 Agnieszka Polak-Iwaniuk
Agnieszka Polak-Iwaniuk Ewa Harasim-Symbor
Ewa Harasim-Symbor Karolina Gołaszewska
Karolina Gołaszewska Adrian Chabowski
Adrian Chabowski- 1Faculty of Health Sciences, Lomza State University of Applied Sciences, Łomża, Poland
- 2Department of Physiology, Medical University of Białystok, Białystok, Poland
Hypertension is one of the most frequently observed cardiovascular diseases, which precedes heart failure in 75% of its cases. It is well-established that hypertensive patients have whole body metabolic complications such as hyperlipidemia, hyperglycemia, decreased insulin sensitivity or diabetes mellitus. Since myocardial metabolism is strictly dependent on hormonal status as well as substrate milieu, the above mentioned disturbances may affect energy generation status in the heart. Interestingly, it was found that hypertension induces a shift in substrate preference toward increased glucose utilization in cardiac muscle, prior to structural changes development. The present work reports advances in the aspect of heart metabolism under high blood pressure conditions, including human and the most common animal models of hypertension.
Introduction
Hypertension is one of the most widespread diseases of civilization, in which chronically elevated blood pressure (BP) induces ventricular hypertrophy and contractile dysfunction together with its clinical manifestations. Interestingly, in 2017 recommended threshold for hypertension diagnosis was set at 130/80 mm Hg (for details see Tables 1, 2) (Whelton et al., 2017) and consequently its prevalence increased in the general population. It was estimated that the number of patients suffering from this disorder is slowly reaching the number of normotensive persons (Whelton et al., 2017). Despite extensive knowledge about factors that promote hypertension (Table 3), determination of its etiology is a significant challenge for clinicians. In more than 90% of patients, the cause of hypertension is unknown (primary hypertension). Nevertheless, it is suspected that genetic predispositions and environmental factors play a crucial role in the development of the primary form of hypertension. In the remaining 10% of patients, high BP is the consequence of other medical conditions (Table 3). If a cause is correctly diagnosed, the secondary form of hypertension may be properly treated (Whelton et al., 2017). A meta-analysis of 61 prospective studies revealed a log-linear relationship between the increase in systolic blood pressure (SBP; from < 115 mm Hg to > 180 mm Hg) as well as diastolic blood pressure (DBP; from < 75 mm Hg to > 105 mm Hg) and increased risk of cardiovascular diseases (CVDs) (Lewington et al., 2002). Importantly, 20 mmHg elevated SBP aguments about 50% risk of heart failure (Haider et al., 2003). Since WHO designated CVDs as the predominant cause of death globally it is not surprising that hypertension is considered to be an important issue.

Table 1. Categories of blood pressure levels (mm Hg) – new classification recommended by the American College of Cardiology (ACC) and American Heart Association (AHA).
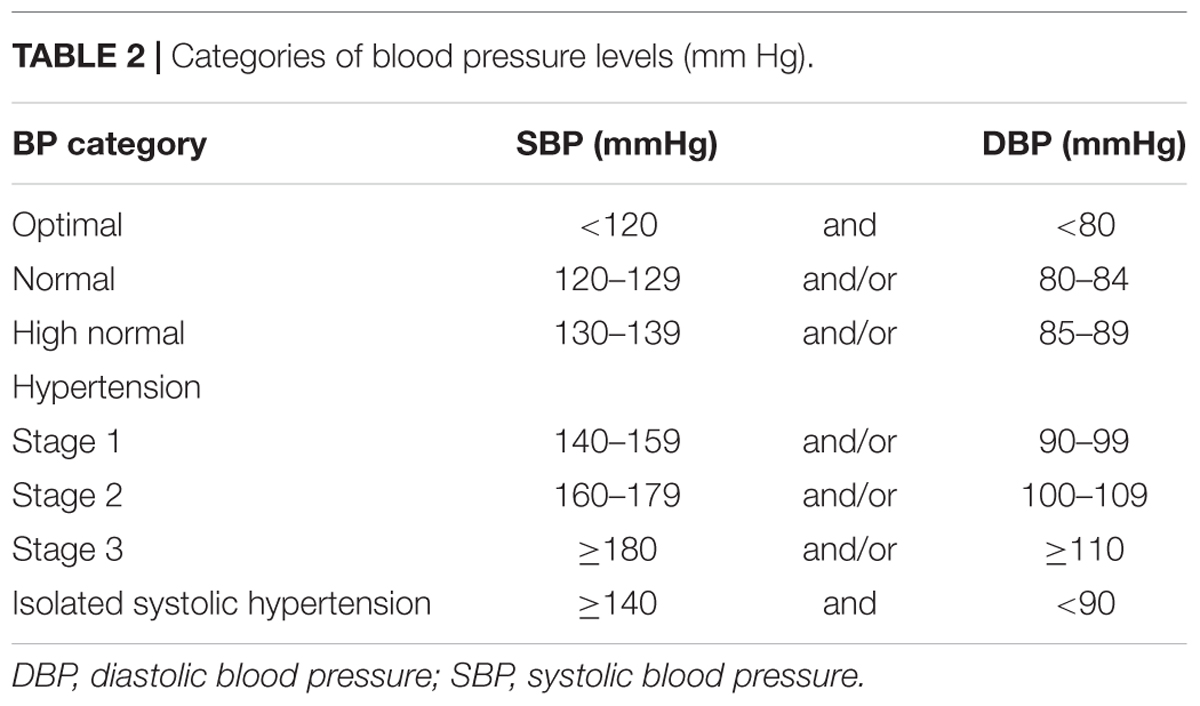
Table 2. Categories of blood pressure levels (mm Hg).
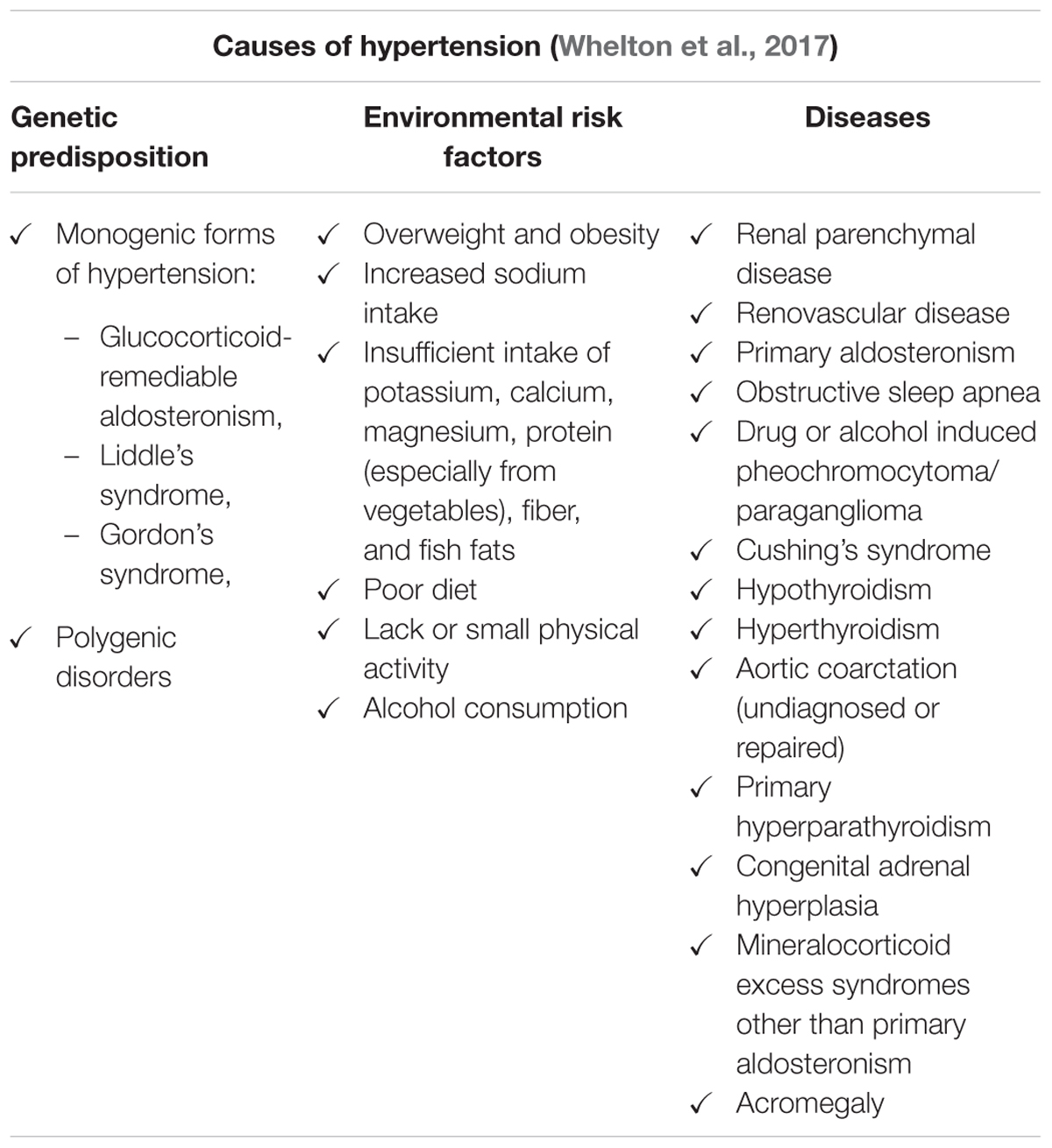
Table 3. Causes of hypertension.
Moreover, it is suggested that approximately 45% of hypertensive patients have metabolic syndrome (Akholkar et al., 2015), indicating the role of hypertension in dysregulation of energy balance. Following this information, we decided to collect and summarize research in the field of metabolism in one of the most important organs affected by hypertensive state. This review describes myocardial energy substrate turnover during physiological state and its changes during increased BP conditions based on the results of tests carried out on humans and animals.
Heart Metabolism
Long-chain fatty acids (LCFAs; 70–80%) and glucose (20–30%) are major energy substrates in the heart under normal, physiological conditions (Gimeno et al., 2003; Stanley et al., 2005). It is well-established that LCFAs are transported across the plasma membrane (PM) by protein-mediated transport (70%) or simple diffusion (30%) (Chabowski et al., 2008). To date, three main groups of LCFA transporters are identified: fatty acid translocase (FAT/CD36), PM-associated fatty acid binding protein (FABPpm) and fatty acid transport proteins 1–6 (FATP1-6; myocardial types 1, 4, and 6) (Bonen et al., 2002; Gimeno et al., 2003; Polak et al., 2016). Upon entering the cardiomyocytes, LCFAs are rapidly bound by the heart-type cytoplasmic fatty acid binding protein (H-FABPC) (Chabowski et al., 2008). Then, the enzyme fatty acyl-CoA synthetase (ACS) catalyzes the conversion of LCFAs into acyl-CoA (Faergeman and Knudsen, 1997). The activated LCFAs are bound by acyl-CoA binding proteins (ACBP) and subsequently they might be transported either into mitochondria for β-oxidation (70–80%) or esterified to distinct lipid pools (20–30%), mainly triacylglycerols (TAGs) (Luiken et al., 1997).
Due to its hydrophilic nature, glucose is unable to pass across the PM by simple, passive diffusion. In this regard, transmembrane uptake of glucose is regulated by special protein carriers (Becker et al., 2001; Szablewski, 2017). There are two families of transporters engaged in glucose uptake by cardiac cells – glucose transporters (GLUTs) and the sodium/glucose cotransporters (SGLTs). Currently, there is little information about SGLTs in the heart. It was found that SGLT1 is highly expressed in cardiac myocytes (Kashiwagi et al., 2015) and predominantly localized in the PM (Banerjee et al., 2009). Moreover, it is suggested that SGLT1 is sensitive to hormone stimuli (Banerjee et al., 2009). However, expression, cellular localization and the role of other SGLTs isoforms in cardiac myocytes is unknown (Szablewski, 2017). Recent data provide evidence for the existence of at least seven transporters belonging to GLUT family in the heart, i.e., GLUT1, GLUT3, GLUT4, GLUT8, GLUT10, GLUT 11, and GLUT 12 (Szablewski, 2017). The most important role in myocardial glucose flux is played by GLUT 1 and 4 (Zorzano et al., 1997; Szablewski, 2017). GLUT1, the isoform, which predominantly occurs in the fetal myocardium, is responsible for basal glucose transport and is mostly located at the PM (Zorzano et al., 1997; Becker et al., 2001). However, mature cardiomyocytes express GLUT4 to a greater extent than GLUT1 (Wang and Hu, 1991; Santalucía et al., 1992). In the resting state, most of the GLUT4 molecules reside in the intracellular, vesicular compartment (Becker et al., 2001). Its translocation to the cell surface is triggered primarily by insulin stimulation or cardiac contractile activity (Zorzano et al., 1997; Becker et al., 2001; Chabowski et al., 2006), which was shown in Figure 1. Activation of insulin pathway (e.g., post-prandial) leads to insulin-induced GLUT4 translocation and glucose influx into cardiomyocytes (Chang et al., 2004; Mora et al., 2004), promoting glycolysis (Depré et al., 1998), as well as myocardial glycogen synthesis (Goodwin et al., 1995). Interestingly, insulin also stimulates fatty acid transporters translocation (Luiken et al., 2002; Chabowski et al., 2004, 2005), mainly FAT/CD36 (Luiken et al., 2002), and concomitantly increases intramyocardial lipid deposition. On the other hand, metabolic demands of the working heart stimulate different cascades among which the major role is played by AMP-activated protein kinase (AMPK) signaling pathway (Luiken et al., 2004). Its activation is known to promote a variety metabolic actions, which in turn intensify glycolysis and fatty acid oxidation (Stanley et al., 2005). Glycolysis (Figure 2) is an oxygen-independent pathway. However, its products (pyruvate and NADH + H+) need oxygen for effective production of ATP (Stanley et al., 2005). When oxygen supply is poor, the pyruvate is converted into lactate in the process called anaerobic glycolysis, which becomes important in the preservation of myocardial energy-yielding process during ischemia or hypoxia incidents (Essop, 2007; Lopaschuk, 2017).
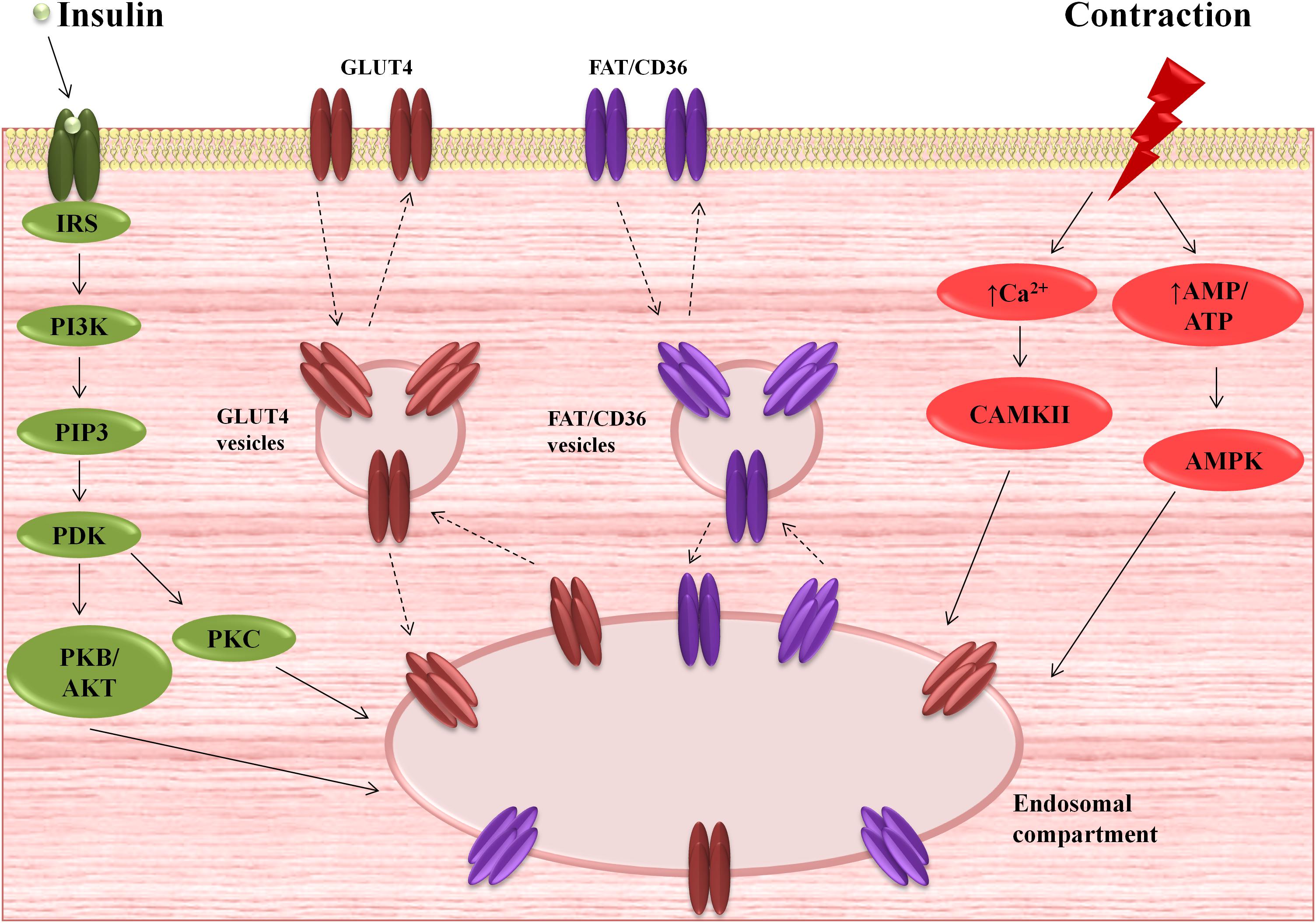
Figure 1. Myocardial activation of insulin and contractile signaling pathways and subsequent translocation of glucose transporter 4 (GLUT4) and fatty acid translocase (FAT/CD36) from the intracellular compartments to the plasma membrane (PM). AMPK, AMP-activated protein kinase; CAMKII, Ca2+/calmodulin-dependent kinase II; IRS, insulin-receptor substrate; PDK, phosphoinositide-dependent kinase; PI3K, phosphatidylinositol 3-kinase; PIP3, phosphatidylinositol 3-phosphates; PKB/AKT, serine/threonine protein kinase B; PKC, protein kinase C.
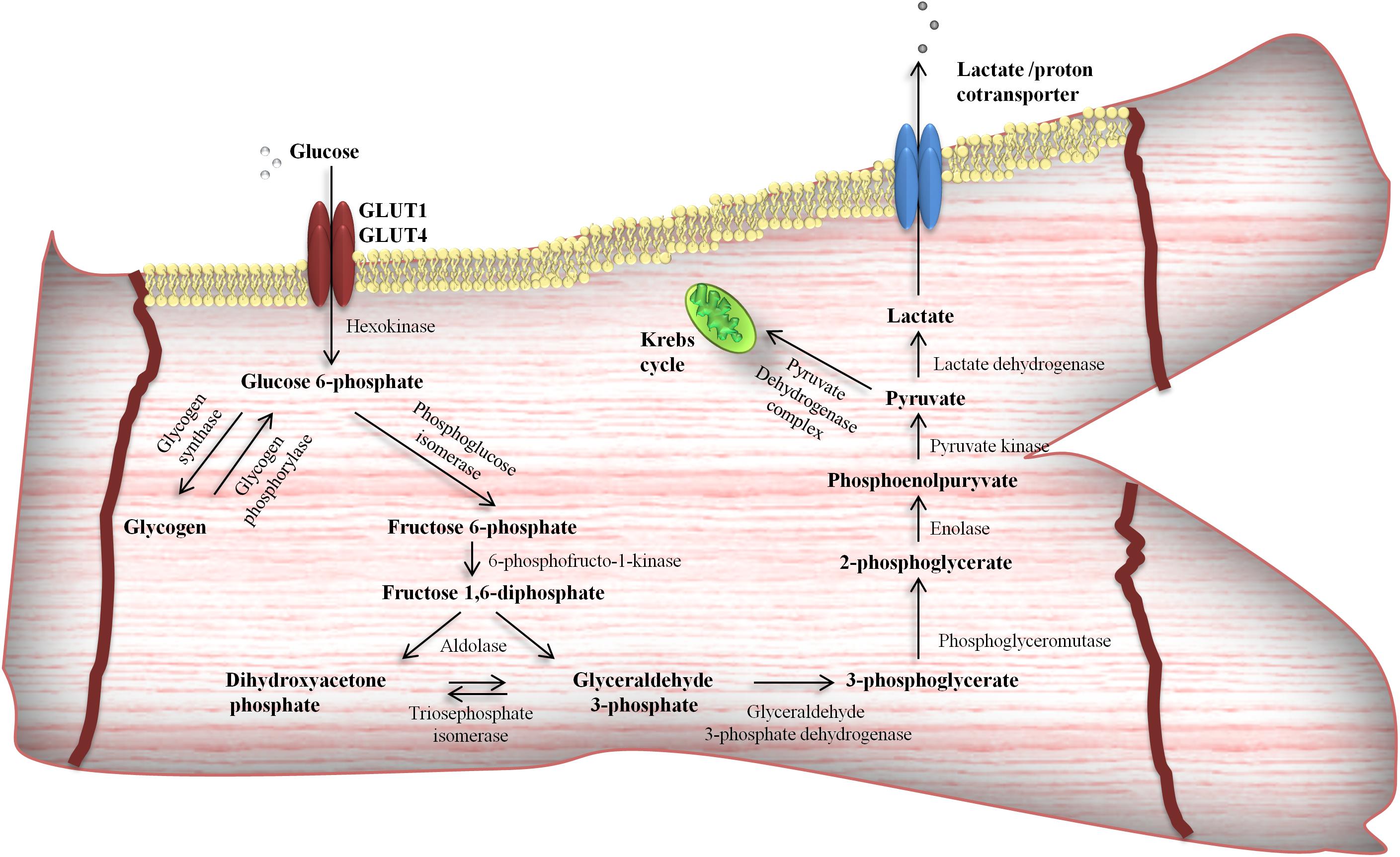
Figure 2. Glycolytic pathway in cardiomyocytes. Glucose after entering the cells via facilitated diffusion [glucose transporter 4 (GLUT4)] can undergo different intramyocardial metabolic pathways and become available for energy production or stored as a glycogen.
Nonetheless, the cardiac metabolic network is a flexible system that adapts to the current environment. Therefore, maintaining a high capacity for ATP production depends on numerous mechanisms. The first type of metabolic control is the classic allosteric regulation of the activity of key enzymes along the metabolic pathway. Allosteric control involves the binding of signaling molecules to specific regulatory sites, which induces conformational changes and alterations in the catalytic activity of enzymes (Kolwicz et al., 2013). For example, inactivation of pyruvate dehydrogenase (PDH) by pyruvate dehydrogenase kinase 4 (PDK4) is associated with a decrease in glucose catabolism and a corresponding rise in fatty acid oxidation in response to increased fatty acid availability during prolonged starvation (Wu et al., 2001; Sugden and Holness, 2003; Zhang et al., 2014). Additionally, a well-known regulator of fatty acid uptake and metabolism, malonyl-CoA, due to allosteric inhibition of carnitine palmitoyltransferase 1 (CPT1) regulates mitochondrial LCFA uptake and β-oxidation (Lopaschuk et al., 2010; Fillmore and Lopaschuk, 2014). A decreased malonyl-CoA level in pathological conditions, such as ischemia or diabetes, increases fatty acid oxidation rates at the expense of glucose metabolism, elevates the oxygen cost of contractility and decreases cardiac efficiency (Fillmore and Lopaschuk, 2014). The second mechanism, post-translational modifications (PTMs), changes the functional diversity of the proteome. The most common PTMs are covalent addition or removal of functional groups, the specific cleavage of precursor proteins and disulfide bonds formation (Lopaschuk, 2017). Accordingly, it was demonstrated that acetylation of mitochondrial enzymes, e.g., LCAD plays an important role in cardiac metabolism regulation. Studies conducted on mice lacking mitochondrial sirtuin 3 (SIRT3) have revealed that increased long-chain acyl coenzyme A dehydrogenase (LCAD) acetylation may be responsible for lower myocardial fatty acid oxidation (Hirschey et al., 2010; Chen et al., 2015). This information is especially important since the downregulation of SIRT3 protein has been implicated in heart failure in Dahl-salt sensitive and spontaneously hypertensive heart failure prone (SHHF) rats (Grillon et al., 2012). Another deacetylase namely SIRT 5, additionally exhibits desuccinylase and demalonylase activity. Additionally, absence of SIRT5 leads to suppression of PDH activity as well as glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in SIRT5-/- compared to wild type mice (Park et al., 2013; Nishida et al., 2015). Moreover, Hershberger et al. (2017) have demonstrated that SIRT5 is required for maintaining cardiac oxidative metabolism in response to cardiac pressure overload. Phosphorylation, as another way of enzyme modification, also plays an important role in the cardiac metabolism regulation. One of the previous studies showed that phosphorylation and subsequent inhibition of both acetyl-CoA carboxylase isoforms, ACC1/ACC2, suppressed acetyl-CoA to malonyl-CoA conversion and increased fatty acid uptake into mitochondria during reperfusion of ischemic rat hearts (Bairwa et al., 2016). Other PTM, such as O-GlcNAcylation may be associated with a significant increase in PM levels of FAT/CD36 and palmitate oxidation, which was observed in rats’ hearts subjected to a high glucosamine concentration. Similar changes reflecting increased fatty acid metabolism are observed in the early stages of diabetes (Laczy et al., 2011). The third mechanism is the regulation of gene transcription in which transduction of specific signal to the cell nucleus affects gene expression. This kind of control includes many of key transcriptional regulators of mitochondrial fuel metabolism and energy production pathways such as the peroxisome proliferator-activated receptors (PPARs) (Leone and Kelly, 2011; Lee et al., 2017), estrogen-related receptors (ERRs) (Wang et al., 2015) or nuclear respiratory factors 1 and 2 (NRF-1 and 2) (Dorn et al., 2015). Transcriptional control has an influence on the level of enzymes expression and acts on a longer time scale. Coordination of these various mechanisms ensures that the energy balance is maintained (Desvergne et al., 2006). The most known regulators of glucose and fatty acids metabolism are summarized in Table 4.
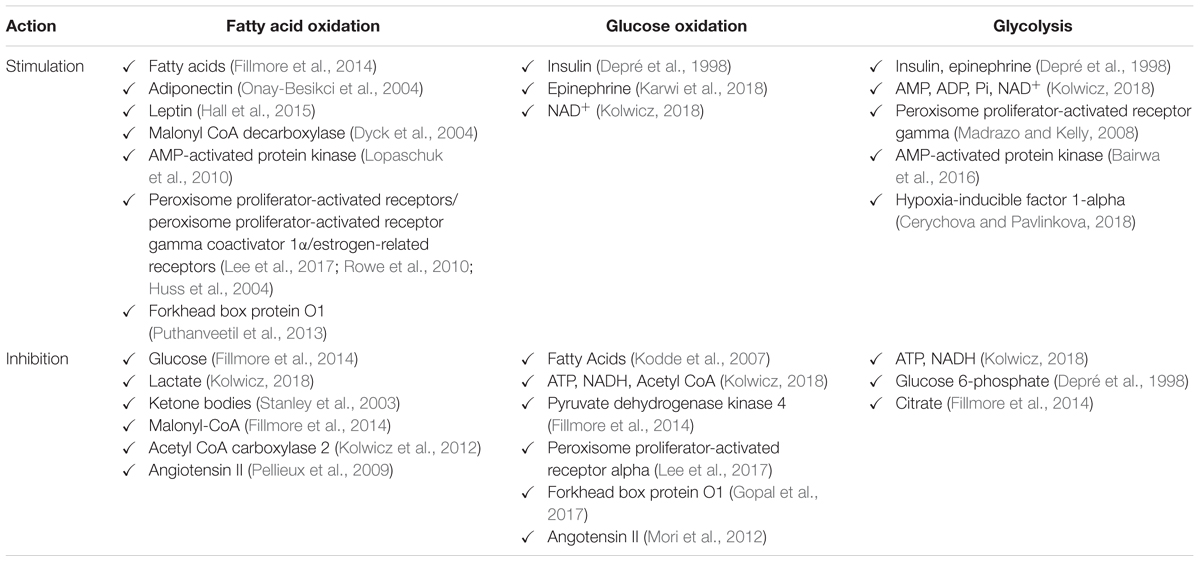
Table 4. Main regulators of glucose and fatty acids metabolism in the heart.
Myocardial Metabolism in Hypertension
Human Hypertension
An abnormal glucose metabolism, including insulin resistance, impaired glucose tolerance or diabetes mellitus as well as dyslipidemia are frequently diagnosed in patients with different types of hypertension (Onat et al., 2005; García-Puig et al., 2006; Chen et al., 2014). However, there are only a few reports investigating cardiac energy homeostasis in such patients. Importantly, de las Fuentes et al. (2003, 2006) demonstrated that patients with hypertension exhibited reduced myocardial fatty acid oxidation. Moreover, it was found this phenomenon preceded an increase in the left ventricle (LV) mass and might have been responsible for decreased myocardial efficiency (de las Fuentes et al., 2006). Presumably, the basis of this metabolic imbalance is complex and includes genomic alterations, e.g., variation in peroxisome proliferator-activated receptor alpha (PPARα) gene (Jamshidi et al., 2002). Taken together, these data support the hypothesis that disturbances in substrate metabolism may be related with morphological and functional abnormalities in the hypertensive heart.
To date, there are two contradictory reports concerning the influence of hypertension on glucose uptake in the human heart. Laine et al. (1999) found no differences in glucose uptake in hypertensive patients with or without LVH in comparison with the normotensive group. Whereas, Hamirani et al. (2016) observed higher myocardial glucose flux in the hypertensive group without LVH compared to the hypertensive patients with LVH and the normotensive group as well. However, myocardial glucose uptake in the hypertensive patients with hypertrophy was significantly reduced in comparison with the normotensive patients in this study (Hamirani et al., 2016). Discrepancies in glucose uptake rate may result from differences in age, body mass index as well as SBP value of patients in the above experiments. Nonetheless, it seems that an increase in glucose utilization results from pressure overload, but not hypertrophy, and occurs only in severe stages of hypertension (over 150 mm Hg SBP) or when the difference in SBP between the examined and control groups is sufficiently high (∼35 mm Hg) (Laine et al., 1999; Hamirani et al., 2016).
Interestingly, it has been shown that myocardial phosphocreatine/ATP ratio, a marker of an energetic state of the heart, was decreased in the hypertensive hypertrophied heart (Lamb et al., 1999). Probably, this resulted from reduced creatine kinase activity and lower total creatine content, which is frequently observed in the hypertrophied human heart (Ingwall et al., 1985). However, it has been proven that myocardial oxygen consumption per weight unit was elevated in the hypertensive patients without LVH, whereas, in those with LVH was normal (Laine et al., 1999). Nonetheless, oxygen consumption in the hypertensive hypertrophied heart at the same level as in the heart from healthy humans occurs to the detriment of cardiac efficiency. This in turn may predispose to heart failure development (Laine et al., 1999).
Animal Models of Hypertension
Primary Hypertension
Spontaneously hypertensive rats (SHRs) introduced by Okamoto and Aoki (1963), are widely used in experimental research as genetic model of hypertension (Polak et al., 2018a). These rats develop hypertension without any physiological, pharmacological or surgical procedures.
Similarly to hypertensive patients, SHRs have been shown to be hyperglycemic and hyperinsulinemic (Gouveia et al., 2000; Daud et al., 2016). Moreover, it is suggested that these animals have reduced peripheral glucose utilization, which also makes them insulin resistant (Hulman et al., 1993; Gouveia et al., 2000; Zecchin et al., 2003). Bearing in mind that both glucose gradient across the PM and insulin action determine the rate of glucose influx and intracellular utilization, systemic abnormalities occurring in SHRs may directly affect myocardial metabolism (Zorzano et al., 1997). Intriguingly, it was demonstrated that the uptake of the glucose tracer 2-[18F]FDG was severalfold increased in SHR hearts (Hajri et al., 2001; Quintana-Villamandos et al., 2013; Wang et al., 2017; Li et al., 2018). Moreover, Hajri et al. (2001) investigation also found impaired myocardial uptake of fatty acid analog [125I]BMIPP. Other studies, which showed diminished palmitate oxidation and moderately elevated glucose oxidation in the perfused hearts, also revealed a shift toward glucose use instead of fatty acid in SHR strain (Christe and Rodgers, 1994, 1995). Furthermore, it was revealed that myocardial level of total carnitine was significantly reduced in SHR (Christe and Rodgers, 1995). Therefore, the above increase in glucose uptake reflects a compensatory response to defective myocardial fatty acid utilization, but not benefits of glucose and insulin abundance.
It should also be mentioned that the second genetic model of hypertension, namely Dahl-salt rats, presents similar to SHRs changes regarding substrate preference in myocardial metabolism (Yonekura et al., 1985; Kato et al., 2010). Why glucose replaces fatty acids, a main source of energy in the heart, is yet not known. It is well-known that about 10% more ATP is generated from glucose than fatty acid oxidation per mole of oxygen (Stanley and Chandler, 2002). Thus, the possible explanation of the switch in fuel type is adaptation to effective energy generation in adverse hemodynamic conditions, which was also reported in other models of the heart hypertrophy (Allard et al., 1994; Barger and Kelly, 1999).
Moreover, it is proposed that impairment in fatty acid metabolic rate intensifies with age in SHRs (Reutter et al., 2011). On the basis of the studies in which 13C-labeled energy substrates were used to determine SHR myocardial metabolism, it may be assumed that cardiac muscle of young (7-week-old) SHRs adopts more effectively to hypertension. It is by the fact that compensational increase in exogenous 13C-glucose use completely replaced reduced exogenous LCFA oxidation in the hearts of 7-week-old SHRs. Moreover, partitioning of exogenous 13C-glucose between glycolysis and oxidation through citric acid cycle was unchanged in the cardiomyocytes of 7-week-old SHRs compared to the normotensive rats (Lauzier et al., 2011). On the contrary, older rats (15-week-old) manifested a reduced contribution of exogenous 13C-labeled LCFA to myocardial energy production, which does not seem to be fully compensated by exogenous carbohydrates but endogenous energy sources. Surprisingly, the authors of these reports imply that these sources are either LCFA released from myocardial TAG stores or amino acids from enhanced proteolysis in the heart (Vincent et al., 2003; Labarthe et al., 2005). In addition, increased lactate release by the heart in 15-week-old SHRs (Vincent et al., 2003) also suggests decreasing capability of cardiac tissue in older animals to maintain its metabolic homeostasis. Despite these alterations, the SHRs display the same as in the normotensive control group myocardial oxygen consumption together with intracellular pH and, surprisingly, unaffected cardiac power and efficiency regardless of age (Vincent et al., 2003; Lauzier et al., 2011).
Interestingly, introduction of short-chain fatty acids (SCFAs) to SHRs’ diet prevented the heart hypertrophy development in spite of persistent hypertension (Hajri et al., 2001). A similar effect was observed in the ex vivo model of perfused SHR heart during medium-chain fatty acid (MCFAs) supplementation (Labarthe et al., 2005), which likewise SCFAs (and in contrast to LCFAs), do not require protein carriers (Schönfeld and Wojtczak, 2016). Two conclusions from these both experiments can be drawn. Firstly, abnormalities regarding myocardial fatty acid metabolism are involved in the development of hypertrophy in the SHR hearts. Secondly, it is possible that the hypertensive state affects fatty acid transport. Interestingly, Aitman et al. (1999) detected a defect in the gene of the major fatty acid transporter, FAT/CD36 in SHRs. This study also demonstrated that the encoded product of mutated FAT/CD36 gene was undetectable in PM of SHRs’ adipocytes, causing doubt whether SHRs are deprived of this protein transporter (Aitman et al., 1999). However, Gotoda et al. (1999) proved that FAT/CD36 mutation in the North American SHR strain used in Aitman et al. (1999) research is absent in the original SHR strains maintained in Japan. Additionally, Bonen et al. (2009) demonstrated that SHR strains express different FAT/CD36 mRNA. This phenomenon was not coupled with the absence of FAT/CD36 expression because its presence was observed in several tissues (including heart) of the North American SHR strain (Bonen et al., 2009). Therefore, the results obtained by Aitman et al. (1999) may be the consequence of inappropriate antibodies used instead of real absence of this protein (Bonen et al., 2009). Nevertheless, it should be pointed out that there was a decreased expression and plasmalemmal content of fatty acid transporters (total FAT/CD36: -26%, total FATP1: -35% and PM FAT/CD36: -36%) in the heart of 15-week-old SHRs (Bonen et al., 2009). Interestingly, the reduced FAT/CD36 protein level in PM and impaired fatty acid utilization occurs also in 7-week-old SHRs, prior to developing LVH (Lauzier et al., 2011). This may imply that the onset of myocardial metabolic disturbances appears during the early stages of hypertension and probably precedes anatomical and functional changes in the heart. Moreover, data indicate susceptibility of FAT/CD36 in the SHR hearts to defective PTMs, specifically to N-glycosylation (Lauzier et al., 2011). The authors of this report pointed out that the above change could be responsible for the lowered plasmalemmal FAT/CD36 content as well as loss of its function, which was followed by reduced capacity for fatty acid transport (Lauzier et al., 2011). Moreover, the studies conducted on SHR-4, a congenic SHR strain with inserted functional FAT/CD36 gene segment (from the Brown Norway strain), revealed that FAT/CD36 mutation in SHR may have influence on LVH development (Klevstig et al., 2011). Interestingly, the alterations in FAT/CD36 level or its function were linked with diminished fatty acid metabolism, elevated cardiovascular risk and heart diseases also in humans (Febbraio and Silverstein, 2007; Kim and Dyck, 2016).
Additionally, there is evidence that hypertension can affect intracellular signaling pathways in the heart which are crucial for maintaining energy substrate transport and myocardial energetic status. Accordingly, it was noted that spontaneous mutation in Pflz gene, occurring in SHR, probably is associated with disturbances in myocardial PPARα and insulin signaling at the genome level (Liška et al., 2017). Furthermore, it was demonstrated that PKCε expression as well as its activation were decreased in the SHR’s cardiac muscle which carries a non-functional variant of the FAT/CD36 (Klevstig et al., 2011). The authors of this report do not exclude the possibility that this phenomenon may be the basis of impaired LCFA transport in the cardiomyocytes (Klevstig et al., 2011). What is more, Dolinsky et al. (2009) showed that phosphorylation of liver kinase B (LKB1) and its direct substrate, AMPK, was significantly reduced in the 15-week (but not in 7-week) SHR hearts compared to the hearts of normotensive rats. Interestingly, this inhibition of the LKB1-AMPK signaling pathway was related to cardiac hypertrophy, but not intracellular energy status because AMP/ATP ratio was unchanged (Dolinsky et al., 2009). Similarly, Christe and Rodgers (1995) also observed that the levels of myocardial high-energy phosphates (ADP, AMP, and ATP) were unaffected in the SHRs, which indicates that the total energy production in myocardium is stable despite of hypertension.
Nonetheless, it should be mentioned that in SHR hearts disorders regarding intracellular energy substrates stores can appear. For example, there is proof for DAG to TAG conversion inhibition, since [125I]BMIPP was incorporated more intensively into DAG than TAG myocardial fraction (Hajri et al., 2001). On the other hand, study conducted by LaPier and Rodnick (2000) revealed that 22- and 14-week-old SHRs exhibited a significantly elevated myocardial TAG content compared to 6-week-old individuals, which proved that TAG accumulation may intensifies with age. The same study also showed that hypertension decreased myocardial glycogen content in all the examined age-matched groups (LaPier and Rodnick, 2000). Moreover, LaPier and Rodnick (2000) demonstrated that hypertensive rats had an elevated plasma glucose availability as well as unchanged myocardial GLUT1 and GLUT4 expression, indicating an unaffected capacity for glucose transport into cardiomyocytes in hypertension. Thus, glycogen depletion probably resulted from its increased use due to pressure overload heart as well as confirmed a shift in substrate preference (LaPier and Rodnick, 2000). It is interesting to note that LaPier and Rodnick (2000) studies have been carried out on female SHRs and observed changes are consistent with those in male SHR hearts, suggesting lack of gender differences in cardiac metabolism in hypertension.
Secondary Hypertension
Renal models
Renal hypertension is induced in animals using surgical procedures, which cause renal parenchyma or renal arteries damages (Polak et al., 2018a). Among all renal hypertension types, the most popular are models introduced by Goldblatt, in which elevation of BP is performed by a constriction of one or both renal arteries using a small clamp. For example, in one-kidney one-clip model (1K1C; one renal artery is constricted and concomitantly the contralateral kidney is removed) mild hypertension causes functional changes without concomitant hypertrophy of the LV, as seen in other animal models (Taegtmeyer and Overturf, 1988). These functional alterations are accompanied by myocardial metabolic changes favoring glucose as a fuel (enhanced glucose utilization and intensified activity of key glycolytic enzymes – hexokinase, phosphofructokinase, and phosphorylase) and decreased ketone bodies metabolism (Taegtmeyer and Overturf, 1988). In this case increased glucose utilization was not coupled with changes in lactate production indicating an increased rate of its oxidation but not the contribution in anaerobic metabolism. However, unchanged activities of two main enzymes of citric acid cycle [citrate synthase (CS) and 2-oxoglutarate dehydrogenase (Taegtmeyer and Overturf, 1988)] reflects similar to observed in SHR an unaltered rate of total energy production in spite of a shift in the myocardial metabolism toward glucose in the 1K1C model of hypertension.
Intriguingly, there are some differences amongst Goldblatt’s hypertension models. The study concerning enzyme activities of the energy-supplying metabolism revealed that myocardial CS activity in two-kidney one-clip rats (2K1C; one renal artery is constricted and the contralateral kidney is left intact) was reduced (Koehler and Medugorac, 1985), which is contradictory to the findings in the 1K1C model (Taegtmeyer and Overturf, 1988). Moreover, the study conducted by Smith et al. (1990) revealed nearly twofold lactate dehydrogenase (LDH) activity elevation after 6 weeks from the surgical procedure in the LV as well as right ventricle (RV) in the 2K1C model. The report of Koehler and Medugorac (1985) also noted that LDH activity in the LV in the initial phase of hypertension induction (4–6 weeks post-operatively) was significantly higher than in the LV of the normotensive group, although the increase was 6%. Nevertheless, this phenomenon was not observed in the further stages of the experiment (8–12 weeks post-operatively), and therefore Koehler and Medugorac (1985) proposed that this initially elevated LDH activity was an artifact of a surgical operation. It is interesting that the more pronounced changes were detected in Smith’s experiment (Smith et al., 1990), which was performed on 4-weeks rats [vs. 8-weeks rats in Koehler investigation (Koehler and Medugorac, 1985)]. A possible explanation may be that induction of hypertension in younger subjects caused immediate return to “fetal” metabolism, in which anaerobic conditions favor use of glucose and lactate (collectively referred to as carbohydrates) as the main energy sources (Taegtmeyer et al., 2010). This suggestion may be confirmed by a decreased activity of adult-type creatine kinase isoenzyme (CK-MM) and concomitantly elevated activity of the fetal-type CK isoenzymes (CK-MB+BB) in the heart of these animals (Smith et al., 1990). Moreover, a decreased total myocardial CK activity in 2K1C model probably indicates similar to hypertensive patients, impaired energy transfer to sarcomeres (Smith et al., 1990). However, a reduced activity of β-hydroxy-acyl-CoA dehydrogenase (β-HADH), which catalyzes the third step of β-oxidation, and increased hexokinase activity (Taegtmeyer and Overturf, 1988), are in line with the findings showing a shift in myocardial metabolism toward increased glucose utilization due to hypertension (Christe and Rodgers, 1994; Hajri et al., 2001).
Endocrinal models
Administration of mineralocorticoids or their synthetic derivatives, such as deoxycorticosterone acetate (DOCA) combined with sodium chloride, in unilateral nephrectomised rats produces volume overload hypertension (Lee et al., 2015). This model represents the form of endocrine hypertension observed in humans with primary aldosteronism. Interestingly, the DOCA-salt rats exhibited reduced insulin concentration in the blood (Dai and McNeill, 1992; Dai et al., 1992; Ndisang and Jadhav, 2010), which probably resulted from destruction of β-cells of pancreatic islets (Ndisang and Jadhav, 2010), similarly to humans with primary aldosteronism (Corry and Tuck, 2003). However, the observed decreased insulin concentration in this model did not affect glycemia (Dai and McNeill, 1992; Dai et al., 1992; Ndisang and Jadhav, 2010). Recent data revealed that DOCA-salt rats exhibit increased tissue insulin sensitivity (Polak et al., 2018b). It is interesting that DOCA-salt rats present similar to other animal models of hypertension a shift toward enhanced carbohydrates utilization in the heart, especially deriving from intramyocardial glycogen (Coprean et al., 1993). Presumably, this process occurred under aerobic conditions since elevated pyruvate dehydrogenase (PDH) expression was observed in the heart of DOCA-salt rats (Polak et al., 2018b). Moreover, recent studies noted unchanged myocardial expression and subcellular redistribution of two main glucose transporters, GLUT1 and GLUT4 in the DOCA-salt rats (Atkins et al., 2001; Polak et al., 2018b). Because glucose transport is regulated by protein carriers (Zorzano et al., 1997), this data imply the notion that glucose influx to cardiomyocytes in this model probably is unaffected by hypertension. In line with previous findings in SHR model, palmitate oxidation was decreased in the heart of DOCA-salt rats in which hypertension was induced already at 6–7 week old (Polak et al., 2017). On the contrary to SHRs, the fatty acid uptake seems to be unaffected, since myocardial palmitate uptake, as well as expression and subcellular redistribution of the primary fatty acid transporters (FAT/CD36, FATP1, FATP4, and FABPpm), were unchanged in these rats in comparison with the normotensive rats (Polak et al., 2017). Moreover, there were no changes in the activity of major proteins in signaling pathways stimulating transmembrane transport of energy substrates, such as AMPK, serine/threonine protein kinase B (AKT) and mitogen-activated protein kinase (MAPK) (Polak et al., 2018b). Nonetheless, there is some evidence that hypertension promotes metabolic changes in an age-dependent manner, also in this model of hypertension. In particular, this was indicated by an increased level of plasma lipids [cholesterol, free fatty acids (FFA), phospholipids (PL), and TAG] and their enhanced accumulation in the heart of DOCA-salt rats in which induction of hypertension was initiated at 11–13 week old (Prahalathan et al., 2012) [vs. unaffected plasma and myocardial lipids content in rats in which hypertension was induced at 6–7 week old (Polak et al., 2017)].
Collectively, DOCA-salt rats have primarily increased carbohydrates metabolism in the heart probably due to enhanced endogenous myocardial sources (glycogen) utilization. On the other hand, disturbances in energy generation from LCFAs are mainly related to their impaired oxidation, but not to transport into cardiomyocytes.
Cardiac Metabolism in the Failing Heart
In hypertensive patients, pressure overload leads to cardiac remodeling including eccentric LV hypertrophy. When this state is sustained, there develops a diastolic dysfunction, decompensation of remodeled LV and finally hypertensive heart failure (Messerli et al., 2017). A growing body of evidence indicates that metabolism in the failing heart is impaired (Doehner et al., 2014). One of the most important alterations is 30–40% decreased ATP production, which results from disturbances in a mitochondrial function and decreased oxidative capacity (Rosca and Hoppel, 2013). Moreover, it is proposed that the failing heart switches from fatty acids oxidation to glucose metabolism to generate energy (Riehle et al., 2011; Kolwicz et al., 2012). However, this switch reflects an increase in glucose uptake and glycolysis instead of mitochondrial oxidative metabolism (Allard et al., 1994; Stanley et al., 2005; Lopaschuk et al., 2010). Uncoupling of glucose metabolism increases lactate and protons production (Folmes et al., 2006; Fillmore et al., 2018; Saleme and Sutendra, 2018). This, in turn, may reduce contractility of the heart, e.g., by decreasing troponin I sensitivity to calcium and inhibiting the slow calcium current (Morimoto and Goto, 2000). Furthermore, the utilization of ATP to eliminate protons and preserve calcium and sodium homeostasis worsens cardiac efficiency and function (Lopaschuk et al., 2010). Additionally, it is suspected that uncoupling glycolysis from glucose oxidation supports development of heart hypertrophy and its progression (Fillmore et al., 2018).
Conclusion
Myocardial energy metabolism is a complex process, which consists of a network of biochemical reactions and pathways. The initial stage is energy substrate utilization due to its uptake and metabolism as well as following participation of intermediates in TCA cycle. Then mitochondrial oxidative phosphorylation enables generation of ATP molecules, and finally, the creatine kinase system transports ATP from mitochondria to the sarcomeres. In the light of the foregoing evidence, metabolism of the heart subjected to pressure overload may be disturbed in all of this areas. In spite of differences among humans and animals, one of the main disorders during hypertension includes a shift from fatty acids toward enhanced carbohydrates utilization in the heart. What is interesting, these abnormalities are connected with impaired myocardial function (Figure 3). Therefore, implementation of strategies aimed at improving cardiac metabolism in BP lowering therapy seems to be extremely important (Swislocki et al., 1999; Prahalathan et al., 2012; Wang et al., 2017; Polak et al., 2018b; Harasim-Symbor et al., 2019).
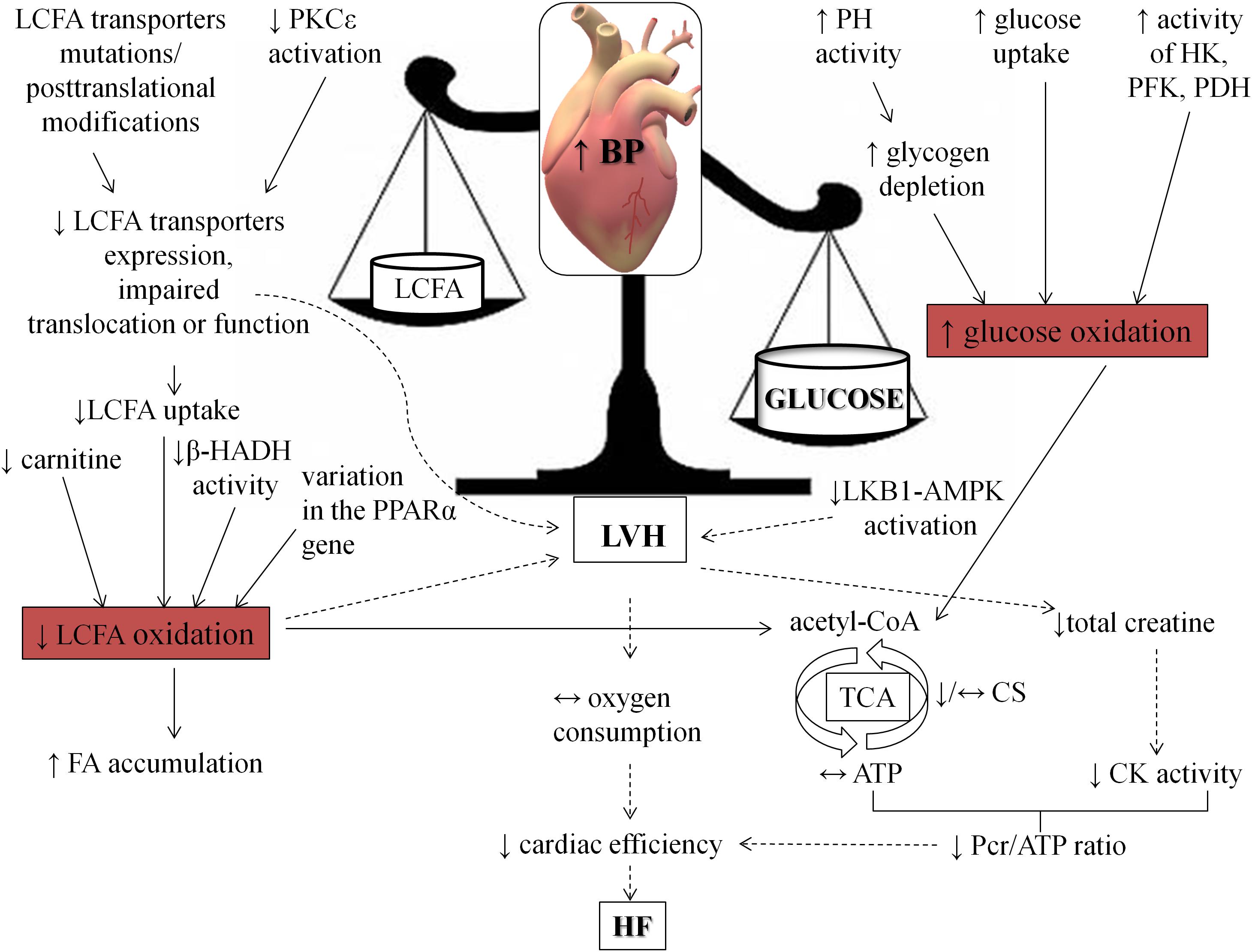
Figure 3. Consequences of hypertension on heart metabolism and function. The figure shows how hypertension shifts fuel preference from fatty acids toward carbohydrates. AMPK, AMP kinase; β-HADH, β-hydroxy-acyl-CoA dehydrogenase; CK, creatine kinase; CS, citrate synthase; HF, heart failure; HK, hexokinase; LKB1, liver kinase B; LVH, left ventricle hypertrophy; Pcr, phosphocreatine; PDH, pyruvate dehydrogenase; PFK, phosphofructokinase; PH, phosphorylase; PKCε, protein kinase C ε; PPARα, peroxisome proliferator-activated receptor alpha.
Author Contributions
AP-I participated in the design of the work, drafted the manuscript, prepared figures and tables and approved final version submitted. EH-S and KG helped to draft the manuscript and approved final version submitted. AC participated in the design of the study, revised manuscript and approved final version submitted. All authors agreed to be accountable for all aspects of the work.
Funding
This work was supported by the National Science Centre of Poland (Grant No. NCN 2017/26/D/N23/01119) and Medical University of Białystok (Grant No. N/ST/ZB/18/002/1118).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aitman, T. J., Glazier, A. M., Wallace, C. A., Cooper, L. D., Norsworthy, P. J., Wahid, F. N., et al. (1999). Identification of Cd36 (Fat) as an insulin-resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat. Genet. 21, 76–83. doi: 10.1038/5013
Akholkar, P. J., Gandhi, A. A., and Shah, C. M. (2015). The metabolic syndrome among hypertensive patients: a cross-sectional study. Int. J. Adv. Med. 2, 188–191. doi: 10.18203/2349-3933.ijam20150542
Allard, M. F., Schönekess, B. O., Henning, S. L., English, D. R., and Lopaschuk, G. D. (1994). Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. 267(2 Pt 2), H742–H750. doi: 10.1152/ajpheart.1994.267.2.h742
Atkins, K. B., Johns, D., Watts, S., Clinton Webb, R., and Brosius, F. C. (2001). Decreased vascular glucose transporter expression and glucose uptake in DOCA-salt hypertension. J. Hypertens. 19, 1581–1587. doi: 10.1016/j.pharep.2015.10.004
Bairwa, S. C., Parajuli, N., and Dyck, J. R. (2016). The role of AMPK in cardiomyocyte health and survival. Biochim. Biophys. Acta 1862, 2199–2210. doi: 10.1016/j.bbadis.2016.07.001
Banerjee, S. K., McGaffin, K. R., Pastor-Soler, N. M., and Ahmad, F. (2009). SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc. Res. 84, 111–118. doi: 10.1093/cvr/cvp190
Barger, P. M., and Kelly, D. P. (1999). Fatty acid utilization in the hypertrophied and failing heart: molecular regulatory mechanisms. Am. J. Med. Sci. 318, 36–42. doi: 10.1097/00000441-199907000-00006
Becker, C., Sevilla, L., Tomàs, E., Palacin, M., Zorzano, A., and Fischer, Y. (2001). The endosomal compartment is an insulin-sensitive recruitment site for GLUT4 and GLUT1 glucose transporters in cardiac myocytes. Endocrinology 142, 5267–5276. doi: 10.1210/endo.142.12.8555
Bonen, A., Han, X. X., Tandon, N. N., Glatz, J. F., Lally, J., Snook, L. A., et al. (2009). FAT/CD36 expression is not ablated in spontaneously hypertensive rats. J. Lipid Res. 50, 740–748. doi: 10.1194/jlr.M800237-JLR200
Bonen, A., Luiken, J. J., and Glatz, J. F. (2002). Regulation of fatty acid transport and membrane transporters in health and disease. Mol. Cell. Biochem. 239, 181–192. doi: 10.1007/978-1-4419-9270-3_23
Cerychova, R., and Pavlinkova, G. (2018). HIF-1, metabolism, and diabetes in the embryonic and adult heart. Front. Endocrinol. 9:460. doi: 10.3389/fendo.2018.00460
Chabowski, A., Coort, S. L., Calles-Escandon, J., Tandon, N. N., Glatz, J. F., Luiken, J. J., et al. (2004). Insulin stimulates fatty acid transport by regulating expression of FAT/CD36 but not FABPpm. Am. J. Physiol. Endocrinol. Metab. 287, E781–E789. doi: 10.1152/ajpendo.00573.2003
Chabowski, A., Coort, S. L., Calles-Escandon, J., Tandon, N. N., Glatz, J. F., Luiken, J. J., et al. (2005). The subcellular compartmentation of fatty acid transporters is regulated differently by insulin and by AICAR. FEBS Lett. 579, 2428–2432. doi: 10.1016/j.febslet.2004.11.118
Chabowski, A., Górski, J., and Bonen, A. (2006). Regulation of fatty acid transport: from transcriptional to posttranscriptional effects. Naunyn Schmiedebergs Arch. Pharmacol. 373, 259–263. doi: 10.1007/s00210-006-0075-0
Chabowski, A., Górski, J., Glatz, J. F., Luiken, J. J., and Bonen, A. (2008). Protein-mediated fatty acid uptake in the heart. Curr. Cardiol. Rev. 4, 12–21. doi: 10.2174/157340308783565429
Chang, L., Chiang, S. H., and Saltiel, A. R. (2004). Insulin signaling and the regulation of glucose transport. Mol. Med. 10, 65–71. doi: 10.2119/2005-00029
Chen, T., Liu, J., Li, N., Wang, S., Liu, H., Li, J., et al. (2015). Mouse SIRT3 attenuates hypertrophy-related lipid accumulation in the heart through the deacetylation of LCAD. PLoS One 10:e0118909. doi: 10.1371/journal.pone.0118909
Chen, W., Li, F., He, C., Zhu, Y., and Tan, W. (2014). Elevated prevalence of abnormal glucose metabolism in patients with primary aldosteronism: a meta-analysis. Ir. J. Med. Sci. 183, 283–291. doi: 10.1007/s11845-013-1007-x
Christe, M. E., and Rodgers, R. L. (1994). Altered glucose and fatty acid oxidation in hearts of the spontaneously hypertensive rat. J. Mol. Cell. Cardiol. 26, 1371–1375. doi: 10.1006/jmcc.1994.1155
Christe, M. E., and Rodgers, R. L. (1995). Cardiac glucose and fatty acid oxidation in the streptozotocin-induced diabetic spontaneously hypertensive rat. Hypertension 25, 235–241. doi: 10.1161/01.hyp.25.2.235
Coprean, D., Roşioru, C., Toma, V., and Tofan, L. (1993). Heart metabolic modifications induced by nifedipine treatment in normo- and hypertensive Wistar rats. Rom. J. Physiol. 30, 109–113.
Corry, D. B., and Tuck, M. L. (2003). The effect of aldosterone on glucose metabolism. Curr. Hypertens. Rep. 5, 106–109. doi: 10.1007/s11906-003-0065-2
Dai, S., Fraser, H., and McNeill, J. H. (1992). Effects of deoxycorticosterone acetate on glucose metabolism in nondiabetic and streptozotocin-diabetic rats. Can. J. Physiol. Pharmacol. 70, 1468–1472. doi: 10.1139/y92-207
Dai, S., and McNeill, J. H. (1992). Myocardial performance of STZ-diabetic DOCA-hypertensive rats. Am. J. Physiol. 263(6 Pt 2), H1798–H1805. doi: 10.1152/ajpheart.1992.263.6.h1798
Daud, D., Badruzzaman, N. A., Sidik, N. J., and Tawang, A. (2016). Phaleria macrocarpa fruits methanolic extract reduces blood pressure and blood glucose in spontaneous hypertensive rats (SHR). J. Appl. Pharm. Sci. 6, 158–161. doi: 10.7324/JAPS.2016.600126
de las Fuentes, L., Herrero, P., Peterson, L. R., Kelly, D. P., Gropler, R. J., and Dávila-Román, V. G. (2003). Myocardial fatty acid metabolism: independent predictor of left ventricular mass in hypertensive heart disease. Hypertension 41, 83–87. doi: 10.1161/01.hyp.0000047668.48494.39
de las Fuentes, L., Soto, P. F., Cupps, B. P., Pasque, M. K., Herrero, P., Gropler, R. J., et al. (2006). Hypertensive left ventricular hypertrophy is associated with abnormal myocardial fatty acid metabolism and myocardial efficiency. J. Nucl. Cardiol. 13, 369–377. doi: 10.1016/j.nuclcard.2006.01.021
Depré, C., Rider, M. H., and Hue, L. (1998). Mechanisms of control of heart glycolysis. Eur. J. Biochem. 258, 277–290. doi: 10.1046/j.1432-1327.1998.2580277.x
Desvergne, B., Michalik, L., and Wahli, W. (2006). Transriptional regulation of metabolism. Physiol. Rev. 86, 465–514. doi: 10.1152/physrev.00025.2005
Doehner, W., Frenneaux, M., and Anker, S. D. (2014). Metabolic impairment in heart failure: the myocardial and systemic perspective. J. Am. Coll. Cardiol. 64, 1388–1400. doi: 10.1016/j.jacc.2014.04.083
Dolinsky, V. W., Chan, A. Y., Robillard Frayne, I., Light, P. E., Des Rosiers, C., and Dyck, J. R. (2009). Resveratrol prevents the prohypertrophic effects of oxidative stress on LKB1. Circulation 119, 1643–1652. doi: 10.1161/CIRCULATIONAHA.108.787440
Dorn, G. W., Vega, R. B., and Kelly, D. P. (2015). Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 29, 1981–1991. doi: 10.1101/gad.269894.115
Dyck, J. R., Cheng, J. F., Stanley, W. C., Barr, R., Chandler, M. P., Brown, S., et al. (2004). Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ. Res. 94, e78–e84. doi: 10.1161/01.RES.0000129255.19569.8f
Essop, M. F. (2007). Cardiac metabolic adaptations in response to chronic hypoxia. J. Physiol. 584(Pt 3), 715–726. doi: 10.1113/jphysiol.2007.143511
Faergeman, N. J., and Knudsen, J. (1997). Role of long-chain fatty acyl-CoA esters in the regulation of metabolism and in cell signalling. Biochem. J. 323(Pt 1), 1–12. doi: 10.1042/bj3230001
Febbraio, M., and Silverstein, R. L. (2007). CD36: implications in cardiovascular disease. Int. J. Biochem. Cell Biol. 39, 2012–2030. doi: 10.1016/j.biocel.2007.03.012
Fillmore, N., Levasseur, J. L., Fukushima, A., Wagg, C. S., Wang, W., Dyck, J. B., et al. (2018). Uncoupling of glycolysis from glucose oxidation accompanies the development of heart failure with preserved ejection fraction. Mol. Med. 24:3. doi: 10.1186/s10020-018-0005-x
Fillmore, N., and Lopaschuk, G. D. (2014). Malonyl CoA: a promising target for the treatment of cardiac disease. IUBMB Life 66, 139–146. doi: 10.1002/iub.1253
Fillmore, N., Mori, J., and Lopaschuk, G. D. (2014). Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 171, 2080–2090. doi: 10.1111/bph.12475
Folmes, C. D., Clanachan, A. S., and Lopaschuk, G. D. (2006). Fatty acids attenuate insulin regulation of 5’-AMP-activated protein kinase and insulin cardioprotection after ischemia. Circ. Res. 99, 61–68. doi: 10.1161/01.res.0000229656.05244.11
García-Puig, J., Ruilope, L. M., Luque, M., Fernández, J., Ortega, R., Dal-Ré, R., et al. (2006). Glucose metabolism in patients with essential hypertension. Am. J. Med. 119, 318–326. doi: 10.1016/j.amjmed.2005.09.010
Gimeno, R. E., Ortegon, A. M., Patel, S., Punreddy, S., Ge, P., Sun, Y., et al. (2003). Characterization of a heart-specific fatty acid transport protein. J. Biol. Chem. 278, 16039–16044. doi: 10.1074/jbc.M211412200
Goodwin, G. W., Arteaga, J. R., and Taegtmeyer, H. (1995). Glycogen turnover in the isolated working rat heart. J. Biol. Chem. 270, 9234–9240. doi: 10.1074/jbc.270.16.9234
Gopal, K., Saleme, B., Al Batran, R., Aburasayn, H., Eshreif, A., and Ho, K. L. (2017). FoxO1 regulates myocardial glucose oxidation rates via transcriptional control of pyruvate dehydrogenase kinase 4 expression. Am. J. Physiol. Heart Circ. Physiol. 313, H479–H490. doi: 10.1152/ajpheart.00191.2017
Gotoda, T., Iizuka, Y., Kato, N., Osuga, J., Bihoreau, M. T., Murakami, T., et al. (1999). Absence of Cd36 mutation in the original spontaneously hypertensive rats with insulin resistance. Nat. Genet. 22, 226–228. doi: 10.1038/10285
Gouveia, L. M., Kettelhut, I. C., and Foss, M. C. (2000). Abnormalities of glucose metabolism in spontaneously hypertensive rats. Braz. J. Med. Biol. Res. 33, 1357–1362. doi: 10.1590/s0100-879x2000001100015
Grillon, J. M., Johnson, K. R., Kotlo, K., and Danziger, R. S. (2012). Non-histone lysine acetylated proteins in heart failure. Biochim. Biophys. Acta 1822, 607–614. doi: 10.1016/j.bbadis.2011.11.016
Haider, A. W., Larson, M. G., Franklin, S. S., and Levy, D. (2003). Systolic blood pressure, diastolic blood pressure, and pulse pressure as predictors of risk for congestive heart failure in the Framingham Heart Study. Ann. Intern. Med. 138, 10–16. doi: 10.5494/wjh.v5.i2.85
Hajri, T., Ibrahimi, A., Coburn, C. T., Knapp, F. F., Kurtz, T., Pravenec, M., et al. (2001). Defective fatty acid uptake in the spontaneously hypertensive rat is a primary determinant of altered glucose metabolism, hyperinsulinemia, and myocardial hypertrophy. J. Biol. Chem. 276, 23661–23666. doi: 10.1074/jbc.M100942200
Hall, M. E., Harmancey, R., and Stec, D. E. (2015). Lean heart: role of leptin in cardiac hypertrophy and metabolism. World J. Cardiol. 26, 511–524. doi: 10.4330/wjc.v7.i9.511
Hamirani, Y. S., Kundu, B. K., Zhong, M., McBride, A., Li, Y., Davogustto, G. E., et al. (2016). Noninvasive detection of early metabolic left ventricular remodeling in systemic hypertension. Cardiology 133, 157–162. doi: 10.1159/000441276
Harasim-Symbor, E., Polak, A., Pȩdzińska-Betiuk, A., Weresa, J., Malinowska, B., Lewandowska, A., et al. (2019). Fatty acid amide hydrolase inhibitor (URB597) as a regulator of myocardial lipid metabolism in spontaneously hypertensive rats. Chem. Phys. Lipids 218, 141–148. doi: 10.1016/j.chemphyslip.2018.12.007
Hershberger, K. A., Abraham, D. M., and Martin, A. S. (2017). Sirtuin 5 is required for mouse survival in response to cardiac pressure overload. J. Biol. Chem. 292, 19767–19781. doi: 10.1074/jbc.M117.809897
Hirschey, M. D., Shimazu, T., Goetzman, E., Jing, E., Schwer, B., Lombard, D. B., et al. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125. doi: 10.1038/nature08778
Hulman, S., Falkner, B., and Freyvogel, N. (1993). Insulin resistance in the conscious spontaneously hypertensive rat: euglycemic hyperinsulinemic clamp study. Metabolism 42, 14–18. doi: 10.1016/0026-0495(93)90165-k
Huss, J. M., Torra, I. P., Staels, B., Giguere, V., and Kelly, D. P. (2004). Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell Biol. 24, 9079–9091. doi: 10.1128/MCB.24.20.9079-9091.2004
Ingwall, J. S., Kramer, M. F., Fifer, M. A., Lorell, B. H., Shemin, R., Grossman, W., et al. (1985). The creatine kinase system in normal and diseased human myocardium. N. Engl. J. Med. 313, 1050–1054. doi: 10.1056/NEJM198510243131704
Jamshidi, Y., Montgomery, H. E., Hense, H. W., Myerson, S. G., Torra, I. P., Staels, B., et al. (2002). Peroxisome proliferator–activated receptor alpha gene regulates left ventricular growth in response to exercise and hypertension. Circulation 105, 950–955. doi: 10.1161/hc0802.104535
Karwi, Q. G., Uddin, G. M., Ho, K. L., and Lopaschuk, G. D. (2018). Loss of metabolic flexibility in the failing heart. Front. Cardiovasc. Med. 5:68. doi: 10.3389/fcvm.2018.00068
Kashiwagi, Y., Nagoshi, T., Yoshino, T., Tanaka, T. D., Ito, K., Harada, T., et al. (2015). Expression of SGLT1 in human hearts and impairment of cardiac glucose uptake by phlorizin during ischemia-reperfusion injury in mice. PLoS One 10:e0130605. doi: 10.1371/journal.pone.0130605
Kato, T., Niizuma, S., Inuzuka, Y., Kawashima, T., Okuda, J., Tamaki, Y., et al. (2010). Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ. Heart Fail. 3, 420–430. doi: 10.1161/CIRCHEARTFAILURE.109.888479
Kim, T. T., and Dyck, J. R. (2016). The role of CD36 in the regulation of myocardial lipid metabolism. Biochim. Biophys. Acta 1861, 1450–1460. doi: 10.1016/j.bbalip.2016.03.018
Klevstig, M. J., Markova, I., Burianova, J., Kazdova, L., Pravenec, M., Novakova, O., et al. (2011). Role of FAT/CD36 in novel PKC isoform activation in heart of spontaneously hypertensive rats. Mol. Cell. Biochem. 357, 163–169. doi: 10.1007/s11010-011-0886-2
Kodde, I. F., van der Stok, J., Smolenski, R. T., and de Jong, J. W. (2007). Metabolic and genetic regulation of cardiac energy substrate preference. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 146, 26–39. doi: 10.1016/j.cbpa.2006.09.014
Koehler, U., and Medugorac, I. (1985). Left ventricular enzyme activities of the energy-supplying metabolism in Goldblatt-II rats. Res. Exp. Med. 185, 299–307. doi: 10.1007/bf01851955
Kolwicz, S. C. (2018). An “Exercise” in cardiac metabolism. Front. Cardiovasc. Med. 5:66. doi: 10.3389/fcvm.2018.00066
Kolwicz, S. C., Olson, D. P., Marney, L. C., Garcia-Menendez, L., Synovec, R. E., and Tian, R. (2012). Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ. Res. 111, 728–738. doi: 10.1161/CIRCRESAHA.112.268128
Kolwicz, S. C., Purohit, S., and Tian, R. (2013). Cardiac metabolism and its interactions with contraction, growth, and survival of the cardiomyocytes. Circ. Res. 113, 603–616. doi: 10.1161/CIRCRESAHA.113.302095
Labarthe, F., Khairallah, M., Bouchard, B., Stanley, W. C., and Des Rosiers, C. (2005). Fatty acid oxidation and its impact on response of spontaneously hypertensive rat hearts to an adrenergic stress: benefits of a medium-chain fatty acid. Am. J. Physiol. Heart Circ. Physiol. 288, H1425–H1436. doi: 10.1152/ajpheart.00722.2004
Laczy, B., Fulop, N., Onay-Besikci, A., Des Rosiers, C., and Chatham, J. C. (2011). Acute regulation of cardiac metabolism by the hexosamine biosynthesis pathway and protein o-glcnacylation. PLoS One 6:e18417. doi: 10.1371/journal.pone.0018417
Laine, H., Katoh, C., Luotolahti, M., Yki-Järvinen, H., Kantola, I., Jula, A., et al. (1999). Myocardial oxygen consumption is unchanged but efficiency is reduced in patients with essential hypertension and left ventricular hypertrophy. Circulation 100, 2425–2430. doi: 10.1161/01.cir.100.24.2425
Lamb, H. J., Beyerbacht, H. P., van der Laarse, A., Stoel, B. C., Doornbos, J., van der Wall, E. E., et al. (1999). Diastolic dysfunction in hypertensive heart disease is associated with altered myocardial metabolism. Circulation 99, 2261–2267. doi: 10.1161/01.cir.99.17.2261
LaPier, T. L., and Rodnick, K. J. (2000). Changes in cardiac energy metabolism during early development of female SHR. Am. J. Hypertens. 13, 1074–1081. doi: 10.1016/s0895-7061(00)00297-1
Lauzier, B., Merlen, C., Vaillant, F., McDuff, J., Bouchard, B., Beguin, P. C., et al. (2011). Post-translational modifications, a key process in CD36 function: lessons from the spontaneously hypertensive rat heart. J. Mol. Cell. Cardiol. 51, 99–108. doi: 10.1016/j.yjmcc.2011.04.001
Lee, L. K., Kim, M. Y., Kim, J. H., Lee, J. U., Park, B. S., Yang, S. M., et al. (2015). A review of deoxycorticosterone acetate-salt hypertension and its relevance for cardiovascular physiotherapy research. J. Phys. Ther. Sci. 27, 303–307. doi: 10.1589/jpts.27.303
Lee, T. W., Bai, K. J., Lee, T. I., Chao, T. F., Kao, Y. H., and Chen, Y. J. (2017). PPARs modulate cardiac metabolism and mitochondrial function in diabetes. J. Biomed. Sci. 24:5. doi: 10.1186/s12929-016-0309-5
Leone, T. C., and Kelly, D. P. (2011). Transcriptional control of cardiac fuel metabolism and mitochondrial function. Cold Spring Harb. Symp. Quant. Biol. 76, 175–182. doi: 10.1101/sqb.2011.76.011965
Lewington, S., Clarke, R., Qizilbash, N., Peto, R., Collins, R., and Prospective Studies Collaboration. (2002). Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 360, 1903–1913. doi: 10.1016/s0140-6736(02)11911-8
Li, J., Huang, Q., Cui, S., Luo, D., Desai, S., Patrie, J., et al. (2018). Metabolic, functional and structural remodeling of spontaneously hypertensive rat hearts over time. J. Nucl. Med. 59(Suppl. 1):366.
Liška, F., Landa, V., Zídek, V., Mlejnek, P., Šilhavý, J., Šimáková, M., et al. (2017). Downregulation of Plzf gene ameliorates metabolic and cardiac traits in the spontaneously hypertensive rat. Hypertension 69, 1084–1091. doi: 10.1161/HYPERTENSIONAHA.116.08798
Lopaschuk, G. D. (2017). Metabolic modulators in heart disease: past, present, and future. Can. J. Cardiol. 33, 838–849. doi: 10.1016/j.cjca.2016.12.013
Lopaschuk, G. D., Ussher, J. R., Folmes, C. D., Jaswal, J. S., and Stanley, W. C. (2010). Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 90, 207–258. doi: 10.1152/physrev.00015.2009
Luiken, J. J., Coort, S. L., Koonen, D. P., Bonen, A., and Glatz, J. F. (2004). Signalling components involved in contraction-inducible substrate uptake into cardiac myocytes. Proc. Nutr. Soc. 63, 251–258. doi: 10.1079/PNS2004333
Luiken, J. J., Dyck, D. J., Han, X. X., Tandon, N. N., Arumugam, Y., Glatz, J. F., et al. (2002). Insulin induces the translocation of the fatty acid transporter FAT/CD36 to the plasma membrane. Am. J. Physiol. Endocrinol. Metab. 282, E491–E495. doi: 10.1152/ajpendo.00419.2001
Luiken, J. J., van Nieuwenhoven, F. A., America, G., van der Vusse, G. J., and Glatz, J. F. (1997). Uptake and metabolism of palmitate by isolated cardiac myocytes from adult rats: involvement of sarcolemmal proteins. J. Lipid Res. 38, 745–758.
Madrazo, J. A., and Kelly, D. P. (2008). The PPAR trio: regulators of myocardial energy metabolism in health and disease. J. Mol. Cell. Cardiol. 44, 968–975. doi: 10.1016/j.yjmcc.2008.03.021
Messerli, F. H., Rimoldi, S. F., and Bangoalore, S. (2017). The transition from hypertension to heart failure: contemporary update. JACC Heart Fail. 5, 543–551. doi: 10.1016/j.jchf.2017.04.012
Mora, A., Komander, D., van Aalten, D. M., and Alessi, D. R. (2004). PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol. 15, 161–170. doi: 10.1016/j.semcdb.2003.12.022
Mori, J., Basu, R., McLean, B. A., Das, S. K., Zhang, L., Patel, V. B., et al. (2012). Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: a metabolic contribution to heart failure with normal ejection fraction. Circ. Heart Fail. 5, 493–503. doi: 10.1161/circheartfailure.112.966705
Morimoto, S., and Goto, T. (2000). Role of troponin I isoform switching in determining the pH sensitivity of Ca2+ regulation in developing rabbit cardiac muscle. Biochem. Biophys. Res. Commun. 267, 912–917. doi: 10.1006/bbrc.1999.2068
Ndisang, J. F., and Jadhav, A. (2010). The heme oxygenase system attenuates pancreatic lesions and improves insulin sensitivity and glucose metabolism in deoxycorticosterone acetate hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R211–R223. doi: 10.1152/ajpregu.91000.2008
Nishida, Y., Rardin, M. J., Carrico, C., He, W., Sahu, A. K., Gut, P., et al. (2015). SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol. Cell 59, 321–332. doi: 10.1016/j.molcel.2015.05.022
Okamoto, K., and Aoki, K. (1963). Development of a strain of spontaneously hypertensive rats. Jpn. Circ. J. 27, 282–293. doi: 10.1253/jcj.27.282
Onat, A., Hergenç, G., Sari, I., Türkmen, S., Can, G., and Sansoy, V. (2005). Dyslipidemic hypertension: distinctive features and cardiovascular risk in a prospective population-based study. Am. J. Hypertens. 18, 409–416. doi: 10.1016/j.amjhyper.2004.10.017
Onay-Besikci, A., Altarejos, J. Y., and Lopaschuk, G. D. (2004). gAd-globular head domain of adiponectin increases fatty acid oxidation in newborn rabbit hearts. J. Biol. Chem. 279, 44320–44326. doi: 10.1074/jbc.M400347200
Park, J., Chen, Y., Tishkoff, D. X., Peng, C., Tan, M., Dai, L., et al. (2013). SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 50, 919–930. doi: 10.1016/j.molcel.2013.06.001
Pellieux, C., Montessuit, C., Papageorgiou, I., and Lerch, R. (2009). Angiotensin II downregulates the fatty acid oxidation pathway in adult rat cardiomyocytes via release of tumour necrosis factor-alpha. Cardiovasc. Res. 82, 341–350. doi: 10.1093/cvr/cvp004
Polak, A., Harasim, E., and Chabowski, A. (2016). Effects of activation of endocannabinoid system on myocardial metabolism. Postepy Hig. Med. Dosw. 70, 542–555. doi: 10.5604/17322693.1202483
Polak, A., Harasim-Symbor, E., and Chabowski, A. (2018a). Animal models of hypertension - revisited. Prog. Health Sci. 8, 167–175. doi: 10.5604/01.3001.0012.1116
Polak, A., Harasim-Symbor, E., Malinowska, B., Kasacka, I., Lewandowska, A., and Chabowski, A. (2018b). The endocannabinoid system affects myocardial glucose metabolism in the DOCA-salt model of hypertension. Cell. Physiol. Biochem. 46, 727–739. doi: 10.1159/000488730
Polak, A., Harasim-Symbor, E., Malinowska, B., Kasacka, I., Pȩdzińska-Betiuk, A., Weresa, J., et al. (2017). The effects of chronic FAAH inhibition on myocardial lipid metabolism in normotensive and DOCA-salt hypertensive rats. Life Sci. 183, 1–10. doi: 10.1016/j.lfs.2017.06.019
Prahalathan, P., Saravanakumar, M., and Raja, B. (2012). The flavonoid morin restores blood pressure and lipid metabolism in DOCA-salt hypertensive rats. Redox Rep. 17, 167–175. doi: 10.1179/1351000212Y.0000000015
Puthanveetil, P., Wan, A., and Rodrigues, B. (2013). FoxO1 is crucial for sustaining cardiomyocyte metabolism and cell survival. Cardiovasc. Res. 97, 393–403. doi: 10.1093/cvr/cvs426
Quintana-Villamandos, B., Delgado-Martos, M. J., Fernandez-Riveira, C., Fernández-Criado, M. C., Martos-Rodríguez, A., Canillas, F., et al. (2013). Can 18F-FDG–PET show differences in myocardial metabolism between Wistar Kyoto rats and spontaneously hypertensive rats? Lab. Anim. 47, 320–323. doi: 10.1177/0023677213495668
Reutter, B. W., Huesman, R. H., Brennan, K. M., Boutchko, R., Hanrahan, S. M., and Gullberg, G. T. (2011). Longitudinal evaluation of fatty acid metabolism in normal and spontaneously hypertensive rat hearts with dynamic MicroSPECT imaging. Int. J. Mol. Imaging 2011:893129. doi: 10.1155/2011/893129
Riehle, C., Wende, A. R., Zaha, V. G., Pires, K. M., Wayment, B., Olsen, C., et al. (2011). PGC-1β deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ. Res. 109, 783–793. doi: 10.1161/circresaha.111.243964
Rosca, M. G., and Hoppel, C. L. (2013). Mitochondrial dysfunction in heart failure. Heart Fail. Rev. 18, 607–622. doi: 10.1007/s10741-012-9340-0
Rowe, G. C., Jiang, A., and Arany, Z. (2010). PGC-1 coactivators in cardiac development and disease. Circ. Res. 107, 825–838. doi: 10.1161/CIRCRESAHA.110.223818
Saleme, B., and Sutendra, G. (2018). A similar metabolic profile between the failing myocardium and tumor could provide alternative therapeutic targets in chemotherapy-induced cardiotoxicity. Front. Cardiovasc. Med. 5:61. doi: 10.3389/fcvm.2018.00061
Santalucía, T., Camps, M., Castelló, A., Muñoz, P., Nuel, A., Testar, X., et al. (1992). Developmental regulation of GLUT-1 (erythroid/Hep G2) and GLUT-4 (muscle/fat) glucose transporter expression in rat heart, skeletal muscle, and brown adipose tissue. Endocrinology 130, 837–846. doi: 10.1210/endo.130.2.1370797
Schönfeld, P., and Wojtczak, L. (2016). Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J. Lipid Res. 57, 943–954. doi: 10.1194/jlr.R067629
Smith, S. H., Kramer, M. F., Reis, I., Bishop, S. P., and Ingwall, J. S. (1990). Regional changes in creatine kinase and myocyte size in hypertensive and nonhypertensive cardiac hypertrophy. Circ. Res. 67, 1334–1344. doi: 10.1161/01.res.67.6.1334
Stanley, W. C., and Chandler, M. P. (2002). Energy metabolism in the normal and failing heart: potential for therapeutic interventions. Heart Fail. Rev. 7, 115–130. doi: 10.1023/A:1015320423577
Stanley, W. C., Meadows, S. R., Kivilo, K. M., Roth, B. A., and Lopaschuk, G. D. (2003). Beta-Hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl-CoA content. Am. J. Physiol. Heart Circ. Physiol. 285, H1626–H1631. doi: 10.1152/ajpheart.00332.2003
Stanley, W. C., Recchia, F. A., and Lopaschuk, G. D. (2005). Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 85, 1093–1129. doi: 10.1152/physrev.00006.2004
Sugden, M. C., and Holness, M. J. (2003). Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am. J. Physiol. Endocrinol. Metab. 284, E855–E862. doi: 10.1152/ajpendo.00526.2002
Swislocki, A. L. M., Kinney LaPier, T. L., Khuu, D. T., Fann, K. Y., Tait, M., and Rodnick, K. J. (1999). Metabolic, hemodynamic, and cardiac effects of captopril in young, spontaneously hypertensive rats. Am. J. Hypertens. 12, 581–589. doi: 10.1016/S0895-7061(99)00012-6
Szablewski, L. (2017). Glucose transporters in healthy heart and in cardiac disease. Int. J. Cardiol. 1, 70–75. doi: 10.1016/j.ijcard.2016.12.083
Taegtmeyer, H., and Overturf, M. L. (1988). Effects of moderate hypertension on cardiac function and metabolism in the rabbit. Hypertension 11, 416–426. doi: 10.1161/01.hyp.11.5.416
Taegtmeyer, H., Sen, S., and Vela, D. (2010). Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann. N. Y. Acad. Sci. 1188, 191–198. doi: 10.1111/j.1749-6632.2009.05100.x
Vincent, G., Khairallah, M., Bouchard, B., and Des Rosiers, C. (2003). Metabolic phenotyping of the diseased rat heart using 13C-substrates and ex vivo perfusion in the working mode. Mol. Cell. Biochem. 242, 89–99. doi: 10.1007/978-1-4757-4712-6_12
Wang, C., and Hu, S. M. (1991). Developmental regulation in the expression of rat heart glucose transporters. Biochem. Biophys. Res. Commun. 177, 1095–1100. doi: 10.1016/0006-291x(91)90651-m
Wang, J., Li, Z., Wang, Y., Zhang, J., Zhao, W., Fu, M., et al. (2017). Qiliqiangxin enhances cardiac glucose metabolism and improves diastolic function in spontaneously hypertensive rats. Evid. Based Complement. Alternat. Med. 2017:3197320. doi: 10.1155/2017/3197320
Wang, T., McDonald, C., Petrenko, N. B., Leblanc, M., Wang, T., Giguere, V., et al. (2015). Estrogen-related receptor α (ERRα) and ERRγ are essential coordinators of cardiac metabolism and function. Mol. Cell Biol. 35, 1281–1298. doi: 10.1128/MCB.01156-14
Whelton, P. K., Carey, R. M., Aronow, W. S., Casey, D. E., Collins, K. J., Dennison Himmelfarb, C., et al. (2017). 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. J. Am. Coll. Cardiol. 71, 1269–1324. doi: 10.1016/j.jacc.2017.11.006
Wu, P., Peters, J., and Harris, R. (2001). Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem. Biophys. Res. Commun. 287, 391–396. doi: 10.1006/bbrc.2001.5608
Yonekura, Y., Brill, A. B., Som, P., Yamamoto, K., Srivastava, S. C., Iwai, J., et al. (1985). Regional myocardial substrate uptake in hypertensive rats: a quantitative autoradiographic measurement. Science 227, 1494–1496. doi: 10.1126/science.3975623
Zecchin, H. G., Bezerra, R. M., Carvalheira, J. B., Carvalho-Filho, M. A., Metze, K., Franchini, K. G., et al. (2003). Insulin signalling pathways in aorta and muscle from two animal models of insulin resistance–the obese middle-aged and the spontaneously hypertensive rats. Diabetologia 46, 479–491. doi: 10.1007/s00125-003-1073-0
Zhang, S., Hulver, M. W., Mcmillan, R. P., Cline, M. A., and Gilbert, E. R. (2014). The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. 11:10. doi: 10.1186/1743-7075-11-10
Keywords: DOCA-salt, FAT/CD36, heart metabolism, hypertension, SHR
Citation: Polak-Iwaniuk A, Harasim-Symbor E, Gołaszewska K and Chabowski A (2019) How Hypertension Affects Heart Metabolism. Front. Physiol. 10:435. doi: 10.3389/fphys.2019.00435
Received: 08 January 2019; Accepted: 29 March 2019;
Published: 16 April 2019.
Edited by:
Tommaso Angelone, University of Calabria, ItalyReviewed by:
Christian Cadeddu Dessalvi, University of Cagliari, ItalyAndrea Gerbino, University of Bari Aldo Moro, Italy
Giulia Borghetti, Temple University, United States
Copyright © 2019 Polak-Iwaniuk, Harasim-Symbor, Gołaszewska and Chabowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agnieszka Polak-Iwaniuk, YWduLnBvbGFrQHdwLnBs