 Anne Tøndervik1
Anne Tøndervik1 Randi Aune1Adelheid Degelmann2Michael Piontek2
Randi Aune1Adelheid Degelmann2Michael Piontek2 Helga Ertesvåg3Gudmund Skjåk-Bræk3Håvard Sletta1*
Helga Ertesvåg3Gudmund Skjåk-Bræk3Håvard Sletta1*- 1Department of Biotechnology and Nanomedicine, SINTEF Industry, Trondheim, Norway
- 2ARTES Biotechnology GmbH, Langenfeld, Germany
- 3Department of Biotechnology and Food Sciences, Trondheim, Norway
Alginates are linear polysaccharides produced by brown algae and some bacteria and are composed of β-D-mannuronic acid (M) and α-L-guluronic acid (G). Alginate has numerous present and potential future applications within industrial, medical and pharmaceutical areas and G rich alginates are traditionally most valuable and frequently used due to their gelling and viscosifying properties. Mannuronan C-5 epimerases are enzymes converting M to G at the polymer level during the biosynthesis of alginate. The Azotobacter vinelandii epimerases AlgE1-AlgE7 share a common structure, containing one or two catalytic A-modules (A), and one to seven regulatory R-modules (R). Despite the structural similarity of the epimerases, they create different M-G patterns in the alginate; AlgE4 (AR) creates strictly alternating MG structures whereas AlgE1 (ARRRAR) and AlgE6 (ARRR) create predominantly G-blocks. These enzymes are therefore promising tools for producing in vitro tailor-made alginates. Efficient in vitro epimerization of alginates requires availability of recombinantly produced alginate epimerases, and for this purpose the methylotrophic yeast Hansenula polymorpha is an attractive host organism. The present study investigates whether H. polymorpha is a suitable expression system for future large-scale production of AlgE1, AlgE4, and AlgE6. H. polymorpha expression strains were constructed using synthetic genes with reduced repetitive sequences as well as optimized codon usage. High cell density cultivations revealed that the largest epimerases AlgE1 (147 kDa) and AlgE6 (90 kDa) are subject to proteolytic degradation by proteases secreted by the yeast cells. However, degradation could be controlled to a large extent either by co-expression of chaperones or by adjusting cultivation conditions. The smaller AlgE4 (58 kDa) was stable under all tested conditions. The results obtained thus point toward a future potential for using H. polymorpha in industrial production of mannuronan C-5 epimerases for in vitro tailoring of alginates.
Introduction
Alginates are linear polysaccharides produced by brown algae and some bacterial species and are composed of the two monomers β-D-mannuronic acid (M) and α-L-guluronic acid (Linker and Jones, 1964; Gorin and Spencer, 1966; Govan et al., 1981; Cote and Krull, 1988). The chemical and physical properties of alginate are determined to a large extent by the sequence and distribution of M and G, which are arranged in consecutive stretches commonly denoted as M-, G-, and MG-blocks in the polymers. Alginate has numerous present and potential future applications within industrial, medical and pharmaceutical areas (Mørch et al., 2007; Hay et al., 2013; Tøndervik et al., 2020; Arlov et al., 2021). G rich alginates are traditionally most valuable and frequently used due to their gelling and viscosifying properties. Mannuronan C-5 epimerases are a family of enzymes responsible for the conversion of M to G at the polymer level during the biosynthesis of alginate (Haug and Larsen, 1969, 1971; Ertesvåg et al., 1995). The alginate producing and nitrogen fixing soil bacterium Azotobacter vinelandii has one periplasmic and seven secreted alginate epimerases, all of them involved at some level in biosynthesis of alginate in the complex life cycle of the organism (Ertesvåg et al., 1994, 1995; Svanem et al., 1999; Høidal et al., 2000). The secreted epimerases AlgE1-AlgE7 share a common modular structure, all containing one or two similar but not identical catalytic A-modules (A), and from one to seven similar regulatory R-modules (R). Similar enzymes have been identified in Azotobacter chroococcum and in Pseudomonas syringae (Bjerkan et al., 2004; Gawin et al., 2020). Intriguingly, despite the structural similarity of the epimerases, they create very different MG patterns in the alginate; AlgE4 being the least complex enzyme (AR) creates alternating MG structure with no G-blocks whereas AlgE1 (ARRRAR) and AlgE6 (ARRR) create predominantly G-blocks, AlgE1 producing longer G-blocks than AlgE6 (Ertesvåg et al., 1995). These different properties make the epimerases very promising tools for producing in vitro tailor-made alginates, i.e., alginates that are structurally defined and optimized for specific purposes (Mørch et al., 2007; Tøndervik et al., 2013; Arlov et al., 2014). The epimerases have previously mainly been produced for characterization purposes using E. coli as expression host (Ertesvåg et al., 1998; Høidal et al., 1999; Ramstad et al., 1999; Svanem et al., 1999), but alternative bacterial hosts like Lactococcus lactis has also been briefly explored (Blatny et al., 2003). Efficient processes for in vitro epimerization of alginate requires recombinantly produced alginate epimerases. For this purpose, the methylotrophic yeast Hansenula polymorpha is an attractive host organism that has been successfully utilized for expression of several recombinant proteins (Moussa et al., 2012; Manfrao-Netto et al., 2019). The main advantages using this host are the generally stable heterologous expression by chromosomal integration of genes of interest, generally high copy numbers of the integrated genes, lack of endotoxin production and the possibility for efficient secretion of recombinant products into the growth medium facilitating easy recovery from the fermentation broth. Additionally, H. polymorpha has status as GRAS (generally regarded as safe) being advantageous from a regulatory point of view. In the present study, our main goal was to investigate whether H. polymorpha is a suitable expression system for future large-scale production of the mannuronan C-5 epimerases AlgE1, AlgE4, and AlgE6. To obtain stable expression, strains were constructed using synthetic genes with reduced repetitive sequences as well as optimized codon usage. Cultivation of the epimerase expression strains at high cell densities revealed that the largest epimerases AlgE1 (147 kDa) and AlgE6 (90 kDa) are subject to proteolytic degradation by proteases secreted by the yeast cells, and different strategies for counteracting such degradation was successfully explored.
Materials and Methods
Strain Constructions
The genes encoding the epimerases AlgE1, AlgE4, and AlgE6 were assembled by de novo chemical synthesis (Geneart, Regensburg), which included adaptation of codon usage as well as reduction of DNA sequence repetitivity in the cases of algE1 and algE6. Importantly, in all cases the amino acid sequence of the enzymes remained unchanged. The synthetic genes (algE1syn, algE6syn, algE4syn) and the algE1 wild-type gene (algE1wt) were each fused in frame to the Saccharomyces cerevisiae prepro MFα1 secretion leader sequence and cloned into the expression vector B14 [containing the formate dehydrogenase (FMD) promoter], a derivative of pFPMT121 devoid of antibiotic resistance genes (Klabunde et al., 2007). Expression plasmids were transformed into H. polymorpha RB11 (ura3) or KLA8-1 (ura3 leu2) and uracil-prototrophic transformants were selected. Individual colonies were grown in test tubes containing YPG (2% Bacto peptone/1% yeast extract/2% glycerol/20 mM CaCl2) for 2 days at 37°C. Cell-free culture supernatants were screened for epimerase production by dot and Western blotting with an anti-epimerase antibody. The generation of epimerase strains overexpressing HpCNE1 was described previously (Klabunde et al., 2007). Overexpression of both HpCNE1 and HpSEB1 was achieved in a similar fashion by introduction of a plasmid containing both chaperone genes. Protease knockout mutants (yps1, yps7, pep4, and 3 additional genes predicted to encode proteases) were generated in strain KLA8-1 by targeted deletion using S. cerevisiae LEU2 as the selectable marker.
High Cell Density Fermentations
The fermentation media used was YSPG (25 g/l glucose, 10 g/l yeast extract, 20 g/l soyton pepton, 55 mM CaCl2) or HCD (10.9 g/l KH2PO4, 5.8 g/l (NH4)2HPO4, 5 g/l (NH4)2SO4, 2.3 g/l KCl, 0.5 g/l NaCl, 0.1 g/l Na-EDTA, 25 g/l glucose, 25 g/l yeast extract, 18 mM MgSO4, 55 mM CaCl2) or combinations of these as described in the text. For both media, 20 ml/l of a vitamin solution (thiamine-HCl 5 g/l, biotin 0.015 g/l) and 10 ml/l of a trace mineral solution (H3BO3 0.05 g/l, (NH4)2Fe(SO4)2 ⋅ 6H2O 10 g/l, CuSO4 ⋅ 5H2O 0.8 g/l, ZnSO4 ⋅ 7H2O 3 g/l, MnSO4 ⋅ H2O 4 g/l, NiCl2 ⋅ 6H2O 0.09 g/l, CoCl2 ⋅ 6H2O 0.1 g/l, Na2MoO4 ⋅ 2H2O 0.1 g/l, KI 0.1 g/l, HCl 50 ml/l) was added. If not otherwise stated, the cultivation temperature used was 37°C, and pH in YSPG and HCD was 6.0 and 5.5, respectively. The feeding solution used was glucose (30 or 60%) with methanol (10% of glucose). The feeding was started at a low rate (3–5 g/L, h) approx.12 h after inoculation and increased to 5–7 g/L, h after approx. 20 h or when the cultures were glucose limited. The rest of the fermentation was then run with glucose limitation. Precultures for fermentations were grown in shake-flasks in a medium containing 5 g/l yeast extract and 20 g/l soytone peptone for 16–18 h before inoculation. At the end of a standard 4-day fermentation process, samples from the cultures were routinely checked for purity by plating on solid medium with the same composition as for the pre-cultivation.
Determination of Epimerase Activity
For determination of epimerase activity, a spectrophotometric assay was developed using mannuronan (polyM) isolated from a mutant strain of Pseudomonas fluorescens NCIMB 10525 as substrate (Gimmestad et al., 2003). Dilutions of epimerase-containing samples, i.e., culture supernatants were mixed with polyM (1 mg/ml) in a buffer composed of 20 mM MOPS, 3.6 mM CaCl2 and 100 mM NaCl and incubated at 37°C for 16–18 h. Alginate lyase AlyA degrading G-M and G-G linkages (Tøndervik et al., 2010) was then added to a final concentration of 0.05 U/ml. Incubation was continued at 25°C for 4 h with recording of absorbance at 230 nm (A230) before (T0) the addition of AlyA and after 4 h (T1) and subtracting the values of a buffer blank. Lyase degradation introduces unsaturated uronic acid residues that can be quantified by A230, and the epimerase activity (i.e., introduction of G residues into polyM) in the samples is thus proportional to the ΔA230 (T1-T0) obtained. One unit of epimerase activity is defined as the amount of enzyme resulting in ΔA230 = 1 under the described conditions.
NMR Analysis of Epimerized Alginate Samples
All experiments were recorded on a BRUKER Avance 600 or DPX 400 spectrometer equipped with a 5 mm cryogenic CP-TCI z-gradient probe and 5 mm z-gradient DUL (C/H) probe, respectively. End-point analysis of epimerized samples were recorded at 363 K. To reduce the viscosity of the alginate samples prior to NMR measurements the samples were depolymerized by mild acid hydrolysis to a final average DPn ∼30 residues. 3-(trimethylsilyl)-propionic-2,2,3,3-d4 acid sodium salt (Aldrich, Milwaukee, WI, United States) was used as internal standard for the chemical shift and triethylenetetra-amine hexa-acetate (Sigma-Aldrich) was added to chelate residual calcium ions in end-point epimerized samples.
Cell Sampling, Isolation of RNA and RT-PCR Analysis
Yeast cells from fermentations were harvested by centrifugation (14,000 rpm, 5 min), supernatants were removed, and pellets frozen immediately in liquid nitrogen before storage at −80°C. RNA was isolated from yeast cells by the RNeasy Mini Kit (QIAGEN) applying mechanical disruption for lysis of cells (0.5 mm glassbeads and MiniBeadBeater). RNA concentration was measured by a NanoDrop Spectrophotometer ND-1000. Potential residual DNA was removed using the DNA-free™ kit from Ambion, and cDNA synthesis was performed using the First-Strand cDNA Synthesis Kit from GE Healthcare. The quantitative PCR was performed using Power SYBR® Green and a 7500 Real Time PCR System from Applied Biosystems. The PCR profile was as follows: stage 1; 50°C, 2 min (1 cycle) stage 2; 95°C, 10 min (1 cycle) stage 3; 95°C, 15 s and 60°C, 1 min (40 cycles). The actin (act) gene of H. polymorpha was used as an endogenous control. The primers used are listed in Table 1. All PCR reactions were performed in triplicates.
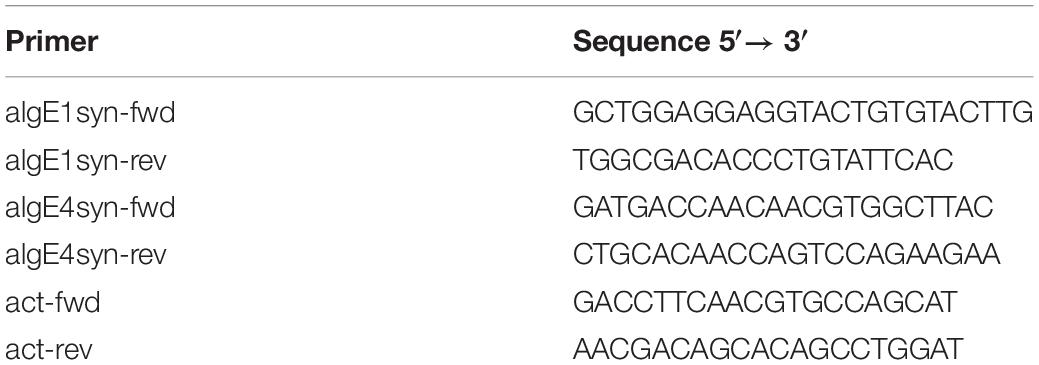
Table 1. Primers used for gene expression analyses of algE1syn and algE4syn.
Other Methods
Deglycosylation of proteins in the culture supernatants before SDS-PAGE analysis was performed by adding Endoglucosidase H (Roche) to supernatant samples (1:45) and incubating at 37°C overnight. N-terminal sequencing of protein fragments from the H. polymorpha supernatant was performed by separating the proteins by SDS-PAGE followed by blotting them on PVDF membranes as described earlier (Ertesvåg et al., 1994). The sequencing was performed by the Biotechnology Centre of Oslo at the University of Oslo. AlgE1 and the shortened derivatives used for the spiking experiments were expressed and purified as previously described (Ertesvåg et al., 1998).
Results and Discussion
Generation and Initial Characterization of Hansenula polymorpha Strains for Expression of Alginate Epimerases AlgE1, AlgE4, and AlgE6
Epimerase production, secretion and activity was first analyzed in small-scale test-tube cultures. In the case of AlgE1, the activity was fully dependent on the presence of excess calcium ions in the growth medium. Furthermore, it was found that only about 10% of the strains transformed with wild type algE1 and screened (approx. 100 transformant cultures) for epimerase activity secreted an active enzyme. The native gene contains several repetitive sequences, and the GC content is high. A striking improvement of AlgE1 production, both in terms of the frequency of expression strains obtained and the level of secreted epimerase, resulted from introduction of a synthetic gene (algE1syn) with less repetitive sequences and a codon usage adapted to H. polymorpha. By comparing the production of AlgE1 from strains expressing wild type (RB11/algE1wt) and synthetic gene (RB11/algE1syn) during fermentation in HCD medium, we found that the epimerase activity detected in the culture medium (U/ml) increased about fourfold by the introduction of an optimized gene (Figure 1A), proving the importance of codon adaptation. Furthermore, it was also found that addition of methanol to the feeding solution (to 10% of the glucose concentration used) led to another twofold increase in the secreted epimerase activity, presumably due to induction of expression from the FMD-promoter under the glucose limiting conditions (Figure 1A). The two strains expressing either wild type or synthetic algE1 showed identical growth kinetics and growth yield in the medium utilized, and these parameters were not influenced by addition of methanol (Figure 1B). In all further fermentation experiments we therefore routinely used methanol in the feeding solution if not stated otherwise. Based on the experiences with expression of algE1 in H. polymorpha, strains for expressing algE4 and algE6 were constructed using synthetic genes only (see Supplementary File 1 for alignments of wt and synthetic genes for algE1, algE4, and algE6). As described below, stability of AlgE1 is a significant issue using H. polymorpha as expression host. To elucidate whether expression of the integrated epimerase genes could be a bottleneck during the fermentations, expression analysis of algE1 and algE4 were performed by RT-PCR using the H. polymorpha actin gene (act) as an endogenous control. The strains tested were RB11/algE1syn and KLA8-1/algE4syn. Fermentations were performed using HCD medium at 37°C, and samples for isolation of RNA and production analysis were taken at 27, 50, 73, and 96 h after inoculation. The expression level of the genes was found to be quite stable during the cultivation period (Figures 2A,B), however with a higher level at the two first timepoints for RB11/algE1syn and the first timepoint for KLA8-1/algE4syn. These data indicate that the main challenges connected to AlgE1 production is related to stability of the produced protein.
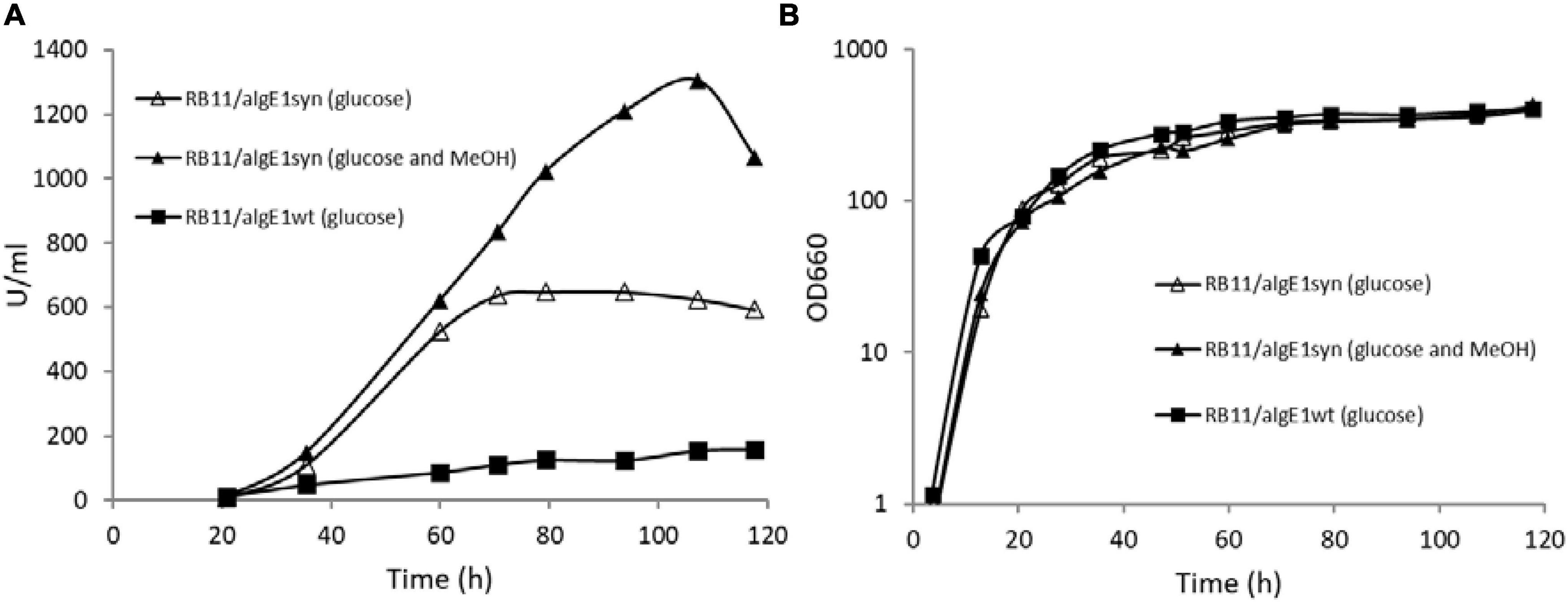
Figure 1. AlgE1 epimerase activity (U/ml) produced (A) and growth (B) of RB11/algE1wt and RB11/algE1syn when grown in fermentors with HCD medium (pH 4.6, 37°C) and 60% glucose or glucose (60%) and methanol (10% of glucose) in the feeding solution.
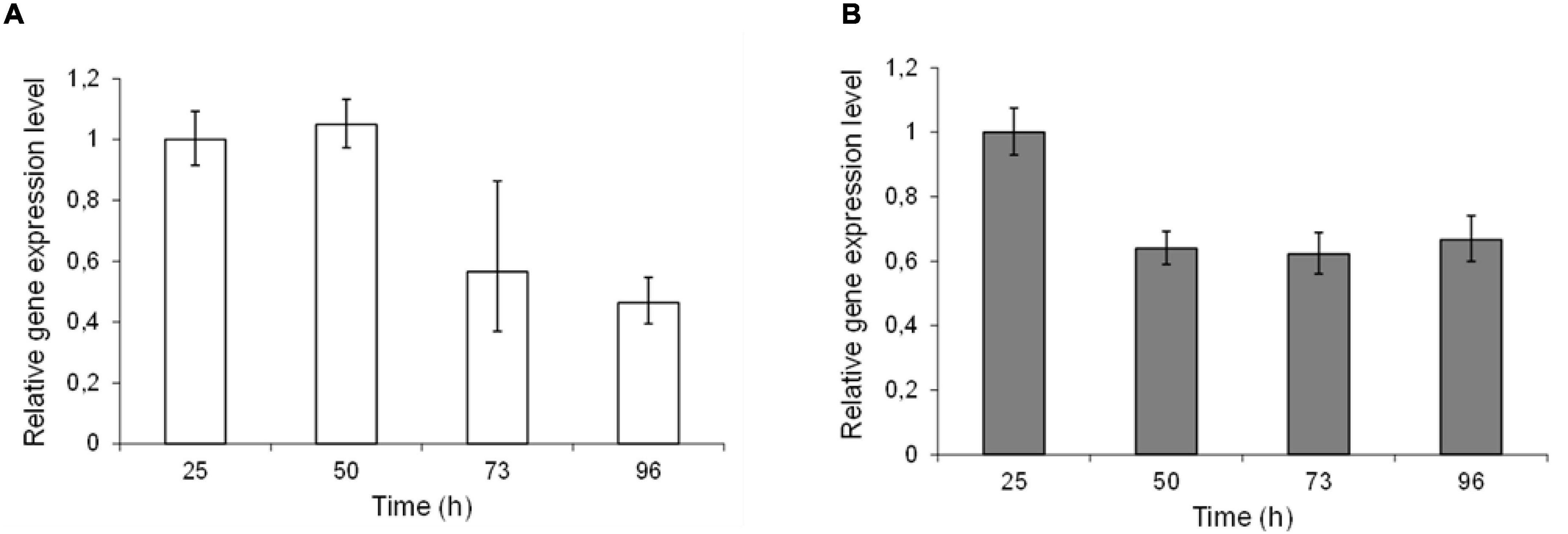
Figure 2. Relative expression levels of epimerase genes in RB11/algE1syn (A) and RB11/algE4syn (B) during cultivation in HCD medium with feeding solution composed of glucose (60%) and methanol (10% of glucose). Samples for DNA isolation were taken during the fermentation at 27, 50, 73, and 96 h after inoculation. For each strain the expression level in sample 1 is set as 1. Quantifications were performed in triplicate and standard deviations from the mean are indicated by error bars.
Recombinant Alginate Epimerase AlgE1 Is Subject to Degradation by Host Proteases During Fermentation
Epimerase producing H. polymorpha strains were initially fermented in HCD medium, and for determining the yield and integrity of the secreted enzymes, samples taken during fermentation were analyzed by protein gel-electrophoresis (SDS-PAGE). Through these analyses it became evident that all three epimerases were heavily N-glycosylated, however glycosylation did not measurably compromise the enzymatic activity (data not shown). It was also found that AlgE1 (147 kDa) was degraded into two main smaller fragments during the fermentation process, and only very faint signals corresponding to full length AlgE1 were visible on the gel (data not shown). Degradation could be partly controlled by growing the strains in a less rich medium (2xYSPG), Figure 3A. However, when a fraction of HCD (to a concentration of 1/3) was added to the medium attempting to increase the biomass yield, this again led to severe degradation. From the SDS-PAGE analyses it appears that degradation is taking place during the entire fermentation period, indicating that this is caused by one or several proteases that are secreted by the yeast cells independently of the growth phase.
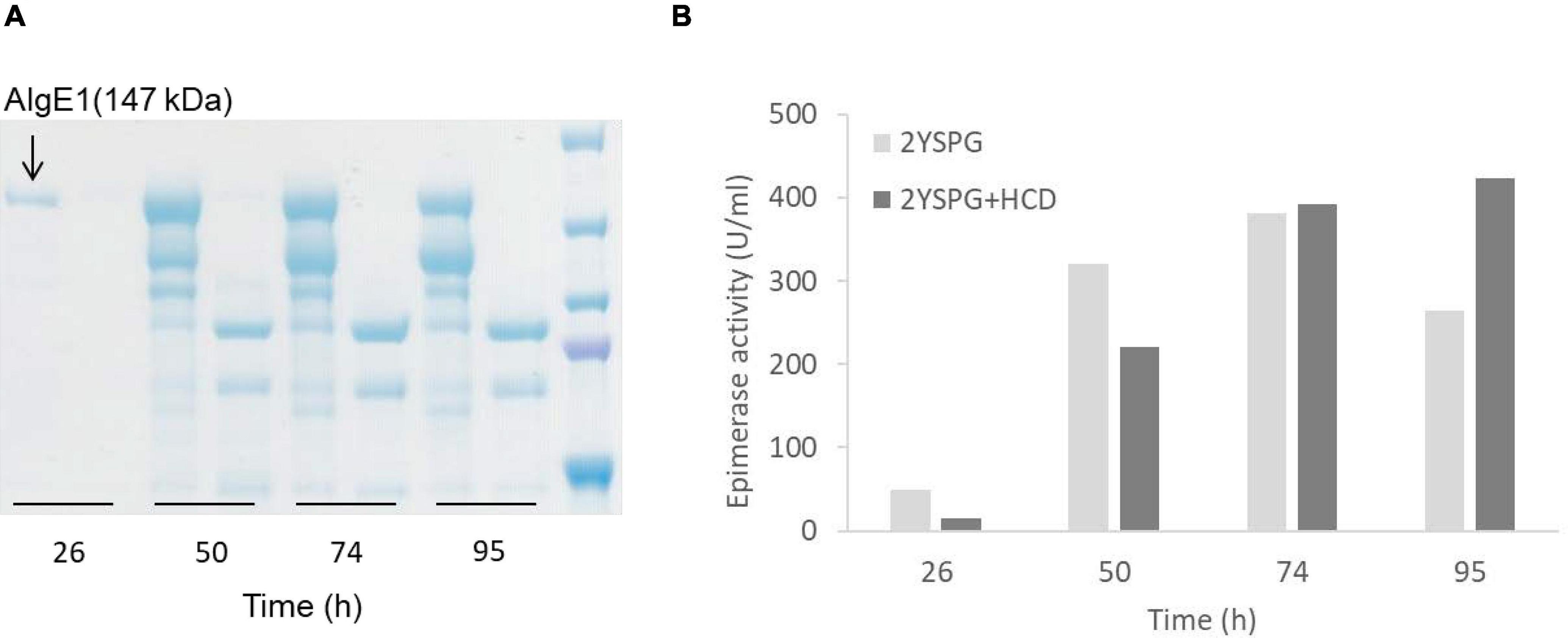
Figure 3. SDS-PAGE analysis of AlgE1 stability during fermentation of RB11/algE1syn in 2xYSPG (37°C, pH 6) and 2xYSPG + 1/3HCD (37°C, pH 6) both with feeding solutions composed of glucose (30%) and methanol (10% of glucose) in samples taken at 26, 50, 74, and 95 h (A), and AlgE1 epimerase activity (U/ml culture) of the same samples (B). The samples on the SDS-PAGE gel are arranged in the same order as in the bar chart shown in panel B of the figure. Size of protein fragments in standard (right); 250, 150 100, 75, and 50 kDa.
Despite substantial degradation, epimerase activity was still detected in the supernatants indicating that the degraded enzyme retained activity (Figure 3B). NMR analysis of mannuronan epimerized with degraded enzyme partially purified from the supernatant showed that the alginate obtained did not contain a high level of G-blocks as is the case for intact AlgE1. The level of G-blocks decreased during the fermentation with and accompanied increase in the level of alternating MG/GM residues (Table 2). This indicated that the residual activity could be caused by an intact A2 module which introduces single G’s and degradation of the A1 module of AlgE1 which is responsible for the block forming properties (Ertesvåg et al., 1998). This was confirmed by obtaining the N-terminal sequence of the protein fragment with the highest Mw (confer Figure 3A). The sequence SGSGQ indicated that the protein was cut after residue 371 in the A1 module of AlgE1 (Figure 4). The remaining part contains all R modules and it has previously been shown that proteins that contain at least the last R module and the second A module of AlgE1 have a specific activity not much less than the wild-type enzyme (Ertesvåg et al., 1998).
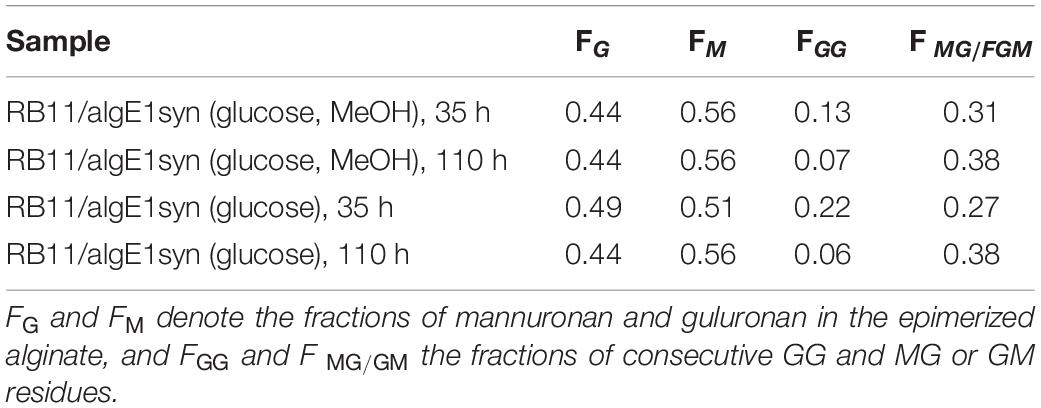
Table 2. NMR analyses of polyM epimerized with AlgE1-containing samples from fermentations of RB11/algE1syn showed in Figure 1.

Figure 4. Alignment of the A-modules of AlgE1, AlgE4, and AlgE6 emphasizing the differences between the first A module of AlgE1 (A1) and the other three A-modules. Dots indicate identical amino acids whereas amino acids differing are shaded. The amino acid identified as the N-terminal in the degraded AlgE1 produced by H. polymorpha is shown in green, while the ones identified by incubation of supernatant from H. polymorpha with purified AlgE1 is marked in red.
Exploring the Use of Protease Deficient Strains to Avoid Epimerase Degradation
In the case of extracellular proteases, it should be possible to use the supernatant from H. polymorpha known to degrade the produced AlgE1 to degrade purified proteins containing only one of the two A-modules of AlgE1 expressed from individual vectors (Ertesvåg et al., 1998). SDS-PAGE showed that the proteins containing the A1 module was degraded into smaller fragments, whereas the A2 module remained intact (not shown). The protein fragments from the A1 module were subjected to N-terminal sequencing, which indicated that the protein firstly is cut close to the end of the A-module (Figure 4). Prolonged incubation resulted in a band with smaller molecular weight consisting of several peptides that could not be resolved by N-terminal sequencing. Based on the effect of protease inhibitors, it seemed probable that more than one protease is involved in the degradation. Heat inactivation of the supernatant by boiling prior to incubation with purified AlgE1 abolished degradation, indeed pointing to an enzymatic degradation reaction, and attempts were therefore made to identify the protease(s).
This information could potentially be used for specific gene inactivation of the relevant targets. The supernatant from H. polymorpha known to degrade AlgE1 was fractionated using a gel-filtration column, and fractions displaying proteolytic activity were separated on SDS-PAGE before transferring proteins to PVDF membranes by western blotting. Proteins were visualized with Coomassie staining and bands excised from the membrane for analysis with MS after trypsination. H. polymorpha proteins were identified by utilizing genome/protein data (Ramezani-Rad et al., 2003), but unfortunately none were recognized as potential proteases. Presumably, the relevant proteases exist in very minor amounts, and there might also be several responsible proteases acting sequentially as vulnerable parts of the epimerase are gradually being exposed to the environment.
Several cellular proteases have previously been found to contribute to degradation of secreted heterologous proteins in yeast. These include proteases of the yapsin (Yps) family and the vacuolar proteinase A (Pep4). We generated mutants in strain KLA8-1 that lacked the H. polymorpha homologs of the genes YPS1, YPS7, PEP4, and three other genes predicted to encode proteases from the genome annotation data (Ramezani-Rad et al., 2003). The effect of the knockouts on epimerase stability was investigated by spiking the fermenter broth in which individual mutants had been grown, or cell suspensions, with purified E. coli-derived AlgE1. However, none of the protease knockouts significantly enhanced the stability of AlgE1 (data not shown). The KLA8-1 protease deficient strains constructed can be of value for similar degradation susceptibility evaluations in future studies.
Increased Calnexin Gene Dosage Increases Secretion of AlgE1 and AlgE4
It has previously been shown that increased gene dosage of the molecular chaperone calnexin (HpCNE1) enhances secretion of AlgE1 in recombinant H. polymorpha production strains when analyzed by cultivation in shake flasks (Klabunde et al., 2007). Here, we show that this is also the case when strains are grown to high cell density in fermentors, resulting in an increased epimerase activity at the end of a standard 4-day fermentation experiment (Figure 5B). However, overexpression of calnexin did not protect the produced AlgE1 from proteolytic degradation, indicating that the host proteases still have access to specific target sites in the protein (Figure 5A).
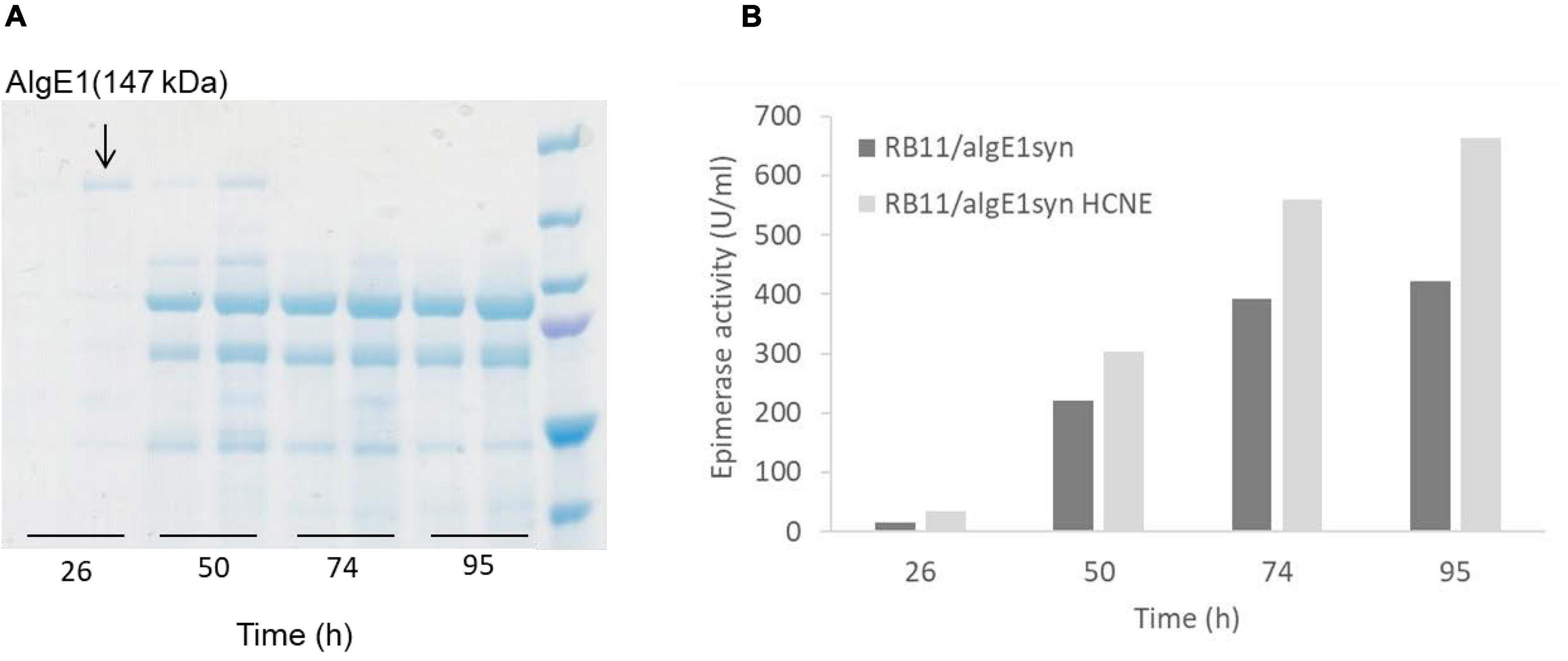
Figure 5. Effect of increased calnexin gene dosage (HpCNE) on AlgE1 stability analyzed by SDS-PAGE (A) and epimerase activity (U/ml culture) (B) in RB11/algE1syn and RB11/algE1syn HCNE. Strains were cultivated in 2xYSPG + 1/3HCD (37°C, pH 6) with feeding solution composed of glucose (60%) and methanol (10% of glucose), and samples were taken at 26, 50, 74, and 95 h. The samples on the SDS-PAGE gel are arranged in the same order as in the bar chart shown in panel B of the figure. Size of protein fragments in standard (right); 250, 150 100, 75, 50, and 37 kDa.
Contrary to AlgE1, we found that the smaller AlgE4 was not subject to degradation when expressed in H. polymorpha, presumably due to absence of the main target amino acid sequence of protease attack identified in AlgE1 (Figure 4). The effect of calnexin dosage on expression on AlgE4 was also explored. Strain KLA8-1/algE4syn HCNE was constructed and found to give approximately twofold increase in the secreted AlgE4 activity compared to the parental strain KLA8-1/algE4syn (Figure 6). Moreover, in order to attempt strengthening the protein folding apparatus further, additional copies of the H. polymorpha SEB1 ortholog (HSEB1) were inserted into KLA8-1/algE4syn HCNE. Overexpression of this subunit of the SEC61 translocon complex has previously been shown to increase secretion of certain heterologous proteins (Boisrame et al., 2002; Toikkanen et al., 2004). However, the expression of HSEB1 apparently counteracted the positive effect exerted by increased calnexin dosage since the specific AlgE4 activity obtained with KLA8-1/algE4syn HCNE HSEB was reduced compared to KLA8-1/algE4syn HCNE (Figure 6). The reason for this is not clear but might be connected to overloading the total protein expression capacity of the H. polymorpha cells.
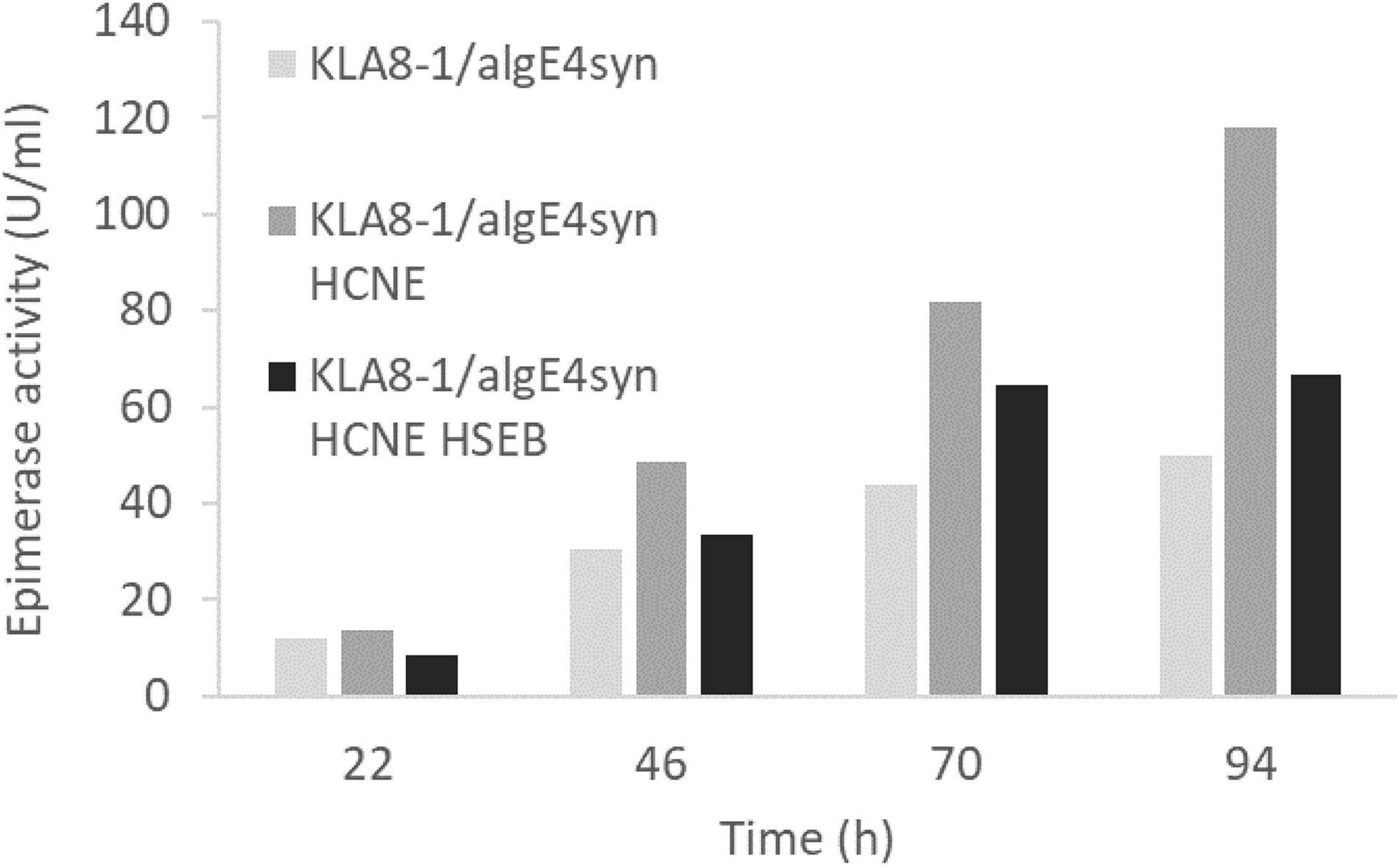
Figure 6. Effect of increased calnexin (HpCNE) and SEB1 (HpSEB) gene dosage on the production of AlgE4 in H. polymorpha. Specific AlgE4 epimerase activity (U/ml culture) obtained with KLA8-1/algE4syn, KLA8-1/algE4syn HCNE, and KLA8-1/algE4syn HCNE HSEB during the fermentation process with samples taken at 22, 46, 70, and 94 h. Strains were cultivated in HCD (37°C, pH 5.5) and with feeding solutions composed of glucose (60%) and methanol (10% of glucose).
Growth on Glycerol Increases the Stability of AlgE6
As with AlgE1, it was found that AlgE6 was degraded to a large extent during the fermentations when strain RB11/algE6syn was cultivated in HCD and in 2xYSPG + 1/3HCD with glucose and methanol in the feeding solution. Basically, no full-length AlgE6 was visible on the gels during the fermentation period (Figure 7A). In the initial strain characterizations carried out in small scale shake-flasks in a medium containing yeast extract, peptone and glycerol stability was however not a problem. Fermentation experiments were therefore performed with 2xYSPG where glycerol was used as a carbon source instead of glucose in the basis medium, and with 60% glycerol as feeding solution. Growth in glycerol medium resulted in significantly lower biomass, i.e., at the end of the cultivation (95 h) OD660 was approx. 80, whereas with glucose the corresponding value was 250. Due to the lower cell mass the activity per ml detected was lower (Figure 7B), however the AlgE6 produced remained mainly intact (Figure 7A).
Figure 7. SDS-PAGE analysis (A) and epimerase activity (U/ml culture) (B) of RB11/algE6syn cultivated in 2xYSPG with glycerol in the basis medium and a feeding solution composed of 60% glycerol, or in 2xYSPG + 1/3 HCD (glucose in basis medium) and feeding solution composed of glucose (60%) and methanol (10% of glucose). Samples for activity analysis was taken at 25, 48, 72, and 95 h. SDS-PAGE show the AlgE6 content in the samples taken at 95 h. Size of protein fragments in standard (right); 250, 150 100, 75, 50, and 37 kDa.
Despite the degradation observed when RB11/algE6syn was fermented in glucose-based medium (2YSPG + 1/3HCD), epimerase activity was detected. This might indicate that the catalytic A-module of AlgE6 is not affected by proteolytic degradation, rather the three C-terminal non-catalytic R-modules are the protease target. The A module of AlgE6 has previously been shown to be active without its associated R-modules (Stanisci et al., 2018). The degradation pattern of AlgE6 was, however, not investigated in more detail. The results obtained with AlgE6 show that by modifying the composition of the cultivation medium it is possible to increase the stability of the epimerase product. Due to the similarity of AlgE6 and AlgE1, these findings will probably also apply equally well to AlgE1, but this needs to be evaluated in future studies.
Data from production of the epimerases by high cell density cultivations of E. coli (OD of approx. 200) show that for AlgE1 higher levels in terms of U/ml culture are obtained in H. polymorpha, whereas for AlgE4 and AlgE6 higher levels are obtained in E. coli (unpublished). Furthermore, in E. coli the epimerases are not subject to degradation.
Conclusion
The potential for using the yeast H. polymorpha for expression of mannuronan C-5 epimerases AlgE1, AlgE4, and AlgE6 was investigated. For construction of epimerase expression strains, synthetic genes were used since wild type genes with several direct repeats, were found to be highly unstable when integrated into the yeast chromosome. Fermentation of the epimerase-expressing strains under conditions giving high cell densities (OD660 > 250) resulted in degradation of AlgE1 and AlgE6, but not AlgE4. AlgE1 which consists of two catalytic A-modules each giving rise to fundamentally different epimerase patterns, was found to be fragmented by cleavage of the N-terminal A1 module, while leaving the A2 module intact and active. Degradation due to host proteases secreted into the medium could be reduced but not avoided for AlgE1 by co-expressing the chaperone protein calnexin. Co-expression of calnexin in AlgE4 producing strains increased the epimerase activity obtained per cell mass, indicating that although not subject to proteolytic degradation AlgE4 stability and activity can be improved by this strategy. Increased stability of AlgE6 was obtained by using glycerol in the feeding medium instead of glucose. Although the secreted epimerases were not purified from the culture supernatants, it could be observed from the SDS-PAGE gels that the recombinant proteins were dominating. Compared to intracellular expression in E. coli which necessitates cell disruption before purification, the down-stream processing from secretory production in H. polymorpha would be simpler. Altogether, the results obtained show that using H. polymorpha for epimerase expression involves challenges related to both the DNA sequence as well as the properties of the recombinant enzymes. However, strategies targeted both to the level of strain construction and process development show that these challenges can be met and large-scale production of epimerases in this GRAS organism are therefore considered as a promising approach.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
AD and MP performed the strain construction and initial characterization. HS and RA performed the fermentation experiments. AT and HE performed the analytical work. AT wrote the first draft of the manuscript. All authors reviewed, edited the manuscript, contributed to the planning, and design of experiments.
Funding
This work was supported by AlgiPharma AS, FMC Biopolymer AS, The Research Council of Norway (Grant No. 139845), and the European FP6 (Grant No. 026515-2).
Conflict of Interest
AD and MP were employed by the ARTES Biotechnology GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
In memory of Edvar Onsøyen, AlgiPharma AS, who passed away during this work. Hanne Jørgensen and Wenche Strand are thanked for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.837891/full#supplementary-material
References
Arlov, O., Aachmann, F. L., Sundan, A., Espevik, T., and Skjåk-Bræk, G. (2014). Heparin-like properties of sulfated alginates with defined sequences and sulfation degrees. Biomacromolecules 15, 2744–2750. doi: 10.1021/bm500602w
Arlov, O., Rutsche, D., Korayem, M. A., Ozturk, E., and Zenobi-Wong, M. (2021). Engineered sulfated polysaccharides for biomedical applications. Adv. Funct. Mat. 31:52.
Bjerkan, T. M., Bender, C. L., Ertesvåg, H., Drabløs, F., Fakhr, M. K., Preston, L. A., et al. (2004). The Pseudomonas syringae genome encodes a combined mannuronan C-5-epimerase and O-acetylhydrolase, which strongly enhances the predicted gel-forming properties of alginates. J. Biol. Chem. 279, 28920–28929. doi: 10.1074/jbc.M313293200
Blatny, J. M., Ertesvåg, H., Nes, I. F., and Valla, S. (2003). Heterologous gene expression in Lactococcus lactis; expression of the Azotobacter vinelandii AlgE6 gene product displaying mannuronan C-5 epimerase activity. FEMS. Microbiol. Lett. 227, 229–235. doi: 10.1016/S0378-1097(03)00685-2
Boisrame, A., Chasles, M., Babour, A., Beckerich, J. M., and Gaillardin, C. (2002). Sbh1p, a subunit of the Sec61 translocon, interacts with the chaperone calnexin in the yeast Yarrowia lipolytica. J. Cell Sci. 115, 4947–4956. doi: 10.1242/jcs.00187
Cote, G. L., and Krull, L. H. (1988). Characterization of the exocellular polysaccharides from Azotobacter chroococcum. Carbohyd. Res. 181, 143–152. doi: 10.1016/0008-6215(88)84030-8
Ertesvåg, H., Doseth, B., Larsen, B., Skjåk-Bræk, G., and Valla, S. (1994). Cloning and expression of an Azotobacter vinelandii mannuronan C-5 epimerase gene. J. Bacteriol. 176, 2846–2853. doi: 10.1128/jb.176.10.2846-2853.1994
Ertesvåg, H., Høidal, H. K., Hals, I. K., Rian, A., Doseth, B., and Valla, S. (1995). A family of modular type mannuronan C-5 epimerase genes controls alginate structure in Azotobacter vinelandii. Mol. Microbiol. 16, 719–731. doi: 10.1111/j.1365-2958.1995.tb02433.x
Ertesvåg, H., Høidal, H. K., Skjåk-Bræk, G., and Valla, S. (1998). The Azotobacter vinelandii mannuronan C-5-epimerase AlgE1 consists of two separate catalytic domains. J. Biol. Chem. 273, 30927–30932. doi: 10.1074/jbc.273.47.30927
Gawin, A., Tietze, L., Aarstad, O. A., Aachmann, F. L., Brautaset, T., and Ertesvåg, H. (2020). Functional characterization of three Azotobacter chroococcum alginate-modifying enzymes related to the Azotobacter vinelandii AlgE mannuronan C-5-epimerase family. Sci. Rep. 10:14. doi: 10.1038/s41598-020-68789-3
Gimmestad, M., Sletta, H., Ertesvåg, H., Bakkevig, K., Jain, S., Suh, S., et al. (2003). The Pseudomonas fluorescens AlgG protein, but not its mannuronan C-5-epimerase activity, is needed for alginate polymer formation. J. Bacteriol. 185, 3515–3523. doi: 10.1128/JB.185.12.3515-3523.2003
Gorin, P. A. J., and Spencer, J. F. T. (1966). Exocellular alginic acid from Azotobacter vinelandii. Can. J. Chem. 44, 993–998. doi: 10.1139/v66-147
Govan, J. R., Fyfe, J. A., and Jarman, T. R. (1981). Isolation of alginate producing mutants of Pseudomonas fluorescens, Pseudomonas putida and Pseudomonas mendocina. J. Gen. Microbiol. 125, 217–220. doi: 10.1099/00221287-125-1-217
Haug, A., and Larsen, B. (1969). Biosynthesis of alginate. Epimerisation of D-mannuronic acid to L-guluronic acid residues in the polymer chain. Biochimica et Biophysica Acta 192, 557–559. doi: 10.1016/0304-4165(69)90414-0
Haug, A., and Larsen, B. (1971). Biosynthesis of alginate. II. Polymannuronic acid C-5-epimerase from Azotobacter vinelandii (Lipman). Carbohyd. Res. 2, 297–308. doi: 10.1016/s0008-6215(00)82537-9
Hay, I. D., Rehman, Z. U., Moradali, M. F., Wang, Y. J., and Rehm, B. H. A. (2013). Microbial alginate production, modification and its applications. Microbial. Biotechnol. 6, 637–650. doi: 10.1111/1751-7915.12076
Høidal, H. K., Ertesvåg, H., Skjåk-Bræk, G., Stokke, B. T., and Valla, S. (1999). The recombinant Azotobacter vinelandii mannuronan C-5-epimerase AlgE4 epimerizes alginate by a nonrandom attack mechanism. J. Biol. Chem. 274, 12316–12322. doi: 10.1074/jbc.274.18.12316
Høidal, H. K., Svanem, B. I. G., Gimmestad, M., and Valla, S. (2000). Mannuronan C-5 epimerases and cellular differentiation of Azotobacter vinelandii. Environ. Microbiol. 2, 27–38. doi: 10.1046/j.1462-2920.2000.00074.x
Klabunde, J., Kleebank, S., Piontek, M., Hollenberg, C. P., Hellwig, S., and Degelmann, A. (2007). Increase of calnexin gene dosage boosts the secretion of heterologous proteins by Hansenula polymorpha. FEMS. Yeast Res. 7, 1168–1180. doi: 10.1111/j.1567-1364.2007.00271.x
Linker, A., and Jones, R. S. (1964). A polysaccharide resembling alginic acid from a Pseudomonas micro-organism. Nature 204, 187–188. doi: 10.1038/204187a0
Manfrao-Netto, J. H. C., Gomes, A. M. V., and Parachin, N. S. (2019). Advances in using Hansenula polymorpha as chassis for recombinant protein production. Front. Bioeng. Biotechnol. 7:13. doi: 10.3389/fbioe.2019.00094
Mørch, Y. A., Donati, I., Strand, B. L., and Skjåk-Bræk, G. (2007). Molecular engineering as an approach to design new functional properties of alginate. Biomacromolecules 8, 2809–2814. doi: 10.1021/bm700502b
Moussa, M., Ibrahim, M., El Ghazaly, M., Rohde, J., Gnoth, S., Anton, A., et al. (2012). Expression of recombinant staphylokinase in the methylotrophic yeast Hansenula polymorpha. BMC Biotechnol. 12:13. doi: 10.1186/1472-6750-12-96
Ramezani-Rad, M., Hollenberg, C. P., Lauber, J., Wedler, H., Griess, E., Wagner, C., et al. (2003). The Hansenula polymorpha (strain CBS4732) genome sequencing and analysis. Fems. Yeast Res. 4, 207–215. doi: 10.1016/S1567-1356(03)00125-9
Ramstad, M. V., Ellingsen, T. E., Josefsen, K. D., Hoidal, H. K., Valla, S., Skjak-Braek, G., et al. (1999). Properties and action pattern of the recombinant mannuronan C-5-epimerase AlgE2. Enzy. Microbial. Technol. 24, 636–646. doi: 10.1016/s0141-0229(98)00148-3
Stanisci, A., Aarstad, O. A., Tøndervik, A., Sletta, H., Dypås, L. B., Skjåk-Bræk, G., et al. (2018). Overall size of mannuronan C5-Epimerases influences their ability to epimerize modified alginates and alginate gels. Carbohyd. Polym. 180, 256–263. doi: 10.1016/j.carbpol.2017.09.094
Svanem, B. I. G., Skjåk-Bræk, G., Ertesvåg, H., and Valla, S. (1999). Cloning and expression of three new Azotobacter vinelandii genes closely related to a previously described gene family encoding mannuronan C-5-epimerases. J. Bacteriol. 181, 68–77. doi: 10.1128/JB.181.1.68-77.1999
Toikkanen, J. H., Sundqvist, L., and Keranen, S. (2004). Kluyveromyces lactis SSOI and SEBI genes are functional in Saccharamyces cerevisiae and enhance production of secreted proteins when overexpressed. Yeast 21, 1045–1055. doi: 10.1002/yea.1151
Tøndervik, A., Aarstad, O. A., Aune, R., Maleki, S., Rye, P. D., Dessen, A., et al. (2020). Exploiting mannuronan C-5 epimerases in commercial alginate production. Mar. Drugs 18:14. doi: 10.3390/md18110565
Tøndervik, A., Klinkenberg, G., Aachmann, F. L., Svanem, B. I. G., Ertesvåg, H., Ellingsen, T. E., et al. (2013). Mannuronan C-5 epimerases suited for tailoring of specific alginate structures obtained by high-throughput screening of an epimerase mutant library. Biomacromolecules 14, 2657–2666. doi: 10.1021/bm4005194
Keywords: Hansenula polymorpha, recombinant expression, mannuronan C-5 epimerases, alginate, HCDC
Citation: Tøndervik A, Aune R, Degelmann A, Piontek M, Ertesvåg H, Skjåk-Bræk G and Sletta H (2022) Strain Construction and Process Development for Efficient Recombinant Production of Mannuronan C-5 Epimerases in Hansenula polymorpha. Front. Plant Sci. 13:837891. doi: 10.3389/fpls.2022.837891
Received: 17 December 2021; Accepted: 08 April 2022;
Published: 06 June 2022.
Edited by:
Elizabeth Ficko-Blean, UMR 8227 Laboratoire de Biologie Intégrative des Modèles Marins, FranceReviewed by:
Tsan-Yu Chiu, Beijing Genomics Institute (BGI), ChinaMarlene Vuillemin, Technical University of Denmark, Denmark
Copyright © 2022 Tøndervik, Aune, Degelmann, Piontek, Ertesvåg, Skjåk-Bræk and Sletta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Håvard Sletta, SGF2YXJkLlNsZXR0YUBzaW50ZWYubm8=