 Kiana M. Schulze1*†
Kiana M. Schulze1*† Andrew G. Horn1†Ramona E. Weber1Bradley J. Behnke1David C. Poole1,2Timothy I. Musch1,2
Andrew G. Horn1†Ramona E. Weber1Bradley J. Behnke1David C. Poole1,2Timothy I. Musch1,2- 1Department of Kinesiology, Kansas State University, Manhattan, KS, United States
- 2Department of Anatomy and Physiology, Kansas State University, Manhattan, KS, United States
Pulmonary hypertension (PH) is characterized by pulmonary vascular remodeling, respiratory muscle and cardiac impairments, and exercise intolerance. Specifically, impaired gas exchange increases work of the diaphragm; however, compromised contractile function precludes the diaphragm from meeting the increased metabolic demand of chronic hyperventilation in PH. Given that muscle contractile function is in part, dependent upon adequate blood flow (
Methods: Sprague-Dawley rats were randomized into healthy (HC, n = 7) or PH (n = 7) groups. PH rats were administered monocrotaline (MCT) while HC rats received vehicle. Disease progression was monitored via echocardiography. Regional and total diaphragm blood flow and vascular conductance at baseline and during 3 min of electrically-stimulated contractions were determined using fluorescent microspheres.
Results: PH rats displayed morphometric and echocardiographic criteria for disease (i.e., acceleration time/ejection time, right ventricular hypertrophy). In all rats, total costal diaphragm
Conclusion: These findings support a redistribution of regional diaphragm perfusion and an impaired medial costal hyperemic response in PH, suggesting that PH alters diaphragm vascular function and oxygen delivery, providing a potential mechanism for PH-induced diaphragm contractile dysfunction.
1 Introduction
Pulmonary hypertension (PH) is a chronic disease characterized by elevated pulmonary arterial pressures and structural remodeling of the pulmonary vasculature resulting in dyspnea, cardiac dysfunction, and premature death. PH can occur idiopathically or as a comorbidity, however, all PH etiologies lower exercise tolerance and quality of life (Halank et al., 2013). The pathological changes in the pulmonary vasculature with PH can induce pulmonary congestion and poor gas exchange, which culminate in an increased ventilatory demand (Sun et al., 2003; Laveneziana et al., 2013) and consequently, increased work of the primary respiratory muscle, the diaphragm. Impaired pulmonary function and ventilatory insufficiency (Sun et al., 2001; Sun et al., 2003) largely contribute to dyspnea at rest and during exercise, therefore, understanding the consequences of heightened work of the diaphragm in PH are integral to improving exercise capacity and quality of life for PH patients. Previous investigations in PH patients (Meyer et al., 2005; Kabitz et al., 2007; Laveneziana et al., 2013; Breda et al., 2014; Spiesshoefer et al., 2019) and animal models of PH (de Man et al., 2011; Manders et al., 2012; Ahn et al., 2013; Himori et al., 2017) demonstrate diaphragm weakness and contractile dysfunction. Presently, the precise mechanisms responsible for PH-induced diaphragm dysfunction remain unknown.
It was initially believed that PH-induced systemic dysfunction occurred solely due to cardiopulmonary pathology. While central dysfunction undoubtedly plays a substantial role, it is becoming evident that pathological changes extend beyond these central manifestations, specifically in skeletal muscle. Impaired strength, oxygenation, and mitochondrial function in peripheral skeletal muscle (Bauer et al., 2007; Mainguy et al., 2010; Schulze et al., 2021; Schulze et al., 2022; Malenfant et al., 2015a; Malenfant et al., 2015b; Dimopoulos et al., 2013) independent of reduced cardiac output support the notion of vascular dysfunction beyond the pulmonary circulation. It has been suggested that diaphragm contractile dysfunction precedes central hemodynamic compromise (Kosmas et al., 2022), however, the effects of PH on bulk and regional diaphragm perfusion have not been investigated directly. In order to sustain diaphragmatic contractile function, there must be a tight coupling of O2 delivery (
Therefore, the overall objective of this study was to investigate the effects of monocrotaline (MCT)-induced PH on bulk and regional diaphragmatic blood flow and vascular conductance. We tested the hypotheses that in MCT rats with moderate severity PH versus healthy control there will be: 1) lower diaphragm vascular conductance and blood flow, and 2) a decreased diaphragm hyperemic response to 1 Hz contractions. Additionally, answering this question will provide insights to the mechanistic bases for the diaphragm dysfunction associated with PH.
2 Materials and methods
2.1 Animals
Fourteen female Sprague-Dawley rats (4–6 months old) were obtained from Charles River Laboratories (Boston, MA, USA) for this investigation. The greater prevalence of PH in females (Badesch et al., 2010; Hester et al., 2019), in combination with the paucity of literature examining females with PH, provides support for our use of female rats herein. Rats were randomized into healthy control (HC, n = 7) and monocrotaline (MCT)-induced pulmonary hypertension animals (MCT, n = 7). The Sprague-Dawley rat was chosen due to the similar properties (e.g., anatomical and physiological) of its diaphragm to the human diaphragm (Metzger et al., 1985; Mizuno, 1991; Poole et al., 1997). The diaphragm is comprised of two distinct muscle regions: the costal (ventral, medial, and dorsal) and the crural diaphragm. We elected to focus on the medial costal region. Given its anatomical position and fiber orientation, the medial costal diaphragm sustains the highest proportion of ventilatory work (Poole et al., 1997) and receives the highest mass specific blood flow within the costal diaphragm (Davis et al., 2012; Horn et al., 2020; Horn et al., 2022; Poole et al., 1997; Poole et al., 2000), therefore serving as the primary driver for inspiration. All procedures were approved by the Kansas State University Institutional Animal Care and Use Committee and complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Upon arrival, animals were housed and maintained in a temperature-controlled (23°C ± 2°C) room with a 12:12-h light-dark cycle with water and rat chow provided ad libitum.
2.2 Monocrotaline-induced pulmonary hypertension
Pulmonary hypertension (PH) was induced using monocrotaline alkaloid (MCT; 50 mg/kg) administered via a single intraperitoneal injection which has been shown to induce progressive PH (Gomez-Arroyo et al., 2012; Schulze et al., 2021; Schulze et al., 2022). MCT (BOC Sciences; Shirley, NY) was dissolved in a solution containing 50% sterile saline and 50% 200-proof ethanol at room temperature and a total of 0.5 mL of fluid was injected into each rat in the experimental group while HC rats received 0.5 mL of vehicle. While maintained on 1%–2% isoflurane, injections were given immediately following pre-injection echoes (see Echocardiography) with a 27 G needle in the lower right quadrant of the abdomen with care taken to avoid internal organs. Importantly, characteristics of PH (i.e., elevations in pulmonary artery pressures, remodeling of small pulmonary arteries, and right ventricular (RV) hypertrophy) occur 3–4 weeks following injection, prior to the development of overt heart failure (Gomez-Arroyo et al., 2012). Animals were monitored weekly via echocardiography along with measures of body weight and body condition scoring.
2.3 Echocardiography
Transthoracic echocardiography was performed using a commercially available system (Logiq S8; GE Health Care, Milwaukee, WI) with a 13 MHz linear transducer (L4-12t) prior to MCT or saline injection. Subsequent transthoracic echocardiography was performed weekly following injection to monitor disease progression. All comparisons were made between the pre-injection measurement and final measurement taken just prior to experimentation. Animals were initially anaesthetized by inhalation of a 5% isofluorane-O2 mixture and maintained on ≤2.0% isofluorane-O2 (Butler Health Supply, Dublin, OH). Core temperature was maintained at ∼37°C, measured via rectal thermometer. 2-D and M-mode images of the left ventricle (LV) were obtained from the parasternal short axis window and analyzed for end-systolic/diastolic volumes, stroke volume, and ejection fraction as previously described (Craig et al., 2019). RV measurements were taken at the level of the aortic valve, just proximal to the pulmonary valve, in the short axis view. Pulsed Doppler ultrasound was used to assess pulmonary artery ejection time (ET), acceleration time (AT), and peak velocity, and the ratio of AT to ET (AT/ET) was calculated and used as a clinical parameter for the confirmation of PH (AT/ET < 0.3) (Jones et al., 2002). Upon verification of PH, (average 23 days post-injection) the following experiments were performed.
2.4 Surgical preparation
All surgical procedures were performed using aseptic techniques. Rats were initially anesthetized with a 5% isoflurane-O2 mixture for 5 min (isoflurane vaporizer; Harvard Apparatus, Cambridge, MA) and subsequently maintained on 3% isoflurane-O2. Body temperature was maintained at 37°C ± 1°C (via rectal thermometer) by use of a water-recirculating heating blanket. An incision was made on the ventral side of the neck and the left carotid artery was isolated and cannulated with PE-10 connected to PE-50 (Intra-Medic polyethylene tubing; Clay Adams Brand, Becton, Dickinson, Sparks, MD) for measurements of mean arterial pressure (MAP; Digi-Med BPA, Micro-Med Inc., Louisville, KY) and infusion of fluorescent microspheres (see below). A second catheter (PE-10 connected to PE-50) was inserted into the caudal artery for the infusion of pentobarbital sodium anesthesia (50 mg/mL) and reference sampling for blood flow determination. Rats were then transitioned to pentobarbital sodium anesthesia (20 mg/kg body wt) given intra-arterially while concentrations of isoflurane were decreased and subsequently discontinued over ∼30 min. The level of anesthesia was regularly monitored via toe pinch and palpebral reflex, with pentobarbital anesthesia supplemented (3.5–7.0 mg/kg) as necessary.
2.5 Diaphragm contractions
Following carotid and tail artery catheterization, animals were tracheotomized and mechanically ventilated (Kent Scientific PhysioSuite, Torrington, CT). The diaphragm was exposed via laparotomy as previously described (Poole et al., 1995; Geer et al., 2002; Davis et al., 2012). Following diaphragm exposure, stainless steel electrodes were sutured to the inferior aspect of the right ventral costal (cathode) and the right dorsal costal (anode) diaphragm. Prior to contractions, fluorescent microspheres were infused (described below) to assess baseline diaphragmatic blood flow in the inactive diaphragm. Thereafter, electrically stimulated twitch contractions were induced at 1 Hz (3–6 V, 2-ms pulse duration) with a Grass S88 stimulator (Quincy, MA) for 180s. This stimulation protocol elicits diaphragm contractions at a frequency similar to that of a spontaneously breathing animal (Geer et al., 2002). Blood flow was measured via fluorescent microspheres (described below) at 180s of stimulation to assess the contracting steady-state diaphragm hyperemia.
2.6 Fluorescent microsphere injection
The fluorescent microsphere technique, as previously described (Deveci and Eggington, 1999; Musch et al., 1986), was used to quantify tissue blood flow in each experimental group. Fluorescent microspheres were infused at two different time points: 1) baseline (10 min following laparotomy) and 2) at 180s of electrically induced contractions. For each measure, a reference blood sample was taken from the caudal artery catheter, using a Harvard withdrawal pump (model 907, Cambridge, MA) that was initiated 30s prior to microsphere infusion at a withdrawal rate of 0.25 mL/min and 2.0–2.5 × 105 fluorescent microspheres (colors: red, scarlet, or blue-green; 15.5 μm diameter, Invitrogen FluoSpheres, Carlsbad, CA) were infused into the aortic arch via the carotid artery catheter. Adequate mixing of microspheres was determined by <20% difference in left and right kidney or left and right soleus muscle blood flows at each time point. Following the final microsphere infusion, rats were euthanized with pentobarbital sodium overdose (>50 mg/kg I.A.) followed by cardiac excision. Thereafter, tissues (diaphragm, soleus, intercostal muscles, and kidneys) were harvested, weighed, and placed in 15 mL screw cap polypropylene conical tubes and then placed in a −80°C freezer for later blood flow analysis. Using the template previously described (Horn et al., 2020; Horn et al., 2022; Poole et al., 1997), the costal diaphragm was sectioned into ventral, medial, and dorsal portions to determine regional distribution of diaphragmatic blood flow, whereas the sum of these portions was used to calculate total costal diaphragm blood flow.
2.7 Calculation of blood flow and vascular resistance
The fluorescent microsphere assay was performed as previously described (Deveci and Eggington, 1999; Horn et al., 2020; Horn et al., 2022). Tissue blood flow was calculated as follows (Deveci and Eggington, 1999):
where
where VC is vascular conductance (ml/mmHg/min/100g),
2.8 Postmortem measurements
The RV, LV, and lungs were dissected and weighed following euthanasia. RV hypertrophy was measured via the Fulton index: RV weight (mg)/LV + septum (S) weight (mg) (Fulton et al., 1952). RV hypertrophy was also expressed as RV/body weight.
2.9 Data analysis
Data were analyzed using GraphPad Prism9 (GraphPad Software, San Deigo, CA). Student paired (within animal) and independent two-sample (between HC and MCT) t-tests were utilized to determine differences in echocardiographic and morphometric measurements. Body mass (g), diaphragm mass (mg), and diaphragm:body mass ratio were analyzed using a one-way ANOVA. Tissue blood flows were analyzed with a mixed-effects model. A Grubb’s outlier test was performed on all data, and 1 outlier was identified and removed from the respective data subset when detected. Post-hoc analyses were performed using a Tukey’s test. All data are presented as means ± SE. Significance was established at p < 0.05.
3 Results
3.1 Morphometric and echocardiographic measurements
All MCT rats displayed characteristics consistent with PH including a decreased AT/ET ratio, RV hypertrophy (RV/LV + S), and an elevated lung weight compared to HC animals (Tables 1,2). Total diaphragm weight did not differ significantly between groups (Table 1). There were no differences in left ventricular ejection fraction, fractional shortening, or stroke volume between HC and MCT rats (Table 2; all p > 0.05]. Furthermore, heart rate, cardiac output, and cardiac index did not differ between groups (Table 3), so we do not believe cardiac factors associated with blood flow to be a limitation to interpretation of the findings herein. MAP was not different between groups, or when comparing baseline to diaphragm contractions (Table 3). Additionally, there were no differences between groups in regional diaphragm masses, nor in total costal diaphragm mass [all p > 0.05].
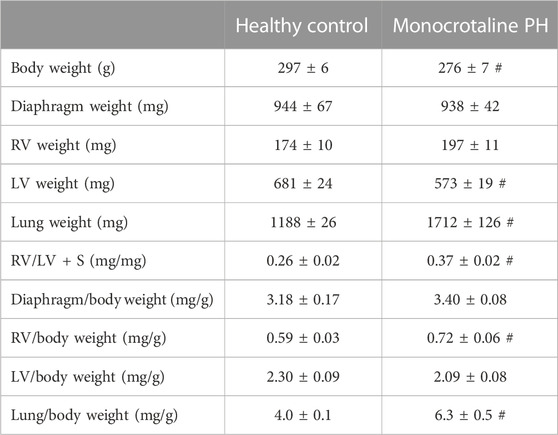
TABLE 1. Morphometric measurements. Data are means ± SE; n, number of rats; n = 7 for all; PH, pulmonary hypertension; RV, right ventricle; LV, left ventricle; LV + S, LV + septum; # p < 0.05 vs. healthy control.
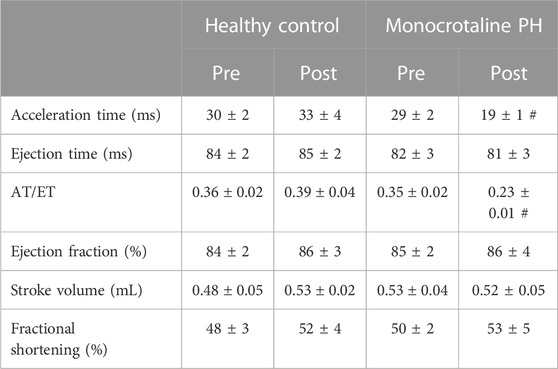
TABLE 2. Echocardiographic measurements. Data are means ± SE; n, number of rats; n = 7 for all; PH, pulmonary hypertension; AT, acceleration time; ET, ejection time; #p < 0.05 vs. all other conditions.
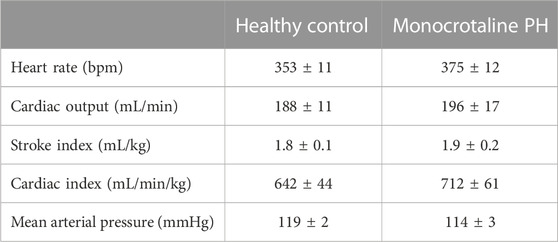
TABLE 3. Central hemodynamics. Data are means ± SE; n, number of rats; n = 7 for all; PH, pulmonary hypertension.
3.2 Blood flow
At baseline (i.e., in the inactive diaphragm), total costal diaphragm blood flow was not different between groups (Figure 1). Despite this, MCT rats had a higher baseline ventral costal blood flow compared to HC and all other costal regions of the diaphragm in MCT and HC rats (Figures 2 and 3A). No regional differences were present between costal portions of the diaphragm in HC rats at baseline (Figure 2A and Figure 3A). In response to 1 Hz contractions, total costal diaphragm blood flow increased in both HC and MCT animals (Figure 1). In HC rats, 1 Hz contractions elicited the greatest blood flow increase to the medial costal diaphragm (Figure 2A); whereas in MCT animals, the ventral costal portion received the highest baseline and contracting blood flow, and the dorsal costal had the largest increase in blood flow compared to baseline (Figure 2B). Therefore, the hyperemic response of the medial costal diaphragm was greater in HC rats, increasing 55% ± 3% compared to baseline versus only 44% ± 4% in MCT rats (Figure 2A and Figure 4; p = 0.04), while the total costal diaphragm hyperemic response was not different between groups (HC, 49% ± 4% vs. MCT, 41% ± 12%, p = 0.55). Kidney blood flows in all conditions fell within the 400–600 mL/min/100 g range expected based on previous investigations from our laboratory (Horn et al., 2020; Horn et al., 2022; Colburn et al., 2020). There were no differences between group or condition in blood flow to the kidneys, soleus, or intercostal muscles (Table 4; all p > 0.05).
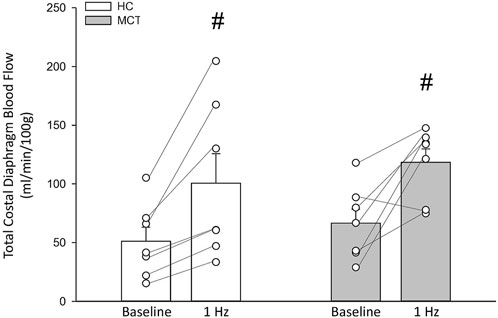
FIGURE 1. Total costal diaphragm blood flow (ml/min/100 g) in healthy control (HC; n = 7) and monocrotaline-induced pulmonary hypertension (MCT; n = 7) animals at baseline and during steady-state 1 Hz contractions. #Significant (p < 0.05) versus baseline.
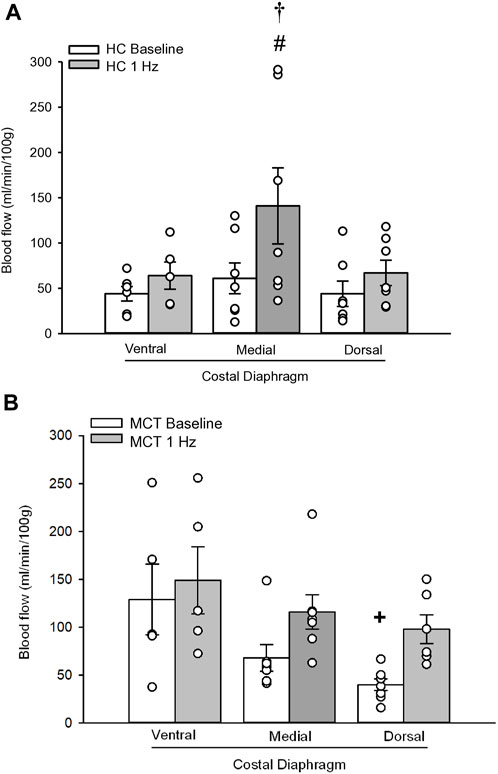
FIGURE 2. Regional diaphragm blood flow (ml/min/100 g) at baseline and during steady-state 1 Hz contractions in healthy control (HC; n = 7) (A) and monocrotaline-induced pulmonary hypertension (MCT; n = 7) (B) animals. #Significant (p < 0.05) versus baseline within diaphragm region. †Significant (p < 0.05) versus other diaphragm regions at 1 Hz +Significant (p < 0.05) versus ventral costal baseline.
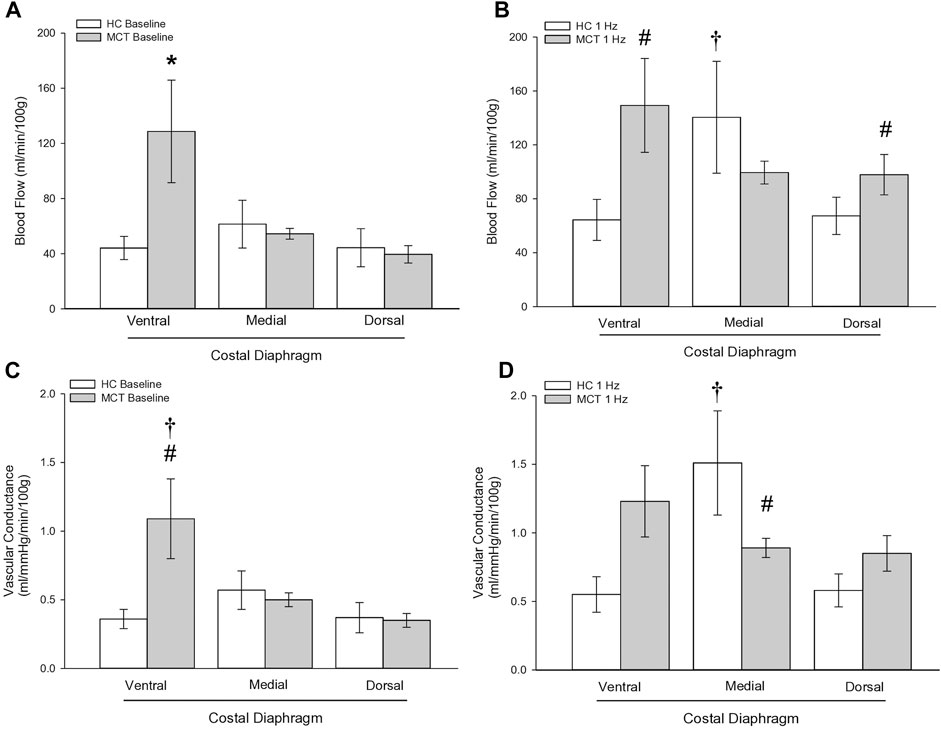
FIGURE 3. Regional diaphragm blood flow at baseline (A) and during steady-state 1 Hz contractions (B) and vascular conductance (ml/mmHg/min/100 g) at baseline (C) and during steady-state 1 Hz contractions (D) in healthy control (HC; n = 7) and monocrotaline-induced pulmonary hypertension (MCT; n = 7) animals. *Significant (p < 0.05) versus all other bars (Figure 3A). #Significant (p < 0.05) versus HC within diaphragm region. †Significant (p < 0.05) versus other diaphragm regions within group (HC or MCT).
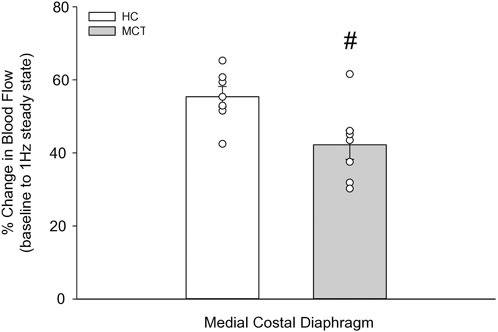
FIGURE 4. The change in medial costal diaphragm blood flow from baseline to steady-state 1 Hz contractions in healthy control (HC; n = 7) and monocrotaline-induced pulmonary hypertension (MCT; n = 7) animals. #Significant (p < 0.05) versus HC.
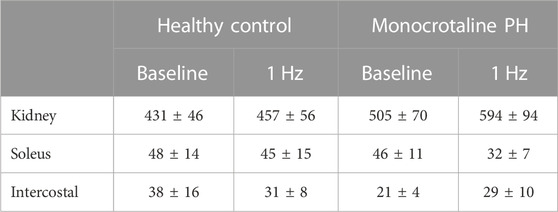
TABLE 4. Blood flow. Data are means ± SE; n, number of rats; n = 7 for all; PH, pulmonary hypertension.
3.3 Vascular conductance
Total costal diaphragm vascular conductance was not different between HC and MCT rats at baseline (0.38 ± 0.1 vs. 0.46 ± 0.1 mL/mmHg/min/100g; p = 0.5) or during 1 Hz steady-state contractions (0.83 ± 0.18 vs. 0.87 ± 0.08 mL/mmHg/min/100g; p = 0.8). At baseline, vascular conductance was highest in the ventral costal portion of the diaphragm in MCT rats (Figure 3C). During contractions, vascular conductance was highest in the medial costal portion for HC rats, and the ventral costal portion for MCT rats (Figure 3D). Vascular conductance did not differ between groups in the kidneys, soleus, or intercostals (all p > 0.05).
4 Discussion
This is the first investigation to our knowledge to demonstrate that PH alters regional diaphragm blood flow distribution and impacts the ability of the diaphragm to augment blood flow with contractile activity. Specifically, PH induced a greater distribution of diaphragm blood flow to the ventral costal diaphragm, a region that in health contributes proportionally less to the work of breathing than the medial and dorsal costal diaphragm. Further, the blunted diaphragm hyperemic response to 1 Hz contractions suggests that PH compromises diaphragm vascular function, potentially via a decreased vasodilatory responsiveness and/or increased tonic vasoconstriction of the diaphragm resistance vasculature. These findings in a female rat model of PH raise the possibility that the pathophysiological consequences of PH extend into the respiratory muscle vasculature and provide a putative mechanism for the diaphragm contractile dysfunction associated with PH.
4.1 Monocrotaline-induced PH
This investigation utilized the MCT model of PH at a dose demonstrated to elicit characteristics consistent with the human pathology within 3–4 weeks including medial wall thickening of the pulmonary arteries, elevated right ventricular systolic pressure, pulmonary congestion, and RV hypertrophy (Gomez Arroyo et al., 2012; Nogueira-Ferreria et al., 2015; Schulze et al., 2021; Schulze et al., 2022). RV hypertrophy is commonly determined using the Fulton Index (RV/LV + S) or RV/body weight. In this investigation, MCT animals were smaller; however, RV mass was not different, demonstrating a compensatory RV hypertrophy in response to the higher expected afterload. Additionally, PH was confirmed in these rats using clinically established echocardiographic measurements (i.e., AT/ET) (Jones et al., 2002; Kitabatake et al., 1983). Lower AT/ET has been demonstrated by Kato et al. to be associated with increased RV myocyte cross sectional area and elevated RV/body weight (Kato et al., 2003). Importantly, this model induces moderate PH, indicating that the alterations in diaphragm blood flow presented herein occur prior to, and independent of, distinct heart failure as indicated by lack of changes in LV ejection fraction (Table 2).
4.2 Diaphragmatic blood flow and vascular conductance
Contrary to our hypothesis, bulk diaphragm blood flow (Figure 1) and vascular conductance were not different in MCT rats versus HC. Given the heterogenous distribution of blood flow in the diaphragm (Brancatisano et al., 1991; Davis et al., 2012; Poole et al., 1997; Sexton and Poole, 1995; Horn et al., 2020), altered distribution of regional diaphragm blood flow may represent significant changes in the diaphragm vasculature with PH that are masked by only investigating whole diaphragm hemodynamics. In the healthy rodent (i.e., rat), the medial costal diaphragm sustains the greatest proportion of diaphragm inspiratory work and therefore commands the highest blood flow (Poole et al., 1997; Sexton and Poole, 1995). The diaphragm contractions protocol herein has been used previously in our laboratory and is expected to produce a stimulus akin to spontaneous breathing (Geer et al., 2002; Davis et al., 2012). Diaphragm blood flow data in the current investigation are consistent with spontaneously breathing animals (Horn et al., 2020; Horn et al., 2022). In HC animals, medial costal diaphragm blood flow was augmented from baseline to steady-state contractions as expected (Figure 2A); however, in MCT animals there was no significant increase in medial costal diaphragm perfusion during steady-state contractions (Figure 2B). Further, medial costal diaphragm vascular conductance during contractions was lower in MCT rats compared to HC (Figure 3D). These data support that PH impairs the hyperemic response, specifically in the medial costal diaphragm (Figure 4). Lower medial costal vascular conductance and the reduced ability to augment blood flow with contractile activity suggest that PH induces vascular dysfunction in the diaphragm. Such vascular impairments in the diaphragm with PH may be due to a reduced vasodilatory responsiveness, enhanced intrinsic vascular tone, endothelial or vascular smooth muscle signaling mechanisms, changes in neurovascular coupling, and/or structural alterations in the diaphragm resistance vasculature that manifest throughout disease progression, though this remains to be determined.
Previous work suggests that diaphragm contractile dysfunction is predicated by an inability to match oxygen delivery to oxygen demand (i.e., upstream vascular dysfunction) (Supinski, 1988; de Man et al., 2011; Manders et al., 2012). Our data demonstrate reduced blood flow, and therefore oxygen delivery, to the medial costal diaphragm. Given that blood flow distribution and functional hyperemia is determined by vascular control mechanisms, the blunted medial costal hyperemic response seen with PH is likely due in large part to impaired resistance vessel (i.e., arterioles) vasodilation. Arteriolar dysfunction has been shown across a range of pathological conditions (i.e., aging, hypertension, heart failure, prolonged mechanical ventilation) in several vascular beds [for rev. Vanhoutte et al., 2017; Poole et al., 2021], including skeletal muscle (Muller-Delp et al., 2002) and the diaphragm (Horn et al., 2019). Impaired endothelial function has been demonstrated in pulmonary arteries (Mathew et al., 1995; Christou et al., 2018), coronary arterioles (Kajiya et al., 2007), and large systemic arteries (Wolff et al., 2007; Peled et al., 2008) with PH. However, there is a paucity of data regarding diaphragm vascular function. In PH, dysregulation of the nitric oxide (NO) pathway and production of excess reactive oxygen species (ROS) (Giaid and Saleh, 1995; Humbert et al., 2004; Hampl and Herget, 2000) may act in concert to reduce endothelium-dependent vasodilation [Poole et al., 1997). Further, the increased levels of endothelin-1 (Rubens et al., 2001) and Angiotensin II (Kumpers et al., 2010) in PH would increase tonic vasoconstriction and blunt NO-mediated vasodilation (Mathew et al., 1995). The degree to which these markers are upregulated is associated with disease severity. In the present study where animals presented with moderate PH preceding heart failure, the extent of the impact of these vasoconstrictors is not known. Additionally, in the hyperactive diaphragm with PH,
Interestingly, ventral costal diaphragm perfusion was higher in MCT rats compared to control both at baseline (Figure 3A) and during 1 Hz steady-state contractions [Figure 3B), suggesting that PH results in altered blood flow distribution. Considering adequate blood flow and thus resistance vessel dilation constitute a major determinant of contractile function (Supinski, 1988; Supinski et al., 1993), an inability to meet respiratory muscle oxygen demand would hasten diaphragm fatigue. Therefore, maintenance of a heightened metabolic demand in the chronically hyperactive diaphragm with PH (Laveneziana et al., 2013) may require recruitment of regions that do not contribute as significantly in health. Such a redistribution of blood flow within the diaphragm may actually serve to better match
4.3 Absence of diaphragm atrophy
Previous investigations in male murine models of PH (Ahn et al., 2013; de Man et al., 2011) demonstrate diaphragm atrophy, however, these studies differ in MCT dosage and time course for PH development (6–8 weeks). In contrast, our MCT rats did not display diaphragm atrophy. While we did not assess cross sectional area of muscle fibers, diaphragm mass and diaphragm mass:body weight were not different between groups (Table 1). The MCT rats in the current investigation are studied prior to development of RV failure in order to assess moderate PH. It is possible that PH-induced diaphragm atrophy is the consequence of late-stage decompensation resulting from the inability to match
4.4 Experimental considerations
The present investigation focused on moderate PH preceding RV failure. A longer time course following MCT injection would allow for development of a more severe form of PH that may elicit diaphragm atrophy not seen herein. Future directions may usefully assess
4.5 Ramifications
The potential consequences of our findings in MCT-induced female rats are that blood flow redistribution toward the ventral costal diaphragm and lack of an increase in medial costal perfusion may be an indication of higher work of a less efficient region of the diaphragm at rest, which would be exacerbated during exercise. This provides a possible mechanism for diaphragm contractile dysfunction as well as dyspnea. Furthermore, the impaired medial costal hyperemic response may also represent an insufficiency to augment blood flow during exercise to this region, which serves as the primary driver of respiration. Importantly, this could contribute to the exercise intolerance reported in PH (Frost et al., 2019). An inability to comfortably be physically active would act to discourage patients from participating in exercise, which has been shown to mitigate symptoms of PH and improve exercise capacity, muscle function, and quality of life in PH (Arena et al., 2015; Becker et al., 2022; Brown et al., 2017; Chan et al., 2013; de Man et al., 2009; Grunig et al., 2011; Mereles et al., 2006). The regional diaphragm blood flow redistribution in the present investigation demonstrates a possible impediment to exercise tolerance in PH, and, notably, exercise training and/or respiratory muscle training may serve as a potential therapeutic approach to improve vascular function and normally distribute diaphragm blood flow, mitigating contractile dysfunction in PH.
4.6 Conclusion
This investigation reports, for the first time, bulk and regional blood flow and the hyperemic response in the diaphragm of PH rats. Our findings demonstrate that PH alters regional diaphragm blood flow distribution and impairs the ability of the medial costal diaphragm to augment blood flow with contractions (Figure 5). Together, these data provide a putative mechanism for PH-induced diaphragm contractile dysfunction. Given that such alterations occurred prior to development of gross diaphragm atrophy, this suggests the presence of vascular dysfunction within the diaphragm which may precede diaphragm fiber weakness. Whether vascular mechanisms associated with the reduced hyperemic response and redistribution of regional diaphragm blood flow are functional and/or structural in nature warrants further scientific investigation.
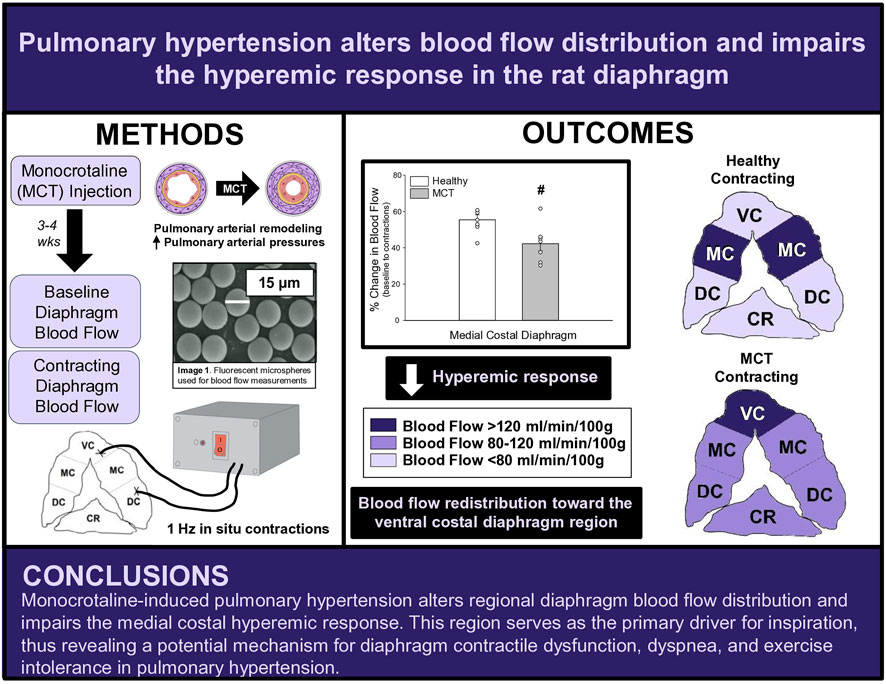
FIGURE 5. Summary Schematic. VC, ventral costal; MC, medial costal; DC, dorsal costal; CR, crural.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by Institutional Animal Care and Use Committee at Kansas State University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
KS: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Writing–original draft, Writing–review and editing. AH: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Writing–original draft, Writing–review and editing. RW: Writing–review and editing. BB: Funding acquisition, Resources, Supervision, Writing–review and editing. DP: Funding acquisition, Resources, Supervision, Writing–review and editing. TM: Funding acquisition, Resources, Supervision, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported in part by the Sustained Momentum for Investigators with Laboratories Established (SMILE) Grants awarded to TM and DP by Kansas State University College of Veterinary Medicine and the National Institute on Aging 1R15AG078060 awarded to BB and DP. AH and KS were financially supported by Ruth L. Kirschstein National Research Service Awards 1F31HL167618-01 and 1F31HL170643-01, respectively.
Acknowledgments
We gratefully acknowledge Dr. Carl J. Ade for his expertise in echocardiography.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahn B., Empinado H. M., Al-Rajhi M., Judge A. R., Ferreira L. F. (2013). Diaphragm atrophy and contractile dysfunction in a murine model of pulmonary hypertension. PLOS ONE 8, e62702. doi:10.1371/journal.pone.0062702
Arena R., Cahalin L. P., Borghi-Silva A., Myers J. (2015). The effect of exercise training on the pulmonary arterial system in patients with pulmonary hypertension. Prog. Cardiovasc. Dis. 57, 480–488. doi:10.1016/j.pcad.2014.03.008
Badesch D. B., Raskob G. E., Elliott C. G., Krichman A. M., Farber H. W., Frost A. E., et al. (2010). Pulmonary arterial hypertension: baseline characteristics from the REVEAL registry. Chest 137, 376–387. doi:10.1378/chest.09-1140
Bauer R., Dehnert C., Schoene P., Filusch A., Bärtsch P., Borst M. M., et al. (2007). Skeletal muscle dysfunction in patients with idiopathic pulmonary arterial hypertension. Respir. Med. 101, 2366–2369. doi:10.1016/j.rmed.2007.06.014
Becker C. U., Sartório C. L., Campos-Carraro C., Siqueira R., Colombo R., Zimmer A., et al. (2022). Exercise training decreases oxidative stress in skeletal muscle of rats with pulmonary arterial hypertension. Archives Physiology Biochem. 128, 1330–1338. doi:10.1080/13813455.2020.1769679
Brancatisano A., Amis T. C., Tully A., Kelly W. T., Engel L. A. (1991). Regional distribution of blood flow within the diaphragm. J. Appl. Physiology 71, 583–589. doi:10.1152/jappl.1991.71.2.583
Breda A. P., Albuquerque A. L. P., Jardim C., Morinaga L. K., Suesada M. M., Fernandes C. J. C., et al. (2014). Skeletal muscle abnormalities in pulmonary arterial hypertension. PLOS ONE 9, e114101. doi:10.1371/journal.pone.0114101
Brown M. B., Neves E., Long G., Graber J., Gladish B., Wiseman A., et al. (2017). High-intensity interval training, but not continuous training, reverses right ventricular hypertrophy and dysfunction in a rat model of pulmonary hypertension. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 312, R197–R210. doi:10.1152/ajpregu.00358.2016
Chan L., Chin L. M. K., Kennedy M., Woolstenhulme J. G., Nathan S. D., Weinstein A. A., et al. (2013). Benefits of intensive treadmill exercise training on cardiorespiratory function and quality of life in patients with pulmonary hypertension. Chest 143, 333–343. doi:10.1378/chest.12-0993
Christou H., Hudalla H., Michael Z., Filatava E. J., Li J., Zhu M., et al. (2018). Impaired pulmonary arterial vasoconstriction and nitric oxide–mediated relaxation underlie severe pulmonary hypertension in the sugen-hypoxia rat model. J. Pharmacol. Exp. Ther. 364, 258–274. doi:10.1124/jpet.117.244798
Colburn T. D., Holdsworth C. T., Craig J. C., Hirai D. M., Montgomery S., Poole D. C., et al. (2020). ATP-sensitive K+ channel inhibition in rats decreases kidney and skeletal muscle blood flow without increasing sympathetic nerve discharge. Respir. Physiology Neurobiol. 278, 103444. doi:10.1016/j.resp.2020.103444
Craig J. C., Colburn T. D., Caldwell J. T., Hirai D. M., Tabuchi A., Baumfalk D. R., et al. (2019). Central and peripheral factors mechanistically linked to exercise intolerance in heart failure with reduced ejection fraction. Am. J. Physiology-Heart Circulatory Physiology 317, H434–H444. doi:10.1152/ajpheart.00164.2019
Davis R. T. I., Bruells C. S., Stabley J. N., McCullough D. J., Powers S. K., Behnke B. J. (2012). Mechanical ventilation reduces rat diaphragm blood flow and impairs oxygen delivery and uptake. Crit. Care Med. 40, 2858–2866. doi:10.1097/CCM.0b013e31825b933a
de Man F. S., Handoko M. L., Groepenhoff H., Hul A.J., Abbink J., Koppers R. J. H., et al. (2009). Effects of exercise training in patients with idiopathic pulmonary arterial hypertension. Eur. Respir. J. 34, 669–675. doi:10.1183/09031936.00027909
de Man F. S., van Hees H. W. H., Handoko M. L., Niessen H. W., Schalij I., Humbert M., et al. (2011). Diaphragm muscle fiber weakness in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 183, 1411–1418. doi:10.1164/rccm.201003-0354OC
Deveci D., Egginton S. (1999). Development of the fluorescent microsphere technique for quantifying regional blood flow in small mammals. Exp. Physiol. 84, 615–630. doi:10.1111/j.1469-445x.1999.01852.x
Dimopoulos S., Tzanis G., Manetos C., Tasoulis A., Mpouchla A., Tseliou E., et al. (2013). Peripheral muscle microcirculatory alterations in patients with pulmonary arterial hypertension: a pilot study. Respir. Care 58, 2134–2141. doi:10.4187/respcare.02113
Dorfmüller P., Perros F., Balabanian K., Humbert M. (2003). Inflammation in pulmonary arterial hypertension. Eur. Respir. J. 22, 358–363. doi:10.1183/09031936.03.00038903
Frost A., Badesch D., Gibbs J. S. R., Gopalan D., Khanna D., Manes A., et al. (2019). Diagnosis of pulmonary hypertension. Eur. Respir. J. 53, 1801904. doi:10.1183/13993003.01904-2018
Fulton R. M., Hutchinson E. C., Jones A. M. (1952). Ventricular Weight In Cardiac Hypertrophy. Br. Heart J. 14, 413–420. doi:10.1136/hrt.14.3.413
Geer C. M., Behnke B. J., McDonough P., Poole D. C. (2002). Dynamics of microvascular oxygen pressure in the rat diaphragm. J. Appl. Physiology 93, 227–232. doi:10.1152/japplphysiol.00735.2001
Giaid A., Saleh D. (1995). Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 333, 214–221. doi:10.1056/NEJM199507273330403
Gomez-Arroyo J. G., Farkas L., Alhussaini A. A., Farkas D., Kraskauskas D., Voelkel N. F., et al. (2012). The monocrotaline model of pulmonary hypertension in perspective. Am. J. Physiology-Lung Cell. Mol. Physiology 302, L363–L369. doi:10.1152/ajplung.00212.2011
Grünig E., Ehlken N., Ghofrani A., Staehler G., Meyer F. J., Juenger J., et al. (2011). Effect of exercise and respiratory training on clinical progression and survival in patients with severe chronic pulmonary hypertension. Respiration 81, 394–401. doi:10.1159/000322475
Halank M., Einsle F., Lehman S., Bremer H., Ewert R., Wilkens H., et al. (2013). Exercise capacity affects quality of life in patients with pulmonary hypertension. Lung 191, 337–343. doi:10.1007/s00408-013-9472-6
Hampl V., Herget J. (2000). Role of nitric oxide in the pathogenesis of chronic pulmonary hypertension. Physiol. Rev. 80, 1337–1372. doi:10.1152/physrev.2000.80.4.1337
Hester J., Ventetuolo C., Lahm T. (2019). Sex, gender, and sex hormones in pulmonary hypertension and right ventricular failure. Compr. Physiol. 10, 125–170. doi:10.1002/cphy.c190011
Himori K., Abe M., Tatebayashi D., Lee J., Westerblad H., Lanner J. T., et al. (2017). Superoxide dismutase/catalase mimetic EUK-134 prevents diaphragm muscle weakness in monocrotalin-induced pulmonary hypertension. PLOS ONE 12, e0169146. doi:10.1371/journal.pone.0169146
Hogan M. C., Arthur P. G., Bebout D. E., Hochachka P. W., Wagner P. D. (1992). Role of O2 in regulating tissue respiration in dog muscle working in situ. J. Appl. Physiology 73, 728–736. doi:10.1152/jappl.1992.73.2.728
Hogan M. C., Richardson R. S., Kurdak S. S. (1994). Initial fall in skeletal muscle force development during ischemia is related to oxygen availability. J. Appl. Physiology 77, 2380–2384. doi:10.1152/jappl.1994.77.5.2380
Horn A. G., Baumfalk D. R., Schulze K. M., Kunkel O. N., Colburn T. D., Weber R. E., et al. (2020). Effects of elevated positive end-expiratory pressure on diaphragmatic blood flow and vascular resistance during mechanical ventilation. J. Appl. Physiology 129, 626–635. doi:10.1152/japplphysiol.00320.2020
Horn A. G., Davis R. T., Baumfalk D. R., Kunkel O. N., Bruells C. S., McCullough D. J., et al. (2019). Impaired diaphragm resistance vessel vasodilation with prolonged mechanical ventilation. J. Appl. Physiology 127, 423–431. doi:10.1152/japplphysiol.00189.2019
Horn A. G., Kunkel O. N., Schulze K. M., Baumfalk D. R., Weber R. E., Poole D. C., et al. (2022). Supplemental oxygen administration during mechanical ventilation reduces diaphragm blood flow and oxygen delivery. J. Appl. Physiology 132, 1190–1200. doi:10.1152/japplphysiol.00021.2022
Humbert M., Morrell N. W., Archer S. L., Stenmark K. R., MacLean M. R., Lang I. M., et al. (2004). Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 43, 13S. doi:10.1016/j.jacc.2004.02.029
Jones J. E., Mendes L., Rudd M. A., Russo G., Loscalzo J., Zhang Y.-Y. (2002). Serial noninvasive assessment of progressive pulmonary hypertension in a rat model. Am. J. Physiology-Heart Circulatory Physiology 283, H364–H371. doi:10.1152/ajpheart.00979.2001
Kabitz H.-J., Schwoerer A., Bremer H.-C., Sonntag F., Walterspacher S., Walker D., et al. (2007). Impairment of respiratory muscle function in pulmonary hypertension. Clin. Sci. 114, 165–171. doi:10.1042/CS20070238
Kajiya M., Hirota M., Inai Y., Kiyooka T., Morimoto T., Iwasaki T., et al. (2007). Impaired NO-mediated vasodilation with increased superoxide but robust EDHF function in right ventricular arterial microvessels of pulmonary hypertensive rats. Am. J. Physiology-Heart Circulatory Physiology 292, H2737–H2744. doi:10.1152/ajpheart.00548.2006
Kanj N. A., Nasser M. G., Medawar W. A., Al Tayeh A. U., Khoury M. Y., Nassar C. F. (1999). Reversal of impaired calcium homeostasis in the rat diaphragm subjected to Monocrotaline–induced pulmonary hypertension. Toxicol. Lett. 105, 177–182. doi:10.1016/S0378-4274(98)00398-1
Kato Y., Iwase M., Kanazawa H., Kawata N., Yoshimori Y., Hashimoto K., et al. (2003). Progressive development of pulmonary hypertension leading to right ventricular hypertrophy assessed by echocardiography in rats. Exp. Anim. 52, 285–294. doi:10.1538/expanim.52.285
Kindig C. A., Poole D. C. (1998). A comparison of the microcirculation in the rat spinotrapezius and diaphragm muscles. Microvasc. Res. 55, 249–259. doi:10.1006/mvre.1998.2075
Kitabatake A., Inoue M., Asao M., Masuyama T., Tanouchi J., Morita T., et al. (1983). Noninvasive evaluation of pulmonary hypertension by a pulsed Doppler technique. Circulation 68, 302–309. doi:10.1161/01.CIR.68.2.302
Kosmas K., Michael Z., Papathanasiou A. E., Spyropoulos F., Adib E., Jasuja R., et al. (2022). Skeletal muscle dysfunction in experimental pulmonary hypertension. Int. J. Mol. Sci. 23, 10912. doi:10.3390/ijms231810912
Kümpers P., Nickel N., Lukasz A., Golpon H., Westerkamp V., Olsson K. M., et al. (2010). Circulating angiopoietins in idiopathic pulmonary arterial hypertension. Eur. Heart J. 31, 2291–2300. doi:10.1093/eurheartj/ehq226
Laveneziana P., Garcia G., Joureau B., Nicolas-Jilwan F., Brahimi T., Laviolette L., et al. (2013). Dynamic respiratory mechanics and exertional dyspnoea in pulmonary arterial hypertension. Eur. Respir. J. 41, 578–587. doi:10.1183/09031936.00223611
Mainguy V., Maltais F., Saey D., Gagnon P., Martel S., Simon M., et al. (2010). Peripheral muscle dysfunction in idiopathic pulmonary arterial hypertension. Thorax 65, 113–117. doi:10.1136/thx.2009.117168
Malenfant S., Potus F., Fournier F., Breuils-Bonnet S., Pflieger A., Bourassa S., et al. (2015a). Skeletal muscle proteomic signature and metabolic impairment in pulmonary hypertension. J. Mol. Med. 93, 573–584. doi:10.1007/s00109-014-1244-0
Malenfant S., Potus F., Mainguy V., Leblanc E., Malenfant M., Ribeiro F., et al. (2015b). Impaired skeletal muscle oxygenation and exercise tolerance in pulmonary hypertension. Med. Sci. Sports Exerc 47, 2273–2282. doi:10.1249/MSS.0000000000000696
Manders E., de Man F. S., Handoko M. L., Westerhof N., van Hees H. W. H., Stienen G. J. M., et al. (2012). Diaphragm weakness in pulmonary arterial hypertension: role of sarcomeric dysfunction. Am. J. Physiology-Lung Cell. Mol. Physiology 303, L1070–L1078. doi:10.1152/ajplung.00135.2012
Martinez-Lemus A., Hill L. A., Bolz M. S., Pohl U., Meininger G. S. (2004). Acute mechanoadaptation of vascular smooth muscle cells in response to continuous arteriolar vasoconstriction: implications for functional remodeling. FASEB J. 18, 708–710. doi:10.1096/fj.03-0634fje
Marzetti E., Calvani R., Cesari M., Buford T. W., Lorenzi M., Behnke B. J., et al. (2013). Mitochondrial dysfunction and sarcopenia of aging: from signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. Mol. Basis Muscle Wasting 45, 2288–2301. doi:10.1016/j.biocel.2013.06.024
Mathew R., Zeballos G. A., Tun H., Gewitz M. H. (1995). Role of nitric oxide and endothelin-1 in monocrotaline-induced pulmonary hypertension in rats. Cardiovasc. Res. 30, 739–746. doi:10.1016/S0008-6363(95)00108-5
Mereles D., Ehlken N., Kreuscher S., Ghofrani S., Hoeper M. M., Halank M., et al. (2006). Exercise and respiratory training improve exercise capacity and quality of life in patients with severe chronic pulmonary hypertension. Circulation 114, 1482–1489. doi:10.1161/CIRCULATIONAHA.106.618397
Metzger J. M., Scheidt K. B., Fitts R. H. (1985). Histochemical and physiological characteristics of the rat diaphragm. J. Appl. Physiology 58, 1085–1091. doi:10.1152/jappl.1985.58.4.1085
Meyer F. J., Lossnitzer D., Kristen A. V., Schoene A. M., Kübler W., Katus H. A., et al. (2005). Respiratory muscle dysfunction in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 25, 125–130. doi:10.1183/09031936.04.00095804
Mizuno M. (1991). Human respiratory muscles: fibre morphology and capillary supply. Eur. Respir. J. 4, 587–601. doi:10.1183/09031936.93.04050587
Muller-Delp J. M., Spier S. A., Ramsey M. W., Delp M. D. (2002). Aging impairs endothelium-dependent vasodilation in rat skeletal muscle arterioles. Am. J. Physiology-Heart Circulatory Physiology 283, H1662–H1672. doi:10.1152/ajpheart.00004.2002
Musch T. I., Moore R. L., Leathers D. J., Bruno A., Zelis R. (1986). Endurance training in rats with chronic heart failure induced by myocardial infarction. Circulation 74, 431–441. doi:10.1161/01.CIR.74.2.431
Nogueira-Ferreira R., Vitorino R., Ferreira R., Henriques-Coelho T. (2015). Exploring the monocrotaline animal model for the study of pulmonary arterial hypertension: a network approach. Pulm. Pharmacol. Ther. 35, 8–16. doi:10.1016/j.pupt.2015.09.007
Peled N., Bendayan D., Shitrit D., Fox B., Yehoshua L., Kramer M. R. (2008). Peripheral endothelial dysfunction in patients with pulmonary arterial hypertension. Respir. Med. 102, 1791–1796. doi:10.1016/j.rmed.2008.06.014
Poole D. C., Behnke B. J., Musch T. I. (2021). The role of vascular function on exercise capacity in health and disease. J. Physiology 599, 889–910. doi:10.1113/JP278931
Poole D. C., Kindig C. A., Behnke B. J. (2001). Effects of emphysema on diaphragm microvascular oxygen pressure. Am. J. Respir. Crit. Care Med. 163, 1081–1086. doi:10.1164/ajrccm.163.5.2008065
Poole D. C., Sexton W. L., Behnke B. J., Ferguson C. S., Hageman K. S., Musch T. I. (2000). Respiratory muscle blood flows during physiological and chemical hyperpnea in the rat. J. Appl. Physiology 88, 186–194. doi:10.1152/jappl.2000.88.1.186
Poole D. C., Sexton W. L., Farkas G. A., Powers S. K., Reid M. B. (1997). Diaphragm structure and function in health and disease. Med. Sci. Sports Exerc 29, 738–754. doi:10.1097/00005768-199706000-00003
Poole D. C., Wagner P. D., Wilson D. F. (1995). Diaphragm microvascular plasma PO2 measured in vivo. J. Appl. Physiology 79, 2050–2057. doi:10.1152/jappl.1995.79.6.2050
Rubens C., Ewert R., Halank M., Wensel R., Orzechowski H.-D., Schultheiss H.-P., et al. (2001). Big endothelin-1 and endothelin-1 plasma levels are correlated with the severity of primary pulmonary hypertension. Chest 120, 1562–1569. doi:10.1378/chest.120.5.1562
Schulze K. M., Musch T. I. (2023). Skeletal muscle blood flow during exercise is reduced in a rat model of pulmonary hypertension. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 324, R772–R773. doi:10.1152/ajpregu.00009.2023
Schulze K. M., Weber R. E., Colburn T. D., Horn A. G., Ade C. J., Hsu W.-W., et al. (2021). The effects of pulmonary hypertension on skeletal muscle oxygen pressures in contracting rat spinotrapezius muscle. Exp. Physiol. 106, 2070–2082. doi:10.1113/EP089631
Schulze K. M., Weber R. E., Horn A. G., Colburn T. D., Ade C. J., Poole D. C., et al. (2022). Effects of pulmonary hypertension on microcirculatory hemodynamics in rat skeletal muscle. Microvasc. Res. 141, 104334. doi:10.1016/j.mvr.2022.104334
Sexton W. L., Poole D. C. (1995). Costal diaphragm blood flow heterogeneity at rest and during exercise. Respir. Physiol. 101, 171–182. doi:10.1016/0034-5687(95)00033-A
Smith J. R., Hageman K. S., Harms C. A., Poole D. C., Musch T. I. (2017). Respiratory muscle blood flow during exercise: effects of sex and ovarian cycle. J. Appl. Physiology 122, 918–924. doi:10.1152/japplphysiol.01007.2016
Spiesshoefer J., Herkenrath S., Mohr M., Randerath W., Tuleta I., Diller G. P., et al. (2019). Diaphragm function does not independently predict exercise intolerance in patients with precapillary pulmonary hypertension after adjustment for right ventricular function. Biosci. Rep. 39, BSR20190392. doi:10.1042/BSR20190392
Sun X.-G., Hansen J. E., Oudiz R. J., Wasserman K. (2001). Exercise Pathophysiology in patients with primary pulmonary hypertension. Circulation 104, 429–435. doi:10.1161/hc2901.093198
Sun X.-G., Hansen J. E., Oudiz R. J., Wasserman K. (2003). Pulmonary function in primary pulmonary hypertension. J. Am. Coll. Cardiol. 41, 1028–1035. doi:10.1016/S0735-1097(02)02964-9
Supinski G. S. (1988). Respiratory muscle blood flow. Clin. Chest Med. 9, 211–223. doi:10.1016/S0272-5231(21)00500-1
Supinski G. S., Stofan D., Nashawati E., DiMarco A. F. (1993). Failure of vasodilator administration to increase blood flow to the fatiguing diaphragm. J. Appl. Physiology 74, 1178–1185. doi:10.1152/jappl.1993.74.3.1178
Vanhoutte P. M., Shimokawa H., Feletou M., Tang E. H. C. (2017). Endothelial dysfunction and vascular disease – a 30th anniversary update. Acta Physiol. 219, 22–96. doi:10.1111/apha.12646
Keywords: perfusion, oxygen transport, vascular dysfunction, monocrotaline, respiratory muscle
Citation: Schulze KM, Horn AG, Weber RE, Behnke BJ, Poole DC and Musch TI (2023) Pulmonary hypertension alters blood flow distribution and impairs the hyperemic response in the rat diaphragm. Front. Physiol. 14:1281715. doi: 10.3389/fphys.2023.1281715
Received: 22 August 2023; Accepted: 07 December 2023;
Published: 22 December 2023.
Edited by:
Zhongjun Du, Shandong First Medical University, ChinaCopyright © 2023 Schulze, Horn, Weber, Behnke, Poole and Musch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kiana M. Schulze, kiana7@ksu.edu
†These authors have contributed equally to this work