 Maame Efua S. Sampah
Maame Efua S. Sampah David J. Hackam
David J. Hackam- Division of Pediatric Surgery, Department of Surgery, Johns Hopkins University School of Medicine , Baltimore, MD, United States
Prior to birth, the neonate has limited exposure to pathogens. The transition from the intra-uterine to the postnatal environment initiates a series of complex interactions between the newborn host and a variety of potential pathogens that persist over the first few weeks of life. This transition is particularly complex in the case of the premature and very low birth weight infant, who may be susceptible to many disorders as a result of an immature and underdeveloped immune system. Chief amongst these disorders is necrotizing enterocolitis (NEC), an acute inflammatory disorder that leads to necrosis of the intestine, and which can affect multiple systems and have the potential to result in long term effects if the infant is to survive. Here, we examine what is known about the interplay of the immune system with the maternal uterine environment, microbes, nutritional and other factors in the pathogenesis of neonatal pathologies such as NEC, while also taking into consideration the effects on the long-term health of affected children.
Introduction
The characteristic underdevelopment of the neonatal immune system predisposes infants to inflammatory disorders, including necrotizing enterocolitis (NEC), an acute inflammatory disease that develops in up to 10% of premature infants (1). NEC is characterized by the sudden development of inflammation and necrosis of the intestine leading to overwhelming sepsis and death in up to half of all patients (2, 3). Unique characteristics of the neonatal immune system that confer a susceptibility to the development of diseases like NEC include the potential exposure of priming antigens within the maternal uterine environment (4), changes in cytokine, growth factor and hormone signaling pathways (5, 6), nutritional effects related to exposure to probiotics and breastmilk (7), as well as patterns of microbial colonization in the gut and other mucosa that are specific to the neonatal period (8). These changes may have long-term effects on the immune system, creating immune activation or tolerance that determine whether pathology will develop (9). Importantly, NEC and other inflammatory neonatal conditions typically also affect the lungs and brain (10–12) resulting in long-term sequelae that may be underappreciated initially. In infants who are fortunate to survive NEC, lifelong problems may develop, including short bowel syndrome (13), nutritional deficiencies impact growth (14), and persistent white matter injury associated with cognitive impairment (10, 15–17) among others.
While many important human clinical studies, as well as preclinical studies in clinically relevant animal models (18), have made important gains in elucidating the complexities of the cellular and biological processes associated with immunological disorders including NEC, many gaps in knowledge in this field remain. We will review available studies that explore the pathogenesis of neonatal gut inflammation and its short- and long-term impacts on various secondary organ systems. We will assess existing research gaps, and the potential future directions for investigations in the field.
Impaired Immunity in the Neonate
Impairment of many components of the immune system is well documented in neonates and is especially exacerbated in preterm infants (19). This is due to a relative lack of antigenic exposure and enhanced self-modulatory immunosuppressive mechanisms that make way for beneficial microbial colonization and prevent potentially harmful inflammation and autoimmunity (19). As a result, preterm infants are exposed to increased risk of infections from a variety of potential pathogens (20, 21). Known deficiencies include decreased physical integrity of the pulmonary and gastrointestinal epithelial barriers (22, 23), reduced numbers of goblet cells and thinner mucus secretions (24, 25), low numbers of Paneth cells with decreased antimicrobial producing function (26), lower levels of circulating maternal IgG with low opsonic activity (27–29), complement protein deficiency in the setting of reduced activity (30–32), and reduction in numbers and function of neutrophils and monocytes (33–36). As a result of these characteristics, multiple levels of susceptibility to bacterial, viral and fungal infection exist. The implications of these impairments in immunity are reflected in data from clinical studies that report up to five-fold higher risk of sepsis in preterm infants compared with their term counterparts (37, 38). Preterm infants diagnosed with sepsis are, in turn, at significantly higher risk of respiratory and neurological complications such as respiratory distress syndrome, severe intraventricular hemorrhage and periventricular leukomalacia (39). These findings contribute considerably to prolonged neonatal hospital stays (40), increased numbers of rehospitalizations and overall mortality compared with term infants (41, 42).
Deficiencies that allow pathogenic invasion from colonized sites such as the respiratory and gastrointestinal tract in preterm infants are compounded by a tendency toward exaggerated immune activity (43–45). Certain components of the immune system have been noted to be reach a heightened degree of activation, resulting in pathogenic inflammation that has severe ramifications for disease pathogenesis. For instance, Th17 cells are abundant lymphocytes at the intestinal mucosa of the premature newborn (46) and contribute to host defense against extracellular microbial pathogens (47, 48). This subset of CD4+ T cells mainly produce IL-17, which has been shown to induce a pro-inflammatory state, leading to infection-induced immunopathology in many human autoimmune diseases, including multiple sclerosis, rheumatoid arthritis, psoriasis and Lyme arthritis (49–51). In the preterm infant, the relative abundance of IL-17 signaling dampens the effect of counter-inflammatory mechanisms such as Foxp3+ Treg cell activity and in fact, downregulates Foxp3+ Treg expression (52). The net result of this skew towards a pro-inflammatory lymphocyte state is the finding of impaired enterocyte tight junctions, increased enterocyte apoptosis, and decreased enterocyte proliferation, culminating in global mucosal injury as observed in necrotizing enterocolitis (46).
Innate Immune Signaling and Downstream Consequences
The exaggerated signaling in response to TLR4 in the premature infant represents an important example of immature immune activation that leads to disease (53). The cell walls of gram-negative bacteria like Escherichia coli and Helicobacter pylori contain endotoxins which mediate host recognition and inflammatory response to infection. Lipopolysaccharide (LPS), which is the main endotoxin encountered, binds to an intramembrane complex made up of TLR4 and CD14 and initiates the recruitment of myeloid differentiation primary response gene 88 (MyD88), signaling an inflammatory response involving the activation of nuclear factor-κB (54). This response is accompanied by cellular stress responses, leading to the release of pro-inflammatory cytokines including IL-6, IL-1β, TNF-α and other cellular stress markers including nitric oxide (54), as well as the expression of co-stimulatory molecules (55). It should be pointed out that other TLR4 ligands exist, including viral proteins, endogenous proteins such as low-density lipoprotein, beta-defensins, and heat shock proteins (56–59). TLR4 signaling may also occur via an alternative adapter protein TRIF (60, 61). In leukocytes, the downstream effects of TLR4 activation are critical for host defense against infection (62, 63) via NF-κB activation and downstream cytokine-mediated responses (63). However, in the gut, TLR4 is notably an essential component for normal intestinal development via the Notch signaling pathway (64, 65). For this reason, TLR4 is expressed at higher levels in the premature (and thus still developing) gut as compared to the full-term gut, which does not result in an inflammatory response given the bacteria-free environment of the developing fetus. By contrast, when the premature infant is born and exposed to the microbiota of the environment, colonization of the intestine occurs, leading to activation of TLR4 by LPS, whereupon TLR4 switches from a developmental to an inflammatory role, leading to the induction of NEC.
In full-term neonates, TLR4 expression in the intestine is notably downregulated around the time of delivery and then slowly increases as the immune system matures (66, 67). It follows that full-term neonates are protected from NEC compared to those that are born preterm, since TLR4 activation in the intestinal epithelium has been shown to be critical for NEC development (68) (Figure 1). TLR4 activation results in increased intestinal epithelial death by both apoptosis (69) and necroptosis (70), the induction of ER stress (71) and enhanced autophagy (72–74). Other critical features of TLR4 activation in the intestine include decreased mucosal healing both in vitro and in vivo (67), reduced goblet cell production and thus loss of a mucin barrier for defense (68), increased expression of proinflammatory cytokines leading to increased barrier permeability, decreased tight junction function, and increased epithelial cell apoptosis (75). These direct effects of TLR4 activation cumulatively promote bacterial translocation (76), which leads to further TLR4 activation on the endothelium, leading to loss of endothelial nitric oxide synthase (eNOS) and associated vasoconstriction (77), which in turn contributes to the development of ischemic necrosis in NEC. The premature intestine, which has increased expression of TLR4, is therefore more susceptible to NEC due to the factors described above (64, 65, 78).
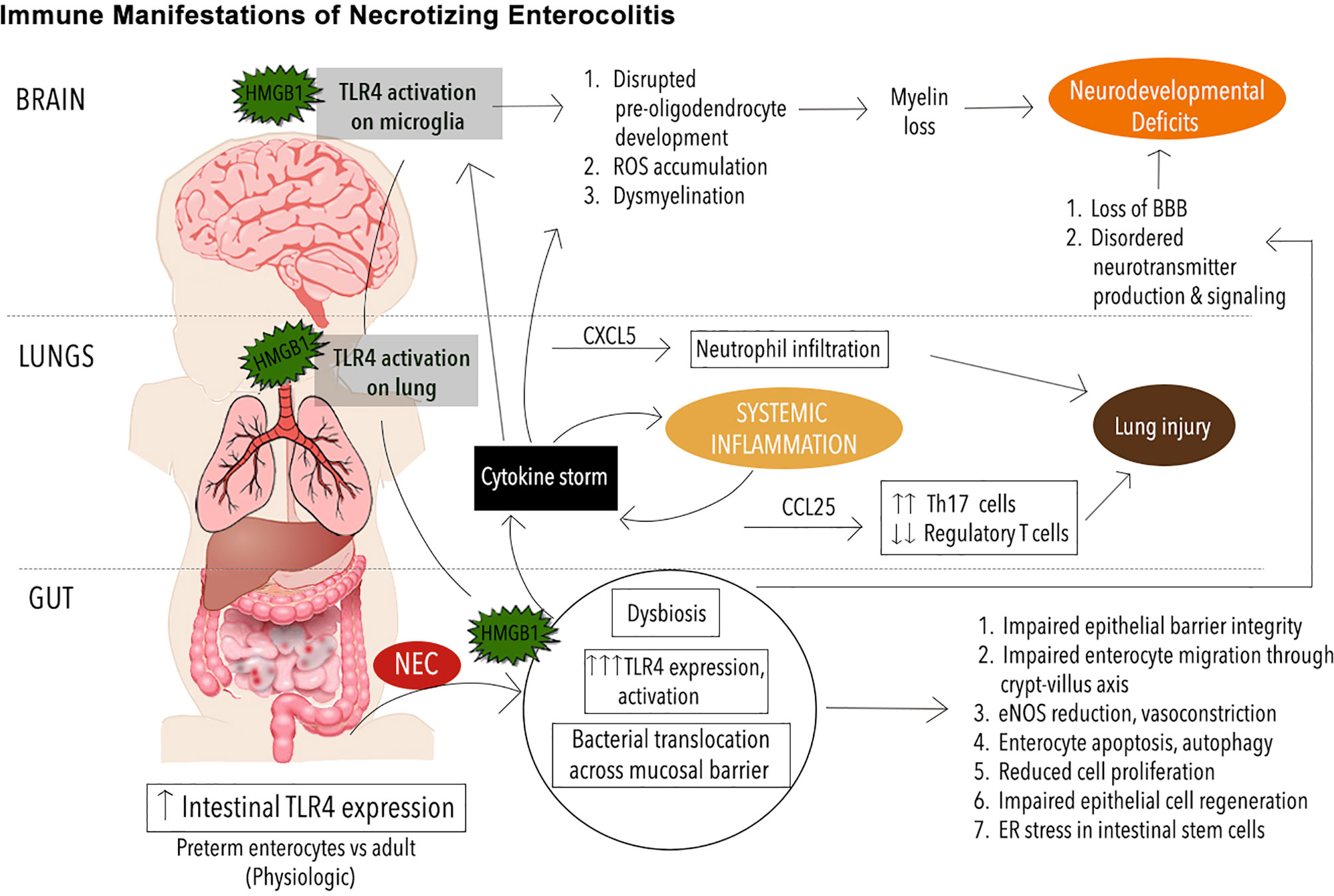
Figure 1 Immune Manifestations in NEC. Schematic illustrating immune signaling involving toll-like receptor 4 (TRL4) in necrotizing enterocolitis (NEC) pathogenesis. The premature immune system leaves the neonate prone to infectious and inflammatory diseases such as NEC. Mediated by exaggerated TLR4 signaling on the intestinal epithelium, the onset of NEC leads to mobilization of an endogenous TLR4 ligand, high mobility group box 1 (HMGB1) from the intestine to the lungs and brain where TLR4 activation on the pulmonary epithelium and microglia respectively leads to phenomena such as neutrophil infiltration, reactive oxygen species (ROS) buildup, and other downstream effects that exacerbate pathology in the lungs, brain and other organ systems.
It is important to note that other TLRs have been implicated in NEC pathogenesis. TLR5 and TLR9, particularly, have been demonstrated to be protective in NEC pathogenesis. On the other hand, TLRs 2 and 8 have been demonstrated to be upregulated in NEC intestinal tissue from experimental animal models, as well as those samples obtained in the clinical setting from infants. However, functional pathways related to the role of these latter TLRs in pathogenesis have not been clearly defined (79, 80). Similarly, other pattern recognition receptors including nucleotide‐binding domain and leucine‐rich repeat containing (NLR) proteins, RIG-I-like receptor and antimicrobial mediators such as mannose-binding lectin are upregulated in NEC and studies are ongoing to elucidate the associated pathways in order to identify viable therapeutic targets (81–84). Interestingly, emerging data suggests that one such mediator, NLR pyrin domain containing 3 (NLRP3) likely acts via a TLR4 dependent pathway (83).
Mucosal Dysbiosis and Associated Disruptions in Immune Function
It has been long established that the intestinal microbiome, consisting of many millions of microbes inhabiting the gut, has a significant impact on disease processes (24). Normally, bacteria that compose the microbiome synthesize several mediators including lipopolysaccharides, peptidoglycans, short-chain fatty acids (SCFA), signaling molecules such as nitric oxide and essential vitamins which influence host physiology by regulating the mucosal immune system (85, 86). Preclinical data derived from studies in germ-free mice have shown that lymphoid follicles in the gut require peptidoglycan from gram negative bacteria for maturation, reflecting the importance of bacterial colonization for immune development (87). Other studies have shown that bacteria-naïve mice have fewer T cells and Paneth cells (88), lower expression of secretory IgA, lower levels of intestinal epithelial cells compared to conventional mice.
From clinical data, premature infants have been described to have an immature microbiome, with amniotic fluid, placenta, and meconium of premature neonates containing variations in microbes compared to full-term infants (89–91). The specific characteristics of the immature microbiome include well-described characteristic progressions from one dominant microbial population to the next (92). However, the preterm microbiome overall remains consistently less diverse compared to the variations in microbial classes observed in full term neonatal stool (92–94). For instance, in one 16S rRNA study, four bacterial classes accounted for more than 90% of the microbial sequence reads derived from preterm infant stool (92, 95). Other studies have verified lower alpha-diversity in preterm stool microbiota compared to full-term controls (96, 97). The immature preterm microbiome has also been demonstrated to impact metabolism and metabolomics, resulting in a metabolic state analogous to fasting, in spite of adequate caloric intake. Hence, metabolic derangements likely present another mediator of the relationship between microbiota disruption in preterm infants and functional immune deficiencies, which may persist and impact long-term health (98). Other factors that modulate the microbiota of premature infant include the use of antibiotics and h2-blockers, the administration of breast milk, and practices within the NICU (98–101).
Microbial dysbiosis is considered to play important roles in the complex etiology of NEC (92, 102, 103). Alterations in the microbiome have the ability to cause disruptions in immune function and result in a shift in balance to pathogenic bacterial colonization (104, 105). Further, activation of TLR4 by LPS present on potentially pathogenic gram-negative bacteria is an important aspect of NEC pathogenesis as discussed earlier. The tendency towards pathogenic microbial colonization results in TLR4 mediated phagocytosis and translocation of these Gram-negative bacteria across the intestinal mucosal barrier (106), resulting in activation of NF-κB and caspases, which triggers inflammation (107).
Overall, similar bacterial species have been identified in infants diagnosed with NEC compared to control infants. However, neonates with NEC have been described to have microbiomes that are temporally dynamic (with more changes in composition over time), but feature lower variations of strains within species (108, 109). Preterm infants with relatively lower alpha-diversity have higher risk of a later propensity for NEC development (95, 97). More specifically, 16S rRNA and metagenomic sequencing studies have reported an association between phylum Proteobacteria overrepresentation and increased NEC incidence (95, 110). Of note, this phylum consists of gram-negative pathogens that express high levels of LPS and are overproducers of SCFA including propionic acid which is thought to exacerbate NEC-associated neurodevelopmental conditions including movement disorders, seizure, and developmental delay (111).
While these data are compelling, reports outlining the constitution of NEC and non-NEC associated microbiota continue to vary, and further studies are needed to shed more light on this field. Ongoing studies examining how specific human milk oligosaccharides (HMOs) exert a protective effect on neonates against development have also focused in part on the microbiome. HMOs may promote the growth of bacteria from phyla Bacteroidetes, the latter of which is mainly represented by the Bifidobacterium genus (112, 113). These bacteria have been demonstrated to have a beneficial effect on immunity and are associated with positive outcomes with respect to NEC, as well as respiratory complications in neonates (112, 114). For this reason, probiotic therapy remains an active research area in NEC therapeutics. The influence of breast milk derived HMOs on the airway microbiome in preterm infants and its potential effects in reducing airway inflammation for instance, also remains an open question under investigation.
In Utero Influences and Immune Tolerance
Various studies reveal that there are important maternal effects on immune system development during the in utero period (103, 115, 116). Ongoing work continues to establish that important events and influences may be exerted during this time that precede post-natal influences such as breastmilk, enteral feeds and antibiotic exposure (117–119). The maternal microbiome and related factors such as maternal diet, antibiotic use and maternal infections all remain active areas of research to find potential mechanisms to guide development of targeted interventions that can impact early colonization patterns in infants to prevent early pathology such as NEC as well as longer term diseases that may persist into adulthood (120).
One such study has demonstrated that transiently exposing germ-free mice to E. coli during pregnancy results in pups with increased intestinal type 3 innate lymphoid cells (ILC3) and F4/80 mononuclear cells (121), an effect that persisted 8 weeks postpartum. This effect was associated with altered intestinal transcriptional profiles and increased production of epithelial antibacterial peptides compared to those offspring from germ-free dams. Similarly, a pig model in which chorioamnionitis was induced by LPS administration resulted in higher intestinal endotoxin and neutrophil/macrophage density, with shorter villi in the offspring, accompanied by upregulation of various innate immune response, neutrophil chemotaxis and antigen processing genes after 5 days (122). Unfortunately, this and other existing studies are limited, in that they do not evaluate longer term outcomes of these alterations in the immune system. These studies also are limited by not accounting for the specific site of LPS signaling (immune versus epithelial). A longer term study has been performed in a murine model of colitis where developing mice were exposed in utero to LPS (123). Subsequent chemical induction in adulthood, at age 5 weeks, revealed protection from development of colitis in those pups exposed to endotoxin during development. Notably, this protection was not present when mice received LPS postnatally at 7 days of age.
While a preponderance of the studies described above involve animals, important data from human infants has also been obtained. Studies that are able to be performed without harm to infants include stool analyses in preterm neonates, which have yielded robust metabolomic data (114) and include those studies that have compared preterm neonates with NEC to healthy controls to demonstrate that NEC does not have a uniform microbial signature. Combined with emerging laboratory tools and approaches that are able to probe regulation of the immune system at the molecular level, it is becoming clearer that the preterm immune system possesses in-built immune tolerance, which is associated with impaired metabolism as described earlier, including minimized glycolytic activity (124). Accordingly, preterm naive CD4+ T cells have been found to have a higher threshold for inducing inflammation compared to adults, with impaired early Th1 differentiation including IFNγ production (125). Instead, Th2 and Th17 polarization are noted following bacterial stimulation, accompanied by low innate antiviral type 1 interferon responses (79, 126, 127). Similarly, IFNγ production by stimulated naive cord blood CD4+ T cells has been measured as 5 to 10-fold less relative to adult CD4+ T cells, resulting in a characterization of the preterm CD4 response as Th2 skewed (128). This allows baseline prioritization of energy for replenishment and maintenance of organ functions over inflammatory responses until a specific threshold is exceeded, as seen in the mucosal and systemic perturbations characteristic of NEC (129). This hyper-inflammatory status has been correlated with the diagnostic phase of NEC and sepsis in preterm infants (114).
Epithelial Barrier Dysfunction
One of the direct consequences of the local inflammation caused by TLR4 activation is destruction of the intestinal epithelial barrier (130, 131). Under physiologic conditions, epithelial cells regulate the penetration of gut lumen contents including bacteria and their components, digestive enzymes and degraded food products such as ions, nutrients and water (132). This is accomplished by the formation of tight junctions containing complexes of proteins which bind the epithelial cells together (133), as well as active transport mechanisms (134, 135). A disruption of these tight junctions allows unregulated traverse of pathogenic antigens which trigger mucosal injury and inflammation leading to disorders such as NEC.
In the premature neonate, control of luminal contents is complicated by a pre-disposition to intestinal epithelial injury (25). It has been shown that proteins that make up the tight junction, including claudins and occludins, show a tissue-specific distribution pattern including those patterns characteristic of GI tract epithelium (136, 137). Further, some tight junction proteins have downregulated expression in preterm infants (138, 139), resulting in alterations in intestinal tight junction function (25, 140, 141). In one animal study, intestinal tissue obtained from germ free mice colonized by microbiota from preterm infants was found to express lower levels of occludin and tight junction associated protein ZO-1 compared to controls, in addition to disorganization in TJ protein assembly (139). Such imbalances of tight junctions are thought to render the epithelium even more susceptible to injury during NEC, during which expression of these tight junction proteins are significantly impaired via alterations to HIF-1 pathway signaling (142).
When TLR4 is activated during NEC, enterocyte migration from the crypt to the villus can also be impaired via modifications to the cell-extracellular matrix interactions (143, 144). Whereas TLR4 activation in the adult leads to increased enterocyte proliferation, in the premature intestine, apoptosis is instead induced, leading to reduced cell proliferation, impaired epithelial regeneration (69, 144, 145), and dysfunction in the epithelial barrier. In addition, autophagy – a mechanism by which cells recycle their intracellular organelles but which can also lead to cell death - is induced in intestinal epithelial cells as a consequence of TLR4 activation during NEC, leading to impaired migration (146). Strategies to block enterocyte autophagy, including epidermal growth factor therapy, have been found to reduce NEC severity in animal models (73, 147).
Interestingly, a unique pathway to epithelial injury mediated by neutrophils has been described in murine models. Neutrophil infiltration has long been noted histologically in murine and human tissue samples as characteristic of NEC pathology (148, 149). While it has been hypothesized that neutrophil recruitment may contribute to epithelial injury in NEC via the release of toxic products and reactive oxygen species, definitive evidence from murine models suggests that the formation of neutrophil extracellular traps (NETosis) are important actors in neutrophil-mediated epithelial damage and pathogenesis (150).
Patient Outcomes Following Inflammatory Disorders of Premature Birth
Gut
The immunopathology of NEC presents clinically as a devastating intestinal disease of high mortality and morbidity, with associated complications including bowel necrosis and perforation, leading to intestinal resections and resulting nutritional disorders such as short bowel syndrome. These sequelae may notably also have effects that last beyond the acute period of illness, affecting nutritional status, susceptibility to other illnesses and developmental delay. Premature infants who survive NEC are at risk of long-term severe growth failure in the years following infection (151). A number of multicenter cohort studies have showed that premature infants that underwent surgery for NEC were more likely to have significant growth delay compared to those without NEC (152, 153). This is thought to be mainly due to impaired nutritional delivery and loss of functional gastrointestinal mass secondary to bowel resection (154). However, it has been demonstrated that neonatal sepsis in premature infants also results in a hypermetabolic state with increased energy expenditure and protein catabolism that persists beyond the acute period of illness (155–157). This is important, considering that even premature infants with medical NEC are at increased risk for growth failure. Importantly, adult and animal studies have demonstrated that TNF-α, IL-1β and IL-6, which are globally elevated in sepsis and are expressed downstream of TLR4 signaling in NEC, are likely modifiers of protein and energy metabolism (158, 159). Hence while the molecular mechanisms that mediate this phenomenon are yet to be fully elucidated, it is clear that the devastating effects of exaggerated TLR4 signaling in the premature infant gut persists beyond the acute period of illness. Notwithstanding, clinical studies reviewing long-term growth impairments and gastrointestinal sequelae of NEC beyond three years are lacking. This is the case for longer term laboratory studies examining longer term immune manifestations, as previously outlined.
Brain
Recent clinical and experimental studies have established that NEC is not only an intestinal condition (160, 161). Its broader sequelae including systemic inflammation, hypoxia, ischemia have the ability to trigger multisystem organ dysfunction, notably in the brain and lungs (162). Systematic reviews have noted that NEC is an independent risk factor for neurodevelopmental delay and poor neurocognitive outcomes in preterm infants (3, 163, 164). Importantly, these deficits may continue to manifest, with children continuing to have poor mental and psychomotor development around 2 years and in a significant proportion, persistent cognitive deficits well into school age (153, 165, 166). Proposed mechanisms for these sequalae suggest a multifactorial etiology. For instance, characteristic changes to the microbiome associated with NEC affect the not only gut, but may also mediate preterm brain development via the modulation of neurotransmitter levels (167). As in the gut, endothelial barrier dysfunction in the brain is a critical aspect of NEC pathophysiology, as the blood-brain-barrier (BBB) has been found to be directly impacted by dysbiosis. In this case, mouse models have demonstrated that low levels of SCFA due to alterations in bacterial composition can cause a permanent increase in BBB permeability due to alterations in tight junction protein expression (168).
Most broadly, downstream intestinal injury and barrier dysfunction discussed in prior sections promote bacterial translocation into systemic circulation, accompanied by the recruitment of inflammatory mediators and cytokines, leading to systemic inflammation and sepsis. The brain’s own immune cells, the microglia and astrocytes, modulate the development of normal brain functions including synaptic pruning, synapse formation and synaptic transmission regulate neurogenesis, neuronal migration, and synaptic plasticity (169). However, multiple animal studies have demonstrated that exposure to inflammatory molecules disrupts brain development. For instance, exposure to IL-1β and TNF-α in one mouse study resulted in long‐lasting disruptions in oligodendrocyte maturation (170). In other animal studies, early exposure to LPS has resulted in reduction in hippocampal volume, disordered neurogenesis, increased microglia populations and activity, axonal injury, and memory impairment (171, 172). Accordingly, increased disease severity, for instance, surgical NEC (173) and increased levels of pro-inflammatory cytokines (160), are associated with worse neurodevelopmental outcomes in preterm infants.
Finally, it is now known that TLR4 signaling is an important part of NEC pathophysiology in the brain (17). Using a clinically relevant murine NEC model, Nino et al. showed that TLR4 activation occurs during NEC, an endogenous TLR4 ligand, high mobility group box 1 (HMGB1) is released from the intestinal and activates TLR4 on microglia, leading to the accumulation of reactive oxygen species (ROS), loss of oligodendrocyte progenitor cells (OPCs), dysmyelination, and cognitive impairments. Importantly, the administration of targeted microglia anti-inflammatory and antioxidant therapy ameliorated the degree of neurological dysfunction, demonstrating a novel therapeutic target in NEC-associated brain injury.
Lung
Extra-intestinal sequelae of NEC are well documented in the pulmonary system. Up to half of premature infants born prior to 36 weeks gestation develop lung injury (174). However, lung injury that occurs in the presence of infectious and inflammatory neonatal conditions such as NEC tend to be more severe and have longer term effects than in matched patients without (12). Overall, approximately 15% of infants with NEC experience lung damage that is characterized by neutrophil infiltration and inflammatory cytokine upregulation.
Interestingly, recent insights into underlying causes of NEC-induced lung injury have revealed a mechanism mediated by TLR4 that is analogous to that seen in brain injury (175, 176). Using a murine NEC model, Jia et al. have shown that TLR4, highly expressed on the pulmonary epithelium in animals with NEC, is activated by TLR4 ligand high-mobility group box 1 (HMGB1) derived from the gut epithelium. This leads to downstream upregulation of CXCL5, a chemoattractant, and subsequent recruitment of neutrophils. Importantly, the aerosolized delivery of a novel TLR4 small molecule inhibitor was sufficient to reverse this inflammatory cascade and prevent the lung injury normally triggered by NEC in this model. Further, in NEC and other diseases with similar histopathology, Th17 skewed CD4 T cells have been shown to drive the cytokine upregulation and immune cell infiltration that constitute the mechanism of inflammation and lung injury (79, 177). The inflammation is typically via upregulation of the chemokine CCL25, which is now known to be upregulated downstream of TLR4 activation in the lung during NEC, a phenomenon that simultaneously depletes the population of protective regulatory T cells (Tregs) present in the lung epithelium (176).
Summary
The premature neonate has multiple levels predispositions to infectious and inflammatory afflictions such as NEC. A dynamic and complicated interface between pathogens, other microbiota, the maternal environment and the immune system mediates such pathology and has severe acute effects locally at the intestinal epithelium, but importantly impacts multiple systems secondary to systemic inflammation, as well as widespread effects of important mediators such as TLR4. Short term disruptions to intestinal cell renewal and homeostasis, metabolism and growth, gut microbial colonization, neurodevelopment, and lung physiology are evident from clinical studies. Longer term data are largely deficient from even clinical literature. Yet, a growing field of NEC and associated research continue to shed light on how the above discussed monumental disruptions in the immune system impact long term immune function and how knowledge gleaned from such phenomena can play a role in prevention and treatment of such neonatal disorders.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
DJH is supported by grants from the National Institutes of Health, USA under award numbers R01 DK1117186 and R01 DK121824.
Conflict of Interest
DH is supported by research grants from Abbott Nutrition and Noveome.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Nino DF, Sodhi CP, Hackam DJ. Necrotizing Enterocolitis: New Insights Into Pathogenesis and Mechanisms. Nat Rev Gastroenterol Hepatol (2016) 13(10):590–600. doi: 10.1038/nrgastro.2016.119
2. Bizzarro MJ, Ehrenkranz RA, Gallagher PG. Concurrent Bloodstream Infections in Infants With Necrotizing Enterocolitis. J Pediatr (2014) 164(1):61–6. doi: 10.1016/j.jpeds.2013.09.020
3. Jones IH, Hall NJ. Contemporary Outcomes for Infants With Necrotizing Enterocolitis—a Systematic Review. J Pediatr (2020) 220:86–92.e3. doi: 10.1016/j.jpeds.2019.11.011
4. Perez-Muñoz ME, Arrieta MC, Ramer-Tait AE, Walter J. A Critical Assessment of the “Sterile Womb” and “in Utero Colonization” Hypotheses: Implications for Research on the Pioneer Infant Microbiome. Microbiome (2017) 28(5):48. doi: 10.1186/s40168-017-0268-4
5. Maheshwari A, Schelonka RL, Dimmitt RA, Carlo WA, Munoz-Hernandez B, Das A, et al. Cytokines Associated With Necrotizing Enterocolitis in Extremely-Low-Birth- Weight Infants. Pediatr Res (2014) 76(1):100–8. doi: 10.1038/pr.2014.48
6. Chan KYY, Leung KT, Tam YH, Lam HS, Cheung HM, Ma TPY, et al. Genome-Wide Expression Profiles of Necrotizing Enterocolitis Versus Spontaneous Intestinal Perforation in Human Intestinal Tissues: Dysregulation of Functional Pathways. Ann Surg (2014) 260(6):1128–37. doi: 10.1097/SLA.0000000000000374
7. Sisk PM, Lovelady CA, Dillard RG, Gruber KJ, O’Shea TM. Early Human Milk Feeding is Associated With a Lower Risk of Necrotizing Enterocolitis in Very Low Birth Weight Infants. J Perinatol (2007) 27(7):428–33. doi: 10.1038/sj.jp.7211826
8. Costello EK, Carlisle EM, Bik EM, Morowitz MJ, Relman DA. Microbiome Assembly Across Multiple Body Sites in Low-Birthweight Infants. MBio (2013) 4(6):e00782–13. doi: 10.1128/mBio.00782-13
9. Dirix V, Vermeulen F, Mascart F. Maturation of CD4+ Regulatory T Lymphocytes and of Cytokine Secretions in Infants Born Prematurely. J Clin Immunol (2013) 33(6):1126–33. doi: 10.1007/s10875-013-9911-4
10. Robinson JR, Kennedy C, van Arendonk KJ, Green A, Martin CR, Blakely ML. Neurodevelopmental Considerations in Surgical Necrotizing Enterocolitis. Semin Pediatr Surg (2018) 27(1):52–6. doi: 10.1053/j.sempedsurg.2017.11.010
11. Chau V, Brant R, Poskitt KJ, Tam EWY, Synnes A, Miller SP. Postnatal Infection is Associated With Widespread Abnormalities of Brain Development in Premature Newborns. Pediatr Res (2012) 71(3):274–9. doi: 10.1038/pr.2011.40
12. Ganapathy V, Hay JW, Kim JH, Lee ML, Rechtman DJ. Long Term Healthcare Costs of Infants Who Survived Neonatal Necrotizing Enterocolitis: A Retrospective Longitudinal Study Among Infants Enrolled in Texas Medicaid. BMC Pediatr (2013) 13:127. doi: 10.1186/1471-2431-13-127
13. Murthy K, Yanowitz TD, Digeronimo R, Dykes FD, Zaniletti I, Sharma J, et al. Short-Term Outcomes for Preterm Infants With Surgical Necrotizing Enterocolitis. J Perinatol (2014) 34(10):736–40. doi: 10.1038/jp.2014.153
14. Soraisham AS, Amin HJ, Al-Hindi MY, Singhal N, Sauve RS. Does Necrotising Enterocolitis Impact the Neurodevelopmental and Growth Outcomes in Preterm Infants With Birthweight ≤1250 G? J Paediatr Child Health (2006) 42(9):499–504. doi: 10.1111/j.1440-1754.2006.00910.x
15. Hickey M, Georgieff M, Ramel S. Neurodevelopmental Outcomes Following Necrotizing Enterocolitis. Semin Fetal Neonatal Med (2018) 23(6):426–32. doi: 10.1016/j.siny.2018.08.005
16. Shah DK, Doyle LW, Anderson PJ, Bear M, Daley AJ, Hunt RW, et al. Adverse Neurodevelopment in Preterm Infants With Postnatal Sepsis or Necrotizing Enterocolitis is Mediated by White Matter Abnormalities on Magnetic Resonance Imaging At Term. J Pediatr (2008) 153(2):P170-5.E1. doi: 10.1016/j.jpeds.2008.02.033
17. Niño DF, Zhou Q, Yamaguchi Y, Martin LY, Wang S, Fulton WB, et al. Cognitive Impairments Induced by Necrotizing Enterocolitis Can be Prevented by Inhibiting Microglial Activation in Mouse Brain. Sci Transl Med (2018) 10(471):eaan0237. doi: 10.1126/scitranslmed.aan0237
18. Lu P, Sodhi CP, Jia H, Shaffiey S, Good M, Branca MF, et al. Animal Models of Gastrointestinal and Liver Diseases. Animal Models of Necrotizing Enterocolitis: Pathophysiology, Translational Relevance, and Challenges. Am J Physiol - Gastrointest Liver Physiol (2014) 306(11):G917–28. doi: 10.1152/ajpgi.00422.2013
19. Dowling DJ, Levy O. Ontogeny of Early Life Immunity. Trends Immunol (2014) 35(7):299–310. doi: 10.1016/j.it.2014.04.007
20. Collins A, Weitkamp JH, Wynn JL. Why are Preterm Newborns At Increased Risk of Infection? Arch Dis Child Fetal Neonatal (2018) 103(4):F391–4. doi: 10.1136/archdischild-2017-313595
21. Battersby AJ, Gibbons DL. The Gut Mucosal Immune System in the Neonatal Period. Pediatr Allergy Immunol (2013) 24(5):414–21. doi: 10.1111/pai.12079
22. Starner TD, Agerberth B, Gudmundsson GH, McCray PB. Expression and Activity of β-Defensins and LL-37 in the Developing Human Lung. J Immunol (2005) 174(3):1608–15. doi: 10.4049/jimmunol.174.3.1608
23. Weaver LT, Laker MF, Nelson R. Intestinal Permeability in the Newborn. Arch Dis Child (1984) 59(3):236–41. doi: 10.1136/adc.59.3.236
24. Salzman NH, Underwood MA, Bevins CL. Paneth Cells, Defensins, and the Commensal Microbiota: A Hypothesis on Intimate Interplay At the Intestinal Mucosa. Semin Immunol (2007) 19(2):70–83. doi: 10.1016/j.smim.2007.04.002
25. Clark JA, Doelle SM, Halpern MD, Saunders TA, Holubec H, Dvorak K, et al. Intestinal Barrier Failure During Experimental Necrotizing Enterocolitis: Protective Effect of EGF Treatment. Am J Physiol - Gastrointest Liver Physiol (2006) 291:G938–49. doi: 10.1152/ajpgi.00090.2006
26. Shen L, Turner JR. Role of Epithelial Cells in Initiation and Propagation of Intestinal Inflammation. Eliminating the Static: Tight Junction Dynamics Exposed. Am J Physiol - Gastrointest Liver Physiol (2006) 290(4):577. doi: 10.1152/ajpgi.00439.2005
27. van den Berg JP, Westerbeek EAM, van der Klis FRM, Berbers GAM, Van Elburg RM. Transplacental Transport of Igg Antibodies to Preterm Infants: A Review of the Literature. Early Hum Dev (2011) 87(2):67–72. doi: 10.1016/j.earlhumdev.2010.11.003
28. Palmeira P, Quinello C, Silveira-Lessa AL, Zago CA, Carneiro-Sampaio M. Igg Placental Transfer in Healthy and Pathological Pregnancies. Clin Dev Immunol (2012) 2012:985646. doi: 10.1155/2012/985646
29. Buckley RH. The Immunologic System and Disorders. Kliegman RM, RE B, HB J, editors. Philadelphia: WB Saunders (2004). p. 681. (Nelson’s textbook of pediatrics).
30. Davis CA, Vallota EH, Forristal J. Serum Complement Levels in Infancy: Age Related Changes. Pediatr Res (1979) 13(9):1043–6. doi: 10.1203/00006450-197909000-00019
31. Johnston RB, Altenburger KM, Atkinson AW, Curry RH. Complement in the Newborn Infant. Pediatrics (1979) 64(5 Suppl.):781–6.
32. McGreal EP, Hearne K, Spiller OB. Off to a Slow Start: Under-Development of the Complement System in Term Newborns is More Substantial Following Premature Birth. Immunobiology (2012) 217(2):176–86. doi: 10.1016/j.imbio.2011.07.027
33. Carr R, Modi N. Haemopoietic Colony Stimulating Factors for Preterm Neonates. Arch Dis Childhood: Fetal Neonatal Edition (1997) 76:F128–33. doi: 10.1136/fn.76.2.F128
34. Raymond SL, Mathias BJ, Murphy TJ, Rincon JC, López MC, Ungaro R, et al. Neutrophil Chemotaxis and Transcriptomics in Term and Preterm Neonates. Transl Res (2017) 190:4–15. doi: 10.1016/j.trsl.2017.08.003
35. Maródi L, Goda K, Palicz A, Szabó G. Cytokine Receptor Signalling in Neonatal Macrophage: Defective STAT-1 Phosphorylation in Response to Stimulation With Ifn-γ. Clin Exp Immunol (2001) 126(3):456–60. doi: 10.1046/j.1365-2249.2001.01693.x
36. Azizia M, Lloyd J, Allen M, Klein N, Peebles D. Immune Status in Very Preterm Neonates. Pediatrics (2012) 129(4):967. doi: 10.1542/peds.2011-1579
37. McIntire DD, Leveno KJ. Neonatal Mortality and Morbidity Rates in Late Preterm Births Compared With Births At Term. Obstet Gynecol (2008) 111(1):35–41. doi: 10.1097/01.AOG.0000297311.33046.73
38. Stoll BJ, Hansen N, Fanaroff AA, Wright LL, Carlo WA, Ehrenkranz RA, et al. Changes in Pathogens Causing Early-Onset Sepsis in Very-Low-Birth-Weight Infants. N Engl J Med (2002) 347(4):240–7. doi: 10.1056/NEJMoa012657
39. Strunk T, Inder T, Wang X, Burgner D, Mallard C, Levy O. Infection-Induced Inflammation and Cerebral Injury in Preterm Infants. Lancet Infect Diseases (2014) 14(8):751–62. doi: 10.1016/S1473-3099(14)70710-8
40. Pulver LS, Denney JM, Silver RM, Young PC. Morbidity and Discharge Timing of Late Preterm Newborns. Clin Pediatr (Phila) (2010) 49(11):1061–7. doi: 10.1177/0009922810376821
41. Escobar GJ, Clark RH, Greene JD. Short-Term Outcomes of Infants Born At 35 and 36 Weeks Gestation: We Need to Ask More Questions. Semin Perinatol (2006) 30(1):28–33. doi: 10.1053/j.semperi.2006.01.005
42. Khashu M, Narayanan M, Bhargava S, Osiovich H. Perinatal Outcomes Associated With Preterm Birth At 33 to 36 Weeks’ Gestation: A Population-Based Cohort Study. Pediatrics (2009) 123(1):109–13. doi: 10.1016/S8756-5005(09)79097-3
43. Nanthakumar NN, Fusunyan RD, Sanderson I, Walker WA. Inflammation in the Developing Human Intestine: A Possible Pathophysiologic Contribution to Necrotizing Enterocolitis. Proc Natl Acad Sci USA (2000) 97(11):6043–8. doi: 10.1073/pnas.97.11.6043
44. Claud EC, Lu L, Anton PM, Savidge T, Walker WA, Cherayil BJ. Developmentally Regulated Iκb Expression in Intestinal Epithelium and Susceptibility to Flagellin-Induced Inflammation. Proc Natl Acad Sci USA (2004) 101(19):7404–8. doi: 10.1073/pnas.0401710101
45. Been JV, Zimmermann LJI, Debeer A, Kloosterboer N, van Iwaarden JF. Bronchoalveolar Lavage Fluid From Preterm Infants With Chorioamnionitis Inhibits Alveolar Epithelial Repair. Respir Res (2009) 10(1):116. doi: 10.1186/1465-9921-10-116
46. Egan CECE, Sodhi CPCP, Good M, Lin J, Jia H, Yamaguchi Y, et al. Toll-Like Receptor 4-Mediated Lymphocyte Influx Induces Neonatal Necrotizing Enterocolitis. J Clin Invest (2016) 126(2):495–508. doi: 10.1172/JCI83356
47. Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, et al. Interleukin-17 and Lung Host Defense Against Klebsiella Pneumoniae Infection. Am J Respir Cell Mol Biol (2001) 25(3):335–40. doi: 10.1165/ajrcmb.25.3.4424
48. Fedele G, Nasso M, Spensieri F, Palazzo R, Frasca L, Watanabe M, et al. Lipopolysaccharides From Bordetella Pertussis and Bordetella Parapertussis Differently Modulate Human Dendritic Cell Functions Resulting in Divergent Prevalence of Th17-Polarized Responses. J Immunol (2008) 181(1):208–16. doi: 10.4049/jimmunol.181.1.208
49. Matusevicius D, Kivisäkk P, He B, Kostulas N, Özenci V, Fredrikson S, et al. Interleukin-17 MRNA Expression in Blood and CSF Mononuclear Cells is Augmented in Multiple Sclerosis. Mult Scler (1999) 5(2):101–4. doi: 10.1177/135245859900500206
50. Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, et al. High Levels of IL-17 in Rheumatoid Arthritis Patients: IL-15 Triggers in Vitro IL-17 Production Via Cyclosporin a-Sensitive Mechanism. J Immunol (2000) 164(5):2832–8. doi: 10.4049/jimmunol.164.5.2832
51. Teunissen MBM, Koomen CW, De Waal Malefyt R, Wierenga EA, Bos JD. Interleukin-17 and Interferon-γ Synergize in the Enhancement of Proinflammatory Cytokine Production by Human Keratinocytes. J Invest Dermatol (1998) 111(4):645–9. doi: 10.1046/j.1523-1747.1998.00347.x
52. Weitkamp JH, Koyama T, Rock MT, Correa H, Goettel JA, Matta P, et al. Necrotising Enterocolitis is Characterised by Disrupted Immune Regulation and Diminished Mucosal Regulatory (FOXP3)/effector (CD4, CD8) T Cell Ratios. Gut (2013) 62(1):73–82. doi: 10.1136/gutjnl-2011-301551
53. Hackam DJ, Sodhi CP, Good M. New Insights Into Necrotizing Enterocolitis: From Laboratory Observation to Personalized Prevention and Treatment. J Pediatr Surg (2019) 54(3):398–404. doi: 10.1016/j.jpedsurg.2018.06.012
54. Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 Signal Transduction Pathway. Cytokine (2008) 42(2):145–51. doi: 10.1016/j.cyto.2008.01.006
55. Rhee SH. Basic and Translational Understandings of Microbial Recognition by Toll-Like Receptors in the Intestine. J Neurogastroenterol Motility (2011) 17(1):28–34. doi: 10.5056/jnm.2011.17.1.28
56. Tang AH, Brunn GJ, Cascalho M, Platt JL. Pivotal Advance: Endogenous Pathway to SIRS, Sepsis, and Related Conditions. J Leukoc Biol (2007) 82(2):282–5. doi: 10.1189/jlb.1206752
57. Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, et al. Toll-Like Receptor 4-Dependent Activation of Dendritic Cells by β-Defensin 2. Sci (80- ) (2002) 298(5595):1025–9. doi: 10.1126/science.1075565
58. Ohashi K, Burkart V, Flohé S, Kolb H. Cutting Edge: Heat Shock Protein 60 is a Putative Endogenous Ligand of the Toll-Like Receptor-4 Complex. J Immunol (2000) 164(2):558–61. doi: 10.4049/jimmunol.164.2.558
59. Singh RK, Haka AS, Asmal A, Barbosa-Lorenzi VC, Grosheva I, Chin HF, et al. TLR4 (Toll-Like Receptor 4)-Dependent Signaling Drives Extracellular Catabolism of LDL (Low-Density Lipoprotein) Aggregates. Arterioscler Thromb Vasc Biol (2020) 40(1):86–102. doi: 10.1161/ATVBAHA.119.313200
60. Takeda K, Akira S. Toll-Like Receptors. Curr Protoc Immunol (2015) 109:14.12.1–14.12.10. doi: 10.1002/0471142735.im1412s109
61. Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, et al. The Repertoire for Pattern Recognition of Pathogens by the Innate Immune System is Defined by Cooperation Between Toll-Like Receptors. Proc Natl Acad Sci U S A (2000) 97(25):13766–71. doi: 10.1073/pnas.250476497
62. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS Signaling in C3H/Hej and C57BL/10ScCr Mice: Mutations in Tlr4 Gene. Sci (80- ) (1998) 282(5396):2085–8. doi: 10.1126/science.282.5396.2085
63. Lakhani SA, Bogue CW. Toll-Like Receptor Signaling in Sepsis. Curr Opin Pediatr (2003) 15(3):278–82. doi: 10.1097/00008480-200306000-00009
64. Niño DF, Sodhi CP, Hackam DJ. Necrotizing Enterocolitis: New Insights Into Pathogenesis and Mechanisms. Nat Rev Gastroenterol Hepatol (2016) 13(10):590–600. doi: 10.1038/nrgastro.2016.119
65. Claud E. Neonatal Necrotizing Enterocolitis – Inflammation and Intestinal Immaturity. Antiinflamm Antiallergy Agents Med Chem (2009) 8(3):248–59. doi: 10.2174/187152309789152020
66. Meng D, Zhu W, Shi HN, Lu L, Wijendran V, Xu W, et al. Toll-Like Receptor-4 in Human and Mouse Colonic Epithelium is Developmentally Regulated: A Possible Role in Necrotizing Enterocolitis. Pediatr Res (2015) 77(3):416–24. doi: 10.1038/pr.2014.207
67. Hackam DJ, Sodhi CP. Toll-Like Receptor-Mediated Intestinal Inflammatory Imbalance in the Pathogenesis of Necrotizing Enterocolitis. Cell Mol Gastroenterol Hepatol (2018) 6(2):229–38.e1. doi: 10.1016/j.jcmgh.2018.04.001
68. Sodhi CP, Neal MD, Siggers R, Sho S, Ma C, Branca MF, et al. Intestinal Epithelial Toll-Like Receptor 4 Regulates Goblet Cell Development and is Required for Necrotizing Enterocolitis in Mice. Gastroenterology (2012) 143(3):708–18.e1-5. doi: 10.1053/j.gastro.2012.05.053
69. Neal MD, Sodhi CP, Jia H, Dyer M, Egan CE, Yazji I, et al. Toll-Like Receptor 4 is Expressed on Intestinal Stem Cells and Regulates Their Proliferation and Apoptosis Via the p53 Up-Regulated Modulator of Apoptosis. J Biol Chem (2012) 287(44):37296–308. doi: 10.1074/jbc.M112.375881
70. Werts AD, Fulton WB, Ladd MR, Saad-Eldin A, Chen YX, Kovler ML, et al. A Novel Role for Necroptosis in the Pathogenesis of Necrotizing Enterocolitis. CMGH (2020) 9(3):403–23. doi: 10.1016/j.jcmgh.2019.11.002
71. Afrazi A, Branca MF, Sodhi CP, Good M, Yamaguchi Y, Egan CE, et al. Toll-Like Receptor 4-Mediated Endoplasmic Reticulum Stress in Intestinal Crypts Induces Necrotizing Enterocolitis. J Biol Chem (2014) 289(14):9584–99. doi: 10.1074/jbc.M113.526517
72. Shiou SR, Yu Y, Chen S, Ciancio MJ, Petrof EO, Sun J, et al. Erythropoietin Protects Intestinal Epithelial Barrier Function and Lowers the Incidence of Experimental Neonatal Necrotizing Enterocolitis. J Biol Chem (2011) 286(14):12123–32. doi: 10.1074/jbc.M110.154625
73. Yu Y, Shiou SR, Guo Y, Lu L, Westerhoff M, Sun J, et al. Erythropoietin Protects Epithelial Cells From Excessive Autophagy and Apoptosis in Experimental Neonatal Necrotizing Enterocolitis. PloS One (2013) 8(7):e69620. doi: 10.1371/journal.pone.0069620
74. Yamoto M, Alganabi M, Chusilp S, Lee D, Yazaki Y, Lee C, et al. Lysosomal Overloading and Necrotizing Enterocolitis. Pediatr Surg Int (2020) 36(10):1157–65. doi: 10.1007/s00383-020-04724-x
75. Guan Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J Immunol Res (2019) 2019:1–16. doi: 10.1155/2019/7247238
76. Sharma R, Tepas JJ, Hudak ML, Mollitt DL, Wludyka PS, Teng RJ, et al. Neonatal Gut Barrier and Multiple Organ Failure: Role of Endotoxin and Proinflammatory Cytokines in Sepsis and Necrotizing Enterocolitis. J Pediatr Surg (2007) 42(3):454–61. doi: 10.1016/j.jpedsurg.2006.10.038
77. Yazji I, Sodhi CP, Lee EK, Good M, Egan CE, Afrazi A, et al. Endothelial TLR4 Activation Impairs Intestinal Microcirculatory Perfusion in Necrotizing Enterocolitis Via ENOS-NO-nitrite Signaling. Proc Natl Acad Sci USA (2013) 110(23):9451–6. doi: 10.1073/pnas.1219997110
78. Hackam DJ, Sodhi CP. Toll-Like Receptor–Mediated Intestinal Inflammatory Imbalance in the Pathogenesis of Necrotizing Enterocolitis. Cmgh (2018) 6(2):229–38.e1. doi: 10.1016/j.jcmgh.2018.04.001
79. Cho SX, Rudloff I, Lao JC, Pang MA, Goldberg R, Bui CB, et al. Characterization of the Pathoimmunology of Necrotizing Enterocolitis Reveals Novel Therapeutic Opportunities. Nat Commun (2020) 11(1):5794. doi: 10.1038/s41467-020-19400-w
80. Zhou W, Li W, Zheng XH, Rong X, Huang LG. Glutamine Downregulates TLR-2 and TLR-4 Expression and Protects Intestinal Tract in Preterm Neonatal Rats With Necrotizing Enterocolitis. J Pediatr Surg (2014) 1057–63. doi: 10.1016/j.jpedsurg.2014.02.078
81. Prencipe G, Azzari C, Moriondo M, Devito R, Inglese R, Pezzullo M, et al. Association Between Mannose-Binding Lectin Gene Polymorphisms and Necrotizing Enterocolitis in Preterm Infants. J Pediatr Gastroenterol Nutr (2012) 55(2):160–5. doi: 10.1097/MPG.0b013e31824e5f7a
82. Auriti C, Prencipe G, Moriondo M, Bersani I, Bertaina C, Mondì V, et al. Mannose-Binding Lectin: Biologic Characteristics and Role in the Susceptibility to Infections and Ischemia-Reperfusion Related Injury in Critically Ill Neonates. J Immunol Res (2017) 2017:7045630. doi: 10.1155/2017/7045630
83. Yu R, Jiang S, Tao Y, Li P, Yin J, Zhou Q. Inhibition of HMGB1 Improves Necrotizing Enterocolitis by Inhibiting NLRP3 Via TLR4 and NF-κb Signaling Pathways. J Cell Physiol (2019) 234(8):13431–8. doi: 10.1002/jcp.28022
84. Lamkanfi M, Dixit VM. Mechanisms and Functions of Inflammasomes. Cell (2014). doi: 10.1016/j.cell.2014.04.007
85. Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut Microbiota in the Pathogenesis of Inflammatory Bowel Disease. Clin J Gastroenterol (2018) 11(1):1–10. doi: 10.1007/s12328-017-0813-5
86. Shen ZH, Zhu CX, Quan YS, Yang ZY, Wu S, Luo WW, et al. Relationship Between Intestinal Microbiota and Ulcerative Colitis: Mechanisms and Clinical Application of Probiotics and Fecal Microbiota Transplantation. World J Gastroenterol (2018) 24(1):5–14. doi: 10.3748/wjg.v24.i1.5
87. Bouskra D, Brézillon C, Bérard M, Werts C, Varona R, Boneca IG, et al. Lymphoid Tissue Genesis Induced by Commensals Through NOD1 Regulates Intestinal Homeostasis. Nature (2008) 456(7221):507–10. doi: 10.1038/nature07450
88. Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic Bacteria Direct Expression of an Intestinal Bactericidal Lectin. Sci (80- ) (2006) 313(5790):1126–30. doi: 10.1126/science.1127119
89. Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The Placenta Harbors a Unique Microbiome. Sci Transl Med (2014) 6(237):237ra65. doi: 10.1126/scitranslmed.3008599
90. Ardissone AN, De La Cruz DM, Davis-Richardson AG, Rechcigl KT, Li N, Drew JC, et al. Meconium Microbiome Analysis Identifies Bacteria Correlated With Premature Birth. PloS One (2014) 9(3):e90784. doi: 10.1371/journal.pone.0090784
91. Vinturache AE, Gyamfi-Bannerman C, Hwang J, Mysorekar IU, Jacobsson B. Maternal Microbiome - a Pathway to Preterm Birth. Semin Fetal Neonatal Med (2016) 21(2):94–9. doi: 10.1016/j.siny.2016.02.004
92. Thänert R, Keen EC, Dantas G, Warner BB, Tarr PI. Necrotizing Enterocolitis and the Microbiome: Current Status and Future Directions. J Infect Dis (2020) jiaa604. doi: 10.1093/infdis/jiaa604
93. La Rosa PS, Warner BB, Zhou Y, Weinstock GM, Sodergren E, Hall-Moore CM, et al. Patterned Progression of Bacterial Populations in the Premature Infant Gut. Proc Natl Acad Sci U S A (2014) 111(34):12522–7. doi: 10.1073/pnas.1409497111
94. Gasparrini AJ, Wang B, Sun X, Kennedy EA, Hernandez-Leyva A, Ndao IM, et al. Persistent Metagenomic Signatures of Early-Life Hospitalization and Antibiotic Treatment in the Infant Gut Microbiota and Resistome. Nat Microbiol (2019) 4(12):2285–97. doi: 10.1038/s41564-019-0550-2
95. Morrow AL, Lagomarcino AJ, Schibler KR, Taft DH, Yu Z, Wang B, et al. Early Microbial and Metabolomic Signatures Predict Later Onset of Necrotizing Enterocolitis in Preterm Infants. Microbiome (2013) 1(1):13. doi: 10.1186/2049-2618-1-13
96. Younge NE, Araújo-Pérez F, Brandon D, Seed PC. Early-Life Skin Microbiota in Hospitalized Preterm and Full-Term Infants. Microbiome (2018) 6(1):98. doi: 10.1186/s40168-018-0486-4
97. Younge NE, Newgard CB, Cotten CM, Goldberg RN, Muehlbauer MJ, Bain JR, et al. Disrupted Maturation of the Microbiota and Metabolome Among Extremely Preterm Infants With Postnatal Growth Failure. Sci Rep (2019) 9(1):8167. doi: 10.1038/s41598-019-44547-y
98. Arboleya S, Sánchez B, Milani C, Duranti S, Solís G, Fernández N, et al. Intestinal Microbiota Development in Preterm Neonates and Effect of Perinatal Antibiotics. J Pediatr (2015) 166(3):538–44. doi: 10.1016/j.jpeds.2014.09.041
99. Clark RH, Bloom BT, Spitzer AR, Gerstmann DR. Reported Medication Use in the Neonatal Intensive Care Unit: Data From a Large National Data Set. Pediatrics (2006) 117(6):1979–87. doi: 10.1542/peds.2005-1707
100. Terrin G, Passariello A, De Curtis M, Manguso F, Salvia G, Lega L, et al. Ranitidine is Associated With Infections, Necrotizing Enterocolitis, and Fatal Outcome in Newborns. Pediatrics (2012) 129(1):e40–5. doi: 10.1542/peds.2011-0796
101. Riskin A, Almog M, Peri R, Halasz K, Srugo I, Kessel A. Changes in Immunomodulatory Constituents of Human Milk in Response to Active Infection in the Nursing Infant. Pediatr Res (2012) 71(2):220–5. doi: 10.1038/pr.2011.34
102. Carlisle EM, Morowitz MJ. The Intestinal Microbiome and Necrotizing Enterocolitis. Curr Opin Pediatr (2013) 25(3):382–7. doi: 10.1097/MOP.0b013e3283600e91
103. Gopalakrishna KP, Macadangdang BR, Rogers MB, Tometich JT, Firek BA, Baker R, et al. Maternal Iga Protects Against the Development of Necrotizing Enterocolitis in Preterm Infants. Nat Med (2019) 25(7):1110–5. doi: 10.1038/s41591-019-0480-9
104. Abegunde AT, Muhammad BH, Bhatti O, Ali T. Environmental Risk Factors for Inflammatory Bowel Diseases: Evidence Based Literature Review. World J Gastroenterol (2016) 22(27):6296–317. doi: 10.3748/wjg.v22.i27.6296
105. Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, et al. Functional Characterization of Human Receptors for Short Chain Fatty Acids and Their Role in Polymorphonuclear Cell Activation. J Biol Chem (2003) 278(28):25481–9. doi: 10.1074/jbc.M301403200
106. Neal MD, Leaphart C, Levy R, Prince J, Billiar TR, Watkins S, et al. Enterocyte TLR4 Mediates Phagocytosis and Translocation of Bacteria Across the Intestinal Barrier. J Immunol (2006) 176(5):3070–9. doi: 10.4049/jimmunol.176.5.3070
107. Neish AS, Gewirtz AT, Zeng H, Young AN, Hobert ME, Karmali V, et al. Prokaryotic Regulation of Epithelial Responses by Inhibition of Iκb-α Ubiquitination. Sci (80- ) (2000) 289(5484):1560–3. doi: 10.1126/science.289.5484.1560
108. Claud EC, Keegan KP, Brulc JM, Lu L, Bartels D, Glass E, et al. Bacterial Community Structure and Functional Contributions to Emergence of Health or Necrotizing Enterocolitis in Preterm Infants. Microbiome (2013) 1(1):20. doi: 10.1186/2049-2618-1-20
109. Mai V, Young CM, Ukhanova M, Wang X, Sun Y, Casella G, et al. Fecal Microbiota in Premature Infants Prior to Necrotizing Enterocolitis. PloS One (2011) 6(6):e20647. doi: 10.1371/journal.pone.0020647
110. Wang Y, Hoenig JD, Malin KJ, Qamar S, Petrof EO, Sun J, et al. 16S RRNA Gene-Based Analysis of Fecal Microbiota From Preterm Infants With and Without Necrotizing Enterocolitis. ISME J (2009) 3(8):944–54. doi: 10.1038/ismej.2009.37
111. Nafday SM, Chen W, Peng L, Babyatsky MW, Holzman IR, Lin J. Short-Chain Fatty Acids Induce Colonic Mucosal Injury in Rats With Various Postnatal Ages. Pediatr Res (2005) 57(2):201–4. doi: 10.1203/01.PDR.0000150721.83224.89
112. Puccio G, Alliet P, Cajozzo C, Janssens E, Corsello G, Sprenger N, et al. Effects of Infant Formula With Human Milk Oligosaccharides on Growth and Morbidity: A Randomized Multicenter Trial. J Pediatr Gastroenterol Nutr (2017) 64(4):624–31. doi: 10.1097/MPG.0000000000001520
113. Jiao X, Fu M, Wang YY, Xue J, Zhang Y. Bifidobacterium and Lactobacillus for Preventing Necrotizing Enterocolitis in Very-Low-Birth-Weight Preterm Infants: A Systematic Review and Meta-Analysis. World J Pediatr (2020) 16(2):135–42. doi: 10.1007/s12519-019-00297-5
114. Stewart CJ, Embleton ND, Marrs ECL, Smith DP, Nelson A, Abdulkadir B, et al. Temporal Bacterial and Metabolic Development of the Preterm Gut Reveals Specific Signatures in Health and Disease. Microbiome (2016) 4(1):67. doi: 10.1186/s40168-016-0216-8
115. Tormo-Badia N, Håkansson Å, Vasudevan K, Molin G, Ahrné S, Cilio CM. Antibiotic Treatment of Pregnant Non-Obese Diabetic Mice Leads to Altered Gut Microbiota and Intestinal Immunological Changes in the Offspring. Scand J Immunol (2014) 80(4):250–60. doi: 10.1111/sji.12205
116. Yan X, Managlia E, Tan X, De Plaen IG. Prenatal Inflammation Impairs Intestinal Microvascular Development Through a TNF-dependent Mechanism and Predisposes Newborn Mice to Necrotizing Enterocolitis. Am J Physiol - Gastrointest Liver Physiol (2019) 317:G57–66. doi: 10.1152/ajpgi.00332.2018
117. Hansen CHF, Krych Ł, Buschard K, Metzdorff SB, Nellemann C, Hansen LH, et al. A Maternal Gluten-Free Diet Reduces Inflammation and Diabetes Incidence in the Offspring of NOD Mice. Diabetes (2014) 63(8):2821–32. doi: 10.2337/db13-1612
118. Neu J. Developmental Aspects of Maternal-Fetal, and Infant Gut Microbiota and Implications for Long-Term Health. Matern Heal Neonatol Perinatol (2015) 1:6. doi: 10.1186/s40748-015-0007-4
119. Ren S, Hui Y, Goericke-Pesch S, Pankratova S, Kot W, Pan X, et al. Gut and Immune Effects of Bioactive Milk Factors in Preterm Pigs Exposed to Prenatal Inflammation. Am J Physiol - Gastrointest Liver Physiol (2019) 317:G67–77. doi: 10.1152/ajpgi.00042.2019
120. Lundgren SN, Madan JC, Emond JA, Morrison HG, Christensen BC, Karagas MR, et al. Maternal Diet During Pregnancy is Related With the Infant Stool Microbiome in a Delivery Mode-Dependent Manner. Microbiome (2018) 6(1):109. doi: 10.1186/s40168-018-0490-8
121. De Agüero MG, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, et al. The Maternal Microbiota Drives Early Postnatal Innate Immune Development. Sci (80- ) (2016) 351:1296–302. doi: 10.1126/science.aad2571
122. Pan X, Zhang D, Nguyen DN, Wei W, Yu X, Gao F, et al. Postnatal Gut Immunity and Microbiota Development is Minimally Affected by Prenatal Inflammation in Preterm Pigs. Front Immunol (2020) 11:420. doi: 10.3389/fimmu.2020.00420
123. Banfield E, Fulton W, Burd I, Kovler ML, Sodhi CP, Hackam DJ. The Role of in Utero Endotoxin Exposure in the Development of Inflammatory Bowel Disease in Mice. Am J Reprod Immunol (2020) 84(4):e13302. doi: 10.1111/aji.13302
124. Harbeson D, Francis F, Bao W, Amenyogbe NA, Kollmann TR. Energy Demands of Early Life Drive a Disease Tolerant Phenotype and Dictate Outcome in Neonatal Bacterial Sepsis. Front Immunol (2018) 9:1918. doi: 10.3389/fimmu.2018.01918
125. Chen L, Cohen AC, Lewis DB. Impaired Allogeneic Activation and T-Helper 1 Differentiation of Human Cord Blood Naive CD4 T Cells. Biol Blood Marrow Transplant (2006) 12(2):160–71. doi: 10.1016/j.bbmt.2005.10.027
126. Lewis DB, Wilson CB. Developmental Immunology and Role of Host Defenses in Fetal and Neonatal Susceptibility to Infection. In: Infectious Diseases of the Fetus and Newborn Infant. Philadephia: W.B. Saunders (2006). p. 87–210. (Infectious Diseases of the Fetus and Newborn Infant). doi: 10.1016/B0-72-160537-0/50006-2
127. Palin AC, Ramachandran V, Acharya S, Lewis DB. Human Neonatal Naive CD4 + T Cells Have Enhanced Activation-Dependent Signaling Regulated by the MicroRNA miR-181a. J Immunol (2013) 190(6):2682–91. doi: 10.4049/jimmunol.1202534
128. Wilson CB, Westall J, Johnston L, Lewis DB, Dower SK, Alpert AR. Decreased Production of Interferon-Gamma by Human Neonatal Cells. Intrinsic and Regulatory Deficiencies. J Clin Invest (1986) 77(3):860–7. doi: 10.1172/JCI112383
129. Kan B, Michalski C, Fu H, Au HHT, Lee K, Marchant EA, et al. Cellular Metabolism Constrains Innate Immune Responses in Early Human Ontogeny. Nat Commun (2018) 9(1):4822. doi: 10.1038/s41467-018-07215-9
130. Gribar SC, Sodhi CP, Richardson WM, Anand RJ, Gittes GK, Branca MF, et al. Reciprocal Expression and Signaling of TLR4 and TLR9 in the Pathogenesis and Treatment of Necrotizing Enterocolitis. J Immunol (2009) 182(1):636–46. doi: 10.4049/jimmunol.182.1.636
131. Good M, Siggers RH, Sodhi CP, Afrazi A, Alkhudari F, Egan CE, et al. Amniotic Fluid Inhibits Toll-Like Receptor 4 Signaling in the Fetal and Neonatal Intestinal Epithelium. Proc Natl Acad Sci U S A (2012) 109(28):11330–5. doi: 10.1073/pnas.1200856109
132. Moore SA, Nighot P, Reyes C, Rawat M, McKee J, Lemon D, et al. Intestinal Barrier Dysfunction in Human Necrotizing Enterocolitis. J Pediatr Surg (2016) 51(12):1907–13. doi: 10.1016/j.jpedsurg.2016.09.011
133. Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. Identification of ZO-1: A High Molecular Weight Polypeptide Associated With the Tight Junction (Zonula Occludens) in a Variety of Epithelia. J Cell Biol (1986) 103(3):755–66. doi: 10.1083/jcb.103.3.755
134. Kellett GL. The Facilitated Component of Intestinal Glucose Absorption. J Physiol (2001) 537:585–95. doi: 10.1111/j.1469-7793.2001.0585h.x
135. Kellett GL, Brot-Laroche E, Mace OJ, Leturque A. Sugar Absorption in the Intestine: The Role of GLUT2. Annu Rev Nutr (2008) 28:35–54. doi: 10.1146/annurev.nutr.28.061807.155518
136. Rahner C, Mitic LL, Anderson JM. Heterogeneity in Expression and Subcellular Localization of Claudins 2, 3, 4, and 5 in the Rat Liver, Pancreas, and Gut. Gastroenterology (2001) 120(2):411–22. doi: 10.1053/gast.2001.21736
137. Fujita H, Chiba H, Yokozaki H, Sakai N, Sugimoto K, Wada T, et al. Differential Expression and Subcellular Localization of Claudin-7, –8, –12, –13, and –15 Along the Mouse Intestine. J Histochem Cytochem (2006) 54(8):933–44. doi: 10.1369/jhc.6A6944.2006
138. Bai M, Lu C, An L, Gao Q, Xie W, Miao F, et al. SIRT1 Relieves Necrotizing Enterocolitis Through Inactivation of Hypoxia-Inducible Factor (HIF)-1a. Cell Cycle (2020) 19(16):2018–27. doi: 10.1080/15384101.2020.1788251
139. Yu Y, Lu L, Sun J, Petrof EO, Claud EC. Preterm Infant Gut Microbiota Affects Intestinal Epithelial Development in a Humanized Microbiome Gnotobiotic Mouse Model. Am J Physiol - Gastrointest Liver Physiol (2016) 311:G521–32. doi: 10.1152/ajpgi.00022.2016
140. Young JC, Pan C, Adams RM, Brooks B, Banfield JF, Morowitz MJ, et al. Metaproteomics Reveals Functional Shifts in Microbial and Human Proteins During a Preterm Infant Gut Colonization Case. Proteomics (2015) 5(20):3463–73. doi: 10.1002/pmic.201400563
141. Liu D, Xu Y, Feng J, Yu J, Huang J, Li Z. Mucins and Tight Junctions are Severely Altered in Necrotizing Enterocolitis Neonates. Am J Perinatol (2020). doi: 10.1055/s-0040-1710558
142. Bein A, Eventov-Friedman S, Arbell D, Schwartz B. Intestinal Tight Junctions are Severely Altered in NEC Preterm Neonates. Pediatr Neonatol (2018) 59(5):464–73. doi: 10.1016/j.pedneo.2017.11.018
143. Dai S, Sodhi C, Cetin S, Richardson W, Branca M, Neal MD, et al. Extracellular High Mobility Group Box-1 (HMGB1) Inhibits Enterocyte Migration Via Activation of Toll-Like Receptor-4 and Increased Cell-Matrix Adhesiveness. J Biol Chem (2010) 285(7):4995–5002. doi: 10.1074/jbc.M109.067454
144. Leaphart CL, Cavallo J, Gribar SC, Cetin S, Li J, Branca MF, et al. A Critical Role for TLR4 in the Pathogenesis of Necrotizing Enterocolitis by Modulating Intestinal Injury and Repair. J Immunol (2007) 179(7):4808–20. doi: 10.4049/jimmunol.179.7.4808
145. Sodhi CP, Shi XH, Richardson WM, Grant ZS, Shapiro RA, Prindle TJ, et al. Toll-Like Receptor-4 Inhibits Enterocyte Proliferation Via Impaired beta-Catenin Signaling in Necrotizing Enterocolitis. Gastroenterology (2010) 138:185–96. doi: 10.1053/j.gastro.2009.09.045
146. Neal MD, Sodhi CP, Dyer M, Craig BT, Good M, Jia H, et al. A Critical Role for TLR4 Induction of Autophagy in the Regulation of Enterocyte Migration and the Pathogenesis of Necrotizing Enterocolitis. J Immunol (2013) 190(7):3541–51. doi: 10.4049/jimmunol.1202264
147. Maynard AA, Dvorak K, Khailova L, Dobrenen H, Arganbright KM, Halpern MD, et al. Epidermal Growth Factor Reduces Autophagy in Intestinal Epithelium and in the Rat Model of Necrotizing Enterocolitis. Am J Physiol - Gastrointest Liver Physiol (2010) 299:G614–22. doi: 10.1152/ajpgi.00076.2010
148. Emami CN, Mittal R, Wang L, Ford HR, Prasadarao NV. Role of Neutrophils and Macrophages in the Pathogenesis of Necrotizing Enterocolitis Caused by Cronobacter Sakazakii. J Surg Res (2012) 172(1):18–28. doi: 10.1016/j.jss.2011.04.019
149. Stefanutti G, Lister P, Smith VV, Peters MJ, Klein NJ, Pierro A, et al. P-Selectin Expression, Neutrophil Infiltration, and Histologic Injury in Neonates With Necrotizing Enterocolitis. J Pediatr Surg (2005) 40:942–7; discussion 947-8. doi: 10.1016/j.jpedsurg.2005.03.027
150. Vincent D, Klinke M, Eschenburg G, Trochimiuk M, Appl B, Tiemann B, et al. NEC is Likely a NETs Dependent Process and Markers of NETosis are Predictive of NEC in Mice and Humans. Sci Rep (2018) 8(1):12612. doi: 10.1038/s41598-018-31087-0
151. Hong CR, Fullerton BS, Mercier CE, Morrow KA, Edwards EM, Ferrelli KR, et al. Growth Morbidity in Extremely Low Birth Weight Survivors of Necrotizing Enterocolitis At Discharge and Two-Year Follow-Up. J Pediatr Surg (2018) 53(6):1197–202. doi: 10.1016/j.jpedsurg.2018.02.085
152. Pike K, Brocklehurst P, Jones D, Kenyon S, Salt A, Taylor D, et al. Outcomes At 7 Years for Babies Who Developed Neonatal Necrotising Enterocolitis: The ORACLE Children Study. Arch Dis Child Fetal Neonatal Ed (2012) 97(5):F318–22. doi: 10.1136/fetalneonatal-2011-300244
153. Stoll BJ, Hansen NI, Adams-Chapman I, Fanaroff AA, Hintz SR, Vohr B, et al. Neurodevelopmental and Growth Impairment Among Extremely Low-Birth-Weight Infants With Neonatal Infection. J Am Med Assoc (2004) 292(19):2357–65. doi: 10.1001/jama.292.19.2357
154. Griffin IJ, Tancredi DJ, Bertino E, Lee HC, Profit J. Postnatal Growth Failure in Very Low Birthweight Infants Born Between 2005 and 2012. Arch Dis Child Fetal Neonatal Ed (2016) 101(1):F50–5. doi: 10.1136/archdischild-2014-308095
155. Ehrenkranz RA, Dusick AM, Vohr BR, Wright LL, Wrage LA, Poole WK. Growth in the Neonatal Intensive Care Unit Influences Neurodevelopmental and Growth Outcomes of Extremely Low Birth Weight Infants. Pediatrics (2006) 117(4):1253–61. doi: 10.1542/peds.2005-1368
156. Bauer J, Hentschel R, Linderkamp O. Effect of Sepsis Syndrome on Neonatal Oxygen Consumption and Energy Expenditure. Pediatrics (2002) 110(6):e69. doi: 10.1542/peds.110.6.e69
157. Mrozek JD, Georgieff MK, Blazar BR, Mammel MC, Schwarzenberg SJ. Effect of Sepsis Syndrome on Neonatal Protein and Energy Metabolism. J Perinatol (2000) 20(2):96–100. doi: 10.1038/sj.jp.7200319
158. Hasselgren PO. Muscle Protein Metabolism During Sepsis. Biochem Soc Trans (1995) 23(4):1019–25. doi: 10.1042/bst0231019
159. Smith IJ, Lecker SH, Hasselgren PO. Calpain Activity and Muscle Wasting in Sepsis. Am J Physiol - Endocrinol Metab (2008) 295(4):E762–71. doi: 10.1152/ajpendo.90226.2008
160. Lodha A, Asztalos E, Moore A. Cytokine Levels in Neonatal Necrotizing Enterocolitis and Long-Term Growth and Neurodevelopment. Acta Paediatr Int J Paediatr (2010) 99(3):338–43. doi: 10.1111/j.1651-2227.2009.01600.x
161. Wang FS, Yu ML, Li WZ, Hong K, Bin X, Wang GH. Intestinal Tract and Parenteral Multi-Organ Sequential Pathological Injury Caused by Necrotizing Enterocolitis. BMC Pediatr (2020) 20(1):418. doi: 10.21203/rs.3.rs-22105/v2
162. Cassir N, Simeoni U, Scola B. Gut Microbiota and the Pathogenesis of Necrotizing Enterocolitis in Preterm Neonates. Future Microbiol (2016) 11(2):273–92. doi: 10.2217/fmb.15.136
163. Schulzke SM, Deshpande GC, Patole SK. Neurodevelopmental Outcomes of Very Low-Birth-Weight Infants With Necrotizing Enterocolitis: A Systematic Review of Observational Studies. Arch Pediatr Adolesc Med (2007) 161(6):583–90. doi: 10.1001/archpedi.161.6.583
164. Rees CM, Pierro A, Eaton S. Neurodevelopmental Outcomes of Neonates With Medically and Surgically Treated Necrotizing Enterocolitis. Arch Dis Child Fetal Neonatal Ed (2007) 92(3):F193–8. doi: 10.1136/adc.2006.099929
165. Roze E, Ta BDP, Van Der Ree MH, Tanis JC, Van Braeckel KNJA, Hulscher JBF, et al. Functional Impairments At School Age of Children With Necrotizing Enterocolitis or Spontaneous Intestinal Perforation. Pediatr Res (2011) 70(6):619–25. doi: 10.1203/PDR.0b013e31823279b1
166. Humberg A, Spiegler J, Fortmann MI, Zemlin M, Marissen J, Swoboda I, et al. Surgical Necrotizing Enterocolitis But Not Spontaneous Intestinal Perforation is Associated With Adverse Neurological Outcome At School Age. Sci Rep (2020) 10(1):2373. doi: 10.1038/s41598-020-58761-6
167. Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, et al. Metabolomics Analysis Reveals Large Effects of Gut Microflora on Mammalian Blood Metabolites. Proc Natl Acad Sci USA (2009) 106(10):3698–703. doi: 10.1073/pnas.0812874106
168. Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Tóth M, et al. The Gut Microbiota Influences Blood-Brain Barrier Permeability in Mice. Sci Transl Med (2014) 6(263):263ra158. doi: 10.1126/scitranslmed.3009759
169. Benarroch EE. Microglia: Multiple Roles in Surveillance, Circuit Shaping, and Response to Injury. Neurology (2013) 81(12):1079–88. doi: 10.1212/WNL.0b013e3182a4a577
170. Favrais G, Van De Looij Y, Fleiss B, Ramanantsoa N, Bonnin P, Stoltenburg-Didinger G, et al. Systemic Inflammation Disrupts the Developmental Program of White Matter. Ann Neurol (2011) 70(4):550–65. doi: 10.1002/ana.22489
171. Wang KC, Fan LW, Kaizaki A, Pang Y, Cai Z, Tien LT. Neonatal Lipopolysaccharide Exposure Induces Long-Lasting Learning Impairment, Less Anxiety-Like Response and Hippocampal Injury in Adult Rats. Neuroscience (2013) 36(2):119–31. doi: 10.1016/j.neuroscience.2012.12.049
172. Smith PLP, Hagberg H, Naylor AS, Mallard C. Neonatal Peripheral Immune Challenge Activates Microglia and Inhibits Neurogenesis in the Developing Murine Hippocampus. Dev Neurosci (2014) 36(2):119–31. doi: 10.1159/000359950
173. Wadhawan R, Oh W, Hintz SR, Blakely ML, Das A, Bell EF, et al. Neurodevelopmental Outcomes of Extremely Low Birth Weight Infants With Spontaneous Intestinal Perforation or Surgical Necrotizing Enterocolitis. J Perinatol (2014) 34(1):64–70. doi: 10.1038/jp.2013.128
174. Jobe AH, Bancalari E. Bronchopulmonary Dysplasia. Am J Respir Crit Care Med (2001) 163(7):1723–9. doi: 10.1164/ajrccm.163.7.2011060
175. Jia H, Sodhi CP, Yamaguchi Y, Lu P, Martin LY, Good M, et al. Pulmonary Epithelial TLR4 Activation Leads to Lung Injury in Neonatal Necrotizing Enterocolitis. J Immunol (2016) 197(3):859–71. doi: 10.4049/jimmunol.1600618
176. Jia H, Sodhi CP, Yamaguchi Y, Lu P, Ladd MR, Werts A, et al. Toll Like Receptor 4 Mediated Lymphocyte Imbalance Induces Nec-Induced Lung Injury. Shock (2018) 52(2):215–23. doi: 10.1097/SHK.0000000000001255
Keywords: NEC = necrotizing enterocolitis, TL4 – Toll-like receptor 4, microbiota (microorganism), prematurity and low birth weight, pediatric sepsis
Citation: Sampah MES and Hackam DJ (2021) Prenatal Immunity and Influences on Necrotizing Enterocolitis and Associated Neonatal Disorders. Front. Immunol. 12:650709. doi: 10.3389/fimmu.2021.650709
Received: 07 January 2021; Accepted: 06 April 2021;
Published: 21 April 2021.
Edited by:
Philipp Henneke, University of Freiburg Medical Center, GermanyReviewed by:
Hailong Song, University of Pennsylvania, United StatesYue Zhang, Harvard University, United States
Copyright © 2021 Sampah and Hackam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David J. Hackam, Dhackam1@jhmi.edu