
- 1 Section of Bacterial Pathogenesis, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Tokyo, Japan
- 2 Department of Medical Genome Sciences, Graduate School of Frontier Sciences, University of Tokyo, Chiba, Japan
Bacteria rapidly change their transcriptional patterns during infection in order to adapt to the host environment. To investigate host–bacteria interactions, various strategies including the use of animal infection models, in vitro assay systems and microscopic observations have been used. However, these studies primarily focused on a few specific genes and molecules in bacteria. High-density tiling arrays and massively parallel sequencing analyses are rapidly improving our understanding of the complex host–bacterial interactions through identification and characterization of bacterial transcriptomes. Information resulting from these high-throughput techniques will continue to provide novel information on the complexity, plasticity, and regulation of bacterial transcriptomes as well as their adaptive responses relative to pathogenecity. Here we summarize recent studies using these new technologies and discuss the utility of transcriptome analysis.
Introduction
The host expresses various defense systems against bacterial infection. At the same time, pathogens attempt to protect themselves from recognition and removal by the host immune system through changes in their gene expression patterns. Thus, multidirectional investigations of bacterial factors for adaptation to host environments are required for understanding the interactions between the host and bacteria as well as to clarify the acquired and innate immune responses against infection. For approaching this issue, various strategies have been used including animal infection models, in vitro assay systems and microscopic observations. Until recently, the majority of these reports have been focused on the roles of a few specific genes of either the pathogen or its host primarily due to technical limitations. These detailed and focused studies greatly affected and strengthened our understanding of only a limited portion of host–bacteria interactions. However, they did not reflect the dynamics of transcriptional regulation at the whole genome level between the host and pathogen upon infection. Comprehensive analyses to reveal the function and regulation of global factors involved in the bacteria–host interactions should be invaluable because such approaches may reveal novel virulence genes or mechanisms, which have not previously been linked to bacterial infection.
Recently whole genomic tiling arrays and massively parallel sequencing approaches have emerged as powerful tools in microbiology. Genomic tiling arrays use a set of overlapping oligonucleotide probes that represent a subset of or the whole genome at very high resolution (Wang et al., 2009). There are two general types of the tiling arrays that are most widely used (Mockler et al., 2005). The first array generally contains relatively short probes (<100-mer) synthesized directly on the surface of a chip by photolithographic method (Fodor et al., 1991; Hughes et al., 2001; Nuwaysir et al., 2002). This type of array can be made with greater than 6 million discrete features, each of which contains millions of copies of a distinct probe. The second array is consisted of mechanically printing probes including amplified PCR products, oligonucleotides or cloned DNA fragments onto the chip. This type of array can hold up to nearly 40,000 features per chip (Mockler et al., 2005).
As massively parallel sequencing approach, three commercial technologies, Roche 454 FLX titanium (Roche Diagnostics, Basel, Switzerland), Illumina Genome Analyzer (Illumina Inc., San Diego, CA, USA) and Life Technologies SOLiD (Applied Biosystems by Life Technologies, CA, USA), are now widely used and can produce millions or a billion of sequences at once. These high-throughput sequencing technologies allow the cost effective DNA sequencing compared with standard dye-terminator Sanger methods. Roche 454 FLX titanium technology is based on pyrosequencing and its advantages are the generation of long sequence reads (400 bp) and the relatively rapid sequencing run (approximately 10 h per run)1. This technology generates a small amount of data (>400 Mbp per run) among the three sequencers and may lead to homopolymer errors because multiple incorporations were provided at a given cycle (Engstrand, 2009). The Illumina GA technology is based on massively parallel sequencing of millions of fragments using a reversible terminator-based sequencing chemistry2. Advantages of Illumina technology are the generation of large amount of data (100 Gbp total per run) and less homopolymer errors compared with Roche 454 technology. However, this sequencing generates relatively short sequence length (100 bp) and takes long time for sequence run (7 days). Finally, the Life Technologies SOLiD technology is based on sequencing by ligation of dye-labeled oligonucleotides3. This technology can deal with many samples using multiple sequence tags in a single run and generate large datasets (>100 Gbp total per run). However, disadvantages are the short read length (50 bp) and the long run times as in case of Illumina technology.
Applications of tiling arrays and massively parallel sequencing include de novo assembly, chromatin immunoprecipitation analyses, genome resequencing, and metagenomics. Here we describe several major applications of both technologies (Figure 1) and briefly introduce their relevance to bacteria–host interactions.
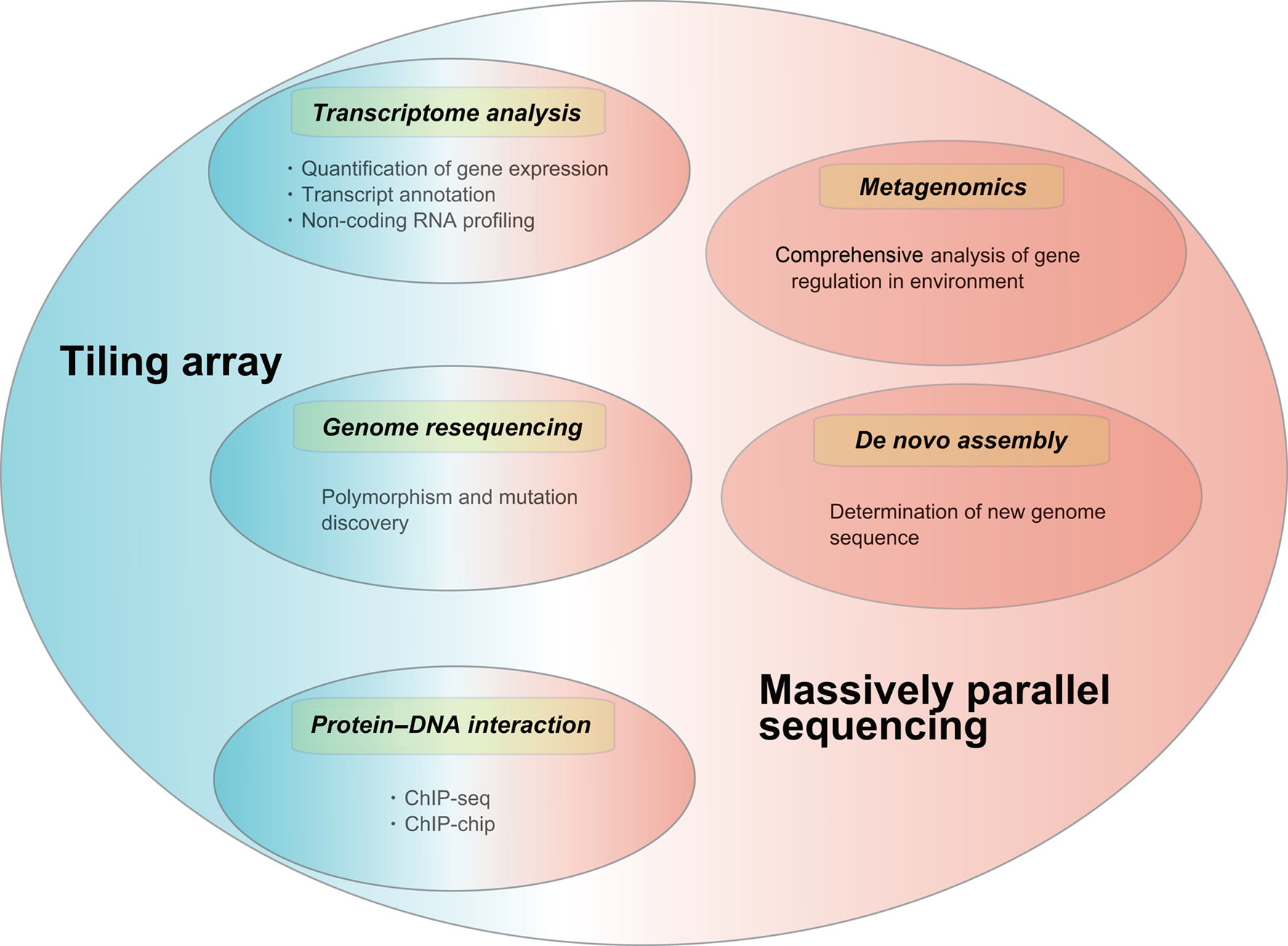
Figure 1. Applications of tiling array and massively parallel sequencing. Transcriptome analysis, genome resequencing and protein–DNA interaction (ChIP-) studies can employ both tiling array and massively parallel sequencing while applications like metagenomic studies and de novo assembly can only be performed using massively parallel sequencing. Tiling array: Blue, Massively parallel sequencing: Red.
De novo genome assembly using massively parallel sequencing and/or Sanger sequencing have been performed for some bacterial genomes including Mycoplasma conjunctive (Calderon-Copete et al., 2009), Brucella microti (Audic et al., 2009), and Helicobacter pylori strain G27 (Baltrus et al., 2009). This approach provides rapid and low cost closure of whole genome assembly and is useful for fine drafts of genome assemblies for other bacteria.
Genome resequencing using both approaches can accurately characterize mutant genomes relative to previously sequenced parental (reference) strains. In this approach, sequence differences such as insertions/deletions or sequential single strand polymorphisms (SNPs) are primarily identified with mutant and reference strains. This approach has been applied with methicillin-resistant Staphylococcus aureus (Kennedy et al., 2008), Chlamydia trachomatis (Kari et al., 2008), Brucella species (Foster et al., 2009), and Salmonella enterica serovar Typhimurium (Holt et al., 2008). These studies have demonstrated the value and importance of genome resequencing to define distinct virulence factors.
Chromatin immunoprecipitation analyses followed by microarrays (ChIP-chip) or sequencing (ChIP-seq) have been developed as powerful methods for the study of genome-wide protein–DNA interactions. These approaches can accurately identify transcriptional factors regulating bacterial pathogenesis at the whole genome level. ChIP-chip analysis using tiling arrays has been performed for Bacillus subtilis (Ishikawa et al., 2007), Escherichia coli (Cho et al., 2008a,b). In addition, ChIP-seq analysis using massively parallel sequencing has been carried out with Mycobacterium tuberculosis (Lun et al., 2009). There are few studies using ChIP-seq up to now; however, since sequencing has become faster and cheaper, ChIP-seq will likely become more available for mapping sites of protein–DNA interactions in the future.
Metagenomics is the genomic analysis of microbial communities by direct extraction of DNA from an assemblage of microorganisms (Handelsman, 2004) and reveals landscapes of bacterial diversity for a wide range of environments. This analysis has been performed with Sanger sequencing earlier (Venter et al., 2004; Kurokawa et al., 2007) but recently has been conducted using massively parallel sequencing. Several projects including the characterization of the soil metagenome (Roesch et al., 2007), the honey bee metagenome (Cox-Foster et al., 2007), the human gut metagenome (McKenna et al., 2008), mouse gut metagenome (Turnbaugh et al., 2006), the mine metagenome (Edwards et al., 2006), and the chicken cecum metagenome (Qu et al., 2008) have been recently completed.
In addition to the above applications, transcriptome analysis is a novel application for a better understanding of host–bacteria interactions. Eukaryotic transcriptome analyses by massively parallel sequencing have been recently carried out because of its effectiveness and power in collecting data (Mardis, 2008; Shendure and Ji, 2008; Wang et al., 2009; Wilhelm and Landry, 2009). For bacteria, these analytical strategies are now available for elucidating the complexity of transcriptomes but only a few applications have been carried out so far.
As well as high-throughput mRNA sequencing (RNA-seq) using massively parallel sequencing, genomic tiling arrays have been used in genome-wide transcriptome analysis approaches. In this review, we summarize recent significant reports in the field of cellular microbiology, in which two powerful tools, RNA-seq and genomic tiling arrays, have been used. The significance of these technologies is also described relative to obtain more knowledge of the transcriptional regulation of pathogenicity.
Use of Tiling Array Technology for Bacterial Transcriptomes
Compared with massively parallel sequencing, tiling arrays do not always require mRNA enrichment, and their experimental protocols are now well established. Despite these obvious advantages, tiling arrays have one major drawback, i.e., transcriptome maps are usually of a lower resolution than the maps produced by RNA-seq. The most optimal candidates for tiling array probes should begin at every single base position in the genome (Sorek and Cossart, 2010). However, most tiling arrays have lower densities mainly because of cost issues. In addition, tiling arrays often produce high backgrounds because of non-specific or cross-hybridization reactions (van Vliet, 2010). Thus, the raw data for tiling arrays must be subjected to extensive normalization. After suitable normalization of the data, tiling arrays reveal dynamic and abundant units of transcription. Conventional open reading frame (ORF) microarrays are designed to detect gene expression with relatively few probes for known or predicted genes. In contrast, tiling arrays can lead to the identification of many novel non-coding RNAs (ncRNAs) since these use probes that span the entire genome. Therefore, use of tiling arrays is the major technique for transcriptome analysis to date and it has been applied to several bacterial transcriptome studies including Bacillus subtilis, Caulobacter crescentus, Halobacterium salinarum, and Mycobacterium leprae (McGrath et al., 2007; Koide et al., 2009; Rasmussen et al., 2009; Akama et al., 2009). In addition, one excellent earlier study and several more recent new studies in cellular microbiology focused on transcriptomes using tiling arrays and are summarized in the following sections (Table 1).
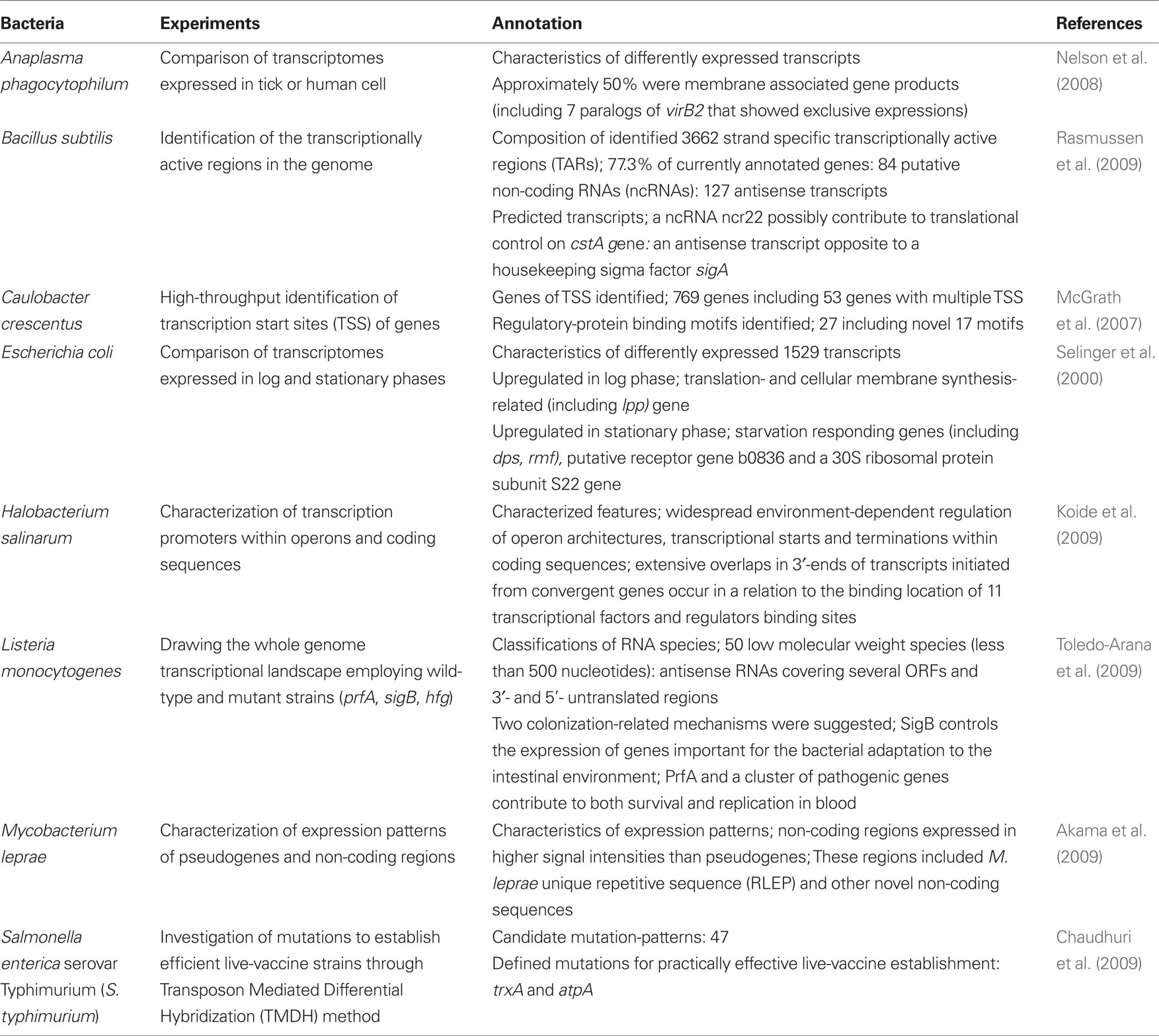
Table 1. Bacterial transcriptome analyses using genomic tiling array.
Escherichia Coli
Ten years ago, the transcriptomes of E. coli in the growth and stationary phases were compared using tiling arrays (Selinger et al., 2000). In this approach, the authors used average 25-mer probes, arranged every 6 bases for the intergenic regions and every 60 bases for ORFs. In nutrient rich medium, transcripts detected in the stationary and log phases covered 97 and 87% of the ORFs, respectively. Under these conditions, the 1529 transcripts showed differential expression. In log phase, proteins involved in translation (rRNA, tRNA, and ribosomal proteins) and the synthesis of cell membrane (lpp) were expressed at higher levels than those of the stationary phase, while genes encoding proteins involved in responding to starvation (such as dps and rmf) were expressed at higher levels in the stationary phase. In addition, putative receptor (b0836) and a 30S ribosomal protein subunit (S22) genes have been revealed to be highly upregulated in stationary phase for the first time (Selinger et al., 2000). In this study, the density of probes was higher than in previous studies, and significant expression of RNAs was clearly detected from antisense strands and intergenic regions. Thus, this study has been recognized as a milestone in the technical development of tiling arrays for prokaryotic transcriptome analyses.
Anaplasma Phagocytophilum
Anaplasma phagocytophilum causes the tick-bone disease human anaplasmosis. A. phagocytophilum can replicate in tick cell line ISE6 (Munderloh et al., 2003) and two human cell lines HL-60 and HMEC-1 which have been used as models of human infection (Ades et al., 1992). Transcriptomes of A. phagocytophilum in ticks (ISE6) and humans (HL-60 and HMEC-1) were compared to obtain clues for life cycle regulation and the pathogenecity of this bacterium (Nelson et al., 2008). As a result, no significant difference was found between bacterial transcriptomes expressed in the two human cell lines, however, distinct differences in transcriptional activities of bacterial genes were observed between the two different host species. Specifically, transcriptional levels of half of the membrane associated protein genes including seven virB2 paralog genes (associated with the bacterial type IV secretion system) were markedly distinct. Moreover, a few paralogs of the major surface protein genes p44/msp2 were newly identified through hybridization between transcripts and hypervariable regions (HVRs) in human cells while this was not found in ISE6. This study indicated the flexibility of the bacterium in adapting and altering its pathogenicity for different hosts by changing its transcriptional patterns.
Salmonella Enterica Serovar Typhimurium
Systemic typhoid fever is one of the important targets for vaccine therapy (Mastroeni and Menager, 2003; Girard et al., 2006). In this study, a novel microarray-based technology designated as transposon mediated differential hybridization (TMDH) (Charles, 2001) was used to identify attenuated transposon mutants of the bacterium which inactivated virulence genes against mice. The authors examined selected genes from the mutants as live vaccine candidates by the TMDH method with tiling arrays (Chaudhuri et al., 2009). In this approach, modified transposons carrying outward-facing T7 and Sp6 promoters were introduced into the bacterium and the mixture of transformants were either infected into the mice or cultured in vitro. Subsequently, genomic DNA libraries from both infecting and cultured bacteria were prepared and subject to in vitro transcription in the presence of isotope-labeled UTP. The DNA/RNA-mixtures isolated were then digested with a restriction endonuclease Rsa I and applied to the tiling arrays for analysis. The data from both infecting and cultured samples were used to evaluate attenuation scores and provided 47 subsets of transposons carrying distinct deleted genes. Among these mutants, subsequent analyses focused on two mutants as candidates for preparing live-vaccination strains; trxA encoding thioredoxin 1, which is known to be important for infection of mice (Bjur et al., 2006) and atpA involved in oxidative phosphorylation (Turner et al., 2003). Eventually, two strains of the bacterium carrying the respective candidate mutations were immunized into mice and the mice were successfully confirmed to have become resistant against infection with wild-type S. typhimurium (Chaudhuri et al., 2009). Thus, the TMDH method with tiling arrays could be applicable to other bacterial species in identification of attenuated virulence genes.
Listeria Monocytogenes
Listeria monocytogenes ubiquitously inhabits many different environments and often causes severe food-bone diseases. Toredo-Arana et al. (2009) used wild-type and mutant (prfA, sigB, hfq) strains to describe the complete operon map of the pathogen. It is known that PrfA controls transcription of virulence genes in the blood (Scortti et al., 2007) and SigB mediates virulence activation in the host intestine (Chakraborty et al., 2000). Hfq is an RNA-binding protein and is involved in stress tolerance and virulence control (Christiansen et al., 2004). In this study, total RNA of the strains was extracted from ex vivo and in vitro cultures and were used with tiling arrays to analyze whole genome transcriptomes. As a result, the presence of a variety of RNA species was observed. These RNAs include 50 low molecular weight species (less than 500 nucleotides) and at least two of them were involved in virulence in mice. Antisense RNAs covering several ORFs and 3′ and 5′ untranslated regions (UTRs) were also detected. Following detailed analysis, a possible role for a riboswitch functioning in the termination of an upstream gene was suggested. In addition this study also described a novel proposal regarding the relevance of SigB in specifically controlling the expression of genes important for the bacterial adaptation to the intestinal environment as well as the involvement of PrfA and a pathogenic gene cluster in survival and replication in the blood. Interestingly, this analysis revealed that changes in transcriptional levels of ncRNAs were similar as for virulence genes in L. monocytogenes although no such changes were observed in non-pathogenic L. innocula. As a consequence, it was suggested that successive and coordinated global transcriptional changes occur during infection (Toledo-Arana et al., 2009). This study suggested significant progress in comprehensive whole-transcriptome analysis of a bacterial species. In addition, this report provided insight into the greater complexity of bacterial transcription than was previously predicted.
Use of Massively Parallel Sequencing for Bacterial Transcriptome Analysis
Approaches for studying pathogenic bacteria with massively parallel sequencing have much improved our knowledge of their pathogenicity, evolution and adaptation to different environments including the host. In order to evaluate this approach, the basic procedures involved will be summarized below.
Important Notes for Sample Preparation
1. Isolated bacterial RNA consists of approximately 80% rRNA and tRNA (Condon, 2007). Therefore, removal of tRNA/rRNA is usually carried out before reverse transcription (Passalacqua et al., 2009; Perkins et al., 2009; Yoder-Himes et al., 2009). Size fractionation of RNA prior to cDNA synthesis has been optionally used for the removal of mRNA and rRNA (Liu et al., 2009).
2. Most bacterial mRNA does not contain a poly-A tail as do eukaryotes and thus immobilized poly-T cannot enrich for mRNA relative to other RNA species following hybridization. As a consequence, cDNA synthesis (reverse transcription) should use one of the following priming methods: use of random hexamers (Passalacqua et al., 2009; Perkins et al., 2009; Yoder-Himes et al., 2009), oligo (dT) priming after polyadenylation of mRNA (Frias-Lopez et al., 2008) or priming after ligation of specific RNA adaptors to mRNA (Sittka et al., 2008; Wurtzel et al., 2009).
3. Sequencers such as Illumina GA/Solexa, ABI SOLiD, or 454 FLX/Titanium are now widely available for high-throughput analyses.
4. The adaptors ligated before cDNA synthesis should be removed followed by mapping the reads to its genome sequence as the first step in information processing.
Several reports that used RNA-seq with the relevant sequencers are summarized (Table 2). Among these studies, several recent excellent studies which have not been referenced in previous reviews (Sorek and Cossart, 2010; van Vliet, 2010) are introduced in the following sections.
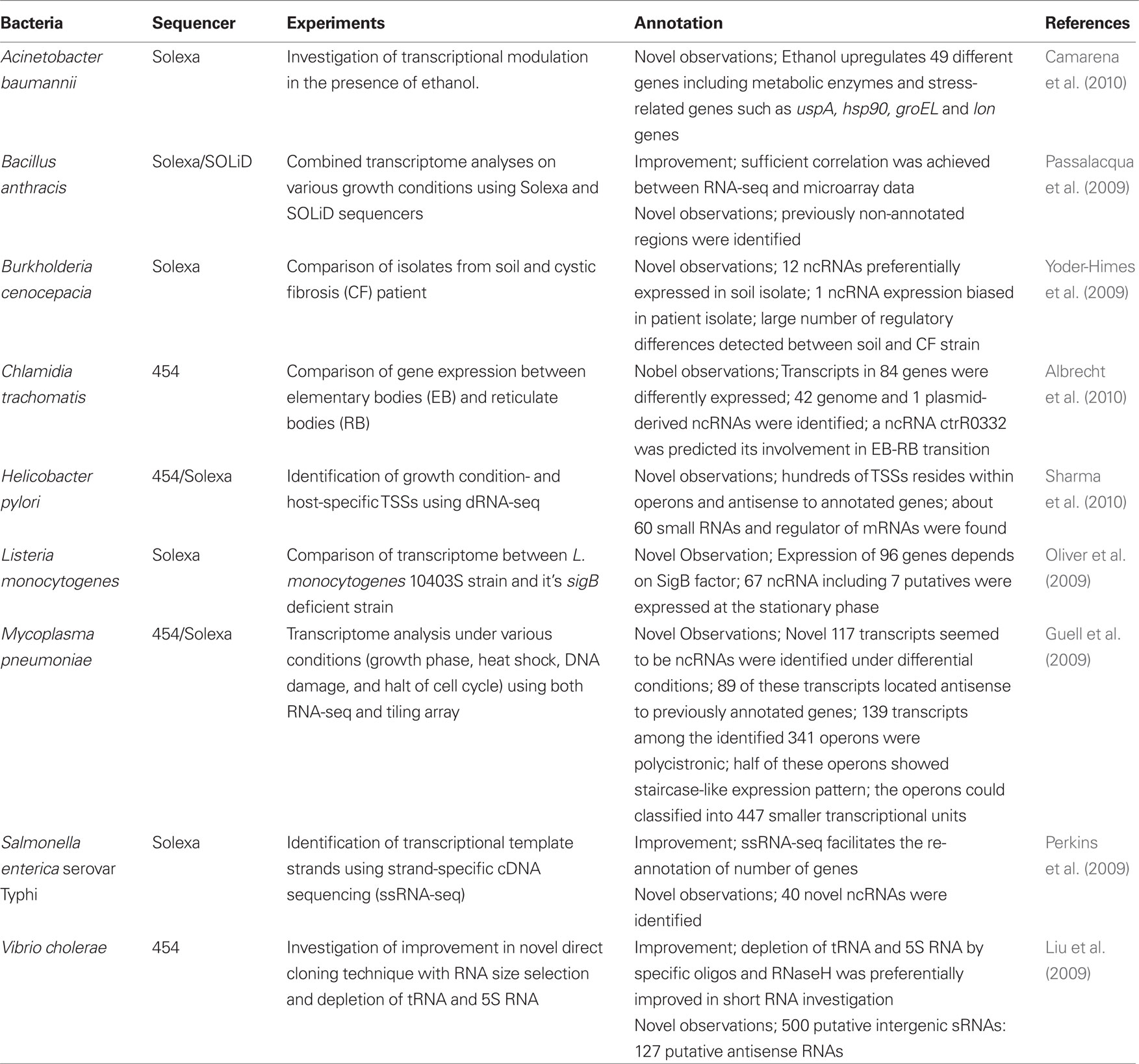
Table 2. Bacterial transcriptome analyses using massively parallel sequencers.
Chlamydia Trachomatis
Chlamydia trachomatis, a causative agent of a sexually transmitted disease and/or a contagious eye infection, was subjected to transcriptome sequencing (Albrecht et al., 2010). The authors compared the transcriptome of C. trachomatis in different states: metabolically inactive elementary bodies (EB) and metabolically active reticulate bodies (RB), which can replicate in vacuoles inside of host cells. In this study, the Roche/454 GS-FLX system was used and the sequences obtained were subjected to determinative analysis of transcriptional start sites (TSS). To identify primary TSS, cDNA libraries from both EB and RB were sequenced; one library was generated from untreated total RNA and the other was constructed following enrichment of primary transcripts by selective enzymatic degradation of “processed RNA species” (see H. pylori section for details). Transcripts of 84 genes revealed distinct expression levels between EB and RB. In addition, 42 genome and 1 plasmid-derived ncRNA were identified, respectively. Among these ncRNAs, ctrR0332 in the genome showed approximately ten times greater expression in EB than that in RB. This result suggests that ctrR0332 plays an important role in the EB-RB transition (Albrecht et al., 2010). The precise identification of TSS should lead to a better understanding of genome organization as well as the control of bacterial behavior.
Acinetobacter Baumannii
Transcriptome analysis of A. baumannii was carried out using Illumina technology (Camarena et al., 2010). In this study, cDNA libraries (obtained from mRNA which were enriched by removal of 23S and 16S rRNA) were prepared from cultures with or without ethanol since previous reports showed that ethanol increases the virulence of the pathogen in both Caenorhabditis elegans and Dictyostelium discoideum (Wanner, 1987; Smith et al., 2004). Sequence data showed that 49 genes were upregulated in the presence of ethanol. Among these genes, some encoded metabolic enzymes including several dehydrogenases for ethanol which were highly induced, suggesting that A. baumannii oxidizes ethanol to acetate by these enzymes. The genes encoding stress proteins including hsp90, groEL, and lon were also detected in the presence of ethanol. These genes are involved in the heat-shock stress response (HSR) in many bacterial species (Asadulghani et al., 2003; Green and Donohue, 2006; Qin et al., 2006; Slamti et al., 2007; Audia et al., 2008; Martinez-Salazar et al., 2009). The HSR is controlled by the rpoH gene encoding sigma factor σ32 (Yura et al., 1993) and has been shown to be required for optimal virulence in some bacteria including Vibrio tapetis and Neisseria gonorrhoeae (Du et al., 2005; Lakhal et al., 2008). In addition, a previous report showed that A. baumannii carrying a transposon insertion in rpoH attenuated virulence in the presence of ethanol (Smith et al., 2007). Therefore, the authors suggested that ethanol could increase the virulence of the bacterium through the induction of heat-shock proteins, such as Hsp90, GroEL and Lon. Furthermore, ethanol-dependent upregulation was also observed in secretory phospholipase C. Since deletion of phospholipase C gene in the bacterium diminished its cytotoxicity in epithelial cells, this gene may be significantly involved in the virulence of the pathogen.
Listeria Monocytogenes
RNA-seq was also used to examine the transcriptome of L. monocytogenes in addition to the tiling array approach noted above. In this method, the authors analyzed the differences in the transcriptome between L. monocytogenes strain 10403S and its sigB deficient strain using Illumina sequencing (Oliver et al., 2009). cDNA libraries were obtained following enrichment of mRNA by removal of 23S and 16S rRNA, and were fractionated into 60-200 nucleotides. The authors identified transcripts of 96 genes which were expressed in a sigB-dependent manner. According to the RNA-seq data, the bacterium expressed 67 ncRNAs including seven novel ncRNAs. Furthermore, a total of 65 putative sigB promoters upstream of 82 of the 96 sigB-dependent genes and upstream of the one sigB-dependent ncRNA were identified. This study provided comprehensive insight into prokaryotic transcriptional regulation following comparison of a mutant devoid of a transcriptional regulator and its parent strain.
Helicobacter Pylori
Sharma et al. (2010) analyzed the transcriptome of H. pylori strain 26695 with the Roche/454 GS-FLX system and Illumina technology. In this study, a new approach named differential RNA-seq (dRNA-seq) was employed to identify primary TSS. Primary transcripts included most precursor mRNAs and small RNAs (sRNAs) carrying a 5′ tri-phosphate (5′PPP) group, whereas processed transcripts include mature rRNA and tRNA harboring a 5′ mono-phosphate (5′P). The authors presented a single-nucleotide resolution map of the primary transcriptome of H. pylori through discrimination of primary transcripts with native 5′ (5′PPP) ends from processed species (5′P) following treatment with a 5′P-dependent exonuclease. Total RNA was extracted from the pathogen in various states such as different growth phases, stressed with acid, and different host cells. After removal of genomic DNA with DNase I and treatment by 5′P-dependent exonuclease, the cDNA libraries were then analyzed with the 454 system and mapped to the H. pylori chromosome to identify TSS. Solexa sequencing for operon mapping was also performed under the same growth conditions. TSSs were identified within operons and antisense sequences to annotated genes. These observations suggested that the major factors for increasing transcriptional complexities in H. pylori were the uncoupling of polycistrons and genome-wide antisense transcription. Approximately 60 small ncRNAs were detected in this study. These ncRNAs included 6S RNA which is a ubiquitous riboregulator of RNA polymerase but is not present in ε-proteobacteria (Barrick et al., 2005; Weinberg et al., 2007). The dRNA-seq could identify TSS at the genome-wide level and uncovered a surprisingly large number of novel ncRNA and antisense transcripts in H. pylori. This approach could be applicable to all bacterial species where native transcripts carry a 5′PPP and should be widespread soon.
Mycoplasma Pneumoniae
A different approach used for the study of the transcriptome of M. pneumoniae (Guell et al., 2009), where three methods: spotted arrays, tiling arrays, and RNA-seq were used in combination. RNA-seq and tiling array data obtained from the bacteria grown under four different conditions (growth phase, heat shock, DNA damage, and interruption of the cell cycle) revealed novel 117 transcripts. Almost all of the novel transcripts appeared not to be structural RNAs but ncRNAs and 89 of these transcripts were antisense with respect to previously annotated genes. Among the 341 operons identified, 139 transcripts were polycistronic and half of the operons showed decay patterns in transcription. This suggests that such staircase-like expression is a widespread phenomenon in bacteria. Comparison of transcriptomes obtained under various growth conditions suggested the possible classification of operons into 447 smaller transcriptional units. In addition, growth condition dependent alternative transcripts were detected as a result of spotted array data. The complexity, as known in eukaryotes, of the bacterial transcriptome is clearly indicated from these studies and was unexpected.
Concluding Remarks
In a very short period of time, bacterial transcriptomics using tiling arrays and massively parallel sequencing has been remarkably improved and has become a powerful tool for understanding host–bacteria interactions. A number of studies revealed that the bacterial transcriptome is much more complicated than previously thought. Like eukaryotes, RNA molecules are key factors in regulating gene expression in prokaryotes (Waters and Storz, 2009). Among these, regulatory RNAs, including antisense RNA and riboswitches, have been shown to modulate pathogenesis (Toledo-Arana et al., 2007), iron metabolism (Masse et al., 2007), quorum sensing (Bejerano-Sagie and Xavier, 2007) through regulation of gene expression. Novel types of RNA molecules found by tiling arrays and massively parallel sequencing are rapidly increasing (Toledo-Arana and Solano, 2010). It is difficult for current bioinformatic algorithms and databases to predict the existence and functions of all of the novel RNA or small proteins detected but several studies have attempted to clarify their functions through validation of their different expression patterns (see reviews: Romby et al., 2006; Sharma and Vogel, 2009; Sorek and Cossart, 2010).
During infection, bacteria colonize in the host environment not as single entities but as communities. Therefore, the elucidation of the transcriptome of bacterial communities is essential for a more complete understanding of host–bacteria interactions. Metatranscriptomics has emerged as an approach for enhancing our understanding of the transcriptome of bacterial communities. Several metatransciptomic studies have recently been performed for microbial communities in the soil or ocean water (Leininger et al., 2006; Frias-Lopez et al., 2008; Gilbert et al., 2008; Urich et al., 2008). In metatranscriptomics, total RNA is extracted from a microbial community, converted into cDNA and sequenced without primers (DeLong, 2009). Moreover, in this approach there is no need to be concerned about the number of genes surveyed and to select specific genes to target (Moran, 2009). Therefore, metatranscriptomics may become one of the most powerful tools for understanding bacterial regulation and adaptation upon infection within complex microbial communities.
Bacterial transcriptomics using tiling arrays and/or massively parallel sequencing will be more frequently utilized in coming years. These high-throughput technologies will continue to further improve through the use of lower amounts of starting samples, longer reads, increasing number of reads, and lower costs. Under these circumstances, when data will be almost overwhelming, new approaches for information management and interpretation will be also developed. Therefore, in the future these technologies will become more convenient and can serve as general tools for bacterial transcriptome analysis due to their valuable contributions to our knowledge base.
Selection of the appropriate technology is an issue for many researchers to perform their purpose. Massively parallel sequencing provides clear advantages over the tilling arrays, since massively parallel sequencing offers both a single-base resolution and a high-mapping resolution (Marguerat et al., 2008; Wang et al., 2009). On the other hand, tiling arrays is inherently biased by the chip design and frequently miss out alternative and antisense transcripts (Wang et al., 2009). However, massively parallel sequencing also has several assignments. Massively parallel sequencing is more expensive than array-based analysis and large data obtained from this technology need highly efficient software with a high performance computer. In contrast, tiling arrays is a good tool for first screening of bacterial transcriptome because it is more cost effective and the data derived from this technology can be analyzed with conventional computers in a laboratory or an individual level. With regard to this, Guell et al. (2009) provided a valuable report, in which they used tiling arrays and massively parallel sequencing to study the transcriptome of M. pneumoniae. They reported that sequencing data alone were insufficient to clearly detect operon boundaries in the case that genes were lowly expressed. They also described that the combination analysis using both technologies provide more accurate landscape of bacterial transcriptome. Taken together, the use of tiling arrays gives a valuable data for the first analysis of bacterial transcriptome. If more detailed data are necessary, for example, to determine the boundary of mRNA, we recommend the addition of massively parallel sequencing data to the tiling array data.
Transcriptome analysis allows the identification or prediction of novel bacterial virulence factors required for adaptation and survival within host environments as well as the enhancement of disease potential. In addition, the combination of transcriptome analyses with clinical or other experimental analyses (i.e. proteomic or metabolic analysis) will enable us to identify novel functions relating to gene expression. This will provide new insights into the molecular mechanism of host–bacteria interactions and also enhance our ability to develop a number of potential targeting molecules more efficiently. Therefore, such comprehensive analyses will continue to increase our understanding of the molecular complexity of host–bacteria interactions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Grants-in-Aids for Scientific Research (#21390487 and #22592032) and the Japan Society for the Promotion of Science, and Japan Science and Technology Agency.
Footnotes
References
Ades, E. W., Candal, F. J., Swerlick, R. A., George, V. G., Summers, S., Bosse, D. C., and Lawley, T. J. (1992). HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J. Invest. Dermatol. 99, 683–690.
Akama, T., Suzuki, K., Tanigawa, K., Kawashima, A., Wu, H., Nakata, N., Osana, Y., Sakakibara, Y., and Ishii, N. (2009). Whole-genome tiling array analysis of Mycobacterium leprae RNA reveals high expression of pseudogenes and noncoding regions. J. Bacteriol. 191, 3321–3327.
Albrecht, M., Sharma, C. M., Reinhardt, R., Vogel, J., and Rudel, T. (2010). Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res. 38, 868–877.
Asadulghani, Suzuki, Y., and Nakamoto, H. (2003). Light plays a key role in the modulation of heat shock response in the cyanobacterium Synechocystis sp PCC 6803. Biochem. Biophys. Res. Commun. 306, 872–879.
Audia, J. P., Patton, M. C., and Winkler, H. H. (2008). DNA microarray analysis of the heat shock transcriptome of the obligate intracytoplasmic pathogen Rickettsia prowazekii. Appl. Environ. Microbiol. 74, 7809–7812.
Audic, S., Lescot, M., Claverie, J. M., and Scholz, H. C. (2009). Brucella microti: the genome sequence of an emerging pathogen. BMC Genomics 10, 352.
Baltrus, D. A., Amieva, M. R., Covacci, A., Lowe, T. M., Merrell, D. S., Ottemann, K. M., Stein, M., Salama, N. R., and Guillemin, K. (2009). The complete genome sequence of Helicobacter pylori strain G27. J. Bacteriol. 191, 447–448.
Barrick, J. E., Sudarsan, N., Weinberg, Z., Ruzzo, W. L., and Breaker, R. R. (2005). 6S RNA is a widespread regulator of eubacterial RNA polymerase that resembles an open promoter. RNA 11, 774–784.
Bejerano-Sagie, M., and Xavier, K. B. (2007). The role of small RNAs in quorum sensing. Curr. Opin. Microbiol. 10, 189–198.
Bjur, E., Eriksson-Ygberg, S., Aslund, F., and Rhen, M. (2006). Thioredoxin 1 promotes intracellular replication and virulence of Salmonella enterica serovar Typhimurium. Infect. Immun. 74, 5140–5151.
Calderon-Copete, S. P., Wigger, G., Wunderlin, C., Schmidheini, T., Frey, J., Quail, M. A., and Falquet, L. (2009). The Mycoplasma conjunctivae genome sequencing, annotation and analysis. BMC Bioinformatics 10(Suppl 6), S7.
Camarena, L., Bruno, V., Euskirchen, G., Poggio, S., and Snyder, M. (2010). Molecular mechanisms of ethanol-induced pathogenesis revealed by RNA-sequencing. PLoS Pathog. 6, e1000834. doi: 10.1371/journal.ppat.1000834.
Chakraborty, T., Hain, T., and Domann, E. (2000). Genome organization and the evolution of the virulence gene locus in Listeria species. Int. J. Med. Microbiol. 290, 167–174.
Charles, I. G., and Maskell, D. J. (2001). Transposon mediated differential hybridisation. International Patent Number WO2001/007651.
Chaudhuri, R. R., Peters, S. E., Pleasance, S. J., Northen, H., Willers, C., Paterson, G. K., Cone, D. B., Allen, A. G., Owen, P. J., Shalom, G., Stekel, D. J., Charles, I. G., and Maskell, D. J. (2009). Comprehensive identification of Salmonella enterica serovar typhimurium genes required for infection of BALB/c mice. PLoS Pathog. 5, e1000529. doi: 10.1371/journal.ppat.1000529.
Cho, B. K., Barrett, C. L., Knight, E. M., Park, Y. S., and Palsson, B. O. (2008a). Genome-scale reconstruction of the Lrp regulatory network in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 105, 19462–19467.
Cho, B. K., Knight, E. M., Barrett, C. L., and Palsson, B. O. (2008b). Genome-wide analysis of Fis binding in Escherichia coli indicates a causative role for A-/AT-tracts. Genome Res. 18, 900–910.
Christiansen, J. K., Larsen, M. H., Ingmer, H., Sogaard-Andersen, L., and Kallipolitis, B. H. (2004). The RNA-binding protein Hfq of Listeria monocytogenes: role in stress tolerance and virulence. J. Bacteriol. 186, 3355–3362.
Condon, C. (2007). Maturation and degradation of RNA in bacteria. Curr. Opin. Microbiol. 10, 271–278.
Cox-Foster, D. L., Conlan, S., Holmes, E. C., Palacios, G., Evans, J. D., Moran, N. A., Quan, P. L., Briese, T., Hornig, M., Geiser, D. M., Martinson, V., vanEngelsdorp, D., Kalkstein, A. L., Drysdale, A., Hui, J., Zhai, J., Cui, L., Hutchison, S. K., Simons, J. F., Egholm, M., Pettis, J. S., and Lipkin, W. I. (2007). A metagenomic survey of microbes in honey bee colony collapse disorder. Science 318, 283–287.
Du, Y., Lenz, J., and Arvidson, C. G. (2005). Global gene expression and the role of sigma factors in Neisseria gonorrhoeae in interactions with epithelial cells. Infect. Immun. 73, 4834–4845.
Edwards, R. A., Rodriguez-Brito, B., Wegley, L., Haynes, M., Breitbart, M., Peterson, D. M., Saar, M. O., Alexander, S., Alexander, E. C., Jr. and Rohwer, F. (2006). Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 7, 57.
Engstrand, L. (2009). How will next-generation sequencing contribute to the knowledge concerning Helicobacter pylori? Clin. Microbiol. Infect. 15, 823–828.
Fodor, S. P., Read, J. L., Pirrung, M. C., Stryer, L., Lu, A. T., and Solas, D. (1991). Light-directed, spatially addressable parallel chemical synthesis. Science 251, 767–773.
Foster, J. T., Beckstrom-Sternberg, S. M., Pearson, T., Beckstrom-Sternberg, J. S., Chain, P. S., Roberto, F. F., Hnath, J., Brettin, T., and Keim, P. (2009). Whole-genome-based phylogeny and divergence of the genus Brucella. J. Bacteriol. 191, 2864–2870.
Frias-Lopez, J., Shi, Y., Tyson, G. W., Coleman, M. L., Schuster, S. C., Chisholm, S. W., and Delong, E. F. (2008). Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. U.S.A. 105, 3805–3810.
Gilbert, J. A., Field, D., Huang, Y., Edwards, R., Li, W., Gilna, P., and Joint, I. (2008). Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities. PLoS ONE 3, e3042. doi: 10.1371/journal.pone.0003042.
Girard, M. P., Steele, D., Chaignat, C. L., and Kieny, M. P. (2006). A review of vaccine research and development: human enteric infections. Vaccine 24, 2732–2750.
Green, H. A., and Donohue, T. J. (2006). Activity of Rhodobacter sphaeroides RpoHII, a second member of the heat shock sigma factor family. J. Bacteriol. 188, 5712–5721.
Guell, M., van Noort, V., Yus, E., Chen, W. H., Leigh-Bell, J., Michalodimitrakis, K., Yamada, T., Arumugam, M., Doerks, T., Kuhner, S., Rode, M., Suyama, M., Schmidt, S., Gavin, A. C., Bork, P., and Serrano, L. (2009). Transcriptome complexity in a genome-reduced bacterium. Science 326, 1268–1271.
Handelsman, J. (2004). Metagenomics: application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 68, 669–685.
Holt, K. E., Parkhill, J., Mazzoni, C. J., Roumagnac, P., Weill, F. X., Goodhead, I., Rance, R., Baker, S., Maskell, D. J., Wain, J., Dolecek, C., Achtman, M., and Dougan, G. (2008). High-throughput sequencing provides insights into genome variation and evolution in Salmonella typhi. Nat. Genet. 40, 987–993.
Hughes, T. R., Mao, M., Jones, A. R., Burchard, J., Marton, M. J., Shannon, K. W., Lefkowitz, S. M., Ziman, M., Schelter, J. M., Meyer, M. R., Kobayashi, S., Davis, C., Dai, H., He, Y. D., Stephaniants, S. B., Cavet, G., Walker, W. L., West, A., Coffey, E., Shoemaker, D. D., Stoughton, R., Blanchard, A. P., Friend, S. H., and Linsley, P. S. (2001). Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat. Biotechnol. 19, 342–347.
Ishikawa, S., Ogura, Y., Yoshimura, M., Okumura, H., Cho, E., Kawai, Y., Kurokawa, K., Oshima, T., and Ogasawara, N. (2007). Distribution of stable DNAA-binding sites on the Bacillus subtilis genome detected using a modified ChIP-chip method. DNA Res. 14, 155–168.
Kari, L., Whitmire, W. M., Carlson, J. H., Crane, D. D., Reveneau, N., Nelson, D. E., Mabey, D. C., Bailey, R. L., Holland, M. J., McClarty, G., and Caldwell, H. D. (2008). Pathogenic diversity among Chlamydia trachomatis ocular strains in nonhuman primates is affected by subtle genomic variations. J. Infect. Dis. 197, 449–456.
Kennedy, A. D., Otto, M., Braughton, K. R., Whitney, A. R., Chen, L., Mathema, B., Mediavilla, J. R., Byrne, K. A., Parkins, L. D., Tenover, F. C., Kreiswirth, B. N., Musser, J. M., and DeLeo, F. R. (2008). Epidemic community-associated methicillin-resistant Staphylococcus aureus: recent clonal expansion and diversification. Proc. Natl. Acad. Sci. U.S.A. 105, 1327–1332.
Koide, T., Reiss, D. J., Bare, J. C., Pang, W. L., Facciotti, M. T., Schmid, A. K., Pan, M., Marzolf, B., Van, P. T., Lo, F. Y., Pratap, A., Deutsch, E. W., Peterson, A., Martin, D., and Baliga, N. S. (2009). Prevalence of transcription promoters within archaeal operons and coding sequences. Mol. Syst. Biol. 5, 285.
Kurokawa, K., Itoh, T., Kuwahara, T., Oshima, K., Toh, H., Toyoda, A., Takami, H., Morita, H., Sharma, V. K., Srivastava, T. P., Taylor, T. D., Noguchi, H., Mori, H., Ogura, Y., Ehrlich, D. S., Itoh, K., Takagi, T., Sakaki, Y., Hayashi, T., and Hattori, M. (2007). Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 14, 169–181.
Lakhal, F., Bury-Mone, S., Nomane, Y., Le Goic, N., Paillard, C., and Jacq, A. (2008). DjlA, a membrane-anchored DnaJ-like protein, is required for cytotoxicity of clam pathogen Vibrio tapetis to hemocytes. Appl. Environ. Microbiol. 74, 5750–5758.
Leininger, S., Urich, T., Schloter, M., Schwark, L., Qi, J., Nicol, G. W., Prosser, J. I., Schuster, S. C., and Schleper, C. (2006). Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442, 806–809.
Liu, J. M., Livny, J., Lawrence, M. S., Kimball, M. D., Waldor, M. K., and Camilli, A. (2009). Experimental discovery of sRNAs in Vibrio cholerae by direct cloning, 5S/tRNA depletion and parallel sequencing. Nucleic Acids Res. 37, e46.
Lun, D. S., Sherrid, A., Weiner, B., Sherman, D. R., and Galagan, J. E. (2009). A blind deconvolution approach to high-resolution mapping of transcription factor binding sites from ChIP-seq data. Genome Biol. 10, R142.
Mardis, E. R. (2008). Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 9, 387–402.
Marguerat, S., Wilhelm, B. T., and Bahler, J. (2008). Next-generation sequencing: applications beyond genomes. Biochem. Soc. Trans. 36, 1091–1096.
Martinez-Salazar, J. M., Sandoval-Calderon, M., Guo, X., Castillo-Ramirez, S., Reyes, A., Loza, M. G., Rivera, J., Alvarado-Affantranger, X., Sanchez, F., Gonzalez, V., Davila, G., and Ramirez-Romero, M. A. (2009). The Rhizobium etli RpoH1 and RpoH2 sigma factors are involved in different stress responses. Microbiology 155, 386–397.
Masse, E., Salvail, H., Desnoyers, G., and Arguin, M. (2007). Small RNAs controlling iron metabolism. Curr. Opin. Microbiol. 10, 140–145.
Mastroeni, P., and Menager, N. (2003). Development of acquired immunity to Salmonella. J. Med. Microbiol. 52, 453–459.
McGrath, P. T., Lee, H., Zhang, L., Iniesta, A. A., Hottes, A. K., Tan, M. H., Hillson, N. J., Hu, P., Shapiro, L., and McAdams, H. H. (2007). High-throughput identification of transcription start sites, conserved promoter motifs and predicted regulons. Nat. Biotechnol. 25, 584–592.
McKenna, P., Hoffmann, C., Minkah, N., Aye, P. P., Lackner, A., Liu, Z., Lozupone, C. A., Hamady, M., Knight, R., and Bushman, F. D. (2008). The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS Pathog. 4, e20. doi: 10.1371/journal.ppat.0040020.
Mockler, T. C., Chan, S., Sundaresan, A., Chen, H., Jacobsen, S. E., and Ecker, J. R. (2005). Applications of DNA tiling arrays for whole-genome analysis. Genomics 85, 1–15.
Moran, M. A. (2009). Metatranscriptomics: eavesdropping on complex microbial communities. Microbe 4, 329–335.
Munderloh, U. G., Tate, C. M., Lynch, M. J., Howerth, E. W., Kurtti, T. J., and Davidson, W. R. (2003). Isolation of an Anaplasma sp. organism from white-tailed deer by tick cell culture. J. Clin. Microbiol. 41, 4328–4335.
Nelson, C. M., Herron, M. J., Felsheim, R. F., Schloeder, B. R., Grindle, S. M., Chavez, A. O., Kurtti, T. J., and Munderloh, U. G. (2008). Whole genome transcription profiling of Anaplasma phagocytophilum in human and tick host cells by tiling array analysis. BMC Genomics 9, 364.
Nuwaysir, E. F., Huang, W., Albert, T. J., Singh, J., Nuwaysir, K., Pitas, A., Richmond, T., Gorski, T., Berg, J. P., Ballin, J., McCormick, M., Norton, J., Pollock, T., Sumwalt, T., Butcher, L., Porter, D., Molla, M., Hall, C., Blattner, F., Sussman, M. R., Wallace, R. L., Cerrina, F., and Green, R. D. (2002). Gene expression analysis using oligonucleotide arrays produced by maskless photolithography. Genome Res. 12, 1749–1755.
Oliver, H. F., Orsi, R. H., Ponnala, L., Keich, U., Wang, W., Sun, Q., Cartinhour, S. W., Filiatrault, M. J., Wiedmann, M., and Boor, K. J. (2009). Deep RNA sequencing of L. monocytogenes reveals overlapping and extensive stationary phase and sigma B-dependent transcriptomes, including multiple highly transcribed noncoding RNAs. BMC Genomics 10, 641.
Passalacqua, K. D., Varadarajan, A., Ondov, B. D., Okou, D. T., Zwick, M. E., and Bergman, N. H. (2009). Structure and complexity of a bacterial transcriptome. J. Bacteriol. 191, 3203–3211.
Perkins, T. T., Kingsley, R. A., Fookes, M. C., Gardner, P. P., James, K. D., Yu, L., Assefa, S. A., He, M., Croucher, N. J., Pickard, D. J., Maskell, D. J., Parkhill, J., Choudhary, J., Thomson, N. R., and Dougan, G. (2009). A strand-specific RNA-Seq analysis of the transcriptome of the typhoid bacillus Salmonella typhi. PLoS Genet. 5, e1000569. doi: 10.1371/journal.pgen.1000569.
Qin, J. H., Sheng, Y. Y., Zhang, Z. M., Shi, Y. Z., He, P., Hu, B. Y., Yang, Y., Liu, S. G., Zhao, G. P., and Guo, X. K. (2006). Genome-wide transcriptional analysis of temperature shift in L. interrogans serovar lai strain 56601. BMC Microbiol. 6, 51.
Qu, A., Brulc, J. M., Wilson, M. K., Law, B. F., Theoret, J. R., Joens, L. A., Konkel, M. E., Angly, F., Dinsdale, E. A., Edwards, R. A., Nelson, K. E., and White, B. A. (2008). Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS ONE 3, e2945. doi: 10.1371/journal.pone.0002945.
Rasmussen, S., Nielsen, H. B., and Jarmer, H. (2009). The transcriptionally active regions in the genome of Bacillus subtilis. Mol. Microbiol. 73, 1043–1057.
Roesch, L. F., Fulthorpe, R. R., Riva, A., Casella, G., Hadwin, A. K., Kent, A. D., Daroub, S. H., Camargo, F. A., Farmerie, W. G., and Triplett, E. W. (2007). Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 1, 283–290.
Romby, P., Vandenesch, F., and Wagner, E. G. (2006). The role of RNAs in the regulation of virulence-gene expression. Curr. Opin. Microbiol. 9, 229–236.
Scortti, M., Monzo, H. J., Lacharme-Lora, L., Lewis, D. A., and Vazquez-Boland, J. A. (2007). The PrfA virulence regulon. Microbes Infect. 9, 1196–1207.
Selinger, D. W., Cheung, K. J., Mei, R., Johansson, E. M., Richmond, C. S., Blattner, F. R., Lockhart, D. J., and Church, G. M. (2000). RNA expression analysis using a 30 base pair resolution Escherichia coli genome array. Nat. Biotechnol. 18, 1262–1268.
Sharma, C. M., Hoffmann, S., Darfeuille, F., Reignier, J., Findeiss, S., Sittka, A., Chabas, S., Reiche, K., Hackermuller, J., Reinhardt, R., Stadler, P. F., and Vogel, J. (2010). The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464, 250–255.
Sharma, C. M., and Vogel, J. (2009). Experimental approaches for the discovery and characterization of regulatory small RNA. Curr. Opin. Microbiol. 12, 536–546.
Sittka, A., Lucchini, S., Papenfort, K., Sharma, C. M., Rolle, K., Binnewies, T. T., Hinton, J. C., and Vogel, J. (2008). Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 4, e1000163. doi: 10.1371/journal.pgen.1000163.
Slamti, L., Livny, J., and Waldor, M. K. (2007). Global gene expression and phenotypic analysis of a Vibrio cholerae rpoH deletion mutant. J. Bacteriol. 189, 351–362.
Smith, M. G., Des Etages, S. G., and Snyder, M. (2004). Microbial synergy via an ethanol-triggered pathway. Mol. Cell. Biol. 24, 3874–3884.
Smith, M. G., Gianoulis, T. A., Pukatzki, S., Mekalanos, J. J., Ornston, L. N., Gerstein, M., and Snyder, M. (2007). New insights into Acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Dev. 21, 601–614.
Sorek, R., and Cossart, P. (2010). Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat. Rev. Genet. 11, 9–16.
Toledo-Arana, A., Dussurget, O., Nikitas, G., Sesto, N., Guet-Revillet, H., Balestrino, D., Loh, E., Gripenland, J., Tiensuu, T., Vaitkevicius, K., Barthelemy, M., Vergassola, M., Nahori, M. A., Soubigou, G., Regnault, B., Coppee, J. Y., Lecuit, M., Johansson, J., and Cossart, P. (2009). The Listeria transcriptional landscape from saprophytism to virulence. Nature 459, 950–956.
Toledo-Arana, A., Repoila, F., and Cossart, P. (2007). Small noncoding RNAs controlling pathogenesis. Curr. Opin. Microbiol. 10, 182–188.
Toledo-Arana, A., and Solano, C. (2010). Deciphering the physiological blueprint of a bacterial cell: revelations of unanticipated complexity in transcriptome and proteome. Bioessays 32, 461–467.
Turnbaugh, P. J., Ley, R. E., Mahowald, M. A., Magrini, V., Mardis, E. R., and Gordon, J. I. (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031.
Turner, A. K., Barber, L. Z., Wigley, P., Muhammad, S., Jones, M. A., Lovell, M. A., Hulme, S., and Barrow, P. A. (2003). Contribution of proton-translocating proteins to the virulence of Salmonella enterica serovars Typhimurium, Gallinarum, and Dublin in chickens and mice. Infect. Immun. 71, 3392–3401.
Urich, T., Lanzen, A., Qi, J., Huson, D. H., Schleper, C., and Schuster, S. C. (2008). Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS ONE 3, e2527. doi: 10.1371/journal.pone.0002527.
van Vliet, A. H. (2010). Next generation sequencing of microbial transcriptomes: challenges and opportunities. FEMS Microbiol. Lett. 302, 1–7.
Venter, J. C., Remington, K., Heidelberg, J. F., Halpern, A. L., Rusch, D., Eisen, J. A., Wu, D., Paulsen, I., Nelson, K. E., Nelson, W., Fouts, D. E., Levy, S., Knap, A. H., Lomas, M. W., Nealson, K., White, O., Peterson, J., Hoffman, J., Parsons, R., Baden-Tillson, H., Pfannkoch, C., Rogers, Y. H., and Smith, H.O. (2004). Environmental genome shotgun sequencing of the Sargasso Sea. Science 304, 66–74.
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63.
Wanner, B. L. (1987). “Phosphate regulation of gene expression in Escherichia coli,” in Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology, Vol. 2, eds F. C. Neidhardt, J. L. Ingraham, K. B. Low, B. Magasanik, M. Schaechter, and H. E. Umbarger (Washington, D.C.: American Society for Microbiology), 1326–1333.
Weinberg, Z., Barrick, J.E., Yao, Z., Roth, A., Kim, J. N., Gore, J., Wang, J. X., Lee, E. R., Block, K. F., Sudarsan, N., Neph, S., Tompa, M., Ruzzo, W. L., and Breaker, R. R. (2007). Identification of 22 candidate structured RNAs in bacteria using the CMfinder comparative genomics pipeline. Nucleic Acids Res. 35, 4809–4819.
Wilhelm, B. T., and Landry, J. R. (2009). RNA-Seq-quantitative measurement of expression through massively parallel RNA-sequencing. Methods 48, 249–257.
Wurtzel, O., Sapra, R., Chen, F., Zhu, Y., Simmons, B. A., and Sorek, R. (2009). A single-base resolution map of an archaeal transcriptome. Genome Res. 20, 133–141.
Yoder-Himes, D. R., Chain, P. S., Zhu, Y., Wurtzel, O., Rubin, E. M., Tiedje, J. M., and Sorek, R. (2009). Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. Proc. Natl. Acad. Sci. U.S.A. 106, 3976–3981.
Keywords: transcriptome, tiling array, massively parallel sequencing
Citation: Aikawa C, Maruyama F and Nakagawa I (2010) The dawning era of comprehensive transcriptome analysis in cellular microbiology. Front. Microbio. 1:118. doi: 10.3389/fmicb.2010.00118
Received: 20 August 2010;
Paper pending published: 11 September 2010;
Accepted: 06 October 2010;
Published online: 05 November 2010.
Edited by:
Adel M. Talaat, University of Wisconsin Madison, USACopyright: © 2010 Aikawa, Maruyama and Nakagawa. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Fumito Maruyama, Section of Bacterial Pathogenesis, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, 1-5-45, Yushima, Bunkyo-Ku, Tokyo 113-8510, Japan. e-mail: fumito-m.bac@tmd.ac.jp