 Peter P. Jones
Peter P. Jones Niall MacQuaide
Niall MacQuaide William E. Louch
William E. Louch- 1Department of Physiology, School of Biomedical Sciences, University of Otago, Dunedin, New Zealand
- 2HeartOtago, University of Otago, Dunedin, New Zealand
- 3Institute of Cardiovascular Sciences, University of Glasgow, Glasgow, United Kingdom
- 4Clyde Biosciences, Glasgow, United Kingdom
- 5Institute for Experimental Medical Research, Oslo University Hospital, University of Oslo, Oslo, Norway
- 6KG Jebsen Center for Cardiac Research, University of Oslo, Oslo, Norway
Contraction of cardiomyocytes is dependent on sub-cellular structures called dyads, where invaginations of the surface membrane (t-tubules) form functional junctions with the sarcoplasmic reticulum (SR). Within each dyad, Ca2+ entry through t-tubular L-type Ca2+ channels (LTCCs) elicits Ca2+ release from closely apposed Ryanodine Receptors (RyRs) in the SR membrane. The efficiency of this process is dependent on the density and macroscale arrangement of dyads, but also on the nanoscale organization of LTCCs and RyRs within them. We presently review accumulating data demonstrating the remarkable plasticity of these structures. Dyads are known to form gradually during development, with progressive assembly of both t-tubules and junctional SR terminals, and precise trafficking of LTCCs and RyRs. While dyads can exhibit compensatory remodeling when required, dyadic degradation is believed to promote impaired contractility and arrythmogenesis in cardiac disease. Recent data indicate that this plasticity of dyadic structure/function is dependent on the regulatory proteins junctophilin-2, amphiphysin-2 (BIN1), and caveolin-3, which critically arrange dyadic membranes while stabilizing the position and activity of LTCCs and RyRs. Indeed, emerging evidence indicates that clustering of both channels enables “coupled gating”, implying that nanoscale localization and function are intimately linked, and may allow fine-tuning of LTCC-RyR crosstalk. We anticipate that improved understanding of dyadic plasticity will provide greater insight into the processes of cardiac compensation and decompensation, and new opportunities to target the basic mechanisms underlying heart disease.
Introduction: Dyadic Organization at the Macroscale and Nanoscale
In mammalian cardiac myocytes, contraction of the cell is elicited by a process known as excitation-contraction coupling. This process is initiated by electrical excitation during the cardiac action potential, which triggers the opening of voltage-gated L-type Ca2+ channels (LTCCs) present in both the surface membrane and within membrane invaginations called the transverse-axial tubule system (t-tubules). Ca2+ influx through LTCCs triggers release of Ca2+ from ryanodine receptors (RyRs) in the sarcoplasmic reticulum (SR), and contraction as this released Ca2+ binds to the myofilaments. Tight control of contractility thus requires efficient crosstalk between LTCCs and RyRs, which is afforded by close apposition of the sarcolemmal and SR membranes at junctions called dyads (Figure 1; Sun et al., 1995; Bers, 2001).
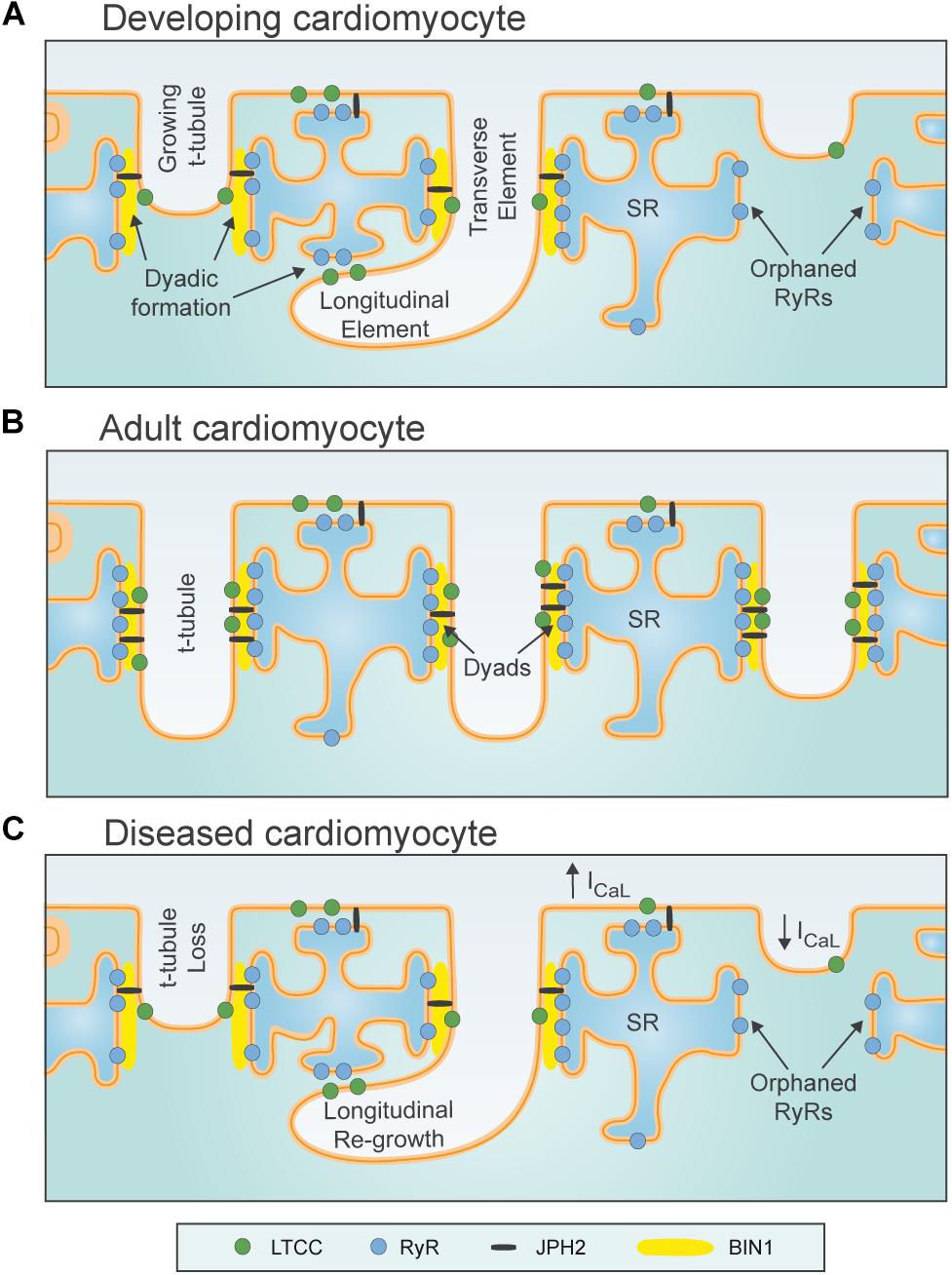
Figure 1. Plasticity of dyadic structure in ventricular cardiomyocytes. (A) Dyads form gradually in developing ventricular cardiomyocytes, as growing t-tubules extend the surface sarcolemma into the cell interior, initially in a largely longitudinal orientation. Rudimentary junctional SR terminals and contained ryanodine receptors (RyRs) are present in advance of t-tubule arrival. Formation of dyadic junctions between L-type Ca2+ channels (LTCCs) and RyRs requires the anchoring protein Junctophilin (JPH2), and the membrane sensing and bending protein BIN1. (B) Dyadic density increases toward adulthood, and assumes a predominantly transverse orientation. (C) During diseases such as heart failure, levels of JPH2 and BIN1 decline, and ventricular cardiomyocytes exhibit loss of t-tubules and SR. However, new dyads in the longitudinal orientation reappear, in resemblance to developing cells. T-tubule function also declines during heart failure, as L-type Ca2+ current (ICaL) is shifted to the surface sarcolemma.
Adult ventricular cardiomyocytes generally have a well-organized network of dyads, with transverse elements predominantly arranged along z-lines at the ends of each sarcomere (Fawcett and McNutt, 1969; Brette and Orchard, 2003; Song et al., 2005; Louch et al., 2010). However, longitudinal or axial dyads are also present at the level of the A-band (between z-lines), where they are oriented along the long axis of the cell (Asghari et al., 2009; Swift et al., 2012; Pinali et al., 2013). Smaller mammalian species with high heart rates such as mice and rats exhibit high densities of dyads in both orientations, while a less dense dyadic network with fewer longitudinal tubules is present in ventricular cardiomyocytes from larger species (Brette and Orchard, 2003; Song et al., 2005; Louch et al., 2010). Atrial cardiomyocytes generally exhibit a lower dyadic density than ventricular cells, although dyadic organization varies across the atria (Lenaerts et al., 2009; Smyrnias et al., 2010; Richards et al., 2011; Dibb et al., 2013; Frisk et al., 2014; Glukhov et al., 2015; Gadeberg et al., 2016; Arora et al., 2017).
Dyadic density and organization have considerable functional implications. A high density of dyads ensures that Ca2+ release occurs evenly across the cell, resulting in a rapid and co-ordinated rise in intracellular Ca2+ concentration ([Ca2+]i) and rapid contraction. Of note, findings from a range of species indicate that RyR organization has greater regularity than the t-tubule network, resulting in the presence of “orphaned” or non-junctional RyRs along z-lines which do not have colocalized t-tubules (Louch et al., 2006; Song et al., 2006; Heinzel et al., 2008). Ca2+ release at these orphaned RyRs is delayed, as it is dependent on the diffusion of Ca2+ released from nearby RyRs. Thus, greater dyssynchrony and slowing of Ca2+ release is promoted by conditions which trigger loss of t-tubules (and dyads) including hyperosmotic shock (Brette et al., 2004, 2005), cell culture (Lipp et al., 1996; Louch et al., 2004), and diseases such as heart failure (Louch et al., 2006; Song et al., 2006; Heinzel et al., 2008).
Beyond macroscale considerations of the local presence or absence of dyads, the nanoscale arrangement of proteins within dyads is also of key importance. Recent studies employing electron microscopy (EM) and super-resolution imaging have indicated that dyads are not completely filled with RyRs, but often contain multiple, smaller RyR clusters (Baddeley et al., 2009; Hayashi et al., 2009; Jayasinghe et al., 2018; Kolstad et al., 2018). These considerations are essential for understanding Ca2+ sparks, the fundamental units of Ca2+ release in cardiomyocytes (Cheng et al., 1993). On the t-tubule side of the dyad, LTCCs are arranged opposite from RyR clusters. Interestingly, recent data suggest that neighboring LTCCs may be clustered and functionally paired (Dixon et al., 2012, 2015), in a manner somewhat reminiscent of groupings of neighboring RyRs (Marx et al., 2001; Sobie et al., 2006; Cabra et al., 2016). The precise mechanisms by which individual or grouped LTCCs may be anchored and apposed from RyRs is unclear, although the dyadic anchor junctophilin-2 (JPH2) has been shown to interact with both proteins (Jiang et al., 2016; Munro et al., 2016; Reynolds et al., 2016). More clear is the role that JPH2 plays in setting a consistent and remarkably narrow dyadic cleft (12–15 nm) (Sun et al., 1995; Takeshima et al., 2000) required for efficient LTCC-RyR crosstalk.
The above discussion has illustrated that considerable progress has been made into understanding dyadic organization and function in healthy adult cardiomyocytes. However, accumulating data indicate that these structures also exhibit remarkable plasticity. Indeed, dyads are known to form gradually during development (Ziman et al., 2010; Louch et al., 2015), and to exhibit compensatory remodeling when required (Kemi et al., 2011; Swift et al., 2012). In contrast, dyadic degradation is widely described during cardiac disease, where it is believed to contribute to impaired contractility and arrythmogenesis (Guo et al., 2013; Orchard et al., 2013; Manfra et al., 2017). In the remainder of this review, we will summarize how such plasticity of dyadic structure/function is attained, with focus on macroscale changes in t-tubule and SR structure, as well as nanoscale regulation of LTCCs and RyRs. Particular attention will be given to an emerging understanding of the drivers of dyadic plasticity, and their potential targeting for novel therapies.
Macroscale Plasticity
T-Tubules During Development and Disease
In small rodents such as mice and rats, t-tubules form after birth, growing from the cell surface into the interior of the ventricular myocyte (Ziman et al., 2010; Louch et al., 2015; Mackova et al., 2017). Initially, this developing t-tubule network is rather disorganized in appearance, and oriented largely along the longitudinal axis of the cell (Figures 1A, 2A). With further maturation, t-tubule density increases and the network becomes predominantly transversely organized along z-lines; a process that continues until surprisingly late periods of adulthood (Ziman et al., 2010; Øyehaug et al., 2013; Louch et al., 2015; Mackova et al., 2017). Recent data indicate that sheep myocytes already exhibit t-tubules in utero (Munro and Soeller, 2016), supporting species-dependent differences in the time course of ventricular myocyte development. In either case, prenatal or postnatal t-tubule maturation coincides with expression of JPH2 (Ziman et al., 2010; Munro and Soeller, 2016), which critically forms dyads by anchoring MORN motifs in the t-tubular membrane to the junctional SR (Takeshima et al., 2000). Indeed, when JPH2 levels are reduced in mice, t-tubules either don’t appear or remain in an immature longitudinal configuration (Chen et al., 2013; Reynolds et al., 2013). Full knockout of JPH2 in mice results in embryonic mortality, consistent with a requirement of the protein to form dyads at the surface of the cell, in advance of t-tubule development (Takeshima et al., 2000; Franzini-Armstrong et al., 2005). The membrane sensing and bending protein Amphyphisin-2 (BIN1) is also reported to play a key role in t-tubule growth (Lee et al., 2002), and the intricate folding of the tubule inner membrane (Hong et al., 2014). Hong et al noted a particularly important role of a cardiac-specific isoform of BIN1 (isoform 13+17) which is capable of initiating t-tubule growth even in non-muscle cells (Hong et al., 2014). Assuming that BIN1 is essential for t-tubule development across a range of species, it is anticipated that this role is prominent at earlier stages in larger mammals which exhibit t-tubule development in utero.
Evidence of t-tubule plasticity is further supported by examinations of cardiac disease. A large number of studies have reported remodeling of t-tubules in left ventricular cardiomyocytes during heart failure with an array of etiologies, spanning myocardial infarction (Louch et al., 2006; Swift et al., 2008; Lyon et al., 2009; Biesmans et al., 2011; Chen et al., 2012; Wagner et al., 2012; Øyehaug et al., 2013; Frisk et al., 2016; Sanchez-Alonso et al., 2016; Figure 2B), aortic stenosis (Wei et al., 2010; Ibrahim et al., 2013; Pinali et al., 2013), tachycardia (He et al., 2001; Balijepalli et al., 2003), hypertension (Song et al., 2006; Singh et al., 2017), chronic ischemia (Heinzel et al., 2008), and diabetes (Stølen et al., 2009; Ward and Crossman, 2014). Despite the range of species and disease models employed in these studies, there is general agreement that overall t-tubule density is reduced, and commonly accompanied by a re-emergence of longitudinally-oriented tubules (Figures 1C, 2B). More detailed analyses have revealed t-tubular swelling in failing myocytes (Wagner et al., 2012; Pinali et al., 2013, 2017; Crossman et al., 2017; Figures 3A,B) and the appearance of abnormal t-tubule “sheets” which may result from fusion of neighboring tubules (Seidel et al., 2017a; Figures 3C,D). Similar changes in t-tubule organization have been observed during right ventricular failure (Xie et al., 2012; Caldwell et al., 2014) and in the atria during heart failure (Dibb et al., 2009) and atrial fibrillation (Lenaerts et al., 2009), suggesting that t-tubular remodeling may be endemic to a variety of cardiac pathologies across the chambers of the heart.
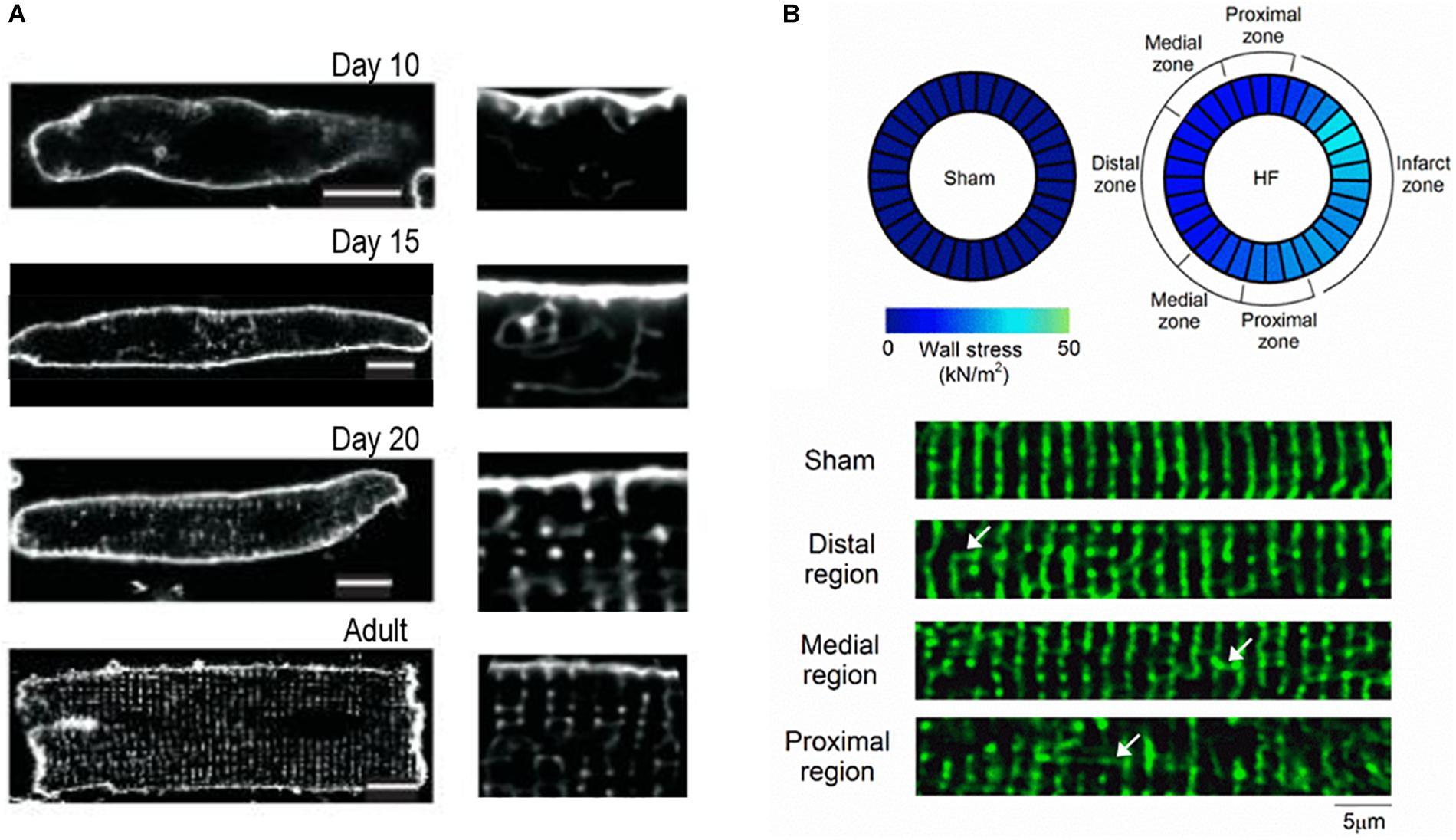
Figure 2. T-tubule plasticity during development and heart failure. (A) Confocal imaging of rat cardiomyocytes isolated at a range of post-natal time points reveals progressive t-tubule growth. T-tubules initially appear as a sparse network which is largely oriented in the longitudinal orientation, before the dense, predominantly transverse network is established in adulthood (whole cell images at left, with enlargements at right; adapted from Ziman et al. (2010); scale bar = 10 μm, copyright permission to reproduce the figure). (B) Typical t-tubule remodeling during heart failure exhibits a return to an immature phenotype, with loss of transverse elements and re-appearance of longitudinal elements (arrows). In a post-infarction rat model of heart failure, it was observed that remodeling is most marked proximal to the infarction scar, where in vivo wall stress is particularly elevated (adapted from Frisk et al. (2016), copyright permission to reproduce the figure). These data contribute to a growing understanding that high workload/wall stress signals t-tubule remodeling in this condition (reviewed in Ibrahim and Terracciano, 2013; Manfra et al., 2017).
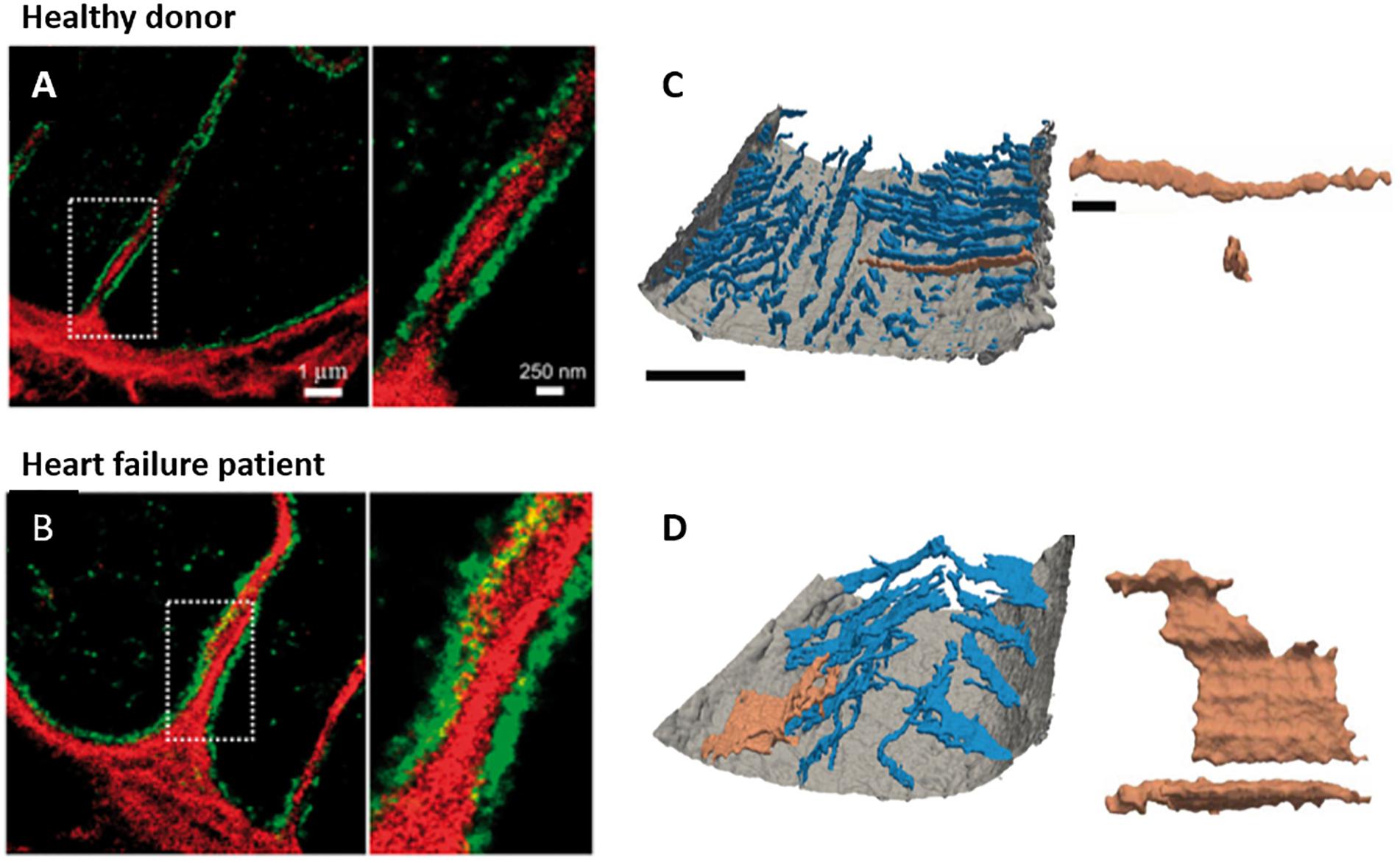
Figure 3. New insights into t-tubule remodeling during human heart failure. In comparison with ventricular tissue obtained from healthy donor hearts (A) tissue from heart failure patients undergoing transplant (B) revealed dilation of t-tubules associated with collagen deposition within the t-tubule lumen. Images were obtained with dSTORM super-resolution microscopy with staining for collagen VI (red) and dystrophin (green); enlargements of the indicated regions are shown at right (adapted from Crossman et al., 2017, copyright permission to reproduce the figure). Other recent work has indicated that in addition to t-tubule loss, cardiomyocytes in heart failure patients exhibit fusion of neighboring t-tubules into sheet-like structures (donor example in C, heart failure patient in D). 3D reconstructions illustrate the surface sarcolemma (gray) and t-tubules (blue), with the indicated tubule enlarged at right (longitudinal and transverse views; left scale bar = 10 μm, right scale bar = 2 μm; adapted from Seidel et al., 2017a, copyright permission to reproduce the figure).
Structural similarities between diseased and developing ventricular myocytes (Figure 1) have implied that pathological t-tubule remodeling may result from re-expression of fetal genes and/or suppression of adult genes in these conditions (Louch et al., 2015). Although the details of these mechanisms are still being elucidated, existing studies have already linked t-tubule remodeling in failing cells to declining expression of JPH2 (Minamisawa et al., 2004; Wei et al., 2010; Landstrom et al., 2011; Frisk et al., 2016), which is reminiscent of developing cells. An important role of JPH2 reduction in disease pathophysiology is supported by the observation that overexpression of this dyadic anchor protects against t-tubule degradation and heart failure development (Guo et al., 2014). Xu et al reported that JPH2 expression may be suppressed during disease by upregulation of microRNA-24 (miR-24), and showed that a miR-24 antagomir protected against changes in t-tubular architecture (Xu et al., 2012). Others have reported that JPH2 may be mislocalized in the failing heart, due to reorganization of microtubules necessary for its delivery to dyads (Zhang et al., 2014; Prins et al., 2016). JPH2 may also be degraded during heart failure by calpain cleavage. Guo et al. (2015) identified four distinct cut sites on JPH2 which resulted in functionally inactive fragments and disrupted dyadic junctions. Finally, recent data have suggested that JPH2’s functionality is dependent on its phosphorylation status. Quick et al. (2017) showed that JPH2 is phosphorylated by Striated Muscle Preferentially Expressed Protein Kinase (SPEG) and that this phosphorylation is reduced in heart failure, with knockout of SPEG also resulting in t-tubule disarray. These findings suggest that there may be several mechanisms which underlie loss of JPH2 expression and/or function during disease leading to pathologic disruption of t-tubule structure. Thus, JPH2 is an exciting potential candidate for future therapies aimed at preserving t-tubule integrity (Røe et al., 2015; Manfra et al., 2017).
BIN1 is another regulator of t-tubule structure which plays key roles in the developing and diseased heart. Indeed, just as BIN1 is believed crucial for the formation of t-tubules during development, so too has t-tubule loss and disarray during disease been linked to its downregulation (Lee et al., 2002; Caldwell et al., 2014; Hong et al., 2014). While the precise role of BIN1 in t-tubule growth and maintenance is unclear, it has been shown to group phosphoinositides allowing dynamin-2 polymerization; steps essential in tubulogenesis (Lee et al., 2002; Picas et al., 2014). Declining BIN1 levels in heart failure are reported to promote not only overall t-tubule loss (Caldwell et al., 2014), but also decreased t-tubule folding (Hong et al., 2014). Based on mathematical modeling studies, Hong et al. (2014) predicted that such loss of fine structure augments ion diffusion within the t-tubule, predisposing for cardiac arrhythmia. These authors have further proposed that continuous turnover of BIN1 from dyads in healthy patients maintains high levels in blood, explaining why decreased BIN1 plasma levels are linked to heart failure in patients and predict- future arrhythmia (Hong et al., 2012). These exciting data suggest that BIN1 may serve as both a biomarker and therapeutic target in heart failure patients.
While new molecular regulators such as JPH2 and BIN1 are emerging, recent work has also linked control of t-tubule structure to upstream mechanical signals. Experiments pioneered by the Terracciano group first indicated that t-tubule loss during heart failure may be directly triggered by the elevated ventricular workload in this condition. They observed that unloading failing hearts by heterotopic transplantation into healthy animals rescued t-tubule structure (Ibrahim et al., 2012a,b). Indeed, other strategies that unload the failing heart either pharmacologically (Chen et al., 2012; Xie et al., 2012; Huang et al., 2016) or via resynchronization therapy (Lichter et al., 2014), are similarly, protective. More recent work by our group indicated that elevated ventricular wall stress, which occurs in the dilated, thin-walled ventricle of the failing heart, may be the specific mechanical signal underlying t-tubule degradation (Frisk et al., 2016; Figure 2B).
How does mechanical overload lead to t-tubule degradation? While the precise mechanisms are unclear, it is important to consider that elevated workload and wall stress regulate not only cardiomyocyte remodeling but also promote changes in the extracellular matrix, including significant fibrosis. An exciting new study by Crossman et al., 2017; has shown striking localization of fibroblast filopodia and collagen within the t-tubular lumen in failing ventricular cardiomyocytes (Figures 3A,B). The authors suggested that such collagen deposition may directly drive t-tubular dilation in this condition, although it may also stiffen the t-tubule membrane, and impair normal mechanosignaling (McNary et al., 2012). Perhaps such changes mark a t-tubule for degradation (Louch and Nattel, 2017). In support of this view, regions of the failing heart with the most pronounced fibrosis, such as those proximal to an infarction, exhibit the most marked t-tubule loss (Frisk et al., 2016; Seidel et al., 2017b; Figure 2B). The Terracciano group has proposed that the stretch-sensitive protein titin cap (TCap) may play a key role in integrating these mechanical signals (Ibrahim and Terracciano, 2013; Ibrahim et al., 2013). With established binding proteins in the t-tubule membrane as well as partners in the cytoskeleton, TCap certainly appears to be well-positioned to serve such a function. Direct manipulation of the cytoskeleton has also been shown to regulate t-tubule structure, as cytoskeletal disruptors inhibit t-tubule loss during culture (Tian et al., 2012; Hodne et al., 2017). Recent data from the Song laboratory have further implicated protein kinase C activation as a critical determinant of cytoskeletal reorganization and t-tubule degradation (Guo et al., 2018). Taken together, these data raise the intriguing possibility that, by sensing local load, the t-tubule can regulate its own structure via signals transmitted from the extracellular matrix to the cytoskeleton.
Not all changes in t-tubule structure appear to be detrimental. At early stages of heart failure, longitudinal tubules appear before transverse elements have disappeared (Louch et al., 2006); changes which are suggested to be compensatory since additional Ca2+ influx at these sites supports the Ca2+ transient (Swift et al., 2012). However, a full understanding of the consequences of t-tubule dynamics for cardiomyocyte function requires detailed knowledge of SR structure, as well as the regulation of LTCCs and RyRs within these membranes. These topics are discussed in the following sections.
Plasticity of SR Structure
In comparison with t-tubule structure, SR structure has, in general, been less extensively studied. This is in part due to the fact that t-tubule structure is rather easily assessed by simple membrane staining and confocal microscopy; techniques which can be extended to 3D with relative ease. Direct staining and fluorescence imaging has not proven to effectively reveal SR structure, although junctional SR localization has been inferred from confocal immunostaining of RyRs (Bootman et al., 2006; Song et al., 2006; Swift et al., 2012) or calsequestrin (Terentyev et al., 2003). Greater detail has been provided by studies employing transmission EM, which revealed that the SR consists of a complex, branching network (Franzini-Armstrong, 1980). 3D structure has more recently been unveiled by serial block-face imaging with scanning EM, showing that the SR network is in fact contiguous between each t-tubule, is in regular contact with the surface membrane, and is variable between species (Pinali et al., 2013; Figure 4A). Importantly, the junctional SR forms dyads not only with the surface sarcolemma and along transversely-oriented t-tubules at z-lines, but also with longitudinally-oriented tubules within the A-band. EM studies have reported that these longitudinal dyads have similar dimensions to their transversely-oriented counterparts, suggesting that the two types of dyads may have similar functionality (Asghari et al., 2009; Pinali et al., 2013; Figures 4B,C).
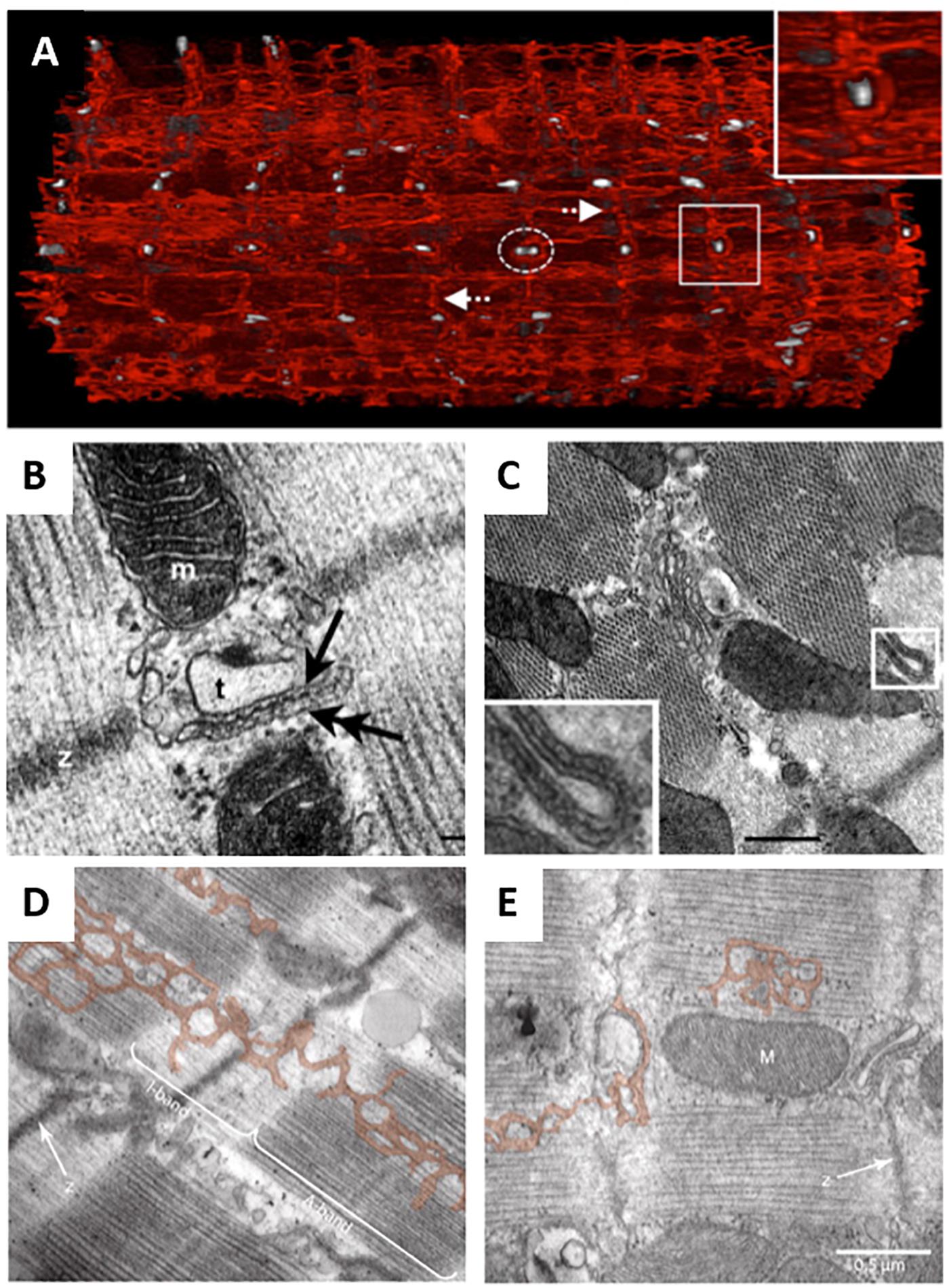
Figure 4. EM imaging of dyadic structure. (A) Block-face scanning EM performed on a sheep cardiomyocyte illustrates the complex, mesh-like nature of 3D SR structure (red), and it’s interrelationship with t-tubules (gray). Both longitudinal and perpendicular elements (arrows) are readily apparent, which converge to engulf t-tubules (enlarged region). Occasional twinning of t-tubules was observed with surrounding junctional SR (dashed elipsoid) (adapted from Pinali et al., 2013, copyright permission to reproduce the figure). Transmission EM imaging of transversely (B) and longitudinally-oriented dyads (C) revealed similar geometries, suggesting similar functionality of these structures (t, t-tubule; m, mitochondrion; double arrow, SR; scale bar in C = 100 nm). Ryanodine receptor heads are readily apparent (single arrow, adapted from Asghari et al., 2009, copyright permission to reproduce the figure). SR degradation during heart failure (Pinali et al., 2013) is suggested to be linked to reduction in SERCA levels, based on observations in the conditional SERCA knockout mouse (D, control cardiomyocyte; E, following SERCA knockout, with SR pseudo-colored; adapted from Swift et al., 2012, copyright permission was not required to reproduce the figure).
Accumulating data suggest that, like t-tubules, SR structure is also malleable. Transmission EM imaging of developing hearts has shown that the junctional SR forms dyads with the surface sarcolemma from early stages of embryonic ventricular development (Franzini-Armstrong et al., 2005). However, rudimentary junctional SR terminals (cisternae) appear at internal sites along z-lines during the late embryonic stage (Korhonen et al., 2010), and remain present during the neonatal period prior to the arrival of growing t-tubules (Ziman et al., 2010). Thus, wavelike Ca2+ release patterns are observed, traveling from the cell membrane toward the cell interior, as Ca2+ release propagates between as yet “orphaned” RyRs. The subsequent arrival of t-tubules and formation of internal dyads synchronizes Ca2+ release across the cell, and has been linked to the presence of the dyadic anchoring protein JPH2, as described above (Chen et al., 2013; Reynolds et al., 2013).
Restructuring of the SR is also apparent during disease. Pinali et al. (2013) reported an overall loss of SR in sheep following tachypacing-induced heart failure, with local patchiness and disorder of SR observed near sites of abnormal mitochondrial clustering. The implications of such remodeling are unclear but imply that there may be disruption of Ca2+ fluxes within the network SR in diseased cells. Results from the SERCA knockout mouse suggest that SR degradation may be driven directly by SERCA loss during heart failure (Swift et al., 2012; Figures 4D,E). However, despite evidence of overall SR loss during heart failure, most report that the junctional SR and associated RyRs remain present along z-lines, at least in a rudimentary arrangement (Song et al., 2006; Swift et al., 2008; Pinali et al., 2013; Frisk et al., 2016). Thus, there is an increased presence of orphaned RyRs in diseased cells reminiscent of the developing heart (Louch et al., 2015). With fewer RyR clusters served by a t-tubule, Ca2+ release becomes desynchronized, as uncoupled CRUs are recruited by diffusion (Louch et al., 2004, 2006; Song et al., 2006; Heinzel et al., 2008). The resulting overall slowing and reduced amplitude of systolic Ca2+ release has been linked to reduced cardiac output in this condition (Bøkenes et al., 2008; Mørk et al., 2009; Guo et al., 2013; Røe et al., 2015). At sites where the junctional SR remains coupled to t-tubules, Wu et al. (2012) reported that there is shortening of the interface in failing cells as the SR terminals are shortened. This implies that there is less available space for LTCCs and RyRs within the dyad, which may impair triggering of Ca2+ release beyond effects associated with loss of t-tubules and reduced Ca2+ release synchrony. Importantly, while there may be some loss of SR structure along z-lines, there appears to be growth or at least specialization of SR within the A-band, allowing the formation of dyads with newly-grown longitudinal t-tubules (Song et al., 2006; Swift et al., 2012). It is hypothesized that these new dyads somewhat counterbalance those lost along z-lines, at least at early stages of disease, to help maintain Ca2+ release (Swift et al., 2012).
The above discussion has illustrated that t-tubule and SR structure exhibit considerable plasticity, which enables a malleable arrangement of dyads important for controlling the synchrony of Ca2+ homeostasis. In the following section we will describe emerging data indicating that there is also impressive plasticity of LTCCs and RyRs within dyads, consistent with nanoscale control of dyadic function.
Nanoscale Plasticity
Plasticity of LTCC Localization and Function
Whilst much is known of how the LTCC localizes to the triad in skeletal muscle, targeting of LTCCs to the cardiac dyad is more poorly understood. In skeletal muscle, LTCC positioning appears to be stabilized by both the presence of STAC3 (Nelson et al., 2013; Polster et al., 2016; Campiglio and Flucher, 2017) and direct physical interaction between the II-III loop of the channel and the skeletal muscle ryanodine receptor (RyR1) (Lu et al., 1994; El-Hayek et al., 1995). These interactions enable the formation of a distinctive tetrad arrangement, with 4 LTCCs apposed from 4 RyRs (Franzini-Armstrong et al., 1998; Takekura et al., 2004). However, cardiac muscle does not express STAC3 or a homologous protein nor does the cardiac isoform of the LTCC have a physical interaction with RyR (c.f. Dulhunty et al., 2005), leading to the question of how the channel is targeted.
At both the surface of cardiomyocytes and within t-tubules, LTCCs are found resident within caveolae. The bulk of channel delivery to these caveloae appears to be dependent on BIN1, which couples the channels to microtubules (Hong et al., 2010). However, the actin filament cytoskeleton is also proposed to play a role in LTCC trafficking, at least in neurons and recombinant cell lines (Hall et al., 2013; Ghosh et al., 2018). Once delivered, LTCCs are maintained within the dyad via links between the caveolae and cytoskeleton (Head et al., 2006; Balijepalli and Kamp, 2008). Interestingly, BIN1 may also help maintain LTCC positioning, as BIN1-induced microfolds within the t-tubule membrane are suggested to prevent lateral movement of the caveolae (Basheer and Shaw, 2016). Further evidence of the importance of BIN1 for LTCC maintenance is provided by Hong et al. (2012), who showed that in human heart failure there is no change in LTCC expression but a significant reduction in dyadic channels. Such loss of dyadic LTCCs correlates well with a reduction in the expression of BIN1, both in failing patients (Hong et al., 2012) and sheep (Caldwell et al., 2014). Finally, evidence from skeletal muscle indicates that JPH2 may also play a role in maintaining LTCCs as part of a dyadic protein complex (Golini et al., 2011), suggesting that JPH2 reduction during heart failure could have complex effects on both LTCC localization and overall dyadic structure. The mechanism by which un-anchored LTCCs are degraded is presently unclear, although indirect evidence showing that dynasore increases surface LTCC expression indicates that channel internalization may occur via dynamin-dependent endocytosis (Hong et al., 2010).
While the above discussion has highlighted an important role of caveolae in clustering LTCCs within dyads, accumulating data suggest that these structural arrangements also critically regulate channel function. Recent studies have shown that Caveolin-3 (Cav-3), which is known to play an important role in the formation of caveolae and t-tubules (Parton et al., 1997), interacts with both LTCCs and protein-kinase A (PKA) to enable PKA-mediated phosphorylation of the channel and augmentation of L-type current (Kamp and Hell, 2000). Indeed, using peptide mimics of the scaffolding domain of Cav-3 to disrupt this interaction, Bryant et al. (2014) observed reduction in both basal L-type function and its response to β-adrenergic stimulation. More recently the same group has gone on to show that in heart failure the loss of t-tubular Cav-3 reduces t-tubular L-type current, despite the continued presence of L-type channels (Bryant et al., 2015, 2018; Figure 1C). Thus, while loss of t-tubules promotes dyssynchronous release in failing ventricular cardiomyocytes, as discussed above, it seems that there is also an important Cav-3-dependent loss of functionality in remaining tubules which further compromises Ca2+ release in this condition. Interestingly, the loss of t-tubular L-type current appears to be paralleled by increased current on the cell surface (Bryant et al., 2015; Sanchez-Alonso et al., 2016), likely explaining why many groups have reported unchanged overall current density in heart failure (Gomez et al., 1997; Benitah et al., 2002; Kamp and He, 2002; Mørk et al., 2009). The Gorelik group has proposed that increased LTCC activity on the surface sarcolemma results from physical movement of channels out of dyads and onto the membrane crests present between Z-grooves (Sanchez-Alonso et al., 2016). They further suggest that the delocalization-induced increase in channel activity promotes instability of myocyte membrane potential and a concomitant increase in arrhythmias often associated with heart failure (Sanchez-Alonso et al., 2016). These findings raise the possibility that nanoscale LTCC localization might be therapeutically targeted in disease.
Finally, exciting recent data indicate that LTCC activity is also critically regulated by the physical clustering of the channels themselves. Using single channel electrophysiology and optical channel recordings in neonatal myocytes, Navedo et al. (2010) found that clustered LTCCs open together more frequently than stochastic opening would predict. The same group went on to show in ventricular myocytes that this functional coupling of LTCCs occurs through the physical interaction of their C-terminal tails (Dixon et al., 2012, 2015). It is likely that such coupling ensures that Ca2+ influx is rapid and large enough to drive efficient RyR opening during Ca2+-induced Ca2+ release. In light of findings described above, it seems plausible that loss of t-tubular L-type current during heart failure (Bryant et al., 2015; Sanchez-Alonso et al., 2016) may, at least in part, result from loss of channel clustering due to downregulation of Cav-3, BIN1, and/or JPH2.
Plasticity of RyR Organization
Inter-Cluster RyR Dynamics
While it was traditionally believed that dyads in ventricular myocytes are uniformly packed with RyRs, more recent super-resolution microscopy studies have indicated that dyads are in fact composed of sub-clusters (Baddeley et al., 2009; Hayashi et al., 2009; Jayasinghe et al., 2018; Kolstad et al., 2018; Figures 5A,B). Neighboring RyR clusters with sufficiently short distances between them (<100 nm in Sobie et al., 2006; <150 nm in Macquaide et al., 2015) are suggested to concertively generate Ca2+ sparks, as released Ca2+ can effectively jump from one cluster to the next. These functional groupings have thus been termed “superclusters” or Ca2+ release units (CRUs) (Baddeley et al., 2009), and may provide opportunities to fine tune spark dynamics (Walker et al., 2014). EM data suggest that RyR clusters, and presumably CRUs, are assembled gradually during development, first at the cell surface and then within the cell interior (Franzini-Armstrong et al., 2005). This process may be reversed during disease, as emerging data from our laboratories indicate that RyR clusters are broken apart (Macquaide et al., 2015; Kolstad et al., 2018; Shen et al., 2018; Figures 5C,D, 6). We have specifically linked dispersion of RyR clusters and CRUs during post-infarction heart failure to slowing of Ca2+ spark kinetics, due to the time lag inherent as multiple clusters are sequentially activated (Louch et al., 2013; Kolstad et al., 2018; Shen et al., 2018). Slowing of Ca2+ sparks in these cells was additionally linked to de-synchronization and slowing of the overall Ca2+ transient (Louch et al., 2013; Kolstad et al., 2018). We observed similar fragmentation of RyR clusters and slowing of Ca2+ spark kinetics during atrial fibrillation (Macquaide et al., 2015). An associated increased fraction of RyRs located between Z-lines was further predicted to augment propagation of pro-arrhythmic Ca2+ waves. Thus, accumulating data indicate that RyR clusters exhibit marked plasticity of their organization and function, and that this malleability has important implications for pathophysiology.
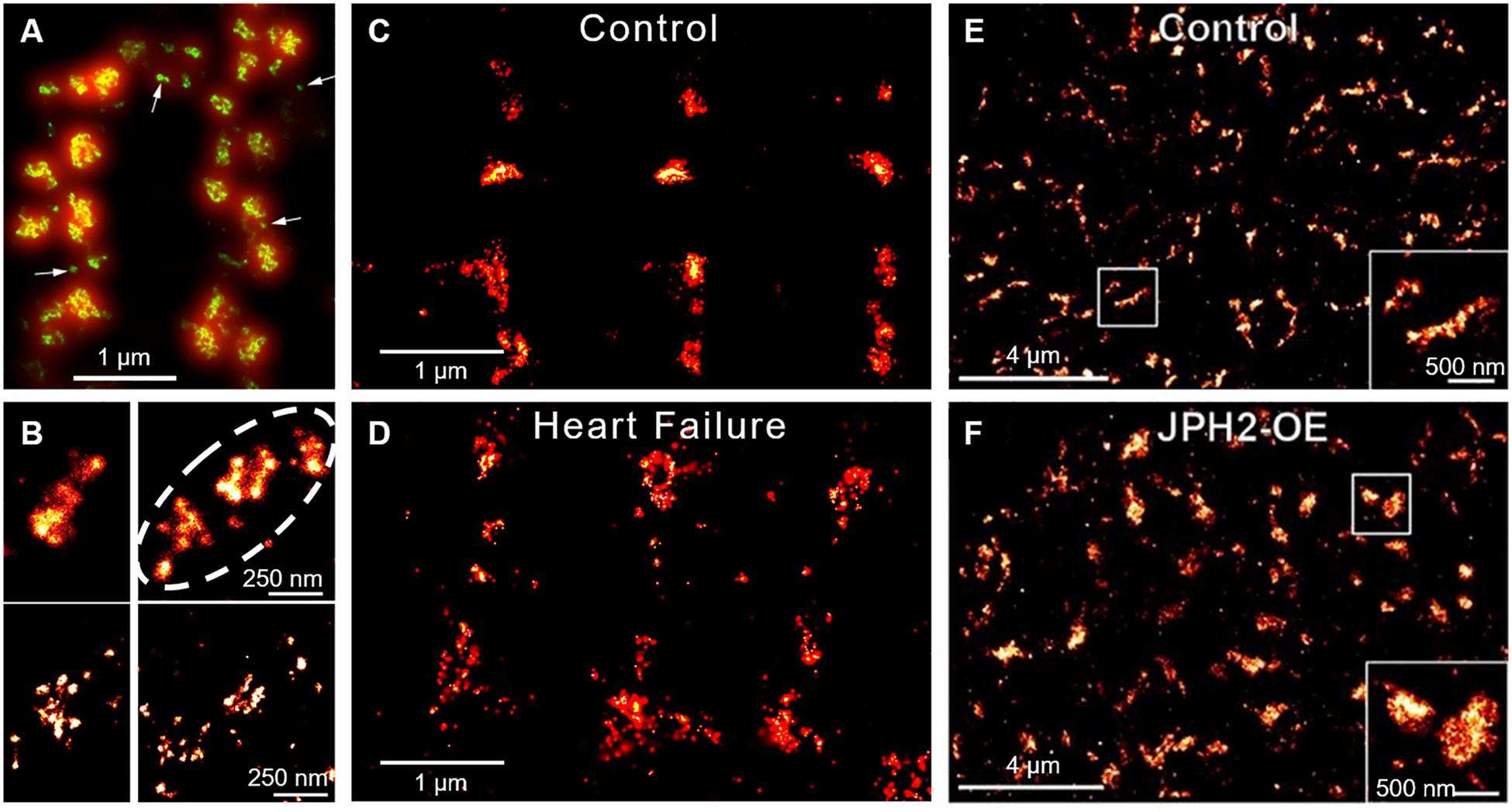
Figure 5. Super-resolution imaging of RyR clusters and plasticity. (A) RyRs on the cell surface of ventricular cardiomyocytes form clusters primarily along either side of the z-lines (double rows of RyRs). The limited resolution of conventional confocal imaging (shown red) is markedly improved using the dSTORM technique (green), which reveals the range of size and morphologies of RyR clusters. dSTORM also allows the visualization of smaller clusters and single RyRs (arrows, from Baddeley et al., 2009, copyright permission was not required to reproduce the figure). (B) Neighboring clusters can form superclusters or Ca2+ release units (CRUs) (dotted line; upper panel). DNA-PAINT reveals that RyRs within clusters are found in various orientations and groupings (lower panel, from Jayasinghe et al., 2018, copyright permission to reproduce the figure). (C,D) During heart failure, RyR clusters are broken apart, resulting in dispersed CRUs (from Kolstad et al., 2018, copyright permission was not required to reproduce the figure). (E,F) In contrast, RyR cluster size is increased in response to JPH2 overexpression (from Munro et al., 2016, copyright permission to reproduce the figure).
What is the timescale of RyR cluster dynamics? Some insight has been provided by a recent study employing direct visualization of RyR clusters using GFP-labeled RyR in live cells (Hiess et al., 2018). The authors observed that clusters close to the periphery of cells can undergo movement either laterally along the cell surface or down toward the interior. This movement altered cluster size as both cluster fusion and fission were observed. Remarkably, over a 12 min period movements of up to 1 μm occurred, which was sufficient for clusters to traverse half a sarcomere. Importantly, not all neighboring clusters repositioned, suggesting that cluster rearrangement was not an artifact due to SR movement (Hiess et al., 2018).
Importantly, inter-cluster dynamics appear to be controlled by external stimuli. Stimulatory conditions such as high Ca2+ were found to promote movement whereas conditions known to suppress RyR function such as low Ca2+ and tetracaine retarded their movement (Hiess et al., 2018). This raises the possibility that hyperactivity of the RyR, a hallmark of diseased cardiomyocytes (Bers, 2014; Dries et al., 2018), directly promotes its migration. However, it has been suggested that RyR localization is also dependent on associated dyadic proteins. BIN1 is reported to traffic RyRs to the t-tubule during β-adrenergic stimulation (Fu et al., 2016), with a time scale that appears to be consistent with the rate of RyR cluster movements reported by Hiess et al. (2018). JPH2 may also play a role in RyR arrangement, as overexpression of the protein was observed to augment RyR cluster size (Munro et al., 2016; Figures 5E,F). Interestingly, in these larger clusters JPH2 expression appears to stabilize RyR activity (van Oort et al., 2011; Beavers et al., 2013; Reynolds et al., 2016). These observations suggest that both dispersion of RyRs and increased channel activity during heart failure and atrial fibrillation could be linked to downregulation of BIN1 and JPH2 in these conditions.
Intra-Cluster RyR Dynamics
Although super-resolution imaging techniques such as dSTORM have yielded a wealth of information about RyR cluster size and distribution, their ability to examine intra-cluster architecture is limited. A recent breakthrough by the Soeller group employed the DNA-paint technique, which yielded <10 nm resolution (Jayasinghe et al., 2018). These measurements allowed the first visualization of single channels using optical means. Jayasinghe et al found that most clusters contained largely disordered channels with a relatively low packing density (Jayasinghe et al., 2018). This finding disagrees with historical assumptions of ventricular cardiomyocyte RyR packing based on EM descriptions of skeletal muscle, which showed a crystalline checkerboard arrangement of RyR1 (Ferguson et al., 1984; Franzini-Armstrong et al., 1999). More recent EM studies have suggested a somewhat structured arrangement with cardiac RyRs predominantly organized in a checkerboard pattern (∼50% of channels) but with many channels present in side-by-side or disordered arrangements (Asghari et al., 2014; Figure 6). Strikingly, as with clusters themselves, the orientation of channels within a cluster appears highly plastic. For example, exposure of cells to high Mg2+ conditions was observed to drive clusters into a side-by-side orientation, whereas low Mg2+ favored checkerboard. Most dramatically, however, was the finding that phosphorylated channels appeared almost exclusively in a checkerboard orientation (Asghari et al., 2014) (Figure 6). These findings suggest that the crystalline array favors greater RyR activity. More recently, the same group has refined their model with data indicating that it is phosphorylation of RyR at S2030 and S2814 that drives the change in cluster orientation (Asghari et al., 2017). Whether other established post-translational modifications of RyR such as oxidation (Waddell et al., 2016) or agonists such as caffeine (Jones et al., 2008) also result in similar changes in orientation remains unknown.
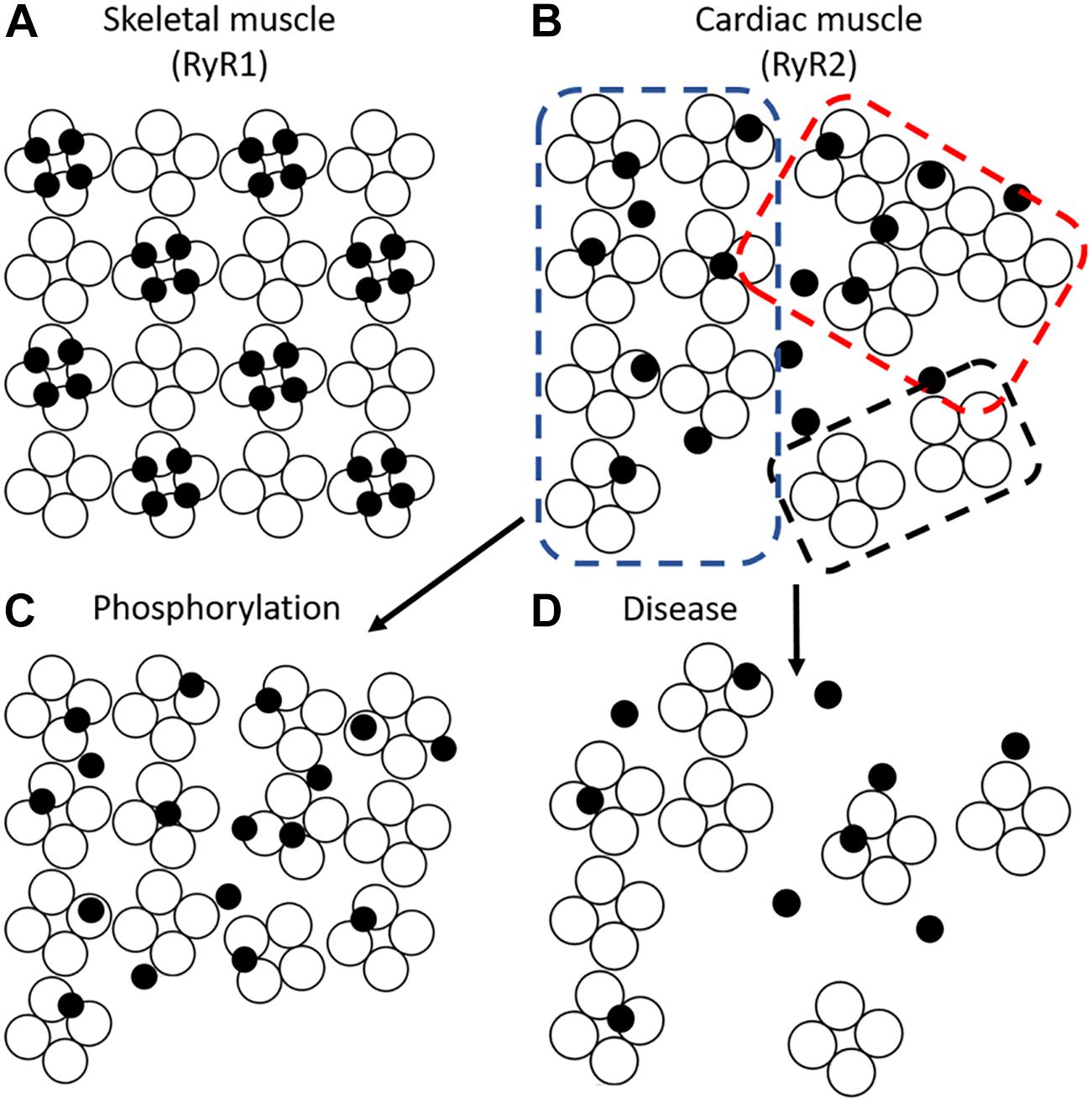
Figure 6. RyR cluster conformations. (A) Skeletal muscle RyRs (RyR1) form a regular crystalline array with 4 four LTCCs (filled circles) apposing alternate RyRs (open circles) to form tetrads. (B) Cardiac muscle RyRs (RyR2) interact in several conformations including crystalline array (blue dotted box), side-by-side (red dotted box), and disordered arrangements (black dotted box). Different stimuli modify the predominant conformation; phosphorylation promotes a crystalline array which may enhance the apposition of LTCCs and RyRs (C). Pathological conditions are reported to promote RyR cluster dispersion (Louch et al., 2013; Macquaide et al., 2015; Kolstad et al., 2018) and mislocalize t-tubules, thus, decreasing the coupling between LTCCs and RyRs (D).
Given that the clamp region of RyR is thought to enable assembly of the crystalline array, and that this region undergoes substantial movement during the transition of a channel to an open state, it is plausible that re-arrangement into the checkerboard pattern enhances coupled gating of channels (Cabra et al., 2016). Coupled gating has been previously proposed based on recordings of RyRs in bilayers, and implies that neighboring RyRs within a cluster exhibit synchronization of their opening and closing (Marx et al., 2001; Sobie et al., 2006). This mechanism has been suggested to be facilitated by FK506-binding protein (FKBP12.6), however, a role for this accessory protein in controlling RyR function remains controversial (Marx et al., 2000, 2001; Zhang et al., 2016; Gonano and Jones, 2017). Regardless of mechanism, an increase in coupled gating during transition to the checkerboard arrangement appears essential, as Walker et al. (2015) suggest that the increased intra-channel spacing in this configuration would otherwise reduce the likelihood of spark occurrence. However, the concept of coupled gating remains contentious as direct evidence linking cluster orientation and single channel activity is lacking (Williams et al., 2018).
In support of the work of the Moore group (Asghari et al., 2014), Samso and co-workers employed an alternative approach to examine RyR cluster formation, namely self-association in solution (Cabra et al., 2016). They found that akin to the patterns observed in cardiac muscle, in vitro RyRs adopt two major arrangements, namely side-by-side (further classified as center to side and adjoining) and checkerboard (further classified as oblique and center-to-corner). At low Ca2+ concentrations the channels were found to be relatively evenly distributed between the two conformations but, consistent with Asghari et al., stimulatory conditions (high Ca2+) favored an oblique and center-to-corner conformation, the basis of the crystalline array (Cabra et al., 2016). Given that stimulatory conditions such as high Ca2+ levels or phosphorylation appear to also increase both intra- and inter-cluster mobility, it would be of interest to examine the relative mobility of checkerboard and side-by-side orientated clusters in future studies.
Plasticity of LTCC-RyR Coupling
The preceding sections have illustrated that there is remarkable plasticity of not only the membranes of the dyad, but also their contained LTCCs and RyRs. The malleable activity of these proteins is mediated through direct changes to single channel function but also through the coordinated movement and organization of the channels which provides another layer of control. However, our knowledge of these phenomena is largely limited to LTCCs and RyRs individually; whether plasticity of the two proteins is coordinated, or indeed if the molecular stimuli are shared, remains less clear. The dynamic clustering observed in response to β-adrenergic stimulation of both channels might not only increase each channel’s activity but also hints at more effective coupling through optimization of LTCC and RyR apposition. Perhaps the phosphorylation-induced transition of RyR to a checkerboard conformation might, similarly, position LTCCs into an arrangement more similar to that found in skeletal muscle. Similarly, it is plausible that the loss of dyadic channels in disease not only reduces their own function but, due to an altered nano-structural arrangement, also results in a further reduction in Ca2+ coupling between the remaining channels. Since molecular players such as JPH2, BIN1, and Cav-3 control the localization of both LTCCs and RyRs, perhaps downregulation of these regulators critically reduces channel alignment in diseased cardiomyocytes. More indirect effects might also be important, as changes to the t-tubule architecture such as dilation or swelling (Wagner et al., 2012; Pinali et al., 2013, 2017; Crossman et al., 2017) could also “misalign” or otherwise disrupt the functional coupling between the channels. This form of remodeling has been hypothesized for more than 20 years, based on an observed reduction in the “gain” of Ca2+-induced Ca2+ release (Gomez et al., 1997; Litwin et al., 2000). However, it is only now with the host of recent technological advances, that we might examine this concept experimentally, by combining gain measurements with live-cell imaging of LTCCs and RyRs.
Conclusion
This review has presented a growing body of evidence illustrating that the concept of a static dyad severely underestimates the complexity of the structure. Rather, it is now fully apparent that there is remarkable plasticity of both t-tubule and SR structure, which enables dynamic dyad formation and degradation. Furthermore, it appears that within these structures there is likely continual regulation of both the positioning and activity of LTCCs and RyRs. This plasticity is postulated to augment dyadic Ca2+ cycling when required, but also to underlie impaired Ca2+ cycling during disease. Thus, greater understanding of dyadic plasticity holds considerable therapeutic potential.
Author Contributions
All authors contributed to the conceptualization, drafting, and editing of the manuscript.
Funding
This work was supported by the European Union’s Horizon 2020 Research and Innovation Programme (Consolidator grant, WEL) under grant agreement No. 647714. Additional support for WEL was provided by The South-Eastern Norway Regional Health Authority, Anders Jahre’s Fund for the Promotion of Science, The Norwegian Institute of Public Health, Oslo University Hospital Ullevål, and the University of Oslo. PPJ was supported by the Marsden Fund administered by the Royal Society of New Zealand (UOO1501) and the Health Research Council of New Zealand (18-232).
Conflict of Interest Statement
NM was employed by Clyde Biosciences (Glasgow, United Kingdom).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Arora, R., Aistrup, G. L., Supple, S., Frank, C., Singh, J., Tai, S., et al. (2017). Regional distribution of T-tubule density in left and right atria in dogs. Heart Rhythm 14, 273–281. doi: 10.1016/j.hrthm.2016.09.022
Asghari, P., Schulson, M., Scriven, D. R., Martens, G., and Moore, E. D. (2009). Axial tubules of rat ventricular myocytes form multiple junctions with the sarcoplasmic reticulum. Biophys. J. 96, 4651–4660. doi: 10.1016/j.bpj.2009.02.058
Asghari, P., Scriven, D. R., Sanatani, S., Gandhi, S. K., Campbell, A. I., and Moore, E. D. (2014). Nonuniform and variable arrangements of ryanodine receptors within mammalian ventricular couplons. Circ. Res. 115, 252–262. doi: 10.1161/CIRCRESAHA.115.303897
Asghari, P., Scriven, D. R., Zhao, Y., Mondragon, R. R., Valdivia, H., Wehrens, X. H., et al. (2017). RyR2 tetramer distributions in ventricular myocytes from phosphomutant mice. Biophys. J. 112:161a. doi: 10.1016/j.bpj.2016.11.886
Baddeley, D., Jayasinghe, I. D., Lam, L., Rossberger, S., Cannell, M. B., and Soeller, C. (2009). Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc. Natl. Acad. Sci. U.S.A. 106, 22275–22280. doi: 10.1073/pnas.0908971106
Balijepalli, R. C., and Kamp, T. J. (2008). Caveolae, ion channels and cardiac arrhythmias. Prog. Biophys. Mol. Biol. 98, 149–160. doi: 10.1016/j.pbiomolbio.2009.01.012
Balijepalli, R. C., Lokuta, A. J., Maertz, N. A., Buck, J. M., Haworth, R. A., Valdivia, H. H., et al. (2003). Depletion of T-tubules and specific subcellular changes in sarcolemmal proteins in tachycardia-induced heart failure. Cardiovasc. Res. 59, 67–77. doi: 10.1016/S0008-6363(03)00325-0
Basheer, W. A., and Shaw, R. M. (2016). Connexin 43 and CaV1.2 ion channel trafficking in healthy and diseased myocardium. Circ. Arrhythm. Electrophysiol. 9:e001357. doi: 10.1161/CIRCEP.115.001357
Beavers, D. L., Wang, W., Ather, S., Voigt, N., Garbino, A., Dixit, S. S., et al. (2013). Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J. Am. Coll. Cardiol. 62, 2010–2019. doi: 10.1016/j.jacc.2013.06.052
Benitah, J. P., Kerfant, B. G., Vassort, G., Richard, S., and Gomez, A. M. (2002). Altered communication between L-type calcium channels and ryanodine receptors in heart failure. Front. Biosci. 7, e263–e275. doi: 10.2741/A922
Bers, D. M. (2001). Excitation-Contraction Coupling and Cardiac Contractile Force, 2nd Edn. Dordrecht: Springer. doi: 10.1007/978-94-010-0658-3
Bers, D. M. (2014). Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 76, 107–127. doi: 10.1146/annurev-physiol-020911-153308
Biesmans, L., Macquaide, N., Heinzel, F. R., Bito, V., Smith, G. L., and Sipido, K. R. (2011). Subcellular heterogeneity of ryanodine receptor properties in ventricular myocytes with low T-tubule density. PLoS One 6:e25100. doi: 10.1371/journal.pone.0025100
Bøkenes, J., Aronsen, J. M., Birkeland, J. A., Henriksen, U. L., Louch, W. E., Sjaastad, I., et al. (2008). Slow contractions characterize failing rat hearts. Basic Res. Cardiol. 103, 328–344. doi: 10.1007/s00395-008-0719-y
Bootman, M. D., Higazi, D. R., Coombes, S., and Roderick, H. L. (2006). Calcium signalling during excitation-contraction coupling in mammalian atrial myocytes. J. Cell Sci. 119, 3915–3925. doi: 10.1242/jcs.03223
Brette, F., Despa, S., Bers, D. M., and Orchard, C. H. (2005). Spatiotemporal characteristics of SR Ca2+ uptake and release in detubulated rat ventricular myocytes. J. Mol. Cell. Cardiol. 39, 804–812. doi: 10.1016/j.yjmcc.2005.08.005
Brette, F., and Orchard, C. (2003). T-tubule function in mammalian cardiac myocytes. Circ. Res. 92, 1182–1192. doi: 10.1161/01.RES.0000074908.17214.FD
Brette, F., Rodriguez, P., Komukai, K., Colyer, J., and Orchard, C. H. (2004). beta-adrenergic stimulation restores the Ca transient of ventricular myocytes lacking t-tubules. J. Mol. Cell. Cardiol. 36, 265–275. doi: 10.1016/j.yjmcc.2003.11.002
Bryant, S., Kimura, T. E., Kong, C. H., Watson, J. J., Chase, A., Suleiman, M. S., et al. (2014). Stimulation of ICa by basal PKA activity is facilitated by caveolin-3 in cardiac ventricular myocytes. J. Mol. Cell. Cardiol. 68, 47–55. doi: 10.1016/j.yjmcc.2013.12.026
Bryant, S. M., Kong, C. H., Watson, J., Cannell, M. B., James, A. F., and Orchard, C. H. (2015). Altered distribution of ICa impairs Ca release at the t-tubules of ventricular myocytes from failing hearts. J. Mol. Cell. Cardiol. 86, 23–31. doi: 10.1016/j.yjmcc.2015.06.012
Bryant, S. M., Kong, C. H. T., Cannell, M. B., Orchard, C. H., and James, A. F. (2018). Loss of caveolin-3-dependent regulation of ICa in rat ventricular myocytes in heart failure. Am. J. Physiol. Heart Circ. Physiol. 314, H521–H529. doi: 10.1152/ajpheart.00458.2017
Cabra, V., Murayama, T., and Samso, M. (2016). Ultrastructural analysis of self-associated RyR2s. Biophys. J. 110, 2651–2662. doi: 10.1016/j.bpj.2016.05.013
Caldwell, J. L., Smith, C. E., Taylor, R. F., Kitmitto, A., Eisner, D. A., Dibb, K. M., et al. (2014). Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN-1). Circ. Res. 115, 986–996. doi: 10.1161/CIRCRESAHA.116.303448
Campiglio, M., and Flucher, B. E. (2017). STAC3 stably interacts through its C1 domain with CaV1.1 in skeletal muscle triads. Sci. Rep. 7:41003. doi: 10.1038/srep41003
Chen, B., Guo, A., Zhang, C., Chen, R., Zhu, Y., Hong, J., et al. (2013). Critical roles of junctophilin-2 in T-tubule and excitation-contraction coupling maturation during postnatal development. Cardiovasc. Res. 100, 54–62. doi: 10.1093/cvr/cvt180
Chen, B., Li, Y., Jiang, S., Xie, Y. P., Guo, A., Kutschke, W., et al. (2012). beta-Adrenergic receptor antagonists ameliorate myocyte T-tubule remodeling following myocardial infarction. FASEB J. 26, 2531–2537. doi: 10.1096/fj.11-199505
Cheng, H., Lederer, W. J., and Cannell, M. B. (1993). Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science 262, 740–744. doi: 10.1126/science.8235594
Crossman, D. J., Shen, X., Jullig, M., Munro, M., Hou, Y., Middleditch, M., et al. (2017). Increased collagen within the transverse tubules in human heart failure. Cardiovasc. Res. 113, 879–891. doi: 10.1093/cvr/cvx055
Dibb, K. M., Clarke, J. D., Eisner, D. A., Richards, M. A., and Trafford, A. W. (2013). A functional role for transverse (t-) tubules in the atria. J. Mol. Cell. Cardiol. 58, 84–91. doi: 10.1016/j.yjmcc.2012.11.001
Dibb, K. M., Clarke, J. D., Horn, M. A., Richards, M. A., Graham, H. K., Eisner, D. A., et al. (2009). Characterization of an extensive transverse tubular network in sheep atrial myocytes and its depletion in heart failure. Circ. Heart Fail. 2, 482–489. doi: 10.1161/CIRCHEARTFAILURE.109.852228
Dixon, R. E., Moreno, C. M., Yuan, C., Opitz-Araya, X., Binder, M. D., Navedo, M. F., et al. (2015). Graded Ca2+/calmodulin-dependent coupling of voltage-gated CaV1.2 channels. eLife 4:e05608. doi: 10.7554/eLife.05608
Dixon, R. E., Yuan, C., Cheng, E. P., Navedo, M. F., and Santana, L. F. (2012). Ca2+ signaling amplification by oligomerization of L-type Cav1.2 channels. Proc. Natl. Acad. Sci. U.S.A. 109, 1749–1754. doi: 10.1073/pnas.1116731109
Dries, E., Santiago, D. J., Gilbert, G., Lenaerts, I., Vandenberk, B., Nagaraju, C. K., et al. (2018). Hyperactive ryanodine receptors in human heart failure and ischemic cardiomyopathy reside outside of couplons. Cardiovasc. Res. 114, 1512–1524. doi: 10.1093/cvr/cvy088
Dulhunty, A. F., Karunasekara, Y., Curtis, S. M., Harvey, P. J., Board, P. G., and Casarotto, M. G. (2005). The recombinant dihydropyridine receptor II-III loop and partly structured ’C’ region peptides modify cardiac ryanodine receptor activity. Biochem. J. 385, 803–813. doi: 10.1042/BJ20041152
El-Hayek, R., Antoniu, B., Wang, J., Hamilton, S. L., and Ikemoto, N. (1995). Identification of calcium release-triggering and blocking regions of the II-III loop of the skeletal muscle dihydropyridine receptor. J. Biol. Chem. 270, 22116–22118. doi: 10.1074/jbc.270.38.22116
Fawcett, D. W., and McNutt, N. S. (1969). The ultrastructure of the cat myocardium. I. Ventricular papillary muscle. J. Cell Biol. 42, 1–45. doi: 10.1083/jcb.42.1.1
Ferguson, D. G., Schwartz, H. W., and Franzini-Armstrong, C. (1984). Subunit structure of junctional feet in triads of skeletal muscle: a freeze-drying, rotary-shadowing study. J. Cell Biol. 99, 1735–1742. doi: 10.1083/jcb.99.5.1735
Franzini-Armstrong, C., Protasi, F., and Ramesh, V. (1998). Comparative ultrastructure of Ca2+ release units in skeletal and cardiac muscle. Ann. N. Y. Acad. Sci. 853, 20–30. doi: 10.1111/j.1749-6632.1998.tb08253.x
Franzini-Armstrong, C., Protasi, F., and Ramesh, V. (1999). Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys. J. 77, 1528–1539. doi: 10.1016/S0006-3495(99)77000-1
Franzini-Armstrong, C., Protasi, F., and Tijskens, P. (2005). The assembly of calcium release units in cardiac muscle. Ann. N. Y. Acad. Sci. 1047, 76–85. doi: 10.1196/annals.1341.007
Frisk, M., Koivumaki, J. T., Norseng, P. A., Maleckar, M. M., Sejersted, O. M., and Louch, W. E. (2014). Variable t-tubule organization and Ca2+ homeostasis across the atria. Am. J. Physiol. Heart Circ. Physiol. 307, H609–H620. doi: 10.1152/ajpheart.00295.2014
Frisk, M., Ruud, M., Espe, E. K., Aronsen, J. M., Røe, A. T., Zhang, L., et al. (2016). Elevated ventricular wall stress disrupts cardiomyocyte t-tubule structure and calcium homeostasis. Cardiovasc. Res. 112, 443–451. doi: 10.1093/cvr/cvw111
Fu, Y., Shaw, S. A., Naami, R., Vuong, C. L., Basheer, W. A., Guo, X., et al. (2016). Isoproterenol promotes rapid ryanodine receptor movement to bridging integrator 1 (BIN1)-organized dyads. Circulation 133, 388–397. doi: 10.1161/CIRCULATIONAHA.115.018535
Gadeberg, H. C., Bond, R. C., Kong, C. H., Chanoit, G. P., Ascione, R., Cannell, M. B., et al. (2016). Heterogeneity of T-tubules in pig hearts. PLoS One 11:e0156862. doi: 10.1371/journal.pone.0156862
Ghosh, D., Nieves-Cintron, M., Tajada, S., Brust-Mascher, I., Horne, M. C., Hell, J. W., et al. (2018). Dynamic L-type CaV1.2 channel trafficking facilitates CaV1.2 clustering and cooperative gating. Biochim. Biophys. Acta 1865, 1341–1355. doi: 10.1016/j.bbamcr.2018.06.013
Glukhov, A. V., Balycheva, M., Sanchez-Alonso, J. L., Ilkan, Z., Alvarez-Laviada, A., Bhogal, N., et al. (2015). Direct evidence for microdomain-specific localization and remodeling of functional L-type calcium channels in rat and human atrial myocytes. Circulation 132, 2372–2384. doi: 10.1161/CIRCULATIONAHA.115.018131
Golini, L., Chouabe, C., Berthier, C., Cusimano, V., Fornaro, M., Bonvallet, R., et al. (2011). Junctophilin 1 and 2 proteins interact with the L-type Ca2+ channel dihydropyridine receptors (DHPRs) in skeletal muscle. J. Biol. Chem. 286, 43717–43725. doi: 10.1074/jbc.M111.292755
Gomez, A. M., Valdivia, H. H., Cheng, H., Lederer, M. R., Santana, L. F., Cannell, M. B., et al. (1997). Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science 276, 800–806. doi: 10.1126/science.276.5313.800
Gonano, L. A., and Jones, P. P. (2017). FK506-binding proteins 12 and 12.6 (FKBPs) as regulators of cardiac Ryanodine Receptors: insights from new functional and structural knowledge. Channels 11, 415–425. doi: 10.1080/19336950.2017.1344799
Guo, A., Chen, R., Wang, Y., Huang, C. K., Chen, B., Kutschke, W., et al. (2018). Transient activation of PKC results in long-lasting detrimental effects on systolic Ca2+i in cardiomyocytes by altering actin cytoskeletal dynamics and T-tubule integrity. J. Mol. Cell. Cardiol. 115, 104–114. doi: 10.1016/j.yjmcc.2018.01.003
Guo, A., Hall, D., Zhang, C., Peng, T., Miller, J. D., Kutschke, W., et al. (2015). Molecular determinants of calpain-dependent cleavage of junctophilin-2 protein in cardiomyocytes. J. Biol. Chem. 290, 17946–17955. doi: 10.1074/jbc.M115.652396
Guo, A., Zhang, C., Wei, S., Chen, B., and Song, L. S. (2013). Emerging mechanisms of T-tubule remodelling in heart failure. Cardiovasc. Res. 98, 204–215. doi: 10.1093/cvr/cvt020
Guo, A., Zhang, X., Iyer, V. R., Chen, B., Zhang, C., Kutschke, W. J., et al. (2014). Overexpression of junctophilin-2 does not enhance baseline function but attenuates heart failure development after cardiac stress. Proc. Natl. Acad. Sci. U.S.A. 111, 12240–12245. doi: 10.1073/pnas.1412729111
Hall, D. D., Dai, S., Tseng, P. Y., Malik, Z., Nguyen, M., Matt, L., et al. (2013). Competition between alpha-actinin and Ca2+-calmodulin controls surface retention of the L-type Ca2+ channel Ca(V)1.2. Neuron 78, 483–497. doi: 10.1016/j.neuron.2013.02.032
Hayashi, T., Martone, M. E., Yu, Z., Thor, A., Doi, M., Holst, M. J., et al. (2009). Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J. Cell Sci. 122, 1005–1013. doi: 10.1242/jcs.028175
He, J., Conklin, M. W., Foell, J. D., Wolff, M. R., Haworth, R. A., Coronado, R., et al. (2001). Reduction in density of transverse tubules and L-type Ca2+ channels in canine tachycardia-induced heart failure. Cardiovasc. Res. 49, 298–307. doi: 10.1016/S0008-6363(00)00256-X
Head, B. P., Patel, H. H., Roth, D. M., Murray, F., Swaney, J. S., Niesman, I. R., et al. (2006). Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. J. Biol. Chem. 281, 26391–26399. doi: 10.1074/jbc.M602577200
Heinzel, F. R., Bito, V., Biesmans, L., Wu, M., Detre, E., von, W. F., et al. (2008). Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ. Res. 102, 338–346. doi: 10.1161/CIRCRESAHA.107.160085
Hiess, F., Detampel, P., Nolla-Colomer, C., Vallmitjana, A., Ganguly, A., Amrein, M., et al. (2018). Dynamic and irregular distribution of RyR2 clusters in the periphery of live ventricular myocytes. Biophys. J. 114, 343–354. doi: 10.1016/j.bpj.2017.11.026
Hodne, K., Lipsett, D. B., and Louch, W. E. (2017). Gene transfer in isolated adult cardiomyocytes. Methods Mol. Biol. 1521, 169–182. doi: 10.1007/978-1-4939-6588-5_11
Hong, T., Yang, H., Zhang, S. S., Cho, H. C., Kalashnikova, M., Sun, B., et al. (2014). Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat. Med. 20, 624–632. doi: 10.1038/nm.3543
Hong, T. T., Cogswell, R., James, C. A., Kang, G., Pullinger, C. R., Malloy, M. J., et al. (2012). Plasma BIN1 correlates with heart failure and predicts arrhythmia in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 9, 961–967. doi: 10.1016/j.hrthm.2012.01.024
Hong, T. T., Smyth, J. W., Gao, D., Chu, K. Y., Vogan, J. M., Fong, T. S., et al. (2010). BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biol. 8:e1000312. doi: 10.1371/journal.pbio.1000312
Huang, C. K., Chen, B. Y., Guo, A., Chen, R., Zhu, Y. Q., Kutschke, W., et al. (2016). Sildenafil ameliorates left ventricular T-tubule remodeling in a pressure overload-induced murine heart failure model. Acta Pharmacol. Sin. 37, 473–482. doi: 10.1038/aps.2016.13
Ibrahim, M., Kukadia, P., Siedlecka, U., Cartledge, J. E., Navaratnarajah, M., and Tokar, S. (2012a). Cardiomyocyte Ca2+ handling and structure is regulated by degree and duration of mechanical load variation. J. Cell. Mol. Med. 16, 2910–2918. doi: 10.1111/j.1582-4934.2012.01611.x
Ibrahim, M., Navaratnarajah, M., Siedlecka, U., Rao, C., Dias, P., Moshkov, A. V., et al. (2012b). Mechanical unloading reverses transverse tubule remodelling and normalizes local Ca2+-induced Ca2+release in a rodent model of heart failure. Eur. J. Heart Fail. 14, 571–580. doi: 10.1093/eurjhf/hfs038
Ibrahim, M., Siedlecka, U., Buyandelger, B., Harada, M., Rao, C., Moshkov, A., et al. (2013). A critical role for Telethonin in regulating t-tubule structure and function in the mammalian heart. Hum. Mol. Genet. 22, 372–383. doi: 10.1093/hmg/dds434
Ibrahim, M., and Terracciano, C. M. (2013). Reversibility of T-tubule remodelling in heart failure: mechanical load as a dynamic regulator of the T-tubules. Cardiovasc. Res. 98, 225–232. doi: 10.1093/cvr/cvt016
Jayasinghe, I., Clowsley, A. H., Lin, R., Lutz, T., Harrison, C., Green, E., et al. (2018). True molecular scale visualization of variable clustering properties of ryanodine receptors. Cell Rep. 22, 557–567. doi: 10.1016/j.celrep.2017.12.045
Jiang, M., Zhang, M., Howren, M., Wang, Y., Tan, A., Balijepalli, R. C., et al. (2016). JPH-2 interacts with Ca2+i-handling proteins and ion channels in dyads: contribution to premature ventricular contraction-induced cardiomyopathy. Heart Rhythm 13, 743–752. doi: 10.1016/j.hrthm.2015.10.037
Jones, P. P., Meng, X., Xiao, B., Cai, S., Bolstad, J., Wagenknecht, T., et al. (2008). Localization of PKA phosphorylation site, Ser (2030), in the three-dimensional structure of cardiac ryanodine receptor. Biochem. J. 410, 261–270. doi: 10.1042/BJ20071257
Kamp, T. J., and He, J. Q. (2002). L-type Ca2+ channels gaining respect in heart failure. Circ. Res. 91, 451–453. doi: 10.1161/01.RES.0000035346.21625.4A
Kamp, T. J., and Hell, J. W. (2000). Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ. Res. 87, 1095–1102. doi: 10.1161/01.RES.87.12.1095
Kemi, O. J., Høydal, M. A., Macquaide, N., Haram, P. M., Koch, L. G., Britton, S. L., et al. (2011). The effect of exercise training on transverse tubules in normal, remodeled, and reverse remodeled hearts. J. Cell. Physiol. 226, 2235–2243. doi: 10.1002/jcp.22559
Kolstad, T. R., van den Brink, J., MacQuaide, N., Lunde, P. K., Frisk, M., Aronsen, J. M. et al. (2018). Ryanodine receptor dispersion disrupts Ca2+ release in failing cardiac myocytes. eLife 7: e39427. doi: 10.7554/eLife.39427
Korhonen, T., Rapila, R., Ronkainen, V. P., Koivumaki, J. T., and Tavi, P. (2010). Local Ca2+ releases enable rapid heart rates in developing cardiomyocytes. J. Physiol. 588, 1407–1417. doi: 10.1113/jphysiol.2009.185173
Landstrom, A. P., Kellen, C. A., Dixit, S. S., van Oort, R. J., Garbino, A., Weisleder, N., et al. (2011). Junctophilin-2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium-handling. Circ. Heart Fail. 4, 214–223. doi: 10.1161/CIRCHEARTFAILURE.110.958694
Lee, E., Marcucci, M., Daniell, L., Pypaert, M., Weisz, O. A., Ochoa, G. C., et al. (2002). Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science 297, 1193–1196. doi: 10.1126/science.1071362
Lenaerts, I., Bito, V., Heinzel, F. R., Driesen, R. B., Holemans, P., D’hooge, J., et al. (2009). Ultrastructural and functional remodeling of the coupling between Ca2+ influx and sarcoplasmic reticulum Ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circ. Res. 105, 876–885. doi: 10.1161/CIRCRESAHA.109.206276
Lichter, J. G., Carruth, E., Mitchell, C., Barth, A. S., Aiba, T., Kass, D. A., et al. (2014). Remodeling of the sarcomeric cytoskeleton in cardiac ventricular myocytes during heart failure and after cardiac resynchronization therapy. J. Mol. Cell. Cardiol. 72, 186–195. doi: 10.1016/j.yjmcc.2014.03.012
Lipp, P., Huser, J., Pott, L., and Niggli, E. (1996). Spatially non-uniform Ca2+ signals induced by the reduction of transverse tubules in citrate-loaded guinea-pig ventricular myocytes in culture. J. Physiol. 497, 589–597. doi: 10.1113/jphysiol.1996.sp021792
Litwin, S. E., Zhang, D., and Bridge, J. H. (2000). Dyssynchronous Ca2+ sparks in myocytes from infarcted hearts. Circ. Res. 87, 1040–1047. doi: 10.1161/01.RES.87.11.1040
Louch, W. E., Bito, V., Heinzel, F. R., Macianskiene, R., Vanhaecke, J., Flameng, W., et al. (2004). Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc. Res. 62, 63–73. doi: 10.1016/j.cardiores.2003.12.031
Louch, W. E., Hake, J., Mørk, H. K., Hougen, K., Skrbic, B., Ursu, D., et al. (2013). Slow Ca2+ sparks de-synchronize Ca2+ release in failing cardiomyocytes: evidence for altered configuration of Ca2+ release units? J. Mol. Cell. Cardiol. 58, 41–52. doi: 10.1016/j.yjmcc.2013.01.014
Louch, W. E., Koivumaki, J. T., and Tavi, P. (2015). Calcium signalling in developing cardiomyocytes: implications for model systems and disease. J. Physiol. 593, 1047–1063. doi: 10.1113/jphysiol.2014.274712
Louch, W. E., Mørk, H. K., Sexton, J., Stromme, T. A., Laake, P., Sjaastad, I., et al. (2006). T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J. Physiol. 574, 519–533. doi: 10.1113/jphysiol.2006.107227
Louch, W. E., and Nattel, S. (2017). T-tubular collagen: a new player in mechanosensing and disease? Cardiovasc. Res. 113, 839–840. doi: 10.1093/cvr/cvx091
Louch, W. E., Sejersted, O. M., and Swift, F. (2010). There goes the neighborhood: pathological alterations in T-tubule morphology and consequences for cardiomyocyte Ca2+ handling. J. Biomed. Biotechnol. 2010:503906. doi: 10.1155/2010/503906
Lu, X., Xu, L., and Meissner, G. (1994). Activation of the skeletal muscle calcium release channel by a cytoplasmic loop of the dihydropyridine receptor. J. Biol. Chem. 269, 6511–6516.
Lyon, A. R., MacLeod, K. T., Zhang, Y., Garcia, E., Kanda, G. K., Lab, M. J., et al. (2009). Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc. Natl. Acad. Sci. U.S.A. 106, 6854–6859. doi: 10.1073/pnas.0809777106
Mackova, K., Zahradnikova, A. Jr., Hotka, M., Hoffmannova, B., Zahradnik, I., and Zahradnikova, A. (2017). Calcium release-dependent inactivation precedes formation of the tubular system in developing rat cardiac myocytes. Eur. Biophys. J. 46, 691–703. doi: 10.1007/s00249-017-1249-z
Macquaide, N., Tuan, H. T., Hotta, J., Sempels, W., Lenaerts, I., Holemans, P., et al. (2015). Ryanodine receptor cluster fragmentation and redistribution in persistent atrial fibrillation enhance calcium release. Cardiovasc. Res. 108, 387–398. doi: 10.1093/cvr/cvv231
Manfra, O., Frisk, M., and Louch, W. E. (2017). Regulation of cardiomyocyte T-tubular structure: opportunities for therapy. Curr. Heart Fail. Rep. 14, 167–178. doi: 10.1007/s11897-017-0329-9
Marx, S. O., Gaburjakova, J., Gaburjakova, M., Henrikson, C., Ondrias, K., and Marks, A. R. (2001). Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ. Res. 88, 1151–1158. doi: 10.1161/hh1101.091268
Marx, S. O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N., et al. (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376. doi: 10.1016/S0092-8674(00)80847-8
McNary, T. G., Spitzer, K. W., Holloway, H., Bridge, J. H., Kohl, P., and Sachse, F. B. (2012). Mechanical modulation of the transverse tubular system of ventricular cardiomyocytes. Prog. Biophys. Mol. Biol. 110, 218–225. doi: 10.1016/j.pbiomolbio.2012.07.010
Minamisawa, S., Oshikawa, J., Takeshima, H., Hoshijima, M., Wang, Y., Chien, K. R., et al. (2004). Junctophilin type 2 is associated with caveolin-3 and is down-regulated in the hypertrophic and dilated cardiomyopathies. Biochem. Biophys. Res. Commun. 325, 852–856. doi: 10.1016/j.bbrc.2004.10.107
Mørk, H. K., Sjaastad, I., Sejersted, O. M., and Louch, W. E. (2009). Slowing of cardiomyocyte Ca2+ release and contraction during heart failure progression in postinfarction mice. Am. J. Physiol. Heart Circ. Physiol. 296, H1069–H1079. doi: 10.1152/ajpheart.01009.2008
Munro, M. L., Jayasinghe, I. D., Wang, Q., Quick, A., Wang, W., Baddeley, D., et al. (2016). Junctophilin-2 in the nanoscale organisation and functional signalling of ryanodine receptor clusters in cardiomyocytes. J. Cell Sci. 129, 4388–4398. doi: 10.1242/jcs.196873
Munro, M. L., and Soeller, C. (2016). Early transverse tubule development begins in utero in the sheep heart. J. Muscle Res. Cell Motil. 37, 195–202. doi: 10.1007/s10974-016-9462-4
Navedo, M. F., Cheng, E. P., Yuan, C., Votaw, S., Molkentin, J. D., Scott, J. D., et al. (2010). Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ. Res. 106, 748–756. doi: 10.1161/CIRCRESAHA.109.213363
Nelson, B. R., Wu, F., Liu, Y., Anderson, D. M., McAnally, J., Lin, W., et al. (2013). Skeletal muscle-specific T-tubule protein STAC3 mediates voltage-induced Ca2+ release and contractility. Proc. Natl. Acad. Sci. U.S.A. 110, 11881–11886. doi: 10.1073/pnas.1310571110
Orchard, C. H., Bryant, S. M., and James, A. F. (2013). Do t-tubules play a role in arrhythmogenesis in cardiac ventricular myocytes? J. Physiol. 591, 4141–4147. doi: 10.1113/jphysiol.2013.254540
Øyehaug, L., Loose, K. O., Jølle, G. F., Røe, A. T., Sjaastad, I., Christensen, G., et al. (2013). Synchrony of cardiomyocyte Ca2+ release is controlled by t-tubule organization, SR Ca2+ content, and ryanodine receptor Ca2+ sensitivity. Biophys. J. 104, 1685–1697. doi: 10.1016/j.bpj.2013.03.022
Parton, R. G., Way, M., Zorzi, N., and Stang, E. (1997). Caveolin-3 associates with developing T-tubules during muscle differentiation. J. Cell Biol. 136, 137–154. doi: 10.1083/jcb.136.1.137
Picas, L., Viaud, J., Schauer, K., Vanni, S., Hnia, K., Fraisier, V., et al. (2014). BIN1/M-Amphiphysin2 induces clustering of phosphoinositides to recruit its downstream partner dynamin. Nat. Commun. 5:5647. doi: 10.1038/ncomms6647
Pinali, C., Bennett, H., Davenport, J. B., Trafford, A. W., and Kitmitto, A. (2013). Three-dimensional reconstruction of cardiac sarcoplasmic reticulum reveals a continuous network linking transverse-tubules: this organization is perturbed in heart failure. Circ. Res. 113, 1219–1230. doi: 10.1161/CIRCRESAHA.113.301348
Pinali, C., Malik, N., Davenport, J. B., Allan, L. J., Murfitt, L., Iqbal, M. M., et al. (2017). Post-myocardial infarction T-tubules form enlarged branched structures with dysregulation of junctophilin-2 and bridging integrator 1 (BIN-1). J. Am. Heart Assoc. 6:e004834. doi: 10.1161/JAHA.116.004834
Polster, A., Nelson, B. R., Olson, E. N., and Beam, K. G. (2016). Stac3 has a direct role in skeletal muscle-type excitation-contraction coupling that is disrupted by a myopathy-causing mutation. Proc. Natl. Acad. Sci. U.S.A. 113, 10986–10991. doi: 10.1073/pnas.1612441113
Prins, K. W., Asp, M. L., Zhang, H., Wang, W., and Metzger, J. M. (2016). Microtubule-mediated misregulation of junctophilin-2 underlies T-tubule disruptions and calcium mishandling in mdx mice. JACC Basic Transl. Sci. 1, 122–130. doi: 10.1016/j.jacbts.2016.02.002
Quick, A. P., Wang, Q., Philippen, L. E., Barreto-Torres, G., Chiang, D. Y., Beavers, D., et al. (2017). SPEG (Striated Muscle Preferentially Expressed Protein Kinase) is essential for cardiac function by regulating junctional membrane complex activity. Circ. Res. 120, 110–119. doi: 10.1161/CIRCRESAHA.116.309977
Reynolds, J. O., Chiang, D. Y., Wang, W., Beavers, D. L., Dixit, S. S., Skapura, D. G., et al. (2013). Junctophilin-2 is necessary for T-tubule maturation during mouse heart development. Cardiovasc. Res. 100, 44–53. doi: 10.1093/cvr/cvt133
Reynolds, J. O., Quick, A. P., Wang, Q., Beavers, D. L., Philippen, L. E., Showell, J., et al. (2016). Junctophilin-2 gene therapy rescues heart failure by normalizing RyR2-mediated Ca2+ release. Int. J. Cardiol. 225, 371–380. doi: 10.1016/j.ijcard.2016.10.021
Richards, M. A., Clarke, J. D., Saravanan, P., Voigt, N., Dobrev, D., Eisner, D. A., et al. (2011). Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am. J. Physiol. Heart Circ. Physiol. 301, H1996–H2005. doi: 10.1152/ajpheart.00284.2011
Røe, A. T., Frisk, M., and Louch, W. E. (2015). Targeting cardiomyocyte Ca2+ homeostasis in heart failure. Curr. Pharm. Des. 21, 431–448. doi: 10.2174/138161282104141204124129
Sanchez-Alonso, J. L., Bhargava, A., O’Hara, T., Glukhov, A. V., Schobesberger, S., Bhogal, N., et al. (2016). Microdomain-specific modulation of l-type calcium channels leads to triggered ventricular arrhythmia in heart failure. Circ. Res. 119, 944–955. doi: 10.1161/CIRCRESAHA.116.308698
Seidel, T., Navankasattusas, S., Ahmad, A., Diakos, N. A., Xu, W. D., Tristani-Firouzi, M., et al. (2017a). Sheet-like remodeling of the transverse tubular system in human heart failure impairs excitation-contraction coupling and functional recovery by mechanical unloading. Circulation 135, 1632–1645. doi: 10.1161/CIRCULATIONAHA.116.024470
Seidel, T., Sankarankutty, A. C., and Sachse, F. B. (2017b). Remodeling of the transverse tubular system after myocardial infarction in rabbit correlates with local fibrosis: a potential role of biomechanics. Prog. Biophys. Mol. Biol. 130, 302–314. doi: 10.1016/j.pbiomolbio.2017.07.006
Shen, X., van den Brink, J., Kolstad, T., Norden, E., Edwards, A. G., Frisk, M., et al. (2018). 3D dSTORM imaging reveals disassembly of ryanodine receptor clusters in failing cardiomyocytes. Biophys. J. 114:621a. doi: 10.1016/j.bpj.2017.11.3357
Singh, J. K., Barsegyan, V., Bassi, N., Marszalec, W., Tai, S., Mothkur, S., et al. (2017). T-tubule remodeling and increased heterogeneity of calcium release during the progression to heart failure in intact rat ventricle. Physiol. Rep. 5:e13540. doi: 10.14814/phy2.13540
Smyrnias, I., Mair, W., Harzheim, D., Walker, S. A., Roderick, H. L., and Bootman, M. D. (2010). Comparison of the T-tubule system in adult rat ventricular and atrial myocytes, and its role in excitation-contraction coupling and inotropic stimulation. Cell Calcium 47, 210–223. doi: 10.1016/j.ceca.2009.10.001
Sobie, E. A., Guatimosim, S., Gomez-Viquez, L., Song, L. S., Hartmann, H., Saleet, J. M., et al. (2006). The Ca2+ leak paradox and rogue ryanodine receptors: SR Ca2+ efflux theory and practice. Prog. Biophys. Mol. Biol. 90, 172–185. doi: 10.1016/j.pbiomolbio.2005.06.010
Song, L. S., Guatimosim, S., Gomez-Viquez, L., Sobie, E. A., Ziman, A., Hartmann, H., et al. (2005). Calcium biology of the transverse tubules in heart. Ann. N. Y. Acad. Sci. 1047, 99–111. doi: 10.1196/annals.1341.009
Song, L. S., Sobie, E. A., McCulle, S., Lederer, W. J., Balke, C. W., and Cheng, H. (2006). Orphaned ryanodine receptors in the failing heart. Proc. Natl. Acad. Sci. U.S.A. 103, 4305–4310. doi: 10.1073/pnas.0509324103
Stølen, T. O., Høydal, M. A., Kemi, O. J., Catalucci, D., Ceci, M., Aasum, E., et al. (2009). Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ. Res. 105, 527–536. doi: 10.1161/CIRCRESAHA.109.199810
Sun, X. H., Protasi, F., Takahashi, M., Takeshima, H., Ferguson, D. G., and Franzini-Armstrong, C. (1995). Molecular architecture of membranes involved in excitation-contraction coupling of cardiac muscle. J. Cell Biol. 129, 659–671. doi: 10.1083/jcb.129.3.659
Swift, F., Birkeland, J. A., Tovsrud, N., Enger, U. H., Aronsen, J. M., Louch, W. E., et al. (2008). Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase alpha2-isoform in heart failure. Cardiovasc. Res. 78, 71–78. doi: 10.1093/cvr/cvn013
Swift, F., Franzini-Armstrong, C., Øyehaug, L., Enger, U. H., Andersson, K. B., Christensen, G., et al. (2012). Extreme sarcoplasmic reticulum volume loss and compensatory T-tubule remodeling following Serca2 knockout. Proc. Natl. Acad. Sci. U.S.A. 109, 3997–4001. doi: 10.1073/pnas.1120172109
Takekura, H., Paolini, C., Franzini-Armstrong, C., Kugler, G., Grabner, M., and Flucher, B. E. (2004). Differential contribution of skeletal and cardiac II-III loop sequences to the assembly of dihydropyridine-receptor arrays in skeletal muscle. Mol. Biol. Cell 15, 5408–5419. doi: 10.1091/mbc.e04-05-0414
Takeshima, H., Komazaki, S., Nishi, M., Iino, M., and Kangawa, K. (2000). Junctophilins: a novel family of junctional membrane complex proteins. Mol. Cell 6, 11–22.
Terentyev, D., Viatchenko-Karpinski, S., Gyorke, I., Volpe, P., Williams, S. C., and Gyorke, S. (2003). Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: mechanism for hereditary arrhythmia. Proc. Natl. Acad. Sci. U.S.A. 100, 11759–11764. doi: 10.1073/pnas.1932318100
Tian, Q., Pahlavan, S., Oleinikow, K., Jung, J., Ruppenthal, S., Scholz, A., et al. (2012). Functional and morphological preservation of adult ventricular myocytes in culture by sub-micromolar cytochalasin D supplement. J. Mol. Cell. Cardiol. 52, 113–124. doi: 10.1016/j.yjmcc.2011.09.001
van Oort, R. J., Garbino, A., Wang, W., Dixit, S. S., Landstrom, A. P., Gaur, N., et al. (2011). Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation 123, 979–988. doi: 10.1161/CIRCULATIONAHA.110.006437
Waddell, H. M. M., Zhang, J. Z., Hoeksema, K. J., McLachlan, J. J., McLay, J. C., and Jones, P. P. (2016). Oxidation of RyR2 has a biphasic effect on the threshold for store overload-induced calcium release. Biophys. J. 110, 2386–2396. doi: 10.1016/j.bpj.2016.04.036
Wagner, E., Lauterbach, M. A., Kohl, T., Westphal, V., Williams, G. S., Steinbrecher, J. H., et al. (2012). Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ. Res. 111, 402–414. doi: 10.1161/CIRCRESAHA.112.274530
Walker, M. A., Kohl, T., Lehnart, S. E., Greenstein, J. L., Lederer, W. J., and Winslow, R. L. (2015). On the adjacency matrix of RyR2 cluster structures. PLoS Comput. Biol. 11:e1004521. doi: 10.1371/journal.pcbi.1004521
Walker, M. A., Williams, G. S. B., Kohl, T., Lehnart, S. E., Jafri, M. S., Greenstein, J. L., et al. (2014). Superresolution modeling of calcium release in the heart. Biophys. J. 107, 3018–3029. doi: 10.1016/j.bpj.2014.11.003
Ward, M. L., and Crossman, D. J. (2014). Mechanisms underlying the impaired contractility of diabetic cardiomyopathy. World J. Cardiol. 6, 577–584. doi: 10.4330/wjc.v6.i7.577
Wei, S., Guo, A., Chen, B., Kutschke, W., Xie, Y. P., Zimmerman, K., et al. (2010). T-tubule remodeling during transition from hypertrophy to heart failure. Circ. Res. 107, 520–531. doi: 10.1161/CIRCRESAHA.109.212324
Williams, A. J., Thomas, N. L., and George, C. H. (2018). The ryanodine receptor: advances in structure, and organization. Curr. Opin. Physiol. 1, 1–6. doi: 10.4081/ejtm.2015.4840
Wu, H. D., Xu, M., Li, R. C., Guo, L., Lai, Y. S., Xu, S. M., et al. (2012). Ultrastructural remodelling of Ca2+ signalling apparatus in failing heart cells. Cardiovasc. Res. 95, 430–438. doi: 10.1093/cvr/cvs195
Xie, Y. P., Chen, B., Sanders, P., Guo, A., Li, Y., Zimmerman, K., et al. (2012). Sildenafil prevents and reverses transverse-tubule remodeling and Ca2+ handling dysfunction in right ventricle failure induced by pulmonary artery hypertension. Hypertension 59, 355–362. doi: 10.1161/HYPERTENSIONAHA.111.180968
Xu, M., Wu, H. D., Li, R. C., Zhang, H. B., Wang, M., Tao, J., et al. (2012). Mir-24 regulates junctophilin-2 expression in cardiomyocytes. Circ. Res. 111, 837–841. doi: 10.1161/CIRCRESAHA.112.277418
Zhang, C., Chen, B., Guo, A., Zhu, Y., Miller, J. D., Gao, S., et al. (2014). Microtubule-mediated defects in junctophilin-2 trafficking contribute to myocyte transverse-tubule remodeling and Ca2+ handling dysfunction in heart failure. Circulation 129, 1742–1750. doi: 10.1161/CIRCULATIONAHA.113.008452
Zhang, J. Z., Waddell, H. M., Wu, E., Dholakia, J., Okolo, C. A., McLay, J. C., et al. (2016). FKBPs facilitate the termination of spontaneous Ca2+ release in wild-type RyR2 but not CPVT mutant RyR2. Biochem. J. 473, 2049–2060. doi: 10.1042/BCJ20160389
Keywords: dyad, t-tubule, sarcoplasmic reticulum, calcium homeostasis, development, disease
Citation: Jones PP, MacQuaide N and Louch WE (2018) Dyadic Plasticity in Cardiomyocytes. Front. Physiol. 9:1773. doi: 10.3389/fphys.2018.01773
Received: 13 August 2018; Accepted: 23 November 2018;
Published: 11 December 2018.
Edited by:
Antonius Baartscheer, University of Amsterdam, NetherlandsReviewed by:
Jin O-Uchi, University of Minnesota Twin Cities, United StatesToon Van Veen, Utrecht University, Netherlands
Copyright © 2018 Jones, MacQuaide and Louch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William E. Louch, w.e.louch@medisin.uio.no