 Cristian Tornador1,2
Cristian Tornador1,2 Edgar Sánchez-Prados3
Edgar Sánchez-Prados3 Beatriz Cadenas4,5,6
Beatriz Cadenas4,5,6 Roberta Russo7,8
Roberta Russo7,8 Veronica Venturi9
Veronica Venturi9 Immacolata Andolfo7,8
Immacolata Andolfo7,8 Ines Hernández-Rodriguez10
Ines Hernández-Rodriguez10 Achille Iolascon7,8
Achille Iolascon7,8 Mayka Sánchez1,9*
Mayka Sánchez1,9*- 1BloodGenetics S.L., Barcelona, Spain
- 2Teresa Moreto Foundation, Barcelona, Spain
- 3Bioinformatics for Health Sciences Master Programme, Universitat Pompeu Fabra, Barcelona, Spain
- 4Whole Genix SL., Barcelona, Spain
- 5Universitat de Vic-Universitat Central de Catalunya, Vic, Spain
- 6Iron Metabolism: Regulation and Diseases Group, Josep Carreras Leukaemia Research Institute, Campus Can Ruti, Barcelona, Spain
- 7Department of Molecular Medicine and Medical Biotechnologies, University of Naples Federico II, Naples, Italy
- 8CEINGE–Biotecnologie Avanzate, Naples, Italy
- 9Iron Metabolism: Regulation and Diseases Group, Department of Basic Sciences, Faculty of Medicine and Health Sciences, Universitat Internacional de Catalunya, Barcelona, Spain
- 10Haematology Service, Hospital Germans Trias i Pujol University Hospital, Oncology Catalan Institute, Barcelona, Spain
Congenital Dyserythropoietic Anemia (CDA) is a heterogeneous group of hematological disorders characterized by chronic hyporegenerative anemia and distinct morphological abnormalities of erythroid precursors in the bone marrow. In many cases, a final diagnosis is not achieved due to different levels of awareness for the diagnosis of CDAs and lack of use of advanced diagnostic procedures. Researchers have identified five major types of CDA: types I, II, III, IV, and X-linked dyserythropoietic anemia and thrombocytopenia (XLDAT). Proper management in CDA is still unsatisfactory, as the different subtypes of CDA have different genetic causes and different but overlapping patterns of signs and symptoms. For this reason, we developed a new telemedicine tool that will help doctors to achieve a faster diagnostic for this disease. Using open access code, we have created a responsive webpage named CoDysAn (Congenital Dyserythropoietic Anemia) that includes practical information for CDA awareness and a step-by-step diagnostic tool based on a CDA algorithm. The site is currently available in four languages (Catalan, Spanish, Italian, and English). This telemedicine webpage is available at http://www.codysan.eu.
Introduction
Congenital Dyserythropoietic Anemia (CDA) is a heterogeneous group of hematological disorders characterized by chronic hyporegenerative anemia and distinct morphological abnormalities of erythroid precursors in the bone marrow. Patients with CDA present congenital and chronic anemia of variable degree with a reticulocytosis not corresponding to the degree of anemia (ineffective erythropoiesis), jaundice and frequently splenomegaly and/or hepatomegaly (Iolascon et al., 2012, 2013).
Five classical types of CDAs (I–II–III–IV and XLTDA) have been defined based on bone marrow morphology. Among all types, CDA type II is the most common and well-known form. Genetically, CDA type Ia (OMIN 224120) and CDA type Ib (OMIM 615631) are caused by mutations in codanin 1 (CDAN1) (chr 15q15.2) and C15orf41 (chr15q14) genes, respectively (Dgany et al., 2002; Babbs et al., 2013). CDA type II (OMIM 224100) is due to pathogenic variants in Sec23 homolog B, coat complex II component (SEC23B) gene (chr20p11.23) (Bianchi et al., 2009; Schwarz et al., 2009). Few patients with CDA type III (OMIM 105600) have been described: they present the same mutation in the Kinesin Family Member 23 (KIF23) gene (chr15q23) (Liljeholm et al., 2013). CDA type IV (OMIM 613673) is due to mutations in the Kruppel Like Factor 1 (KLF1) gene (chr19p13.13) (Arnaud et al., 2010; Jaffray et al., 2013). Finally, X-linked dyserythropoietic anemia and thrombocytopenia (XLDAT) (OMIM 300367) is caused by mutations in transcription factor GATA Binding Protein 1 (GATA1) gene (chr Xp11.23) (Nichols et al., 2000; Del Vecchio et al., 2005). CDA types I and II are inherited in an autosomal recessive manner, CDA type III and IV present an autosomal dominant inheritance pattern and X-linked dyserythropoietic anemia with thrombocytopenia has an X-linked mode of inheritance.
Depending on the type of CDA, different treatments have been established. Allogenic bone marrow transplantation has been successfully employed in a few severe cases of CDAI and CDAII. CDA III patients may require a transfusion only during times of extreme anemia e.g., pregnancy or surgery. Treatment focuses on hemoglobin normalization with the administration of interferon (IFN) alpha is used with success in CDA I patients with CDAN1 mutations; however, patients bearing a mutation in a different gene i.e., C15ORF41 were unresponsive to this same treatment. Severe cases of fetal anemia associated with CDAI, CDAII, and XLTDA may require intrauterine transfusions. Blood iron levels should be closely monitored in CDA I, CDAII, and other CDA patients undergoing regular transfusions. In these cases, morbidity may be severe due to iron overload complications that can be fatal if left untreated (Gambale et al., 2016; Palmer et al., 2018); therefore, it is imperative to monitor iron overload and induce iron depletion, when needed, by iron chelation. This working classification of CDA is still in use in clinical practice; however, the identification of the mutated genes involved in the majority of CDA subgroups will improve the diagnostic possibilities and allow a better classification of CDA patients. At present, in many cases, a final diagnosis is not achieved due to different levels of awareness for the diagnosis of CDAs and lack of use of advanced diagnostic procedures. In addition, there are families that fulfill the general definition of CDAs, but do not conform to any of the classical CDA variants. Therefore, it is very plausible that new forms of CDA may exist. These new forms will be possibly identified if a proper diagnosed is achieved in each patient suspected with CDA. Toward this goal, we have developed a new telemedicine tool named CoDysAn (Congenital Dyserythropoietic Anemia) for the management and diagnosis of patients with this disease.
The aim of CoDysAn webpage is to provide a freely accessible website where general public, patients and medical doctors can better understand and learn more about this disease. Moreover, CoDysAn web page includes a diagnosis algorithm tool to ease the classification and diagnostic of CDA types.
Methods
Patients and Validation
CoDysAn web algorithm has been developed with a set of 24 patients genetically diagnosed of different types of CDA (18 CDA type II, 1 CDA type Ib, 4 CDA type Ia, and 1 XLTDA) and with a set of 19 additional patients genetically diagnosed of non-CDA hereditable anemias including eight hereditary spherocytosis, four patients with pyruvate kinase defects, one patient with pyruvate kinase defect and a beta thalassemia trait, one patient with defects in hemolytic anemia due to adenylate kinase deficiency (AK1) gene, one patient with X-linked sideroblastic anemia, three patients with dehydrated hereditary stomatocytosis type 1 (DHS1) and one patient with dehydrated hereditary stomatocytosis type 2 (DHS2). A different set of 23 CDAII patients was utilized to independently validate the algorithm. Patients were previously reported (Iolascon et al., 2009; Schwarz et al., 2009; Russo et al., 2010, 2011, 2013, 2014, 2016, 2018; Unal et al., 2014; Andolfo et al., 2015, 2018; Di Pierro et al., 2015) and diagnosed at the Medical Genetics Unit of A.O.U. Federico II, CEINGE–Biotecnologie Avanzate (Napoli).
Design of Web Server
CoDysAn is implemented in PHP, HTML5, CSS, and Javascript. The web server is executed in a XAMPP. Network visualization and interactive exploration modules are based on several open-source projects: Bootstrap, jQuery and Filezilla. The source code of the diagnostic tool algorithm is implemented in php at http://www.codysan.eu/diagnostics-tool.html. It is integrated within this web page between lines 661 and 1,204 in four steps corresponding to the four steps of the form. The code can be checked by typing in a browser: “view-source: http://www.codysan.eu/diagnostics-tool.html.”
Implementation
CoDysAn algorithm is based on the diagnostic workflow previously proposed (Iolascon et al., 2012; Gambale et al., 2016). This algorithm is based on hematological parameters depending on age and gender (Table 1). Age is split in three groups: from 0 to 6 months old; from 6 months to 12 years old; and older than 12 years. Hematological tested parameters include: hemoglobin levels, mean corpuscular volume (MCV), reticulocytes count and platelets count. Exclusion of other possible causes of anemia is also considered in the final step of the algorithm. References values for hematological data are adapted from general hematological reference books (Rabinovitch, 1990; Wakeman et al., 2007; Hoffman et al., 2018).
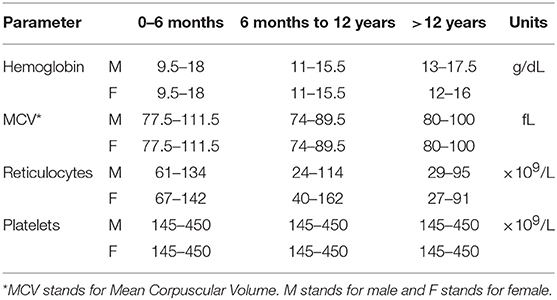
Table 1. Parameter thresholds used by the diagnostic CoDysAn algorithm.
Results
CoDySan Scope
Following previous experience of the group in developing telemedicine tools for management and diagnosis of patients (HIGHFERRITIN Web server http://highferritin.imppc.org/ tool) (Altes et al., 2014), we have developed CoDySan web tool. CoDySan is a user-friendly webpage for a better awareness on the rare hereditary hematological diseases, congenital dyserythropoietic anemias (CDAs). CoDySan webpage includes a step-by-step diagnostic algorithm based on Figure 1. The site is freely available at URL http://www.codysan.eu in four languages (Catalan, Spanish, Italian, and English).
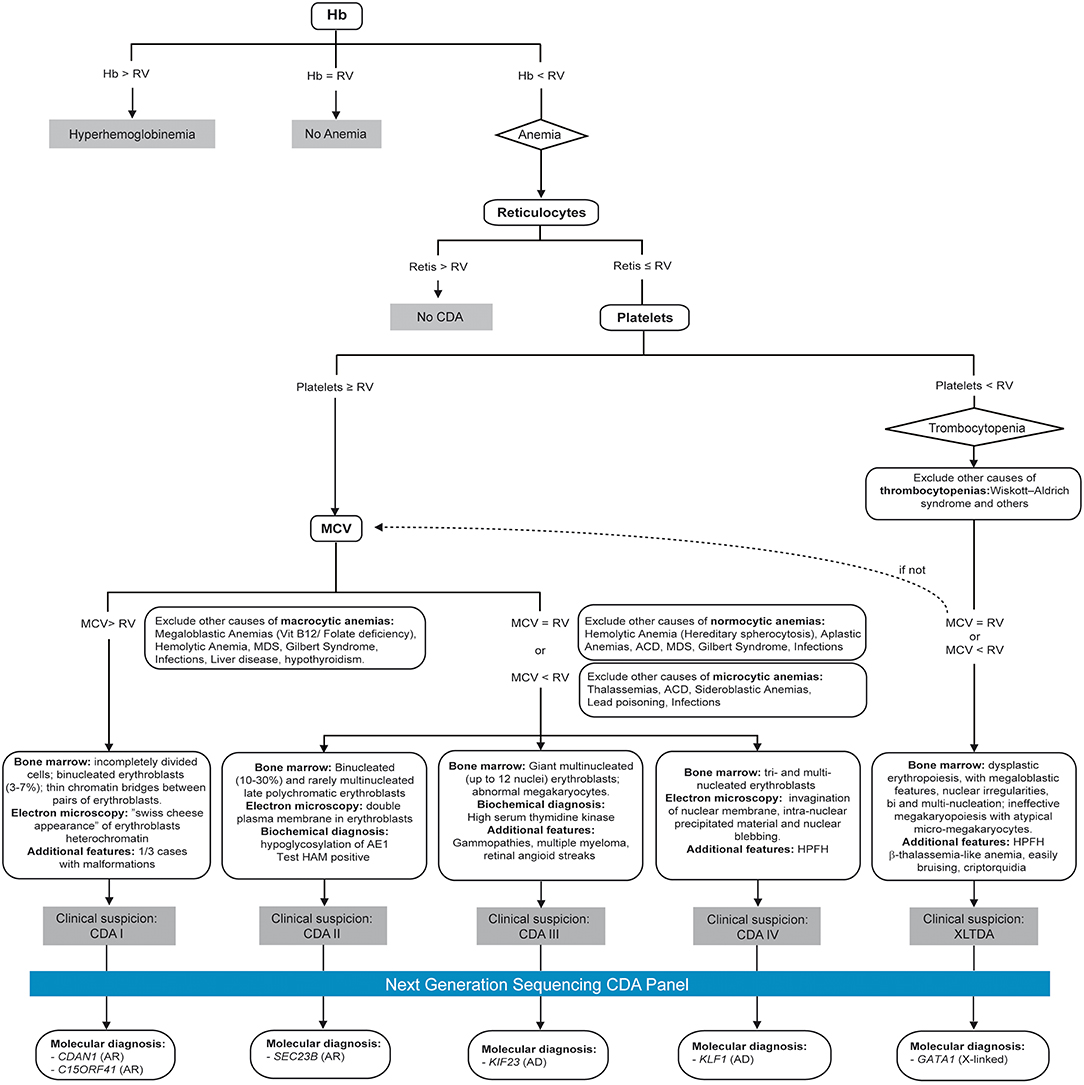
Figure 1. Diagnostic algorithm used by the CoDysAn telemedicine tool. Hb, hemoglobin; RV, reference value; MCV, mean corpuscular volume; HPFH, hereditary persistence of fetal hemoglobin; AR, autosomal recessive; AD, autosomal dominant.
Webpage Structure and Design
The CoDysAn website is currently containing seven sections (see Supplementary Figure 1 to visualize several screenshots of different sections of CoDysAn webpage): A home or main webpage section including links to other sections of the site to make information more accessible; The CoDysAn section, where one can found information about congenital dyserythropoietic anemia (CDA) disease, the CoDysAn project as a whole research project, the privacy policy, cookies policy and medical disclaimer reminding that, as any other telemedicine project, CoDysAn diagnostic tool is a preliminary diagnostic test and expert medical doctors should be contacted for a conclusive diagnosis; The diagnostic section, including the CDA algorithm flowchart (Figure 1) and a step-by-step diagnostic tool with specific instructions on how to use it; The collaborators section, including links to the contributors for the CoDysAn project, patient associations and links to similar web tools, such as HIGHFERRITIN web server; A resource section, including news on the CoDysAn project, bibliographical references and reference values used for the diagnostic algorithm (see also Table 1); An opinion section containing a Google form that allows users to express their opinion and degree of satisfaction with the website; A contact section, where users can directly contact CoDysAn developers to address any doubt regarding the webpage.
Diagnostic Telemedicine Tool
The diagnostic algorithm used for setting up the CoDysAn diagnostic tool is depicted in Figure 1. A step-by-step and user-friendly form will progressively ask relevant patient information; in the first stage, age, gender and hemoglobin levels should be provided to discern if the patient has hyperhemoglobinemia (high hemoglobin values in regards to the reference value), anemia or if the values are inside the normal range, in which case the web tool will return a text indicating that there is no anemia.
Due to fluctuations in the hematological parameters, the algorithm correlates to the reference values for the hematological provided data (hemoglobin level, reticulocytes, platelets, etc.) according to age and gender, see Table 1. To simplify the algorithm, we have only considered three different age ranges, from 0 to 6 months, from 6 to 12 years, and older than 12 years old.
If anemia is detected, i.e., the hemoglobin levels are below the normal values for the indicated gender and age range, the algorithm will ask for three additional hematological parameters: mean corpuscular volume (MCV), reticulocytes count, and platelets count.
Users can change the input units of the provided hematological parameter. These values are converted to the international system of reference units and the value is used to check if the parameters are within range for their given thresholds (see Table 1).
Depending on the data provided, a new form will appear asking to exclude specific possible causes of macrocytic, normocytic or microcytic anemia. At least one alternative cause of anemia should be excluded to proceed with the diagnostic tool. In the following step, the user is asked to select additional patient's clinical or biochemical factors, such as binucleated erythroblasts, malformations or electron microscopy features.
Finally, depending on the provided information, CoDySan tool will return a result of clinical suspicion (any of the CDA types) if it applies, or a brief explanation if there is no clinical suspicion of CDA.
If a clinical suspicion of CDA is indicated, the user has the option to search for world-wide genetic laboratories that provide clinical diagnostic tests for a particular CDA gene via the button “Search Lab.” The list of world-wide genetic laboratories is taken from the NCBI's Genetic Testing Registry (GTR) webpage (Rubinstein et al., 2013). There is also the possibility to refresh the webpage and perform a new diagnostic test via the button “New diagnostic.”
Validation
The CodysAn algorithm has been designed with 43 patients with hereditable anemia, including 24 patients genetically diagnosed with different types of CDA (18 CDA type II, 1 CDA type Ib, 4 CDA type IIa, and 1 XLTDA) and 19 additional patients genetically diagnosed with non-CDA hereditable anemias. The algorithm achieved a specificity of 89.5% and a sensibility of 87.5%. An additional set of 23 patients (all CDA II) was utilized to validate the algorithm, which returned a specificity of 87%.
Discussion
Telemedicine webpages and tools are significantly changing the way medical doctors and patients approach health care and diagnosis (Dinesen et al., 2016). CoDysAn telemedicine tool is a webpage intended to increase awareness about the rare disease CDA as, currently, patients suffering from this disease are under-diagnosed (Russo et al., 2014). The content of the webpage serves as an informative and training resource for the general public, patients and medical doctors. The use of this tool presents limits: patients should be considered as a whole entity and multiple biochemical determinations are needed due to daily parameters' variability within the same subject. Although hematological reference ranges are useful in results interpretation and in clinical decision-making, it should be borne in mind that variations within the population may affect some outcomes. CoDysAn incorporates a diagnostic algorithm that proved to be useful for a preliminary diagnostic. It will help medical doctors to know which molecular diagnostics they should request, reducing time and effort necessary for the diagnostic of CDA and allowing a direct implementation of a proper treatment once reached a definitive molecular diagnosis. Few reference centers are now offering genetic diagnostic panels screening the six known genes causing CDA. CoDySan algorithm is connected to the NCBI Genetic Testing Registry (GTR) in a way to inform medical doctors about the existence of these accredited diagnostic centers to perform a complete genetic test, if required. This telemedicine tool aims to inform the general public and aid in the diagnosis of CDA. It is not intended as an attempt to practice medicine or provide specific medical advice and it should not be used to replace or overrule a qualified health care provider's judgment. Users should not rely upon this website for self-medication. We believe that CoDysAn webpage will positively contribute to improve medical and scientific communication on the anemia field.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Author Contributions
MS designed the webpage, designed the study, and wrote the manuscript. ES-P, CT, and BC created the webpage and wrote the diagnostic algorithm. BC designed Figure 1. IA and RR translated the CoDysAn webpage to Italian. AI, IA, and RR provided the patients data to test CoDysAn algorithm. VV wrote and revised the manuscript. IH-R helped with reference values and Table 1. All authors read and approved the final version of the manuscript.
Funding
The CoDysAn project was supported by a research grant from Ramón Areces Foundation (Reference CIVP18A1857) to MS and AI, and by a collaborative agreement between the patient associations Pablo Ugarte Association (APU) and Patient and Relative Association of Congenital Dyserythropoietic Anemia (ADISCON).
Conflict of Interest Statement
CT was employed by company BloodGenetics SL. BC was employed by company Whole Genix SL.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Nuria Centeno from UPF for Master Project advisor tasks and Francisco Fuster from Josep Carreras Leukaemia Research Institute for assistance with the opinion questionnaire. This work was performed in the context of the Experimental Sciences and Technology Ph.D. program of the University of Vic (UVic).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2019.01063/full#supplementary-material
References
Altes, A., Perez-Lucena, M. J., Bruguera, M., and Grp Iberico, F. (2014). Systematic approach to the diagnosis of hyperferritinemia. Med. Clin. (Barc). 142, 412–417. doi: 10.1016/j.medcli.2013.06.010
Andolfo, I., Russo, R., Manna, F., Shmukler, B. E., Gambale, A., Vitiello, G., et al. (2015). Novel Gardos channel mutations linked to dehydrated hereditary stomatocytosis (xerocytosis). Am. J. Hematol. 90, 921–926. doi: 10.1002/ajh.24117
Andolfo, I., Russo, R., Rosato, B. E., Manna, F., Gambale, A., Brugnara, C., et al. (2018). Genotype-phenotype correlation and risk stratification in a cohort of 123 hereditary stomatocytosis patients. Am. J. Hematol. 93, 1509–1517. doi: 10.1002/ajh.25276
Arnaud, L., Saison, C., Helias, V., Lucien, N., Steschenko, D., Giarratana, M. C., et al. (2010). A Dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia. Am. J. Hum. Genet. 87, 721–727. doi: 10.1016/j.ajhg.2010.10.010
Babbs, C., Roberts, N. A., Sanchez-Pulido, L., McGowan, S. J., Ahmed, M. R., Brown, J. M., et al. (2013). Homozygous mutations in a predicted endonuclease are a novel cause of congenital dyserythropoietic anemia type I. Haematologica 98, 1383–1387. doi: 10.3324/haematol.2013.089490
Bianchi, P., Fermo, E., Vercellati, C., Boschetti, C., Barcellini, W., Iurlo, A., et al. (2009). Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Hum Mutat. 30, 1292–1298. doi: 10.1002/humu.21077
Del Vecchio, G. C., Giordani, L., De Santis, A., and De Mattia, D. (2005). Dyserythropoietic anemia and thrombocytopenia due to a novel mutation in GATA-1. Acta Haematol. 114, 113–116. doi: 10.1159/000086586
Dgany, O., Avidan, N., Delaunay, J., Krasnov, T., Shalmon, L., Shalev, H., et al. (2002). Congenital dyserythropoietic anemia type I is caused by mutations in codanin-1. Am. J. Hum. Genet. 71, 1467–1474. doi: 10.1086/344781
Di Pierro, E., Russo, R., Karakas, Z., Brancaleoni, V., Gambale, A., Kurt, I., et al. (2015). Congenital erythropoietic porphyria linked to GATA1-R216W mutation: challenges for diagnosis. Eur. J. Haematol. 94, 491–497. doi: 10.1111/ejh.12452
Dinesen, B., Nonnecke, B., Lindeman, D., Toft, E., Kidholm, K., Jethwani, K., et al. (2016). Personalized telehealth in the future: a global research agenda. J. Med. Internet Res. 18:e53. doi: 10.2196/jmir.5257
Gambale, A., Iolascon, A., Andolfo, I., and Russo, R. (2016). Diagnosis and management of congenital dyserythropoietic anemias. Expert Rev. Hematol. 9, 283–296. doi: 10.1586/17474086.2016.1131608
Hoffman, R., Benz, E. J. Jr., Silberstein, L. E., Heslop, H., Weitz, J., and Anastasi, J. (2018). Hematology: Basic Principles and Practice, 7th Edn. Philadelphia, PA: Elsevier Saunders.
Iolascon, A., Esposito, M. R., and Russo, R. (2012). Clinical aspects and pathogenesis of congenital dyserythropoietic anemias: from morphology to molecular approach. Haematologica 97, 1786–1794. doi: 10.3324/haematol.2012.072207
Iolascon, A., Heimpel, H., Wahlin, A., and Tamary, H. (2013). Congenital dyserythropoietic anemias: molecular insights and diagnostic approach. Blood 122, 2162–2166. doi: 10.1182/blood-2013-05-468223
Iolascon, A., Russo, R., Esposito, M. R., Asci, R., Piscopo, C., Perrotta, S., et al. (2009). Molecular analysis of 42 patients with congenital dyserythropoietic anemia type II: new mutations in the SEC23B gene and a search for a genotype-phenotype relationship. Haematologica 95, 708–715. doi: 10.3324/haematol.2009.014985
Jaffray, J. A., Mitchell, W. B., Gnanapragasam, M. N., Seshan, S. V., Guo, X., Westhoff, C. M., et al. (2013). Erythroid transcription factor EKLF/KLF1 mutation causing congenital dyserythropoietic anemia type IV in a patient of Taiwanese origin: review of all reported cases and development of a clinical diagnostic paradigm. Blood Cells Mol. Dis. 51, 71–75. doi: 10.1016/j.bcmd.2013.02.006
Liljeholm, M., Irvine, A. F., Vikberg, A. L., Norberg, A., Month, S., Sandström, H., et al. (2013). Congenital dyserythropoietic anemia type III (CDA III) is caused by a mutation in kinesin family member, KIF23. Blood 121, 4791–4799. doi: 10.1182/blood-2012-10-461392
Nichols, K. E., Crispino, J. D., Poncz, M., White, J. G., Orkin, S. H., Maris, J. M., et al. (2000). Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat. Genet. 24, 266–270. doi: 10.1038/73480
Palmer, W. C., Vishnu, P., Sanchez, W., Aqel, B., Riegert-Johnson, D., Seaman, L. A. K., et al. (2018). Diagnosis and management of genetic iron overload disorders. J. Gen. Intern. Med. 33, 2230–2236. doi: 10.1007/s11606-018-4669-2
Rubinstein, W. S., Maglott, D. R., Lee, J. M., Kattman, B. L., Malheiro, A. J., Ovetsky, M., et al. (2013). The NIH genetic testing registry: a new, centralized database of genetic tests to enable access to comprehensive information and improve transparency. Nucleic Acids Res. 41, D925–D935. doi: 10.1093/nar/gks1173
Russo, R., Andolfo, I., Manna, F., De Rosa, G., De Falco, L., Gambale, A., et al. (2016). Increased levels of ERFE-encoding FAM132B in patients with congenital dyserythropoietic anemia type II. Blood 128, 1899–1902. doi: 10.1182/blood-2016-06-724328
Russo, R., Andolfo, I., Manna, F., Gambale, A., Marra, R., Rosato, B. E., et al. (2018). Multi-gene panel testing improves diagnosis and management of patients with hereditary anemias. Am. J. Hematol. 93, 672–682. doi: 10.1002/ajh.25058
Russo, R., Esposito, M. R., Asci, R., Gambale, A., Perrotta, S., Ramenghi, U., et al. (2010). Mutational spectrum in congenital dyserythropoietic anemia type II: identification of 19 novel variants in SEC23B gene. Am. J. Hematol. 85, 915–920. doi: 10.1002/ajh.21866
Russo, R., Gambale, A., Esposito, M. R., Serra, M. L., Troiano, A., De Maggio, I., et al. (2011). Two founder mutations in the SEC23B gene account for the relatively high frequency of CDA II in the Italian population. Am. J. Hematol. 86, 727–732. doi: 10.1002/ajh.22096
Russo, R., Gambale, A., Langella, C., Andolfo, I., Unal, S., and Iolascon, A. (2014). Retrospective cohort study of 205 cases with congenital dyserythropoietic anemia type II: definition of clinical and molecular spectrum and identification of new diagnostic scores. Am. J. Hematol. 89, E169–E175. doi: 10.1002/ajh.23800
Russo, R., Langella, C., Esposito, M. R., Gambale, A., Vitiello, F., Vallefuoco, F., et al. (2013). Hypomorphic mutations of SEC23B gene account for mild phenotypes of congenital dyserythropoietic anemia type II. Blood Cells Mol. Dis. 51, 17–21. doi: 10.1016/j.bcmd.2013.02.003
Schwarz, K., Iolascon, A., Verissimo, F., Trede, N. S., Horsley, W., Chen, W., et al. (2009). Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat. Genet. 41, 936–940. doi: 10.1038/ng.405
Unal, S., Russo, R., Gumruk, F., Kuskonmaz, B., Cetin, M., Sayli, T., et al. (2014). Successful hematopoietic stem cell transplantation in a patient with congenital dyserythropoietic anemia type II. Pediatr. Transplant. 18, E130–E133. doi: 10.1111/petr.12254
Keywords: telemedicine tool, congenital dyserythropoietic anemia, diagnosis, algorithm, hematological disease
Citation: Tornador C, Sánchez-Prados E, Cadenas B, Russo R, Venturi V, Andolfo I, Hernández-Rodriguez I, Iolascon A and Sánchez M (2019) CoDysAn: A Telemedicine Tool to Improve Awareness and Diagnosis for Patients With Congenital Dyserythropoietic Anemia. Front. Physiol. 10:1063. doi: 10.3389/fphys.2019.01063
Received: 28 March 2019; Accepted: 02 August 2019;
Published: 13 September 2019.
Edited by:
Richard Van Wijk, Utrecht University, NetherlandsReviewed by:
Anna Rita Migliaccio, Icahn School of Medicine at Mount Sinai, United StatesPedro Cabrales, University of California, San Diego, United States
Copyright © 2019 Tornador, Sánchez-Prados, Cadenas, Russo, Venturi, Andolfo, Hernández-Rodriguez, Iolascon and Sánchez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mayka Sánchez, msanchezfe@uic.es