 Barbara Gasse1Megana Prasad2
Barbara Gasse1Megana Prasad2 Sidney Delgado1
Sidney Delgado1 Mathilde Huckert2,3Marzena Kawczynski3,4
Mathilde Huckert2,3Marzena Kawczynski3,4 Annelyse Garret-Bernardin3,5
Annelyse Garret-Bernardin3,5 Serena Lopez-Cazaux6Isabelle Bailleul-Forestier7Marie-Cécile Manière3,4
Serena Lopez-Cazaux6Isabelle Bailleul-Forestier7Marie-Cécile Manière3,4 Corinne Stoetzel2
Corinne Stoetzel2 Agnès Bloch-Zupan3,4,8,9,10
Agnès Bloch-Zupan3,4,8,9,10 Jean-Yves Sire1*
Jean-Yves Sire1*- 1Institut de Biologie Paris-Seine, UMR 7138-Evolution Paris-Seine, Sorbonne Universités, Université Pierre et Marie Curie, Paris, France
- 2Laboratoire de Génétique Médicale, Institut National de la Santé et de la Recherche Médicale UMRS_1112, Institut de Génétique Médicale d'Alsace, FMTS, Université de Strasbourg, Strasbourg, France
- 3Faculté de Chirurgie Dentaire, Université de Strasbourg, Strasbourg, France
- 4Pôle de Médecine et Chirurgie Bucco-Dentaires, Centre de Référence des Manifestations Odontologiques des Maladies Rares, O-Rares, Hôpitaux Universitaires de Strasbourg, Strasbourg, France
- 5Unit of Dentistry, IRCCS, Bambino Gesù Children's Hospital, Rome, Italy
- 6Faculté de Chirurgie Dentaire, Département d'Odontologie Pédiatrique, Centre de Compétences Maladies Rares, CHU Hôtel Dieu, Service d'odontologie Conservatrice et Pédiatrique, Nantes, France
- 7Faculté de Chirurgie Dentaire, CHU de Toulouse, Centre de Compétences Maladies Rares, Odontologie Pédiatrique, Université Paul Sabatier, Toulouse, France
- 8Centre Européen de Recherche en Biologie et en Médecine, Centre National de la Recherche Scientifique UMR7104, Institut National de la Santé et de la Recherche Médicale U964, Institut de Génétique et de Biologie Moléculaire and Cellulaire, Université de Strasbourg, Illkirch, France
- 9Institut d'Etudes Avancées, Université de Strasbourg, USIAS, Strasbourg, France
- 10Eastman Dental Institute, University College London, London, United Kingdom
Amelogenesis imperfecta (AI) designates a group of genetic diseases characterized by a large range of enamel disorders causing important social and health problems. These defects can result from mutations in enamel matrix proteins or protease encoding genes. A range of mutations in the enamel cleavage enzyme matrix metalloproteinase-20 gene (MMP20) produce enamel defects of varying severity. To address how various alterations produce a range of AI phenotypes, we performed a targeted analysis to find MMP20 mutations in French patients diagnosed with non-syndromic AI. Genomic DNA was isolated from saliva and MMP20 exons and exon-intron boundaries sequenced. We identified several homozygous or heterozygous mutations, putatively involved in the AI phenotypes. To validate missense mutations and predict sensitive positions in the MMP20 sequence, we evolutionarily compared 75 sequences extracted from the public databases using the Datamonkey webserver. These sequences were representative of mammalian lineages, covering more than 150 million years of evolution. This analysis allowed us to find 324 sensitive positions (out of the 483 MMP20 residues), pinpoint functionally important domains, and build an evolutionary chart of important conserved MMP20 regions. This is an efficient tool to identify new- and previously-identified mutations. We thus identified six functional MMP20 mutations in unrelated families, finding two novel mutated sites. The genotypes and phenotypes of these six mutations are described and compared. To date, 13 MMP20 mutations causing AI have been reported, making these genotypes and associated hypomature enamel phenotypes the most frequent in AI.
Introduction
Amelogenesis imperfecta (AI) describes a group of genetic diseases producing a large range of alterations in enamel structure. Defects include hypoplastic, hypomineralized, or hypomature enamel structure and altered enamel appearance -including rough, pitted, banded, or discolored teeth. These alterations produce severe health problems and impair normal social interactions. Defects can result from mutations either of enamel matrix protein encoding genes [amelogenin (AMELX), ameloblastin (AMBN), amelotin (AMTN), and enamelin (ENAM)], or proteases [matrix metalloproteinase-20 (MMP20), kallikrein-related peptidase 4 (KLK4))]—often displaying specific roles in tooth—and hence at the current stage of our knowledge do not display disorders elsewhere in the body. Other genes encoding proteins having functions in cell attachment, ionic transport, and mineralization processes, when mutated are responsible for AI phenotypes associated with various abnormalities in syndromes (Crawford et al., 2007; reviewed in Bloch-Zupan et al., 2012).
The prevalence of AI can vary from 1:700 to 1:14,000 depending on the country. Although many candidate genes were identified to date, many AI-producing mutations remain to be identified for approximately 50% of diagnosed patients.
Here, we focused on the matrix metalloproteinase-20 gene (MMP20), an interesting candidate gene given the numerous identified AI causing mutations (Kim et al., 2005, 2016a; Ozdemir et al., 2005; Papagerakis et al., 2008; Lee et al., 2010; Gasse et al., 2013; Wang et al., 2013; Seymen et al., 2015; Prasad et al., 2016a).
MMPs function in plants, invertebrates, and vertebrates, by their active center possessing a catalytic zinc domain (Gomis-Ruth, 2009; Fanjul-Fernandez et al., 2010). In vertebrates, matrix metalloproteinases (MMPs, also called matrixins) consist of a large family (24 members identified in humans) of endopeptidases, which are phylogenetically related (Fanjul-Fernandez et al., 2010). MMPs were initially thought involved in the degradation and turnover of the extracellular matrix, but recent studies indicate important biological roles regulating cell behavior and signaling pathways (Rodríguez et al., 2010).
MMP20 (also termed enamelysin) is only present in vertebrates. Its origin dates certainly back to an ancestral gnathostome, before the divergence of actinopterygians and sarcopterygians more than 450 million years ago (Kawasaki and Suzuki, 2011). In amniotes, MMP20 is an enamel specific protease. This gene is absent in every species lacking either teeth or enamel (e.g., in turtles, birds, and various mammals -Meredith et al., 2011, 2013).
In toothed mammals, MMP20 was identified first in porcine enamel (Bartlett et al., 1996; Moradian-Oldak et al., 1996) then characterized in humans (Llano et al., 1997). It is expressed both by ameloblasts and odontoblasts, and acts from the enamel secretory to the maturation stages through proteolysis of the enamel organic matrix, required for correct mineralization (Bègue-Kirn et al., 1998). MMP20 cleaves AMEL (Llano et al., 1997; Ryu et al., 1999; Nagano et al., 2009), the enamel matrix protein (EMP) representing more than 90% of the forming enamel matrix in mammals (Fincham et al., 1989). MMP20 also cleaves AMBN, and probably also ENAM, two EMPs with critical functions during enamel mineralization (Iwata et al., 2007; Chun et al., 2010).
Mutations in the MMP20 gene have been associated with autosomal recessive type 2 amelogenesis imperfecta (AI2A2, MIM #612529, ORPHA100033) also called hypomature AI. In affected patients, enamel displays a normal thickness but is pigmented and hypomineralized as demonstrated by the lack of radio-opacity contrast with dentin (Witkop, 1989).
To find mutations on MMP20 in French patients diagnosed for AI, we sequenced and identified several new homozygous or heterozygous missense mutations. To validate the potential role of these mutations, hence to predict sensitive positions in MMP20, we analyzed a large set of representative mammalian lineages sequences, covering over 150 million years of evolution. This type of evolutionary analysis has been shown to be an efficient method to validate and predict disease-associated missense mutations (Delgado et al., 2007; Al Hashimi et al., 2009; Bardet et al., 2010; Silvent et al., 2014). This method is termed phylomedicine (Kumar et al., 2011), which is complementary to existing genetic diagnosis.
The aims of the present study were to document MMP20 evolutionary analysis to pinpoint where newly identified mutations act in this evolutionary chart, hence identifying sensitive positions. Collectively this is an efficient tool to functionally validate MMP20 mutations identified to date.
Materials and Methods
Evolutionary Analysis
Data Set
Mammalian MMP20 sequences were extracted from public databases, NCBI [http://www.ncbi.nlm.nih.gov] and Ensembl [http://www.ensembl.org]. A total of 75 sequences representative of the main mammalian lineages (55 families distributed within 19 orders) were retained for our analyses (Supplementary Table 1). Identical sequences (such as species from the same genus) were not included in our dataset. MMP20 being enamel specific the sequences of species lacking either teeth or enamel (i.e., Xenarthra, Pholidota, Mysticeta, and Tubulidentata) were not included in our study because they display various mutations.
Among the selected MMP20 sequences only five were published in GenBank. Of the 70 other sequences, 65 were computer-predicted from sequenced genomes and six were obtained using Basic Local Alignment Search Tool (BLAST) of the whole genome shotgun (WGS) repository sequences in NCBI (Supplementary Table 1). The coding sequences were traduced into amino acid sequences and unpublished sequences were validated through alignment to published ones using Se-Al v.2.0a11 software [http://tree.bio.ed.ac.uk/software/seal]. The intron-exon boundaries were also carefully checked. The dataset of the 75 MMP20 sequences is available in Supplementary Data 1.
Our final alignment consisted of 483 positions, and no insertions were needed (Supplementary Figure 1). A few residues were missing in some, uncompletely sequenced genomic DNA (i.e., 726 nucleotides, nt, representing <0.7% of the data), and the corresponding positions were treated as “unknown data”. In addition, we aligned 21 nucleotides of the intronic region located on both sides of the exons from 12 MMP20 sequences representative of the main mammalian lineages, known to be important for correct intron splicing (Supplementary Table 2).
Analyses
The putative signal peptide sequence and its cleavage site were predicted using SignalP 3.0 server (http://www.cbs.dtu.dk/services/SignalP).
Single Likelihood Ancestor Counting (SLAC) analysis was performed using the Datamonkey webserver (http://www.datamonkey.org/; Delport et al., 2010) to identify amino acids subjected either to purifying or to positive selection, as previously described (Silvent et al., 2014). Biologically significant amino acids (i.e., site-specific selections) in MMP20 were identified in our alignment for the 483 positions and displayed on the human sequence. The analysis was performed according to the substitution preferences of amino acids, i.e., favoring property conservation (see Silvent et al., 2014). We defined three levels of selection throughout mammalian evolution (i.e., 180 Ma): conserved (i.e., unchanged residues), conservative (i.e., substituted residues having similar properties) and variable positions (i.e., substitution with various residues).
Mutation Analyses
Patients
The patients and their families were selected from the pool of patients participating in the French Ministry of Health National Program for Clinical Research, PHRC 2008 HUS (Strasbourg University Hospital) N°4266, Amelogenesis Imperfecta, AI (for further details see Gasse et al., 2013) and in the INTERREG IV Offensive Sciences A27 “Orodental manifestation of rare diseases” EU funded (ERDF) project.
These patients came to The Reference Centre for Orodental Manifestations of Rare Diseases (CRMR Strasbourg, France) or other affiliated Competence Centres (CCMR) for clinical diagnosis and management. Dentists specializing in rare diseases diagnosed amelogenesis imperfecta.
The oral phenotypes were documented using the D[4]/phenodent registry, a Diagnosing Dental Defects Database [see www.phenodent.org, to access assessment form], which is approved by CNIL (French National commission for informatics and liberty, number 908416). This clinical study is registered at https://clinicaltrials.gov: NCT01746121 and NCT02397824, and with the MESR (French Ministry of Higher Education and Research) Bioethics Commission as a biological collection “Orodental Manifestations of Rare Diseases” DC-2012-1677 within DC-2012-1002 and was acknowledged by the CPP (person protection committee) Est IV on the 11 Dec 2012.
Affected and unaffected family members gave informed written consents both for the D4/phenodent registry and for genetic analyses performed on the salivary samples included in the biological collection.
In this study, we selected patients from unrelated families suffering from non-syndromic AI, some of them displaying clinical diagnoses matching possible MMP20 mutations.
Analyses
Patients spit into an Oragene kit (Oragene DNA®, DNA Genotek, Canada) and genomic DNA was then isolated from saliva according to the manufacturer's protocol. We used previously defined primers (see Gasse et al., 2013). Mutational analysis was performed for the 10 exons of MMP20 including exon-intron boundaries. PCR products were sent to GATC Biotech for purification and sequencing in both directions in order to minimize sequencing artifacts. The sequences were aligned manually with the reference human MMP20 sequence NG_012151.1 using Se-Al v2.0a11 software.
When necessary the sequences were analyzed for splicing site prediction using the NetGene2 server (http://www.cbs.dtu.dk/services/NetGene2/) and MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html). The NetGene2 server is a service producing neural network predictions of splice sites in human genes (Hebsgaard et al., 1996) and MaxEntScan is based on the “Maximum Entropy Principle” and generalizes most previous probabilistic models of sequence motifs such as weight matrix models and inhomogeneous Markov models (Yeo and Burge, 2004).
SNPs known to date in the human MMP20 sequence were found at http://www.ncbi.nlm.nih.gov/snp and at https://www.ncbi.nlm.nih.gov/variation/tools/1000 genomes/(the 1000 genomes project).
Results and Discussion
Evolutionary Analysis of Functional Constraints in MMP20 Sequence
The alignment of the 75 MMP20 amino acid (aa) sequences indicated a highly conserved protein structure throughout more than 180 million years of mammalian evolution, a finding demonstrating the importance of many regions of this protease as found for the alkaline phosphatase, ALPL (Silvent et al., 2014). The 10 exons encoding the protein do not show insertions and only a limited number of rodent-specific deletions, in which seven MMP20 lack either one codon in exon 1 (Mus, Rattus, Cricetulus, Mesocricetus, Microtus, and Chinchilla) or two codons in exon 2 (Octodon). The MMP20 sequences were therefore mostly composed of 483 residues, from the methionine M1, encoded by exon 1, to the cysteine C483 encoded by exon 10 and preceding the stop codon (Supplementary Figure 1). At the first glance our alignment indicated that many positions and large domains were conserved throughout the sequence. This is particularly obvious in the large regions encoded by the 3' end of exon 2, by exons 4 and 5, by the 3′ region of exon 6, by most of exons 7 and 8, and by many positions of exons 9 and 10 (Supplementary Figure 1). The alignment of the 21 untranslated nucleotides on both sides of the coding exons indicates that the acceptor sites are either tag or cag, and the donor sites are mostly gta (with the exception of gtt for intron 1 and gtg for intron 9 and in a few other sequences; Supplementary Table 2). This finding is in accordance with the splice site consensus sequences for introns. The other positions are somewhat variable with the exception of the 21 nucleotides of the 5′ region of intron 4 that are unchanged in all species studied (i.e., for 180 MA). We do not know why this region was unchanged during evolution but its conservation suggests functionally important domains otherwise some nucleotide substitutions would have occurred at random. We question whether this intron region could be involved in a regulatory process or was previously encoding region.
The evolutionary analysis using SLAC selected several positions, notably in the N-terminal region, and, in contrast indicated that the MMP20 sequence is globally under strong purifying selection (Figure 1). The detailed analysis of each position confirmed the numerous fonctional or structural constraints acting on many amino acids along the sequence (Figure 2, Supplementary Figure 2). Out of the 483 amino acids composing the human MMP20 sequence, more than a half (324 aa, 67.08%) were identified as sensitive positions, i.e., that were either conserved (i.e., unchanged residues) (243 aa, 50.31%) or conservative (i.e., substituted with residues having similar properties) (81 aa, 16.77%) during 180 million years of mammalian evolution (Figure 2). In contrast, 159 positions (32.92%) were identified as variable (i.e., substituted with various aa). This large number of sensitive positions revealed by our analysis is similar to the values obtained for ALPL (Silvent et al., 2014). These unchanged, conservative and variable positions were reported on the human sequence, resulting in the chart of sensitive positions of human MMP20 (Figure 2, Supplementary Figure 2). We predict that any substitution of one of the 243 unchanged positions, or of one of the 81 conservative positions with a residue having a different property, would disrupt MMP20 function and would lead to enamel defects described as amelogenesis imperfecta in patients who unfortunately possess such a mutation in both DNA alleles (homozygous mutation) or an additional mutation in the other allele (counpound heterozygous mutation). Indeed, all missense mutations validated until now in various proteins were located always at conserved positions (Delgado et al., 2007; Kumar et al., 2011).
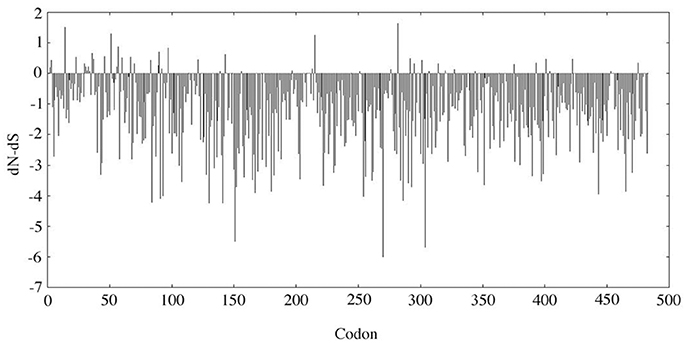
Figure 1. SLAC analysis. The dN-dS value was analyzed along the MMP20 codon sequence. dN, non-synonymous substitution rate; dS, synonymous substitution rate. When dN-dS < 0, the codon is subjected to purifying selection. When dN-dS > 0 (i.e., dN > dS), the codon is considered positively selected.
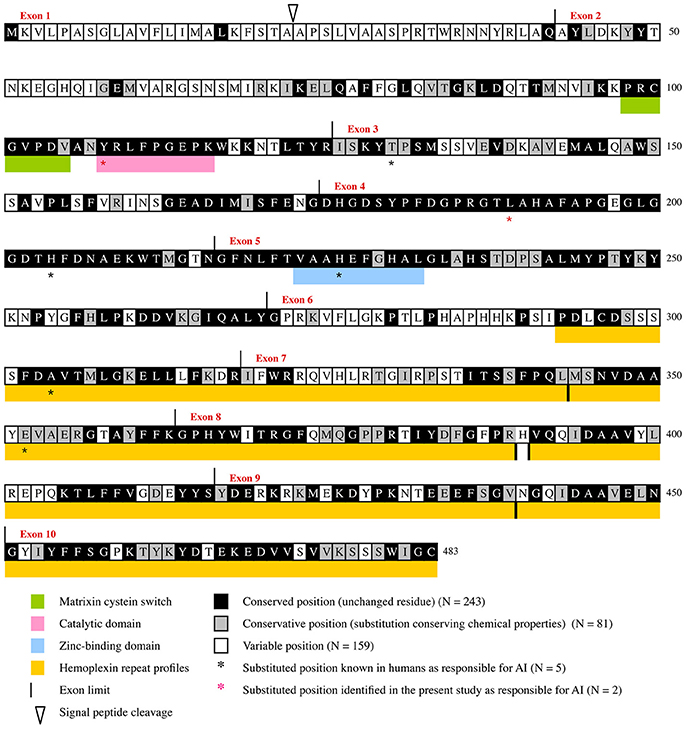
Figure 2. Evolutionary chart of MMP20. This chart was obtained from the alignment of 75 sequences representative of the main mammalian lineages (approx. 180 million years of evolution) and was deduced from the results obtained when dN/dS was calculated at each codon of MMP20 by Consurf (see Supplementary Figures 1, 2). Human sequence is used as a reference. Positions subjected to purifying selecton are on black (conserved positions) and gray (conservative positions) background. Variable positions on white background.
The boundaries of three, already known, functional domains are also better defined when considering the number of conserved amino acids: the matrixin cystein switch extends from aa98 to aa104, the catalytic domain from aa108 to aa116, and the zinc-binding domain from aa223 to aa232. Similar, accurate definition of the boundaries of functional domains was obtain in various proteins through evolutionary analysis (Al Hashimi et al., 2009; Silvent et al., 2013, 2014). Aside these crucial regions our study highlighted many other evolutionary-conserved residues and domains that have probably a strong functional or structural importance for MMP20. One of these large domains, encoded by the 3′ end of exon 3, exon 4, and most of exon 5, is composed of 109 residues, out of them 102 (93.58%) are either unchanged or conservative. Several, but shorter domains are also encoded by the 3′ region of exons 6 and 7, and by most of the sequence of exons 8–10 (Figure 2, Supplementary Figure 2). The only 70 aa located at the N-terminal region are subjected to low selective constraints with 15.71% of sensitive positions detected.
It is worth noting that the percentage of purifying selection is high along the protein sequence, and varies from 7.14% in the region encoded by exon 1–95.24% in the region encoded by exon 4. More precisely, our evolutionary analysis (i) confirmed and accurately defined the boundaries of already known important domains of the protein, (ii) highlighted many sensitive residues, and (iii) revealed various domains having putative important roles that should be experimentally studied in the future (Figure 2).
Mutation Analyses
Genotypes
Among our patients displaying non-syndromic AI, mutations in the MMP20 coding gene were diagnosed in six, unrelated families. Clinical diagnoses were confirmed through sequencing as described below. Sequencing DNA of patients 1 and 2 revealed new mutations in the MMP20 sequence, which are validated as being responsible for the AI phenotype by means of evolutionary analysis. The pedigrees and DNA sequencing chromatograms are presented in Figure 3. Moreover, patient 4 displayed two, already reported mutations, but in a new, compound heterozygous context. In patient 3, the compound heterozygous mutations were already described by Prasad et al. (2016a) but not illustrated (see below). Eventually, patients 5 and 6 possessed an already described homozygous mutation. In addition, in these six families we identified several SNPs that change the amino acid but these mutations are not validated by our evolutionary analysis as they occurred in variable positions.
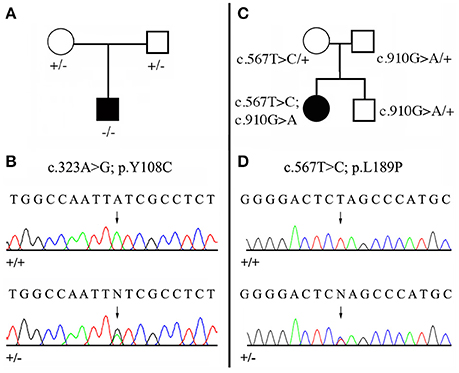
Figure 3. Mutational analysis of the two new MMP20 mutations. (A,C) Pedigree of the AI kindred; (B,D) DNA sequencing chromatograms of control (+/+) and of heterozygous (+/−) mutations. Arrows point to the mutation sites. See Figure 2, Supplementary Figure 1, Supplementary Table 2 for the validation of these mutations by means of evolutionary analyses. (A,B) Patient 1 with the homozygous mutation (c.323 A>G; p.Y108C). (C,D) Patient 2 with the compound heterozygous mutation (c.567 T>C; p.L189P / c.910 G>A; p.A304T). The second mutation was already reported as homozygous MMP20 mutation (Lee et al., 2010).
Patient 1: homozygous mutation c.323 A>G
We identified a missense, homozygous mutation in exon 2 of MMP20 of this male proband. The mutation was not previously reported and is referred to as g.8,470 A>G, c.323 A>G, p.Y108C. Both unaffected parents were heterozygous (Figures 3A,B). The mutation occurred at a tyrosine residue of the catalytic domain, a position that is unchanged in the 75 MMP20 mammalian sequences studied and surrounded by a number of conserved residues of this domain (Figure 2, Supplementary Figures 1, 2). This finding indicates a putative important function for this amino acid and validates this homozygous mutation as being responsible for the AI phenotype.
A second missense, homozygous mutation was identified in exon 1, p.K18T. This frequent mutation in the human population occurred in a variable region of MMP20 and is not responsible for the enamel disorder.
Patient 2: compound heterozygous mutation c.567 T>C and c.910 G>A
In this female proband, we identified two missense, heterozygous mutations that were validated as responsible for AI phenotype in a compound genotype. The mutation on allele 1 is located in exon 6 and referred to as g.18,755 G>A, c.910 G>A, p.A304T. This MMP20 mutation was already reported in the literature as responsible for the AI phenotype in a patient homozygous for the mutation (Lee et al., 2010). The patient 2 was heterozygous for this mutation, as well as his unaffected father and his brother (Figure 3C). The second mutation, on allele 2 was not previously reported and is located in exon 4 and referred to as g.15,345 T>C, c.567 T>C, p.L189P. The patient 2 was heterozygous for this mutation, as well as her unaffected mother (Figures 3C,D). The substitution of a leucine with a proline occurred at a conserved position, unchanged in mammals, and surrounded by numerous conserved residues. The substitution of this putative, functionally, or structurally important amino acid validates the mutation as involved in the AI phenotype.
In addition, the MMP20 sequence of patient 2 displayed four other missense mutations of various functional weight when considering our evolutionary analysis (Figure 2, Supplementary Figures 1, 2): (i) An uncommon missense mutation, on allele 2, was found in exon 3 and referred to as g.13,560 A>C, c.505 A>C, p.I169L. The mother is heterozygous for this mutation. The substitution of the isoleucine with a leucine occurred at a conservative position, at which isoleucine is substituted with the only valine in a few mammalian species but not with leucine (Figure 2, Supplementary Figures 1, 2). These amino acids have, however, similar properties. Also, this mutation occurred in a position surrounded by many conserved residues. The missense mutation p.I169L is present in 3% of the human population (the 1,000 genomes project). The involvement of this missense mutation in the AI phenotype is therefore quite doubtful; (ii) three, common, missense mutations were identified in exon 1, p.K18T (as in patient 1) and p.P31L, and in exon 6, p.T281N, also located in a variable region. These mutations are frequent in the human population occurred in variable regions of MMP20 and are not responsible for the enamel disorder.
The compound heterozygous mutation c.567 T>C (p.L189P) and c.910 G>A (p.A304T) is validated by our evolutionary analysis as the two mutations are located in conserved domains of MMP20, indicating putative important functions for these two amino acids.
Patient 3: compound heterozygous mutation c.126+6 t>g and c.954-2 a>t
The compound heterozygous mutation of MMP20 in intronic splicing sites identified in this male proband was recently reported in the literature (Prasad et al., 2016a) but was not detailed and not documented with pictures (see below).
The mutation on allele 1 is located at the splicing acceptor site of intron 6 (3′ splice site) and referred to as g.30574 a>t, c.954-2 a>t, p.I319fs338X. This MMP20 mutation disturbing the splice site consensus sequence for introns was already reported in the literature as responsible for the AI phenotype, but in a homozygous context (Kim et al., 2005). Patient 3 was heterozygous for the mutation, as well as his unaffected mother. The mutation on allele 2 is located at the splicing donor site of intron 1 (5′ splice site) and referred to as g.145 t>g, c.126+6 t>g. The male proband was heterozygous for the mutation as well as his unaffected father. This substitution did not occur during 180 million years of mammalian evolution (Supplementary Table 2) and this position is therefore considered of importance for correct intron 1 splicing. In addition, (i) the position +6 belongs to the splice site consensus sequence of the 5′ splice site for introns (A/CAG | gta/gagt), (ii) Netgen2 server predicted that the splice donor site in intron 1 does not exist in the mutant sequence and MaxEntScan analysis showed a reduced score of the splicing signal in the mutant compared to the wild sequence (Supplementary Data 2), and (iii) this mutation is not present in the human population (1,000 genomes project).
In addition, three missense mutations, common in the human population (p.K18T, V275A, and p.N281T) were also identified in this patient but are not responsible for the enamel disorder.
Patient 4: compound heterozygous mutation c.389 C>T and c.954-2 a>t<
We identified two already reported mutations in this male proband. These mutations were, however, described as homozygous MMP20 mutations in two, unrelated patients, and are reported here to occur as a compound heterozygous mutation in the same patient. The first mutation found on allele 1 and located in exon 3 is referred to as: g.13444 C>T, c.389 C>T, p.T130I (Gasse et al., 2013). Patient 4 was heterozygous for the mutation as well as his unaffected father. The second mutation on allele 2 occurred at the splicing acceptor site of intron 6 and was referred to as g.30574 a>t, c.954-2 a>t, p.I319fs338X (Kim et al., 2005). The male proband was heterozygous for the mutation as well as his unaffecte mother.
Patients 5 and 6: homozygous mutation c.954-2 a>t
Patient 5: The homozygous mutation at the splicing acceptor site of intron 6 identified also in patients 3 and 4 was identified in two sisters of a family, in which the mother, the father, two sisters and a brother were unaffected as heterozygous for thed mutation.
Patient 6: The same mutation was also found in two sisters of this family.
All patients sharing the c.954-2 a>t mutation were from unrelated families, a finding that could indicate a high frequency of this mutation in the human population in an heterozygous context.
Clinical Phenotypes
Here below is a brief description of the features for the six patients displaying non-syndromic hypomature AI and diagnosed as possessing mutations in the MMP20 coding gene (Figure 4).
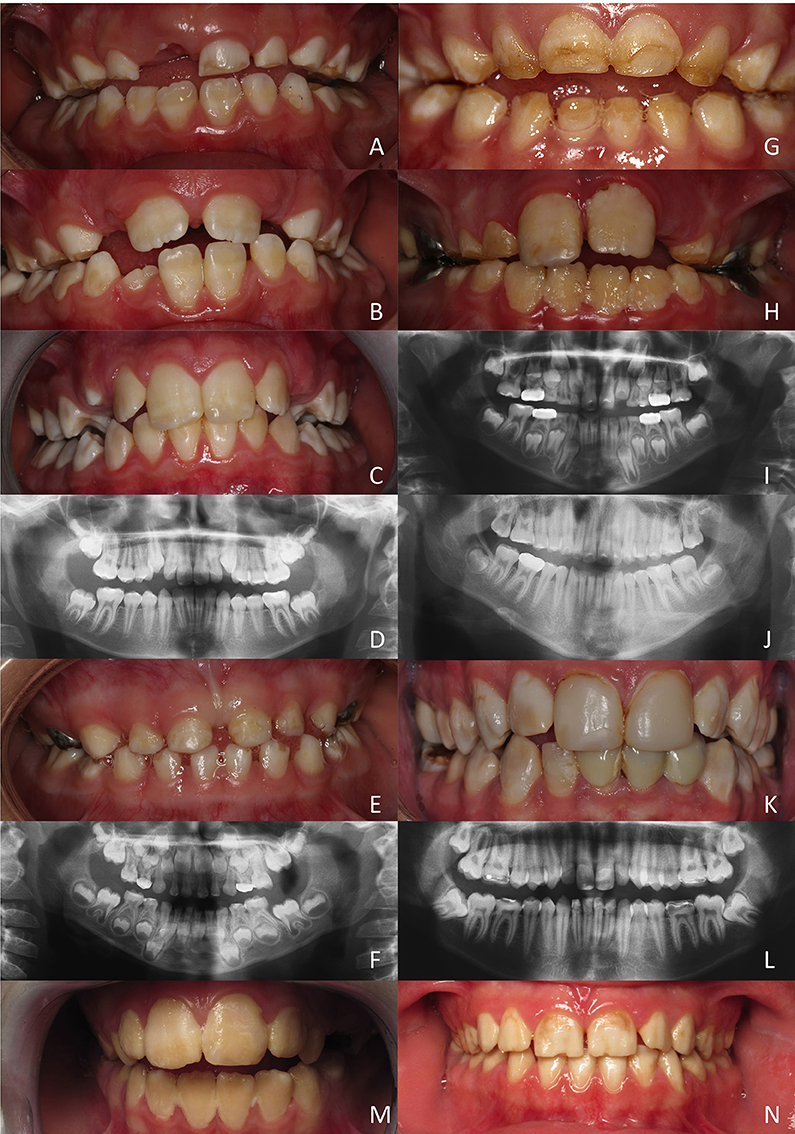
Figure 4. Hypomature amelogenesis imperfecta encountered in patients with diverse MMP20 mutations. (A–D): Patient 1 (c. 323 A>G). Intraoral clinical views and panoramic radiograph of primary, mixed (A: 6 years old; B: 7 years old) and permanent (C,D: 11 year old) dentitions. Note the limited contrast between enamel and dentine on X-rays. (E,F): Patient 2 (c.567 T>C + c.910 G>A). Primary dentition of a 5 year old girl. No enamel or very thin enamel was visible on X-rays. (G–J): Patient 3 (c.126+6 t>g + c.954-2 a>t). A young boy at 5 (G,I: primary teeth) and then at 8 years (H,J: permanent teeth). Limited radio-opaque enamel, if none, was seen on X-rays. (K, L): Patient 4 (c.389 C>T + c.954-2 a>t). Permanent teeth of a 20-year old man. Hypomature enamel is clearly visible on X-rays. (M,N): Patient 5 and patient 6 (c.954-2 a>t). Two girls displaying the same mutation leading to similar phenotypes with hypomature amelogenesis imperfecta.
Patient 1 (Figures 4A–D)
All teeth of this young boy were affected and parents reported damaged teeth since eruption. Enamel was chalky white and opaque. In primary teeth, enamel was either hypoplastic and/or was prematurely shed and worn through mastication and occlusal forces. The panoramic radiograph revealed the poor contrast of enamel compared to dentine, confirming the under-mineralization of enamel.
Patient 2 (Figures 4E,F)
This 5 years old girl was in her primary dentition. The parents reported that primary tooth eruption was delayed as no teeth were present at 1 year. As soon as teeth erupted they showed more opaque enamel and it crumbled. Teeth were small microdont and numerous diastema separated them. Enamel was white, orangy and wore off. On panoramic radiograph, no or very thin enamel was visible on primary teeth. In non-erupted permanent teeth, enamel seemed thicker and more mineralized with a stronger differential contrast with dentine, at least on the first permanent molar germs.
Patient 3 (Figures 4G–J)
In this 5 years old boy, the enamel of primary teeth was more opaque and was prone to disintegration, leaving areas of dentine apparent. In permanent teeth, enamel was also colored and opaque. Erupting teeth were sensitive and the patient experienced difficulties to brush teeth. Dental plaque and gingivitis were clearly visible. On panoramic radiographs limited radiopaque enamel if none was visible, and no contrast existed between enamel and dentine. This patient displayed a severe phenotype.
Patient 4 (Figures 4K,L)
In this 20 year old man, all permanent teeth demonstrated colored, opaque white brownish teeth with hypomature enamel. On the panoramic radiograph enamel is thin and the contrast with dentine is hardly visible.
Patient 5 and patient 6 (Figures 4M,N)
These two girls from unrelated families shared the same mutation and displayed similar phenotypes with colored hypomature amelogenesis imperfecta. The overall tooth contour was respected and enamel chipping was visible at the incisal edge.
Phenotype Comparison
Patient 1 (p.Y108C) was the least affected patient, while the most severe case was patient 3 displaying two mutations in splicing sites, leading to porous enamel and very sensitive teeth. Patient 2′s phenotype was slightly different as it presented additional quantitative defect associated to hypoplastic AI (smaller teeth). The enamel of patients 4, 5, and 6 showed similar mottled appearance of the enamel with more or less irregular staining. In patients 5 and 6 enamel looked rather opaque and uniformly colored.
The comparison of our case series with patients' phenotypes published in the literature led us to the following observations: Patient 1 phenotype was close to the one described for patient 1 by Gasse et al. (2013), characterized by a stronger contrast between enamel and dentine on X-rays. The enamel phenotype of patient 4 was similar to the affected individual described by Kim et al. (2005, Figure 1), to patient 2 described by Gasse et al. (2013), and to our patients 5 and 6. However, an anterior openbite was not present. Taurodontism was seen in molars especially in the upper permanent molars.
MMP20 Mutations Known to Date and Validation Using Evolutionary Analysis
When including the two new mutations reported in the present study, there are now 13 different MMP20 mutations leading to AI reported in the literature. Eight of them are simply missense mutations leading to the only substitution of an amino acid (Figures 2, 5). This finding demonstrates the crucial importance of these residues for the correct function of this protease. With the exception of p.Arg35Arg, which concerns the substitution of a nucleotide only (see discussion in Prasad et al., 2016a), the seven other residue substitutions in MMP20 are validated by our evolutionary analysis as occurring either on a conserved (unchanged residue: 5 cases) or on a conservative (2 cases) position.
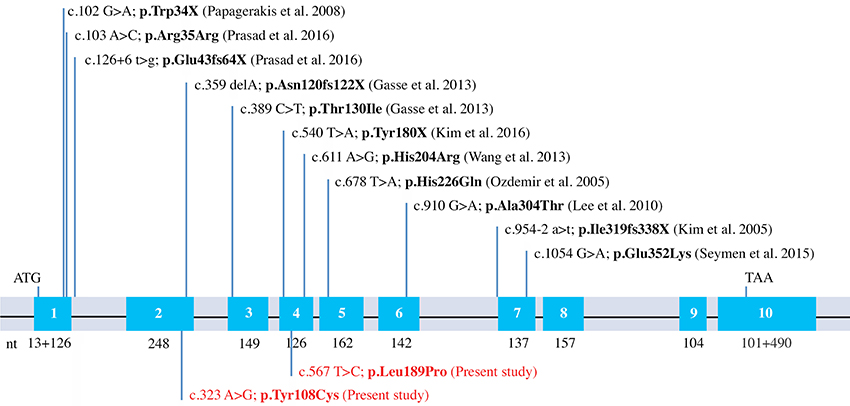
Figure 5. MMP20 mutations known to date. Schematical representation of the human MMP20 gene structure with indication of the previously reported mutations and the two new mutations identified in this study (in red).
To date, 18 other genes have been shown causing non-syndromic AI: AMELX (16 different mutations; Kim et al., 2017), FAM83H (14, Pourhashemi et al., 2014), WDR72 (10, Hentschel et al., 2016), ENAM (4; Pavlic et al., 2007; Seymen et al., 2014), ITGB6/4 (4; Poulter et al., 2014; Wang et al., 2014b), SLC24A4 (3; Wang et al., 2014b), LAMA3 (3, Gostyńska et al., 2016), GPR68 (3, Parry et al., 2016b), LTBP3 (3; Huckert et al., 2015), AMBN (2; Prasad et al., 2016a), KLK4 (2; Lu et al., 2008; Wang et al., 2013), DLX3 (2; Kim et al., 2016b), STIM1 (2; Wang et al., 2014b; Parry et al., 2016a), COL17A1 (1; Prasad et al., 2016b), C4orf26 (1; Prasad et al., 2016b), LAMB3 (1; Kim et al., 2016c), ACPT (1, Seymen et al., 2016), and AMTN (1, Smith et al., 2016). A total of 13 different mutations on the gene sequence causing AI places MMP20 among the top three sensitive proteins involved in non-syndromic AI when mutated.
Providing a clear genetic diagnosis linking genotype and phenotype on the basis of a missense variant can be challenging. A mutation present in a region that is highly conserved in evolution suggests that the amino acid is functionally important. The validated evolutionary analysis is crucial to address these important conserved positions and to facilitate variant analysis leading to disease diagnosis.
Author Contributions
BG, MP, SD, AB, and JS have substantially contributed to the conception, design of the work and interpretation of data for the work; MH, MK, AG, SL, IB, and MM have substantially contributed to the acquisition or analysis of data. BG, AB, and JS have drafted the work and MP, SD, MH, MK, AG, SL, IB, and MM revised it critically for important intellectual content. All authors finally approved the version to be published; they all agree to be accountable for all aspects of the work and ensure that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer EB and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
We would like to thank the family members for their invaluable contribution and generous help throughout the investigations. We are grateful to colleagues and general dental practitioners (Drs J. M. Schweitzer, A. Froehly, F. Charton, P. Basson) for their collaboration with the Strasbourg Reference Centre for orodental manifestations of rare diseases. This work was supported by grants from the French Ministry of Health (National Program for Clinical Research, PHRC 2008 N°4266 Amelogenesis imperfecta), the EU-funded project (ERDF) A27 “Oro-dental manifestations of rare diseases,” supported by the RMT-TMO Offensive Sciences initiative, INTERREG IV Upper Rhine program and the INTERREG V RARENET program, the grant ANR-10-LABX-0030-INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame programme Investissements d'Avenir labeled ANR-10-IDEX-0002-02. This research was funded by the University of Strasbourg Institute for Advanced Study (USIAS) as part of a USIAS Fellowship granted to AB. We would like to thank also Dr K. Niederreither for critical reading and English language editing of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fphys.2017.00398/full#supplementary-material
References
Al Hashimi, N., Sire, J. Y., and Delgado, S. (2009). Evolutionary analysis of mammalian enamelin, the largest enamel protein, supports a crucial role for the 32 kDa peptide and reveals selective adaptation in rodents and primates. J. Mol. Evol. 69, 635–656. doi: 10.1007/s00239-009-9302-x
Bardet, C., Delgado, S., and Sire, J. Y. (2010). MEPE evolution in mammals reveals regions and residues of prime functional importance. Cell. Mol. Life Sci. 67, 305–320. doi: 10.1007/s00018-009-0185-1
Bartlett, J. D., Simmer, J. P., Xue, J., Margolis, H. C., and Moreno, E. C. (1996). Molecular cloning and mRNA tissue distribution of a novel matrix metalloproteinase isolated from porcine enamel organ. Gene 183, 123–128. doi: 10.1016/S0378-1119(96)00525-2
Bègue-Kirn, C., Krebsbach, P. H., Bartlett, J. D., and Butler, W. T. (1998). Dentin sialoprotein, dentin phosphoprotein, enamelysin and ameloblastin: tooth-specific molecules that are distinctively expressed during murine dental differentiation. Eur. J. Oral Sci. 106, 963–970. doi: 10.1046/j.0909-8836.1998.eos106510.x
Bloch-Zupan, A., Sedano, H., and Scully, C. (2012). Dento/Oro/Craniofacial Anomalies and Genetics, 1st Edn. Boston, MA: Elsevier.
Chun, Y. H., Yamakoshi, Y., Yamakoshi, F., Fukae, M., Hu, J. C., Bartlett, J. D., et al. (2010). Cleavage site specificity of MMP-20 for secretory-stage ameloblastin. J. Dent. Res. 89, 785–790. doi: 10.1177/0022034510366903
Crawford, P. J., Aldred, M., and Bloch-Zupan, A. (2007). Amelogenesis imperfecta. Orphanet J. Rare Dis. 2:17. doi: 10.1186/1750-1172-2-17
Delgado, S., Ishiyama, M., and Sire, J. Y. (2007). Validation of amelogenesis imperfecta inferred from amelogenin evolution. J. Dent. Res. 86, 326–330. doi: 10.1177/154405910708600405
Delport, W., Scheffler, K., Botha, G., Gravenor, M. B., Muse, S. V., and Kosakovsky Pond, S. L. (2010). CodonTest: modeling amino acid substitution preferences in coding sequences. PLoS Comput. Biol. 6:e1000885. doi: 10.1371/journal.pcbi.1000885
Fanjul-Fernandez, M., Folgueras, A. R., Cabrera, S., and Lopez-Otin, C. (2010). Matrix metalloproteinases: evolution, gene regulation and functional analysis in mouse models. Biochim. Biophys. Acta 1803, 3–19. doi: 10.1016/j.bbamcr.2009.07.004
Fincham, A. G., Hu, Y., Pavlova, Z., Slavkin, H. C., and Snead, M. L. (1989). Human amelogenins: sequences of “TRAP” molecules. Calcif. Tissue Int. 45, 243–250. doi: 10.1007/BF02556044
Gasse, B., Karayigit, E., Mathieu, E., Jung, S., Garret, A., Huckert, M., et al. (2013). Homozygous and compound heterozygous MMP20 mutations in amelogenesis imperfecta. J. Dent. Res. 92, 598–603. doi: 10.1177/0022034513488393
Gomis-Ruth, F. X. (2009). Catalytic domain architecture of metzincin metalloproteases. J. Biol. Chem. 284, 15353–15357. doi: 10.1074/jbc.R800069200
Gostyńska, K. B., Yan Yuen, W., Pasmooij, A. M., Stellingsma, C., Pas, H. H., Lemmink, H., et al. (2016). Carriers with functional null mutations in LAMA3 have localized enamel abnormalities due to haploinsufficiency. Eur. J. Hum. Genet. 25, 94–99. doi: 10.1038/ejhg.2016.136
Hebsgaard, S. M., Korning, P. G., Tolstrup, N., Engelbrecht, J., Rouze, P., and Brunak, S. (1996). Splice site prediction in Arabidopsis thaliana DNA by combining local and global sequence information. Nucl. Acid Res. 24, 3439–3452. doi: 10.1093/nar/24.17.3439
Hentschel, J., Tatun, D., Parkhomchuk, D., Kurth, I., Schimmel, B., Heinrich-Weltzien, R., et al. (2016). Identification of the first multi-exonic WDR72 deletion in isolated amelogenesis imperfecta, and generation of a WDR72-specific copy number screening tool. Gene 590, 1–4. doi: 10.1016/j.gene.2016.05.040
Huckert, M., Stoetzel, C., Morkmued, S., Laugel-Haushalter, V., Geoffroy, V., Muller, J., et al. (2015). Mutations in the latent TGF-beta binding protein 3 (LTBP3) gene cause brachyolmia with amelogenesis imperfecta. Hum. Mol. Genet. 24, 3038–3049. doi: 10.1093/hmg/ddv053
Iwata, T., Yamakoshi, Y., Hu, J. C., Ishikawa, I., Bartlett, J. D., Krebsbach, P. H., et al. (2007). Processing of ameloblastin by MMP-20. J. Dent. Res. 86, 153–157. doi: 10.1177/154405910708600209
Kawasaki, K., and Suzuki, T. (2011). Molecular evolution of matrix metalloproteinase 20. Eur. J. Oral Sci. 119, 247–253. doi: 10.1111/j.1600-0722.2011.00898.x
Kim, J. W., Simmer, J. P., Hart, T. C., Hart, P. S., Ramaswami, M. D., Bartlett, J. D., et al. (2005). MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J. Med. Genet. 42, 271–275. doi: 10.1136/jmg.2004.024505
Kim, Y. J., Kang, J., Seymen, F., Koruyuku, M., Gencay, K., Hu, J. C., et al. (2016a). “MMP20 mutations causing enamel hypomaturation in two families,” in Enamel 9 Conference, (Harrogate, UK), Abstract.
Kim, Y. J., Seymen, F., Koruyucu, M., Kasimoglu, Y., Gencay, K., Shin, T. J., et al. (2016b). Unexpected identification of a recurrent mutation in the DLX3 gene causing amelogenesis imperfecta. Oral Dis. 22, 297–302. doi: 10.1111/odi.12439
Kim, Y. J., Shin, T. J., Hyun, H. K., Lee, S. H., Lee, Z. H., and Kim, J. W. (2016c). A novel de novo mutation in LAMB3 causes localized hypoplastic enamel in the molar region. Eur. J. Oral Sci. 124, 403–435. doi: 10.1111/eos.12280
Kim, Y. J., Kim, Y. J., Kang, J., Shin, T. J., Hyun, H. K., Lee, S. H., et al. (2017). A novel AMELX mutation causes hypoplastic amelogenesis imperfecta. Arch. Oral Biol. 76, 61–65. doi: 10.1016/j.archoralbio.2017.01.004
Kumar, S., Dudley, J. T., Filipski, A., and Liu, L. (2011). Phylomedicine: an evolutionary telescope to explore and diagnose the universe of disease mutations. Trends Genet. 27, 377–386. doi: 10.1016/j.tig.2011.06.004
Lee, S. K., Seymen, F., Kang, H. Y., Lee, K. E., Gencay, K., Tuna, B., et al. (2010). MMP20 hemopexin domain mutation in amelogenesis imperfecta. J. Dent. Res. 89, 46–50. doi: 10.1177/0022034509352844
Llano, E., Pendas, A. M., Knauper, V., Sorsa, T., Salo, T., Salido, E., et al. (1997). Identification and structural and functional characterization of human enamelysin (MMP-20). Biochemistry 36, 15101–15108. doi: 10.1021/bi972120y
Lu, Y., Papagerakis, P., Yamakoshi, Y., Hu, J. C., Bartlett, J. D., and Simmer, J. P. (2008). Functions of KLK4 and MMP-20 in dental enamel formation. Biol. Chem. 389, 695–700. doi: 10.1515/BC.2008.080
Meredith, R. W., Gatesy, J., Cheng, J., and Springer, M. S. (2011). Pseudogenization of the tooth gene enamelysin (MMP20) in the common ancestor of extant baleen whales. Proc. Biol. Sci. 278, 993–1002. doi: 10.1098/rspb.2010.1280
Meredith, R. W., Gatesy, J., and Springer, M. S. (2013). Molecular decay of enamel matrix protein genes in turtles and other edentulous amniotes. BMC Evol. Biol. 13:e20. doi: 10.1186/1471-2148-13-20
Moradian-Oldak, J., Leung, W., Simmer, J. P., Zeichner-David, M., and Fincham, A. G. (1996). Identification of a novel proteinase (ameloprotease-I) responsible for the complete degradation of amelogenin during enamel maturation. Biochem. J. 318, 1015–1021. doi: 10.1042/bj3181015
Nagano, T., Kakegawa, A., Yamakoshi, Y., Tsuchiya, S., Hu, J. C., Gomi, K., et al. (2009). Mmp-20 and Klk4 cleavage site preferences for amelogenin sequences. J. Dent. Res. 88, 823–828. doi: 10.1177/0022034509342694
Ozdemir, D., Hart, P. S., Ryu, O. H., Choi, S. J., Ozdemir-Karatas, M., Firatli, E., et al. (2005). MMP20 active-site mutation in hypomaturation amelogenesis imperfecta. J. Dent. Res. 84, 1031–1035. doi: 10.1177/154405910508401112
Papagerakis, P., Lin, H. K., Lee, K. Y., Hu, Y., Simmer, J. P., Bartlett, J. D., et al. (2008). Premature stop codon in MMP20 causing amelogenesis imperfecta. J. Dent. Res. 87, 56–59. doi: 10.1177/154405910808700109
Parry, D. A., Holmes, T. D., Gamper, N., El-Sayed, W., Hettiarachchi, N. T., Ahmed, M., et al. (2016a). A homozygous STIM1 mutation impairs store-operated calcium entry and natural killer cell effector function without clinical immunodeficiency. J. Allergy Clin. Immun. 137, 955–957. doi: 10.1016/j.jaci.2015.08.051
Parry, D. A., Smith, C. E., El-Sayed, W., Poulter, J. A., Shore, R. C., Logan, C. V., et al. (2016b). Mutations in the pH-sensing G-protein-coupled receptor GPR68 cause amelogenesis imperfecta. Am. J. Hum. Genet. 99, 984–990. doi: 10.1016/j.ajhg.2016.08.020
Pavlic, A., Petelin, M., and Battelino, T. (2007). Phenotype and enamel ultrastructure characteristics in patients with ENAM gene mutations g.13185-13186insAG and 8344delG. Arch. Oral Biol. 52, 209–217. doi: 10.1016/j.archoralbio.2006.10.010
Poulter, J. A., El-Sayed, W., Shore, R. C., Kirkham, J., Inglehearn, C. F., and Mighell, A. J. (2014). Whole-exome sequencing, without prior linkage, identifies a mutation in LAMB3 as a cause of dominant hypoplastic amelogenesis imperfecta. Eur. J. Hum. Genet. 22, 132–135. doi: 10.1038/ejhg.2013.76
Pourhashemi, S. J., Ghandehari Motlagh, M., Meighani, G., Ebrahimi Takaloo, A., Mansouri, M., Mohandes, F., et al. (2014). Missense mutation in Fam83H gene in Iranian patients with amelogenesis imperfecta. Iran J. Public Health 43, 1680–1687.
Prasad, M. K., Geoffroy, V., Vicaire, S., Jost, B., Dumas, M., Le Gras, S., et al. (2016a). A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. J. Med. Genet. 53, 98–110. doi: 10.1136/jmedgenet-2015-103302
Prasad, M. K., Laouina, S., El Alloussi, M., Dollfus, H., and Bloch-Zupan, A. (2016b). Amelogenesis Imperfecta: 1 family, 2 phenotypes, and 2 mutated genes. J. Dent. Res. 95, 1457–1463. doi: 10.1177/0022034516663200
Rodríguez, D., Morrison, C. J., and Overall, C. M. (2010). Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta 1803, 39–54. doi: 10.1016/j.bbamcr.2009.09.015
Ryu, O. H., Fincham, A. G., Hu, C. C., Zhang, C., Qian, Q., Bartlett, J. D., et al. (1999). Characterization of recombinant pig enamelysin activity and cleavage of recombinant pig and mouse amelogenins. J. Dent. Res. 78, 743–750. doi: 10.1177/00220345990780030601
Seymen, F., Kim, Y. J., Lee, Y. J., Kang, J., Kim, T. H., Choi, H., et al. (2016). Recessive mutations in ACPT, encoding testicular acid phosphatase, cause hypoplastic amelogenesis imperfecta. Am. J. Hum. Genet. 99, 1199–1205. doi: 10.1016/j.ajhg.2016.09.018
Seymen, F., Lee, K.-E., Koruyucu, M., Gencay, K., Bayram, M., Tuna, E. B., et al. (2014). ENAM mutations with incomplete penetrance. J. Dent. Res. 93, 988–992. doi: 10.1177/0022034514548222
Seymen, F., Park, J. C., Lee, K. E., Lee, H. K., Lee, D. S., Koruyucu, M., et al. (2015). Novel MMP20 and KLK4 mutations in amelogenesis imperfecta. J. Dent. Res. 94, 1063–1069. doi: 10.1177/0022034515590569
Silvent, J., Gasse, B., Mornet, E., and Sire, J. Y. (2014). Molecular evolution of the tissue-nonspecific alkaline phosphatase allows prediction and validation of missense mutations responsible for hypophosphatasia. J. Biol. Chem. 289, 24168–24179. doi: 10.1074/jbc.M114.576843
Silvent, J., Sire, J. Y., and Delgado, S. (2013). The dentin matrix acidic phosphoprotein 1 (DMP1) in the light of mammalian evolution. J. Mol. Evol. 76, 59–70. doi: 10.1007/s00239-013-9539-2
Smith, C. E., Murillo, G., Brookes, S. J., Poulter, J. A., Silva, S., Kirkham, J., et al. (2016). Deletion of amelotin exons 3-6 is associated with amelogenesis imperfecta. Hum. Mol. Genet. 25, 3578–3587. doi: 10.1093/hmg/ddw203
Wang, S., Choi, M., Richardson, A. S., Reid, B. M., Seymen, F., Yildirim, M., et al. (2014b). STIM1 and SLC24A4 are critical for enamel maturation. J. Dent. Res. 93, 94S–100S. doi: 10.1177/0022034514527971
Wang, S. K., Hu, Y., Simmer, J. P., Seymen, F., Estrella, N. M., Pal, S., et al. (2013). Novel KLK4 and MMP20 mutations discovered by whole-exome sequencing. J. Dent. Res. 92, 266–271. doi: 10.1177/0022034513475626
Witkop, C. J. (1989). Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J. Oral Pathol. 17, 547–553. doi: 10.1111/j.1600-0714.1988.tb01332.x
Keywords: MMP20, mutations, amelogenesis imperfecta, evolution, phenotype
Citation: Gasse B, Prasad M, Delgado S, Huckert M, Kawczynski M, Garret-Bernardin A, Lopez-Cazaux S, Bailleul-Forestier I, Manière M-C, Stoetzel C, Bloch-Zupan A and Sire J-Y (2017) Evolutionary Analysis Predicts Sensitive Positions of MMP20 and Validates Newly- and Previously-Identified MMP20 Mutations Causing Amelogenesis Imperfecta. Front. Physiol. 8:398. doi: 10.3389/fphys.2017.00398
Received: 10 March 2017; Accepted: 26 May 2017;
Published: 14 June 2017.
Edited by:
Alexandre Rezende Vieira, University of Pittsburgh, United StatesReviewed by:
Elia Beniash, University of Pittsburgh, United StatesRafaela Scariot De Moraes, Universidade Positivo, Brazil
Copyright © 2017 Gasse, Prasad, Delgado, Huckert, Kawczynski, Garret-Bernardin, Lopez-Cazaux, Bailleul-Forestier, Manière, Stoetzel, Bloch-Zupan and Sire. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Yves Sire, jean-yves.sire@upmc.fr