 Rachel M. Handy
Rachel M. Handy Graham P. Holloway
Graham P. Holloway- Department of Human Health and Nutritional Science, University of Guelph, Guelph, ON, Canada
While impairments in peripheral tissue insulin signalling have a well-characterized role in the development of insulin resistance and type 2 diabetes (T2D), the specific mechanisms that contribute to these impairments remain debatable. Nonetheless, a prominent hypothesis implicates the presence of a high-lipid environment, resulting in both reactive lipid accumulation and increased mitochondrial reactive oxygen species (ROS) production in the induction of peripheral tissue insulin resistance. While the etiology of insulin resistance in a high lipid environment is rapid and well documented, physical inactivity promotes insulin resistance in the absence of redox stress/lipid-mediated mechanisms, suggesting alternative mechanisms-of-action. One possible mechanism is a reduction in protein synthesis and the resultant decrease in key metabolic proteins, including canonical insulin signaling and mitochondrial proteins. While reductions in mitochondrial content associated with physical inactivity are not required for the induction of insulin resistance, this could predispose individuals to the detrimental effects of a high-lipid environment. Conversely, exercise-training induced mitochondrial biogenesis has been implicated in the protective effects of exercise. Given mitochondrial biology may represent a point of convergence linking impaired insulin sensitivity in both scenarios of chronic overfeeding and physical inactivity, this review aims to describe the interaction between mitochondrial biology, physical (in)activity and lipid metabolism within the context of insulin signalling.
Introduction
Type 2 diabetes (T2D) remains one of the most significant medical challenges of the 21st century. While the earliest reports of the disease extend back centuries, modern day technical revolutions have resulted in low physical activity combined with the overconsumption of inexpensive, calorically dense, and inadequately satiating food, leading to extraordinary increases in obesity and the incidence of metabolic disorders (Wilmot et al., 2012; Boden et al., 2015). Indeed, in 1995 it was estimated that 135 million adults worldwide had diabetes, a population that was projected to double by 2025 (King et al., 1998). However, more recent reports estimate that 537 million adults were living with diabetes in 2021, and predict a further rise to 783 million by the year 2045 (IDF Diabetes Atlas, 2023), highlighting the disproportional increase in the incidence of metabolic disorders over recent years. While the specific underlying mechanisms that contribute to the development of T2D remain largely ambiguous, it is broadly accepted that excessive caloric intake, especially dietary fat, negatively affects insulin action in key tissues, including white adipose tissue (WAT), liver and muscle (Figure 1). The accumulation of lipids in peripheral tissues contributes to the induction of insulin resistance, defined as an inability of insulin to adequately promote peripheral glucose disposal, suppress lipolysis, and decrease hepatic glucose output. Importantly, the idea that chronic overfeeding may be involved in the development of peripheral insulin resistance originated in 1963 when Randle and colleagues first suggested that a greater availability of substrates (specifically fatty acids) is fundamental to the impaired insulin sensitivity observed in diabetic animals (Randle et al., 1963). Given that post-prandial regulation of whole-body glucose homeostasis is dominated by skeletal muscle and liver function, accumulation of excess lipids in these tissues is believed to be a primary contributor to impairments in glucose tolerance (Hulver et al., 2003; Loomba et al., 2021). Importantly, the excessive presence of lipids within these tissues has been shown to directly (via diacylglycerol (DAG)/ceramide-induced protein kinase C (PKC) signaling) and indirectly (via lipid-mediated mitochondrial reactive oxygen species (ROS) emission) induce insulin resistance. While chronic overfeeding has a well-established role in the development of insulin resistance, it should be acknowledged that physical inactivity represents an independent risk factor for the development of the disease (Wilmot et al., 2012), and occurs in the absence of cellular redox stress and increased skeletal muscle lipid accumulation (Dirks et al., 2016; 2020). As a result, the downstream effects of chronic overfeeding and physical inactivity are likely synergistic with respect to the pathophysiology of insulin resistance in many scenarios.
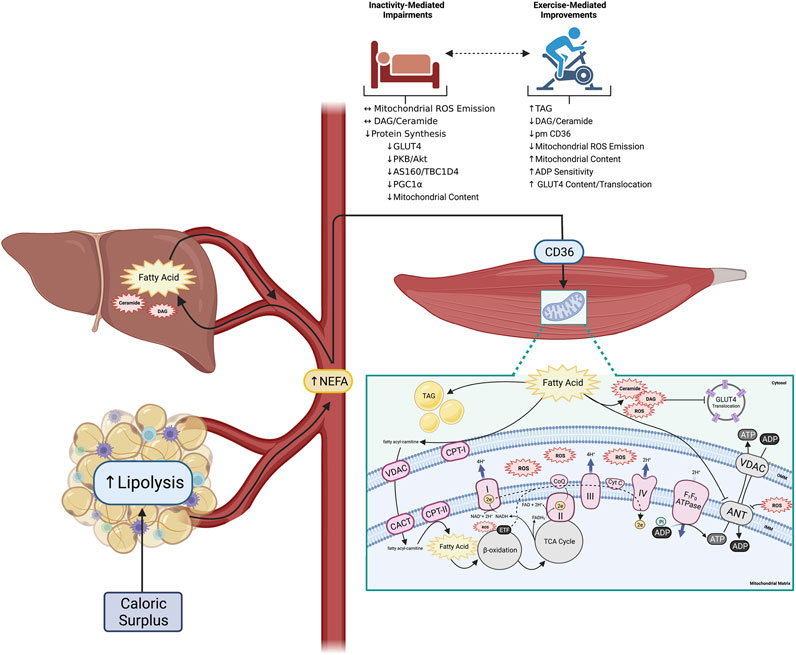
FIGURE 1. Altered peripheral tissue lipid metabolism initiates skeletal muscle reactive lipid accretion and ROS emission, a mechanism linked to peripheral insulin resistance. Obesity is associated with increased lipid availability (from both an impaired ability to regulate adipose tissue lipolysis, and increased hepatic de novo lipogenesis) resulting in increased flux of fatty acids through plasma membrane CD36 in skeletal muscle and the accumulation of various fatty acid species. While some excess fatty acids are converted to TAG for neutral storage, others support the accumulation of reactive lipids (DAG and ceramide). Accumulation of reactive lipids potentiate mitochondrial ROS emission due to increased CPT-I dependent fatty acid transport into the mitochondria, which directly attenuates ADP transport at the level of ANT. Importantly physical inactivity can exacerbate the induction of peripheral insulin resistance, while physical activity can mediate many of the outlined perturbations (created with BioRender.com). Abbreviations: Adenine nucleotide translocase (ANT), adenosine diphosphate (ADP), adenosine triphosphate (ATP), carnitineacylcarnitine translocase (CACT), carnitine palmitoyltransferase-I, -II (CPT-I, -II), cytochrome C (Cyt C), diacylglycerol (DAG), electron (e−), electron transport chain complexes I—IV (I—IV), electron transfer flavoprotein (ETF), fatty acid translocase (CD36), flavin adenine dinucleotide (FADH2), glucose transporter 4 (GLUT4), inner mitochondrial membrane (IMM), nicotinamide adenine dinucleotide (NADH), non-esterified fatty acid (NEFA), outer mitochondrial membrane (OMM), reactive oxygen species (ROS), triacylglycerol (TAG), tricarboxylic acid cycle (TCA), voltage-dependent anion channel (VDAC).
The regulation of lipid metabolism has been a focus of preventative approaches for decades, and given their primary role in fatty acid oxidation, mitochondria have a considerable impact on the accumulation/metabolism of both neutral (triacylglycerol (TAG)) and reactive (DAG/ceramides) lipids (Koves et al., 2008). In addition to their regulation of substrate metabolism, these organelles also play a significant role in the maintenance of cellular redox homeostasis given their prominent superoxide/ROS production (Brand, 2010). While pharmaceutical interventions that target mitochondrial bioenergetic efficiency have been identified, serious and potentially fatal side effects have rendered many treatment options inadequate (Cutting et al., 1933; Tainter et al., 1933; Cariou et al., 2012). It is now well recognized that exercise is a potent stimulus to induce various insulin sensitising processes, and multiple pathways that produce robust phenotypic changes in the mitochondrial milieu to improve the quantity and quality of the organelle network. Consequently, the inclusion of physical activity to a healthy lifestyle represents an important treatment/prevention strategy for individuals at risk for the development of insulin resistance and T2D.
While several mechanisms have been proposed to regulate mitochondrial quality, including gene transcription, mitophagy and reticulum formation, the importance of these mechanisms are premised fundamentally on affecting mitochondrial bioenergetics. As a result, this review will emphasize the relationship between mitochondrial ‘function’ and insulin signalling before discussing possible mechanisms that negatively influence mitochondrial bioenergetics. Within this framework, we highlight that physical activity has a robust influence on a range of mitochondrial characteristics and can be used as a viable treatment strategy for insulin resistance and T2D.
Lipid-mediated impairments in peripheral tissue insulin signaling
The accumulation of lipids in non-adipose tissues (namely skeletal muscle and liver) is a distinguished event in various models of insulin resistance (Randle et al., 1963; Marceau et al., 1999; Marchesini et al., 1999; Kim et al., 2000; Ryysy et al., 2000; Boden et al., 2001). Importantly, analysis of the temporal development of whole-body glucose intolerance demonstrate that impairments in skeletal muscle insulin signalling occur secondary to the rapid induction of insulin resistance in WAT following high-fat diet feeding in rodents (Turner et al., 2013). In line with this, impaired suppression of lipolysis in insulin resistant WAT, has been directly linked to increased circulation of non-esterified fatty acids (NEFA) (Boden et al., 1994; Paglialunga et al., 2015), which ultimately supports increased fatty acid uptake into both skeletal muscle (Luiken et al., 2001; Bonen et al., 2004; Han et al., 2007; Holloway et al., 2008) and liver (Westerbacka et al., 2007; Greco et al., 2008; Miquilena-Colina et al., 2011). Importantly, this increase in circulating NEFA appears to be suffucient for the development of whole-body glucose intolerance, evidenced by acute lipid oversupply experiments utilizing intralipid and heparin infusions in humans (Boden et al., 1994; Dresner et al., 1999; Belfort et al., 2005) and rodents (Yu et al., 2002). More recently, reductions in whole-body and hepatic insulin sensitivity following a single dose of palm oil (saturated fat) has been associated with increased rates of hepatic gluconeogenesis and glycogen phosphorylase flux in humans, and differential regulation of hepatic genes involved in canonical insulin signalling in rodents (Hernández et al., 2017). These findings support the classical dogma that increasing lipid supply, directly modulates peripheral tissue insulin signalling and results in the induction of insulin resistance. However, it is likely that the accumulation of neutral lipids (i.e. TAG) is relatively inert. Accordingly, an abundance of evidence demonstrates that the esterification of neutral lipids (i.e., the production of reactive lipid intermediates), in addition to the pathological production of mitochondrial-derived ROS independently contribute to impairments in insulin signalling (Summers, 2006; Liu et al., 2007; Timmers et al., 2008; Anderson et al., 2009; Dubé et al., 2011).
Reactive lipids have been proposed to attenuate insulin signalling through several mechanisms. Within skeletal muscle, DAGs have been demonstrated to activate PKC-θ and PKC-ε in both human and rodent models (Itani et al., 2002; Yu et al., 2002; Szendroedi et al., 2014; Lee et al., 2017). PKC-mediated serine phosphorylation of the insulin receptor substrate (IRS-1) leads to decreased insulin stimulated glucose transport (Yu et al., 2002; Li et al., 2004). The importance of DAG-mediated PKC activation in this pathway has been evidenced with the ablation of PKC-θ in murine skeletal muscle which protects mice from high-fat diet-induced insulin resistance (Kim et al., 2004), as does muscle-specific alanine substitutions on IRS-1 at various serine phosphorylation sites (Morino et al., 2008). In addition to DAGs, the accumulation of ceramides in skeletal muscle may also contribute to insulin resistance by attenuating phosphorylation of protein kinase B (PKB/Akt) (Schmitz-Peiffer et al., 1999; Bruce et al., 2012), a step required for insulin-mediated phosphorylation of Rab-GAP activating proteins (AS160/TBC1D4) and the induction of glucose transporter (GLUT4) translocation to the plasma membrane. Likewise, excessive reactive lipid accumulation within the liver is strongly associated with hepatic insulin resistance. In the liver, DAG-mediated activation of PKC-ε stimulates threonine phosphorylation of the insulin receptor kinase (IRK) at its catalytic subunit (Petersen et al., 2016), which decreases Akt phosphorylation and inhibits insulin signalling. Additionally, impaired insulin-stimulated Akt activity results in decreased insulin-stimulated glycogen synthesis through a reduction of glycogen synthase activity, and decreased insulin inhibition of gluconeogenesis (Samuel et al., 2004; 2007; Petersen et al., 2016). Collectively, DAG inhibits the direct effect of insulin on supressing hepatic glucose production and peripheral tissue glucose disposal, which combined contributes to whole-body glucose intolerance.
The presence of excess lipids and the subsequent production of reactive lipid intermediates is a direct reflection of an imbalance between peripheral tissue lipid supply and intracellular utilization. Given that intracellular lipid utilization occurs via mitochondrial oxidative phosphorylation, an increase in substrate (lipid) delivery/transport to the mitochondrial matrix, beyond what can be readily oxidized, results in the rapid accumulation of reactive lipid intermediates (Itani et al., 2002; Koves et al., 2008) and stimulation of mitochondrial ROS emission (Anderson et al., 2009). While multiple lines of evidence have demonstrated that basal fatty acid oxidation rates (Kelley et al., 1999) and the capacity of skeletal muscle to oxidize lipids (Hulver et al., 2003) are lower in obese/diabetic humans, a compensatory increase in mitochondrial fatty acid oxidation has also been demonstrated at the onset of obesity to buffer the accumulation of reactive lipid intermediates (Helge and Kiens, 1997). From a bioenergetic perspective, an increase in fatty acid oxidation, in the absence of an increase in energy expenditure, increases mitochondrial membrane potential and consequently increases the rate of ROS production (Murphy, 2009; Smith et al., 2021). Further, mitochondrial lipid transport via carnitine palmitoyltransferase-I (CPT-I), which is regarded as the rate limiting step in mitochondrial fatty acid oxidation, is stimulated by substrate provision, as increased lipid concentration has been shown to override the inhibitory effect usually exerted by malonyl-CoA (Smith et al., 2012). Since matrix located enzymes involved in β-oxidation are near-equilibrium, including the electron transfer flavoprotein, which is a potent site of electron leakage to produce ROS (Seifert et al., 2010), combined with the excessive production of NADH, an increase in lipid availability has a high propensity to produce mitochondrial ROS. Importantly, within skeletal muscle this process is unique to lipid provision as carbohydrate oxidation requires flux through the non-equilibrium enzyme pyruvate-dehydrogenase (PDH), which in the absence of allosteric activators/inhibitors of specific phosphatase/kinases is maintained ‘off’ with limited flux (Sugden and Holness, 1994), thus the oxidation of carbohydrates is not controlled by intracellular substrate (i.e. pyruvate) concentration. Essentially at rest, PDH is allosterically inhibited by PDH-kinase (PDK) (Spriet and Heigenhauser, 2002). Given that both PDK content and activity are increased following the acute consumption of a high-fat/low-carbohydrate diet in humans (Peters et al., 2001), and long-term high-fat diet feeding in rodents (Orfali et al., 1993), the ability of carbohydrates to drive mitochondrial ROS production is limited. In support of this, research in murine models indicate that inhibition of reactive lipid synthesis protects against high-fat diet-induced insulin resistance (Ussher et al., 2010; Kurek et al., 2015), while pharmacological (Anderson et al., 2009) and genetic (Lee et al., 2010; Boden et al., 2012) inhibition of mitochondrial ROS emission preserves insulin sensitivity.
While an abundance of evidence suggests that mitochondrial-derived ROS have a primary role in the etiology of insulin resistance, the specific underlying mechanism has yet to be delineated. Nonetheless, it has been proposed that mitochondrial-ROS activate the nuclear factor-κβ pathway (NF-κβ/Iκβ/IKKβ pathway) resulting in serine phosphorylation of IRS-1, and attenuation of insulin signalling in a similar manner to DAGs (Yuan et al., 2001; Sinha et al., 2004). Additionally, both isolated adipocytes and myocytes exposed to H2O2 demonstrate impaired PKB activation (Tirosh et al., 1999; JeBailey et al., 2007) and markers of insulin-signaling (Rudich et al., 1997; Tirosh et al., 2001; JeBailey et al., 2007), while conversely the use of antioxidants (e.g. lipoic acid or N-acetylcysteine) prevent the induction of insulin resistance (Rudich et al., 1999; JeBailey et al., 2007). Though seemingly paradoxical, it should be noted that while chronically elevated mitochondrial H2O2 emission is detrimental to cellular homeostasis (Anderson et al., 2009), overexpression of the cytosolic H2O2 antioxidant glutathione peroxidise leads to the development of insulin resistance in mice (McClung et al., 2004). Additionally, neither chronic mitochondrial targeted antioxidant treatment nor enhanced mitochondrial superoxide scavenging prevent HFD-induced insulin resistance despite preserving redox balance (Paglialunga et al., 2012; Lark et al., 2015). Together, these findings indicate that the development of insulin resistance is not exclusively determined by an increase in redox stress, which highlights the complexity surrounding the role of ROS in cellular homeostasis. Indeed, insulin itself has been demonstrated to stimulate the generation of localized ROS, which plays a role in facilitating downstream insulin signalling (Goldstein et al., 2005; Loh et al., 2009; Contreras-Ferrat et al., 2014). In line with this, inhibition of insulin-stimulated H2O2 production inhibits PI3K activity which corresponds with a ∼50% reduction of insulin-stimulated Akt activation (Mahadev et al., 2001), highlighting the requirement of these molecules for the maintenance of whole-body glucose homeostasis. Collectively, these data emphasize that while low levels of cytosolic ROS are required for the regulation of insulin signalling, in the context of a high-lipid environment (as is observed with chronic caloric overconsumption) both reactive lipid accumulation and mitochondrial ROS production link excessive lipids to many contemporary mechanisms of insulin resistance. However, there remains considerable debate within the scientific community if an intrinsic mitochondrial dysfunction exists, or alternatively that the provision of excess lipid substrates to the mitochondria is inherently problematic as suggested by Randle in 1963.
Contributions of mitochondrial biology in peripheral tissue insulin resistance
There is an abundance of evidence to suggest that the accumulation of reactive lipids within peripheral tissues can impair various aspects of mitochondrial bioenergetics and is associated with the induction of insulin resistance. Since the first description of mitochondrial dysfunction in the context of glucose intolerance more than 40 years ago (Yamada et al., 1975), mitochondria have been placed as a key organelle in the development of peripheral insulin resistance. While the term ‘mitochondrial dysfunction’ is rather equivocal, it is often used to describe a reduction in the maximal rate of mitochondrial fatty acid oxidation as a result of decreased mitochondrial content and/or activity of key enzymes involved in oxidative phosphorylation. In support of this, an abundance of evidence demonstrates reduced mitochondrial content in insulin resistant/obese individuals (Kelley et al., 2002; Morino et al., 2005; Ritov et al., 2005; Holloway et al., 2007), while genetically increasing mitochondrial content in rodents attenuates lipid-mediated insulin resistance (Koves et al., 2005; Benton et al., 2010; Consitt et al., 2010). In theory, a reduction in mitochondrial fatty acid oxidation would promote accumulation of reactive lipids and increase mitochondrial ROS emission, to ultimately impair peripheral insulin signalling. However, given that obesity is associated with increased plasma free fatty acid concentrations (Randle et al., 1963; Boden, 2003), plasma membrane fatty acid transport (Bonen et al., 2004; 2015), and accumulation of intracellular lipids (Pan et al., 1997; Krssak et al., 2004), the suggestion of reduced mitochondrial fatty acid oxidation cannot be explained by lack of availability and/or tissue delivery. Additionally, the induction of insulin resistance can occur in the absence of reductions in mitochondrial content (Bruce et al., 2005; Holloszy, 2013) or reductions in isolated mitochondrial fatty acid oxidation (Holloway et al., 2007; 2009; Mogensen et al., 2007). These findings in combination with evidence that high-fat diet feeding rapidly increases mitochondrial content alongside the induction of insulin resistance in rodents (Garcia-Roves et al., 2007; Hancock et al., 2008; Miotto et al., 2018), demonstrates that reductions in mitochondrial content are not required in the etiology of insulin resistance. Taken together, there is little evidence to support the hypothesis that an intrinsic impairment in the ability of mitochondria to oxidize fatty acids accounts for increased lipid accretion in peripheral tissues associated with insulin resistance.
While the aforementioned data collectively challenge the relationship between ‘mitochondrial dysfunction’ and insulin resistance, it should be noted that molecular approaches to evaluate mitochondrial function are often performed in the presence of saturating ADP concentrations, which do not reflect the true biological environment. In this regard, it is well-supported that increased mitochondrial biogenesis following aerobic exercise training, results in an enhanced sensitivity of mitochondria to ADP (Holloszy and Coyle, 1984; Dudley et al., 1987), demonstrating that submaximal responses to ADP is likely a key process in regulating cellular homeostasis. Functionally, an increase in mitochondrial ADP sensitivity elicits a reduction in the concentration of ADP required to drive the same absolute respiration (JO2), which ultimately lowers cytosolic free ADP (Phillips et al., 1996; Perry et al., 2008). While ADP binds to F1F0 ATPase to reduce membrane potential and ultimately supress mitochondrial ROS emission (Anderson and Neufer, 2006; Anderson et al., 2009), a reduction in ADP transport can attenuate aerobic metabolism and increase mitochondrial ROS emission (Esposito et al., 1999). Interestingly, insulin can enhance submaximal-ADP supported respiration in permeabilized skeletal muscle fibres from both healthy and insulin resistant mice, while the presence of high concentrations of palmitoyl-CoA (P-CoA; to model a high lipid environment) prevented this acute effect (Brunetta et al., 2021). These findings highlight the importance of removing and/or attenuating accumulation of excess lipids to optimize mitochondrial bioenergetics. In line with this, ADP/ATP exchange by adenine nucleotide translocase (ANT) is impaired in the presence of high concentrations of P-CoA, providing proof of principle that lipids can directly impair ADP sensitivity (Ho and Pande, 1974; Ludzki et al., 2015). This concept is supported with the evaluation of submaximal ADP kinetics in various murine models of obesity and T2D, where marked reductions in mitochondrial respiration across a range of submaximal/physiological ADP concentrations is consistently observed (Smith et al., 2013; Miotto et al., 2018). These findings align with what is speculated to occur with the induction of mitochondrial ROS-mediated insulin resistance in a high-lipid environment—whereby the presence of reactive lipids override the inhibition of CPT-I by malonyl-CoA (M-CoA), ‘pushing’ lipids into the mitochondrial matrix, while simultaneously inhibiting ADP transport and submaximal ADP-supported respiration (Smith et al., 2012), both of which would markedly increase mitochondrial ROS emission. However, an avenue of research that has yet to be clearly articulated, is the role of mitophagy in this phenomenon.
Mitochondria are highly dynamic organelles that readily adapt their morphology to the cellular environment—the physiological consequence of overabundant and/or dysfunctional mitochondria is the processes of fission and mitochondrial clearance (mitophagy) to re-establish metabolic homeostasis. While the importance of mitophagy in regulating cellular homeostasis has been previously reviewed (Palikaras et al., 2018), the functional consequence of alterations in mitophagy in the context of insulin resistance is ambiguous. While it may seem logical that reduced mitophagy (i.e., reduced clearance of damaged mitochondrial fragments) may contribute to the enhanced mitochondrial ROS emission and redox stress observed with insulin resistance, recent work has demonstrated contradictory findings. Indeed, high-fat-diet-induced insulin resistance in rodents is associated with increased markers of mitophagy (Ehrlicher et al., 2021), while ablation of the key mitophagy regulating protein FUNDC1 within skeletal muscle does not impair mitochondrial bioenergetics or induce insulin resistance within this tissue (Fu et al., 2018). Thus, it remains unknown whether alterations in mitophagy processes are a cause or consequence of altered mitochondrial bioenergetics. Nonetheless, it could be speculated that the observed increase in mitophagy is a compensatory effort to maintain cellular homeostasis, which may align with alterations in ADP/ATP exchange in insulin resistant tissue (Garcia et al., 2019). Given that ANT is the most abundantly expressed mitochondrial protein (Brand et al., 2005), it is likely susceptible to enhanced redox damage which would account for increased markers of mitophagy alongside alterations in submaximal ADP kinetics observed with chronic obesity/high-fat feeding and prolonged insulin resistance (Brand et al., 2005; Miotto et al., 2018). Taken together, a contemporary model suggests that redox damage on ANT and attenuated ADP/ATP exchange is likely an integral perturbation in the etiology of insulin resistance, caused by the increased availability and transport of fatty acids into the mitochondria.
Physical inactivity and lipid-independent induction of insulin resistance
Impairments in insulin sensitivity have long been known as a physiological consequence of sedentary behaviour and even short-term inactivity (Smith et al., 2016). This ideology has been evidenced by various step-reduction studies (i.e., <1000–1500 steps/day) which demonstrate negative effects on whole-body glucose homeostasis in as little as 14 days (Knudsen et al., 2012; Breen et al., 2013; McGlory et al., 2018). At the molecular level, this impairment in glucose homeostasis has been associated with a significant decrease in insulin-stimulated skeletal muscle Akt phosphorylation, independent of increased inflammatory markers including TNF and IL-6 (Krogh-Madsen et al., 2010). Further, 7 days of step reduction has been demonstrated to significantly alter the expression of key genes involved in metabolic homeostasis and insulin action (upregulation of SREBP-1C, FAS, GLUT4; downregulation of PDK4, IRS2, HSL) in adipose tissue (Walhin et al., 2013). This is in line with impaired post-prandial glucose control throughout 3 days of reduced (<5,000 steps) physical activity (Mikus et al., 2012). While the specific time-course through which these impairments in insulin action occur remain unknown, a single day of limited physical activity (∼260 steps) has been demonstrated to be sufficient to impair insulin action, evidenced by a ∼39% reduction in the rate of whole-body glucose disappearance in physically active young men and women (Stephens et al., 2011). Importantly, these impairments in glucose homeostasis observed following restriction of physical activity are not rescued within a day of return to normal activity (Reynolds et al., 2015). While the mechanism underlying the induction of insulin resistance following physical inactivity remains unclear, it would appear to be disparate from what is observed following overfeeding.
The exact mechanism(s) causing insulin resistance during acute periods of inactivity remain debatable, as there are inconsistent reports of reactive lipid accumulation (i.e., DAGs and ceramides) (Reidy et al., 2018; Appriou et al., 2019; McKenzie et al., 2020; Kakehi et al., 2021). While substantial increases in DAG concentration have been reported in as little as 3 h of soleus muscle denervation (Turinsky et al., 1990), in humans 7-day bedrest, and 1 day of unilateral limb suspension, induces insulin resistance in the absence of significant changes in skeletal muscle reactive lipid content (Dirks et al., 2016; Bilet et al., 2020). Collectively, these data suggest a dissociation between reactive lipids and the development of insulin resistance within the context of physical inactivity in humans. However, it has been suggested that specific reactive lipids that are not abundant may play an important role in signal transduction (Sowell et al., 1991), which aligns with marginal increases in specific DAG and free fatty acid (FFA) species observed during bedrest-induced insulin resistance (Dirks et al., 2016). Additionally, while this work provides no insight on the subcellular location of lipids, the use of transmission electron microscopy (TEM) has suggested an inverse relationship between lipid accumulation in the SS region and insulin sensitivity in human T2D subjects (Nielsen et al., 2010). Despite the abundance of contradictory findings, it could be speculated that in scenarios of severe physical inactivity, incremental increases in intramyocellular lipid droplets within the SS region influence skeletal muscle insulin signalling, or alternatively that the increase in FFA inhibits mitochondrial ANT/ADP transport and mitochondrial bioenergetics in this specific region of the muscle.
An alternative hypothesis implicates attenuated transcriptional/translational processes and reductions in protein synthesis during the development of skeletal muscle disuse-induced insulin resistance. While an abundance of evidence clearly suggests that a reduction in ambulatory activity results in skeletal muscle atrophy and is often associated with impairments in insulin action and glucose tolerance (reviewed in Oikawa et al., 2019), the underlying mechanism initiating these alterations remain relatively ambiguous. Important to this discussion is the recognition that acute (i.e. 24 h) reductions in ambulatory activity have also been demonstrated to impair glucose homeostasis (Stephens et al., 2011), which would occur independent of significant skeletal muscle atrophy. Thus, impaired synthesis of contractile proteins may not be a plausible explanation for the induction of insulin resistance, at least in an acute model. Alternatively, it could be speculated that attenuated synthesis of proteins involved in canonical insulin signalling and mitochondrial biogenesis have a more significant role in the induction of disuse-mediated insulin resistance over a longer duration of time. Indeed, various studies have associated muscle disuse with a decline in mitochondrial content (determined by citrate synthase activity and oxidative phosphorylation (OXPHOS) protein content) (Alibegovic et al., 2010; Ringholm et al., 2011; Dirks et al., 2016). While rodent models of inactivity are not without limitations (Reidy et al., 2021), these data are supported by reductions in mitochondrial function and content at both the mRNA and protein levels observed following skeletal muscle inactivity (Ljubicic and Hood, 2009a; 2009b; Cartee et al., 2016). Further, bedrest (Biensø et al., 2012; Dirks et al., 2020), and physical inactivity in both humans (Vukovich et al., 1996) and rodents (Host et al., 1998) demonstrate decreased content of key proteins involved in canonical insulin signaling events, including hexokinase (HK), GLUT4 protein, PKB/Akt and AS160/TBC1D4, likely contributing to reduced skeletal muscle glucose transport. In line with this, rapid decreases in expression of peroxisome proliferator-activated gamma coactivator 1-α (PGC-1α), a transcriptional co-activator responsible for the upregulation of widespread gene programs including those involved in insulin signalling and mitochondrial biogenesis, are observed following muscle inactivity (Betik et al., 2009; Cartee et al., 2016). Given that PGC-1α is also known as the master regulator of mitochondrial biogenesis (Puigserver et al., 1998; Wu et al., 1999; Lin et al., 2002), reductions in this protein align with reduced mitochondrial protein content and ADP sensitivity following disuse (Miotto et al., 2019; Dirks et al., 2020; Zuccarelli et al., 2021). This is in contrast to maintained OXPHOS protein content observed in obese and insulin resistant rodents (Garcia-Roves et al., 2007; Hancock et al., 2008; Miotto et al., 2018) which appears to be caused by mitochondrial ROS-mediated ryanodine receptor calcium leak induced gene transcription (Jain et al., 2014). Further, experiments modelling muscle disuse in rodents through denervation or hind limb suspension have demonstrated a significant reduction in the mitochondrial fusion: fission ratio and fragmentation of the mitochondrial reticulum (Iqbal et al., 2013). More recent work has also demonstrated that SS mitochondria from denervated muscle exhibits greater flux in mitophagy markers than the IMF subfraction (Triolo et al., 2022), suggesting that SS mitochondria are likely more sensitive to disuse (Powers et al., 2012b). Importantly, immobilization in humans appears to decrease respiratory capacity faster than declines in OXPHOS protein content (Miotto et al., 2019), suggesting increased mitophagy as a secondary mechanism to remove damaged mitochondrial proteins.
Mechanistically, it has been widely reported that prolonged skeletal muscle disuse results in the accumulation of oxidatively modified proteins and lipids leading to the initiation of various atrophic pathways (Powers et al., 2012a). While mitochondria have been implicated as a key source of inactivity-mediated ROS production in rodent models (Kavazis et al., 2009; Min et al., 2011; Powers et al., 2011), it should be noted that there is an important distinction between the metabolic rate of rodents and humans which may influence the translatability of rodent data. Indeed, rodents demonstrate a ∼8-fold higher metabolic rate than humans (Hudson and Franklin, 2002; Dutta and Sengupta, 2016), which likely contributes to the greater magnitude of ROS production and skeletal muscle atrophy observed in rodent models. Importantly, this paradigm is premised on a significant increase in muscle protein breakdown (MPB) as opposed to a decrease in muscle protein synthesis (MPS), which may not translate to humans. In support of this, physical inactivity is associated with significant reductions in protein synthesis and lean mass (Dirks et al., 2016; McGlory et al., 2018) in the absence of changes in redox stress or mitochondrial ROS emission in humans (Dirks et al., 2016; 2020; Miotto et al., 2019).
Acute improvements in insulin sensitivity following exercise and influence of training modality
Within 5 years of the discovery of insulin by Banting and Best in 1921, the first reports of exercise-induced improvements in insulin action were published (Lawrence, 1926). Skeletal muscle responds to contraction/exercise with a rapid insulin-independent increase in glucose uptake (reviewed in Richter and Hargreaves, 2013). Notably, exercise also enhances the sensitivity of skeletal muscle to subsequent insulin stimulation (Devlin et al., 1987; Richter et al., 1989; 1989; Cartee et al., 1989) via increased insulin-stimulated translocation of GLUT4 to the muscle membrane following exercise (Hansen et al., 1998). Additionally, the regulation of skeletal muscle membrane permeability to glucose is a key component of exercise-stimulated improvements in whole-body glucose homeostasis. Various studies have demonstrated that the increase in muscle glucose uptake immediately following exercise is mediated by PI3K-independent translocation of GLUT4 to the plasma membrane (Douen et al., 1990; Lee et al., 1995). Importantly, this mechanism is not dependent on insulin, as muscle-specific insulin receptor knockout mice demonstrate increased 2-deoxy-glucose uptake following an acute exercise bout (Wojtaszewski et al., 1999). Collectively, these data provide evidence for an insulin-independent mechanism in the regulation of whole-body glucose homeostasis immediately following exercise. In the 24–72 h following exercise, insulin-dependent pathways, specifically activation of PI3K (Zhou and Dohm, 1997), are upregulated resulting in improved insulin sensitivity during this time. While the specific mechanism leading to PI3K activation remains unclear, some evidence suggests that this occurs independently of IRS-1 (Howlett et al., 2002). This is supported by human data, which demonstrate that improved insulin sensitivity following acute exercise is not caused by increased activity of proximal signalling molecules in the insulin signaling cascade, as downstream kinases appear to be activated first (Wojtaszewski et al., 2000). In line with this, acute exercise has been reported to alleviate lipid-induced insulin resistance in human skeletal muscle by increasing interaction at the level of TBC1D4 (Pehmøller et al., 2012). While the specific mechanism through which TBC1D4 phosphorylation occurs in this scenario is not clear, an insulin-independent mechanism could be speculated given that TBC1D4 contains a phosphorylation domain for both Akt and AMPK. Interestingly, TBC1D4 phosphorylation is maintained for several hours after cessation of exercise, and is further potentiated with insulin stimulation (Sjøberg et al., 2017; Hingst et al., 2018). Additionally, as originally proposed by Holloszy’s group in 2006, the insulin sensitizing effects of exercise are also a function of GLUT4 relocation from the sarcolemma and T-tubules, towards cellular locations that are more accessible for recruitment following insulin stimulation (Geiger et al., 2006). Collectively, these studies demonstrate that acute exercise contributes to whole-body glucose homeostasis via increased glucose transport and transient improvements in insulin sensitivity—both of which provide evidence for acute exercise as a viable therapeutic for insulin resistant and T2D patients, as insulin sensitivity following exercise is conserved in these individuals (Devlin et al., 1987).
Despite clear evidence for the improvement in insulin sensitivity and glycaemic control following exercise, intensity and duration of physical activity may elicit a differential response on peripheral insulin sensitivity. The most consistent evidence for exercise-induced improvements in insulin sensitivity stem from the study of moderate intensity aerobic exercise. Various reports describe improvements in insulin resistance (measured via HOMA-IR), fasting plasma insulin and fasting glucose following 25–60 min of walking or running at 50%–60% VO2 MAX (i.e., low to moderate intensity) three times per week (Damirchi et al., 2014; Herzig et al., 2014; Motahari-Tabari et al., 2014). Insulin sensitivity has also been demonstrated to have a dose-dependent response to aerobic exercise duration, whereby longer exercise duration (i.e., >1900 kcal/week) is associated with further increases in insulin sensitivity and β-cell function in prediabetic individuals (Malin et al., 2013). While longer duration exercise appears to be desirable, the additional improvements in insulin sensitivity when comparing 300 v. 600 kcal/day are marginal (Reichkendler et al., 2014), which is likely an important consideration given that compliance to exercise programs in this population is often low (Ford and Herman, 1995; Troiano et al., 2008; Colley et al., 2011). Notably, the improvements in insulin sensitivity observed with exercise training were diminished within 6 weeks of detraining (Damirchi et al., 2014), indicating the importance of repeated exercise in this response.
The variable of exercise intensity, and the use of high intensity interval training (HIIT) and/or sprint interval training (SIT) in the treatment of metabolic disorders, has gained significant popularity in recent years. HIIT leverages repeated, brief bouts of vigorous exercise separated by short recovery periods, to elicit the metabolic effect of moderate intensity aerobic exercise in a condensed duration. An abundance of literature suggests that HIIT elicits a greater improvement in VO2 max (a parameter linked to all-cause mortality) and transient glycaemic control when compared with moderate-intensity, longer duration exercise (Earnest et al., 2013; Racil et al., 2013; Mann et al., 2014; Mitranun et al., 2014). However there remains considerable debate regarding the efficacy of this training program given that a majority of research in this area quantifies acute metabolic responses to HIIT compared with volume-matched lower-intensity endurance exercise, which out of necessity is short in duration and less frequent. However, a recent report utilizing a single-leg, 4-week training program to compare endurance and HIIT adaptations within an individual suggests endurance training elicits greater mitochondrial biogenesis (Skelly et al., 2023), and when not volume matched, endurance exercise also appears to be favorable for improvements in VO2 max and post-prandial lipid tolerance when compared to SIT (Petrick et al., 2021). This is in line with the earlier finding that a single bout of endurance exercise prevents impairments in insulin sensitivity following an 18 h lipid infusion (Schenk et al., 2005). Prevention of lipid-induced insulin resistance after endurance exercise has been demonstrated to be accompanied by enhanced skeletal muscle protein expression of key lipogenic enzymes and increases in muscle TAG synthesis (Schenk and Horowitz, 2007), as well as a reduction in the key plasma membrane transporter CD36 abundance in rodents (Smith et al., 2007). Importantly, the partitioning of more fatty acids towards TAG synthesis within muscle following exercise is associated with reduced accumulation of reactive lipid intermediates and decreased phosphorylation/activation of proinflammatory signalling pathways (Schenk and Horowitz, 2007). Conversely, it has been suggested that daily adherence to HIIT elicits an overtraining response and impairs glucose tolerance in trained individuals (Flockhart et al., 2021). However, timing of HIIT exercise may be an important consideration, given evidence to suggest that morning HIIT has an acute, deleterious effect on blood glucose homeostasis, while afternoon HIIT appears to be more efficacious at improving blood glucose in men with T2D (Savikj et al., 2019). While speculative, these observations are likely a function of increases in lipid oxidation occurring in the recovery period as opposed to during the high-intensity exercise bout (as is observed with endurance training). Given that circulating lipids increase overnight (during an approximate 8 h fasting period), it may be logical that this post-exercise increase in lipid oxidation is advantageous. Alternatively, HIIT has been demonstrated to elevate circulating epinephrine (Talanian et al., 2007), which may be problematic in the morning when plasma epinephrine concentrations are naturally elevated (Dodt et al., 1997), resulting in increased hepatic glucose output, thus attenuating the potential benefits of acute exercise on glucose tolerance. Altogether, given the clear importance of daily exercise and the opportunistic timing required for the metabolic benefits of HIIT, these data appear to support endurance exercise as a superior aerobic training modality with respect to regulation of whole-body glucose homeostasis.
While much of the early research on exercise-mediated regulation of insulin sensitivity focused on aerobic exercise training, the use of resistance exercise as a training modality to improve insulin sensitivity should not be overlooked. Primary outcomes in studies evaluating the effects of resistance training in insulin resistant individuals have demonstrated a 10%–15% improvement in insulin sensitivity (Gordon et al., 2009). Similar to aerobic exercise, resistance training increases energy expenditure, which is important for excess substrate oxidation, however, unique to this training modality is skeletal muscle hypertrophy (Maughan et al., 1983), which alleviates reactive lipid accumulation and the induction of insulin resistance (Haines et al., 2020). Importantly this increase in muscle mass is not associated with analogous increases in mitochondrial content (MacDougall et al., 1979), which further divorces the relationship between absolute mitochondrial content and insulin resistance, as mentioned previously. An additional consideration is a combination of moderate intensity aerobic exercise with resistance training which has been demonstrated to significantly increase insulin-mediated glucose uptake compared to individuals performing aerobic exercise alone (Cuff et al., 2003). These findings suggest that combined training for patients with T2D may augment insulin sensitivity via differing mechanisms and importantly, that combined training protocols may elicit additive benefits, however, this could simply be related to a greater volume of physical activity, once again highlighting the importance of regular exercise for metabolic health.
Molecular mechanisms for chronic exercise-induced changes in insulin sensitivity
While a single bout of exercise causes a transient improvement in insulin sensitivity as outlined above, the molecular mechanisms accounting for improvements in insulin sensitivity induced by chronic exercise training are complex and incomplete. Nonetheless, it is generally accepted that repeated bouts of regular exercise upregulate PGC-1α (Russell et al., 2003; Wright et al., 2007; Perry et al., 2010). While it is well known that PGC-1α upregulates an abundance of genetic programs, including those involved in the canonical insulin signalling cascade (Chibalin et al., 2000; Ryder et al., 2001), it may also be central in the regulation of lipid metabolism. Notably, PGC-1α abundance is positively correlated with enhanced fatty acid oxidation (Huang et al., 2017), which associates training induced improvements in peripheral lipid handling. Specifically, co-activation of peroxisome proliferator-activated receptor (PPAR) by PGC-1α upregulates the TAG synthesizing enzymes DAG acyltransferase 1 (DGAT1) (Summermatter et al., 2010); and perilipin (Arimura et al., 2004), both of which promote lipid storage as neutral TAG. This is evidenced by enhanced TAG synthesis and reduced DAG and ceramide accumulation in skeletal muscle following adherence to chronic training in humans and rodents (Goodpaster et al., 2001; Holloway et al., 2014). However, the consideration of subcellular location of intramuscular lipid droplets is important, given the relationship between lipid accumulation in the SS region and insulin resistance in both humans and rodents (Nielsen et al., 2010; Lally et al., 2012). Importantly, intramyocellular lipid droplets stored in the SS region are used to a greater extent during exercise (Stellingwerff et al., 2007), a finding that extends to T2D subjects who demonstrate decreased SS lipids following aerobic exercise training (Nielsen et al., 2010). Despite the clear metabolic improvements observed with chronic exercise training, repeated exercise is paramount to these responses. In support of this concept, many models demonstrate that the absence of training for 3 to 5 days is sufficient to mitigate the benefits of chronic training (Roberts et al., 2013). This aligns with original research in this area which demonstrates a 50% reduction in the activities of key mitochondrial enzymes, citrate synthase and succinate dehydrogenase, 12 days following the cessation of prolonged intense endurance training (Coyle et al., 1984). As such, an evolving area of research has focused on the induction of mitophagy within skeletal muscle in response to exercise and detraining.
While general apoptosis is not linked to exercise (Quadrilatero et al., 2010), our understanding of the potential role of mitophagy in response to exercise is still emerging. Recent work suggests that the energy sensor AMPK is required for the increase in mitophagy that is observed 6 hours following acute treadmill exercise (Laker et al., 2017) and elicits an increase in Parkin localization to muscle mitochondria (Vainshtein et al., 2015; Chen et al., 2018). Importantly, an increase in mitochondrial Parkin localization following acute treadmill exercise, aligns with an increase in mitophagy flux (measured via LC3-II, p62, and ubiquitin) which remains elevated post-exercise (Vainshtein et al., 2015; Chen et al., 2018). Further, the observed increase in mitophagy flux were not present in Parkin-knockout animals (Chen et al., 2018), suggesting that Parkin is required for the mitophagy response to endurance exercise. While these studies have focused on an acute response to exercise, other research has demonstrated that basal mitophagy is elevated in response to 5 weeks of swimming (Dun et al., 2017) or 4 weeks of voluntary wheel running (Lira et al., 2013), evidenced by an increased LC3-II:LC3-I ratio, decreased p62, and elevated BNIP following both interventions. Additionally, endurance trained mice have elevated Parkin protein expression and mitochondrial localization, as well as a blunted mitophagy response to acute exercise (Chen et al., 2018), while studies in humans have demonstrated that endurance training results in elevated phosphorylation of Pink1 and Parkin (Tarpey et al., 2017), both of which suggest enhanced specificity of mitophagy in endurance trained muscle. Given that classical training adaptations confer increases in mitochondrial content, an improved sensitivity of various enzymes involved in metabolic signalling (i.e., AMPK, p38, CaMK) would be expected (Dudley et al., 1987; Ljubicic and Hood, 2009b) and aligns with reductions in mitophagy following acute physical activity in endurance trained individuals (Chen et al., 2018). While speculative, it is likely that these alterations to mitophagy flux initiate remodelling of the existing mitochondrial milieu, which may be one mechanism through which exercise modulates mitochondrial dynamics and improves peripheral insulin sensitivity.
Conclusion
In this review, we aimed to highlight the central role of mitochondrial biology in various mechanisms thought to underly the etiology of peripheral insulin resistance. The accumulation of lipids in non-adipose tissues is a distinguished event in various models of insulin resistance, resulting in both direct (via reactive lipids) and indirect (via mitochondrial ROS emission) impairments in peripheral insulin signalling. While several mechanisms have been proposed to regulate mitochondrial quality, including gene transcription, mitophagy and reticulum formation, the importance of these mechanisms are fundamentally premised on affecting mitochondrial biogenesis. However, clear evidence for the presence of impaired mitochondrial oxidative capacity in the induction of insulin resistance does not exist, and redox stress and reactive lipid accretion appears to be specific to scenarios of chronic overfeeding. Alternatively, it appears that lipids can competitively and/or allosterically inhibit mitochondrial ADP transport as a key event negatively affecting mitochondrial bioenergetics. In contrast to a high lipid environment, extreme physical inactivity attenuates protein synthesis and reduces mitochondrial and canonical insulin signaling proteins as alternative mechanisms-of-action to limit insulin action. While the effects of lipid-induced metabolic perturbations and physical inactivity are therefore likely additive with respect to the pathophysiology of insulin resistance, since exercise stimulates robust phenotypic changes in lipid homeostasis, mitochondrial bioenergetics and protein synthesis/gene transcription, the inclusion of physical activity represents an important treatment/prevention strategy for individuals at risk of the development of insulin resistance and T2D.
Author contributions
RMH and GPH drafted the manuscript and approved the final version.
Funding
GPH holds a Natural Sciences and Engineering Research Council (NSERC) of Canada Grant (400362).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alibegovic, A. C., Sonne, M. P., Højbjerre, L., Bork-Jensen, J., Jacobsen, S., Nilsson, E., et al. (2010). Insulin resistance induced by physical inactivity is associated with multiple transcriptional changes in skeletal muscle in young men. Am. J. Physiology-Endocrinology Metabolism 299, E752–E763. doi:10.1152/ajpendo.00590.2009
Anderson, E. J., Lustig, M. E., Boyle, K. E., Woodlief, T. L., Kane, D. A., Lin, C. T., et al. (2009). Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investigation 119, 573–581. doi:10.1172/JCI37048
Anderson, E. J., and Neufer, P. D. (2006). Type II skeletal myofibers possess unique properties that potentiate mitochondrial H2O2 generation. Am. J. Physiology-Cell Physiology 290, C844–C851. doi:10.1152/ajpcell.00402.2005
Appriou, Z., Nay, K., Pierre, N., Saligaut, D., Lefeuvre-Orfila, L., Martin, B., et al. (2019). Skeletal muscle ceramides do not contribute to physical-inactivity-induced insulin resistance. Appl. Physiol. Nutr. Metab. 44, 1180–1188. doi:10.1139/apnm-2018-0850
Arimura, N., Horiba, T., Imagawa, M., Shimizu, M., and Sato, R. (2004). The peroxisome proliferator-activated receptor γ regulates expression of the perilipin gene in adipocytes. J. Biol. Chem. 279, 10070–10076. doi:10.1074/jbc.M308522200
Belfort, R., Mandarino, L., Kashyap, S., Wirfel, K., Pratipanawatr, T., Berria, R., et al. (2005). Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes 54, 1640–1648. doi:10.2337/diabetes.54.6.1640
Benton, C. R., Holloway, G. P., Han, X.-X., Yoshida, Y., Snook, L. A., Lally, J., et al. (2010). Increased levels of peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1alpha) improve lipid utilisation, insulin signalling and glucose transport in skeletal muscle of lean and insulin-resistant obese Zucker rats. Diabetologia 53, 2008–2019. doi:10.1007/s00125-010-1773-1
Betik, A. C., Thomas, M. M., Wright, K. J., Riel, C. D., and Hepple, R. T. (2009). Exercise training from late middle age until senescence does not attenuate the declines in skeletal muscle aerobic function. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 297, R744–R755. doi:10.1152/ajpregu.90959.2008
Biensø, R. S., Ringholm, S., Kiilerich, K., Aachmann-Andersen, N.-J., Krogh-Madsen, R., Guerra, B., et al. (2012). GLUT4 and glycogen synthase are key players in bed rest–induced insulin resistance. Diabetes 61, 1090–1099. doi:10.2337/db11-0884
Bilet, L., Phielix, E., van de Weijer, T., Gemmink, A., Bosma, M., Moonen-Kornips, E., et al. (2020). One-leg inactivity induces a reduction in mitochondrial oxidative capacity, intramyocellular lipid accumulation and reduced insulin signalling upon lipid infusion: A human study with unilateral limb suspension. Diabetologia 63, 1211–1222. doi:10.1007/s00125-020-05128-1
Boden, G., Chen, X., Ruiz, J., White, J. V., and Rossetti, L. (1994). Mechanisms of fatty acid-induced inhibition of glucose uptake. J. Clin. Invest. 93, 2438–2446. doi:10.1172/JCI117252
Boden, G. (2003). Effects of free fatty acids on gluconeogenesis and glycogenolysis. Life Sci. 72, 977–988. doi:10.1016/S0024-3205(02)02350-0
Boden, G., Homko, C., Barrero, C. A., Stein, T. P., Chen, X., Cheung, P., et al. (2015). Excessive caloric intake acutely causes oxidative stress, GLUT4 carbonylation, and insulin resistance in healthy men. Sci. Transl. Med. 7, 304re7. doi:10.1126/scitranslmed.aac4765
Boden, G., Lebed, B., Schatz, M., Homko, C., and Lemieux, S. (2001). Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes 50, 1612–1617. doi:10.2337/diabetes.50.7.1612
Boden, M. J., Brandon, A. E., Tid-Ang, J. D., Preston, E., Wilks, D., Stuart, E., et al. (2012). Overexpression of manganese superoxide dismutase ameliorates high-fat diet-induced insulin resistance in rat skeletal muscle. Am. J. Physiology-Endocrinology Metabolism 303, E798–E805. doi:10.1152/ajpendo.00577.2011
Bonen, A., Jain, S. S., Snook, L. A., Han, X. X., Yoshida, Y., Buddo, K. H., et al. (2015). Extremely rapid increase in fatty acid transport and intramyocellular lipid accumulation but markedly delayed insulin resistance after high fat feeding in rats. Diabetologia 58, 2381–2391. doi:10.1007/s00125-015-3691-8
Bonen, A., Parolin, M. L., Steinberg, G. R., Calles-Escandon, J., Tandon, N. N., Glatz, J. F. C., et al. (2004). Triacylglycerol accumulation in human obesity and type 2 diabetes is associated with increased rates of skeletal muscle fatty acid transport and increased sarcolemmal FAT/CD36. FASEB J. 18, 1144–1146. doi:10.1096/fj.03-1065fje
Brand, M. D., Pakay, J. L., Ocloo, A., Kokoszka, J., Wallace, D. C., Brookes, P. S., et al. (2005). The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem. J. 392, 353–362. doi:10.1042/BJ20050890
Brand, M. D. (2010). The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 45, 466–472. doi:10.1016/j.exger.2010.01.003
Breen, L., Stokes, K. A., Churchward-Venne, T. A., Moore, D. R., Baker, S. K., Smith, K., et al. (2013). Two weeks of reduced activity decreases leg lean mass and induces “anabolic resistance” of myofibrillar protein synthesis in healthy elderly. J. Clin. Endocrinol. Metabolism 98, 2604–2612. doi:10.1210/jc.2013-1502
Bruce, C. R., Mertz, V. A., Heigenhauser, G. J. F., and Dyck, D. J. (2005). The stimulatory effect of globular adiponectin on insulin-stimulated glucose uptake and fatty acid oxidation is impaired in skeletal muscle from obese subjects. Diabetes 54, 3154–3160. doi:10.2337/diabetes.54.11.3154
Bruce, C. R., Risis, S., Babb, J. R., Yang, C., Kowalski, G. M., Selathurai, A., et al. (2012). Overexpression of sphingosine kinase 1 prevents ceramide accumulation and ameliorates muscle insulin resistance in high-fat diet-fed mice. Diabetes 61, 3148–3155. doi:10.2337/db12-0029
Brunetta, H. S., Petrick, H. L., Vachon, B., Nunes, E. A., and Holloway, G. P. (2021). Insulin rapidly increases skeletal muscle mitochondrial ADP sensitivity in the absence of a high lipid environment. Biochem. J. 478, 2539–2553. doi:10.1042/BCJ20210264
Cariou, B., Charbonnel, B., and Staels, B. (2012). Thiazolidinediones and PPARγ agonists: Time for a reassessment. Trends Endocrinol. Metabolism 23, 205–215. doi:10.1016/j.tem.2012.03.001
Cartee, G. D., Hepple, R. T., Bamman, M. M., and Zierath, J. R. (2016). Exercise promotes healthy aging of skeletal muscle. Cell Metab. 23, 1034–1047. doi:10.1016/j.cmet.2016.05.007
Cartee, G. D., Young, D. A., Sleeper, M. D., Zierath, J., Wallberg-Henriksson, H., and Holloszy, J. O. (1989). Prolonged increase in insulin-stimulated glucose transport in muscle after exercise. Am. J. Physiol. 256, E494–E499. doi:10.1152/ajpendo.1989.256.4.E494
Chen, C. C. W., Erlich, A. T., and Hood, D. A. (2018). Role of Parkin and endurance training on mitochondrial turnover in skeletal muscle. Skelet. Muscle 8, 10–14. doi:10.1186/s13395-018-0157-y
Chibalin, A. V., Yu, M., Ryder, J. W., Song, X. M., Galuska, D., Krook, A., et al. (2000). Exercise-induced changes in expression and activity of proteins involved in insulin signal transduction in skeletal muscle: Differential effects on insulin-receptor substrates 1 and 2. Proc. Natl. Acad. Sci. 97, 38–43. doi:10.1073/pnas.97.1.38
Colley, R. C., Garriguet, D., Janssen, I., Craig, C. L., Clarke, J., and Tremblay, M. S. (2011). Physical activity of Canadian children and youth: Accelerometer results from the 2007 to 2009 Canadian health measures survey. Health Rep. 22, 15–23.
Consitt, L. A., Bell, J. A., Koves, T. R., Muoio, D. M., Hulver, M. W., Haynie, K. R., et al. (2010). Peroxisome proliferator-activated receptor-gamma coactivator-1alpha overexpression increases lipid oxidation in myocytes from extremely obese individuals. Diabetes 59, 1407–1415. doi:10.2337/db09-1704
Contreras-Ferrat, A., Llanos, P., Vásquez, C., Espinosa, A., Osorio-Fuentealba, C., Arias-Calderon, M., et al. (2014). Insulin elicits a ROS-activated and an IP₃-dependent Ca²⁺ release, which both impinge on GLUT4 translocation. J. Cell Sci. 127, 1911–1923. doi:10.1242/jcs.138982
Coyle, E. F., Martin, W. H., Sinacore, D. R., Joyner, M. J., Hagberg, J. M., and Holloszy, J. O. (1984). Time course of loss of adaptations after stopping prolonged intense endurance training. J. Appl. Physiol. Respir. Environ. Exerc Physiol. 57, 1857–1864. doi:10.1152/jappl.1984.57.6.1857
Cuff, D. J., Meneilly, G. S., Martin, A., Ignaszewski, A., Tildesley, H. D., and Frohlich, J. J. (2003). Effective exercise modality to reduce insulin resistance in women with type 2 diabetes. Diabetes Care 26, 2977–2982. doi:10.2337/diacare.26.11.2977
Cutting, W. C., Mehrtens, H. G., and Tainer, M. L. (1933). Actions and uses of dinitrophenol: Promising metabolic applications. J. Am. Med. Assoc. 101, 193–195. doi:10.1001/jama.1933.02740280013006
Damirchi, A., Tehrani, B. S., Alamdari, K. A., and Babaei, P. (2014). Influence of aerobic training and detraining on serum BDNF, insulin resistance, and metabolic risk factors in middle-aged men diagnosed with metabolic syndrome. Clin. J. Sport Med. 24, 513–518. doi:10.1097/JSM.0000000000000082
Devlin, J. T., Hirshman, M., Horton, E. D., and Horton, E. S. (1987). Enhanced peripheral and splanchnic insulin sensitivity in NIDDM men after single bout of exercise. Diabetes 36, 434–439. doi:10.2337/diab.36.4.434
Dirks, M. L., Miotto, P. M., Goossens, G. H., Senden, J. M., Petrick, H. L., van Kranenburg, J., et al. (2020). Short-term bed rest-induced insulin resistance cannot be explained by increased mitochondrial H2O2 emission. J. Physiology 598, 123–137. doi:10.1113/JP278920
Dirks, M. L., Wall, B. T., van de Valk, B., Holloway, T. M., Holloway, G. P., Chabowski, A., et al. (2016). One week of bed rest leads to substantial muscle atrophy and induces whole-body insulin resistance in the absence of skeletal muscle lipid accumulation. Diabetes 65, 2862–2875. doi:10.2337/db15-1661
Dodt, C., Breckling, U., Derad, I., Fehm, H. L., and Born, J. (1997). Plasma epinephrine and norepinephrine concentrations of healthy humans associated with nighttime sleep and morning arousal. Hypertension 30, 71–76. doi:10.1161/01.HYP.30.1.71
Douen, A. G., Ramlal, T., Rastogi, S., Bilan, P. J., Cartee, G. D., Vranic, M., et al. (1990). Exercise induces recruitment of the “insulin-responsive glucose transporter”. Evidence for distinct intracellular insulin- and exercise-recruitable transporter pools in skeletal muscle. J. Biol. Chem. 265, 13427–13430. doi:10.1016/s0021-9258(18)77362-6
Dresner, A., Laurent, D., Marcucci, M., Griffin, M. E., Dufour, S., Cline, G. W., et al. (1999). Effects of free fatty acids on glucose transport and IRS-1–associated phosphatidylinositol 3-kinase activity. J. Clin. Invest. 103, 253–259. doi:10.1172/JCI5001
Dubé, J. J., Amati, F., Toledo, F. G. S., Stefanovic-Racic, M., Rossi, A., Coen, P., et al. (2011). Effects of weight loss and exercise on insulin resistance, and intramyocellular triacylglycerol, diacylglycerol and ceramide. Diabetologia 54, 1147–1156. doi:10.1007/s00125-011-2065-0
Dudley, G. A., Tullson, P. C., and Terjung, R. L. (1987). Influence of mitochondrial content on the sensitivity of respiratory control. J. Biol. Chem. 262, 9109–9114. doi:10.1016/S0021-9258(18)48054-4
Dun, Y., Liu, S., Zhang, W., Xie, M., and Qiu, L. (2017). Exercise combined with rhodiola sacra supplementation improves exercise capacity and ameliorates exhaustive exercise-induced muscle damage through enhancement of mitochondrial quality control. Oxid. Med. Cell Longev. 2017, 8024857. doi:10.1155/2017/8024857
Dutta, S., and Sengupta, P. (2016). Men and mice: Relating their ages. Life Sci. 152, 244–248. doi:10.1016/j.lfs.2015.10.025
Earnest, C. P., Lupo, M., Thibodaux, J., Hollier, C., Butitta, B., Lejeune, E., et al. (2013). Interval training in men at risk for insulin resistance. Int. J. Sports Med. 34, 355–363. doi:10.1055/s-0032-1311594
Ehrlicher, S. E., Stierwalt, H. D., Newsom, S. A., and Robinson, M. M. (2021). Short-term high-fat feeding does not alter mitochondrial lipid respiratory capacity but triggers mitophagy response in skeletal muscle of mice. Front. Endocrinol. 12. Available at: https://www.frontiersin.org/articles/10.3389/fendo.2021.651211 (Accessed January 4, 2023).
Esposito, L. A., Melov, S., Panov, A., Cottrell, B. A., and Wallace, D. C. (1999). Mitochondrial disease in mouse results in increased oxidative stress. Proc. Natl. Acad. Sci. U. S. A. 96, 4820–4825. doi:10.1073/pnas.96.9.4820
Flockhart, M., Nilsson, L. C., Tais, S., Ekblom, B., Apró, W., and Larsen, F. J. (2021). Excessive exercise training causes mitochondrial functional impairment and decreases glucose tolerance in healthy volunteers. Cell metab. 33, 957–970.e6. doi:10.1016/j.cmet.2021.02.017
Ford, E. S., and Herman, W. H. (1995). Leisure-time physical activity patterns in the U.S. Diabetic population: Findings from the 1990 national health interview survey—health promotion and disease prevention supplement. Diabetes Care 18, 27–33. doi:10.2337/diacare.18.1.27
Fu, T., Xu, Z., Liu, L., Guo, Q., Wu, H., Liang, X., et al. (2018). Mitophagy directs muscle-adipose crosstalk to alleviate dietary obesity. Cell Rep. 23, 1357–1372. doi:10.1016/j.celrep.2018.03.127
Garcia, G. C., Bartol, T. M., Phan, S., Bushong, E. A., Perkins, G., Sejnowski, T. J., et al. (2019). Mitochondrial morphology provides a mechanism for energy buffering at synapses. Sci. Rep. 9, 18306. doi:10.1038/s41598-019-54159-1
Garcia-Roves, P., Huss, J. M., Han, D.-H., Hancock, C. R., Iglesias-Gutierrez, E., Chen, M., et al. (2007). Raising plasma fatty acid concentration induces increased biogenesis of mitochondria in skeletal muscle. Proc. Natl. Acad. Sci. 104, 10709–10713. doi:10.1073/pnas.0704024104
Geiger, P. C., Han, D. H., Wright, D. C., and Holloszy, J. O. (2006). How muscle insulin sensitivity is regulated: Testing of a hypothesis. Am. J. Physiol. Endocrinol. Metab. 291, E1258–E1263. doi:10.1152/ajpendo.00273.2006
Goldstein, B. J., Mahadev, K., Wu, X., Zhu, L., and Motoshima, H. (2005). Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxidants Redox Signal. 7, 1021–1031. doi:10.1089/ars.2005.7.1021
Goodpaster, B. H., He, J., Watkins, S., and Kelley, D. E. (2001). Skeletal muscle lipid content and insulin resistance: Evidence for a paradox in endurance-trained athletes. J. Clin. Endocrinol. Metabolism 86, 5755–5761. doi:10.1210/jcem.86.12.8075
Gordon, B. A., Benson, A. C., Bird, S. R., and Fraser, S. F. (2009). Resistance training improves metabolic health in type 2 diabetes: A systematic review. Diabetes Res. Clin. Pract. 83, 157–175. doi:10.1016/j.diabres.2008.11.024
Greco, D., Kotronen, A., Westerbacka, J., Puig, O., Arkkila, P., Kiviluoto, T., et al. (2008). Gene expression in human NAFLD. Am. J. Physiology-Gastrointestinal Liver Physiology 294, G1281–G1287. doi:10.1152/ajpgi.00074.2008
Haines, M. S., Dichtel, L. E., Santoso, K., Torriani, M., Miller, K. K., and Bredella, M. A. (2020). Association between muscle mass and insulin sensitivity independent of detrimental adipose depots in young adults with overweight/obesity. Int. J. Obes. (Lond) 44, 1851–1858. doi:10.1038/s41366-020-0590-y
Han, X.-X., Chabowski, A., Tandon, N. N., Calles-Escandon, J., Glatz, J. F. C., Luiken, J. J. F. P., et al. (2007). Metabolic challenges reveal impaired fatty acid metabolism and translocation of FAT/CD36 but not FABPpm in obese Zucker rat muscle. Am. J. Physiology-Endocrinology Metabolism 293, E566–E575. doi:10.1152/ajpendo.00106.2007
Hancock, C. R., Han, D. H., Chen, M., Terada, S., Yasuda, T., Wright, D. C., et al. (2008). High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc. Natl. Acad. Sci. U. S. A. 105, 7815–7820. doi:10.1073/PNAS.0802057105
Hansen, P. A., Nolte, L. A., Chen, M. M., and Holloszy, J. O. (1998). Increased GLUT-4 translocation mediates enhanced insulin sensitivity of muscle glucose transport after exercise. J. Appl. Physiol. 85, 1218–1222. doi:10.1152/jappl.1998.85.4.1218
Helge, J. W., and Kiens, B. (1997). Muscle enzyme activity in humans: Role of substrate availability and training. Am. J. Physiol. 272, R1620–R1624. doi:10.1152/ajpregu.1997.272.5.R1620
Hernández, E. Á., Kahl, S., Seelig, A., Begovatz, P., Irmler, M., Kupriyanova, Y., et al. (2017). Acute dietary fat intake initiates alterations in energy metabolism and insulin resistance. J. Clin. Invest. 127, 695–708. doi:10.1172/JCI89444
Herzig, K.-H., Ahola, R., Leppäluoto, J., Jokelainen, J., Jämsä, T., and Keinänen-Kiukaanniemi, S. (2014). Light physical activity determined by a motion sensor decreases insulin resistance, improves lipid homeostasis and reduces visceral fat in high-risk subjects: PreDiabEx study RCT. Int. J. Obes. 38, 1089–1096. doi:10.1038/ijo.2013.224
Hingst, J. R., Bruhn, L., Hansen, M. B., Rosschou, M. F., Birk, J. B., Fentz, J., et al. (2018). Exercise-induced molecular mechanisms promoting glycogen supercompensation in human skeletal muscle. Mol. Metab. 16, 24–34. doi:10.1016/j.molmet.2018.07.001
Ho, C. H., and Pande, S. V. (1974). On the specificity of the inhibition of adenine nucleotide translocase by long chain acyl-coenzyme a esters. Biochimica Biophysica Acta (BBA) - Lipids Lipid Metabolism 369, 86–94. doi:10.1016/0005-2760(74)90195-7
Holloszy, J. O., and Coyle, E. F. (1984). Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J. Appl. Physiol. Respir. Environ. Exerc Physiol. 56, 831–838. doi:10.1152/jappl.1984.56.4.831
Holloszy, J. O. (2013). “Deficiency” of mitochondria in muscle does not cause insulin resistance. Diabetes 62, 1036–1040. doi:10.2337/db12-1107
Holloway, G. P., Benton, C. R., Mullen, K. L., Yoshida, Y., Snook, L. A., Han, X.-X., et al. (2009). In obese rat muscle transport of palmitate is increased and is channeled to triacylglycerol storage despite an increase in mitochondrial palmitate oxidation. Am. J. Physiology-Endocrinology Metabolism 296, E738–E747. doi:10.1152/ajpendo.90896.2008
Holloway, G. P., Han, X. X., Jain, S. S., Bonen, A., and Chabowski, A. (2014). Chronic muscle stimulation improves insulin sensitivity while increasing subcellular lipid droplets and reducing selected diacylglycerol and ceramide species in obese Zucker rats. Diabetologia 57, 832–840. doi:10.1007/s00125-014-3169-0
Holloway, G. P., Luiken, J. J. F. P., Glatz, J. F. C., Spriet, L. L., and Bonen, A. (2008). Contribution of FAT/CD36 to the regulation of skeletal muscle fatty acid oxidation: An overview. Acta Physiol. 194, 293–309. doi:10.1111/j.1748-1716.2008.01878.x
Holloway, G. P., Thrush, A. B., Heigenhauser, G. J. F., Tandon, N. N., Dyck, D. J., Bonen, A., et al. (2007). Skeletal muscle mitochondrial FAT/CD36 content and palmitate oxidation are not decreased in obese women. Am. J. Physiol. Endocrinol. Metab. 292, E1782–E1789. doi:10.1152/ajpendo.00639.2006
Host, H. H., Hansen, P. A., Nolte, L. A., Chen, M. M., and Holloszy, J. O. (1998). Rapid reversal of adaptive increases in muscle GLUT-4 and glucose transport capacity after training cessation. J. Appl. Physiol. 84, 798–802. doi:10.1152/jappl.1998.84.3.798
Howlett, K. F., Sakamoto, K., Hirshman, M. F., Aschenbach, W. G., Dow, M., White, M. F., et al. (2002). Insulin signaling after exercise in insulin receptor substrate-2-deficient mice. Diabetes 51, 479–483. doi:10.2337/diabetes.51.2.479
Huang, T.-Y., Zheng, D., Houmard, J. A., Brault, J. J., Hickner, R. C., and Cortright, R. N. (2017). Overexpression of PGC-1α increases peroxisomal activity and mitochondrial fatty acid oxidation in human primary myotubes. Am. J. Physiol. Endocrinol. Metab. 312, E253–E263. doi:10.1152/ajpendo.00331.2016
Hudson, N. J., and Franklin, C. E. (2002). Maintaining muscle mass during extended disuse: Aestivating frogs as a model species. J. Exp. Biol. 205, 2297–2303. doi:10.1242/jeb.205.15.2297
Hulver, M. W., Berggren, J. R., Cortright, R. N., Dudek, R. W., Thompson, R. P., Pories, W. J., et al. (2003). Skeletal muscle lipid metabolism with obesity. Am. J. Physiology-Endocrinology Metabolism 284, E741–E747. doi:10.1152/ajpendo.00514.2002
IDF Diabetes Atlas (2023). Tenth Edition (n.d.). Available at: https://diabetesatlas.org/ (Accessed March 7, 2023).
Iqbal, S., Ostojic, O., Singh, K., Joseph, A.-M., and Hood, D. A. (2013). Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle & Nerve 48, 963–970. doi:10.1002/mus.23838
Itani, S. I., Ruderman, N. B., Schmieder, F., and Boden, G. (2002). Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 51, 2005–2011. doi:10.2337/diabetes.51.7.2005
Jain, S. S., Paglialunga, S., Vigna, C., Ludzki, A., Herbst, E. A., Lally, J. S., et al. (2014). High-fat diet-induced mitochondrial biogenesis is regulated by mitochondrial-derived reactive oxygen species activation of CaMKII. Diabetes 63, 1907–1913. doi:10.2337/db13-0816
JeBailey, L., Wanono, O., Niu, W., Roessler, J., Rudich, A., and Klip, A. (2007). Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes 56, 394–403. doi:10.2337/db06-0823
Kakehi, S., Tamura, Y., Ikeda, S.-I., Kaga, N., Taka, H., Ueno, N., et al. (2021). Short-term physical inactivity induces diacylglycerol accumulation and insulin resistance in muscle via lipin1 activation. Am. J. Physiol. Endocrinol. Metab. 321, E766–E781. doi:10.1152/ajpendo.00254.2020
Kavazis, A. N., Talbert, E. E., Smuder, A. J., Hudson, M. B., Nelson, W. B., and Powers, S. K. (2009). Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radic. Biol. Med. 46, 842–850. doi:10.1016/j.freeradbiomed.2009.01.002
Kelley, D. E., Goodpaster, B., Wing, R. R., and Simoneau, J.-A. (1999). Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am. J. Physiology-Endocrinology Metabolism 277, E1130–E1141. doi:10.1152/ajpendo.1999.277.6.E1130
Kelley, D. E., He, J., Menshikova, E. V., and Ritov, V. B. (2002). Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51, 2944–2950. doi:10.2337/diabetes.51.10.2944
Kim, J.-Y., Hickner, R. C., Cortright, R. L., Dohm, G. L., and Houmard, J. A. (2000). Lipid oxidation is reduced in obese human skeletal muscle. Am. J. Physiology-Endocrinology Metabolism 279, E1039–E1044. doi:10.1152/ajpendo.2000.279.5.E1039
Kim, J. K., Fillmore, J. J., Sunshine, M. J., Albrecht, B., Higashimori, T., Kim, D.-W., et al. (2004). PKC-θ knockout mice are protected from fat-induced insulin resistance. J. Clin. Invest. 114, 823–827. doi:10.1172/JCI22230
King, H., Aubert, R. E., and Herman, W. H. (1998). Global burden of diabetes, 1995–2025: Prevalence, numerical estimates, and projections. Diabetes Care 21, 1414–1431. doi:10.2337/diacare.21.9.1414
Knudsen, S. H., Hansen, L. S., Pedersen, M., Dejgaard, T., Hansen, J., Hall, G. V., et al. (2012). Changes in insulin sensitivity precede changes in body composition during 14 days of step reduction combined with overfeeding in healthy young men. J. Appl. Physiology 113, 7–15. doi:10.1152/japplphysiol.00189.2011
Koves, T. R., Li, P., An, J., Akimoto, T., Slentz, D., Ilkayeva, O., et al. (2005). Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J. Biol. Chem. 280, 33588–33598. doi:10.1074/jbc.M507621200
Koves, T. R., Ussher, J. R., Noland, R. C., Slentz, D., Mosedale, M., Ilkayeva, O., et al. (2008). Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7, 45–56. doi:10.1016/j.cmet.2007.10.013
Krogh-Madsen, R., Thyfault, J. P., Broholm, C., Mortensen, O. H., Olsen, R. H., Mounier, R., et al. (2010). A 2-wk reduction of ambulatory activity attenuates peripheral insulin sensitivity. J. Appl. Physiology 108, 1034–1040. doi:10.1152/japplphysiol.00977.2009
Krssak, M., Brehm, A., Bernroider, E., Anderwald, C., Nowotny, P., Man, C. D., et al. (2004). Alterations in postprandial hepatic glycogen metabolism in type 2 diabetes. Diabetes 53, 3048–3056. doi:10.2337/diabetes.53.12.3048
Kurek, K., Mikłosz, A., Łukaszuk, B., Chabowski, A., Górski, J., and Żendzian-Piotrowska, M. (2015). Inhibition of ceramide de novo synthesis ameliorates diet induced skeletal muscles insulin resistance. J. Diabetes Res. 2015, e154762. doi:10.1155/2015/154762
Laker, R. C., Drake, J. C., Wilson, R. J., Lira, V. A., Lewellen, B. M., Ryall, K. A., et al. (2017). Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 8, 548. doi:10.1038/s41467-017-00520-9
Lally, J. S. V., Snook, L. A., Han, X. X., Chabowski, A., Bonen, A., and Holloway, G. P. (2012). Subcellular lipid droplet distribution in red and white muscles in the obese Zucker rat. Diabetologia 55, 479–488. doi:10.1007/s00125-011-2367-2
Lark, D. S., Kang, L., Lustig, M. E., Bonner, J. S., James, F. D., Neufer, P. D., et al. (2015). Enhanced mitochondrial superoxide scavenging does not improve muscle insulin action in the high fat-fed mouse. PLOS ONE 10, e0126732. doi:10.1371/journal.pone.0126732
Lawrence, R. D. (1926). The effect of exercise on insulin action in diabetes. Br. Med. J. 1, 648–650. doi:10.1136/bmj.1.3406.648
Lee, A. D., Hansen, P. A., and Holloszy, J. O. (1995). Wortmannin inhibits insulin-stimulated but not contraction-stimulated glucose transport activity in skeletal muscle. FEBS Lett. 361, 51–54. doi:10.1016/0014-5793(95)00147-2
Lee, H.-Y., Choi, C. S., Birkenfeld, A. L., Alves, T. C., Jornayvaz, F. R., Jurczak, M. J., et al. (2010). Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 12, 668–674. doi:10.1016/j.cmet.2010.11.004
Lee, H.-Y., Lee, J. S., Alves, T., Ladiges, W., Rabinovitch, P. S., Jurczak, M. J., et al. (2017). Mitochondrial-targeted catalase protects against high-fat diet–induced muscle insulin resistance by decreasing intramuscular lipid accumulation. Diabetes 66, 2072–2081. –2081. doi:10.2337/db16-1334
Li, Y., Soos, T. J., Li, X., Wu, J., Degennaro, M., Sun, X., et al. (2004). Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101). J. Biol. Chem. 279, 45304–45307. doi:10.1074/jbc.C400186200
Lin, J., Wu, H., Tarr, P. T., Zhang, C.-Y., Wu, Z., Boss, O., et al. (2002). Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418, 797–801. doi:10.1038/nature00904
Lira, V. A., Okutsu, M., Zhang, M., Greene, N. P., Laker, R. C., Breen, D. S., et al. (2013). Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 27, 4184–4193. doi:10.1096/fj.13-228486
Liu, L., Zhang, Y., Chen, N., Shi, X., Tsang, B., and Yu, Y.-H. (2007). Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. J. Clin. Invest. 117, 1679–1689. doi:10.1172/JCI30565
Ljubicic, V., and Hood, D. A. (2009a). Diminished contraction-induced intracellular signaling towards mitochondrial biogenesis in aged skeletal muscle. Aging Cell 8, 394–404. doi:10.1111/j.1474-9726.2009.00483.x
Ljubicic, V., and Hood, D. A. (2009b). Specific attenuation of protein kinase phosphorylation in muscle with a high mitochondrial content. Am. J. Physiology-Endocrinology Metabolism 297, E749–E758. doi:10.1152/ajpendo.00130.2009
Loh, K., Deng, H., Fukushima, A., Cai, X., Boivin, B., Galic, S., et al. (2009). Reactive oxygen species enhance insulin sensitivity. Cell Metab. 10, 260–272. doi:10.1016/j.cmet.2009.08.009
Loomba, R., Friedman, S. L., and Shulman, G. I. (2021). Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184, 2537–2564. doi:10.1016/j.cell.2021.04.015
Ludzki, A., Paglialunga, S., Smith, B. K., Herbst, E. A. F., Allison, M. K., Heigenhauser, G. J., et al. (2015). Rapid repression of ADP transport by palmitoyl-CoA is attenuated by exercise training in humans: A potential mechanism to decrease oxidative stress and improve skeletal muscle insulin signaling. Diabetes 64, 2769–2779. doi:10.2337/db14-1838
Luiken, J. J. F. P., Arumugam, Y., Dyck, D. J., Bell, R. C., Pelsers, M. M. L., Turcotte, L. P., et al. (2001). Increased rates of fatty acid uptake and plasmalemmal fatty acid transporters in obese zucker rats. J. Biol. Chem. 276, 40567–40573. doi:10.1074/jbc.M100052200
MacDougall, J. D., Sale, D. G., Moroz, J. R., Elder, G. C., Sutton, J. R., and Howald, H. (1979). Mitochondrial volume density in human skeletal muscle following heavy resistance training. Med. Sci. Sports 11, 164–166.
Mahadev, K., Wu, X., Zilbering, A., Zhu, L., Lawrence, J. T. R., and Goldstein, B. J. (2001). Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J. Biol. Chem. 276, 48662–48669. doi:10.1074/jbc.M105061200
Malin, S. K., Solomon, T. P. J., Blaszczak, A., Finnegan, S., Filion, J., and Kirwan, J. P. (2013). Pancreatic β-cell function increases in a linear dose-response manner following exercise training in adults with prediabetes. Am. J. Physiology-Endocrinology Metabolism 305, E1248–E1254. doi:10.1152/ajpendo.00260.2013
Mann, S., Beedie, C., Balducci, S., Zanuso, S., Allgrove, J., Bertiato, F., et al. (2014). Changes in insulin sensitivity in response to different modalities of exercise: A review of the evidence. Diabetes/Metabolism Res. Rev. 30, 257–268. doi:10.1002/dmrr.2488
Marceau, P., Biron, S., Hould, F.-S., Marceau, S., Simard, S., Thung, S. N., et al. (1999). Liver pathology and the metabolic syndrome X in severe obesity. J. Clin. Endocrinol. Metabolism 84, 1513–1517. doi:10.1210/jcem.84.5.5661
Marchesini, G., Brizi, M., Morselli-Labate, A. M., Bianchi, G., Bugianesi, E., McCullough, A. J., et al. (1999). Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 107, 450–455. doi:10.1016/S0002-9343(99)00271-5
Maughan, R. J., Watson, J. S., and Weir, J. (1983). Strength and cross-sectional area of human skeletal muscle. J. Physiol. 338, 37–49. doi:10.1113/jphysiol.1983.sp014658
McClung, J. P., Roneker, C. A., Mu, W., Lisk, D. J., Langlais, P., Liu, F., et al. (2004). Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc. Natl. Acad. Sci. 101, 8852–8857. doi:10.1073/pnas.0308096101
McGlory, C., von Allmen, M. T., Stokes, T., Morton, R. W., Hector, A. J., Lago, B. A., et al. (2018). Failed recovery of glycemic control and myofibrillar protein synthesis with 2 wk of physical inactivity in overweight, prediabetic older adults. J. Gerontol. A Biol. Sci. Med. Sci. 73, 1070–1077. doi:10.1093/gerona/glx203
McKenzie, A. I., Reidy, P. T., Nelson, D. S., Mulvey, J. L., Yonemura, N. M., Petrocelli, J. J., et al. (2020). Pharmacological inhibition of TLR4 ameliorates muscle and liver ceramide content after disuse in previously physically active mice. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 318, R503–R511. doi:10.1152/ajpregu.00330.2019
Mikus, C. R., Oberlin, D. J., Libla, J. L., Taylor, A. M., Booth, F. W., and Thyfault, J. P. (2012). Lowering physical activity impairs glycemic control in healthy volunteers. Med. Sci. Sports Exerc 44, 225–231. doi:10.1249/MSS.0b013e31822ac0c0
Min, K., Smuder, A. J., Kwon, O., Kavazis, A. N., Szeto, H. H., and Powers, S. K. (2011). Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol. 111, 1459–1466. doi:10.1152/japplphysiol.00591.2011
Miotto, P. M., LeBlanc, P. J., and Holloway, G. P. (2018). High-fat diet causes mitochondrial dysfunction as a result of impaired ADP sensitivity. Diabetes 67, 2199–2205. doi:10.2337/db18-0417
Miotto, P. M., Mcglory, C., Bahniwal, R., Kamal, M., Phillips, S. M., and Holloway, G. P. (2019). Supplementation with dietary ω-3 mitigates immobilization-induced reductions in skeletal muscle mitochondrial respiration in young women. FASEB J. 33, 8232–8240. doi:10.1096/FJ.201900095R
Miquilena-Colina, M. E., Lima-Cabello, E., Sanchez-Campos, S., Garcia-Mediavilla, M. V., Fernandez-Bermejo, M., Lozano-Rodriguez, T., et al. (2011). Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut 60, 1394–1402. doi:10.1136/gut.2010.222844
Mitranun, W., Deerochanawong, C., Tanaka, H., and Suksom, D. (2014). Continuous vs interval training on glycemic control and macro- and microvascular reactivity in type 2 diabetic patients. Scand. J. Med. Sci. Sports 24, e69–e76. doi:10.1111/sms.12112
Mogensen, M., Sahlin, K., Fernström, M., Glintborg, D., Vind, B. F., Beck-Nielsen, H., et al. (2007). Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 56, 1592–1599. doi:10.2337/db06-0981
Morino, K., Neschen, S., Bilz, S., Sono, S., Tsirigotis, D., Reznick, R. M., et al. (2008). Muscle-specific IRS-1 Ser→Ala transgenic mice are protected from fat-induced insulin resistance in skeletal muscle. Diabetes 57, 2644–2651. doi:10.2337/db06-0454
Morino, K., Petersen, K. F., Dufour, S., Befroy, D., Frattini, J., Shatzkes, N., et al. (2005). Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Invest. 115, 3587–3593. doi:10.1172/JCI25151
Motahari-Tabari, N., Shirvani, M. A., Shirzad-e-Ahoodashty, M., Yousefi-Abdolmaleki, E., and Teimourzadeh, M. (2014). The effect of 8 Weeks aerobic exercise on insulin resistance in type 2 diabetes: A randomized clinical trial. Glob. J. Health Sci. 7, p115–p121. doi:10.5539/gjhs.v7n1p115
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi:10.1042/BJ20081386
Nielsen, J., Mogensen, M., Vind, B. F., Sahlin, K., Højlund, K., Schrøder, H. D., et al. (2010). Increased subsarcolemmal lipids in type 2 diabetes: Effect of training on localization of lipids, mitochondria, and glycogen in sedentary human skeletal muscle. Am. J. Physiology-Endocrinology Metabolism 298, E706–E713. doi:10.1152/ajpendo.00692.2009
Oikawa, S. Y., Holloway, T. M., and Phillips, S. M. (2019). The impact of step reduction on muscle health in aging: Protein and exercise as countermeasures. Front. Nutr. 6. Available at: https://www.frontiersin.org/articles/10.3389/fnut.2019.00075 (Accessed March 28, 2023).
Orfali, K. A., Fryer, L. G., Holness, M. J., and Sugden, M. C. (1993). Long-term regulation of pyruvate dehydrogenase kinase by high-fat feeding. Experiments in vivo and in cultured cardiomyocytes. FEBS Lett. 336, 501–505. doi:10.1016/0014-5793(93)80864-q
Paglialunga, S., Ludzki, A., Root-McCaig, J., and Holloway, G. P. (2015). In adipose tissue, increased mitochondrial emission of reactive oxygen species is important for short-term high-fat diet-induced insulin resistance in mice. Diabetologia 58, 1071–1080. doi:10.1007/s00125-015-3531-x
Paglialunga, S., van Bree, B., Bosma, M., Valdecantos, M. P., Amengual-Cladera, E., Jörgensen, J. A., et al. (2012). Targeting of mitochondrial reactive oxygen species production does not avert lipid-induced insulin resistance in muscle tissue from mice. Diabetologia 55, 2759–2768. doi:10.1007/s00125-012-2626-x
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1013–1022. doi:10.1038/s41556-018-0176-2
Pan, D. A., Lillioja, S., Kriketos, A. D., Milner, M. R., Baur, L. A., Bogardus, C., et al. (1997). Skeletal muscle TMglyceride levels are inversely related to insulin action. Diabetes 46 (6), 983–988. doi:10.2337/diab.46.6.983
Pehmøller, C., Brandt, N., Birk, J. B., Høeg, L. D., Sjøberg, K. A., Goodyear, L. J., et al. (2012). Exercise alleviates lipid-induced insulin resistance in human skeletal muscle–signaling interaction at the level of TBC1 domain family member 4. Diabetes 61, 2743–2752. doi:10.2337/db11-1572
Perry, C. G. R., Heigenhauser, G. J. F., Bonen, A., and Spriet, L. L. (2008). High-intensity aerobic interval training increases fat and carbohydrate metabolic capacities in human skeletal muscle. Appl. Physiol. Nutr. Metab. 33, 1112–1123. doi:10.1139/H08-097
Perry, C. G. R., Lally, J., Holloway, G. P., Heigenhauser, G. J. F., Bonen, A., and Spriet, L. L. (2010). Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J. Physiology 588, 4795–4810. doi:10.1113/jphysiol.2010.199448
Peters, S. J., Harris, R. A., Wu, P., Pehleman, T. L., Heigenhauser, G. J. F., and Spriet, L. L. (2001). Human skeletal muscle PDH kinase activity and isoform expression during a 3-day high-fat/low-carbohydrate diet. Am. J. Physiology-Endocrinology Metabolism 281, E1151–E1158. doi:10.1152/ajpendo.2001.281.6.E1151
Petersen, M. C., Madiraju, A. K., Gassaway, B. M., Marcel, M., Nasiri, A. R., Butrico, G., et al. (2016). Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J. Clin. Invest. 126, 4361–4371. doi:10.1172/JCI86013
Petrick, H. L., King, T. J., Pignanelli, C., Vanderlinde, T. E., Cohen, J. N., Holloway, G. P., et al. (2021). Endurance and sprint training improve glycemia and V˙O2peak but only frequent endurance benefits blood pressure and lipidemia. Med. Sci. Sports Exerc 53, 1194–1205. doi:10.1249/MSS.0000000000002582
Phillips, S. M., Green, H. J., Tarnopolsky, M. A., Heigenhauser, G. J. F., and Grant, S. M. (1996). Progressive effect of endurance training on metabolic adaptations in working skeletal muscle. Am. J. Physiology - Endocrinol. Metabolism 270, E265–E272. doi:10.1152/AJPENDO.1996.270.2.E265
Powers, S. K., Hudson, M. B., Nelson, W. B., Talbert, E. E., Min, K., Szeto, H. H., et al. (2011). Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit. Care Med. 39, 1749–1759. doi:10.1097/CCM.0b013e3182190b62
Powers, S. K., Smuder, A., and Judge, A. (2012a). Oxidative stress and disuse muscle atrophy: Cause or consequence? Curr. Opin. Clin. Nutr. Metab. Care 15, 240–245. doi:10.1097/MCO.0b013e328352b4c2
Powers, S. K., Wiggs, M. P., Duarte, J. A., Zergeroglu, A. M., and Demirel, H. A. (2012b). Mitochondrial signaling contributes to disuse muscle atrophy. Am. J. Physiol. Endocrinol. Metab. 303, E31–E39. doi:10.1152/ajpendo.00609.2011
Puigserver, P., Wu, Z., Park, C. W., Graves, R., Wright, M., and Spiegelman, B. M. (1998). A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839. doi:10.1016/S0092-8674(00)81410-5
Quadrilatero, J., Bombardier, E., Norris, S. M., Talanian, J. L., Palmer, M. S., Logan, H. M., et al. (2010). Prolonged moderate-intensity aerobic exercise does not alter apoptotic signaling and DNA fragmentation in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 298, E534–E547. doi:10.1152/ajpendo.00678.2009
Racil, G., Ben Ounis, O., Hammouda, O., Kallel, A., Zouhal, H., Chamari, K., et al. (2013). Effects of high vs. moderate exercise intensity during interval training on lipids and adiponectin levels in obese young females. Eur. J. Appl. Physiol. 113, 2531–2540. doi:10.1007/s00421-013-2689-5
Randle, P. J., Garland, P. B., Hales, C. N., and Newsholme, E. A. (1963). The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 281, 785–789. doi:10.1016/S0140-6736(63)91500-9
Reichkendler, M. h., Rosenkilde, M., Auerbach, P. l., Agerschou, J., Nielsen, M. b., Kjaer, A., et al. (2014). Only minor additional metabolic health benefits of high as opposed to moderate dose physical exercise in young, moderately overweight men. Obesity 22, 1220–1232. doi:10.1002/oby.20226
Reidy, P. T., McKenzie, A. I., Mahmassani, Z., Morrow, V. R., Yonemura, N. M., Hopkins, P. N., et al. (2018). Skeletal muscle ceramides and relationship with insulin sensitivity after 2 weeks of simulated sedentary behaviour and recovery in healthy older adults. J. Physiol. 596, 5217–5236. doi:10.1113/JP276798
Reidy, P. T., Monnig, J. M., Pickering, C. E., Funai, K., and Drummond, M. J. (2021). Preclinical rodent models of physical inactivity-induced muscle insulin resistance: Challenges and solutions. J. Appl. Physiology 130, 537–544. doi:10.1152/japplphysiol.00954.2020
Reynolds, L. J., Credeur, D. P., Holwerda, S. W., Leidy, H. J., Fadel, P. J., and Thyfault, J. P. (2015). Acute inactivity impairs glycemic control but not blood flow to glucose ingestion. Med. Sci. Sports Exerc 47, 1087–1094. doi:10.1249/MSS.0000000000000508
Richter, E. A., and Hargreaves, M. (2013). Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 93, 993–1017. doi:10.1152/physrev.00038.2012
Richter, E. A., Mikines, K. J., Galbo, H., and Kiens, B. (1989). Effect of exercise on insulin action in human skeletal muscle. J. Appl. Physiol. 66, 876–885. doi:10.1152/jappl.1989.66.2.876
Ringholm, S., Biensø, R. S., Kiilerich, K., Guadalupe-Grau, A., Aachmann-Andersen, N. J., Saltin, B., et al. (2011). Bed rest reduces metabolic protein content and abolishes exercise-induced mRNA responses in human skeletal muscle. Am. J. Physiology-Endocrinology Metabolism 301, E649–E658. doi:10.1152/ajpendo.00230.2011
Ritov, V. B., Menshikova, E. V., He, J., Ferrell, R. E., Goodpaster, B. H., and Kelley, D. E. (2005). Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 54, 8–14. doi:10.2337/diabetes.54.1.8
Roberts, C. K., Little, J. P., and Thyfault, J. P. (2013). Modification of insulin sensitivity and glycemic control by activity and exercise. Med. Sci. Sports Exerc. 45, 1868–1877. doi:10.1249/MSS.0b013e318295cdbb
Rudich, A., Kozlovsky, N., Potashnik, R., and Bashan, N. (1997). Oxidant stress reduces insulin responsiveness in 3T3-L1 adipocytes. Am. J. Physiology-Endocrinology Metabolism 272, E935–E940. doi:10.1152/ajpendo.1997.272.5.E935
Rudich, A., Tirosh, A., Potashnik, R., Khamaisi, M., and Bashan, N. (1999). Lipoic acid protects against oxidative stress induced impairment in insulin stimulation of protein kinase B and glucose transport in 3T3-L1 adipocytes. Diabetologia 42, 949–957. doi:10.1007/s001250051253
Russell, A. P., Feilchenfeldt, J., Schreiber, S., Praz, M., Crettenand, A., Gobelet, C., et al. (2003). Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes 52, 2874–2881. doi:10.2337/diabetes.52.12.2874
Ryder, J. w., Chibalin, A. v., and Zierath, J. r. (2001). Intracellular mechanisms underlying increases in glucose uptake in response to insulin or exercise in skeletal muscle. Acta Physiol. Scand. 171, 249–257. doi:10.1046/j.1365-201x.2001.00827.x
Ryysy, L., Häkkinen, A. M., Goto, T., Vehkavaara, S., Westerbacka, J., Halavaara, J., et al. (2000). Hepatic fat content and insulin action on free fatty acids and glucose metabolism rather than insulin absorption are associated with insulin requirements during insulin therapy in type 2 diabetic patients. Diabetes 49, 749–758. doi:10.2337/diabetes.49.5.749
Samuel, V. T., Liu, Z.-X., Qu, X., Elder, B. D., Bilz, S., Befroy, D., et al. (2004). Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 279, 32345–32353. doi:10.1074/jbc.M313478200
Samuel, V. T., Liu, Z.-X., Wang, A., Beddow, S. A., Geisler, J. G., Kahn, M., et al. (2007). Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Invest. 117, 739–745. doi:10.1172/JCI30400
Savikj, M., Gabriel, B. M., Alm, P. S., Smith, J., Caidahl, K., Björnholm, M., et al. (2019). Afternoon exercise is more efficacious than morning exercise at improving blood glucose levels in individuals with type 2 diabetes: A randomised crossover trial. Diabetologia 62, 233–237. doi:10.1007/s00125-018-4767-z
Schenk, S., Cook, J. N., Kaufman, A. E., and Horowitz, J. F. (2005). Postexercise insulin sensitivity is not impaired after an overnight lipid infusion. Am. J. Physiology-Endocrinology Metabolism 288, E519–E525. doi:10.1152/ajpendo.00401.2004
Schenk, S., and Horowitz, J. F. (2007). Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J. Clin. Investigation 117, 1690–1698. doi:10.1172/JCI30566
Schmitz-Peiffer, C., Craig, D. L., and Biden, T. J. (1999). Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J. Biol. Chem. 274, 24202–24210. doi:10.1074/jbc.274.34.24202
Seifert, E. L., Estey, C., Xuan, J. Y., and Harper, M. E. (2010). Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J. Biol. Chem. 285, 5748–5758. doi:10.1074/JBC.M109.026203
Sinha, S., Perdomo, G., Brown, N. F., and O’Doherty, R. M. (2004). Fatty acid-induced insulin resistance in L6 myotubes is prevented by inhibition of activation and nuclear localization of nuclear factor κB. J. Biol. Chem. 279, 41294–41301. doi:10.1074/jbc.M406514200
Sjøberg, K. A., Frøsig, C., Kjøbsted, R., Sylow, L., Kleinert, M., Betik, A. C., et al. (2017). Exercise increases human skeletal muscle insulin sensitivity via coordinated increases in microvascular perfusion and molecular signaling. Diabetes 66, 1501–1510. doi:10.2337/db16-1327
Skelly, L. E., MacInnis, M. J., Bostad, W., McCarthy, D. G., Jenkins, E. M., Archila, L. R., et al. (2023). Human skeletal muscle mitochondrial responses to single-leg intermittent or continuous cycle exercise training matched for absolute intensity and total work. Scand. J. Med. Sci. Sports n/a. doi:10.1111/sms.14332
Smith, A. C., Mullen, K. L., Junkin, K. A., Nickerson, J., Chabowski, A., Bonen, A., et al. (2007). Metformin and exercise reduce muscle FAT/CD36 and lipid accumulation and blunt the progression of high-fat diet-induced hyperglycemia. Am. J. Physiol. Endocrinol. Metab. 293, E172–E181. doi:10.1152/ajpendo.00677.2006
Smith, A. D., Crippa, A., Woodcock, J., and Brage, S. (2016). Physical activity and incident type 2 diabetes mellitus: A systematic review and dose–response meta-analysis of prospective cohort studies. Diabetologia 59, 2527–2545. doi:10.1007/s00125-016-4079-0
Smith, B. K., Perry, C. G. R., Herbst, E. A. F., Ritchie, I. R., Beaudoin, M. S., Smith, J. C., et al. (2013). Submaximal ADP-stimulated respiration is impaired in ZDF rats and recovered by resveratrol. J. Physiology 591, 6089–6101. doi:10.1113/jphysiol.2013.259226
Smith, B. K., Perry, C. G. R., Koves, T. R., Wright, D. C., Smith, J. C., Neufer, P. D., et al. (2012). Identification of a novel malonyl-CoA IC50 for CPT-I: Implications for predicting in vivo fatty acid oxidation rates. Biochem. J. 448, 13–20. doi:10.1042/BJ20121110
Smith, C. D., Lin, C.-T., McMillin, S. L., Weyrauch, L. A., Schmidt, C. A., Smith, C. A., et al. (2021). Genetically increasing flux through β-oxidation in skeletal muscle increases mitochondrial reductive stress and glucose intolerance. Am. J. Physiology-Endocrinology Metabolism 320, E938–E950. doi:10.1152/ajpendo.00010.2021
Sowell, M. O., Boggs, K. P., Robinson, K. A., Dutton, S. L., and Buse, M. G. (1991). Effects of insulin and phospholipase C in control and denervated rat skeletal muscle. Am. J. Physiol. 260, E247–E256. doi:10.1152/ajpendo.1991.260.2.E247
Spriet, L. L., and Heigenhauser, G. J. F. (2002). Regulation of pyruvate dehydrogenase (PDH) activity in human skeletal muscle during exercise. Exerc. Sport Sci. Rev. 30, 91–95. doi:10.1097/00003677-200204000-00009
Stellingwerff, T., Boon, H., Jonkers, R. A. M., Senden, J. M., Spriet, L. L., Koopman, R., et al. (2007). Significant intramyocellular lipid use during prolonged cycling in endurance-trained males as assessed by three different methodologies. Am. J. Physiology-Endocrinology Metabolism 292, E1715–E1723. doi:10.1152/ajpendo.00678.2006
Stephens, B. R., Granados, K., Zderic, T. W., Hamilton, M. T., and Braun, B. (2011). Effects of 1 day of inactivity on insulin action in healthy men and women: Interaction with energy intake. Metabolism - Clin. Exp. 60, 941–949. doi:10.1016/j.metabol.2010.08.014
Sugden, M. C., and Holness, M. J. (1994). Interactive regulation of the pyruvate dehydrogenase complex and the carnitine palmitoyltransferase system. FASEB J. 8, 54–61. doi:10.1096/fasebj.8.1.8299890
Summermatter, S., Baum, O., Santos, G., Hoppeler, H., and Handschin, C. (2010). Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) promotes skeletal muscle lipid refueling in vivo by activating de novo lipogenesis and the pentose phosphate pathway. J. Biol. Chem. 285, 32793–32800. doi:10.1074/jbc.M110.145995
Summers, S. A. (2006). Ceramides in insulin resistance and lipotoxicity. Prog. Lipid Res. 45, 42–72. doi:10.1016/j.plipres.2005.11.002
Szendroedi, J., Yoshimura, T., Phielix, E., Koliaki, C., Marcucci, M., Zhang, D., et al. (2014). Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. U. S. A. 111, 9597–9602. doi:10.1073/pnas.1409229111
Tainter, M. L., Stockton, A. B., and Cutting, W. C. (1933). Use of dinitrophenol in obesity and related conditions: A progress report. J. Am. Med. Assoc. 101, 1472–1475. doi:10.1001/jama.1933.02740440032009
Talanian, J. L., Galloway, S. D. R., Heigenhauser, G. J. F., Bonen, A., and Spriet, L. L. (2007). Two weeks of high-intensity aerobic interval training increases the capacity for fat oxidation during exercise in women. J. Appl. Physiology 102, 1439–1447. doi:10.1152/japplphysiol.01098.2006
Tarpey, M. D., Davy, K. P., McMillan, R. P., Bowser, S. M., Halliday, T. M., Boutagy, N. E., et al. (2017). Skeletal muscle autophagy and mitophagy in endurance-trained runners before and after a high-fat meal. Mol. Metab. 6, 1597–1609. doi:10.1016/j.molmet.2017.10.006
Timmers, S., Schrauwen, P., and de Vogel, J. (2008). Muscular diacylglycerol metabolism and insulin resistance. Physiology Behav. 94, 242–251. doi:10.1016/j.physbeh.2007.12.002
Tirosh, A., Potashnik, R., Bashan, N., and Rudich, A. (1999). Oxidative stress disrupts insulin-induced cellular redistribution of insulin receptor substrate-1 and phosphatidylinositol 3-kinase in 3T3-L1 adipocytes. A putative cellular mechanism for impaired protein kinase B activation and GLUT4 translocation. J. Biol. Chem. 274, 10595–10602. doi:10.1074/jbc.274.15.10595
Tirosh, A., Rudich, A., Potashnik, R., and Bashan, N. (2001). Oxidative stress impairs insulin but not platelet-derived growth factor signalling in 3T3-L1 adipocytes. Biochem. J. 355, 757–763. doi:10.1042/bj3550757
Triolo, M., Slavin, M., Moradi, N., and Hood, D. A. (2022). Time-dependent changes in autophagy, mitophagy and lysosomes in skeletal muscle during denervation-induced disuse. J. Physiology 600, 1683–1701. doi:10.1113/JP282173
Troiano, R. P., Berrigan, D., Dodd, K. W., Mâsse, L. C., Tilert, T., and McDowell, M. (2008). Physical activity in the United States measured by accelerometer. Med. Sci. Sports Exerc 40, 181–188. doi:10.1249/mss.0b013e31815a51b3
Turinsky, J., O’Sullivan, D. M., and Bayly, B. P. (1990). 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J. Biol. Chem. 265, 16880–16885. doi:10.1016/s0021-9258(17)44844-7
Turner, N., Kowalski, G. M., Leslie, S. J., Risis, S., Yang, C., Lee-Young, R. S., et al. (2013). Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 56, 1638–1648. doi:10.1007/s00125-013-2913-1
Ussher, J. R., Koves, T. R., Cadete, V. J. J., Zhang, L., Jaswal, J. S., Swyrd, S. J., et al. (2010). Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes 59, 2453–2464. doi:10.2337/db09-1293
Vainshtein, A., Tryon, L. D., Pauly, M., and Hood, D. A. (2015). Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am. J. Physiology-Cell Physiology 308, C710–C719. doi:10.1152/ajpcell.00380.2014
Vukovich, M. D., Arciero, P. J., Kohrt, W. M., Racette, S. B., Hansen, P. A., and Holloszy, J. O. (1996). Changes in insulin action and GLUT-4 with 6 days of inactivity in endurance runners. J. Appl. Physiol. 80, 240–244. doi:10.1152/jappl.1996.80.1.240
Walhin, J.-P., Richardson, J. D., Betts, J. A., and Thompson, D. (2013). Exercise counteracts the effects of short-term overfeeding and reduced physical activity independent of energy imbalance in healthy young men. J. Physiol. 591, 6231–6243. doi:10.1113/jphysiol.2013.262709
Westerbacka, J., Kolak, M., Kiviluoto, T., Arkkila, P., Sirén, J., Hamsten, A., et al. (2007). Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 56, 2759–2765. doi:10.2337/db07-0156
Wilmot, E. G., Edwardson, C. L., Achana, F. A., Davies, M. J., Gorely, T., Gray, L. J., et al. (2012). Sedentary time in adults and the association with diabetes, cardiovascular disease and death: Systematic review and meta-analysis. Diabetologia 55, 2895–2905. doi:10.1007/s00125-012-2677-z
Wojtaszewski, J. F., Hansen, B. F., Gade, J., Kiens, B., Markuns, J. F., Goodyear, L. J., et al. (2000). Insulin signaling and insulin sensitivity after exercise in human skeletal muscle. Diabetes 49, 325–331. doi:10.2337/diabetes.49.3.325
Wojtaszewski, J. F., Higaki, Y., Hirshman, M. F., Michael, M. D., Dufresne, S. D., Kahn, C. R., et al. (1999). Exercise modulates postreceptor insulin signaling and glucose transport in muscle-specific insulin receptor knockout mice. J. Clin. Invest. 104, 1257–1264. doi:10.1172/JCI7961
Wright, D. C., Han, D.-H., Garcia-Roves, P. M., Geiger, P. C., Jones, T. E., and Holloszy, J. O. (2007). Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1alpha expression. J. Biol. Chem. 282, 194–199. doi:10.1074/jbc.M606116200
Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124. doi:10.1016/S0092-8674(00)80611-X
Yamada, T., Ida, T., Yamaoka, Y., Ozawa, K., Takasan, H., and Honjo, I. (1975). Two distinct patterns of glucose intolerance in icteric rats and rabbits. Relationship to impaired liver mitochondria function. J. Lab. Clin. Med. 86, 38–45.
Yu, C., Chen, Y., Cline, G. W., Zhang, D., Zong, H., Wang, Y., et al. (2002). Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 277, 50230–50236. doi:10.1074/jbc.M200958200
Yuan, M., Konstantopoulos, N., Lee, J., Hansen, L., Li, Z.-W., Karin, M., et al. (2001). Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 293, 1673–1677. doi:10.1126/science.1061620
Zhou, Q., and Dohm, G. L. (1997). Treadmill running increases phosphatidylinostol 3-kinase activity in rat skeletal muscle. Biochem. Biophysical Res. Commun. 236, 647–650. doi:10.1006/bbrc.1997.7028
Keywords: insulin resistance, metabolism, mitochondria, bioenergetics, skeletal muscle
Citation: Handy RM and Holloway GP (2023) Insights into the development of insulin resistance: Unraveling the interaction of physical inactivity, lipid metabolism and mitochondrial biology. Front. Physiol. 14:1151389. doi: 10.3389/fphys.2023.1151389
Received: 26 January 2023; Accepted: 07 April 2023;
Published: 20 April 2023.
Edited by:
Lauren Sparks, AdventHealth, United StatesReviewed by:
Dominik H. Pesta, German Aerospace Center (DLR), GermanyPaul Timothy Reidy, Miami University, United States
Copyright © 2023 Handy and Holloway. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rachel M. Handy, rhandy@uoguelph.ca; Graham P. Holloway, ghollowa@uoguelph.ca