 Diwen Ye
Diwen Ye Yongjiao Zhang1,2
Yongjiao Zhang1,2 Sumei Lu
Sumei Lu- 1Department of Laboratory Medicine, The First Affiliated Hospital of Shandong First Medical University, Jinan, China
- 2School of Medical Laboratory, Weifang Medical University, Weifang, Shandong, China
Metabolic syndromes are characterized by various complications caused by disrupted glucose and lipid metabolism, which are major factors affecting the health of a population. However, existing diagnostic and treatment strategies have limitations, such as the lack of early diagnostic and therapeutic approaches, variability in patient responses to treatment, and cost-effectiveness. Therefore, developing alternative solutions for metabolic syndromes is crucial. N6-methyladenosine (m6A) is one of the most abundant modifications that determine the fate of RNA. m6A modifications are closely associated with metabolic syndrome development and present novel prospects for clinical applications. Aberrant m6A modifications have been detected during inflammatory infiltration, apoptosis, autophagy, iron sagging, necrosis, and scorching during metabolic syndrome pathogenesis and progression. However, few reviews have systematically described the correlation between m6A modifications and these factors concerning metabolic syndrome pathogenesis and progression. This study summarizes the m6A methylation regulators and their roles in metabolic syndrome development, highlighting the potential of m6A modification as a biomarker in metabolic disorders.
Highlights
1. m6A RNA regulators are closely associated with metabolic syndrome development and present novel prospects for clinical applications.
2. m6A modification-induced inflammatory responses may potentially be valuable in metabolic syndromes.
3. Programmed cell death controlled by m6A regulates metabolic syndromes.
1 Introduction
Metabolic sydromes are a group of disorders caused by glucolipid metabolism dysregulation, with symptoms such as obesity, hyperglycemia, hyperlipidemia, and nonalcoholic fatty liver severely affecting physical health. Furthermore, these symptoms are interrelated metabolic risk factors directly contributing to atherosclerotic cardiovascular disease development and increasing the risk of type 2 diabetes mellitus (T2DM) and its complications (Wang et al., 2015; Shi et al., 2017). Recently, research into the pathogenic mechanisms of metabolic sydromes has increasingly focused on excessive lipid accumulation, inflammatory responses, autophagy, apoptosis and epigenetic modifications (Shi et al., 2017; Huang et al., 2018; Wu et al., 2020).
Several epigenetic modifications exist, including deoxyribonucleic acid (DNA) methylation, histone modifications, messenger RNA (mRNA), and noncoding RNA chemical modifications. Methylated RNA is reportedly functional in pathophysiological processes at all stages of life. Adenylate methylation modifications account for over half of the total methylated ribonucleotides in cellular RNA and 0.1%–0.4% of all adenosines (Goldberg et al., 2007). N6-methyladenosine (m6A), a widespread trans-epigenetic adenylate modification, has become relevant to the specific mechanisms of metabolic sydrome pathogenesis (Li et al., 2017; Wu et al., 2020). m6A modification is a dynamic and reversible biological progress owing to the interaction between “writers” and “erasers.” The “writers” mean methyltransferase, such as methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14). The “erasers” mean demethylase, such as fat mass and obesity-associated protein (FTO) (Jia et al., 2011) and ALKB homolog 5 (ALKBH5). Also, “readers” are the third critical factor which recognizes m6A modifications and ultimately paly function.
m6A primarily controls post-transcriptional gene expression and is involved in DNA repair, cellular reprogramming, cell differentiation, cellular stress responses, and programmed cell death (PCD) (Wu et al., 2020). In addition, m6A is highly conserved in the 3′untranslated region (UTR) and the consensus motif RRACH in the coding region (Desrosiers et al., 1974; Wu et al., 2020). m6A methylation regulates almost all aspects of mRNA metabolism through “readers,” including nuclear export (Roundtree et al., 2017a; Roundtree et al., 2017b), stability (Wang et al., 2015), translation (Huang et al., 2018), and pre-mRNA processing to mRNA decay (Li et al., 2017; Shi et al., 2017). As reported, m6A methylation has been a hot research field in metabolic sydromes. In high-fat diet-induced fatty liver in mice, m6A hyper-methylated genes were significantly enriched in processes and pathways associated with lipid metabolism, such as fatty acid synthesis, triglyceride metabolism, and the PPAR signaling pathways (Zhong et al., 2020). m6A methylation controls the insulin IGF1-AKT-PDX1 pathway and T2DM is exacerbated by targeting METTL3 or METTL14 to reduce m6A levels and decrease AKT phosphorylation and PDX1 protein levels (De Jesus et al., 2019). YTHDF2 accelerates JAK2 mRNA degradation and attenuates JAK2-STAT3-C/EBPβ signaling. In our previous studies, we found that m6A ultimately contributes to the development of NAFLD and insulin resistance by regulating the expression of CYP2B6 (Li et al., 2023). Existing literature indicated the potential value of m6A methylation as novel targets in metabolic sydromes treatment.
In the present study, we review the relationship between m6A and metabolic syndromes and summarize the role of RNA m6A modification in metabolic syndrome pathogenesis and progression. We first introduce the role of m6A methylation “writers,” “erasers,” and “readers,” then the association of m6A methylation modification with inflammatory response, PCD, and other essential biological functions in metabolic syndromes. For the literature review, we searched the PubMed database of the NCBI Homepage using the keywords “m6A and inflammation”, “m6A and autophagy”, “m6A and programmed cell death,” and “m6A and metabolic syndrome” to filter the key publications related to metabolic syndromes and categorize and extract the content of interest. This study can provide evidence for the clinical application of m6A methylation in preventing and treating metabolic syndromes.
2 m6A-related component modulation
We summarized the components of m6A methylation process in Figure 1. The primary molecules involved in m6A are methyltransferases that methylate RNA, called the “writers”; demethylases that remove m6A, called the “erasers”; and “readers” which recognize m6A modifications and ultimately play functions. Thus, m6A modification is dynamically and reversibly regulated at various cellular stages (Roundtree et al., 2017b; Zaccara et al., 2019). Methyltransferase is a multi-component protein primarily comprising methyltransferase-like 3 (METTL3), −14 (METTL14), and Wilms tumor 1-associated protein (WTAP). KIAA1429, RBM15, RBM15B, ZC3H13, and CBLL1. m6A demethylation is primarily achieved by two enzymes: fat mass and obesity-associated protein (FTO) (Jia et al., 2011) and ALKB homolog 5 (ALKBH5) (Zhang et al., 2017). Generally, m6A “readers” are divided into three classes: the YTH family proteins (YTHDF1/2/3, YTHDC1/2), m6A conversion proteins (hnRNPC, hnRNPG, and hnRNPA2B1), and RNA-binding proteins (IGF2BP 1/2/3 and hnRNPA2B1) (Zaccara et al., 2019). Additionally, eIF3 and ELAVL1 are reported as “readers.” Several other m6A RNA binding proteins have been identified, such as LRPPRC, FMR1, FMRP, and SND1 (Chen et al., 2011; Darnell et al., 2011; Arguello et al., 2017).
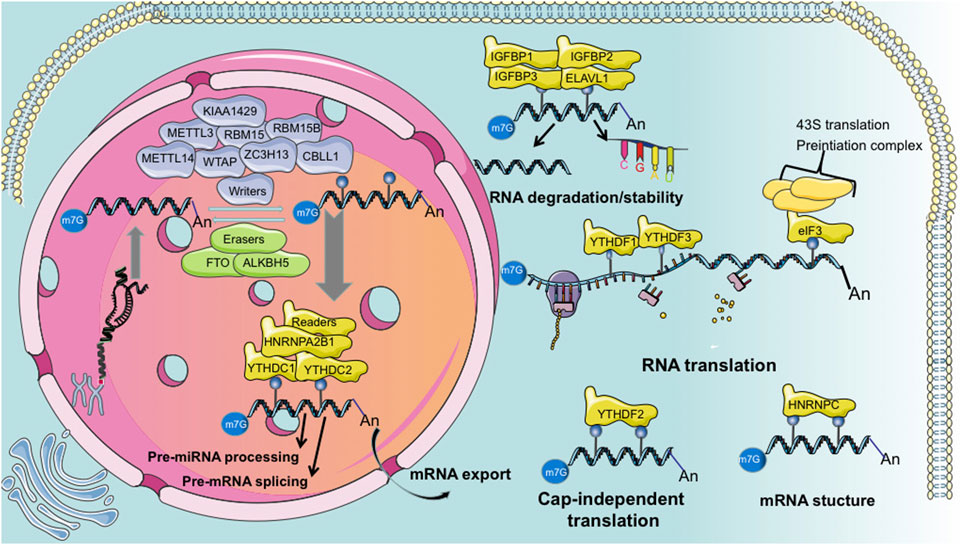
Figure 1. Summarized schematic of “writer,” “eraser,” and “reader” functions during m6A modification.
3 Association of m6A with the inflammatory responses in metabolic syndromes
Metabolic syndromes are primarily associated with inflammatory responses during their development. For example, Day and James proposed the second-strike theory of nonalcoholic fatty liver disease (NAFLD). The “first strike” triggers a metabolic derailment of the mitochondria, endoplasmic reticulum stress, and hepatocyte peroxisomes. Reactive oxygen species (ROS) and further lipid peroxidation, cytokine production, and lipid accumulation promote inflammation and fibrosis. In atherosclerosis pathogenesis, oxidized low-density lipoprotein (ox-LDL) is absorbed by endothelial macrophages to form foam cells, causing inflammatory lesions in the endothelium and leading to atherosclerosis. Diabetes mellitus pathogenesis also involves the interleukin (IL)-1β inflammatory factor that damages islet cells and causes insufficient insulin secretion. Therefore, we focus on the impact of m6A methylation on inflammatory-related responses in metabolic syndromes.
3.1 Role of m6A on lipogenesis
The immune response induced by chronic inflammation is involved in T2DM, NAFLD, and atherosclerosis progression (Yvan-Charvet et al., 2019; Qin et al., 2021). Guo’s study on m6A methylation transcriptional profiles between the normal state and acute inflammation induced by lipopolysaccharide (LPS) in chicken liver potentially identified how m6A regulates acute inflammation and abnormal lipid metabolism (Guo et al., 2022). Similarly, METTL3 influences the uptake of long-chain fatty acids in the intestinal epithelium by affecting the inflammatory response induced by tumor necrosis factor receptor-associated factor 6 (Zong et al., 2019).
Macrophage-related research has also identified the regulatory functions of m6A modifications in inflammation. For example, lipid deposition causes atherosclerosis and NAFLD, which can be attributed to the metabolic reprogramming induced by macrophages due to the inflammatory response. Macrophages absorb and metabolize excess ox-LDL, producing esterified cholesterol in the cytoplasm and producing foam cells (Yvan-Charvet et al., 2019). Macrophage scavenger receptor 1 (MSR1) and cluster determinant 36 (CD36) are highly expressed on macrophage surfaces. These are the primary receptors for the binding, uptake, and removal of cholesterol. Ox-LDL induces dead box protein 5 (DDX5) expression, promoting MSR1 expression in macrophages and inhibiting the methyltransferase METTL3 activity in MSR1 and CD36 knockout mice. Subsequently, MSR1 mRNA stability is enhanced, and lipid uptake is promoted (Kunjathoor et al., 2002; Yvan-Charvet et al., 2019).
In addition, lipogenesis-related gene mRNAs undergo m6A methylation. Activating the transcription factor sterol regulatory element binding protein-1C and lipid-responsive nuclear receptor liver X receptor through m6A in NAFLD animal models results in lipid accumulation in the liver (Salisbury et al., 2021). This lipid accumulation exacerbates NAFLD progression to nonalcoholic steatohepatitis (NASH) (Salisbury et al., 2021). m6A modification stabilizes ATP citrate lyase and stearoyl-CoA desaturase one mRNA, increasing protein expression in a NAFLD DM2 mouse model (Yang et al., 2022) (Figure 2). After that, these mRNAs exacerbate fat acid synthesis and lipid accumulation, leading to excessive compensatory cell proliferation in the liver and NAFLD and hepatocellular cancer progression (Yang et al., 2022). The m6A reader YTHDC2 is also critical for regulating hepatic adipogenesis and triglyceride homeostasis in NAFLD and NASH, which may provide a potential target to treat obesity-associated NAFLD (Zhou et al., 2021a). It has also been shown that Mettl3-IGFBP2-mediated changes in m6A levels increase HDAC1 mRNA stability to affect FGF21 expression, leading to liver injury and insulin resistance caused by hepatic steatosis and ultimately the development of metabolic syndrome (Chen et al., 2023).
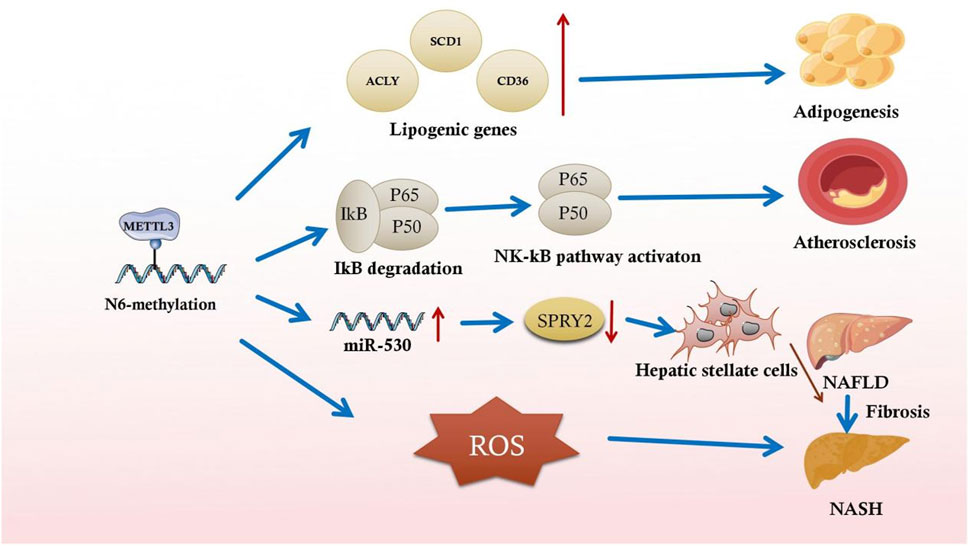
Figure 2. Partial illustration of the association between m6A methylation and inflammatory responses in metabolic syndromes.
These studies demonstrate the critical role of the m6A-mediated inflammatory response in lipogenesis, indicating the potential effects (28) of m6A modification on abnormal lipid metabolism.
3.2 Role of m6A on fibrosis
Recent studies have indicated the underlying function of m6A-mediated inflammation in fibrosis. Serum LPS concentrations are increased in high-fat diet-induced rats with NASH, and global m6A methylation upregulation is closely associated with this increase. LPS-activated Kupffer cells aggravate NAFLD progression to NASH and exacerbate fibrosis in chronically inflamed liver (Yang et al., 2022). In the early stages of liver fibrosis, the local inflammatory response activates acid-sensing ion channel 1a (ASIC1a), leading to stellate cell activation and proliferation, aggravating liver fibrosis (Feng et al., 2021). By altering the specific RNA m6A methylation mechanism, ASIC1a regulates miR-350 through METTL3-dependent m6A modification. By increasing miR-350 levels, ASIC1a suppresses the target gene sprouty receptor tyrosine kinase signal antagonist two expression, further mediating PI3K/AKT and ERK pathway activation and facilitating hepatic stellate cell activation (Figure 2) (Zhu et al., 2020).
Additionally, the immune response and apoptosis are critical factors in liver fibrosis regression. m6A methylation regulates oxidative stress and cytoplasmic metabolism in liver fibrosis mouse models related to hepatocyte immune responses and apoptosis, respectively (Cui et al., 2020). Increased m6A in RNA was also observed in the failing hearts of humans, pigs, and mice. In failing mammalian hearts and hypoxic cardiomyocytes, increased m6A levels reduce FTO levels. FTO overexpression decreases cardiac fibrosis in mice with myocardial infarction and demethylates the contractile transcript SERCA2a (Mathiyalagan et al., 2019). Cardiac fibroblast activation parallels high METTL3 expression, whereas silencing METTL3 substantially attenuates cardiac fibrosis and reduces collagen-related gene expression (Li et al., 2021a). Similarly, METTL3 deletion restricts the conversion of lung fibroblasts to myofibroblasts in vitro and in vivo; however, the regulatory mechanism was not related to m6A modification.
Therefore, further research on m6A modification-related inflammation and fibrosis is required. A better understanding of this topic will provide new insights into fibrotic disease treatment.
3.3 Role of m6A on metabolic-related cell signaling pathways
m6A modification influences the inflammatory state at the cellular level through signaling pathways in an established macrophage polarization system using RAW264.7 macrophages and bone-marrow-derived macrophages (BMDMs) (Liu et al., 2019a; Gu et al., 2020). Macrophages are the primary cell types involved in inflammation. METTL3 knockdown suppresses inflammatory cytokine production and expression of various genes, primarily by altering the phosphorylation levels of relevant signaling pathways in BMDMs (Liu et al., 2019b). For example, in atherosclerosis, myeloid differentiation factor 88 (My88) expression is induced by LPS METTL14 upregulates the My88 phosphorylation level and exacerbates vascular endothelial inflammation, suggesting that METTL14-related m6A is involved in atherosclerotic inflammation (Zheng et al., 2022). METTL3 potentially exerts a pro-inflammatory effect through m6A-derived macrophage polarization to the pro-inflammatory M1 type (Liu et al., 2019b; Wang et al., 2019). Nevertheless, in LPS-induced macrophages, METTL3 upregulation substantially attenuated NF-κB signaling pathway-dependent inflammatory responses in METTL3 knockout mice (Wang et al., 2019; Yu et al., 2019). Additionally, inhibiting demethylases affected the phosphorylation of essential proteins, including IKK α/β, IκBα, and p65, in the NF-κB signaling pathway. Demethylase inhibition also decreases STAT1 and PPAR-γ mRNA stability and thus hinders macrophage polarization (Gu et al., 2020). In contrast, METTL3 increases critical transcription factor expression, such as STAT1, to initiate pro-inflammatory macrophage polarization and induce M1 macrophages in various inflammatory diseases (Liu et al., 2019b).
Regarding metabolic disorders, m6A modification-regulated inflammation-related signaling pathways have been elucidated. METTL3 was highly expressed in type 1 and 2 diabetic nephropathy. Increased m6A levels further enhance TIMP2 stability through an IGF2BP2-dependent mechanism, leading to cellular inflammation and apoptosis via Notch3/4 pathway activation (Jiang et al., 2022). Furthermore, m6A activates macrophages and reprograms cellular metabolism during NAFLD progression. METTL3-mediated m6A in Kupffer cells downregulates DNA damage-inducible transcript 4, leading to NF-κB pathway activation and upregulation of inflammation in the liver. This inflammation ultimately increases lipogenesis and obesity and facilitates the inflammatory progression of NAFLD (Qin et al., 2021).
In summary, the m6A-mediated inflammatory response to cell signaling pathways is critical in metabolic syndromes. However, the exact effect of m6A on inflammation in various metabolic syndromes remains unclear. Therefore, further studies are required to better elucidate the m6A-related pathogenesis of metabolic syndromes.
4 m6A regulates metabolic syndromes through PCD
PCD, in addition to typical cell death modalities such as autophagy, ferroptosis, necrosis, and thermoproteolysis, is a programmed mechanism that eliminates abnormal cells to maintain the balance of the internal environment. Autophagy is an evolutionarily conserved degradation pathway that is primarily free of unnecessary and senescent organelles and proteins and is tightly regulated by autophagy-associated proteins and transcription factors. Autophagic dysregulation is associated with many diseases, such as neurodegenerative syndromes, cardiovascular diseases, and cancer, owing to its several cytoplasmic targets. Furthermore, autophagy is a normal cell function in metabolic syndrome development and progression. Therefore, we focus on the impact of m6A methylation in PCD in metabolic syndromes.
4.1 Association of m6A methylation and autophagy
Several autophagy-related genes are involved in the mRNA processing of m6A modifications. Post-transcriptional ATG1 and ATG5/ATG7 regulation can be altered by m6A modification, thereby inhibiting autophagy (Figure 3) (Wang et al., 2020). Furthermore, many studies have demonstrated the role of m6A modification in autophagic machinery. Recent data suggest that m6A is critical for regulating autophagy (Li et al., 2020). Some m6A modifications inhibit autophagy directly (Song et al., 2019), which may also affect autophagosome formation, thereby dysregulating autophagy in Ftoflox/flox and Fabp4-Cre transgenic mice (Wang et al., 2020). m6A modifications may also promote autophagy initiation (Feng et al., 2021). Moreover, the effect of m6A modification on autophagy is related to the disease. As m6A modification and autophagy play critical roles in regulating health status, a better understanding of this topic is crucial to developing therapeutic strategies.
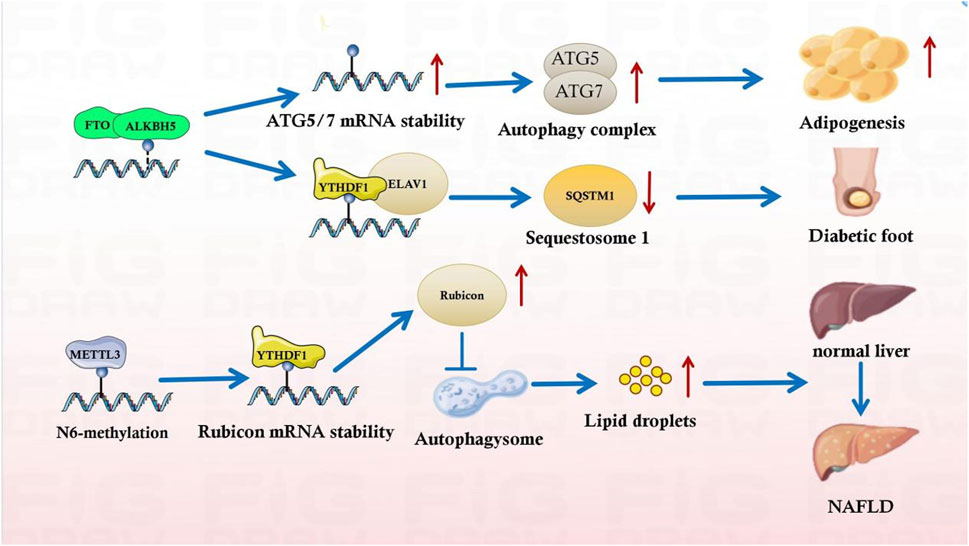
Figure 3. m6A influences the metabolic syndrome progression by regulating autophagy.
m6A harmonizes metabolic syndrome progression by affecting autophagy. METTL3 inhibition promoted hepatic autophagy and lipid droplet (LD) clearance. METTL3 directly mediate m6A modification of Rubicon mRNA, thereby promoting Rubicon mRNA stability. Consequently, Rubicon reduces autophagosome-lysosome fusion, further impairing LDs clearance and leading to liver lipid deposition, hepatic steatosis, and progression to NASH (Peng et al., 2022). Similarly, in a glioblastoma (GBM) stem cells (GSCs) related work, platelet-derived growth factor (PDGF) ligand stimulate early growth response 1 (EGR1) transcription to induce METTL3 to promote GSC proliferation and self-renewal. Targeting the PDGF-METTL3 axis inhibits mitophagy by regulating m6A modification of optineurin (OPTN) (Lv et al., 2022). Also, the adriamycin (ADR) mechanism in diabetic nephropathy treatment is related to m6A-regulated autophagy in ADR nephropathy in adult male C57BL/6 J mice (Lu et al., 2021). METTL14 downregulation reduces SIRT1 degradation in the presence of ADR. SIRT1 upregulates autophagy in podocytes; this upregulation initiates a stress-protective mechanism in podocytes, effectively alleviating podocyte damage and delaying diabetic nephropathy progression in METTL14 knockout mice (Lu et al., 2021). m6A-binding proteins also perform separate functions in regulating autophagy. In a high-glucose-induced diabetic retinopathy model, circFA T1 overexpression upregulated autophagy in retinal pigment epithelium (RPE) cells. By binding to the m6A reader YTHDF2, circFA T1 alleviates diabetic retinopathy (Huang et al., 2022). Notably, a similar protective mechanism was observed in pancreatic islet β-cells. Hypoxia-inducible factor 1-alpha upregulated ATG5, ATG2A, and ATG14 in a YTHDF1-dependent manner, triggering protective autophagy and ameliorating hypoxia-induced cytotoxicity (Fang et al., 2022). Therefore, m6A aggravates metabolic syndromes by slowing down autophagy and cell damage in metabolic-related diseases by initiating cytoprotective autophagy mechanisms.
m6A modification harmonizes metabolic syndromes through the autophagic pathway. A recent study on adipocytes reported that FTO deletion decreased ATG5 and ATG7 expression, the mechanism of which is related to the m6A modification downregulation (Wang et al., 2020). ATG7 undergoes ATG12/ATG5 covalent binding to a ubiquitin-like mechanism. The ATG12-ATG5 homodimer attaches to ATG16L and facilitates the autophagosome extension in BALB/c nude mice and non-small cell lung cancer cell lines PC9 and HCC827 (Liu et al., 2020). FTO deprivation reduces ATG12-ATG5 covalent binding, impedes ternary complex development, and impairs autophagy activation (Wu et al., 2020). Autophagy reduction mediated by FTO and ATG gene expression suggests an association between m6A modification and autophagy. Furthermore, SQSTM1 is a multifunctional protein vital to autophagy (Moscat and Diaz-Meco, 2009). As a critical signaling center, SQSTM1 activates the mechanistic target of rapamycin kinase complex 1, kelch-like ECH-associated protein 1-nuclear factor, and the erythroid 2-like two pathway in addition to selective autophagy (Katsuragi et al., 2015). YTHDC1 cooperatively regulates SQSTM1 expression in keratinocytes. In patients with diabetes, YTHDC1 downregulation induces decreased SQSTM1 expression through accelerated SQSTM1 nuclear mRNA decay, leading to disturbed autophagic flux and keratinocyte migration, thus delaying wound healing (Figure 3) (Moscat and Diaz-Meco, 2009; Katsuragi et al., 2015).
4.2 m6A regulates metabolic syndromes through apoptosis
m6A levels were significantly reduced in the total RNA of mouse islet cells treated with hydrogen peroxide (H2O2) to mimic the ROS environment. H2O2 treatment significantly reduced METTL3 and METTL14 expression, whereas METTL3 interference enhanced cleaved caspase-3 protein and the pro-apoptotic protein Bim expression. This finding suggests that the ROS environment simulated by H2O2 may reduce m6A methylation in pancreatic cells, resulting in apoptosis. Deleting islet cell-specific METTL3 (Mettl3flox/flox, Rip-Cre) is related to cell death and dysfunction, indicating that METTL3 is vital for β-cell survival (Li et al., 2021b). Therefore, METTL3 may be more critical than METTL14 in regulating pancreatic β-cell function. Furthermore, the blood glucose level significantly increased in METTL3 knockout mice (Mettl3flox/flox, Rip-Cre) compared to METTL14 knockout mice (Liu et al., 2019c; Li et al., 2021b). Similarly, the m6A-binding protein YTHDF1 inhibits β-cell apoptosis, suggesting that m6A-related proteins are critical in β-cell death (Fang et al., 2022). Furthermore, myocardial lipid accumulation promotes myocardial apoptosis (Lee et al., 2013). Cleaved caspase-3 protein expression in the myocardium of obese rats increases, indicating increased apoptosis (Sun and Zhang, 2021). FTO mRNA and protein expression are upregulated in obese rats and are associated with disturbed lipid metabolism in LO2 cells (Guo et al., 2013). Furthermore, restricting high-fat foods reduced cleaved caspase-3 protein expression, inhibiting high-fat diet-induced apoptosis in the hearts of obese rats. Similarly, cardiac lipid deposition significantly increases cardiac apoptotic cell death, and dietary intervention reverses these effects. It has been shown that YTHDF2-mediated SIRT3 increases vascular endothelial cell apoptosis and ultimately contributes to the development of diabetic atherosclerosis (Zhang et al., 2023). High glucose-treatment regulates PINK1 expression in response to Mettl3-YTHDF2 in renal tubular epithelial cells, which in turn causes renal tubular epithelial cell apoptosis. This study may reveal that m6A-mediated apoptosis contributes to the pathogenesis of diabetic nephropathy (Wang et al., 2023a).
5 m6A regulated metabolic syndrome through pyroptosis
Inflammatory vesicle-mediated pyroptosis leads to cell death characterized by activating multiple caspases, including caspase-1, in an immortalized mouse podocyte cell-5 line (Liu et al., 2021). m6A is essential in regulating intracellular pyroptosis based on the human RPE cell line ARPE-19 (Zha et al., 2020). Nod-like receptor protein 3 (NLRP3) is a crucial inflammatory vesicle component that causes pyroptosis and increases pro-inflammatory cytokine levels (Yu et al., 2019). High sugar levels induce the increased expression of heat degradation-related proteins (caspase-1, gasdermin D, NLRP3, IL-1β, and IL-18), leading to pyroptosis (Mulay, 2019). These proteins are less expressed when METTL3 is overexpressed and cellular damage is reduced. In contrast, the damage is exacerbated in human peripheral blood mononuclear cell-derived macrophages when METTL3 is knocked down (Guo et al., 2020). A previous study investigated how interferon regulatory factor-1 (IRF-1) facilitates macrophage scorching among patients with acute coronary syndrome, and elevated m6A and METTL3 levels were observed in macrophages (Kunjathoor et al., 2002). IRF-1 overexpression increases m6A and METTL3 levels and promotes acute coronary syndrome (Liu et al., 2019c). According to a study on disc degeneration, METTL14 specifically induces the NLRP3 mRNA m6A modification and increases NLRP3 protein expression in the human osteosarcoma cell line U2OS (Yuan et al., 2021). Therefore, we hypothesized that METTL3/14 plays a joint role in pyroptosis death by regulating NLRP3, thus influencing metabolic syndrome development.
Ferroptosis, a new type of programmed cell death, involves iron-dependent lipid peroxidation and glutathione peroxidase 4, and mitochondrial membrane loss. m6A is vital in ferroptosis as a novel post-transcriptional regulatory mechanism (Shen et al., 2021). YTHDF1 enhanced the BECN1 stability by binding to the BECN1 m6A site and activating hepatic autophagic vesicle formation in primary hepatic stellate cells from ICR mice. Autophagic vesicles trigger ferroptosis in stellate cells and attenuate liver inflammation (Shen et al., 2021). METTL3/ASK1-p38 pathway-induced ferroptosis might be one of the leading causes of diabetic osteoporosis induced by high sugar and a high-fat diet (Lin et al., 2022). m6A modification induces diabetic erectile dysfunction by regulating ferroptosis in penile cells in a diabetes mellitus with erectile dysfunction model in SD rats (Wang et al., 2023b). ALKBH5-mediated m6 A demethylation was reported to lead to the posttranscriptional inhibition of NFE2L2/NRF2, which is crucial for the regulation of antioxidant molecules in cells. Knocking down ALKBH5 subsequently increased the expression of NFE2L2/NRF2 and increased the resistance of HPSCC cells to ferroptosis (Ye et al., 2022). In a diabetic cataract (DC) research, RBM15, WTAP, ALKBH5, FTO, and YTHDF1-were upregulated in DC samples, the mechanism of which is related with the ferroptosis pathway (Cai et al., 2023).
Glucolipotoxicity is toxicity caused by elevated glucose and fatty acid levels, frequently occurring during diabetes development due to hyperglycemia and hyperlipidemia. Autophagy is activated as an adaptive response and provides a possible protective mechanism for β-cells to eliminate damaged mitochondria, unwanted proteins, or both to avoid dysfunction and apoptosis (Meijer and Codogno, 2007). However, glucolipotoxicity induces excessive autophagy in β-cells, leading to cell death. RNA demethylation of critical autophagy pathway genes is an essential factor of β-cells dysfunction and the pathophysiology of diabetes. FTO-m6A demethylation is responsible for NR3C1-stimulated ATG gene expression under glucolipotoxic conditions, triggering excessive autophagy and β-cell death (Wu et al., 2023).
m6A methylation exacerbates metabolic syndrome progression by affecting programmed cell death. In diabetes and its complications, m6A-mediated apoptosis, ferroptosis, and excessive autophagy-induced-cell and osteoblast damage are particularly important. Similarly, markers related to programmed cell death indicate that m6A is a potential emerging target for detecting and treating metabolic syndromes.
To summarize, we included Table 1 to present the m6A-related proteins that play essential roles in modulating metabolic syndrome development. Table 1 also summarizes the target genes related to these proteins. These essential proteins will likely become vital targets for m6A in clinical applications.

Table 1. m6A-related proteins in metabolic disease.
6 Discussion
RNA methylation is one of the most common post-transcriptional modifications. m6A regulates transcription, translocation, splicing, and translation. m6A modifications are commonly detected in biological processes using RNA-seq and methylation RIP-seq (or miCLIP-seq) to explore the role of m6A modifications in metabolic syndromes. m6A modification is reversible using m6A regulators (methylesterases, demethylases, and m6A-RNA binding proteins).
Comparative observations across disease conditions have revealed that the m6A modification levels and their associated regulators vary in T2DM, NAFLD, and atherosclerosis. However, further studies are needed to identify the exact association between m6A modification and the pathogenesis of metabolic syndrome-mediated regulatory factors. In addition, m6A RNA modification is tissue-specific, and whether it is specific to the disease stage remains unclear. Currently, treating metabolic syndromes involves exercise and controlling a high-calorie diet. Although weight-loss surgery effectively slows the progression of metabolic syndromes, it is not widely used because it harms patients. Studying the m6A levels in peripheral blood samples from patients with metabolic syndromes can help identify early biomarkers. Although only confirmed in cellular experiments and animal tests in many studies, these m6A-related markers can become therapeutic alternatives to weight loss surgery in the future. This potential therapeutic alternative can treat patients with metabolic syndromes without the adverse events of surgery.
We summarized the Abbreviation list in Table 2 to better understand the present review. m6A-related proteins, especially METTL3, are critical in regulating m6A in inflammation, autophagy, and cell death. We believe that targeting m6A-related proteins may be ideal for developing metabolic syndrome treatments (Yankova et al., 2021). For example, in cellular and animal studies, STM2457, a small molecule active inhibitor of METTL3, inhibits acute leukemia development caused by elevated m6A levels. Drugs such as these inhibitors may shed new light on treating metabolic syndromes at the epigenetic level. In recent years, some m6A-related clinical trials have been progressively reported (Luo et al., 2020; Cai et al., 2021; Yang et al., 2021). We concluded the information in Table 3. Such as, it has been shown that SNP rs34269950 in the RRACH genome, located in the 3′UTR of RUNX1T1, is significantly regulated by FTO in rural populations in the Caucasus region of Australia, providing important clinical evidence for the association of m6A with metabolic syndrome (Zhou et al., 2021b).
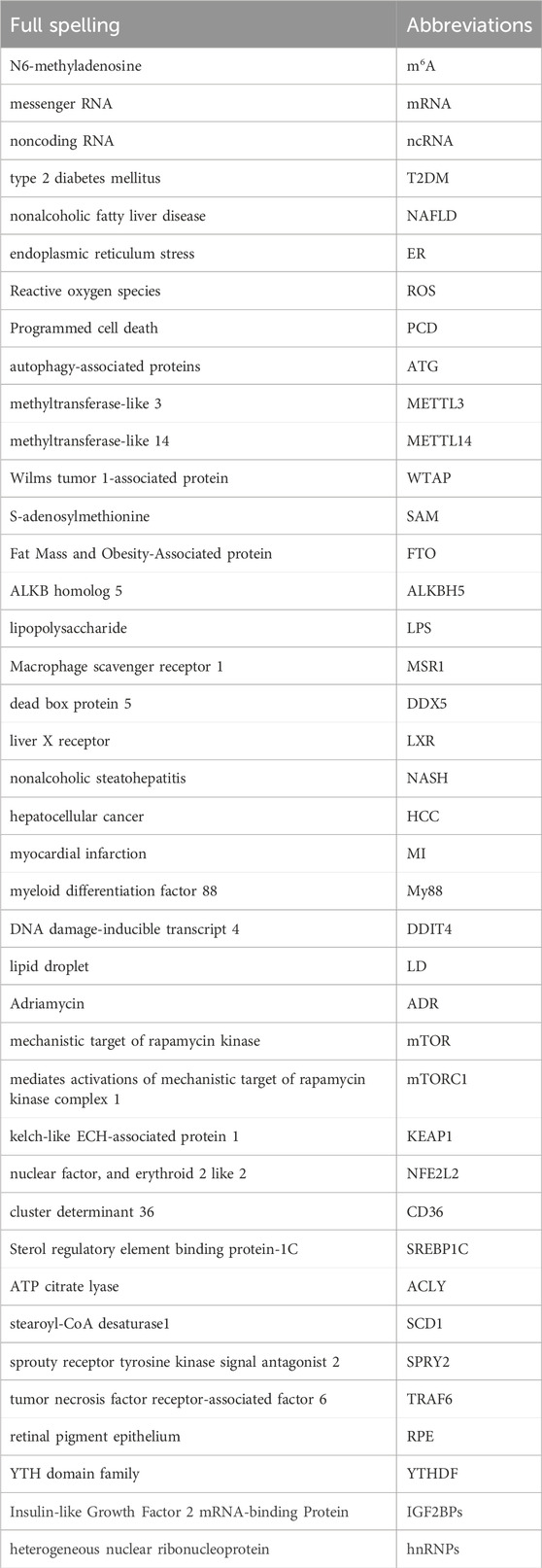
Table 2. Abbreviations.
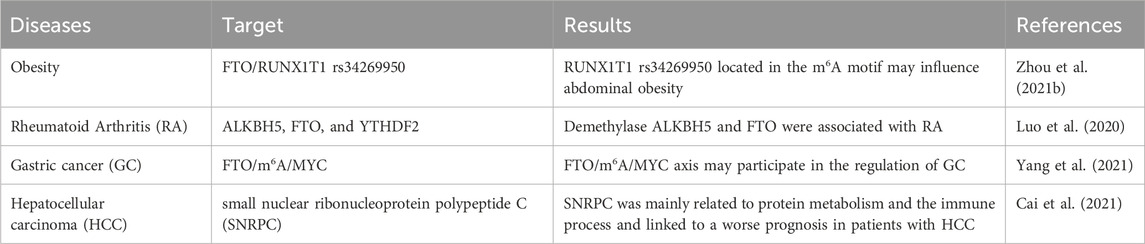
Table 3. Reports of clinical trials.
In conclusion, m6A RNA modification regulates many factors involved in the pathogenesis of metabolic syndromes and may play a vital role in metabolic syndrome development and progression. However, many studies are still needed to answer multiple outstanding questions, such as whether specific inhibitors possess clinical functions in metabolic disorders, how to achieve tissue-specific drug delivery, and whether side effects exist during m6A RNA modification regulation. Clarifying these research questions will provide a new frontier in epigenetic modification and metabolic syndrome research.
7 Conclusion and future direction
In the future, the detection of m6A and its regulatory molecules will also be developed to be more convenient, with a wider range of samples and higher sensitivity, and closer and closer to bedside detection. As basic research on m6A and metabolic sydromes intensifies, more and more basic findings will be confirmed in clinical trials. More convenient detection Methods will facilitate these clinical trials, and clinical trials will provide evidence for drug development targeting m6A regulators. With the maturation of screening technology and the development of artificial intelligence (AI), more and more drugs targeting m6A regulatory molecules will be discovered. The study of Chinese medicinal decoction for m6A regulatory factors will also become a direction for future drug research on m6A-related metabolic sydromes.
Author contributions
DY: Writing–original draft. YZ: Writing–original draft. BZ: Writing–review and editing. JL: Writing–review and editing. TW: Writing–review and editing. SL: Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by Shandong First Medical University (Shandong Academy of Medical Sciences) Youth Science Foundation Funding Program (grant No. 202201-083), the Natural Science Foundation of Shandong Province, China (grant No. ZR2023MH031).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Arguello A. E., DeLiberto A. N., Kleiner R. E. (2017). RNA chemical proteomics reveals the N6-methyladenosine (m6A)-Regulated protein-RNA interactome. J. Am. Chem. Soc. 139 (48), 17249–17252. doi:10.1021/jacs.7b09213
Cai J., Zhou M., Xu J. (2021). N6-methyladenosine (m6A) RNA methylation regulator SNRPC is a prognostic biomarker and is correlated with immunotherapy in hepatocellular carcinoma. World J. Surg. Oncol. 19 (1), 241. doi:10.1186/s12957-021-02354-8
Cai L., Han X.-Y., Li D., Ma D.-M., Shi Y.-M., Lu Y., et al. (2023). Analysis of N6-methyladenosine-modified mRNAs in diabetic cataract. World J. Diabetes 14 (7), 1077–1090. doi:10.4239/wjd.v14.i7.1077
Chen C., Nott T. J., Jin J., Pawson T. (2011). Deciphering arginine methylation: tudor tells the tale. Nat. Rev. Mol. Cell Biol. 12 (10), 629–642. doi:10.1038/nrm3185
Chen Y., Cai K., Du Y., Liu Z., Gong Y. (2023). HDAC1 overexpression promoted by METTL3-IGF2BP2 inhibits FGF21 expression in metabolic syndrome-related liver injury. Biochem. Cell Biol. 101 (1), 52–63. doi:10.1139/bcb-2022-0314
Cui Z., Huang N., Liu L., Li X., Li G., Chen Y., et al. (2020). Dynamic analysis of m6A methylation spectroscopy during progression and reversal of hepatic fibrosis. Epigenomics 12 (19), 1707–1723. doi:10.2217/epi-2019-0365
Darnell J. C., Van Driesche S. J., Zhang C., Hung K. Y. S., Mele A., Fraser C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146 (2), 247–261. doi:10.1016/j.cell.2011.06.013
De Jesus D. F., Zhang Z., Kahraman S., Brown N. K., Chen M., Hu J., et al. (2019). m6A mRNA methylation regulates human β-cell biology in physiological states and in type 2 diabetes. Nat. Metab. 1 (8), 765–774. doi:10.1038/s42255-019-0089-9
Desrosiers R., Friderici K., Rottman F. (1974). Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. U. S. A. 71 (10), 3971–3975. doi:10.1073/pnas.71.10.3971
Fang J., Chen Z., Lai X., Yin W., Guo Y., Zhang W., et al. (2022). Mesenchymal stem cells-derived HIF-1α-overexpressed extracellular vesicles ameliorate hypoxia-induced pancreatic β cell apoptosis and senescence through activating YTHDF1-mediated protective autophagy. Bioorg Chem. 129, 106194. doi:10.1016/j.bioorg.2022.106194
Feng Y., Dong H., Sun B., Hu Y., Yang Y., Jia Y., et al. (2021). METTL3/METTL14 transactivation and m6A-dependent TGF-β1 translation in activated kupffer cells. Cell Mol. Gastroenterol. Hepatol. 12 (3), 839–856. doi:10.1016/j.jcmgh.2021.05.007
Goldberg A. D., Allis C. D., Bernstein E. (2007). Epigenetics: a landscape takes shape. Cell 128 (4), 635–638. doi:10.1016/j.cell.2007.02.006
Gu X., Zhang Y., Li D., Cai H., Cai L., Xu Q. (2020). N6-methyladenosine demethylase FTO promotes M1 and M2 macrophage activation. Cell Signal 69, 109553. doi:10.1016/j.cellsig.2020.109553
Guo F., Zhang Y., Ma J., Yu Y., Wang Q., Gao P., et al. (2022). m6A mRNA methylation was associated with gene expression and lipid metabolism in liver of broilers under lipopolysaccharide stimulation. Front. Genet. 13, 818357. doi:10.3389/fgene.2022.818357
Guo J., Ren W., Li A., Ding Y., Guo W., Su D., et al. (2013). Fat mass and obesity-associated gene enhances oxidative stress and lipogenesis in nonalcoholic fatty liver disease. Dig. Dis. Sci. 58 (4), 1004–1009. doi:10.1007/s10620-012-2516-6
Guo M., Yan R., Ji Q., Yao H., Sun M., Duan L., et al. (2020). IFN regulatory Factor-1 induced macrophage pyroptosis by modulating m6A modification of circ_0029589 in patients with acute coronary syndrome. Int. Immunopharmacol. 86, 106800. doi:10.1016/j.intimp.2020.106800
Huang C., Qi P., Cui H., Lu Q., Gao X. (2022). CircFAT1 regulates retinal pigment epithelial cell pyroptosis and autophagy via mediating m6A reader protein YTHDF2 expression in diabetic retinopathy. Exp. Eye Res. 222, 109152. doi:10.1016/j.exer.2022.109152
Huang H., Weng H., Sun W., Qin X., Shi H., Wu H., et al. (2018). Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20 (3), 285–295. doi:10.1038/s41556-018-0045-z
Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., et al. (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7 (12), 885–887. doi:10.1038/nchembio.687
Jiang L., Liu X., Hu X., Gao L., Zeng H., Wang X., et al. (2022). METTL3-mediated m6A modification of TIMP2 mRNA promotes podocyte injury in diabetic nephropathy. Mol. Ther. 30 (4), 1721–1740. doi:10.1016/j.ymthe.2022.01.002
Katsuragi Y., Ichimura Y., Komatsu M. (2015). p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 282 (24), 4672–4678. doi:10.1111/febs.13540
Kunjathoor V. V., Febbraio M., Podrez E. A., Moore K. J., Andersson L., Koehn S., et al. (2002). Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 277 (51), 49982–49988. doi:10.1074/jbc.M209649200
Lee S. D., Shyu W. C., Cheng I. S., Kuo C. H., Chan Y. S., Lin Y. M., et al. (2013). Effects of exercise training on cardiac apoptosis in obese rats. Nutr. Metab. Cardiovasc Dis. 23 (6), 566–573. doi:10.1016/j.numecd.2011.11.002
Li A., Chen Y.-S., Ping X.-L., Yang X., Xiao W., Yang Y., et al. (2017). Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 27 (3), 444–447. doi:10.1038/cr.2017.10
Li B., Jiang J., Assaraf Y. G., Xiao H., Chen Z.-S., Huang C. (2020). Surmounting cancer drug resistance: new insights from the perspective of N6-methyladenosine RNA modification. Drug Resist Updat 53, 100720. doi:10.1016/j.drup.2020.100720
Li T., Zhuang Y., Yang W., Xie Y., Shang W., Su S., et al. (2021a). Silencing of METTL3 attenuates cardiac fibrosis induced by myocardial infarction via inhibiting the activation of cardiac fibroblasts. FASEB J. 35 (2), e21162. doi:10.1096/fj.201903169R
Li X., Jiang Y., Sun X., Wu Y., Chen Z. (2021b). METTL3 is required for maintaining β-cell function. Metabolism 116, 154702. doi:10.1016/j.metabol.2021.154702
Li Y., Zhang D., Gao Y., Wang P., Wang Z., Zhang B., et al. (2023). METTL3 exacerbates insulin resistance in hepatocytes by regulating m6A modification of cytochrome P450 2B6. Nutr. Metab. (Lond) 20 (1), 40. doi:10.1186/s12986-023-00762-z
Lin Y., Shen X., Ke Y., Lan C., Chen X., Liang B., et al. (2022). Activation of osteoblast ferroptosis via the METTL3/ASK1-p38 signaling pathway in high glucose and high fat (HGHF)-induced diabetic bone loss. FASEB J. 36 (3), e22147. doi:10.1096/fj.202101610R
Liu B.-H., Tu Y., Ni G.-X., Yan J., Yue L., Li Z.-L., et al. (2021). Total flavones of abelmoschus manihot ameliorates podocyte pyroptosis and injury in high glucose conditions by targeting METTL3-dependent m6A modification-mediated NLRP3-inflammasome activation and PTEN/PI3K/akt signaling. Front. Pharmacol. 12, 667644. doi:10.3389/fphar.2021.667644
Liu J., Luo G., Sun J., Men L., Ye H., He C., et al. (2019c). METTL14 is essential for β-cell survival and insulin secretion. Biochim. Biophys. Acta Mol. Basis Dis. 1865 (9), 2138–2148. doi:10.1016/j.bbadis.2019.04.011
Liu Q., Li M., Jiang L., Jiang R., Fu B. (2019a). METTL3 promotes experimental osteoarthritis development by regulating inflammatory response and apoptosis in chondrocyte. Biochem. Biophys. Res. Commun. 516 (1), 22–27. doi:10.1016/j.bbrc.2019.05.168
Liu S., Li Q., Li G., Zhang Q., Zhuo L., Han X., et al. (2020). The mechanism of m6A methyltransferase METTL3-mediated autophagy in reversing gefitinib resistance in NSCLC cells by β-elemene. Cell Death Dis. 11 (11), 969. doi:10.1038/s41419-020-03148-8
Liu Y., Liu Z., Tang H., Shen Y., Gong Z., Xie N., et al. (2019b). The N6-methyladenosine (m6A)-forming enzyme METTL3 facilitates M1 macrophage polarization through the methylation of STAT1 mRNA. Am. J. Physiol. Cell Physiol. 317 (4), C762–C75. doi:10.1152/ajpcell.00212.2019
Lu Z., Liu H., Song N., Liang Y., Zhu J., Chen J., et al. (2021). METTL14 aggravates podocyte injury and glomerulopathy progression through N6-methyladenosine-dependent downregulating of Sirt1. Cell Death Dis. 12 (10), 881. doi:10.1038/s41419-021-04156-y
Luo Q., Gao Y., Zhang L., Rao J., Guo Y., Huang Z., et al. (2020). Decreased ALKBH5, FTO, and YTHDF2 in peripheral blood are as risk factors for rheumatoid arthritis. Biomed. Res. Int. 2020, 5735279. doi:10.1155/2020/5735279
Lv D., Gimple R. C., Zhong C., Wu Q., Yang K., Prager B. C., et al. (2022). PDGF signaling inhibits mitophagy in glioblastoma stem cells through N6-methyladenosine. Dev. Cell 57 (12), 1466–1481.e6. doi:10.1016/j.devcel.2022.05.007
Mathiyalagan P., Adamiak M., Mayourian J., Sassi Y., Liang Y., Agarwal N., et al. (2019). FTO-dependent N6-methyladenosine regulates cardiac function during remodeling and repair. Circulation 139 (4), 518–532. doi:10.1161/CIRCULATIONAHA.118.033794
Meijer A. J., Codogno P. (2007). Macroautophagy: protector in the diabetes drama? Autophagy 3 (5), 523–526. doi:10.4161/auto.4449
Moscat J., Diaz-Meco M. T. (2009). p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 137 (6), 1001–1004. doi:10.1016/j.cell.2009.05.023
Mulay S. R. (2019). Multifactorial functions of the inflammasome component NLRP3 in pathogenesis of chronic kidney diseases. Kidney Int. 96 (1), 58–66. doi:10.1016/j.kint.2019.01.014
Peng Z., Gong Y., Wang X., He W., Wu L., Zhang L., et al. (2022). METTL3-m6A-Rubicon axis inhibits autophagy in nonalcoholic fatty liver disease. Mol. Ther. 30 (2), 932–946. doi:10.1016/j.ymthe.2021.09.016
Qin Y., Li B., Arumugam S., Lu Q., Mankash S. M., Li J., et al. (2021). m6A mRNA methylation-directed myeloid cell activation controls progression of NAFLD and obesity. Cell Rep. 37 (6), 109968. doi:10.1016/j.celrep.2021.109968
Roundtree I. A., Evans M. E., Pan T., He C. (2017a). Dynamic RNA modifications in gene expression regulation. Cell 169 (7), 1187–1200. doi:10.1016/j.cell.2017.05.045
Roundtree I. A., Luo G.-Z., Zhang Z., Wang X., Zhou T., Cui Y., et al. (2017b). YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. Elife 6, e31311. doi:10.7554/eLife.31311
Salisbury D. A., Casero D., Zhang Z., Wang D., Kim J., Wu X., et al. (2021). Transcriptional regulation of N6-methyladenosine orchestrates sex-dimorphic metabolic traits. Nat. Metab. 3 (7), 940–953. doi:10.1038/s42255-021-00427-2
Shen M., Li Y., Wang Y., Shao J., Zhang F., Yin G., et al. (2021). N6-methyladenosine modification regulates ferroptosis through autophagy signaling pathway in hepatic stellate cells. Redox Biol. 47, 102151. doi:10.1016/j.redox.2021.102151
Shi H., Wang X., Lu Z., Zhao B. S., Ma H., Hsu P. J., et al. (2017). YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 27 (3), 315–328. doi:10.1038/cr.2017.15
Song H., Feng X., Zhang H., Luo Y., Huang J., Lin M., et al. (2019). METTL3 and ALKBH5 oppositely regulate m6A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy 15 (8), 1419–1437. doi:10.1080/15548627.2019.1586246
Sun M., Zhang X. (2021). Epigenetic regulation of N6-methyladenosine modifications in obesity. J. Diabetes Investig. 12 (8), 1306–1315. doi:10.1111/jdi.13571
Wang F., Bai J., Zhang X., Wang D., Zhang X., Xue J., et al. (2023a). METTL3/YTHDF2 m6A axis mediates the progression of diabetic nephropathy through epigenetically suppressing PINK1 and mitophagy. J. Diabetes Investig. doi:10.1111/jdi.14113
Wang J., Yan S., Lu H., Wang S., Xu D. (2019). METTL3 attenuates LPS-induced inflammatory response in macrophages via NF-κB signaling pathway. Mediat. Inflamm. 2019, 3120391. doi:10.1155/2019/3120391
Wang X., Wu R., Liu Y., Zhao Y., Bi Z., Yao Y., et al. (2020). m6A mRNA methylation controls autophagy and adipogenesis by targeting Atg5 and Atg7. Autophagy 16 (7), 1221–1235. doi:10.1080/15548627.2019.1659617
Wang X., Zhao B. S., Roundtree I. A., Lu Z., Han D., Ma H., et al. (2015). N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161 (6), 1388–1399. doi:10.1016/j.cell.2015.05.014
Wang Y., Zhang X., Chen Y., Zhu B., Xing Q. (2023b). Identification of hub biomarkers and exploring the roles of immunity, M6A, ferroptosis, or cuproptosis in rats with diabetic erectile dysfunction. Andrology 11 (2), 316–331. doi:10.1111/andr.13265
Wu J., Frazier K., Zhang J., Gan Z., Wang T., Zhong X. (2020). Emerging role of m6 A RNA methylation in nutritional physiology and metabolism. Obes. Rev. 21 (1), e12942. doi:10.1111/obr.12942
Wu T., Shao Y., Li X., Wu T., Yu L., Liang J., et al. (2023). NR3C1/Glucocorticoid receptor activation promotes pancreatic β-cell autophagy overload in response to glucolipotoxicity. Autophagy 19, 2538–2557. doi:10.1080/15548627.2023.2200625
Yang Y., Cai J., Yang X., Wang K., Sun K., Yang Z., et al. (2022). Dysregulated m6A modification promotes lipogenesis and development of non-alcoholic fatty liver disease and hepatocellular carcinoma. Mol. Ther. 30 (6), 2342–2353. doi:10.1016/j.ymthe.2022.02.021
Yang Z., Jiang X., Zhang Z., Zhao Z., Xing W., Liu Y., et al. (2021). HDAC3-dependent transcriptional repression of FOXA2 regulates FTO/m6A/MYC signaling to contribute to the development of gastric cancer. Cancer Gene Ther. 28 (1-2), 141–155. doi:10.1038/s41417-020-0193-8
Yankova E., Blackaby W., Albertella M., Rak J., De Braekeleer E., Tsagkogeorga G., et al. (2021). Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593 (7860), 597–601. doi:10.1038/s41586-021-03536-w
Ye J., Chen X., Jiang X., Dong Z., Hu S., Xiao M. (2022). RNA demethylase ALKBH5 regulates hypopharyngeal squamous cell carcinoma ferroptosis by posttranscriptionally activating NFE2L2/NRF2 in an m6 A-IGF2BP2-dependent manner. J. Clin. Lab. Anal. 36 (7), e24514. doi:10.1002/jcla.24514
Yu R., Li Q., Feng Z., Cai L., Xu Q. (2019). m6A reader YTHDF2 regulates LPS-induced inflammatory response. Int. J. Mol. Sci. 20 (6), 1323. doi:10.3390/ijms20061323
Yuan Y., Yan G., He M., Lei H., Li L., Wang Y., et al. (2021). ALKBH5 suppresses tumor progression via an m6A-dependent epigenetic silencing of pre-miR-181b-1/YAP signaling axis in osteosarcoma. Cell Death Dis. 12 (1), 60. doi:10.1038/s41419-020-03315-x
Yvan-Charvet L., Bonacina F., Guinamard R. R., Norata G. D. (2019). Immunometabolic function of cholesterol in cardiovascular disease and beyond. Cardiovasc Res. 115 (9), 1393–1407. doi:10.1093/cvr/cvz127
Zaccara S., Ries R. J., Jaffrey S. R. (2019). Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 20 (10), 608–624. doi:10.1038/s41580-019-0168-5
Zha X., Xi X., Fan X., Ma M., Zhang Y., Yang Y. (2020). Overexpression of METTL3 attenuates high-glucose induced RPE cell pyroptosis by regulating miR-25-3p/PTEN/Akt signaling cascade through DGCR8. Aging (Albany NY) 12 (9), 8137–8150. doi:10.18632/aging.103130
Zhang S., Zhao B. S., Zhou A., Lin K., Zheng S., Lu Z., et al. (2017). m6A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation Program. Cancer Cell 31 (4), 591–606. doi:10.1016/j.ccell.2017.02.013
Zhang Y., Wang R., Tan H., Wu K., Hu Y., Diao H., et al. (2023). Fufang Zhenzhu Tiaozhi (FTZ) capsule ameliorates diabetes-accelerated atherosclerosis via suppressing YTHDF2-mediated m6A modification of SIRT3 mRNA. J. Ethnopharmacol. 317, 116766. doi:10.1016/j.jep.2023.116766
Zheng Y., Li Y., Ran X., Wang D., Zheng X., Zhang M., et al. (2022). Mettl14 mediates the inflammatory response of macrophages in atherosclerosis through the NF-κB/IL-6 signaling pathway. Cell Mol. Life Sci. 79 (6), 311. doi:10.1007/s00018-022-04331-0
Zhong H., Tang H.-F., Kai Y. (2020). N6-methyladenine RNA modification (m6A): an emerging regulator of metabolic diseases. Curr. Drug Targets 21 (11), 1056–1067. doi:10.2174/1389450121666200210125247
Zhou B., Liu C., Xu L., Yuan Y., Zhao J., Zhao W., et al. (2021a). N6 -methyladenosine reader protein YT521-B homology domain-containing 2 suppresses liver steatosis by regulation of mRNA stability of lipogenic genes. Hepatology 73 (1), 91–103. doi:10.1002/hep.31220
Zhou Y., Hambly B. D., Simmons D., McLachlan C. S. (2021b). RUNX1T1 rs34269950 is associated with obesity and metabolic syndrome. QJM 114 (8), 553–558. doi:10.1093/qjmed/hcaa208
Zhu Y., Pan X., Du N., Li K., Hu Y., Wang L., et al. (2020). ASIC1a regulates miR-350/SPRY2 by N6 -methyladenosine to promote liver fibrosis. FASEB J. 34 (11), 14371–14388. doi:10.1096/fj.202001337R
Keywords: m 6 A, nonalcoholic fatty liver disease, diabetes, atherosclerosis, inflammatory response, autophagy, programmed cell death, metabolic syndrome
Citation: Ye D, Zhang Y, Zhang B, Liu J, Wei T and Lu S (2024) The regulatory role of m6A methylation modification in metabolic syndrome pathogenesis and progression. Front. Physiol. 15:1271874. doi: 10.3389/fphys.2024.1271874
Received: 05 August 2023; Accepted: 29 February 2024;
Published: 14 March 2024.
Edited by:
Abdulrahim Sajini, Khalifa University, United Arab EmiratesReviewed by:
Mohammad Imran Khan, King Faisal Specialist Hospital and Research Centre, Saudi ArabiaDeguan Lv, University of Pittsburgh Medical Center, United States
Copyright © 2024 Ye, Zhang, Zhang, Liu, Wei and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sumei Lu, lsmqianyi@126.com