 Concha Nieto1*
Concha Nieto1* Rafael Bragado2Cristina Municio3Elena Sierra-Filardi1Bárbara Alonso1
Rafael Bragado2Cristina Municio3Elena Sierra-Filardi1Bárbara Alonso1 María M. Escribese1Jorge Domínguez-Andrés4Carlos Ardavín4Antonio Castrillo5,6Miguel A. Vega1
María M. Escribese1Jorge Domínguez-Andrés4Carlos Ardavín4Antonio Castrillo5,6Miguel A. Vega1 Amaya Puig-Kröger3†
Amaya Puig-Kröger3† Angel L. Corbí1†
Angel L. Corbí1†
- 1Centro de Investigaciones Biológicas, Consejo Superior de Investigaciones Científicas (CSIC), Madrid, Spain
- 2Instituto de Investigación Sanitaria, Fundación Jiménez Díaz, Madrid, Spain
- 3Instituto de Investigación Sanitaria Gregorio Marañón, Hospital General Universitario Gregorio Marañón, Madrid, Spain
- 4Centro Nacional de Biotecnología, Consejo Superior de Investigaciones Científicas (CSIC), Madrid, Spain
- 5Instituto Investigaciones Biomédicas “Alberto Sols” (IIBM), and Centro Mixto Consejo Superior de Investigaciones Científicas y Universidad Autónoma de Madrid (ICSIC-UAM), Madrid, Spain
- 6Unidad de Biomedicina (Unidad Asociada al CSIC), IIBM-Universidad Las Palmas de Gran Canaria (ULPGC), and Instituto Universitario de Investigaciones Biomédicas y Sanitarias (IUIBS), Universidad Las Palmas de Gran Canaria (ULPGC), Las Palmas de Gran Canaria, Spain
GM-CSF promotes the functional maturation of lung alveolar macrophages (A-MØ), whose differentiation is dependent on the peroxisome proliferator-activated receptor gamma (PPARγ) transcription factor. In fact, blockade of GM-CSF-initiated signaling or deletion of the PPARγ-encoding gene PPARG leads to functionally defective A-MØ and the onset of pulmonary alveolar proteinosis. In vitro, macrophages generated in the presence of GM-CSF display potent proinflammatory, immunogenic and tumor growth-limiting activities. Since GM-CSF upregulates PPARγ expression, we hypothesized that PPARγ might contribute to the gene signature and functional profile of human GM-CSF-conditioned macrophages. To verify this hypothesis, PPARγ expression and activity was assessed in human monocyte-derived macrophages generated in the presence of GM-CSF [proinflammatory GM-CSF-conditioned human monocyte-derived macrophages (GM-MØ)] or M-CSF (anti-inflammatory M-MØ), as well as in ex vivo isolated human A-MØ. GM-MØ showed higher PPARγ expression than M-MØ, and the expression of PPARγ in GM-MØ was found to largely depend on activin A. Ligand-induced activation of PPARγ also resulted in distinct transcriptional and functional outcomes in GM-MØ and M-MØ. Moreover, and in the absence of exogenous activating ligands, PPARγ knockdown significantly altered the GM-MØ transcriptome, causing a global upregulation of proinflammatory genes and significantly modulating the expression of genes involved in cell proliferation and migration. Similar effects were observed in ex vivo isolated human A-MØ, where PPARγ silencing led to enhanced expression of genes coding for growth factors and chemokines and downregulation of cell surface pathogen receptors. Therefore, PPARγ shapes the transcriptome of GM-CSF-dependent human macrophages (in vitro derived GM-MØ and ex vivo isolated A-MØ) in the absence of exogenous activating ligands, and its expression is primarily regulated by activin A. These results suggest that activin A, through enhancement of PPARγ expression, help macrophages to switch from a proinflammatory to an anti-inflammatory polarization state, thus contributing to limit tissue damage and restore homeostasis.
Introduction
Tissue-resident macrophages in homeostasis, as well as monocyte-derived macrophages within inflamed tissues, exhibit a huge functional diversity which derives from their exquisite sensitivity to extracellular cues (1, 2). GM-CSF and M-CSF drive macrophage differentiation and survival (3). However, M-CSF is required for the generation of most tissue macrophages (4, 5) while GM-CSF is needed for development and maintenance of pulmonary alveolar macrophages (A-MØ) (6). Besides its role in myeloid cell differentiation, GM-CSF is a central mediator of tissue inflammation (7) and its neutralization has been proposed as a therapeutic strategy for inflammatory disorders (8). As a consequence, both colony-stimulating factors promote the generation of functionally distinct macrophages (9): GM-CSF-conditioned human monocyte-derived macrophages (GM-MØ) produce large amounts of proinflammatory cytokines in response to stimulation, whereas M-CSF-dependent monocyte-derived macrophages (M-MØ) primarily produce anti-inflammatory factors upon activation (9–11). At the transcriptional level, while GM-MØ are characterized by the expression of a “Proinflammatory gene set” (11–13) also detected in macrophages under inflammatory conditions in vivo, M-MØ specifically express an “Anti-inflammatory gene set” and resemble macrophages from homeostatic/anti-inflammatory settings (14, 15). Interestingly, the GM-MØ-specific gene signature is critically determined by activin A both in vivo (15) and in vitro (11). In this regard, we have previously demonstrated that GM-MØ produce large amounts of activin A, a member of the TGFβ family (16, 17) that regulates inflammatory responses (18), modulates cytokine release (19, 20) and myeloid cell differentiation (21), and whose functional blockade in GM-MØ skews cells toward the acquisition of an anti-inflammatory signature (11).
The contribution of GM-CSF to differentiation of lung macrophages relies on the GM-CSF-dependent expression of peroxisome proliferator-activated receptor gamma (PPARγ) (22–24), a nuclear receptor that regulates gene transcription through ligand binding (25–28), antagonism of other transcription factors (e.g., NFκB, AP-1) (29, 30) and recruitment of repressor complexes in the absence of ligands (31). As a critical regulator of inflammatory processes (32–34), PPARγ inhibits human and murine macrophage responses to proinflammatory stimuli (35, 36), contributes to IL-4-driven polarization of human and murine macrophages (37, 38) and determines the acquisition of the metabolic disease-specific phenotype of human macrophages (39). In human cells, the ubiquitously expressed PPARγ1 derives from the PPARG1 and PPARG3 mRNA splicing isoforms, while the PPARG2 mRNA isoform codes for PPARγ2, whose expression is restricted to adipocytes (40). Mouse A-MØ exhibit much higher expression of PPARγ than other macrophages in the steady-state (34), and its GM-CSF-dependent expression is essential for their differentiation and maturation from fetal monocytes (24). In fact, PPARγ expression in A-MØ is lost in GM-CSF-deficient mice and in patients with pulmonary alveolar proteinosis (PAP), a pathology derived from a defective expression or activity of GM-CSF (23, 41) and associated with suppressed activin A expression (42). However, it is currently unknown whether PPARγ is required for maintenance of A-MØ throughout adult life (43).
Upon tissue injury, monocyte-derived macrophages modulate inflammation and also promote tissue repair. In the specific case of lung inflammation, monocyte-derived mouse A-MØ are the major drivers of fibrosis and become similar to tissue-resident A-MØ over time (44). Since GM-CSF-conditioned monocyte-derived human macrophages exhibit potent proinflammatory functions upon stimulation (9, 11), and in spite of the intrinsic anti-inflammatory functions of PPARγ, we hypothesized that PPARγ might contribute to the gene signature and functional profile of human GM-CSF-conditioned macrophages. To address this hypothesis, we evaluated the extent of the PPARγ contribution to the gene signature and functional profile of human GM-CSF-dependent macrophages. We now report the activin A-dependent expression and activity of PPARγ in GM-CSF-conditioned human macrophages, and demonstrate that PPARγ displays polarization-dependent activities and significantly shapes the gene signature of proinflammatory monocyte-derived GM-MØ and human A-MØ in the absence of exogenous ligands. The activin A-dependent expression of PPARγ in GM-MØ and in A-MØ also suggests a role for activin A in promoting inflammation resolution.
Experimental Procedures
Generation of Human Monocyte-Derived Macrophages In Vitro and Ex Vivo Isolation of A-MØ
Buffy coats were obtained from healthy blood donors, as anonymously provided by the Comunidad de Madrid blood Bank. Ethical approvals for all blood sources and processes used in this study were approved by the Centro de Investigaciones Biológicas Ethics Committee. All experiments were carried out in accordance with the approved guidelines and regulations. Human PBMCs were isolated from buffy coats over a Lymphoprep™ gradient (#1114545, Axis-Shield PoC AS) according to standard procedures. Monocytes were purified from PBMCs by magnetic cell sorting using human CD14 microbeads (#130-050-201, Miltenyi Biotech). Monocytes (95% CD14+ cells) were cultured at 0.5 × 106 cells/ml for 7 days in RPMI 1640 (#21875-034, Gibco) supplemented with 10% inactivated fetal calf serum (FCS) (#S1810-500, Biowest) (complete medium), at 37°C in a humidified atmosphere with 5% CO2, and containing 1,000 U/ml human GM-CSF (#11343125, Immunotools GmbH) or 10 ng/ml human M-CSF (#11343115, Immunotools GmbH), to generate GM-MØ or M-MØ, respectively. Cytokines were added every 2 days. Blocking anti-activin A Ab (100 ng/ml) (#MAB3381, clone 69403, R&D Systems) or the inhibitors of ALK4, ALK5, and ALK7, SB431542 (10 µM) (#S4317, Sigma-Aldrich) or A-83 (1 µM) (#2039, Tocris) were added every 24 h. Finally, polarized macrophages were treated with ultrapure Escherichia coli 0111:B4 strain LPS (10 ng/ml) (#tlrl-3pelps, Invivogen) for 14–16 h. Exposure to recombinant human activin A (25 ng/ml) (#120-14P, Preprotech) was done for 24 h (monocytes and THP-1 cells) or 48 h (M-MØ). The acute monocytic leukemia cell line THP-1, obtained from ATCC® (#TIB-202™), was cultured in complete medium at 37°C in a humidified atmosphere with 5% CO2. A-MØ were obtained from patients undergoing bronchoalveolar lavage (BAL) following the Fundación Jiménez Díaz Medical Ethics committee procedures and after written informed consent from all subjects, in accordance with the Declaration of Helsinki. BAL procedure was performed with a flexible bronchoscope with a total volume of 200 ml of sterile isotonic saline solution at 37°C. BAL fluid fractions were maintained at 4°C and cellular debris removed using a 40 µm cell strainer (45). BAL cells were washed with PBS, centrifuged and resuspended in complete medium containing 100 U/mL penicillin and 100 µg/mL streptomycin (#15140-122, Gibco), 50 µg/ml gentamicin (#G1397, Sigma-Aldrich), and 2.5 µg/ml amphotericin B (#A2942, Sigma-Aldrich). The cells were seeded at 6–8 × 105 cells per well in 12-well plates for 1 h and washed extensively to remove non-adherent cells. Finally, 2 ml of complete medium with antibiotics was added to each well and the adherent cells incubated for 16–18 h before transfection. More than 95% of adherent BAL cells were identified as macrophages according to morphology and phenotypic analysis.
Generation of Murine Bone Marrow-Derived Macrophages In Vitro
All experiments on mice were conducted according to the Spanish and European regulations on care and protection of laboratory animals and were approved by the Centro de Investigaciones Biológicas animal facility and the Consejo Superior de Investigaciones Científicas Ethics Committee. Bone marrow-derived GM-MØ or M-MØ were obtained by flushing the femurs of 6–10-week-old C57BL/6 mice (provided by the Animal facility at the Centro de Investigaciones Biológicas), and culturing cells during 7 days in DMEM (#41966-029, Gibco) supplemented with 10% FCS and 50 mM 2-ME, containing either murine GM-CSF (1,000 U/ml) (#315-03, PreProtech) or human M-CSF (25 ng/ml) (#11343115, Immunotools GmbH), respectively (46, 47). Cytokines were added every 2 days.
Flow Cytometry
Mouse monoclonal antibodies specific for human CD14 (Alexa Fluor-647-labeled antihuman CD14, #301818, clone M5E2, Biolegend) and human CD163 (PerCP-labeled antihuman CD163, #333625, clone GHI/61, Biolegend) were used. Isotype-matched PerCP-labeled Mouse IgG1 (κ Isotype Ctrl Antibody, #400147, clone MOPC-21, Biolegend) and Alexa Fluor-647 Mouse IgG2a (κ Isotype Ctrl Antibody, #400234, clone MOPC-173, Biolegend) were included as negative controls.
Quantitative Real Time RT-PCR
Total RNA was extracted using the total RNA and protein isolation kit (Macherey-Nagel). RNA samples were retrotranscribed with the High-Capacity cDNA Reverse Transcription kit (AB), and individually amplified cDNA was quantified using the Universal Human Probe Roche library (Roche Diagnostics). Oligonucleotides for selected genes were designed according to the Roche software for quantitative real-time PCR (qRT-PCR), and their sequence is indicated in Table S1 in Supplementary Material. qRT-PCR was performed on a LightCycler® 480 (Roche Diagnostics). Assays were made in triplicates, and results were normalized according to the expression levels of TBP mRNA or/and GAPDH mRNA (for qRT-PCR) or to the mean of the expression level of endogenous reference genes HPRT1, TBP and RPLP0 (for microfluidic gene cards). Results were expressed using the ΔΔCT (cycle threshold) method for quantification.
ELISA
Macrophage supernatants were tested for the presence of cytokines using commercially available ELISA sets for human TNFα (BD OptEIA Human TNF ELISA set, #555212, BD Biosciences), CCL2 (BD OptEIA Human MCP-1 ELISA set, #555179, BD Biosciences), IL-10 (ELISA MAX Standard set, #430601, BioLegend), IL-6 (ELISA MAX Standard set, #430501, BioLegend), and activin A (DuoSet, #DY338, R&D Systems), following the protocols supplied by the manufacturers.
Cell Transfection and Reporter Gene Assays
HEK293-T cells, provided by the Cell culture facility at the Centro de Investigaciones Biológicas, were transfected with an expression vector for PPARγ2 (pBABE-PPARγ2, Addgene) or an empty vector using Superfect transfection reagent (#301305, Qiagen). Human GM-MØ or M-MØ (1 × 106 cells) were transfected using the Human Macrophage Nucleofector® Kit (#VPA-1008, Lonza) with 1 µg of PPAR reporter DNA mixture (#CCS-3026L, Cignal PPAR Reporter assay kit, Qiagen). This mixture contains a PPAR-responsive firefly luciferase construct and a constitutively expressing Renilla luciferase (40:1) The PPAR-dependent construct encodes the firefly luciferase gene under the control of a minimal CMV promoter and tandem repeats of the PPAR responsive element (PPRE). Firefly and Renilla luciferase activities were determined by using the Dual-Luciferase® Reporter Assay System (#E1910, Promega).
Western Blot Assay
Cell lysates (40 µg) and nuclear extracts (30 µg) were subjected to SDS-PAGE and transferred onto an Immobilon polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA). After blocking the unoccupied sites with 5% nonfat dry milk, protein detection was carried out with a goat polyclonal against PPARγ2 (G-18, #sc-22020, Santa Cruz Biotechnology), a goat affinity purified polyclonal antibody against Sp1 (PEP2, #sc-59-G, Santa Cruz Biotechnology), or a monoclonal antibody against GAPDH (6C5, #sc-32233, Santa Cruz Biotechnology), and using the SuperSignal West Pico Chemiluminescent system (#34081, Thermo Fisher Scientific).
Small Interfering Ribonucleic Acid (siRNA) Transfection
To silence PPARG gene expression, human GM-MØ, M-MØ (1 × 106 cells) or A-MØ (6–8 × 105 cells) were transfected with a PPARG-specific siRNA (siPPARG) (50 nM) (#s10888, Thermo Fisher Scientific), using HiPerFect transfection reagent (#301705, Qiagen). A negative control siRNA from the same company was used as a transfection control (siControl) (#4390843, Thermo Fisher Scientific). After 6 h of transfection, cells were allowed to recover from transfection in RPMI 1640 medium with 10% FCS and the cells were treated with GW7845 (1 µM) (kindly provided by Jon Collins, Glaxo SmithKline, USA) or DMSO for 18–24 h before assessing for PPARγ markers.
Microarray Analysis
Global gene expression analysis was performed on RNA obtained from three independent samples of GM-MØ that had been transfected with siPPARG or siControl for 48 h, and using a whole human genome microarray from Agilent Technologies (Palo Alto, CA, USA). Only probes with signal values >60% quantile in at least one condition were considered for the differential expression and statistical analysis. Statistical analysis for differential gene expression was carried out using empirical Bayes moderated t test implemented in the limma package1 and using paired t-test. All the above procedures were coded in R.2 Microarray data were deposited in the Gene Expression Omnibus3 under accession no. GSE88768. The differentially expressed genes were analyzed for annotated gene sets enrichment using the online tool ENRICHR4 (48, 49). Enrichment terms were considered significant when they had a Benjamini-Hochberg-adjusted p value < 0.05. For gene set enrichment analysis (GSEA) (50), the previously defined “Proinflammatory gene set” and “Anti-inflammatory gene set” (12), which contain the top and bottom 150 probes from the GM-MØ versus M-MØ limma analysis of the microarray data in GSE68061 (ranked on the basis of the value of the t statistic), were used.
Statistical Analysis
Statistical analysis was performed using paired Student’s t-test, and p < 0.05 was considered significant (*p < 0.05, **p < 0.01, and ***p < 0.001).
Results
PPARγ Activation Has Different Transcriptional and Functional Outcomes in Human GM-MØ and M-MØ
To initially assess the PPARγ activation-dependent transcriptional profile of GM-MØ and M-MØ, both human macrophage subtypes were exposed for 24 h to the PPARγ agonist GW7845 and the expression of the GM-MØ-specific “Proinflammatory gene set” and M-MØ-specific “Anti-inflammatory gene set” (derived from the data contained in the Gene Expression Omnibus GSE68061) (11, 12) was determined. PPARγ activation upregulated the paradigmatic PPARγ target genes CD36 and FABP4, and downregulated FLT1 and CSF1 expression, in both macrophage subtypes (Figure 1A). However, GW7845 downregulated IL6, IL10, CCL2, HAMP, and CCR2 and enhanced THBS1, exclusively in GM-MØ (Figure 1A). These GW7845-triggered gene expression changes were dependent on PPARγ activation as they were significantly impaired upon siRNA-mediated knockdown of PPARG mRNA (Figures 1B,C). Specifically, PPARG mRNA knockdown inhibited the GW7845-mediated modulation of CD36 and CSF1 expression in M-MØ (Figure 1B) and significantly impaired the GW7845-mediated modulation of CD36, CSF1, FLT1, CCL2, CCR2, IL10, and HAMP in GM-MØ (Figure 1C). Analogous findings were observed in murine bone marrow-derived macrophages, where Pparγ activation modified the expression of a common set of genes in both macrophage subtypes but significantly diminished the expression of Csf1 and Ccr2 only in GM-MØ (Figure 2). Therefore, although PPARγ activation alters the expression of known PPARγ targets in both GM-MØ and M-MØ, it also promotes human macrophage subtype-dependent transcriptional changes because the expression of CCR2, IL10, CCL2, and HAMP is downregulated by GW7845 only in proinflammatory GM-MØ.
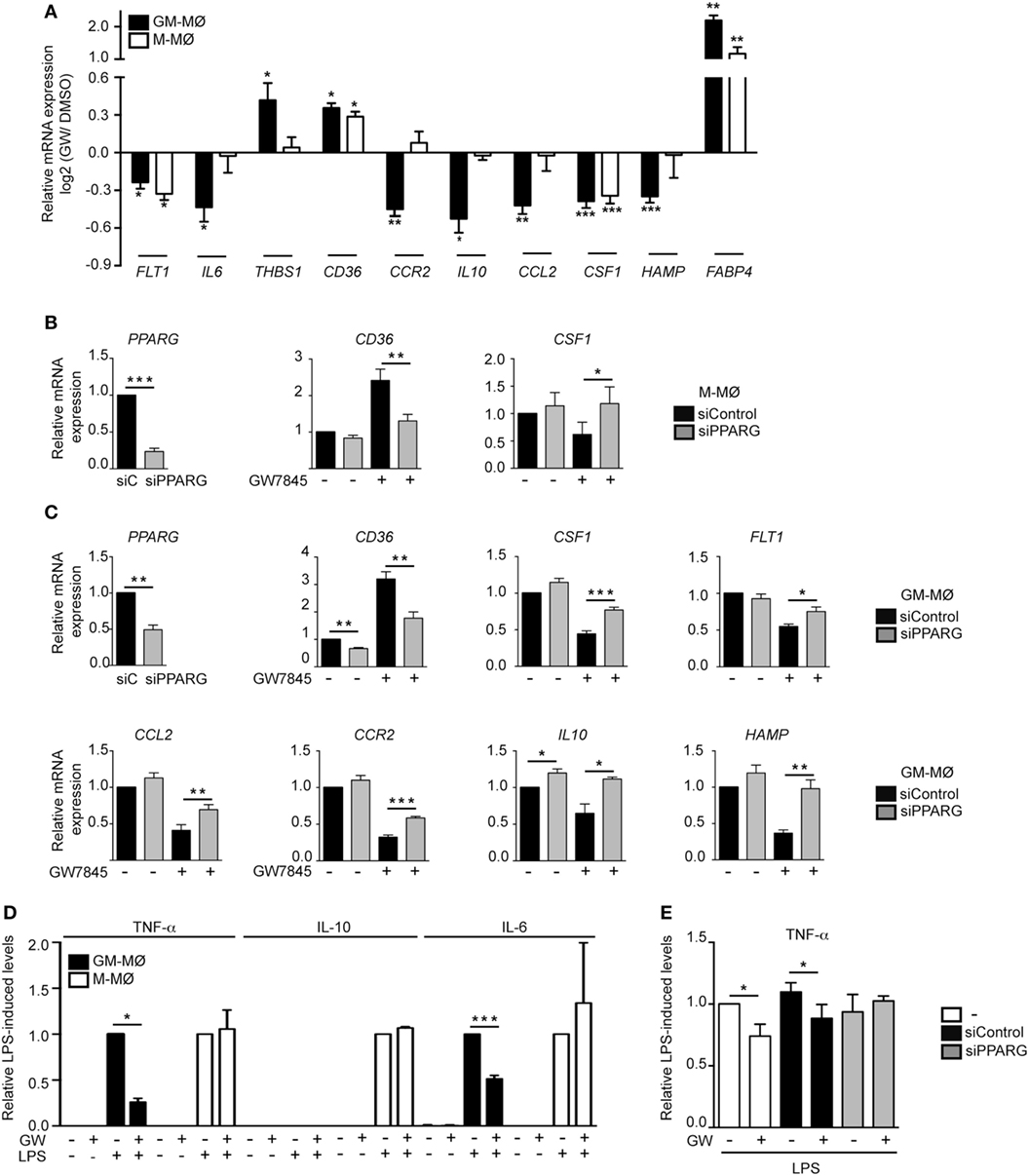
Figure 1. Peroxisome proliferator-activated receptor gamma (PPARγ) mediates the differential effect of GW7845 on the gene and cytokine profile of human GM-CSF-conditioned monocyte-derived macrophages (GM-MØ) and M-CSF-dependent monocyte-derived macrophages (M-MØ). (A) Expression of the indicated genes in GM-MØ and M-MØ exposed for 24 h to either GW7845 (GW, 1 µM) or vehicle (DMSO), as determined by quantitative real-time PCR assay using microfluidic gene cards. Results are indicated as the expression of each gene after GW7845 treatment relative to its expression in the presence of DMSO. Each experiment was performed in triplicate, and mean and SEM of three independent experiments is shown (*p < 0.05; **p < 0.01; ***p < 0.001). (B,C) Expression of the indicated genes in M-MØ (B) or GM-MØ (C) transfected with either siPPARG or siControl (siC), and treated with GW7845 (1 µM) or DMSO for 24 h. Relative mRNA expression indicates the expression of each gene in the different conditions and relative to its expression in DMSO-treated siC-transfected cells (arbitrarily set to 1). (Left panels) PPARG mRNA expression in siPPARG-transfected cells relative to the PPARG mRNA level in siC-transfected cells (arbitrarily set to 1). Mean and SEM of four independent experiments are shown (*p < 0.05; **p < 0.01; ***p < 0.001). (D) TNFα, IL-10, and IL-6 production in LPS-treated (24 h) GM-MØ and M-MØ that had been preexposed (4 h) to DMSO or GW7845 (GW, 1 µM). Results indicate the concentration of each cytokine for each condition relative to the cytokine levels detected in cells treated with DMSO and LPS (arbitrarily set to 1). Mean and SEM of three independent experiments are shown (*p < 0.05; ***p < 0.001). (E) TNFα production in LPS-treated (24 h) untransfected (−), siControl-transfected or siPPARG-transfected GM-MØ that had been preexposed (4 h) to DMSO or GW7845 (GW, 1 µM). Results indicate the concentration of TNFα for each condition relative to the cytokine levels detected in untransfected cells treated with DMSO and LPS (arbitrarily set to 1). Mean and SEM of four independent experiments are shown (*p < 0.05).
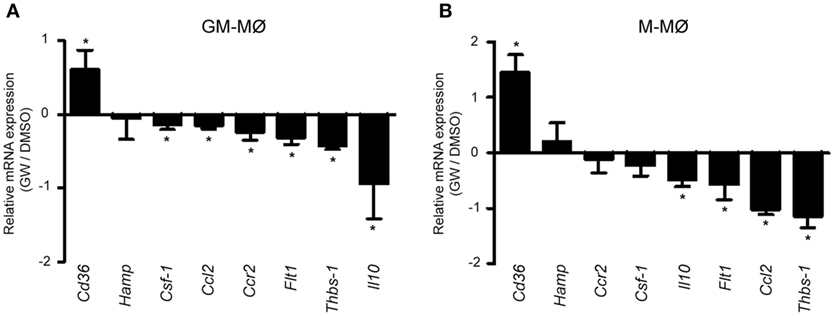
Figure 2. Differential effect of peroxisome proliferator-activated receptor gamma activation on mouse GM-CSF-conditioned bone marrow-derived macrophages (GM-MØ) and M-CSF-dependent bone marrow-derived macrophages (M-MØ). Relative expression of the indicated genes in murine bone marrow-derived GM-MØ (A) and M-MØ (B) exposed to either DMSO or GW7845 (1 µM) for 24 h, as determined by quantitative real-time PCR. Results are expressed as the expression of each gene in the presence of GW7845 relative to its expression in the presence of vehicle (DMSO). Mean and SD of three independent experiments is shown (*p < 0.05).
To determine whether the distinct transcriptional effects of PPARγ activation in GM-MØ and M-MØ had a functional correlate, the LPS-induced cytokine-producing ability of both macrophage subtypes was evaluated in the presence of GW7845. As expected, LPS stimulation of GM-MØ caused the preferential production of the proinflammatory cytokines TNFα and IL-6, whereas LPS-stimulated M-MØ primarily released IL-10 (9–11) (Figure 1D). In line with the transcriptional results, GW7845 significantly reduced the LPS-induced production of TNFα and IL6 from GM-MØ, but had no effect on the LPS-induced cytokine release from M-MØ (Figure 1D). Importantly, the inhibitory effect of GW7845 on the LPS-induced TNFα production of GM-MØ was PPARγ-dependent, as it was reduced upon PPARγ knockdown (Figure 1E). Therefore, agonist-mediated activation of PPARγ exclusively modulates the LPS-induced cytokine production from proinflammatory human monocyte-derived GM-MØ, further arguing for a polarization-dependent effect of PPARγ in human macrophages.
PPARγ Is Preferentially Expressed by Proinflammatory GM-CSF-Dependent Human Macrophages
Given the different effect of PPARγ on GM-MØ and M-MØ, we next determined PPARγ expression and function in both human macrophage subtypes. Transfection of a PPRE reporter construct in both macrophage subtypes revealed that global PPAR-dependent transcriptional activity is higher in GM-MØ than in M-MØ (Figure 3A), thus suggesting that GM-MØ are endowed with a stronger PPARγ-dependent transcriptional activity. Regarding expression, GM-MØ contained higher levels of PPARG1/3 (encoding the ubiquitous PPARγ1 isoform) and PPARG2 (coding for the PPARγ2 isoform) mRNAs than M-MØ (Figure 3B). In fact, the adipocyte-restricted PPARG2 mRNA (40) was barely detectable in M-MØ (Figure 3B). The preferential expression of the PPARγ2-encoding mRNA was also observed in murine bone marrow-derived GM-MØ, whereas mouse M-MØ exhibited significantly higher Pparg1 expression than mouse GM-MØ (Figure 3C), in agreement with a previous report (51) and in line with the distinct gene profiles of monocyte-derived human M-MØ and bone marrow-derived mouse M-MØ (13, 52). Kinetic analysis revealed that PPARG2 mRNA is upregulated in human monocytes exposed to GM-CSF for 3, 5, and 7 days (Figure 3D). Although PPARG1 is expressed at higher levels than PPARG2 mRNA (25-fold approx.), PPARγ2 protein could be detected in whole cell and nuclear extracts from GM-MØ (Figures 3E,F). Therefore, GM-CSF-conditioned proinflammatory human macrophages exhibit a higher expression of PPARγ (PPARγ1 and PPARγ2) than M-CSF-conditioned anti-inflammatory human macrophages.
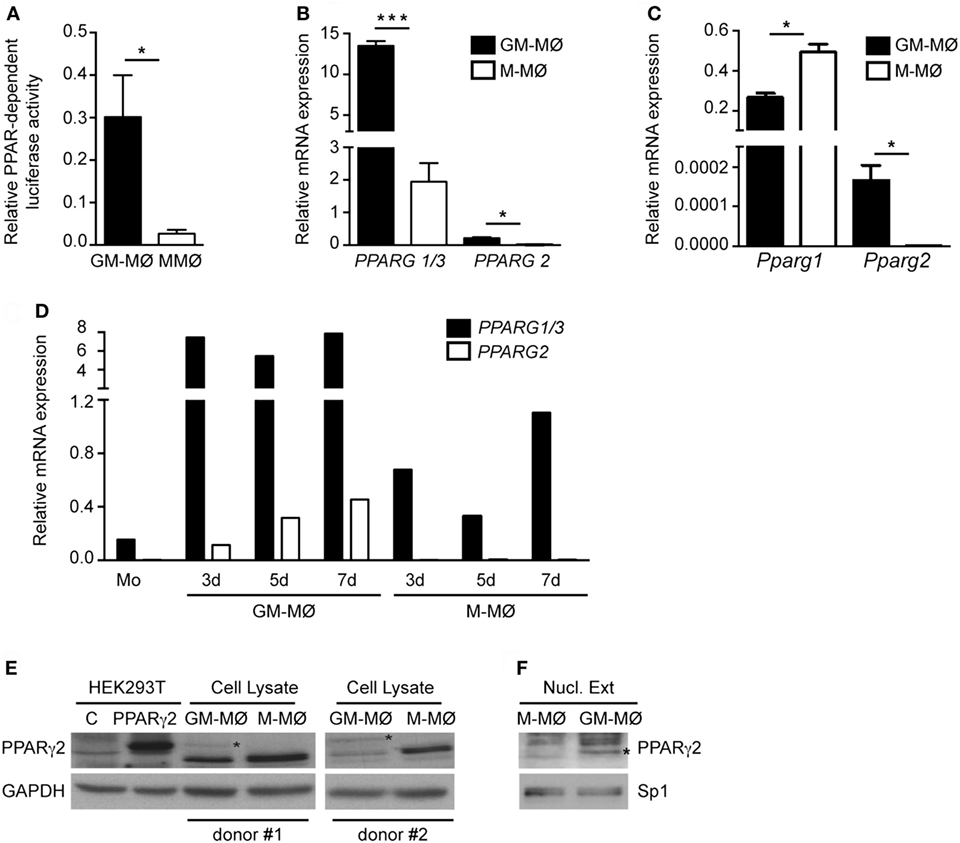
Figure 3. Expression of peroxisome proliferator-activated receptor gamma (PPARγ) isoforms in human and mouse GM-CSF-conditioned macrophages (GM-MØ) and M-CSF-dependent macrophages (M-MØ). (A) Basal PPAR-dependent transcriptional activity in GM-MØ and M-MØ. Mean and SEM of the relative PPAR-dependent luciferase activity (compared to Renilla luciferase activity) of seven independent experiments is shown (*p < 0.05). (B) PPARG1/3 and PPARG2 mRNA expression levels in GM-MØ and M-MØ, as determined by quantitative real-time PCR (qRT-PCR) and relative to TBP mRNA levels. Mean and SEM of three independent experiments is shown (*p < 0.05; ***p < 0.001). (C) Pparg1 and Pparg2 mRNA expression in bone marrow-derived murine GM-MØ and M-MØ, as determined by qRT-PCR and relative to Tbp mRNA levels. Mean and SD of three independent samples is shown (*p < 0.05). (D) PPARG1/3 and PPARG2 mRNA expression levels along GM-MØ and M-MØ differentiation, as determined by qRT-PCR and relative to TBP mRNA levels. A representative experiment is shown. (E,F) PPARγ2 protein levels in whole cell (E) or nuclear lysates (Nucl. Ext.) (F) from mock-transfected (C) or human PPARγ2-transfected HEK293T cells (E) and two independent samples of GM-MØ and M-MØ (donor #1 and donor #2). GAPDH (E) and Sp1 (F) protein levels were detected in parallel as protein loading controls. The band corresponding to PPARγ2 protein is indicated by an asterisk.
Activin A Controls PPARγ Expression in GM-CSF-Dependent Macrophages
The GM-CSF-dependent expression of PPARγ is essential for the differentiation of A-MØ (24). Since proinflammatory human GM-MØ polarization is dependent on the autocrine/paracrine action of activin A (11), we next questioned whether activin A contributes to the preferential expression of PPARG mRNAs in GM-MØ. Activin A significantly elevated PPARG1/3 and PPARG2 mRNA levels in M-MØ, monocytes and THP-1 myeloid cells (Figure 4A). Moreover, inhibition of activin A-initiated Smad signaling by either SB431542 (Figure 4B) or A-83 (Figure 4C), or blockade of activin A with an anti-activin neutralizing antibody (Figure 4D), significantly reduced PPARG1/3 and PPARG2 mRNA levels in GM-CSF-dependent proinflammatory GM-MØ. In line with these results, generation of GM-MØ in the presence of A-83 resulted in significantly reduced expression of the PPARγ target gene ABCA1, a gene whose expression is responsive to PPARγ-LXR activation in human macrophages (Figure 4E). Further, analysis of ex vivo isolated human A-MØ revealed the constitutive expression of activin A (Figure 4F), and that the expression of PPARG1/3 and PPARG2 mRNA, as well as the expression of the PPARγ target ABCA1 mRNA, were significantly reduced in the presence of the A-83 Smad signaling inhibitor (Figure 4G). Altogether, these results indicate that activin A is a positive regulator of PPARγ expression and activity in GM-CSF-conditioned macrophages both in vitro and in vivo.
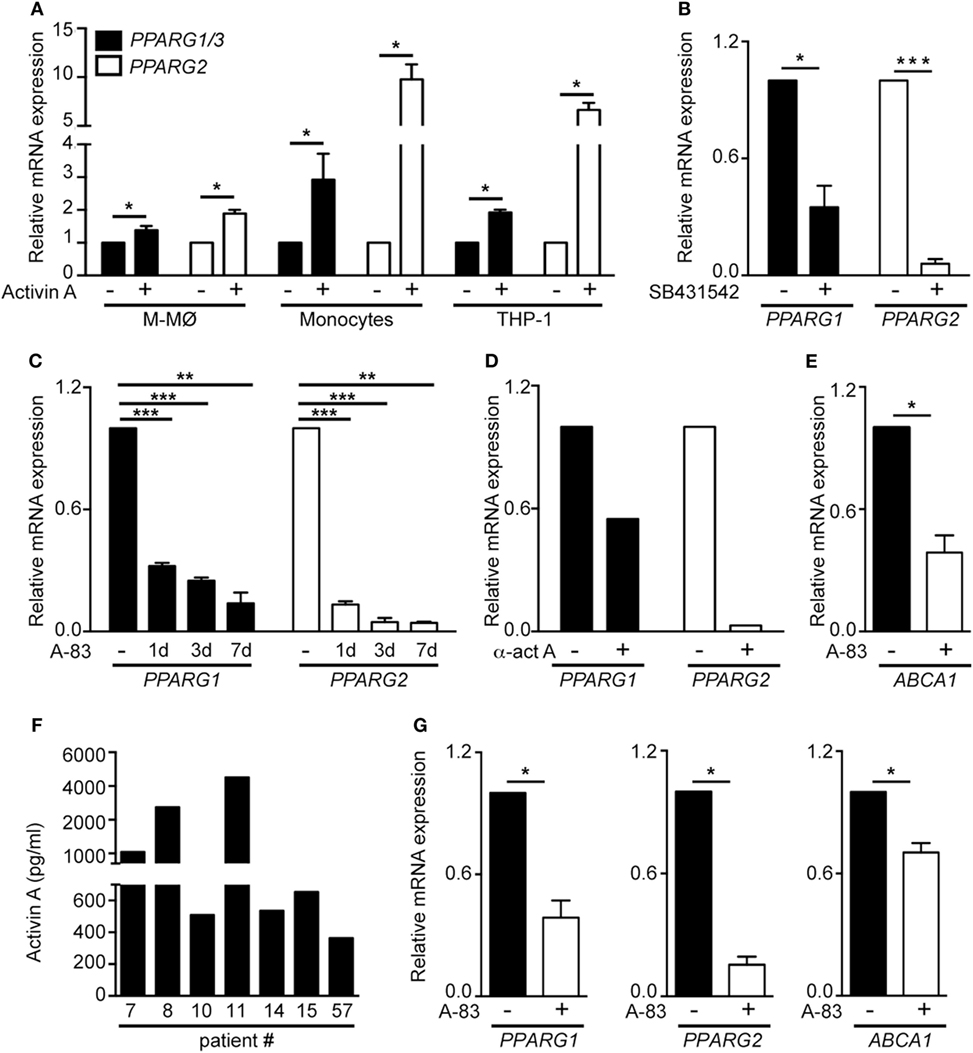
Figure 4. The activin A/Smad signaling pathway determines the differential expression of PPARG1 and PPARG2 in human GM-CSF-conditioned monocyte-derived macrophages (GM-MØ) and M-CSF-dependent monocyte-derived macrophages (M-MØ). (A) PPARG1 and PPARG2 expression in untreated or activin A-treated M-MØ, monocytes and THP-1 cells. Cells were treated with 25 ng/ml recombinant human activin A for 24 h (monocytes and THP-1 cells) or 48 h (M-MØ). Mean and SEM of three independent experiments is shown (*p < 0.05). (B) PPARG1 and PPARG2 gene expression in GM-MØ generated in the presence of either DMSO or the Smad signaling inhibitor SB431542 (10 µM). Mean and SEM of three independent experiments is shown (*p < 0.05; ***p < 0.001). (C) PPARG1 and PPARG2 gene expression in GM-MØ generated in the presence of either DMSO or the Smad signaling inhibitor A-83 (1 µM) for 1, 3, or 7 days. Mean and SEM of three independents experiments is shown (*p < 0.05; **p < 0.01; ***p < 0.001). (D) PPARG1 and PPARG2 gene expression in GM-MØ generated in the presence of a neutralizing antiactivin A antibody (α-ActA) or an isotype-matched antibody (−). One representative experiment is shown. In (A–D), results are referred to the PPARG1 or PPARG2 mRNA levels detected in untreated cells (arbitrarily set to 1). (E) ABCA1 gene expression in GM-MØ generated in the presence of either DMSO or the Smad signaling inhibitor A-83 (1 µM) for seven days. Results are referred to the ABCA1 mRNA level in DMSO-treated cells. Mean and SEM of three independents experiments is shown (*p < 0.05). (F) Activin A expression levels in seven independent samples of human alveolar macrophages kept in culture for 24 h after isolation. (G) PPARG1, PPARG2, and ABCA1 mRNA levels in human alveolar macrophages cultured for 24 h after isolation in the presence of either DMSO or the Smad signaling inhibitor A-83 (1 µM). Results are referred to the mRNA levels of each gene in DMSO-treated cells (arbitrarily set to 1). Means and SEM of three independents samples is shown (*p < 0.05).
Identification of the PPARγ-Dependent Gene Profile in GM-CSF-Conditioned Proinflammatory Human Macrophages
Given the transcriptional effects of PPARγ knockdown (Figure 1C), and to more thoroughly address the role of PPARγ in in vitro generated GM-CSF-conditioned macrophages, we determined the PPARγ-dependent transcriptional profile of GM-MØ in the absence of exogenous agonists. siRNA-mediated PPARγ knockdown significantly modified the transcriptome of GM-MØ, altering the expression of 314 probes (283 annotated genes) (p < 0.003, Table S2 in Supplementary Material). Specifically, PPARγ knockdown led to downregulation of 139 genes and upregulation of 144 genes in GM-MØ (Figure 5A). Twenty-five percent of the genes downregulated by siPPARG (36 out of 139) had been previously predicted as PPAR targets (53), including 20 genes upregulated by long-term rosiglitazone treatment of human monocyte-derived dendritic cells (54) and two genes whose expression is also diminished in mouse Pparγ−/− macrophages (CD36 and GPD1) (24) (Figure 5B). Similarly, the set of genes upregulated upon PPARγ knockdown contained 19 genes predicted as PPAR targets (53) (Figure 5B), including 5 genes upregulated by rosiglitazone in human dendritic cells (54) and CCL2 and CCL7, whose orthologous genes are overexpressed in murine Pparγ−/− macrophages (24). Conversely, siPPARG downregulated the expression of MSR1, whose mouse ortholog is overexpressed in Pparγ−/− macrophages (24). The PPARγ-regulated gene set also included genes whose expression distinguishes A-MØ from other tissue-resident mouse macrophages (KRT79, BCAR3, MAFF, WWTR1) (55) or have been defined as human A-MØ-enriched genes (EDN1, CXCL1, TNFAIP6, IL7R) (56) (Table S2 in Supplementary Material). Therefore, PPARγ knockdown in GM-CSF-conditioned human macrophages allowed the identification of a large set of genes (Table S2 in Supplementary Material) whose expression is specifically modulated by PPARγ in the absence of an exogenous agonist. Besides, and in agreement with the divergent transcriptional profiles of functionally similar human and mouse macrophages (57), the human macrophage PPARγ-dependent gene set in human macrophages only partially overlaps with the list PPARγ-regulated genes previously identified in mouse macrophages.
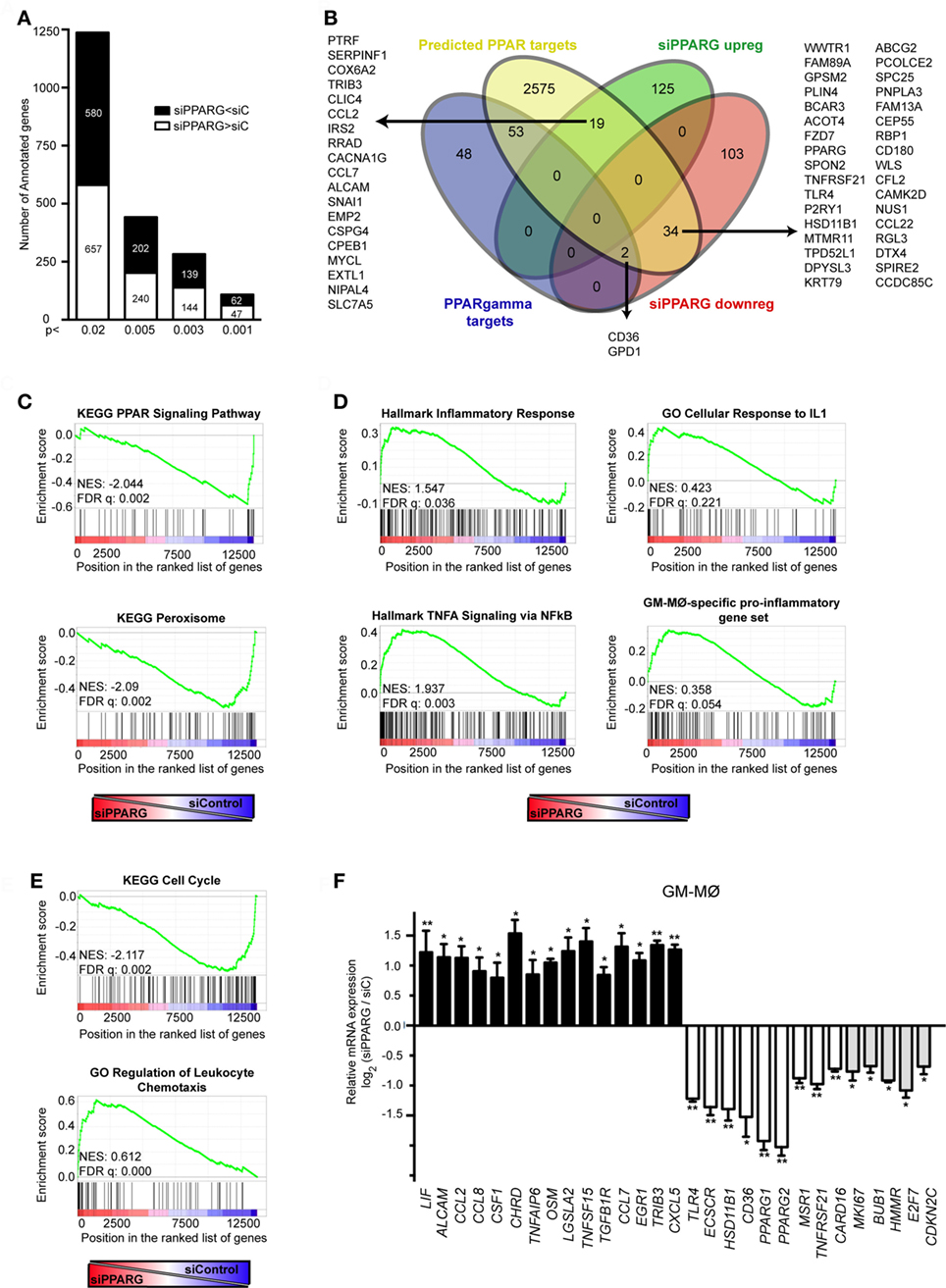
Figure 5. Peroxisome proliferator-activated receptor gamma (PPARγ) controls the global transcriptional signature of GM-CSF-conditioned human monocyte-derived macrophages (GM-MØ). (A) Number of annotated genes whose expression is higher or lower in siPPARG-transfected than in siControl-transfected (siC) GM-MØ at the indicated p-values. (B) Venn diagram analysis of the genes differentially expressed in siPPARG-transfected and siControl-transfected GM-MØ compared to experimentally verified PPARγ target genes (PPARγ targets) and computationally predicted PPAR target genes (predicted PPAR targets), as reported in the PPARgene database (53). (C–E) Gene set enrichment analysis on the “t statistic-ranked” list of genes obtained from the siPPARG-GM-MØ versus siControl-GM-MØ limma analysis, using the indicated gene set. In (D), the previously defined GM-MØ-specific “Proinflammatory gene set” (12) was also used. (F) Expression of the indicated genes in siPPARG-transfected and siControl-transfected (siC) GM-MØ, as determined by quantitative real-time PCR on three to five independent GM-MØ samples. Results are indicated as the mRNA levels of each gene in siPPARG-transfected relative to the levels in siControl-transfected GM-MØ (n = 3–5; *p < 0.05; **p < 0.01).
To gain formation on the biological processes significantly affected after PPARγ knockdown in human macrophages, functional enrichment analysis was performed using GSEA (50). Confirming the validity of the results, PPARγ knockdown led to a very significant reduction in the expression of genes associated with the terms “KEGG_PPAR_Signaling_Pathway” and “KEGG_Peroxisome” (Figure 5C). In line with its known anti-inflammatory function (32, 58), reduction of PPARγ expression in GM-MØ caused a significant increase in the expression of genes within the “Hallmark_Inflammatory Response,” “Hallmark_TNFA signaling via NFKB,” and “GO_Cellular Response to IL1” gene sets (Figure 5D). Also in agreement with the anti-inflammatory activity of PPARγ (32, 58), PPARγ knockdown promoted a significant global upregulation of the GM-MØ-specific “Proinflammatory gene set” (12) (Figure 5D), and specially of two GM-MØ-specific genes like ECSCR and HSD11B1 (11) (Table S2 in Supplementary Material). Unexpectedly, PPARγ knockdown resulted in a very significant increase in the expression of genes within gene sets directly involved cell cycle and proliferation like “KEGG Cell Cycle,” “Reactome Mitotic M G1 Phases,” “Reactome G1 S Transition,” “Hallmark G2M Checkpoint,” “GO Cell Division,” and others (all with FDR q = 0.000) (Figure 5E and not shown). Besides, reduction of PPARγ caused a very significant increase in the expression of genes involved in chemotaxis (Figure 5D), suggesting that PPARγ has a negative regulatory effect on leukocyte mobility. All these results were further supported after ENRICHR analysis (see text footnote 4) (48, 49), which also revealed that PPARγ knockdown specifically impairs the expression of genes whose expression is controlled by a known PPARγ ligand (rosiglitazone, adjusted p = 1.4 × 10−10) and a transcription factor (FOXM1, adjusted p = 8.47 × 10−18) that collaborates with PPARγ in pulmonary inflammation (59, 60) (Table S2 in Supplementary Material and data not shown).
To further validate the correlations found with GSEA and ENRICHR, a representative number of transcriptional changes were assessed after PPARγ knockdown on independent GM-MØ preparations (Figure 5F). PPARγ knockdown significantly reduced the expression of genes encoding PAMP/DAMP receptors (TLR4, CD36), NFκB activators (TNFRSF21, CARD16), the scavenger receptors SR-A1 (MSR1), known PPARγ targets like HSD11B1, and the GM-MØ-specific gene ECSCR (11) (Figure 5F). Conversely, reduction of PPARγ levels increased the expression of genes encoding cytokines (CCL2, CCL7, CCL8, CXCL5), again pointing toward a negative regulatory effect of PPARγ on the expression of genes associated with cell chemotaxis. PPARγ knockdown also enhanced expression of genes encoding various growth-promoting factors (CSF1, TNFSF15, OSM, LIF) (Figure 5F). Moreover, downregulating PPARγ expression led to a significant reduction in the expression of genes that contribute to the significant enrichment signal (“leading-edge”) found after GSEA on gene sets related to cell cycle and proliferation (namely MKI67, BUB1, HMMR, E2F7, and CDKN2C) (Figure 5F, gray-filled bars) and that code for proteins involved in cell cycle regulation like Ki67, E2F7, and CDKN2C. Therefore, and in agreement with the GSEA correlations (Figure 5E), expression of PPARγ in GM-CSF-conditioned macrophages has a positive impact on the expression of genes that directly regulate and mark cell proliferation.
PPARγ-Dependent Gene Profile in Human A-MØ
To assess the physiological relevance of the above findings, we next analyzed the functional and transcriptional consequences of siRNA-mediated PPARγ knockdown in human A-MØ isolated from BAL fluids. In agreement with previous reports (61, 62), isolated A-MØ expressed CD163 and exhibited very low levels of cell surface CD14 (Figure 6A). Regarding PPARγ expression, A-MØ exhibited a level of PPARG1/3 and PPARG2 mRNA expression similar to that found in GM-MØ and considerably higher than the expression seen in M-MØ (Figure 6B), and responded to the presence of the PPARγ agonists GW7845 by enhancing the expression of CD36 (Figure 6C). At the functional level, PPARγ knockdown in A-MØ did not significantly modify the LPS-induced production of TNFα or IL-6 (Figure 6D), thus indicating that PPARγ does not regulate the LPS-induced production of proinflammatory cytokines by A-MØ in the absence of an exogenous agonist, a result also seen in monocyte-derived GM-MØ (Figure 1G). By contrast, PPARγ knockdown significantly enhanced CCL2 production by human A-MØ both under basal and LPS-stimulated conditions (Figure 6D). Regarding the transcriptional role of PPARγ in ex vivo isolated human A-MØ, PPARγ silencing in A-MØ yielded similar effects to those previously observed in GM-MØ (Figure 6E). Specifically, PPARγ knockdown caused a significant upregulation of CCL2, CCL8, CSF1, TNFSF15, OSM, and LIF, and a significant downregulation of TLR4, ECSCR, CD36, MSR1, TNFRSF21, and CARD16 (Figure 6E). However, and in contrast with its effects on GM-MØ, downregulation of PPARγ in human A-MØ had no effect on the expression of genes encoding proteins involved in cell cycle regulation (Figure 6E). Altogether, these results indicate that PPARγ significantly contributes to the transcriptional signature of GM-CSF-conditioned human macrophages (either proinflammatory monocyte-derived GM-MØ or ex vivo isolated A-MØ) in the absence of exogenous agonists.
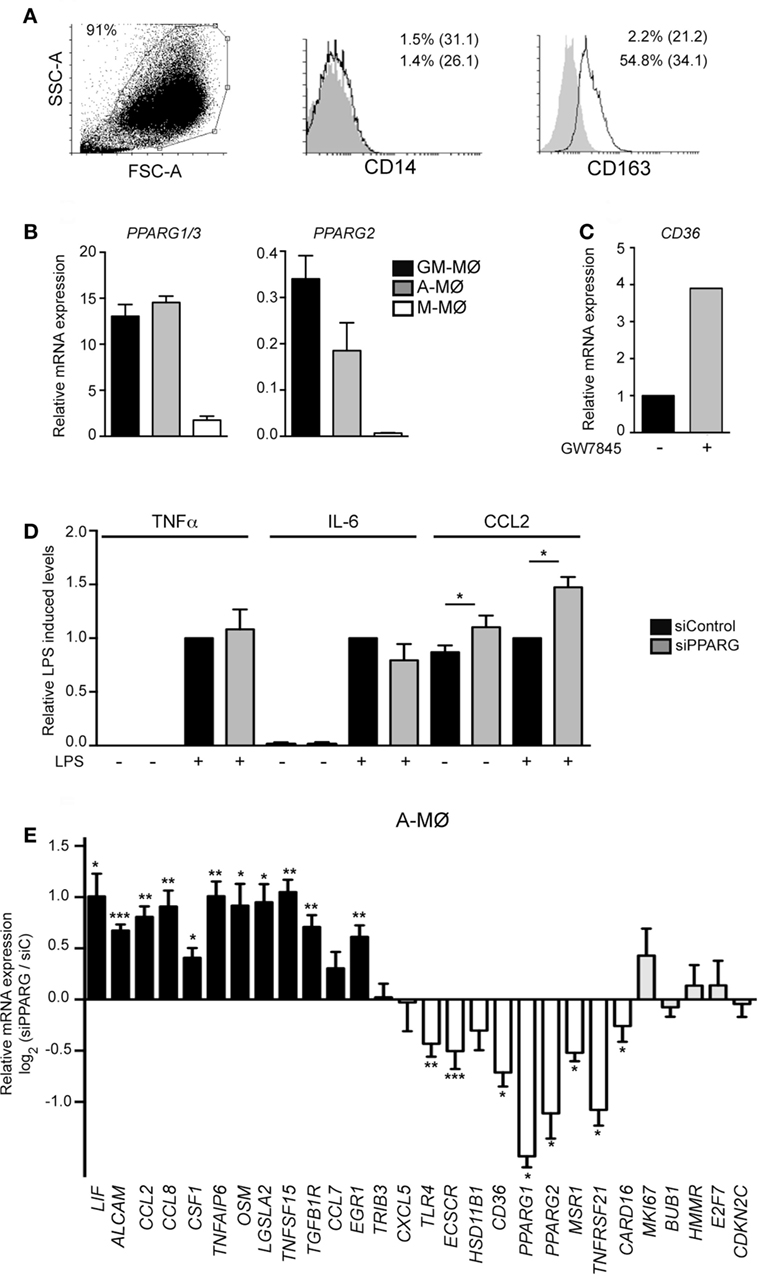
Figure 6. Peroxisome proliferator-activated receptor gamma (PPARγ) controls the transcriptome of human alveolar macrophages. (A) Cell surface expression of CD14 and CD163 in Alveolar macrophage (A-MØ) isolated from a representative bronchoalveolar lavage, as determined by flow cytometry. Background fluorescence was determined using isotype-matched antibodies (gray histograms). Forward scatter (FSC)/Side scatter (SSC) analysis of isolated A-MØ is shown in the left panel. (B) PPARG1/3 and PPARG2 mRNA expression in siControl-transfected A-MØ, GM-MØ, and M-CSF-dependent monocyte-derived macrophages (M-MØ), as determined by quantitative real-time PCR (qRT-PCR) and relative to TBP mRNA levels. Mean and SEM of three independent experiments is shown. (C) CD36 mRNA expression in A-MØ cultured for 24 h after isolation in the presence of GW7845 (1 µM) or DMSO (−), as determined by qRT-PCR. Results are indicated relative to the CD36 mRNA expression in the presence of DMSO (arbitrarily set to 1). One representative experiment is shown. (D) TNFα, IL-6, and CCL2 production in LPS-treated (24 h) siPPARG-transfected and siControl-transfected A-MØ. Results indicate the concentration of each cytokine for each condition relative to the cytokine levels detected in siControl-transfected A-MØ (arbitrarily set to 1). Mean and SEM of three independent experiments are shown (*p < 0.05). (E) Expression of the indicated genes in siPPARG-transfected and siControl-transfected (siC) A-MØ, as determined by qRT-PCR. Results are indicated as the mRNA levels of each gene in siPPARG-transfected relative to the levels in siControl-tranfected (siC) A-MØ (n = 6–7; *p < 0.05; **p < 0.01; ***p < 0.001).
Discussion
Macrophages adapt to changes in the extracellular environment very efficiently and alter their phenotype and effector functions according to their surrounding milieu. Although GM-CSF-conditioned monocyte-derived macrophages (GM-MØ) produce large amounts of proinflammatory cytokines upon TLR stimulation, they express high levels of PPARγ, whose ligand-induced activation down-modulates macrophage proinflammatory responses (35, 36), skews monocytes toward an anti-inflammatory phenotype (38, 63, 64), and limits inflammation in metabolically activated macrophages (39). Assessment of the function of PPARγ in human macrophages has now revealed that (1) the range of PPARγ target genes differs between proinflammatory (GM-MØ) and anti-inflammatory (M-MØ) monocyte-derived macrophages; (2) PPARG1/3 and PPARG2 are preferentially expressed by human GM-MØ; (3) activin A mediates the GM-CSF-induced expression of PPARγ in in vitro generated GM-MØ and ex vivo isolated A-MØ; and (4) PPARγ shapes the gene signature of GM-CSF-conditioned human macrophages in the absence of exogenous agonists. The involvement of activin A in PPARγ expression in GM-CSF-dependent human macrophages points toward a connection between Smad2/3 activation and PPARG gene expression, whose existence has been already suggested in PAP patients, which exhibit a deficiency in PPARγ and a severe reduction in Activin A expression and secretion (42). Thus, our results provide a molecular explanation for such a correlation, and support the existence of a functional GM-CSF/activin A/PPARγ axis in human macrophages.
The higher PPARγ expression exhibited by human proinflammatory GM-MØ is reminiscent of the differential PPARγ levels seen in mouse Ly-6Chi (low PPARγ) and peripheral blood monocytes (34). From this point of view, and since PPARγhigh Ly-6Clo monocytes are thought to become “M2 polarized” macrophages within tissues (and PPARγlow Ly-6Chi monocytes are thought to give rise to “M1 polarized macrophages”), the expression of PPARγ might mark macrophages with distinct inflammatory activities in mice and humans. A possible explanation for this discrepancy could derive from the fact that PPARγlow Ly-6Chi monocytes are precursors of PPARγhigh Ly-6Clo monocytes (65), and that exposure of M-MØ (low PPARγ) to GM-CSF leads to enhanced expression of PPARγ. Therefore, it is tempting to speculate that high PPARγ expression marks macrophages (murine and human) that have been already exposed to an activating/proinflammatory stimulus. Moreover, the preferential expression of PPARG1 and PPARG2 in proinflammatory GM-MØ is in apparent contradiction with the correlation between PPARγ expression and the presence of M2/anti-inflammatory markers in macrophages from human carotid atherosclerotic lesions (63), and with the contribution of PPARγ to the IL-4- and STAT6-dependent M2 macrophage polarization (38). The higher levels of PPARG seen in GM-MØ might be related to the acquisition of the ability to halt proinflammatory responses in a fast and efficient manner, allowing macrophages to rapidly switch from a proinflammatory into an anti-inflammatory polarization state to avoid excessive tissue damage before restoring homeostasis. This explanation is compatible with the preferential expression of other anti-inflammatory/immunosuppressive genes like VDR or HSD11B1 (11) in GM-MØ (GSE27792) and with the ability of PPARγ to limit inflammation in macrophages metabolically activated by glucose, insulin and palmitate (39).
Apart from the polarization-dependent expression of PPARγ (Figure 3) and the distinct cytokine responsiveness of PPARG expression in GM-MØ and M-MØ (data not shown), we have found that the range of genes specifically modulated by the PPARγ agonist GW7845 differs between proinflammatory GM-MØ and anti-inflammatory M-MØ (Figure 1). Although some of the differential PPARγ target genes had been shown to be modulated by PPARγ in various cell types (66–70), the distinct transcriptional consequence of PPARγ activation in human GM-MØ or M-MØ was, to our knowledge, so far unknown. Importantly, PPARγ activation also has different functional consequences in both macrophage subtypes, because the PPARγ agonist GW7845 significantly inhibit the LPS-induced production of proinflammatory cytokines (IL-6, TNFα) in GM-MØ, but has no effect on M-MØ. Therefore, our results indicate that PPARγ activation leads to distinct outcomes in human macrophages exhibiting opposite transcriptional and functional profiles (GM-MØ and M-MØ). This result agrees with those reported by Bouhlel et al. (63), who found that PPARγ activation exclusively modulates CD163 and CD206 in IL-4-polarized macrophages, and supports the polarization-dependent activity of PPARγ in macrophages. Our results on human macrophages are also in line with the divergent PPARγ binding landscape reported in human and mouse macrophages (71), as CCL2 and IL10 mRNA levels, exclusively downregulated by GW7845 in GM-MØ, were diminished in both murine macrophage subtypes upon PPARγ activation, whereas THBS1 mRNA, whose levels were increased in human GM-MØ, were diminished in murine GM-MØ in response to GW7845.
The definition of the PPARγ-dependent transcriptome in GM-MØ also provides evidences to support that PPARγ is transcriptionally active in human macrophages not exposed to exogenous pharmacological PPARγ agonists. The relevance of this finding is further reinforced by the effect of PPARγ knockdown on the gene profile of ex vivo isolated A-MØ. Therefore, down-modulation of PPARγ expression suffices to alter the transcriptome of GM-CSF-conditioned human macrophages (in vitro generated GM-MØ and ex vivo isolated A-MØ) but does not influence their LPS-induced proinflammatory cytokine production. This feature suggests that PPARγ has a distinct role in resting and activated (e.g., LPS-exposed) macrophages. In the former, PPARγ shapes the macrophage transcriptome, positively regulating genes that encode Th2 cytokine-induced chemokines (CCL13, CCL22) and TLR4, and downregulating genes that code for NFκB-regulated monocyte-attracting chemokines (CCL2, CCL8). Both consequences are compatible with the function of PPARγ+ A-MØ, that remove airborne particles and pathogens while avoiding lung inflammatory responses, and indicate a prominent role for the PPARγ transcriptional activating ability in non-activated human macrophages. Conversely, the ability of PPARγ to impair proinflammatory cytokine production after LPS stimulation is only observed after agonist-induced activation, thus suggesting that the anti-inflammatory ability of PPARγ is displayed in full only upon macrophage activation.
In summary, we report that the functions of PPARγ in human macrophages are polarization-dependent, that activin A positively regulates PPARγ expression in GM-CSF-dependent macrophages, and that PPARγ shapes the transcriptome of GM-CSF-conditioned human macrophages in the absence of exogenous agonists. Regarding the latter, the large set of potential novel PPARγ target genes now identified in human macrophages, which code for molecules involved in PAMP and DAMP recognition, inflammatory cell migration, proliferation promotion and cell cycle progression, is indicative of the role of PPARγ in regulation of inflammatory responses and in defense against pathogens, and further supports its contribution to maintenance of lung homeostasis.
Ethics Statement
Buffy coats were obtained from healthy blood donors, as anonymously provided by the Comunidad de Madrid blood Bank. Ethical approvals for all blood sources and processes used in this study were approved by the Centro de Investigaciones Biológicas Ethics Committee. All experiments were carried out in accordance with the approved guidelines and regulations. All experiments on mice were conducted according to the Spanish and European regulations on care and protection of laboratory animals and were approved by the Centro de Investigaciones Biológicas animal facility and the Consejo Superior de Investigaciones Científicas Ethics Committee.
Author Contributions
CN, RB, CM, ES-F, BA, ME, JD-A and CA designed research, performed research, and analyzed data; AC and MAV designed research and analyzed data; CN, AP-K, and ALC conceived the study, designed research, analyzed data, and wrote the article. All authors had final approval of the version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from Ministerio de Economía y Competitividad (SAF2014-54423-R and SAF2017-83785-R to MAV and ALC, SAF2014-56819-R and SAF2015-71878-REDT to AC, and SAF2015-69905 to CA), Comunidad Autónoma de Madrid/FEDER (RAPHYME S2010/BMD-2350 to ALC, AP-K and AC), Instituto de Salud Carlos III (PI14/00075, PI17/00037 and Red de Investigación en Enfermedades Reumáticas, RIER RD16/0012/0007 to AP-K), and cofinanced by the European Regional Development Fund “A way to achieve Europe” (ERDF).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fimmu.2018.00031/full#supplementary-material.
Footnotes
References
1. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol (2010) 11:889–96. doi:10.1038/ni.1937
2. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi:10.1016/j.immuni.2014.06.008
3. Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol (2008) 8:533–44. doi:10.1038/nri2356
4. Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood (2002) 99:111–20. doi:10.1182/blood.V99.1.111
5. Wiktor-Jedrzejczak W, Bartocci A, Ferrante AW Jr, Ahmed-Ansari A, Sell KW, Pollard JW, et al. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc Natl Acad Sci U S A (1990) 87:4828–32. doi:10.1073/pnas.87.12.4828
6. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med (2013) 210:1977–92. doi:10.1084/jem.20131199
7. Becher B, Tugues S, Greter M. GM-CSF: from growth factor to central mediator of tissue inflammation. Immunity (2016) 45:963–73. doi:10.1016/j.immuni.2016.10.026
8. Wicks IP, Roberts AW. Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol (2016) 12:37–48. doi:10.1038/nrrheum.2015.161
9. Verreck FA, de Boer T, Langenberg DM, Hoeve MA, Kramer M, Vaisberg E, et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci U S A (2004) 101:4560–5. doi:10.1073/pnas.0400983101
10. Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol (2007) 178:5245–52. doi:10.4049/jimmunol.178.8.5245
11. Sierra-Filardi E, Puig-Kroger A, Blanco FJ, Nieto C, Bragado R, Palomero MI, et al. Activin A skews macrophage polarization by promoting a proinflammatory phenotype and inhibiting the acquisition of anti-inflammatory macrophage markers. Blood (2011) 117:5092–101. doi:10.1182/blood-2010-09-306993
12. Gonzalez-Dominguez E, Dominguez-Soto A, Nieto C, Flores-Sevilla JL, Pacheco-Blanco M, Campos-Pena V, et al. Atypical activin A and IL-10 production impairs human CD16+ monocyte differentiation into anti-inflammatory macrophages. J Immunol (2016) 196:1327–37. doi:10.4049/jimmunol.1501177
13. Lacey DC, Achuthan A, Fleetwood AJ, Dinh H, Roiniotis J, Scholz GM, et al. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J Immunol (2012) 188:5752–65. doi:10.4049/jimmunol.1103426
14. Gonzalez-Dominguez E, Samaniego R, Flores-Sevilla JL, Campos-Campos SF, Gomez-Campos G, Salas A, et al. CD163L1 and CLEC5A discriminate subsets of human resident and inflammatory macrophages in vivo. J Leukoc Biol (2015) 98:453–66. doi:10.1189/jlb.3HI1114-531R
15. Soler Palacios B, Estrada-Capetillo L, Izquierdo E, Criado G, Nieto C, Municio C, et al. Macrophages from the synovium of active rheumatoid arthritis exhibit an activin A-dependent pro-inflammatory profile. J Pathol (2015) 235:515–26. doi:10.1002/path.4466
16. Phillips DJ, de Kretser DM, Hedger MP. Activin and related proteins in inflammation: not just interested bystanders. Cytokine Growth Factor Rev (2009) 20:153–64. doi:10.1016/j.cytogfr.2009.02.007
17. Sozzani S, Musso T. The yin and yang of Activin A. Blood (2011) 117:5013–5. doi:10.1182/blood-2011-03-342691
18. Jones KL, Mansell A, Patella S, Scott BJ, Hedger MP, de Kretser DM, et al. Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc Natl Acad Sci U S A (2007) 104:16239–44. doi:10.1073/pnas.0705971104
19. Robson NC, Phillips DJ, McAlpine T, Shin A, Svobodova S, Toy T, et al. Activin-A: a novel dendritic cell-derived cytokine that potently attenuates CD40 ligand-specific cytokine and chemokine production. Blood (2008) 111:2733–43. doi:10.1182/blood-2007-03-080994
20. Salogni L, Musso T, Bosisio D, Mirolo M, Jala VR, Haribabu B, et al. Activin A induces dendritic cell migration through the polarized release of CXC chemokine ligands 12 and 14. Blood (2009) 113:5848–56. doi:10.1182/blood-2008-12-194597
21. Musso T, Scutera S, Vermi W, Daniele R, Fornaro M, Castagnoli C, et al. Activin A induces Langerhans cell differentiation in vitro and in human skin explants. PLoS One (2008) 3:e3271. doi:10.1371/journal.pone.0003271
22. Ditiatkovski M, Toh BH, Bobik A. GM-CSF deficiency reduces macrophage PPAR-gamma expression and aggravates atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol (2006) 26:2337–44. doi:10.1161/01.ATV.0000238357.60338.90
23. Bonfield TL, Farver CF, Barna BP, Malur A, Abraham S, Raychaudhuri B, et al. Peroxisome proliferator-activated receptor-gamma is deficient in alveolar macrophages from patients with alveolar proteinosis. Am J Respir Cell Mol Biol (2003) 29:677–82. doi:10.1165/rcmb.2003-0148OC
24. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol (2014) 15:1026–37. doi:10.1038/ni.3005
25. Szanto A, Nagy L. The many faces of PPARgamma: anti-inflammatory by any means? Immunobiology (2008) 213:789–803. doi:10.1016/j.imbio.2008.07.015
26. Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med (2002) 53:409–35. doi:10.1146/annurev.med.53.082901.104018
27. Xu HE, Lambert MH, Montana VG, Plunket KD, Moore LB, Collins JL, et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc Natl Acad Sci U S A (2001) 98:13919–24. doi:10.1073/pnas.241410198
28. Kiss M, Czimmerer Z, Nagy L. The role of lipid-activated nuclear receptors in shaping macrophage and dendritic cell function: from physiology to pathology. J Allergy Clin Immunol (2013) 132:264–86. doi:10.1016/j.jaci.2013.05.044
29. Ricote M, Glass CK. PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta (2007) 1771:926–35. doi:10.1016/j.bbalip.2007.02.013
30. Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol (2010) 10:365–76. doi:10.1038/nri2748
31. Torchia J, Glass C, Rosenfeld MG. Co-activators and co-repressors in the integration of transcriptional responses. Curr Opin Cell Biol (1998) 10:373–83. doi:10.1016/S0955-0674(98)80014-8
32. Chawla A. Control of macrophage activation and function by PPARs. Circ Res (2010) 106:1559–69. doi:10.1161/CIRCRESAHA.110.216523
33. Szeles L, Torocsik D, Nagy L. PPARgamma in immunity and inflammation: cell types and diseases. Biochim Biophys Acta (2007) 1771:1014–30. doi:10.1016/j.bbalip.2007.02.005
34. Gautier EL, Chow A, Spanbroek R, Marcelin G, Greter M, Jakubzick C, et al. Systemic analysis of PPARgamma in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity. J Immunol (2012) 189:2614–24. doi:10.4049/jimmunol.1200495
35. Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature (1998) 391:82–6. doi:10.1038/34184
36. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature (1998) 391:79–82. doi:10.1038/34178
37. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature (2007) 447:1116–20. doi:10.1038/nature05894
38. Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity (2010) 33:699–712. doi:10.1016/j.immuni.2010.11.009
39. Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab (2014) 20:614–25. doi:10.1016/j.cmet.2014.08.010
40. Fajas L, Fruchart JC, Auwerx J. PPARgamma3 mRNA: a distinct PPARgamma mRNA subtype transcribed from an independent promoter. FEBS Lett (1998) 438:55–60. doi:10.1016/S0014-5793(98)01273-3
41. Trapnell BC, Carey BC, Uchida K, Suzuki T. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr Opin Immunol (2009) 21:514–21. doi:10.1016/j.coi.2009.09.004
42. Bonfield TL, Barna BP, John N, Malur A, Culver DA, Kavuru MS, et al. Suppression of activin A in autoimmune lung disease associated with anti-GM-CSF. J Autoimmun (2006) 26:37–41. doi:10.1016/j.jaut.2005.10.004
43. Ginhoux F. Fate PPAR-titioning: PPAR-gamma ‘instructs’ alveolar macrophage development. Nat Immunol (2014) 15:1005–7. doi:10.1038/ni.3011
44. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med (2017) 214:2387–404. doi:10.1084/jem.20162152
45. Heron M, Grutters JC, ten Dam-Molenkamp KM, Hijdra D, van Heugten-Roeling A, Claessen AM, et al. Bronchoalveolar lavage cell pattern from healthy human lung. Clin Exp Immunol (2012) 167:523–31. doi:10.1111/j.1365-2249.2011.04529.x
46. Rathinam C, Poueymirou WT, Rojas J, Murphy AJ, Valenzuela DM, Yancopoulos GD, et al. Efficient differentiation and function of human macrophages in humanized CSF-1 mice. Blood (2011) 118:3119–28. doi:10.1182/blood-2010-12-326926
48. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics (2013) 14:128. doi:10.1186/1471-2105-14-128
49. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res (2016) 44:W90–7. doi:10.1093/nar/gkw377
50. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A (2005) 102:15545–50. doi:10.1073/pnas.0506580102
51. Ricote M, Huang J, Fajas L, Li A, Welch J, Najib J, et al. Expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc Natl Acad Sci U S A (1998) 95:7614–9. doi:10.1073/pnas.95.13.7614
52. Fleetwood AJ, Dinh H, Cook AD, Hertzog PJ, Hamilton JA. GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J Leukoc Biol (2009) 86:411–21. doi:10.1189/jlb.1108702
53. Fang L, Zhang M, Li Y, Liu Y, Cui Q, Wang N. PPARgene: a database of experimentally verified and computationally predicted PPAR target genes. PPAR Res (2016) 2016:6042162. doi:10.1155/2016/6042162
54. Szatmari I, Torocsik D, Agostini M, Nagy T, Gurnell M, Barta E, et al. PPARgamma regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood (2007) 110:3271–80. doi:10.1182/blood-2007-06-096222
55. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol (2012) 13:1118–28. doi:10.1038/ni.2419
56. Tomlinson GS, Booth H, Petit SJ, Potton E, Towers GJ, Miller RF, et al. Adherent human alveolar macrophages exhibit a transient pro-inflammatory profile that confounds responses to innate immune stimulation. PLoS One (2012) 7:e40348. doi:10.1371/journal.pone.0040348
57. Schultze JL, Freeman T, Hume DA, Latz E. A transcriptional perspective on human macrophage biology. Semin Immunol (2015) 27:44–50. doi:10.1016/j.smim.2015.02.001
58. Corona JC, Duchen MR. PPARgamma as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic Biol Med (2016) 100:153–63. doi:10.1016/j.freeradbiomed.2016.06.023
59. Balli D, Ren X, Chou FS, Cross E, Zhang Y, Kalinichenko VV, et al. Foxm1 transcription factor is required for macrophage migration during lung inflammation and tumor formation. Oncogene (2012) 31:3875–88. doi:10.1038/onc.2011.549
60. Ren X, Shah TA, Ustiyan V, Zhang Y, Shinn J, Chen G, et al. FOXM1 promotes allergen-induced goblet cell metaplasia and pulmonary inflammation. Mol Cell Biol (2013) 33:371–86. doi:10.1128/MCB.00934-12
61. Lensmar C, Katchar K, Eklund A, Grunewald J, Wahlstrom J. Phenotypic analysis of alveolar macrophages and lymphocytes following allergen inhalation by atopic subjects with mild asthma. Respir Med (2006) 100:918–25. doi:10.1016/j.rmed.2005.08.014
62. Ferrari-Lacraz S, Nicod LP, Chicheportiche R, Welgus HG, Dayer JM. Human lung tissue macrophages, but not alveolar macrophages, express matrix metalloproteinases after direct contact with activated T lymphocytes. Am J Respir Cell Mol Biol (2001) 24:442–51. doi:10.1165/ajrcmb.24.4.4008
63. Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab (2007) 6:137–43. doi:10.1016/j.cmet.2007.06.010
64. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi:10.1146/annurev-physiol-021909-135846
65. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity (2013) 38:79–91. doi:10.1016/j.immuni.2012.12.001
66. Dasu MR, Park S, Devaraj S, Jialal I. Pioglitazone inhibits toll-like receptor expression and activity in human monocytes and db/db mice. Endocrinology (2009) 150:3457–64. doi:10.1210/en.2008-1757
67. Bonfield TL, Thomassen MJ, Farver CF, Abraham S, Koloze MT, Zhang X, et al. Peroxisome proliferator-activated receptor-Î3 regulates the expression of alveolar macrophage macrophage colony-stimulating factor. J Immunol (2008) 181:235–42. doi:10.4049/jimmunol.181.1.235
68. Neri T, Armani C, Pegoli A, Cordazzo C, Carmazzi Y, Brunelleschi S, et al. Role of NF-κB and PPAR-γ in lung inflammation induced by monocyte-derived microparticles. Eur Respir J (2011) 37:1494–502. doi:10.1183/09031936.00023310
69. McClelland S, Cox C, O’Connor R, de Gaetano M, McCarthy C, Cryan L, et al. Conjugated linoleic acid suppresses the migratory and inflammatory phenotype of the monocyte/macrophage cell. Atherosclerosis (2010) 211:96–102. doi:10.1016/j.atherosclerosis.2010.02.003
70. Hounoki H, Sugiyama E, Mohamed SG-K, Shinoda K, Taki H, Abdel-Aziz HO, et al. Activation of peroxisome proliferator-activated receptor Î3 inhibits TNF-α-mediated osteoclast differentiation in human peripheral monocytes in part via suppression of monocyte chemoattractant protein-1 expression. Bone (2008) 42:765–74. doi:10.1016/j.bone.2007.11.016
Keywords: transcription factor, macrophage, peroxisome proliferator-activated receptor, inflammation, innate immunity
Citation: Nieto C, Bragado R, Municio C, Sierra-Filardi E, Alonso B, Escribese MM, Domínguez-Andrés J, Ardavín C, Castrillo A, Vega MA, Puig-Kröger A and Corbí AL (2018) The Activin A-Peroxisome Proliferator-Activated Receptor Gamma Axis Contributes to the Transcriptome of GM-CSF-Conditioned Human Macrophages. Front. Immunol. 9:31. doi: 10.3389/fimmu.2018.00031
Received: 26 July 2017; Accepted: 04 January 2018;
Published: 29 January 2018
Edited by:
Alexandre Corthay, Oslo University Hospital, NorwayReviewed by:
Ana Rosa Pérez, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaCopyright: © 2018 Nieto, Bragado, Municio, Sierra-Filardi, Alonso, Escribese, Domínguez-Andrés, Ardavín, Castrillo, Vega, Puig-Kröger and Corbí. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Concha Nieto, Y25pZXRvQGNpYi5jc2ljLmVz
†These authors have contributed equally to this work.