa is a major pathway of Na+ influx into cardiac myocytes, although NCX plays the most dominant role both at rest and in contracting cells [see Figure 1 and (Despa and Bers, 2013)]. In normal myocytes INaL contribution to Na+ entry is limited, but when INaL is enhanced in diseased conditions (such as cardiac hypertrophy and HF) it carries as much Na+ as the fast INa transient, thus increasing total cellular Na+ influx during a cardiac cycle and potentially contributing to Na+ overload (Makielski and Farley, 2006; Despa and Bers, 2013). Indeed, intracellular Na+ concentration ([Na+]i) is increased in myocytes from failing hearts compared to non-failing myocytes by 2–6 mM (Pogwizd et al., 2003; Shryock et al., 2013), but the precise mechanism(s) remains unclear (Figure 3). It has been suggested that CaMKII and enhanced INaL contribute to [Na+]i elevation in HF, as CaMKIIδC overexpressing mice with HF exhibit prominent INaL and have increased [Na+]i (Wagner et al., 2006).
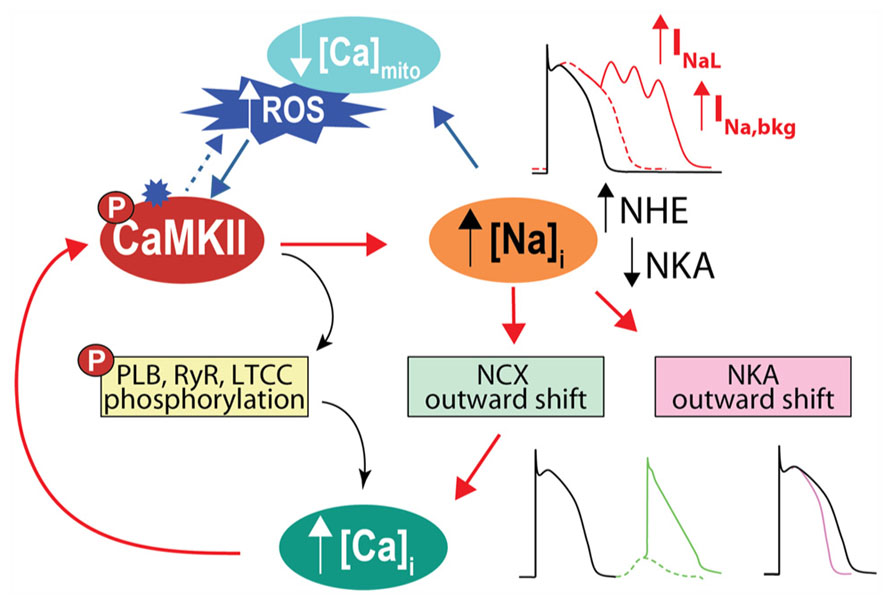
FIGURE 3. CaMKII activation and Na+ homeostasis are intimately related in the regulation of cellular Ca2+, contractility and electrical stability. With CaMKII hyperactivity, increase in late INa favors AP prolongation and EADs. Cellular Na+ loading increases Ca2+, via outward shift in NCX, which further activates CaMKII, fueling a vicious cycle that favors spontaneous SR Ca2+ release and predisposes to Ca2+-related arrhythmia. Elevated [Na+]i can also influence ROS production by affecting mitochondrial [Ca2+]i. A less appreciated observation is that high [Na+]i by causing outward shifts in both NCX and NKA will tend to shorten the AP, as predicted by computational AP models (Grandi et al., 2010, 2011). We speculate that the increased [Na+]i in HF may limit AP prolongation caused by reduced K+ channel conductance and increased INaL.
While the increase in INaL may play a role in the observed [Na+]i loading in these transgenic mice (Wagner et al., 2006), we demonstrated that CaMKII-dependent enhancement of INaL is not quantitatively sufficient to account for the [Na+]i elevation observed in HF (Grandi et al., 2007; Wagner et al., 2011; Moreno et al., 2013; Morotti et al., 2014). Additionally, in a rabbit model of pressure- and volume-overload-induced HF, Despa et al. (2002) reported an increased Na+ influx resulting primarily from a TTX-sensitive pathway, which accounted for a gain in [Na+]i of ~3 mM, and was present both in resting and electrically stimulated cells. This supports the existence of a diastolic Na+ influx in failing myocytes responsible for [Na+]i elevation, although it is currently unclear whether this diastolic influx is carried by NaVs [whether they be of cardiac, neuronal, or skeletal muscle isoforms (Biet et al., 2012; Yang et al., 2012)] or by which gating mechanism. Increases of Na+ window and/or background current can potentially contribute to increased Na+ influx in failing vs. normal myocytes [and could be modified by drugs affecting the voltage dependence of Na+ channel gating (Shryock et al., 2013)]. We showed that simulation of an increased sarcolemmal Na+ leak current allows recapitulating the [Na+]i gain seen in HF (Wagner et al., 2011; Moreno et al., 2013; Morotti et al., 2014). Baartscheer et al. (2003) using the same pressure- and volume-overload rabbit HF model in Despa et al. (2002) found that increased Na+ influx via NHE was the largest contributor to the elevated Na+ influx rate and [Na+]i in paced myocytes. Interestingly, CaMKII activates NHE (Vila-Petroff et al., 2010). Reduced Na+ extrusion could also contribute to the observed intracellular Na+ gain during HF. Despite reduced expression, NKA function is unchanged in HF (Despa et al., 2002; Baartscheer et al., 2003), which is possibly due to higher NKA function secondary to reduced relative expression and elevated phosphorylation of phosholemman (Bossuyt et al., 2005).
Consequences of Na+ Loading on Excitation-Contraction Coupling
[Na+]i elevation in HF is expected to limit Ca2+ extrusion via forward mode NCX, and could even favor Ca2+ entry via reverse mode NCX (Weber et al., 2003b; Figures 1 and 3). Slowing Ca2+ extrusion via NCX will tend to elevate diastolic [Ca2+]i, thereby contributing to diastolic dysfunction. The slowed [Ca2+]i decline and elevated diastolic [Ca2+]i will also tend to increase SR Ca2+ content, thus exerting a positive inotropic effect and enhancing contractility. This explains the efficacy of cardiac glycosides in the treatment of congestive HF. These compounds promote inotropy by selectively inhibiting NKA and thereby impairing Na+ extrusion and weakening the NCX Ca2+ extrusion gradient. However, they are well known for having undesired arrhythmic consequences (Altamirano et al., 2006) by increasing the propensity for spontaneous SR Ca2+ release and delayed after-depolarizations (DADs). DADs arise from a transient inward current Iti through forward mode NCX, which is evoked by the sudden increase in [Ca2+]i. It has been recently proposed that CaMKII is mechanistically involved in glycoside-induced arrhythmogenesis, as ouabain increased CaMKII activity, and CaMKII inhibition significantly reduced ouabain-induced spontaneous contractile activity and Ca2+ waves (Gonano et al., 2011). CaMKII overexpression exacerbated ouabain-induced spontaneous contractile activity (Gonano et al., 2011), possibly by favoring spontaneous SR Ca2+ release and DADs, as demonstrated in a recent computational mouse model (Morotti et al., 2014). Ouabain-induced [Na+]i loading has also been shown to result in apoptosis (Sapia et al., 2010), mediated by reverse mode NCX-dependent activation of CaMKII. Thus, CaMKII inhibition may have potential therapeutic benefit during glycoside treatment to prevent arrhythmia and cell death while minimally impacting the positive inotropic effect. Analogously, the Na+ channel opener ATX-II induces spontaneous diastolic Ca2+ release from the SR and DADs in myocytes (Song et al., 2008) and arrhythmia in Langendorff perfused hearts (Yao et al., 2011), which is attenuated with CaMKII or INaL inhibition.
Our recent model of the failing human ventricular myocyte confirmed that a 20% increase in [Na+]i in HF compared to control conditions slows forward mode inward NCX (Ca2+ extrusion) and enhances reverse mode NCX (Ca2+ entry, at the beginning of the AP; Moreno et al., 2013). This, coupled with AP prolongation due to extensive ionic remodeling in HF (including enhanced INaL), increased diastolic [Ca2+]i while maintaining adequate SR Ca2+ load and Ca2+ transient despite reduced SR Ca2+-ATPase (SERCA) function. However, [Na+]i-induced Ca2+ enhancement, in combination with hyperphosphorylated RyRs, causes diastolic SR Ca2+ release, Iti, and triggered APs [Figure 3, also favored by a more depolarized resting membrane potential (Em) in HF, due to decreased IK1 and increased background Na+ current]. Simulations showed that by targeting pathological late Na+ current and diastolic Na+ influx, ranolazine (1) limits [Na+]i thus restoring normal NCX forward mode that speeds up Ca2+ extrusion, (2) shortens APD, thus further limiting Ca2+ entry, and (3) hyperpolarizes the Em, which elevates the threshold of triggered APs. These simulation results are consistent with recent experimental data in human failing myocytes from hypertrophic cardiomyopathy samples (Coppini et al., 2013). Similarly, CaMKII-dependent [Na+]i elevation (normalized by ranolazine) has been associated with diastolic dysfunction and arrhythmias in CaMKIIδC overexpressing mice with HF (Sossalla et al., 2011). Taken together, these observations suggest that limiting [Na+]i overload may be an appropriate antiarrhythmic therapeutic for the prevention of diastolic tension and arrhythmia triggers driven by [Ca2+]i loading.
Arrhythmogenic CaMKII–Na+–Ca2+–CaMKII Feedback
It has been proposed that [Ca2+]i loading caused by elevated [Na+]i in HF may positively feed back to further activate CaMKII (and enhance target phosphorylation) thus creating an arrhythmogenic vicious circle. This positive feedback from Na+ to Ca2+ to CaMKII to Na+ has been qualitatively described for Na+ loading induced by ATX-II or accompanying a LQT3 mutation associated with increased INaL (Yao et al., 2011). Both of these conditions increased [Ca2+]i, induced CaMKII activation, increased CaMKII-dependent phosphorylation of phosholamban and RyRs, and favored arrhythmias. Although a CaMKII-dependent increase in NaV phosphorylation was not directly confirmed in that study, a [Ca2+]i-dependent increase in INaL involving both CaMKII and PKC has previously been demonstrated (Ma et al., 2012). Accordingly, blockade of INa or INaL with TTX or ranolazine reversed all these effects, as did CaMKII inhibition via AIP or KN-93 (Yao et al., 2011). Our recently developed mouse myocyte model showed that alterations in Na+ handling accompanying transgenic CaMKII overexpression (namely, increase in systolic INaL and diastolic Na+ leak) can increase intracellular Na+ gain to initiate Ca2+ overload and CaMKII activation. Furthermore, this mechanism proved to be quantitatively sufficient to further disrupt Ca2+ (and Na+) homeostasis and promote cellular arrhythmias (Morotti et al., 2014). The simulated effect of Na+ loading to fuel CaMKII-Na+-Ca2+-CaMKII feedback was even more striking when CaMKII was further activated by isoproterenol. This is consistent with the observation that β-adrenergic stimulation of myocytes isolated from mice overexpressing CaMKII (Sag et al., 2009) or subjected to TAC-induced HF (Toischer et al., 2013) increased the number of DADs, which were largely prevented by either ranolazine or AIP (Toischer et al., 2013).
Consequences of [Na+]i Loading on Cardiac Energetics
[Na+]i is also important in regulating cardiac myocyte bioenergetics, by controlling mitochondrial [Ca2+] via the mitochondrial NCX, and critically regulating the production of mitochondrial reactive oxygen species (ROS, Figure 1). High [Na+]i in HF may negatively affect cardiac metabolism during rapid pacing, as elevated [Na+]i has been shown to impair frequency-induced mitochondrial [Ca2+] accumulation thereby decreasing NADH/NAD+ redox potential, and increasing H2O2 formation in myocytes from failing hearts (Liu and O’Rourke, 2008). Notably, higher oxidative stress may further exacerbate [Na+]i loading by enhancing INaL through direct effects on NaV1.5 or secondarily by activating CaMKII (Wagner et al., 2011). This suggests the interesting notion that ROS, like Na+, is both a cause and consequence of CaMKII activation. Not only does oxidative stress activate CaMKII directly (Erickson et al., 2008), but CaMKII-induced [Na+]i loading can induce an increase in ROS (Figures 1 and 3). This parallels recent evidence indicating that CaMKII regulates ROS production (Pandey et al., 2011; Sepulveda et al., 2013; Zhu et al., 2014). Li et al. (2012) put forward an intriguing model-based hypothesis that Na+ accumulation in the SERCA knock-out mouse, which occurs secondary to marked NCX upregulation and intracellular acidosis, might play a role in the development of HF in these animals. This mechanism acts by initiating a reinforcing cycle involving a mismatch between ATP supply and demand, increasingly compromised metabolism, decreased intracellular pH, and further elevation of [Na+]i due to NHE, which may shift the time of transition from compensated to decompensated function.
Concluding Remarks
CaMKII-dependent loss and gain of function effects on NaV1.5 gating are pro-arrhythmic and functionally phenocopy SCN5A inherited mutations causing LQTS and BrS. Analogous to BrS mutations, CaMKII-dependent loss of Na+ channel function and consequent reduction in Na+ channel availability slows cardiac conduction and increases the propensity for conduction block and re-entry. Evidence points to the I–II loop phosphorylation hot spot as a likely mediator of these loss-of-function effects. Conversely, CaMKII enhancement of INaL, while not as well understood, may prolong the APD and predispose to lethal ventricular tachyarrhythmia (as in LQTS). Further study of these mechanisms may provide important advances for rational design of novel antiarrhythmic therapeutics. Additionally, experimental data and quantitative findings support the intriguing hypothesis that drugs designed to correct aberrant Na+ channel gating behavior, causing INaL and cellular Na+ loading, may act importantly by reducing Ca2+ overload. Growing evidence suggests that prevention of Na+-dependent Ca2+ overload is a key mechanism of action for these compounds (Sossalla et al., 2011; Yao et al., 2011). Inhibition of intracellular Na+ loading can contribute to normalizing Ca2+ and Em homeostasis in HF, but should not be achieved at the expense of systolic function. Inhibition of CaMKII or its targets may be an attractive means of facilitating these outcomes.
Author Contributions
Eleonora Grandi and Anthony W. Herren wrote the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the Fondation Leducq Transatlantic CaMKII Alliance and National Institutes of Health training grant T32-GM-099608 (to Anthony W. Herren). We are grateful to Drs. Donald M. Bers and Daniel C. Bartos for critical review of the manuscript.
References
Abriel, H. (2010). Cardiac sodium channel Na(v)1.5 and interacting proteins: physiology and pathophysiology. J. Mol. Cell. Cardiol. 48, 2–11. doi: 10.1016/j.yjmcc.2009.08.025
Altamirano, J., Li, Y., Desantiago, J., Piacentino, V. III, Houser, S. R., and Bers, D. M. (2006). The inotropic effect of cardioactive glycosides in ventricular myocytes requires Na+-Ca2+ exchanger function. J. Physiol. 575, 845–854. doi: 10.1113/jphysiol.2006.111252
Anderson, M. E., Brown, J. H., and Bers, D. M. (2011). CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell. Cardiol. 51, 468–473. doi: 10.1016/j.yjmcc.2011.01.012
Ashpole, N. M., Herren, A. W., Ginsburg, K. S., Brogan, J. D., Johnson, D. E., Cummins, T. R., et al. (2012). Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J. Biol. Chem. 287, 19856–19869. doi: 10.1074/jbc.M111.322537
Baartscheer, A., Schumacher, C. A., Van Borren, M. M., Belterman, C. N., Coronel, R., and Fiolet, J. W. (2003). Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc. Res. 57, 1015–1024. doi: 10.1016/S0008-6363(02)00809-X
Bayer, K. U., Lebel, E., Mcdonald, G. L., O’Leary, H., Schulman, H., and De Koninck, P. (2006). Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J. Neurosci. 26, 1164–1174. doi: 10.1523/JNEUROSCI.3116-05.2006
Beltran-Alvarez, P., Pagans, S., and Brugada, R. (2011). The cardiac sodium channel is post-translationally modified by arginine methylation. J. Proteome Res. 10, 3712–3719. doi: 10.1021/pr200339n
Berry, R. G., Despa, S., Fuller, W., Bers, D. M., and Shattock, M. J. (2007). Differential distribution and regulation of mouse cardiac Na+/K+-ATPase alpha1 and alpha2 subunits in T-tubule and surface sarcolemmal membranes. Cardiovasc. Res. 73, 92–100. doi: 10.1016/j.cardiores.2006.11.006
Bers, D. M., and Grandi, E. (2009). Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J. Cardiovasc. Pharmacol. 54, 180–187. doi: 10.1097/FJC.0b013e3181a25078
Bezzina, C., Veldkamp, M. W., Van Den Berg, M. P., Postma, A. V., Rook, M. B., Viersma, J. W., et al. (1999). A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ. Res. 85, 1206–1213. doi: 10.1161/01.RES.85.12.1206
Biet, M., Barajas-Martinez, H., Ton, A. T., Delabre, J. F., Morin, N., and Dumaine, R. (2012). About half of the late sodium current in cardiac myocytes from dog ventricle is due to non-cardiac-type Na(+) channels. J. Mol. Cell. Cardiol. 53, 593–598. doi: 10.1016/j.yjmcc.2012.06.012
Bossuyt, J., Ai, X., Moorman, J. R., Pogwizd, S. M., and Bers, D. M. (2005). Expression and phosphorylation of the na-pump regulatory subunit phospholemman in heart failure. Circ. Res. 97, 558–565. doi: 10.1161/01.RES.0000181172.27931.c3
Brette, F., and Orchard, C. H. (2006). Density and sub-cellular distribution of cardiac and neuronal sodium channel isoforms in rat ventricular myocytes. Biochem. Biophys. Res. Commun. 348, 1163–1166. doi: 10.1016/j.bbrc.2006.07.189
Camors, E., Mohler, P. J., Bers, D. M., and Despa, S. (2012). Ankyrin-B reduction enhances Ca spark-mediated SR Ca release promoting cardiac myocyte arrhythmic activity. J. Mol. Cell. Cardiol. 52, 1240–1248. doi: 10.1016/j.yjmcc.2012.02.010
Carmeliet, E. (1992). A fuzzy subsarcolemmal space for intracellular Na+ in cardiac cells? Cardiovasc. Res. 26, 433–442. doi: 10.1093/cvr/26.5.433
Clancy, C. E., and Rudy, Y. (2002). Na(+) channel mutation that causes both Brugada and long-QT syndrome phenotypes: a simulation study of mechanism. Circulation 105, 1208–1213. doi: 10.1161/hc1002.105183
Coppini, R., Ferrantini, C., Yao, L., Fan, P., Del Lungo, M., Stillitano, F., et al. (2013). Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 127, 575–584. doi: 10.1161/CIRCULATIONAHA.112.134932
Coyan, F., Burel, S., Lichti, C. F., Brown, J. H., Charpentier, F., Nerbonne, J. M., et al. (2014). Phosphoproteomic identification of CaMKII- and heart failure-dependent phosphorylation sites on the native cardiac Nav1.5 channel protein. Biophys. J. 106, 37a.
Davis, R. P., Casini, S., Van Den Berg, C. W., Hoekstra, M., Remme, C. A., Dambrot, C., et al. (2012). Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation 125, 3079–3091. doi: 10.1161/CIRCULATIONAHA.111.066092
DeGrande, S., Nixon, D., Koval, O., Curran, J. W., Wright, P., Wang, Q., et al. (2012). CaMKII inhibition rescues proarrhythmic phenotypes in the model of human ankyrin-B syndrome. Heart Rhythm 9, 2034–2041. doi: 10.1016/j.hrthm.2012.08.026
Despa, S., and Bers, D. M. (2007). Functional analysis of Na+/K+-ATPase isoform distribution in rat ventricular myocytes. Am. J. Physiol. Cell Physiol. 293, C321-327. doi: 10.1152/ajpcell.00597.2006
Despa, S., and Bers, D. M. (2013). Na(+) transport in the normal and failing heart – Remember the balance. J. Mol. Cell. Cardiol. 61, 2–10. doi: 10.1016/j.yjmcc.2013.04.011
Despa, S., Islam, M. A., Weber, C. R., Pogwizd, S. M., and Bers, D. M. (2002). Intracellular Na(+) concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 105, 2543–2548. doi: 10.1161/01.CIR.0000016701.85760.97
Edwards, A. G., Hake, J., Heller Brown, J., and McCulloch, A. D. (2012). βAr-stimulation causes EADs but not DADs in pre-failure CAMKIIδC transgenic myocytes. Biophys. J. 102, 98a. doi: 10.1016/j.bpj.2011.11.559
Erickson, J. R., Joiner, M. L., Guan, X., Kutschke, W., Yang, J., Oddis, C. V., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. doi: 10.1016/j.cell.2008.02.048
Gonano, L. A., Sepulveda, M., Rico, Y., Kaetzel, M., Valverde, C. A., Dedman, J., et al. (2011). Calcium-calmodulin kinase II mediates digitalis-induced arrhythmias. Circ. Arrhythm. Electrophysiol. 4, 947–957. doi: 10.1161/CIRCEP.111.964908
Grandi, E., Pandit, S. V., Voigt, N., Workman, A. J., Dobrev, D., Jalife, J., et al. (2011). Human atrial action potential and Ca2+ model: sinus rhythm and chronic atrial fibrillation. Circ. Res. 109, 1055–1066. doi: 10.1161/CIRCRESAHA.111.253955
Grandi, E., Pasqualini, F. S., and Bers, D. M. (2010). A novel computational model of the human ventricular action potential and Ca transient. J. Mol. Cell. Cardiol. 48, 112–121. doi: 10.1016/j.yjmcc.2009.09.019
Grandi, E., Puglisi, J. L., Wagner, S., Maier, L. S., Severi, S., and Bers, D. M. (2007). Simulation of Ca-calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials. Biophys. J. 93, 3835–3847. doi: 10.1529/biophysj.107.114868
Grant, A. O., Carboni, M. P., Neplioueva, V., Starmer, C. F., Memmi, M., Napolitano, C., et al. (2002). Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J. Clin. Invest. 110, 1201–1209. doi: 10.1172/JCI0215570
Herren, A. W., Bers, D. M., and Grandi, E. (2013). Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 305, H431–H445. doi: 10.1152/ajpheart.00306.2013
Hund, T. J., Koval, O. M., Li, J., Wright, P. J., Qian, L., Snyder, J. S., et al. (2010). A beta(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Invest. 120, 3508–3519. doi: 10.1172/JCI43621
Jayasinghe, I. D., Cannell, M. B., and Soeller, C. (2009). Organization of ryanodine receptors, transverse tubules, and sodium-calcium exchanger in rat myocytes. Biophys. J. 97, 2664–2673. doi: 10.1016/j.bpj.2009.08.036
Kapa, S., Tester, D. J., Salisbury, B. A., Harris-Kerr, C., Pungliya, M. S., Alders, M., et al. (2009). Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation 120, 1752–1760. doi: 10.1161/CIRCULATIONAHA.109.863076
Kapplinger, J. D., Tester, D. J., Alders, M., Benito, B., Berthet, M., Brugada, J., et al. (2010). An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7, 33–46. doi: 10.1016/j.hrthm.2009.09.069
Koval, O. M., Snyder, J. S., Wolf, R. M., Pavlovicz, R. E., Glynn, P., Curran, J., et al. (2012). Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation 126, 2084–2094. doi: 10.1161/CIRCULATIONAHA.112.105320
Li, L., Louch, W. E., Niederer, S. A., Aronsen, J. M., Christensen, G., Sejersted, O. M., et al. (2012). Sodium accumulation in SERCA knockout-induced heart failure. Biophys. J. 102, 2039–2048. doi: 10.1016/j.bpj.2012.03.045
Lin, X., Liu, N., Lu, J., Zhang, J., Anumonwo, J. M., Isom, L. L., et al. (2011). Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm 8, 1923–1930. doi: 10.1016/j.hrthm.2011.07.016
Litwin, S. E., Li, J., and Bridge, J. H. (1998). Na-Ca exchange and the trigger for sarcoplasmic reticulum Ca release: studies in adult rabbit ventricular myocytes. Biophys. J. 75, 359–371. doi: 10.1016/S0006-3495(98)77520-4
Liu, T., and O’Rourke, B. (2008). Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ. Res. 103, 279–288. doi: 10.1161/CIRCRESAHA.108.175919
Ma, J., Luo, A., Wu, L., Wan, W., Zhang, P., Ren, Z., et al. (2012). Calmodulin kinase II and protein kinase C mediate the effect of increased intracellular calcium to augment late sodium current in rabbit ventricular myocytes. Am. J. Physiol. Cell Physiol. 302, C1141–C1151. doi: 10.1152/ajpcell.00374.2011
Makielski, J. C., and Farley, A. L. (2006). Na(+) current in human ventricle: implications for sodium loading and homeostasis. J. Cardiovasc. Electrophysiol. 17(Suppl. 1), S15–S20. doi: 10.1111/j.1540-8167.2006.00380.x
Maltsev, V. A., Kyle, J. W., and Undrovinas, A. (2009). Late Na+ current produced by human cardiac Na+ channel isoform Nav1.5 is modulated by its beta1 subunit. J. Physiol. Sci. 59, 217–225. doi: 10.1007/s12576-009-0029-7
Marionneau, C., Lichti, C. F., Lindenbaum, P., Charpentier, F., Nerbonne, J. M., Townsend, R. R., et al. (2012). Mass spectrometry-based identification of native cardiac Nav1.5 channel alpha subunit phosphorylation sites. J. Proteome Res. 11, 5994–6007. doi: 10.1021/pr300702
Mohler, P. J., Rivolta, I., Napolitano, C., Lemaillet, G., Lambert, S., Priori, S. G., et al. (2004). Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 101, 17533–17538. doi: 10.1073/pnas.0403711101
Mohler, P. J., Schott, J. J., Gramolini, A. O., Dilly, K. W., Guatimosim, S., Dubell, W. H., et al. (2003). Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421, 634–639. doi: 10.1038/nature01335
Moreno, J. D., Yang, P. C., Bankston, J. R., Grandi, E., Bers, D. M., Kass, R. S., et al. (2013). Ranolazine for congenital and acquired late INa linked arrhythmias: in silico pharmacologic screening. Circ. Res. 113, e50–e61. doi: 10.1161/CIRCRESAHA.113.301971
Morotti, S., Edwards, A. G., McCulloch, A. D., Bers, D. M., and Grandi, E. (2014). A novel computational model of mouse myocyte electrophysiology to assess the synergy between Na+ loading and CaMKII. J. Physiol. doi: 10.1113/jphysiol.2013.266676 [Epub ahead of print].
Murphy, B. J., Rogers, J., Perdichizzi, A. P., Colvin, A. A., and Catterall, W. A. (1996). cAMP-dependent phosphorylation of two sites in the alpha subunit of the cardiac sodium channel. J. Biol. Chem. 271, 28837–28843. doi: 10.1074/jbc.271.46.28837
Pandey, D., Gratton, J. P., Rafikov, R., Black, S. M., and Fulton, D. J. (2011). Calcium/calmodulin-dependent kinase II mediates the phosphorylation and activation of NADPH oxidase 5. Mol. Pharmacol. 80, 407–415. doi: 10.1124/mol.110.070193
Petitprez, S., Zmoos, A. F., Ogrodnik, J., Balse, E., Raad, N., El-Haou, S., et al. (2011). SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ. Res. 108, 294–304. doi: 10.1161/CIRCRESAHA.110.228312
Pogwizd, S. M., Sipido, K. R., Verdonck, F., and Bers, D. M. (2003). Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis. Cardiovasc. Res. 57, 887–896. doi: 10.1016/S0008-6363(02)00735-6
Priori, S. G., Napolitano, C., Giordano, U., Collisani, G., and Memmi, M. (2000). Brugada syndrome and sudden cardiac death in children. Lancet 355, 808–809. doi: 10.1016/S0140-6736(99)05277-0
Remme, C. A., Wilde, A. A., and Bezzina, C. R. (2008). Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc. Med. 18, 78–87. doi: 10.1016/j.tcm.2008.01.002
Sag, C. M., Wadsack, D. P., Khabbazzadeh, S., Abesser, M., Grefe, C., Neumann, K., et al. (2009). Calcium/calmodulin-dependent protein kinase II contributes to cardiac arrhythmogenesis in heart failure. Circ. Heart Fail. 2, 664–675. doi: 10.1161/CIRCHEARTFAILURE.109.865279
Sapia, L., Palomeque, J., Mattiazzi, A., and Petroff, M. V. (2010). Na+/K+-ATPase inhibition by ouabain induces CaMKII-dependent apoptosis in adult rat cardiac myocytes. J. Mol. Cell. Cardiol. 49, 459–468. doi: 10.1016/j.yjmcc.2010.04.013
Sato, P. Y., Coombs, W., Lin, X., Nekrasova, O., Green, K. J., Isom, L. L., et al. (2011). Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ. Res. 109, 193–201. doi: 10.1161/CIRCRESAHA.111.247023
Sepulveda, M., Gonano, L. A., Back, T. G., Chen, S. R., and Vila Petroff, M. (2013). Role of CaMKII and ROS in rapid pacing-induced apoptosis. J. Mol. Cell. Cardiol. 63, 135–145. doi: 10.1016/j.yjmcc.2013.07.013
Shryock, J. C., Song, Y., Rajamani, S., Antzelevitch, C., and Belardinelli, L. (2013). The arrhythmogenic consequences of increasing late INa in the cardiomyocyte. Cardiovasc. Res. 99, 600–611. doi: 10.1093/cvr/cvt145
Song, Y., Shryock, J. C., and Belardinelli, L. (2008). An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 294, H2031–H2039. doi: 10.1152/ajpheart.01357.2007
Sossalla, S., Maurer, U., Schotola, H., Hartmann, N., Didie, M., Zimmermann, W. H., et al. (2011). Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIdelta(C) can be reversed by inhibition of late Na(+) current. Basic Res. Cardiol. 106, 263–272. doi: 10.1007/s00395-010-0136-x
Tan, H. L., Bink-Boelkens, M. T., Bezzina, C. R., Viswanathan, P. C., Beaufort-Krol, G. C., Van Tintelen, P. J., et al. (2001). A sodium-channel mutation causes isolated cardiac conduction disease. Nature 409, 1043–1047. doi: 10.1038/35059090
Tester, D. J., Valdivia, C., Harris-Kerr, C., Alders, M., Salisbury, B. A., Wilde, A. A., et al. (2010). Epidemiologic, molecular, and functional evidence suggest A572D-SCN5A should not be considered an independent LQT3-susceptibility mutation. Heart Rhythm 7, 912–919. doi: 10.1016/j.hrthm.2010.04.014
Tester, D. J., Will, M. L., Haglund, C. M., and Ackerman, M. J. (2005). Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2, 507–517. doi: 10.1016/j.hrthm.2005.01.020
Toischer, K., Hartmann, N., Wagner, S., Fischer, T. H., Herting, J., Danner, B. C., et al. (2013). Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J. Mol. Cell. Cardiol. 61, 111–122. doi: 10.1016/j.yjmcc.2013.03.021
Veldkamp, M. W., Viswanathan, P. C., Bezzina, C., Baartscheer, A., Wilde, A. A., and Balser, J. R. (2000). Two distinct congenital arrhythmias evoked by a multidysfunctional Na(+) channel. Circ. Res. 86, E91–E97. doi: 10.1161/01.RES.86.9.e91
Vila-Petroff, M., Mundina-Weilenmann, C., Lezcano, N., Snabaitis, A. K., Huergo, M. A., Valverde, C. A., et al. (2010). Ca(2+)/calmodulin-dependent protein kinase II contributes to intracellular pH recovery from acidosis via Na(+)/H(+) exchanger activation. J. Mol. Cell. Cardiol. 49, 106–112. doi: 10.1016/j.yjmcc.2009.12.007
Viswanathan, P. C., Benson, D. W., and Balser, J. R. (2003). A common SCN5A polymorphism modulates the biophysical effects of an SCN5A mutation. J. Clin. Invest. 111, 341–346. doi: 10.1172/JCI200316879
Wagner, S., Dybkova, N., Rasenack, E. C., Jacobshagen, C., Fabritz, L., Kirchhof, P., et al. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138. doi: 10.1172/JCI26620
Wagner, S., Ruff, H. M., Weber, S. L., Bellmann, S., Sowa, T., Schulte, T., et al. (2011). Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ. Res. 108, 555–565. doi: 10.1161/CIRCRESAHA.110.221911
Wan, X., Chen, S., Sadeghpour, A., Wang, Q., and Kirsch, G. E. (2001). Accelerated inactivation in a mutant Na(+) channel associated with idiopathic ventricular fibrillation. Am. J. Physiol. Heart Circ. Physiol. 280, H354–H360.
Weber, C. R., Ginsburg, K. S., and Bers, D. M. (2003a). Cardiac submembrane [Na+] transients sensed by Na+-Ca2+ exchange current. Circ. Res. 92, 950–952. doi: 10.1161/01.RES.0000071747.61468.7F
Weber, C. R., Piacentino, V. III, Houser, S. R., and Bers, D. M. (2003b). Dynamic regulation of sodium/calcium exchange function in human heart failure. Circulation 108, 2224–2229. doi: 10.1161/01.CIR.0000095274.72486.94
Wehrens, X. H., Rossenbacker, T., Jongbloed, R. J., Gewillig, M., Heidbuchel, H., Doevendans, P. A., et al. (2003). A novel mutation L619F in the cardiac Na+ channel SCN5A associated with long-QT syndrome (LQT3): a role for the I-II linker in inactivation gating. Hum. Mutat. 21, 552. doi: 10.1002/humu.9136
Wilde, A. A., and Brugada, R. (2011). Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ. Res. 108, 884–897. doi: 10.1161/CIRCRESAHA.110.238469
Xi, Y., Wu, G., Yang, L., Han, K., Du, Y., Wang, T., et al. (2009). Increased late sodium currents are related to transcription of neuronal isoforms in a pressure-overload model. Eur. J. Heart Fail. 11, 749–757. doi: 10.1093/eurjhf/hfp092
Yang, P., Kanki, H., Drolet, B., Yang, T., Wei, J., Viswanathan, P. C., et al. (2002). Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation 105, 1943–1948. doi: 10.1161/01.CIR.0000014448.19052.4C
Yang, T., Atack, T. C., Stroud, D. M., Zhang, W., Hall, L., and Roden, D. M. (2012). Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ. Res. 111, 322–332. doi: 10.1161/CIRCRESAHA.112.265173
Yao, L., Fan, P., Jiang, Z., Viatchenko-Karpinski, S., Wu, Y., Kornyeyev, D., et al. (2011). Nav1.5-dependent persistent Na+ influx activates CaMKII in rat ventricular myocytes and N1325S mice. Am. J. Physiol. Cell Physiol. 301, C577–C586. doi: 10.1152/ajpcell.00125.2011
Zhou, J., Yi, J., Hu, N., George, A. L. Jr., and Murray, K. T. (2000). Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ. Res. 87, 33–38. doi: 10.1161/01.RES.87.1.33
Keywords: CaMKII, Na+ channel, DADs, Na+ overload, arrhythmia
Citation: Grandi E and Herren AW (2014) CaMKII-dependent regulation of cardiac Na+ homeostasis. Front. Pharmacol. 5:41. doi: 10.3389/fphar.2014.00041
Received: 16 January 2014; paper pending published: 03 February 2014;
Accepted: 21 February 2014; Published online: 10 March 2014.
Edited by:
Andrew G. Edwards, Oslo University Hospital, NorwayReviewed by:
William Louch, Oslo University Hospital, NorwayMartin Vila Petroff, Centro de Investigaciones Cardiovasculares, Argentina
Copyright © 2014 Grandi and Herren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eleonora Grandi, Department of Pharmacology, University of California at Davis, 451 Health Sciences Drive, GBSF room 3502, Davis, CA 95616, USA e-mail: ele.grandi@gmail.com