 Anusha Acharya1
Anusha Acharya1 Fenghua Bian1
Fenghua Bian1 Jose Gomez-Arroyo1,2
Jose Gomez-Arroyo1,2 Kimberly A. Wagner1
Kimberly A. Wagner1 Vladimir V. Kalinichenko3,4
Vladimir V. Kalinichenko3,4 Tanya V. Kalin1,3*
Tanya V. Kalin1,3*- 1Division of Pulmonary Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States
- 2Division of Pulmonary and Critical Care and Sleep Medicine, Department of Internal Medicine, University of Cincinnati, Cincinnati, OH, United States
- 3Phoenix Children’s Health Research Institute, University of Arizona College of Medicine—Phoenix, Phoenix, AZ, United States
- 4Division of Neonatology, Phoenix Children’s Hospital, Phoenix, AZ, United States
Introduction: Forkhead Box F1 (FOXF1) transcription factor plays a critical role in lung angiogenesis during embryonic development and lung repair after injury. FOXF1 expression is decreased in endothelial cells after lung injury; however, molecular mechanisms responsible for the FOXF1 transcript changes in injured lung endothelium remain unknown.
Methods: We used immunostaining of injured mouse lung tissues, FACS-sorted lung endothelial cells from hypoxia-treated mice, and data from patients diagnosed with hypoxemic respiratory failure to demonstrate that hypoxia is associated with decreased FOXF1 expression. Endothelial cell cultures were used to induce hypoxia in vitro and identify the upstream molecular mechanism through which hypoxia inhibits FOXF1 gene expression.
Results: Bleomycin-induced lung injury induced hypoxia in the mouse lung tissue which was associated with decreased Foxf1 expression. Human FOXF1 mRNA was decreased in the lungs of patients diagnosed with hypoxemic respiratory failure. Mice exposed to hypoxia exhibited reduced Foxf1 expression in the lung tissue and FACS-sorted lung endothelial cells. In vitro, hypoxia (1% of O2) or treatment with cobalt (II) chloride increased HIF-1α protein levels but inhibited FOXF1 expression in three endothelial cell lines. Overexpression of HIF-1α in cultured endothelial cells was sufficient to inhibit Foxf1 expression. siRNA-mediated depletion of HIF-1α prevented the downregulation of Foxf1 gene expression after hypoxia or cobalt (II) chloride treatment.
Conclusion: Hypoxia inhibits FOXF1 expression in endothelial cells in a HIF-1α dependent manner. Our data suggest that endothelial cell-specific inhibition of HIF-1α via gene therapy can be considered to restore FOXF1 and improve lung repair in patients with severe lung injury.
Introduction
The lung is a well-oxygenated organ; however, multiple pathological conditions exist that result in the tissue becoming hypoxic (Shimoda and Semenza, 2011). These include acute respiratory distress syndrome (ARDS), acute lung injury (ALI), COVID-19 and influenza viral infections, bacterial pneumonia, chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF), cystic fibrosis, bronchopulmonary dysplasia and lung cancer (Carbone et al., 2005; Naeije, 2005; Stenmark and Abman, 2005; Strange and Highland, 2005; Wigley et al., 2005; Ball et al., 2014). Additionally, hypoxia frequently correlates with disease severity and it is associated with poor outcomes in patients with IPF and COPD (Tzouvelekis et al., 2007; Bodempudi et al., 2014; Weng et al., 2014; Aquino-Gálvez et al., 2019).
The transcription factor Forkhead Box F1 (FOXF1) is an evolutionarily conserved member of the Forkhead Box (FOX) family of transcription factors. During embryonic development, FOXF1 functions as an essential mediator of angiogenesis (Mahlapuu et al., 2001a; Mahlapuu et al., 2001b; Claxton et al., 2008; Ren et al., 2014). Heterozygous deletions and loss-of-function point mutations in the FOXF1 gene locus are associated with alveolar capillary dysplasia with misalignment of pulmonary veins (ACDMPV). ACDMPV is a rare congenital disease occurring in neonates and infants which is characterized by impaired development of the alveolar capillary network, misalignment of pulmonary veins, and severe pulmonary hypertension (Bishop et al., 2011). Global deletion of Foxf1 (Foxf1−/−) in mice is embryonically lethal, whereas mice with heterozygous deletion of Foxf1 (Foxf1+/−) present with alveolar capillary dysplasia along with developmental defects in the lung, intestine, and liver (Mahlapuu et al., 2001a; Mahlapuu et al., 2001b; Kalinichenko et al., 2002a; Kalinichenko et al., 2002b; Kalinichenko et al., 2003; Claxton et al., 2008). Conditional deletion of Foxf1 specifically in endothelial cells results in embryonic lethality due to impaired VEGF signaling in the yolk sac, placenta, and lung (Ren et al., 2014). FOXF1 stimulates the expression of genes critical for endothelial barrier function and plays an important role in maintaining normal lung homeostasis and lung repair after injury (Cai et al., 2016).
We previously showed that pulmonary endothelial cells isolated from bleomycin-treated mice and human IPF lungs exhibited low levels of FOXF1 (Bian et al., 2023). Further, employing a transgenic mouse model with endothelial-specific deficiency of Foxf1, we demonstrated that loss of Foxf1 exacerbated the fibrotic phenotype. FOXF1 has previously been reported as an anti-fibrotic factor preventing the reprogramming of lung fibroblasts into myofibroblasts in mouse and human lungs (Black et al., 2018). While the FOXF1 expression is decreased after lung injury, molecular mechanisms through which FOXF1 gene expression changes occur remain unknown.
In our previous studies, the downregulation of FOXF1 in endothelial cells corresponded with diminished arterial oxygenation and hypoxemia in mouse lung tissue (Cai et al., 2016; Pradhan et al., 2019; Ren et al., 2019; Bolte et al., 2020; Wang et al., 2021; Wang et al., 2022). However, the direct impact of hypoxia on the expression of FOXF1 in pulmonary endothelial cells is unclear.
In the present study, we aimed to determine if the hypoxic microenvironment plays a role in the downregulation of FOXF1 in injured lung endothelial cells in vitro and in vivo. We showed that hypoxia inhibits FOXF1 in endothelial cells in a HIF-1α dependent manner. Our data suggest that specific inhibition of HIF-1α in endothelial cells via gene therapy can be considered to increase FOXF1 and improve lung repair in patients with severe lung injury.
Materials and methods
Bleomycin-induced lung injury
To induce pulmonary fibrosis, mice were administered 2 or 2.5 U/kg of bleomycin sulfate (EMD Biosciences) intratracheally (I.T.) once. The control group of mice (Uninjured) was administered PBS I.T. the same way as bleomycin.
Detection of hypoxia in bleomycin model of lung injury
3 days post-induction of bleomycin injury, hypoxia was assessed in the mouse lungs as previously described (Cantelmo et al., 2016). Briefly, mice were injected with 60 mg/kg pimonidazole hydrochloride (Hypoxyprobe™ kit, cat#HP1) 1 hour prior to sacrifice. To detect the formation of pimonidazole adducts, paraffin sections were immunostained with hypoxyprobe-1-mAb1 following the manufacturer’s instructions.
In vivo model of hypoxia
Wild-type C57BL/6 mice were purchased from the Jackson Laboratory. Mice were housed in specific pathogen-free animal facilities at the Cincinnati Children’s Hospital Medical Center. Experiments were conducted on 8–12 weeks old age-matched mice. For hypoxia exposure, mice were housed inside a nitrogen dilution hypoxia chamber set to a fraction of inspired oxygen of 10%. The oxygen concentration was monitored and controlled with a ProOx model 350 unit (BioSpherix) by infusion of Nitrogen. In parallel, a separate set of wild-type mice were kept in normal room air (Normoxia). On day 3, day 7 and day 10 of hypoxia exposure, mice were euthanized, and the lungs were perfused with PBS and harvested. All methods and protocols were approved by the Animal Care and Use Committee of the Cincinnati Children’s Hospital Medical Center.
Cell lines and reagents
Human endothelial HUVEC cells (Lonza, Cat# C2519A) were cultured in EGM2/EBM2 (Lonza) growth medium, and Human Pulmonary Artery Endothelial Cells (HPAEC, Cat# CC2530) were cultured in ECM Medium (ScienCell). HUVEC and HPAEC cells were from pooled donors. Mouse endothelial MFLM-91U cells (Seven Hills, Cat# AMFLM-91U) were cultured in DMEM (Gibco).
In vitro models of hypoxia
For hypoxia exposure, cells were grown to 70% confluency and were changed to fresh complete media before hypoxia treatment using a finely controlled Whitley H35 Hypoxystation (Don Whitley Scientific). The oxygen concentration in the chamber was maintained at 1% with a residual gas mixture composed of 5% carbon dioxide and balanced nitrogen. Normoxia-treated cells used as a control were cultured in 95% atmospheric air and 5% carbon dioxide and 21% oxygen concentration. For a chemically induced model of hypoxia, cells were grown to 70% confluency and changed to fresh complete media with a final concentration of 150 µM of cobalt (II) chloride (Sigma Aldrich, Cat# 60818).
Flow cytometry
Flow cytometry was performed on single cell suspensions from enzyme-digested whole lungs as described previously (Bian et al., 2023). Briefly, whole lungs were dissociated with Liberase, washed with PBS, filtered through a 70-um filter and counted to get the final cell count of the single cell suspension. Live cells were detected with 7-amino actinomycin D (7-AAD) (BioLegend). Hematopoietic cells were detected using CD45 antibody (Biolegend, Cat# 103140). The CD45− population was then assessed for expression of CD31 (eBioscience, Cat# 48-0311-82). Endothelial cells that were identified as CD31+ and CD45− were FACS-sorted using the FACSAria II or FACSymphony S6, five-laser cell sorter (BD Biosciences). After FACS-sorting, the CD45−CD31+ endothelial cell population is >95% pure and lacks fibroblasts and other mesenchymal and epithelial cells as previously described (Ren et al., 2014; Bolte et al., 2017; Sun et al., 2021; Wen et al., 2021; Kolesnichenko et al., 2023).
qRT- PCR and Western blot
For the knockdown of Hif1α, MFLM-91U cells were transfected with siHif1α (Horizon Discovery, Cat#M-040638-00-0010) using Dharmafect transfection reagent 1 according to the manufacturer’s protocol (Dharmacon). A cocktail of non-targeting siRNAs was used as a control (Horizon, Cat# D-001810-01-20). For overexpression of HIF-1α, cells were transfected with CMV-HIF-1α (Origene, Cat# MG210895). Cells were lysed for RNA and protein using lysis buffer (Qiagen, cat#74104) and RIPA buffer (Cell Signaling Technology, Cat#9806) respectively, as per the manufacturer’s protocol. qRT-PCR was performed as previously described (Kalin et al., 2008) using TaqMan probes for mouse Foxf1 (Mm00487497_m1), mouse Hif1α (Mm00468869_m1), mouse ActB (Mm00607939_s1), human FOXF1 (Hs00230962_m1), human HIF1α (Hs00153153_m1) and human ACTB (Hs99999903_m1). Protein extracts were prepared as described (Bolte et al., 2015; Milewski et al., 2017). The following antibodies were used for western blots: anti-HIF-1α (Novus Biologicals, Cat# NB100-449), anti-FOXF1 (RnD systems, Cat#AF4798) and anti-Beta Actin (Santacruz, Cat# sc47778).
Human RNA seq
Human samples analyzed were obtained as part of a larger dataset deposited from the Pulmonary Hypertension Breakthrough Initiative (PHBI) Biobank (Stearman et al., 2018). All samples utilized in this study have been previously characterized, processed and de-identified accordingly, as part of the PHBI Biobank. We have obtained permission to use samples, as well as clinical data for research and publication as per the most recent MTA. Patient enrolment, tissue-processing protocol, RNA isolation and library preparation for all PHBI samples have been previously described (Stacher et al., 2012; Stearman et al., 2018). Only datasets from controls were used for analysis. Paired-end 75 base-pair RNA sequencing (RNA-seq) was performed on all available lung samples using an Illumina sequencer. Samples were sequenced in two batches. The sequencing depth was 20–25 million reads per sample in one batch and 15–20 million reads per sample in the other batch. Reads were pseudo-aligned and quantified using an index transcriptome version of the GRCh38.p14 human genome (RefSeq GCF_000001405.40) using Kallisto with standard settings (Bray et al., 2016). Raw counts were normalized using DESeq2 (Love et al., 2014). Normalized counts were compared between groups using non-parametric testing with GraphPad PRISM v.10.
Statistical analysis
The Student t-test (two-tailed), non-parametric Mann-Whitney U test and one-way ANOVA followed by Dunnett’s multiple comparison test were used to determine statistical significance. p-values < 0.05 were considered significant. Values were shown as mean
Study approval
All animal studies were approved by the Cincinnati Children’s Research Foundation Institutional Animal Care and Use Committee and covered under our animal protocol (IACUC 2022-0041). The Cincinnati Children’s Research Foundation Institutional Animal Care and Use Committee is an AAALAC and NIH accredited institution (NIH Insurance #8310801).
Results
Hypoxia is associated with decreased FOXF1 expression in injured mouse and human lungs
Our published studies demonstrated that bleomycin-induced lung injury decreases the expression of Foxf1 in pulmonary endothelial cells in a time-dependent manner (Bian et al., 2023). This decrease was observed at a time point as early as 3 days after the bleomycin treatment (Bian et al., 2023). Since hypoxia is frequently associated with lung injury, we tested if hypoxia was induced in bleomycin-injured mouse lungs. To determine whether the lung injury affected the oxygen availability in the lung tissue 3 days after the bleomycin treatment, we injected control (PBS injected) and bleomycin-injured mice with pimonidazole, a chemical sensor for hypoxia (Cantelmo et al., 2016). Bleomycin injury increased the hypoxic area in the lung tissue compared to control lungs (Figures 1A, B). Thus, the hypoxia in bleomycin-injured mouse lungs directly correlates with reduced Foxf1 expression shown previously (Bian et al., 2023).
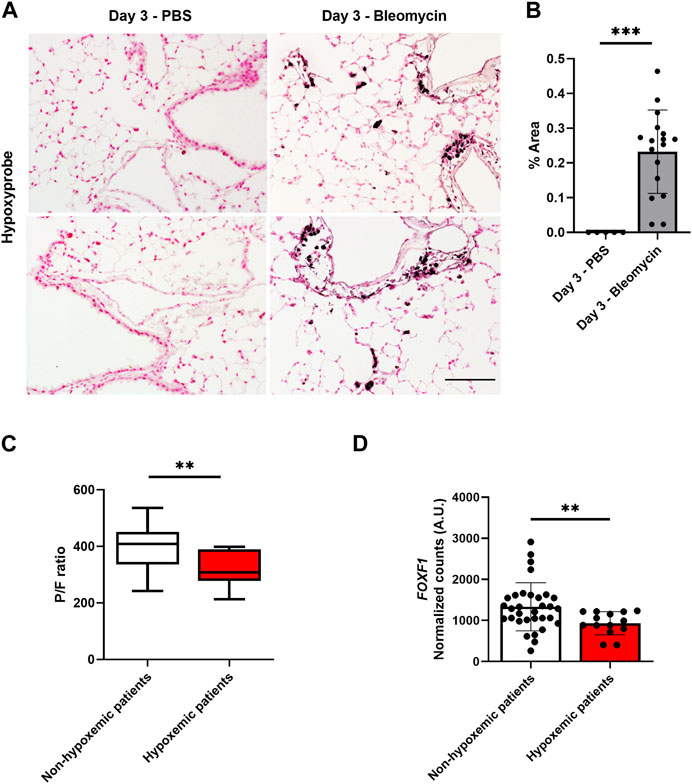
FIGURE 1. Hypoxia is associated with decreased Foxf1 expression in injured mouse and human lungs. (A,B) Immunohistochemical staining of mouse lung sections for hypoxyprobe-1-mAb1 shows bleomycin-injured lungs have increased hypoxia at day 3 post-injury induction. Hypoxyprobe area was calculated as % of hypoxyprobe-1-mAb1 positive area per field in 5 random fields using NIS elements software version 4.5. Each dot represents a single field. N = 3 mice per group. Both male and female mice were included. Scale bar = 50 µm. Values are shown as mean ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001 from Student’s t test (two-tailed). (C) Hypoxemic patients had a P/F value of less than 400. The P/F ratio of patients was calculated as the partial pressure of oxygen in blood divided by the ratio of the inspired fraction of oxygen (P/F ratio) provided by the mechanical ventilator. N = total 46 patients. Values are shown as mean ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001 from non-parametric Mann-Whitney U test. (D) Hypoxemic patients have decreased FOXF1 expression compared to non-hypoxemic patients. Normalized counts of FOXF1 expression were obtained from the RNA seq data set. N = total 46 patients. Each dot represents a patient. Values are shown as mean ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001 from non-parametric Mann-Whitney U test.
Next, we examined if hypoxia is associated with decreased FOXF1 expression in human lungs. Explanted lung tissue from failed donor patients was evaluated. Briefly, these patients became unsuitable to donate after tissue procurement for various reasons including hypoxemic respiratory failure, ABO blood type incompatibility, hemodynamic instability, etc., First, we separated patients by their capacity to oxygenate blood directly assessed by the partial pressure of oxygen in blood divided by the ratio of the inspired fraction of oxygen (P/F ratio) provided by the mechanical ventilator (Figure 1C). P/F values below 400 indicate hypoxemia. This ratio is clinically used to assess for and classify the severity of hypoxemic respiratory failure (Ranieri et al., 2012). Using this dataset, we then analyzed FOXF1 expression in the patients who were hypoxemic before transplantation compared to non-hypoxemic patients. The FOXF1 expression in hypoxemic patients was significantly decreased compared to the patients without hypoxemia (Figure 1D). Thus, the correlation between hypoxemia and decreased FOXF1 expression in human and mouse lungs prompted us to investigate whether FOXF1 expression is regulated by oxygen levels.
Hypoxia inhibits Foxf1 in lung endothelial cells in vivo
Since downregulation of Foxf1 expression coincided with hypoxia in bleomycin-injured mouse lungs, we examined if hypoxia alone is sufficient to decrease the Foxf1 expression in pulmonary endothelial cells. To investigate this possibility, we exposed age-matched adult wild-type C57BL/6 mice to continuous hypoxia (10% oxygen) or normoxia (21% oxygen) for 3, 7, and 10 days (Figure 2A). Foxf1 mRNA was measured in whole lung RNA obtained from mice exposed to either hypoxia or normoxia (control). Foxf1 mRNA was decreased in the lungs of hypoxic mice as early as day 3 and remained decreased at day 7 and day 10 post hypoxic exposure (Figure 2B). Since in the adult lung expression of Foxf1 is the highest in endothelial cells (Kalucka et al., 2020; Paik et al., 2020), we measured Foxf1 mRNA in purified lung endothelial cells obtained from hypoxic and normoxic mice. FACS sorting of CD31+/CD45− lung endothelial cells was performed at different time points after hypoxic and normoxic exposure (Figure 2C). Endothelial Foxf1 mRNA was measured by qRT-PCR. Similar to the total lung RNA data (Figure 2B), Foxf1 mRNA in FACS-sorted lung endothelial cells was reduced starting at day 3 after the initiation of hypoxia exposure (Figure 2D). Western blot analysis was consistent with reduced FOXF1 protein amounts in the lung tissue after hypoxia (Figure 2E). Thus, hypoxia inhibits Foxf1 expression in mouse lung endothelial cells in vivo.
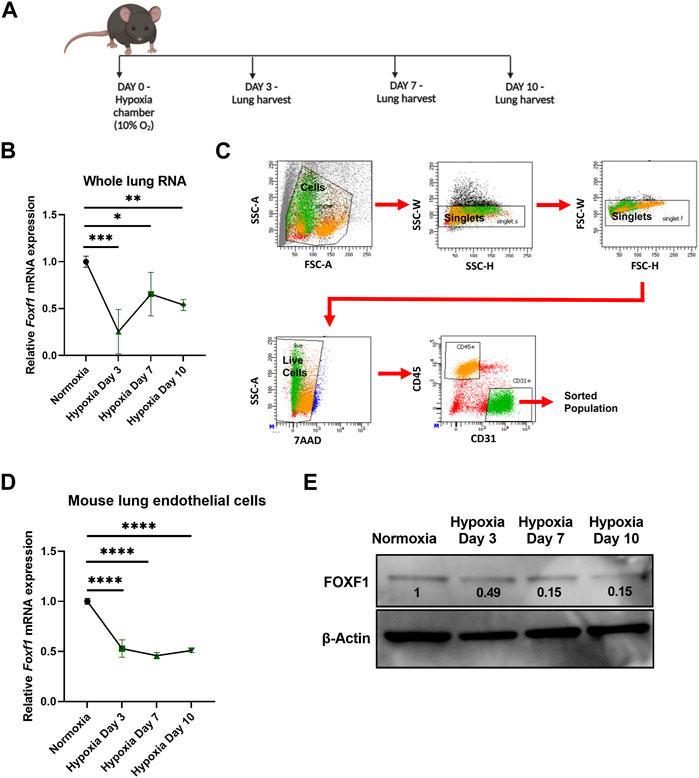
FIGURE 2. Hypoxia inhibits Foxf1 in lung endothelial cells in vivo (A) Experimental schematic showing timeline of exposure of 8–12 weeks old Wild-type C57BL/6 mice to hypoxia or normoxia and lung harvest. (B) Foxf1 mRNA expression was decreased in mouse lungs at day 3, day 7 and day 10 after exposure to hypoxia. qRT-PCR was performed on total RNA isolated from harvested lung tissue. β-Actin (Actb) was used as the housekeeping gene. N = 4 mice per group. (C) The gating strategy utilized to FACS sort CD45-/CD31+ endothelial cells from mouse lungs exposed to hypoxia or normoxia. (D) Foxf1 mRNA expression was decreased in CD45−/CD31+ endothelial cells FACS sorted from mouse lungs at day 3, day 7 and day 10 post hypoxic exposure. qRT-PCR analysis was performed in triplicate on pooled RNA samples from each group. β-Actin (Actb) was used as the housekeeping gene. N = 4 mice per group. (E) FOXF1 protein expression was decreased in CD45-/CD31+ endothelial cells FACS sorted from mouse lungs at day 3, day 7 and day 10 post hypoxic exposure. Values on the western blot image represent FOXF1 protein expression relative to normoxic condition. Densitometric analysis was done using ImageJ software. Values were normalized to housekeeping protein β-Actin. Western blot was performed on pooled protein samples from each group. N = 4 mice per group. All values in this figure are shown as mean ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001 from one-way ANOVA followed by Dunnett’s test.
Hypoxia inhibits Foxf1 in lung endothelial cells in vitro
Next, we tested if hypoxia directly inhibits Foxf1 in vitro. We performed time-course studies using 3 different endothelial cell lines, including embryonic mouse lung MFLM-91U endothelial cells, human umbilical vein endothelial cells (HUVECs) and human pulmonary artery endothelial cells (HPAECs). Cells were subjected to hypoxia (1% oxygen) for 8 hours, 12 hours, and 24 hours. Controls included cells exposed to normoxia (21% oxygen). Foxf1 mRNA was measured using qRT-PCR. Foxf1 mRNA was significantly decreased at 8 hours of hypoxia and remained downregulated until 24 hours in all 3 cell lines (Figures 3A, B). Hypoxia inducible factor 1-α (HIF-1α) is the master regulator of hypoxic responses. Under normoxic conditions, it is degraded via the ubiquitin-proteosome pathway, whereas under hypoxic conditions its degradation is inhibited and the HIF-1α protein is accumulated in the cell (Rhim et al., 2013; Vriend and Reiter, 2016) Therefore, we examined if the HIF-1α protein is increased at the time points at which we observed hypoxia-mediated downregulation of Foxf1. Western blot analysis for HIF-1α was performed in whole endothelial cell lysates at each time point after hypoxia. As seen in Figures 3C, D; Supplementary Figure S1A–C, HIF-1α protein was increased in all 3 endothelial cell lines at 8, 12, and 24 hours after the initiation of hypoxic treatment.
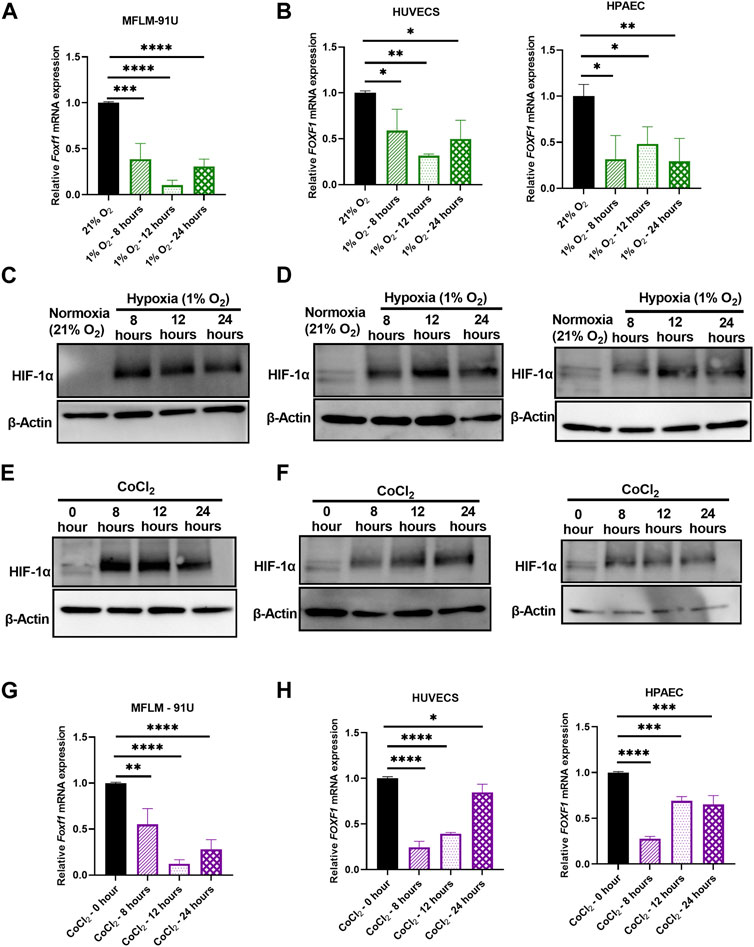
FIGURE 3. Hypoxia inhibits Foxf1 in lung endothelial cells in vitro. (A) Foxf1 mRNA expression was decreased in mouse endothelial MFLM 91U cells under hypoxic exposure (1% O2) for 8 hours, 12 hours, and 24 hours (B) FOXF1 mRNA expression was decreased in human endothelial cells HUVEC (left) and HPAEC (right) under hypoxic exposure (1% O2) for 8 hours, 12 hours, and 24 hours (C) HIF-1α protein was induced in MFLM 91U cells under hypoxia (1% O2) from 8 hours to 24 hours (D) HIF-1α protein was induced in HUVEC (left) and HPAEC (right) under hypoxia (1% O2) from 8 hours to 24 hours (E) HIF-1α protein was induced in MFLM 91U cells upon treatment with 150 µM of cobalt (II) chloride for 8 hours, 12 hours, and 24 hours (F) HIF-1α was induced in HUVEC (left) and HPAEC (right) upon treatment with 150 µM of cobalt (II) chloride for 8 hours, 12 hours and 24 hours (G) Foxf1 mRNA expression was decreased in MFLM 91U upon treatment with 150 µM of cobalt (II) chloride for 8 hours, 12 hours and 24 hours (H) FOXF1 mRNA expression was decreased in HUVEC (left) and HPAEC (right) upon treatment with 150 µM of cobalt (II) chloride for 8 hours, 12 hours, and 24 hours. All qRT-PCR analyses employed β-Actin (Actb) as the housekeeping gene. Experiments were conducted three times. All values in this figure are shown as mean ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001 from one-way ANOVA followed by Dunnett’s test.
To further confirm our observations from the in vitro study of hypoxia, we utilized a chemical model of hypoxia. Cobalt (II) chloride is a widely used hypoxia mimetic (Muñoz-Sánchez and Chánez-Cárdenas, 2018). It is currently accepted that Co2+ substitutes Fe2+ in the prolyl hydroxylase (PHD) enzyme and inhibits it. Subsequently, proteasomal degradation of HIF-1α under normoxic conditions is inhibited, thus leading to its stabilization. To mimic the study in the hypoxia chamber, we treated endothelial MFLM-91U, HUVEC, and HPAEC cells with 150 µM of cobalt (II) chloride for 8, 12, and 24 hours to mimic hypoxia. Untreated cells were used as controls. To confirm if HIF-1α was increased after the cobalt (II) chloride treatment, we performed a Western blot analysis. HIF-1α protein expression was low in untreated cells, but it increased after 8, 12, and 24 hours after the cobalt (II) chloride treatment in all 3 endothelial cell lines (Figures 3E, F; Supplementary Figures 1D–F). Next, we measured Foxf1 mRNA levels in these cells by qRT-PCR. MFLM-91U cells exhibited a maximum reduction in Foxf1 mRNA at 12 hours under 1% oxygen (Figure 3A) and similar results were observed after the cobalt (II) chloride treatment (Figure 3G). HUVECs under 1% oxygen showed the maximum Foxf1 reduction at 12 hours; however, under cobalt (II) chloride treatment the maximum reduction was observed at 8 hours but sustained until 12 hours (Figures 3B, H). HPAECs under 1% oxygen demonstrate comparable levels of Foxf1 mRNA at all time points, while under cobalt (II) chloride treatment, the maximum reduction was observed at 8 hours (Figures 3B, H). Taken together, these data demonstrate that hypoxia directly inhibits Foxf1 expression in endothelial cells in vitro.
Hypoxia represses Foxf1 expression in lung endothelial cells through HIF-1α
Next, we investigated the molecular mechanism whereby hypoxia inhibits Foxf1 expression in endothelial cells. Observations from the in vitro and in vivo studies indicated a relationship between hypoxia, increased HIF-1α protein levels, and reduced Foxf1 gene expression (Figures 1–3). Therefore, we tested if HIF-1α plays a role in the repression of endothelial Foxf1 gene after hypoxia. First, we overexpressed HIF-1α in endothelial MFLM-91U cells under normoxic conditions using transient transfection with CMV-HIF-1α plasmid. Overexpression of HIF-1α led to the accumulation of HIF-1α protein (Figures 4A, B), which was associated with decreased FOXF1 mRNA and protein as determined by qRT-PCR and western blot respectively (Figures 4C–E). Thus, overexpression of HIF-1α was sufficient to inhibit Foxf1 even under normoxic conditions in endothelial cell cultures.
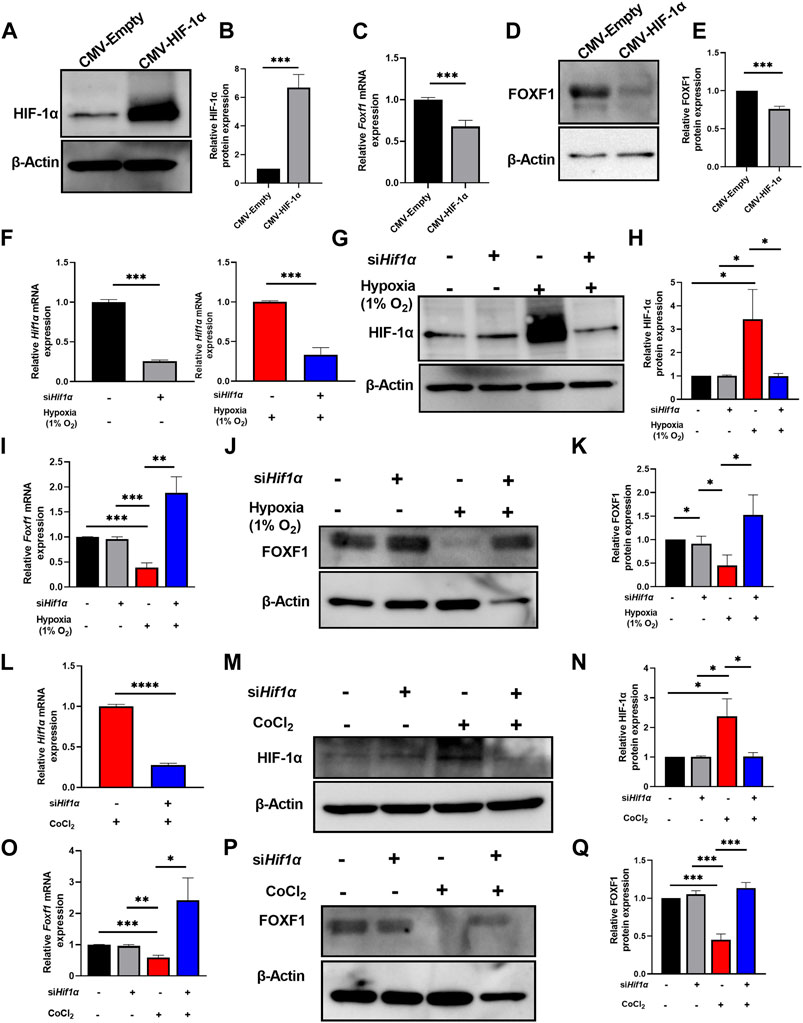
FIGURE 4. Hypoxia represses Foxf1 expression in lung endothelial cells through HIF-1α. (A,B) HIF-1α protein was overexpressed in MFLM 91U cells transfected with CMV-HIF-1α under normoxic conditions. Protein expression is represented relative to control cells transfected with a CMV-empty vector. (C) Foxf1 mRNA expression was decreased in MFLM-91U cells with overexpression of HIF-1α under normoxic conditions. (D,E) FOXF1 protein expression was decreased in MFLM-91U cells with overexpression of HIF-1α under normoxic conditions. Protein expression is represented relative to control cells transfected with a CMV-empty vector. (F) Hif1α was knocked down at an efficiency of 75% under normoxic (left) and 67% under hypoxic (right) conditions. MFLM-91U cells were transfected with non-targeting siRNA (siNT) or siHif1α and subsequently exposed to normoxia (21% O2) or hypoxia (1% O2). (G,H) Knockdown of Hif1α prevents induction of HIF-1α protein under hypoxia (1% O2). MFLM-91U cells transfected with non-targeting siRNA (siNT) or siHif1α were exposed to normoxia (21% O2) or hypoxia (1% O2) for 12 hours. Protein expression is represented relative to control cells transfected with non-targeting siRNA (siNT) in normoxia. (I) Foxf1 mRNA expression is restored under hypoxic conditions upon knockdown of Hif1α. qRT-PCR was performed on MFLM-91U cells transfected with non-targeting siRNA (siNT) or siHif1α and exposed to normoxia (21% O2) or hypoxia (1% O2) for 12 h (J,K) FOXF1 protein expression is restored under hypoxic conditions upon knockdown of Hif1α. Protein expression is represented relative to control cells transfected with non-targeting siRNA (siNT) in normoxia. (L) Hif1α was knocked down at an efficiency of 73% under cobalt (II) chloride treatment. MFLM-91U cells were transfected with non-targeting siRNA (siNT) or siHif1α and subsequently treated with cobalt (II) chloride. (M,N) Knockdown of Hif1α prevents induction of HIF-1α protein under cobalt (II) chloride treatment. MFLM-91U cells were transfected with non-targeting siRNA (siNT) or siHif1α followed by treatment with cobalt (II) chloride for 12 h. Protein expression is represented relative to control cells transfected with non-targeting siRNA (siNT) and no cobalt (II) chloride treatment. (O) Foxf1 mRNA expression is restored under cobalt (II) chloride treatment upon knockdown of Hif1α. qRT-PCR was performed on MFLM-91U cells transfected with non-targeting siRNA (siNT) or siHif-1α and subsequently treated with cobalt (II) chloride for 12 h (P,Q) FOXF1 protein expression is restored under cobalt (II) chloride treatment upon knockdown of Hif1α. Protein expression is represented relative to control cells transfected with non-targeting siRNA (siNT) and no cobalt (II) chloride treatment. All qRT-PCR analyses employed β-Actin (Actb) as the housekeeping gene. Western blot quantifications were performed using densitometric analysis on ImageJ software. Experiments were conducted three times. All values in this figure are shown as mean ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001 from Student’s t test (two-tailed).
Second, to determine if inhibition of HIF-1α can restore the FOXF1 expression, we depleted Hif1α in MFLM-91U cells using Hif1α-specific siRNA (siHif1α). Hif1α knockdown efficiency was approximately 75% as shown by qRT-PCR (Figure 4F). After Hif1α knockdown, we exposed the cells to hypoxia (1% oxygen) for 12 hours. After harvesting the cells, we measured HIF-1α and FOXF1. HIF-1α protein and mRNA were decreased in Hif1α-deficient cells compared to cells transfected with non-targeting siRNA (control) (Figures 4F–H). Based on FOXF1 mRNA and protein expression measurements, there was no difference in FOXF1 expression between control and siHif1α-treated cells grown under normoxic conditions (Figures 4I–K). In contrast, siHif1α-treated cells grown under hypoxia showed an increase in FOXF1 mRNA and protein (Figures 4I–K). Next, we mimicked these experiments using cobalt (II) chloride instead of 1% oxygen. We observed a similar rescue of FOXF1 mRNA and protein in siHif1α-transfected cells exposed to cobalt (II) chloride (Figures 4L–Q). Taken together, our results indicate that hypoxia inhibits Foxf1 gene expression in endothelial cells, at least in part, through HIF-1α dependent mechanism.
Discussion
Maintenance of endothelial barrier function is crucial for the maintenance of normal lung homeostasis and its repair following injury (Mehta and Malik, 2006). Pulmonary endothelial cells are important mediators in the exchange of gases, water and macromolecules between blood and alveolar tissue. They function as regulators of homeostasis and inflammation by secreting various cytokines and chemokines. Additionally, they regulate vascular tone and interact with other vascular cell types and inflammatory cells. Endothelial barrier function is known to be maintained by various factors such as angiopoietin-1 and sphingosine-1 phosphate (Jr et al., 1969; Gale and Yancopoulos, 1999). Cai et al. examined the function of transcription factor Forkhead box F1 (FOXF1) in quiescent endothelial cells of adult mouse lungs and identified it to be essential for the maintenance of lung homeostasis and prevention of edema after lung injury (Cai et al., 2016). They show that FOXF1 is a critical transcriptional regulator of endothelial cells and maintains endothelial barrier function by transcriptionally activating the S1pr1 promoter. Additionally, using heterozygous Pdgfb-iCreER/Foxf1+/− mice, they demonstrate that deletion of one allele of Foxf1 was enough to increase the susceptibility of these mice to acute lung injury (ALI). In a recent study focusing on fibrosis-associated endothelial cells, our group found that Foxf1 is one of the most downregulated genes in the bleomycin-injured mouse model of pulmonary fibrosis (Bian et al., 2023). Thus, restoring or increasing Foxf1 levels in endothelial cells should be considered to alleviate pulmonary fibrosis. Additionally, Foxf1 plays an anti-fibrotic role by preventing the transition of fibroblasts into myofibroblasts by inhibiting the CDH2-CDH11 cadherin switch (Black et al., 2018). Taken together, these studies indicate that Foxf1 plays a crucial role in lung injury and repair. However, what causes the downregulation of endothelial Foxf1 upon lung injury is still not well understood.
In the present study, we demonstrate that hypoxia plays a role in the downregulation of FOXF1 in endothelial cells both in vivo and in vitro. Using in vitro studies, we identified that this downregulation is at least in part through HIF-1α. In our previously published study by Bian et al.; we detected Foxf1 expression in arterial, venous, aCap and gCap sub-clusters of normal lungs and its expression was decreased in all the sub-clusters upon bleomycin-induced lung injury. Therefore, it is possible that the HIF-1a/FOXF1 regulatory mechanism is active in several types of pulmonary endothelial cells. Consistent with this hypothesis, we observed similar effects in different cell lines with high FOXF1 expression levels: MFLM-91U (fetal microvascular endothelial cells), HUVECS (venous endothelial cells) and HPAECS (arterial endothelial cells).
Despite the lung being a well-oxygenated organ, hypoxia is a characteristic of several lung injuries that can be induced chemically or mechanically. Pathological conditions that cause hypoxia include pneumonia, smoke inhalation, lung cancer and influenza or other viruses such as SARS-CoV-2. Hypoxia is also a prominent feature of pulmonary fibrosis (Senavirathna et al., 2018) and patients with interstitial lung diseases and pulmonary fibrosis suffer from varying degrees of dyspnoea. Further, the progression of fibrotic disease correlates with increasing hypoxia (Flaherty et al., 2006).
Herein, we demonstrate that one dose of intratracheal instillation of bleomycin was sufficient to induce hypoxia in the lungs of mice 3 days post bleomycin injury. While induction of hypoxia upon bleomycin injury has been reported before (Akahori et al., 2022), this is the first study showing that it is induced within 3 days. Interestingly, day 3 was also the time point at which we have previously detected a decrease in Foxf1 mRNA expression (Bian et al., 2023) using the same model of injury. Thus, indicating that hypoxia could be a microenvironmental factor responsible for the downregulation of endothelial Foxf1 upon injury. Moreover, several studies have demonstrated the role of hypoxia in the development and progression of pulmonary fibrosis (Tzouvelekis et al., 2007; Mizuno et al., 2009; Weng et al., 2014; Choi et al., 2015; Senavirathna et al., 2018). Interestingly, exposure of mice to hypoxia only resulted in a decrease in endothelial FOXF1 expression compared to mice placed in room air. Exposure of mouse and human endothelial cell lines to a hypoxic environment or chemically induced hypoxia reiterated the observations in vivo. Intriguingly, analysis of the RNA seq dataset from the lungs of deceased patients who were hypoxemic also revealed a decreased expression of FOXF1. Taken together, all these data strengthened our observation that hypoxia initiates a downregulation in endothelial FOXF1.
Adjusting to altering oxygen availability is essential for the survival of all organisms. Hypoxia inducible factors (HIFs) are master regulators of response to hypoxia (Semenza, 2007; Semenza, 2011; Shimoda and Semenza, 2011; Jiang et al., 2019) and are known to be involved in disease pathogenesis (Semenza, 2007; Semenza, 2011). HIF-1α is vital for oxygen homeostasis and it shows a ubiquitous pattern of expression.
Interestingly, the present study reveals that in vitro downregulation of FOXF1 by hypoxia in endothelial cells occurs in HIF-1α dependent manner. Cobalt (II) chloride has been reported to chemically induce hypoxia in vitro by stabilizing HIF-1α (Dai et al., 2012). Utilizing the treatment of cells with cobalt (II) chloride and by knockdown and overexpression of HIF-1α, we demonstrated that FOXF1 is downregulated via HIF-1α. Although HIF-1α has been widely known as a transcriptional activator, it has recently been demonstrated to be both an activator as well as a repressor of gene expression (Manalo et al., 2005). Genes encoding Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), and adenosine kinase (AK) in endothelial cells have been reported to be repressed via HIF-1α (Morote-Garcia et al., 2008; Zheng et al., 2009).
Several limitations are present in the current study. First, at present, we do not know the molecular mechanism by which HIF-1α represses endothelial FOXF1. The repression could be direct or indirect. HIF-1α has been previously reported to repress peroxisome proliferators–activated receptor alpha (PPAR-α) and the ENT1 gene through hypoxia response element (HRE) present on the antisense strand (Narravula and Colgan, 2001; Eltzschig et al., 2005). This indicates potential directionality in transcriptional activity. However, Manalo et al. did a comparison of transcriptionally repressed genes by hypoxia and constitutively active HIF-1α and did not find any unique patterns (Manalo et al., 2005). Another potential mechanism could be that HIF-1α activates a transcriptional repressor, which then represses FOXF1. Thus, more work will be required to define the nature of HIF-1α mediated repression. Secondly, we do not know if HIF-1α represses the Foxf1 gene in vivo. Endothelial-specific HIF-1α knockdown or delivering HIF1-α inhibitor to endothelial cells could be utilized to assess if HIF-1α inhibition can restore endothelial FOXF1 and subsequently rescue the phenotype of lung injury.
In summary, the present studies indicate that hypoxia plays a role in the repression of endothelial FOXF1 post-lung injury. HIF-1α knockdown and overexpression studies point towards the role of HIF-1α coordinating this response.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117261. Gene Expression Omnibus (GEO) accession number GSE117261.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. The animal study was approved by Cincinnati Children’s Research Foundation Institutional Animal Care and Use Committee and covered under our animal protocol (IACUC2020-0041). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
AA: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Writing–original draft, Writing–review and editing. FB: Data curation, Formal Analysis, Methodology, Writing–review and editing. JG-A: Data curation, Formal Analysis, Methodology, Writing–review and editing. KW: Data curation, Methodology, Writing–review and editing. VK: Writing–review and editing. TK: Conceptualization, Formal Analysis, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the NIH grants R01 HL132849 (TK), R01 HL158659 (TK), R01 HL141174 (VK), R01 HL149631 (VK), R01 HL152973 (VK/TK).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2023.1309155/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Exposure of cells to hypoxia (1% oxygen) or cobalt (II) chloride induces HIF-1α protein expression. HIF-1α protein expression is induced under hypoxic exposure (1% O2) from 8 hours to 24 hours in mouse endothelial MFLM-91U cells (B) HUVECS (C) HPAECS. Protein expression is represented relative to control cells cultured under normoxia. HIF-1α protein expression is induced upon treatment with 150 µM cobalt (II) chloride from 8 hours to 24 hours in (D) mouse endothelial MFLM-91U cells (E) HUVECS (F) HPAECS. Protein expression is represented relative to control cells cultured under cobalt (II) chloride treatment for 0 hours. Experiments were conducted three times. Western blot quantifications were performed using densitometric analysis on ImageJ software. All values in this figure are shown as mean ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001 from one-way ANOVA followed by Dunnett’s test.
References
Akahori D., Inui N., Inoue Y., Yasui H., Hozumi H., Suzuki Y., et al. (2022). Effect of hypoxia on pulmonary endothelial cells from bleomycin-induced pulmonary fibrosis model mice. Int. J. Mol. Sci. 23, 8996. doi:10.3390/ijms23168996
Aquino-Gálvez A., González-Ávila G., Jiménez-Sánchez L. L., Maldonado-Martínez H. A., Cisneros J., Toscano-Marquez F., et al. (2019). Dysregulated expression of hypoxia inducible factors augments myofibroblasts differentiation in idiopathic pulmonary fibrosis. Respir. Res. 20, 130. doi:10.1186/s12931-019-1100-4
Ball M. K., Waypa G. B., Mungai P. T., Nielsen J. M., Czech L., Dudley V. J., et al. (2014). Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1a. Am. J. Respir. Crit. Care Med. 189, 314–324. doi:10.1164/rccm.201302-0302OC
Bian F., Lan Y.-W., Zhao S., Deng Z., Shukla S., Acharya A., et al. (2023). Lung endothelial cells regulate pulmonary fibrosis through FOXF1/R-Ras signaling. Nat. Commun. 14, 2560. doi:10.1038/s41467-023-38177-2
Bishop N. B., Stankiewicz P., Steinhorn R. H. (2011). Alveolar capillary dysplasia. Am. J. Respir. Crit. Care Med. 184, 172–179. doi:10.1164/rccm.201010-1697CI
Black M., Milewski D., Le T., Ren X., Xu Y., Kalinichenko V. V., et al. (2018). FOXF1 inhibits pulmonary fibrosis by preventing CDH2-CDH11 cadherin switch in myofibroblasts. Cell. Rep. 23, 442–458. doi:10.1016/j.celrep.2018.03.067
Bodempudi V., Hergert P., Smith K., Xia H., Herrera J., Peterson M., et al. (2014). miR-210 promotes IPF fibroblast proliferation in response to hypoxia. Am. J. Physiology Lung Cell. Mol. Physiology 307, L283–L294. doi:10.1152/ajplung.00069.2014
Bolte C., Flood H. M., Ren X., Jagannathan S., Barski A., Kalin T. V., et al. (2017). FOXF1 transcription factor promotes lung regeneration after partial pneumonectomy. Sci. Rep. 7, 10690. doi:10.1038/s41598-017-11175-3
Bolte C., Herring B. P., Ren X., Tomley T., Ustiyan V., Pradhan A., et al. (2015). Forkhead box F2 regulation of platelet-derived growth factor and myocardin/serum response factor signaling is essential for intestinal development. J. Biol. Chem. 290, 7563–7575. doi:10.1074/jbc.M114.609487
Bolte C., Ustiyan V., Ren X., Dunn A. W., Pradhan A., Wang G., et al. (2020). Nanoparticle delivery of proangiogenic transcription factors into the neonatal circulation inhibits alveolar simplification caused by hyperoxia. Am. J. Respir. Crit. Care Med. 202 (1), 100–111. doi:10.1164/rccm.201906-1232OC
Bray N. L., Pimentel H., Melsted P., Pachter L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. doi:10.1038/nbt.3519
Cai Y., Bolte C., Le T., Goda C., Xu Y., Kalin T. V., et al. (2016). FOXF1 maintains endothelial barrier function and prevents edema after lung injury. Sci. Signal. 9, ra40. doi:10.1126/scisignal.aad1899
Cantelmo A. R., Conradi L.-C., Brajic A., Goveia J., Kalucka J., Pircher A., et al. (2016). Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell. 30, 968–985. doi:10.1016/j.ccell.2016.10.006
Carbone R., Bossone E., Bottino G., Monselise A., Rubenfire M. (2005). Secondary pulmonary hypertension – diagnosis and management. Eur. Rev. Med. Pharmacol. Sci. 9, 331–342.
Choi S.-H., Hong Z.-Y., Nam J.-K., Lee H.-J., Jang J., Yoo R. J., et al. (2015). A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin. Cancer Res. 21, 3716–3726. doi:10.1158/1078-0432.CCR-14-3193
Claxton S., Kostourou V., Jadeja S., Chambon P., Hodivala-Dilke K., Fruttiger M. (2008). Efficient, inducible cre-recombinase activation in vascular endothelium. Genesis 46, 74–80. doi:10.1002/dvg.20367
Dai Z.-J., Gao J., Ma X.-B., Yan K., Liu X.-X., Kang H.-F., et al. (2012). Up-regulation of hypoxia inducible factor-1a by cobalt chloride correlates with proliferation and apoptosis in PC-2 cells. J. Exp. Clin. Cancer Res. 31, 28. doi:10.1186/1756-9966-31-28
Eltzschig H. K., Abdulla P., Hoffman E., Hamilton K. E., Daniels D., Schönfeld C., et al. (2005). HIF-1–dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J. Exp. Med. 202, 1493–1505. doi:10.1084/jem.20050177
Flaherty K. R., Andrei A.-C., Murray S., Fraley C., Colby T. V., Travis W. D., et al. (2006). Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am. J. Respir. Crit. Care Med. 174, 803–809. doi:10.1164/rccm.200604-488OC
Gale N. W., Yancopoulos G. D. (1999). Growth factors acting via endothelial cell-specific receptor tyrosine kinases: VEGFs, Angiopoietins, and ephrins in vascular development. Genes. Dev. 13, 1055–1066. doi:10.1101/gad.13.9.1055
Jiang X., Tian W., Tu A. B., Pasupneti S., Shuffle E., Dahms P., et al. (2019). Endothelial hypoxia-inducible factor-2α is required for the maintenance of airway microvasculature. Circulation 139, 502–517. doi:10.1161/CIRCULATIONAHA.118.036157
Jr M. A. G., Aster R. H., Cotran R. S., Corkery J., Jandl J. H., Folkman J. (1969). Preservation of vascular integrity in organs perfused in vitro with a platelet-rich medium. Nature 222, 33–36. doi:10.1038/222033a0
Kalin T. V., Meliton L., Meliton A. Y., Zhu X., Whitsett J. A., Kalinichenko V. V. (2008). Pulmonary mastocytosis and enhanced lung inflammation in mice heterozygous null for the Foxf1 gene. Am. J. Respir. Cell. Mol. Biol. 39, 390–399. doi:10.1165/rcmb.2008-0044OC
Kalinichenko V. V., Bhattacharyya D., Zhou Y., Gusarova G. A., Kim W., Shin B., et al. (2003). Foxf1 +/- mice exhibit defective stellate cell activation and abnormal liver regeneration following CCl4 injury. Hepatology 37, 107–117. doi:10.1053/jhep.2003.50005
Kalinichenko V. V., Zhou Y., Bhattacharyya D., Kim W., Shin B., Bambal K., et al. (2002a). Haploinsufficiency of the mouse forkhead box f1 gene causes defects in gall bladder development. J. Biol. Chem. 277, 12369–12374. doi:10.1074/jbc.M112162200
Kalinichenko V. V., Zhou Y., Shin B., Stolz D. B., Watkins S. C., Whitsett J. A., et al. (2002b). Wild-type levels of the mouse Forkhead Box f1 gene are essential for lung repair. Am. J. Physiol. Lung Cell. Mol. Physiol. 282, L1253–L1265. doi:10.1152/ajplung.00463.2001
Kalucka J., Rooij L. P. M. H. d., Goveia J., Rohlenova K., Dumas S. b. J., Meta E., et al. (2020). Single-cell transcriptome atlas of murine endothelial cells. Cell. Rep. 180, 764–779. doi:10.1016/j.cell.2020.01.015
Kolesnichenko O. A., Flood H. M., Zhang Y., Ustiyan V., Jimenez H. K. C., Kalin T. V., et al. (2023). Endothelial progenitor cells derived from embryonic stem cells prevent alveolar simplification in a murine model of bronchopulmonary dysplasia. Front. Cell. Dev. Biol. 11, 1209518. doi:10.3389/fcell.2023.1209518
Love M. I., Huber W., Anders a. S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–571. doi:10.1186/s13059-014-0550-8
Mahlapuu M., Enerbäck S., Carlsson P. (2001a). Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development 128, 2397–2406. doi:10.1242/dev.128.12.2397
Mahlapuu M., Ormestad M., Enerbäck S., Carlsson P. (2001b). The forkhead transcription factor Foxf1 is required for differentiation of extraembryonic and lateral plate mesoderm. Development 128, 155–166. doi:10.1242/dev.128.2.155
Manalo D. J., Rowan A., Lavoie T., Natarajan L., Kelly B. D., Ye S. Q., et al. (2005). Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105, 659–669. doi:10.1182/blood-2004-07-2958
Mehta D., Malik A. B. (2006). Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 86, 279–367. doi:10.1152/physrev.00012.2005
Milewski D., Pradhan A., Wang X., Cai Y., Turpin B., Kalinichenko V., et al. (2017). FoxF1 and FoxF2 transcription factors synergistically promote rhabdomyosarcoma carcinogenesis by repressing transcription of p21Cip1 CDK inhibitor. Oncogene 36, 850–862. doi:10.1038/onc.2016.254
Mizuno S., Bogaard H. J., Voelkel N. F., Umeda Y., Kadowaki M., Ameshima S., et al. (2009). Hypoxia regulates human lung fibroblast proliferation via p53-dependent and -independent pathways. Respir. Res. 10, 17. doi:10.1186/1465-9921-10-17
Morote-Garcia J. C., Rosenberger P., Kuhlicke J., Eltzschig H. K. (2008). HIF-1–dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood 111, 5571–5580. doi:10.1182/blood-2007-11-126763
Muñoz-Sánchez J., Chánez-Cárdenas M. E. (2018). The use of cobalt chloride as a chemical hypoxia model. J. Appl. Toxicol. 39, 556–570. doi:10.1002/jat.3749
Naeije R. (2005). Pulmonary hypertension and right heart failure in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2, 20–22. doi:10.1513/pats.200407-037MS
Narravula S., Colgan S. P. (2001). Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor alpha expression during hypoxia. J. Immunol. 166, 7543–7548. doi:10.4049/jimmunol.166.12.7543
Paik D. T., Tian L., Williams I. M., Rhee S., Zhang H., Liu C., et al. (2020). Single-cell RNA sequencing unveils unique transcriptomic signatures of organ-specific endothelial cells. Circulation 142, 1848–1862. doi:10.1161/CIRCULATIONAHA.119.041433
Pradhan A., Dunn A., Ustiyan V., Bolte C., Wang G., Whitsett J. A., et al. (2019). The S52F FOXF1 mutation inhibits STAT3 signaling and causes alveolar capillary dysplasia. Am. J. Respir. Crit. Care Med. 200 (8), 1045–1056. doi:10.1164/rccm.201810-1897OC
Ranieri V. M., Rubenfeld G. D., Thompson B. T., Ferguson N. D., Caldwell E., Fan E., et al. (2012). Acute respiratory distress syndrome: the Berlin Definition. J. Am. Med. Assoc. 307, 2526–2533. doi:10.1001/jama.2012.5669
Ren X., Ustiyan V., Guo M., Wang G., Bolte C., Zhang Y., et al. (2019). Postnatal alveologenesis depends on FOXF1 signaling in c-kit+ endothelial progenitor cells. Am. J. Respir. Crit. Care Med. 200 (9), 1164–1176. doi:10.1164/rccm.201812-2312OC
Ren X., Ustiyan V., Pradhan A., Cai Y., Havrilak J. A., Bolte C. S., et al. (2014). FOXF1 transcription factor is required for formation of embryonic vasculature by regulating VEGF signaling in endothelial cells. Circulation Res. 115, 709–720. doi:10.1161/CIRCRESAHA.115.304382
Rhim T., Lee D. Y., Lee M. (2013). Hypoxia as a target for tissue specific gene therapy. J. Control. Release 172, 484–494. doi:10.1016/j.jconrel.2013.05.021
Semenza G. L. (2011). Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 365, 537–547. doi:10.1056/NEJMra1011165
Senavirathna L. K., Huang C., Yang X., Munteanu M. C., Sathiaseelan R., Xu D., et al. (2018). Hypoxia induces pulmonary fibroblast proliferation through NFAT signaling. Sci. Rep. 8, 2709. doi:10.1038/s41598-018-21073-x
Shimoda L. A., Semenza G. L. (2011). HIF and the lung: role of hypoxia-inducible factors in pulmonary development and disease. Am. J. Respir. Crit. Care Med. 183, 152–156. doi:10.1164/rccm.201009-1393PP
Stacher E., Graham B. B., Hunt J. M., Gandjeva A., Groshong S. D., McLaughlin V. V., et al. (2012). Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 186, 261–272. doi:10.1164/rccm.201201-0164OC
Stearman R. S., Bui Q. M., Speyer G., Handen A., Cornelius A. R., Graham B. B., et al. (2018). Systems analysis of the human pulmonary arterial hypertension lung transcriptome. Am. J. Respir. Cell. Mol. Biol. 60, 637–649. doi:10.1165/rcmb.2018-0368OC
Stenmark K. R., Abman S. H. (2005). Lung vascular development: implications for the pathogenesis ofBronchopulmonary dysplasia. Annu. Rev. Physiology 67, 623–661. doi:10.1146/annurev.physiol.67.040403.102229
Strange C., Highland K. B. (2005). Pulmonary hypertension in interstitial lung disease. Curr. Opin. Pulm. Med. 11, 452–455. doi:10.1097/01.mcp.0000174250.38188.6d
Sun F., Wang G., Pradhan A., Xu K., Gomez-Arroyo J., Zhang Y., et al. (2021). Nanoparticle delivery of STAT3 alleviates pulmonary hypertension in a mouse model of alveolar capillary dysplasia. Circulation 144, 539–555. doi:10.1161/CIRCULATIONAHA.121.053980
Tzouvelekis A., Harokopos V., Paparountas T., Oikonomou N., Chatziioannou A., Vilaras G., et al. (2007). Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis. Am. J. Respir. Crit. Care Med. 176, 1108–1119. doi:10.1164/rccm.200705-683OC
Vriend J., Reiter R. J. (2016). Melatonin and the von Hippel–Lindau/HIF-1 oxygen sensing mechanism: a review. Biochimica Biophysica Acta 1865, 176–183. doi:10.1016/j.bbcan.2016.02.004
Wang G., Wen B., Deng Z., Zhang Y., Kolesnichenko O. A., Ustiyan V., et al. (2022). Endothelial progenitor cells stimulate neonatal lung angiogenesis through FOXF1-mediated activation of BMP9/ACVRL1 signaling. Nat. Commun. 13, 2080. doi:10.1038/s41467-022-29746-y
Wang G., Wen B., Ren X., Li E., Zhang Y., Guo M., et al. (2021) Generation of pulmonary endothelial progenitor cells for cell-based
Wen B., Li E., Ustiyan V., Wang G., Guo M., Na C.-L., et al. (2021). In vivo generation of lung and thyroid tissues from embryonic stem cells using blastocyst complementation. Am. J. Respir. Crit. Care Med. 203 (4), 471–483. doi:10.1164/rccm.201909-1836OC
Wen B., Ren X., Zhang Y., Guo M., et al. (2022). Generation of pulmonary endothelial progenitor cells for cell-based therapy using interspecies mouse-rat chimeras. Am. J. Respir. Crit. Care Med. 204, 326–338. doi:10.1164/rccm.202003-0758OC
Weng T., Poth J. M., Karmouty-Quintana H., Garcia-Morales L. J., Melicoff E., Luo F., et al. (2014). Hypoxia-induced deoxycytidine kinase contributes to epithelial proliferation in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 190, 1402–1412. doi:10.1164/rccm.201404-0744OC
Wigley F. M., Lima J. A. C., Mayes M., McLain D., Chapin J. L., Ward-Able C. (2005). The prevalence of undiagnosed pulmonary arterial hypertension in subjects with connective tissue disease at the secondary health Care level of community-based rheumatologists (the UNCOVER study). Am. Coll. Rheumatology 52, 2125–2132. doi:10.1002/art.21131
Keywords: lung, endothelial cells, hypoxia, FOXF1, HIF-1α
Citation: Acharya A, Bian F, Gomez-Arroyo J, Wagner KA, Kalinichenko VV and Kalin TV (2024) Hypoxia represses FOXF1 in lung endothelial cells through HIF-1α. Front. Physiol. 14:1309155. doi: 10.3389/fphys.2023.1309155
Received: 07 October 2023; Accepted: 28 December 2023;
Published: 11 January 2024.
Edited by:
Yu Ru Kou, Hualien Tzu Chi Hospital, TaiwanReviewed by:
Sanda Predescu, Rush University Medical Center, United StatesP. Padmini S. J. Khedoe, Leiden University Medical Center (LUMC), Netherlands
Tzong-Shyuan Lee, National Taiwan University, Taiwan
Copyright © 2024 Acharya, Bian, Gomez-Arroyo, Wagner, Kalinichenko and Kalin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tanya V. Kalin, tatianakalin@arizona.edu