 Andrew Chao1,2
Andrew Chao1,2

- 1 Laboratory of Neuroendocrinology, Department of Integrative Biology and Physiology, Brain Research Institute, University of California Los Angeles, Los Angeles, CA, USA
- 2 Neuroscience and Behavior Program, Center for Neuroendocrine Studies, University of Massachusetts, Amherst, MA, USA
Accuracy in quantifying brain-derived steroid hormones (“neurosteroids”) has become increasingly important for understanding the modulation of neuronal activity, development, and physiology. Relative to other neuroactive compounds and classical neurotransmitters, steroids pose particular challenges with regard to isolation and analysis, owing to their lipid solubility. Consequently, anatomical studies of the distribution of neurosteroids have relied primarily on the expression of neurosteroid synthesis enzymes. To evaluate the distribution of synthesis enzymes vis-à-vis the actual steroids themselves, traditional steroid quantification assays, including radioimmunoassays, have successfully employed liquid extraction methods (e.g., ether, dichloromethane, or methanol) to isolate steroids from microdissected brain tissue. Due to their sensitivity, safety, and reliability, the use of commercial enzyme-immunoassays (EIA) for laboratory quantification of steroids in plasma and brain has become increasingly widespread. However, EIAs rely on enzymatic reactions in vitro, making them sensitive to interfering substances in brain tissue and thus producing unreliable results. Here, we evaluate the effectiveness of a protocol for combined, two-stage liquid/solid-phase extraction (SPE) as compared to conventional liquid extraction alone for the isolation of estradiol (E2) from brain tissue. We employ the songbird model system, in which brain steroid production is pronounced and linked to neural mechanisms of learning and plasticity. This study outlines a combined liquid–SPE protocol that improves the performance of a commercial EIA for the quantification of brain E2 content. We demonstrate the effectiveness of our optimized method for evaluating the region specificity of brain E2 content, compare these results to established anatomy of the estrogen synthesis enzyme and estrogen receptor, and discuss the nature of potential EIA interfering substances.
Introduction
The unequivocal identification and quantification of steroid levels in the central nervous system (CNS) is critical for understanding how steroids control physiology, brain function, and behavior. By acting within the brain, steroids can regulate long-term (weeks–months) and short-term (seconds–hours) changes in structural morphology, synaptic physiology, and behavior. Although steroid production has been well characterized for peripheral endocrine glands such as the gonads and adrenals, the role of steroid hormones synthesized de novo in the CNS, termed “neurosteroids” is less well understood (Baulieu, 1991, 1998; London et al., 2006). As we continue to gain insight on the profound effects of steroids in the brain on neuronal activity and physiology (Compagnone and Mellon, 2000; Woolley, 2007; Garcia-Segura, 2008; Remage-Healey et al., 2010a), it has become increasingly important to refine and reevaluate current methodology for the analysis of neurosteroids.
Prior to analytical measurement, steroids must be isolated by means of extraction to maximize the accuracy and precision of the assay. To determine the most effective means of extraction, it is important to consider both the nature of the tissue and the type of assay. Steroid assays typically involve a competition binding of ligand for a specific antibody, such as in the case of radioimmunoassay (RIA) or enzyme-immunoassay (EIA), which can be sensitive to pH, temperature as well as the presence of interfering proteins.
Steroid hormones are lipophylic molecules, necessitating organic extraction from largely aqueous biological tissues and fluids. The extraction of steroids from vertebrate plasma is the most widely used, typically involving a single liquid–liquid extraction, which partitions steroids into an organic solvent, traditionally diethyl ether, or dichloromethane (in both birds and mammals: Stone et al., 1971; Wingfield and Farner, 1975; Ball and Wingfield, 1987; Schlinger et al., 1992; Fenske, 1995).
To isolate steroids from more viscous and lipid-rich samples such as brain tissue, simple liquid–liquid extractions often afford only partial separation of hydrophobic (e.g., lipids) and aqueous (e.g., proteins) compounds. More elaborate forms of extraction are necessary. For example, the purification of sex hormones from yolk has been performed via a series of liquid–liquid extractions, followed by solid-phase extraction (SPE, using diatomaceous earth microcolumns; Schwabl, 1993). Likewise, the extraction of gonadal steroids has in some cases required the combination of high-performance liquid chromatography (HPLC) and SPE (Cochran and Ewing, 1979).
Brain tissue presents a serious challenge to conventional techniques because of its exceptionally heterogeneous aqueous–lipid composition. Residual lipids have the ability to interfere with subsequent analysis (Rash et al., 1979; Lepage et al., 1993) and hence necessitate removal using extraction. Moreover, neurosteroids often exist in very low concentrations, which necessitate a clean extraction with minimal loss of target antigen.
Traditional measures of neurosteroid levels have utilized RIA (Abraham, 1974; Corpechot et al., 1981; Wang et al., 1997). Although highly sensitive, immunoassays rely on antibodies with variable cross-reactivity to steroids and non-steroidal molecules. In assaying brain samples, the potential for non-specific binding and protein and/or proteoglycan interference is quite high vs. more homogeneous aqueous samples like plasma. Most importantly, these effects can lead to inaccurate and unreliable assay reporting of steroid levels in the brain (see Schumacher et al., 2008).
The advent of SPE offers a low-cost and safe solution to purify samples prior to immunoassay. Indeed, reverse-phase extractions using coated silica cartridges (C18) have been successfully implemented in the extraction of steroids from plasma (Stone et al., 1971; Heikkinen et al., 1981; Fenske, 1995), urine (Shackleton and Whitney, 1980; Heikkinen et al., 1981; Lee and Goeger, 1998), human prostate (Higashi et al., 2005), rat fetal tissues (Samtani and Jusko, 2007), and brain (Mathur et al., 1993; Wang et al., 1997; Serra et al., 2000).
Songbirds have become an extremely useful model for the study of brain-derived steroid production and action (Schlinger and Brenowitz, 2002; London et al., 2006; Remage-Healey et al., 2010b). The anatomical distribution of the estrogen–production enzyme aromatase in particular has been extensively studied in the zebra finch brain (Shen et al., 1995; Metzdorf et al., 1999; Saldanha et al., 2000; Pinaud et al., 2006). The degree of correspondence of the expression of aromatase (as determined using immunohistochemical methods) with region-specific brain estrogen content (as determined via neural tissue extraction) is crucial to understanding the regional bioavailability of estrogens in the CNS. The use of SPE on songbird brain tissue, for quantification via RIA, has been validated recently (Newman et al., 2008). However whether SPE is in fact a reliable means to quantify neurosteroids via EIA is unclear for any model system. This is particularly important since often EIA can yield increased sensitivity as compared to RIA for assaying steroidal molecules in the sub nanomolar range, such as estrogens. Secondly, EIAs are becoming commonly used in laboratory research, due to their safety, reliability, and cost. Importantly, the photochemical reactions that drive EIAs rely on specific temperature and pH ranges, and can be subject to interference from residual lipids and proteins in the biological tissue sample. Therefore, a straightforward detailed extraction protocol is needed for brain steroid content using EIA.
Here we assess the effectiveness of combined liquid and SPE for the isolation of 17-β-estradiol (E2) from songbird brain tissue. We address this issue in two ways: by measuring the effect of SPE on (1) the standard displacement curve, as measured by a commercially available EIA, and (2) the recovery rate, as measured independently using radioinert and radioisotopic methods. We then employ these newly validated methods to describe the regional specificity of estrogen content in brain, as well as the estrogen content in brain relative to circulating plasma levels.
Materials and Methods
Subjects
All procedures were approved by the UCLA institutional animal care and use committee. All brain tissue and plasma were collected from captive adult zebra finches. Within <2 min of disturbance (to lessen effects of stress on steroid levels) birds were rapidly decapitated and the whole brain was removed, dissected into brain macroareas (see below), and stored at −80°C until homogenization. Blood was collected from the trunk and centrifuged at 10,000 rpm for 2 min. The resulting plasma was stored at −80°C until extraction.
Estradiol Injections
We initially tested the ability of the commercial assay to detect E2 in brain homogenates using conventional liquid (organic) extraction methods with diethyl ether. Adult female zebra finches were isolated for 24 h prior to estradiol or saline injection into the pectoralis muscle. Estradiol treated birds (n = 3) were given 20 μl of 300 μg/ml E2 solution in saline (Sigma). Saline treated birds (n = 3) were given 20 μl of saline (0.9% w/vol). Injections were administered 1 h prior to tissue collection, whereupon the brain was collected and immediately dissected onto iced aluminum foil into functional regions using established landmarks (Remage-Healey et al., 2009): anterior telencephalon (AT), posterior telencephalon (PT), hippocampus (HP), mediobasal telencephalon, which contains aromatase-rich nucleus taeniae (MBT), anterior hypothalamus (AH), posterior hypothalamus (PH), optic tectum (OT), and cerebellum (CB). Brain tissues were weighed immediately after dissection and frozen at −80°C until further processing.
Prior to assaying, samples were thawed, homogenized in SPO4 buffer on ice and ether extracted (see below). Samples were then reconstituted in 120 μl of EIA buffer and assayed on an E2 EIA kit (Cayman Chemical) for measurements of E2 levels.
Ether Extraction Protocol
Whole homogenates were prepared in 2 ml of SPO4 buffer on ice via a homogenizer (Tissue Tearor, Biospec) until complete dispersion of tissue into the buffer. Each sample was then ether extracted by first adding 3 ml of diethyl ether, vortexing for 30 s, and centrifuging at 2100 rpm for 5 min at 40°C. The resulting mixture was then incubated in a MeOH/dry ice bath to solidify the aqueous phase (bottom) and the organic phase was eluted into a new tube. Ether extraction was repeated twice more on the thawed aqueous phase of each sample. Following elution, the organic phase was dried under air in a water bath at 50°C prior to being resuspended in 120 μl EIA buffer for either EIA directly or SPE.
Solid-Phase Extraction Protocol
We utilized a 12-port Visiprep™ SPE vacuum manifold (Supelco) with endcapped C18-SD 3 ml cartridges (Empore). Initial experiments showed that extracting homogenized brain tissue with SPE alone (i.e., without prior organic extraction) severely curtailed recovery of E2 to as much as 10% (preliminary data, not shown), due to homogenized tissue obstructing the flow of eluent through the vacuum manifold. Therefore, we performed a two-stage extraction (ether extraction as above, followed by SPE) on brain tissue and plasma samples to determine the effectiveness of this dual extraction method at reducing EIA assay interference.
Prior to eluting samples, C18 columns were conditioned with 250 μl of 100% MeOH and equilibrated twice with 250 μl ddH2O under vacuum pressure. Dried ether extracted samples were resuspended in 120 μl of EIA buffer, loaded, and eluted through the columns under vacuum pressure. After this initial elution, columns were eluted with 250 μl of ddH2O twice to wash out hydrophilic polar compounds. A third and fourth wash with 250 μl of 90% MeOH then allowed for the elution of relatively hydrophobic compounds, including steroids (e.g., Newman et al., 2008). Columns were dried under vacuum after each loading. For each sample, eluent derived from the 90% MeOH elution was evaporated to dryness under a steady stream of air in a water bath at 50°C and stored at −20°C. Prior experiments using gas chromatography (GC)/mass spectrometry (MS) confirmed that the 90% MeOH fraction fully eluted estradiol (Remage-Healey and Schlinger, unpublished observations). Samples were resuspended with 120 μl of EIA buffer immediately prior to assaying.
Enzyme-Immunoassay
Previously, we optimized the usage of the E2 EIA (Cayman Chemicals) for the quantification of E2 levels in microdialysate samples, validated with independent confirmation of E2 using GC/MS; Remage-Healey et al., 2008). Here we assess the ability of EIA to accurately measure a gradient of exogenous E2 concentrations in whole brain homogenate following two extraction protocols: extraction with ether only and extraction with ether followed by SPE.
Homogenized adult zebra finch brain tissue in SPO4 buffer (pH – 7.4–6, 159.7 mg/ml) was separated into eight 500 μl aliquots. Each aliquot was spiked with radioinert E2 to obtain one of eight concentrations to approximate the physiological range of E2 levels in zebra finch tissue (4000, 1600, 640, 256, 102, 41, 16, 7 pg/ml) and in the dynamic range of the commercial EIA. Samples were ether extracted and then reconstituted in 250 μl of EIA buffer. Each sample was then split into two equal 120 μl aliquots to form two sets of eight 120 μl aliquots, and SPE was conducted on one set (as described above).
Ether and ether +SPE extracted samples were assayed on E2 EIA along with unextracted standards of equivalent E2 concentration (4000, 1600, 640, 256, 102, 41, 16, 7 pg/ml) to determine the effectiveness of the two extraction protocols in reducing assay interference for the displacement curve. We estimated the recovery of E2 after ether extraction alone vs. ether extraction plus SPE. Recovery was estimated in two ways: (1) the extraction recovery of radiolabeled E2 as determined via radioisotopic decay using a liquid scintillation counter and (2) the extraction recovery of radioinert E2 as determined via EIA. We added 10,000 cpm of tritium-labeled E2 (48.9 nM, [3H]-Estradiol; New England Nuclear) into a set of 2 ml SPO4 aliquots in duplicate. Both sets were ether extracted and one set was resuspended with 120 μl of EIA buffer prior to SPE. Extracted aliquots were then reconstituted with 100 μl of methanol and vortexed for scintillation counting alongside unextracted but equivalently spiked samples. Radioinert E2 was added to a separate set of 2 ml SPO4 aliquots in quadruplicate. Both sets were ether extracted and one set was resuspended with 120 μl of EIA buffer prior to SPE. Extracted aliquots were then reconstituted with 2 ml of SPO4 buffer and assayed via EIA alongside unextracted but similarly spiked samples. A similar extraction recovery was estimated for homogenized brain tissue in a separate experiment.
Combined Liquid and Solid-Phase Extraction
Using the optimized combined liquid and SPE procedure we measured differences in E2 levels of dissected macroareas of the adult male zebra finch brain. Adult males (n = 12) were placed in sound isolation chambers 24 h prior to exposure to a stimulus consisting of two females placed in an adjacent cage inside the chamber. Immediately after 30 min of exposure to females, males were decapitated and whole blood was collected, centrifuged, and plasma was stored at −80°C. Brains were dissected on ice into functional groups, according to established procedures for isolating macroareas based on primary landmarks (see above; Remage-Healey et al., 2009). Immediately after dissection, tissue, and plasma wet weight was recorded and frozen at −80°C. Prior to assay, tissues were thawed, homogenized in SPO4 buffer on ice, ether extracted, and solid phase extracted (as above). Finally, samples were reconstituted in 120 μl of EIA buffer and assayed on an E2 EIA kit.
Results
Estradiol Injections
Compared to saline treated birds, we observed a clear elevation in overall E2 levels across ether extracted brains of E2 treated birds (df = 1,28 F = 30.686, p < 0.0001; Figure 1). Mann–Whitney post hoc tests revealed that E2 levels were significantly elevated following E2-injection in HP, AH, OT, nucleus taeniae, and PT (Z > 1.96; p < 0.05 for all comparisons) but not in AT or CB (p > 0.13). As expected, substantial variation in E2 levels occurs across brain regions regardless of treatment (df = 6,28 F = 8.899, p < 0.0001). Therefore, the commercial EIA demonstrated that endogenous and exogenous E2 was detectable in songbird brain tissue following ether extraction alone.
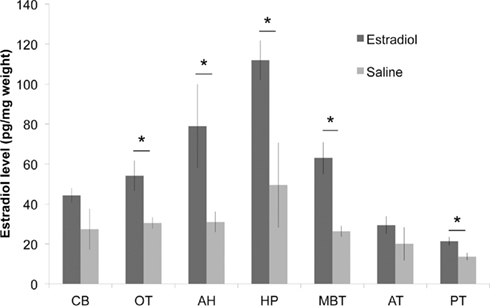
Figure 1. Injection of 17β-estradiol (E2) into the pectoralis muscle produces elevated brain E2 content, relative to saline-injected controls (n = 3 each; *p < 0.05 for between group comparisons for each brain region). CB, cerebellum; OT, optic tectum; AH, anterior hypothalamus; HP, hippocampus; MBT, mediobasal telencephalon (containing avian amygdala); AT, anterior telencephalon; PT, posterior telencephalon.
Combined Ether and Solid-Phase Extraction of Brain Tissue
Recovery of estrogens following our extraction procedures was estimated using both radiolabeled and radioinert estrogens in separate extraction experiments. In SPO4 solution, we observed a 93.4 and 86.2% recovery of radiolabeled and radioinert E2, respectively, due to ether extraction alone. With ether extraction followed by SPE, we observed a total net recovery of 76 and 89.5% of radiolabeled and radioinert E2, respectively (Figure 3). In a separate experiment, the total recovery of E2 following ether +SPE extractions (as measured by EIA) from brain homogenate was 60.09 ± 4.02% (in quadruplicate). The results presented below for extracted samples does not account for recovery.
We compared the slopes of displacement curves that were extracted with ether alone vs. ether +SPE (Figure 2). The slope of the ether extracted dilution curve was indistinguishable from zero (F = −3.946, p = 0.121), and therefore is a poor predictor of brain E2 content, particularly at lower levels. A multiple linear regression model indicated that the ether extracted curve was indeed not parallel to the standard curve (t = 9.708, p = 1.41e-08; Figure 2: box data labels). Our results strongly suggest that ether alone yields a particular poor extraction.
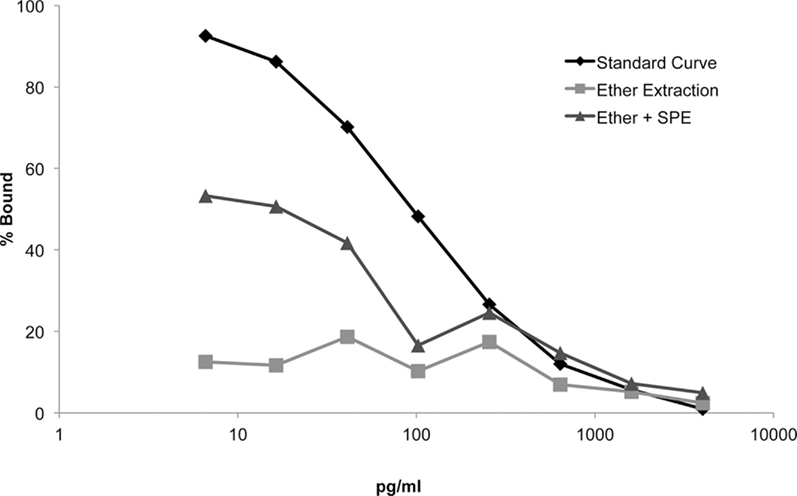
Figure 2. Displacement curves for each extraction method reveal that combined liquid (ether) and solid-phase extraction (SPE) method improves E2 assay sensitivity at lower concentrations (i.e., <640 pg/ml).
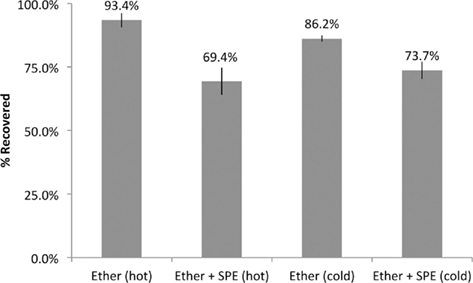
Figure 3. Percent recovery of E2 using two primary extraction methods. Recovery was estimated using both radioisotopic (hot) and radioinert (cold) E2 in extracted samples vs. equivalent unextracted samples.
Enhancing extraction by combining ether followed by SPE, demonstrated a significant improvement in the slope of the displacement curve compared to the slope of the ether only extraction (t = 4.356, p = 0.0038). Taken together, the displacement curve extracted with ether +SPE demonstrated a significant improvement in dynamic sensitivity as compared to the displacement curve extracted with ether alone. Despite this improvement, the use of ether +C18 columns for the extraction of serially diluted E2 spiked brain homogenate yielded a curve that was not strictly parallel with the standard curve (t = 5.352, p = 4.36e-05; Figure 2: triangle data labels). Importantly, however, the commercial EIA was better able to discriminate between differences in E2 levels at lower concentrations (<650 pg/ml) with the addition of the SPE. This is particularly important since E2 concentrations in zebra finch plasma and brain predominantly occur within this critical range.
E2 in Male Zebra Finch Brain
Using our optimized ether +SPE protocol, EIA was used to measure brain E2 levels in adult male zebra finches following 30 min of exposure to adult females (n = 12; Figure 4). There were significant regional differences in E2 levels (df = 8,90 F = 3.025, p = 0.0047). MBT levels were highest, and were significantly greater than those found in telencephalon (Fisher’s PLSD: vs. AT: p = 0.0211, vs. PT: p = 0.028) and hypothalamus (vs. AH: p = 0.0081, vs. PH: p = 0.0057). MBT (containing aromatase-rich nucleus taeniae) and the HP both had E2 levels significantly higher than plasma (MBT: p = 0.0011, HP: p = 0.0499), OT (MBT: p = 0.0003, HP: p = 0.0178), and CB (MBT: p = 0.0002, HP: p = 0.0152).
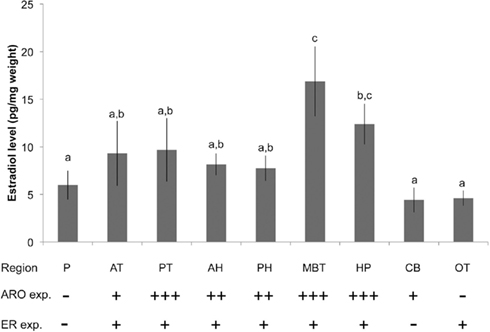
Figure 4. Brain and plasma (P) E2 content in adult male zebra finches (n = 12) using the optimized combined liquid–solid-phase extraction procedure. Elevated brain E2 content demonstrated here corresponds to functional and region-specific differences in aromatase (ARO) and estrogen receptor (ER) expression in the zebra finch brain. Plus signs indicate elevated expression of ARO and ER for each brain region; minus signs indicate little to no expression (after Shen et al., 1995; Metzdorf et al., 1999; Saldanha et al., 2000). Abbreviations as in Figure 1. P, plasma. Letters indicate significant differences among brain regions (p < 0.05).
Discussion
Estrogens are implicated in a vast number of functions in the vertebrate brain, including actions derived from local and acute synthesis within the brain itself (Balthazart et al., 1990; Baulieu, 1998; Forlano et al., 2006). We describe here the challenges of measuring E2 in brain homogenate using liquid extraction alone, and outline a reliable, low-cost means of serial liquid–SPE steps that together improve quantification of brain E2 content with EIA.
Ether Extraction Alone
Our results show that the commercial E2 EIA assay used here is capable of detecting exogenous increases in brain E2 concentration. Further experiments showed that EIA is capable of detecting changes in ether extracted brain E2 content induced by the potent aromatase inhibitor Fadrozole (35–37% decrease in brain E2 content in telencephalon and HP; data not shown) and that ether extracted brain estrogens are detectable and unequivocally confirmed using GC/MS (see Results). These initial experiments confirmed the capacity of the commercial EIA to detect brain E2 levels, and changes in brain E2 content induced via endogenous and exogenous treatments. Importantly, however, the experiment with exogenous E2-injection (Figure 1) alerted us to the potential for inaccuracies in steroid measurements at biological relevant levels using liquid extraction alone.
Validating SPE
We tested the possibility that a two-stage extraction procedure was required to eliminate interfering substances from the EIA reaction plate. SPE, particularly reverse-phase extraction using C18 coated silica, has gained popularity for a variety of applications as a method of purifying samples for steroid analysis (see Introduction). In particular, SPE has been useful in optimizing the detection and quantification of brain E2 content as measured by RIA (Newman et al., 2008). Here we provide an independent confirmation of the validity of this approach for brain E2 content, and we demonstrate SPE’s potential to improve the performance of the EIA for the quantification of brain E2, particularly at the low, subnanomolar concentrations.
Extraction prior to assaying plays an integral part in minimizing possible interference. Indeed, we observe that a limited liquid–liquid extraction of brain homogenate hinders EIA’s ability to detect variation of sample E2 levels at the lower range of steroid concentrations, effectively “flattening” the displacement curve and rendering any measurement below approximately 600 pg/ml unreliable (Figure 2). Subsequent elution of samples through SPE greatly increased the assay’s sensitivity at the lower end of the curve in comparison to a liquid–liquid only extraction.
The most likely explanation for the improvements seen here with SPE is the elimination of substances that interfere with the EIA’s enzyme reaction(s). Assay interference can originate from a number of sources and can occur at different stages of the immunoassay. EIAs rely on the highly specific association between the primary antibody and the target antigen. In the case of competitive EIAs, the resulting antibody–antigen complex must also bind with high affinity to a plate-bound antibody IgG. The degree to which the extraction step minimizes cross-reactivity and non-specific binding during plate development likely directly impacts the accuracy and precision of the resulting colorimetric reaction step.
A number of substances may disrupt proper ligand–antibody competition, which can lead to the distortion of the displacement curve. While highly sensitive, the E2-antibody used here has cross-reactivity for estradiol-3-glucuronide (14%), estrone (12%), and estradiol-17-glucuronide (10%). These and estrogenic derivatives and conjugates, such as E2 fatty acid esters, have similar chemical properties to estradiol, and may remain in significant quantities after a simple ether extraction. Although little is known of the presence of these E2 metabolites in birds, there is evidence that E2 fatty acid esters are synthesized in the rat brain (Xu et al., 2002) and exert estrogenic effects on a variety of rat brain tissue (MacLusky et al., 1989). The presence of these conjugates may inflate the measurement of unconjugated E2 levels by directly competing with free E2 for antibody binding sites. In addition, E2 fatty acid esters are known to associate with lipoproteins during circulation (Larner et al., 1987), granting them both lipophilic and hydrophilic properties that further complicate liquid extraction. These and other substances may persist in the saponified layer between the aqueous and organic phase during liquid (organic) extraction alone, and subsequently provide a source of interference during EIA. Residual non-estrogenic lipids, likely composed of free fatty acids and glycolipids, can prevent binding indirectly, by sequestering steroids or their antibodies within micelles (Rash et al., 1979). The forebrain of the zebra finch also contains robust expression of estrogen receptors (ERs; Gahr and Metzdorf, 1999) and unextracted ER and brain-derived E2 binding proteins may all compete with the assay’s E2-antibody for both free and conjugated E2. The current findings are consistent with the hypothesis that the two-stage liquid/SPE protocol improves the separation of E2 from such sources of assay interference.
Despite it’s safety and cost advantages, EIA may in fact be more sensitive to interfering substances than RIA. The quantification of bound antigen using EIA is achieved by first washing away free reagents, and then measuring the rate of a photochemical reaction (e.g., hydrolysis of acetylcholine by acetylcholinesterase) with a spectrophotometer. This process is susceptible to the presence of non-specific proteases that may significantly reduce the reactive properties of the EIA.
Using this optimized two-stage liquid/SPE protocol, the current results demonstrate that regional differences exist in brain E2 content. The addition of SPE yielded results that are consistent with known patterns of aromatase activity and ER expression in the songbird brain (Saldanha et al., 2000). Areas with established aromatase expression showed higher concentration of E2 in males, including mediobasal telencephalon containing nucleus taeniae, hypothalamus, HP, and telencephalon, in contrast to the CB and OT, which contain little to no known aromatase in the uninjured brain (Saldanha et al., 2000). The region-dependent ER expression throughout the zebra finch brain (Metzdorf et al., 1999) is also very likely to contribute to region differences in brain estrogen content as measured using these and similar methods, due to local accumulation and sequestration of neurally and peripherally derived estrogens. These findings for the region specificity of brain estrogen content, and the correspondence to brain aromatase and ER expression are presented in Figure 4. Lastly, the relative paucity of E2 in plasma compared to brain regions such as the MBT and HP is consistent with the hypothesis that the brain is the primary source of estrogens in the male songbird (Schlinger and Arnold, 1991; Taves et al., 2010).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Stephanie Dong and Anahid Mirzatoni for technical assistance and advice, and Jesse McClure for help on the statistical methods. Support for the study was provided by NINDS F32NS058009, K99/R00 NS066179, and NIMH 061994.
References
Abraham, G. E. (1974). Radioimmunoassay of steroids in biological-materials. Acta Endocrinol. 183, 7–42.
Ball, G. F., and Wingfield, J. C. (1987). Changes in plasma-levels of luteinizing-hormone and sex steroid-hormones in relation to multiple-broodedness and nest-site density in male starlings. Physiol. Zool. 60, 191–199.
Balthazart, J., Foidart, A., Surlemont, C., Vockel, A., and Harada, N. (1990). Distribution of aromatase in the brain of the Japanese-quail, ring dove, and zebra finch – an immunocytochemical study. J. Comp. Neurol. 301, 276–288.
Baulieu, E. E. (1998). Neurosteroids: a novel function of the brain. Psychoneuroendocrinology 23, 963–987.
Cochran, R. C., and Ewing, L. L. (1979). Celite column chromatography followed by reversed-phase high-performance liquid-chromatography – simple, 2-step method for separating 14 testicular-steroids. J. Chromatogr. 173, 175–181.
Compagnone, N. A., and Mellon, S. H. (2000). Neurosteroids: biosynthesis and function of these novel neuromodulators. Front. Neuroendocrinol. 21, 1–56.
Corpechot, C., Robel, P., Axelson, M., Sjovall, J., and Baulieu, E. E. (1981). Characterization and measurement of dehydroepiandrosterone sulfate in rat-brain. Proc. Natl. Acad. Sci. U.S.A. 78, 4704–4707.
Fenske, M. (1995). Rapid and efficient method for extraction and separation of glucocorticosteroids and sex steroids from urines. J. Chromatogr. B Biomed. Appl. 670, 373–375.
Forlano, P. M., Schlinger, B. A., and Bass, A. H. (2006). Brain aromatase: new lessons from non-mammalian model systems. Front. Neuroendocrinol. 27:247–274. doi: 10.1016/j.yfrne.2006.05.002
Gahr, M., and Metzdorf, R. (1999). The sexually dimorphic expression of androgen receptors in the song nucleus hyperstriatalis ventrale pars caudale of the zebra finch develops independently of gonadal steroids. J. Neurosci. 19, 2628–2636.
Garcia-Segura, L. M. (2008). Aromatase in the brain: not just for reproduction anymore. J. Neuroendocrinol. 20, 705–712.
Heikkinen, R., Fotsis, T., and Adlercreutz, H. (1981). Reversed-phase C18-cartridge for extraction of estrogens from urine and plasma. Clin. Chem. 27, 1186–1189.
Higashi, T., Yamauchi, A., Shimada, K., Koh, E., Mizokami, A., and Namiki, M. (2005). Determination of prostatic androgens in 10 mg of tissue using liquid chromatography-tandem mass spectrometry with charged derivatization. Anal. Bioanal. Chem. 382, 1035–1043.
Larner, J. M., Rosner, W., and Hochberg, R. B. (1987). Binding of estradiol-17-fatty acid esters to plasma proteins. Endocrinology 121, 738–744.
Lee, C., and Goeger, D. E. (1998). Interference of 6 beta-hydroxycortisol in the quantitation of urinary free cortisol by immunoassay and its elimination by solid phase extraction. Clin. Biochem. 31, 229–233.
Lepage, N., Roberts, K. D., and Langlais, J. (1993). Interference of lysophosphatidylcholine in hormone radioimmunoassays. Clin. Chem. 39, 865–869.
London, S. E., Monks, D. A., Wade, J., and Schlinger, B. A. (2006). Widespread capacity for steroid synthesis in the avian brain and song system. Endocrinology 147, 5975–5987.
MacLusky, N. J., Larner, J. M., and Hochberg, R. B. (1989). Actions of an estradiol-17-fatty acid ester in estrogen target tissues of the rat: comparison with other C-17 metabolites and a pharmacological C-17 ester. Endocrinology 124, 318–324.
Mathur, C., Prasad, V. V., Raju, V. S., Welch, M., and Lieberman, S. (1993). Steroids and their conjugates in the mammalian brain. Proc. Natl. Acad. Sci. U.S.A. 90, 85–88.
Metzdorf, R., Gahr, M., and Fusani, L. (1999). Distribution of aromatase, estrogen receptor, and androgen receptor mRNA in the forebrain of songbirds and nonsongbirds. J. Comp. Neurol. 4071, 115–129.
Newman, A. E., Chin, E. H., Schmidt, K. L., Bond, L., Wynne-Edwards, K. E., and Soma, K. K. (2008). Analysis of steroids in songbird plasma and brain by coupling solid phase extraction to radioimmunoassay. Gen. Comp. Endocrinol. 155, 503–510.
Pinaud, R., Fortes, A. F., Lovell, P., and Mello, C. V. (2006). Calbindin-positive neurons reveal a sexual dimorphism within the songbird analogue of the mammalian auditory cortex. J. Neurobiol. 66, 182–195.
Rash, J. M., Jerkunica, I., and Sgoutas, D. (1979). Mechanisms of interference of nonesterified fatty-acids in radioimmunoassays of steroids. Clin. Chim. Acta 93, 283–294.
Remage-Healey, L., Coleman, M. J., Oyama, R. K., and Schlinger, B. A. (2010a). Brain estrogens rapidly strengthen auditory encoding and guide song preference in a songbird. Proc. Natl. Acad. Sci. U.S.A. 107, 3852–3857.
Remage-Healey, L., London, S. E., and Schlinger, B. A. (2010b). Birdsong and the neural production of steroids. J. Chem. Neuroanat. 39, 72–81.
Remage-Healey, L., Maidment, N. T., and Schlinger, B. A. (2008). Forebrain steroid levels fluctuate rapidly during social interactions. Nat. Neurosci. 11, 1327–1334.
Remage-Healey, L., Oyama, R. K., and Schlinger, B. A. (2009). Elevated aromatase activity in forebrain synaptic terminals during song. J. Neuroendocrinol. 21, 191–199.
Saldanha, C. J., Tuerk, M. J., Kim, Y. H., Fernandes, A. O., Arnold, A. P., and Schlinger, B. A. (2000). Distribution and regulation of telencephalic aromatase expression in the zebra finch revealed with a specific antibody. J. Comp. Neurol. 423, 619–630.
Samtani, M. N., and Jusko, W. J. (2007). Quantification of dexamethasone and corticosterone in rat biofluids and fetal tissue using highly sensitive analytical methods: assay validation and application to a pharmacokinetic study. Biomed. Chromatogr. 21, 585–597.
Schlinger, B. A., and Arnold, A. P. (1991). Brain is the major site of estrogen synthesis in a male songbird. Proc. Natl. Acad. Sci. U.S.A. 88, 4191–4194.
Schlinger, B. A., and Brenowitz, E. A. (2002). “Neural and hormonal control of birdsong,” in Hormones, Brain and Behavior, ed. D. W. Pfaff (Amsterdam: Elsevier), 799–838.
Schlinger, B. A., Slotow, R. H., and Arnold, A. P. (1992). Plasma estrogens and brain aromatase in winter white-crowned sparrows. Ornis Scand. 23, 292–297.
Schumacher, M., Liere, P., Akwa, Y., Rajkowski, K., Griffiths, W., Bodin, K., Sjovall, J., and Baulieu, E. E. (2008). Pregnenolone sulfate in the brain: a controversial neurosteroid. Neurochem. Int. 52, 522–540.
Schwabl, H. (1993). Yolk is a source of maternal testosterone for developing birds. Proc. Natl. Acad. Sci. U.S.A. 90, 11446–11450.
Serra, M., Pisu, M. G., Littera, M., Papi, G., Sanna, E., Tuveri, F., Usala, L., Purdy, R. H., and Biggio, G. (2000). Social isolation-induced decreases in both the abundance of neuroactive steroids and GABA(A) receptor function in rat brain. J. Neurochem. 75, 732–740.
Shackleton, C. H., and Whitney, J. O. (1980). Use of Sep-pak cartridges for urinary steroid extraction – evaluation of the method for use prior to gas-chromatographic analysis. Clin. Chim. Acta 107, 231–243.
Shen, P., Schlinger, B. A., Campagnoni, A. T., and Arnold, A. P. (1995). An atlas of aromatase messenger-Rna expression in the zebra finch brain. J. Comp. Neurol. 360, 172–184.
Stone, S., Kharma, K. M., Nakamura, R. M., Mishell, D. R., and Thorneycroft, I. H. (1971). Technique for Assay of 17-hydroxyprogesterone (17- hydroxy-4-pregnene-3,20-dione) in serum using celite column chromatography and competitive protein binding. Steroids 18, 161–173.
Taves, M. D., Schmidt, K. L., Ruhr, I. M., Kapusta, K., Prior, N. H., and Soma, K. K. (2010). Steroid concentrations in plasma, whole blood and brain: effects of saline perfusion to remove blood contamination from brain. PLoS ONE 5, e15727. doi: 10.1371/journal.pone.0015727
Wang, M. D., Wahlstrom, G., and Backstrom, T. (1997). The regional brain distribution of the neurosteroids pregnenolone and pregnenolone sulfate following intravenous infusion. J. Steroid Biochem. 62, 299–306.
Wingfield, J. C., and Farner, D. S. (1975). Determination of 5 steroids in avian plasma by radioimmunoassay and competitive-protein-binding. Steroids 26, 311–327.
Woolley, C. S. (2007). Acute effects of estrogen on neuronal physiology. Annu. Rev. Pharmacol. Toxicol. 47, 657–680.
Keywords: estrogen, neurosteroid, ELISA, protocol
Citation: Chao A, Schlinger BA and Remage-Healey L (2011) Combined liquid and solid-phase extraction improves quantification of brain estrogen content. Front. Neuroanat. 5:57. doi: 10.3389/fnana.2011.00057
Received: 05 June 2011; Accepted: 16 August 2011;
Published online: 01 September 2011.
Edited by:
Emmanuel Valjent, Institut de Génomique Fonctionnelle, INSERM U661, UMR 5203 CNRS, Université Montpellier 1&2, FranceReviewed by:
Noritaka Ichinohe, Hirosaki University Graduate School of Medicine, JapanFiorenzo Conti, Universita Politecnica delle Marche, Italy
Copyright: © 2011 Chao, Schlinger and Remage-Healey. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Luke Remage-Healey, Neuroscience and Behavior Program, Center for Neuroendocrine Studies, University of Massachusetts, Amherst, MA 01003, USA. e-mail:aGVhbGV5QGNucy51bWFzcy5lZHU=