 Derya Kabacaoglu
Derya Kabacaoglu Katrin J. Ciecielski
Katrin J. Ciecielski Dietrich A. Ruess
Dietrich A. Ruess Hana Algül
Hana Algül- Internal Medicine II, Klinikum rechts der Isar, Technische Universität München, Munich, Germany
Pancreatic ductal adenocarcinoma (PDAC), as the most frequent form of pancreatic malignancy, still is associated with a dismal prognosis. Due to its late detection, most patients are ineligible for surgery, and chemotherapeutic options are limited. Tumor heterogeneity and a characteristic structure with crosstalk between the cancer/malignant cells and an abundant tumor microenvironment (TME) make PDAC a very challenging puzzle to solve. Thus far, targeted therapies have failed to substantially improve the overall survival of PDAC patients. Immune checkpoint inhibition, as an emerging therapeutic option in cancer treatment, shows promising results in different solid tumor types and hematological malignancies. However, PDAC does not respond well to immune checkpoint inhibitors anti-programmed cell death protein 1 (PD-1) or anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) alone or in combination. PDAC with its immune-privileged nature, starting from the early pre-neoplastic state, appears to escape from the antitumor immune response unlike other neoplastic entities. Different mechanisms how cancer cells achieve immune-privileged status have been hypothesized. Among them are decreased antigenicity and impaired immunogenicity via both cancer cell-intrinsic mechanisms and an augmented immunosuppressive TME. Here, we seek to shed light on the recent advances in both bench and bedside investigation of immunotherapeutic options for PDAC. Furthermore, we aim to compile recent data about how PDAC adopts immune escape mechanisms, and how these mechanisms might be exploited therapeutically in combination with immune checkpoint inhibitors, such as PD-1 or CTLA-4 antibodies.
Introduction
Pancreatic ductal adenocarcinoma (PDAC), as one of the most fatal malignancies in the world, is the fourth leading cause of cancer-related deaths among both men and women in developed countries (1). Its mortality almost equals its incidence: for 2018 alone, 55,440 diagnoses of pancreatic cancer are projected for the United States with 44,330 associated deaths in the same year (2). At the time of diagnosis, only a minority of patients have localized, resectable tumors (10%); while most patients display locally advanced disease (29%) and/or distant metastasis (52%), and the remainder are not even staged (2). The 5-year survival rate of pancreatic cancer is only around 7–8% in the United States, which is likely due to late stage diagnosis (2, 3). The high number of estimated pancreatic cancer-related deaths can be hypothesized to be due to several factors: first, late and unspecific symptoms, as well as a lack of PDAC-specific markers or screening resources result in late diagnosis at an advanced stage. Second, delayed diagnosis leads to low resection rates, since most of the tumor patients present with locally advanced or metastatic disease. And third, PDAC is characterized by a low response to radiotherapy (RT) and chemotherapy, which, at least in part, is due to its dense desmoplastic stroma impairing drug delivery (4–6). Also, targeted therapies including small drug inhibitors of key molecular signaling pathways associated with PDAC progression showed almost none (i.e., MEK and PI3K) or only mild benefits (e.g., EGFR) with a moderate increase in overall survival (7–12). Recent advances in chemotherapeutic options for PDAC appear to provide a survival benefit that will likely not be sufficient to decrease mortality rates (13–15). Hence, in developed countries, PDAC is expected to be the second leading cause of cancer-related deaths by 2030 (1, 16). Impaired efficacy of chemotherapy or targeted therapies in cancer was associated with innate and acquired resistance through genetic and epigenetic instability of cancer cells (17, 18). Immunotherapy offers great potential for the treatment of tumors displaying such resistance. Especially T cells with their ability to generate receptors recognizing the heterogeneous and specific repertoire of tumor-related antigens provide great promise in cancer therapeutics. The adaptive immune response might even form an immunological memory providing long-term tumor control. Studies revealing how T cells function under pathophysiological conditions led to development of immune checkpoint inhibitors, which have been successfully translated into the clinic. Thus far, immune checkpoint inhibition (ICI) has shown promising results for the treatment of solid tumors, including melanomas (19–22), as well as lung (23–27), renal (28, 29), bladder (30–32), and head and neck cancers (33, 34), as well as in hematological malignancies, such as Hodgkin’s disease (35, 36) and follicular or diffuse-large B-cell lymphoma (37). Although single-agent treatment with immune checkpoint inhibitors showed great promise with many solid tumors, their effect on PDAC has been quite disappointing (38, 39).
Here, we want to discuss the unique characteristics of PDAC immune evasion and why PDAC is unresponsive toward checkpoint inhibition. First, we will provide details concerning immune checkpoint inhibitors and their mechanism of action. Second, the immune-privileged nature of PDAC will be examined. Then, the antigenic and immunogenic attributes of PDAC and how tumor cell-intrinsic and -extrinsic factors within the tumor microenvironment (TME) regulate immunogenicity will be comprehensively discussed, including options for pharmacological modulation of these mechanisms to increase ICI therapy response in the clinic.
Immune Checkpoint Inhibitors
T cells with their various subsets are involved in the regulation of immune responses in autoimmune diseases, but also against infections and cancer. In TME or in tumor-resident lymph nodes, professional antigen-presenting cells (APCs) such as dendritic cells (DCs) display tumor-specific antigens to naïve T cells through major histocompatibility complexes (MHCs) in a process called priming (40). Antigen presentation through MHC-class II acts on naïve CD4+ T cells, giving rise to Th1, Th2, and Foxp3+ regulatory T cell (Treg) subtypes, which are all important for immune response orchestration (41): Th1 polarization induces cytokines (characterized mainly by IFNγ production) further augmenting MHC expression in APCs (42) and antitumor T cell and macrophage cytotoxic activity (43). Th2 polarized cells are characterized by IL-4 and IL-13 production, leading to exhaustion of T cells and enhancement of other tumor-promoting responses (44, 45). Tregs get activated once the effector T cell activation reaches a threshold. With the release of immunosuppressive cytokines (TGFβ and IL-10) Tregs negatively regulate T cell effector function (46). On the other hand, antigen presentation through MHC-class I leads to differentiation of naïve CD8+ T cells into cytotoxic T lymphocytes (CTLs), which are directly able to kill antigen-expressing cancer cells (41). Upon MHC:antigen engagement, activated T cells clonally expand in secondary lymphoid organs, and traffic into the inflammatory sites to execute their functions and release intermediary cytokines and ligands to provoke helper immune cells for further support (40).
T cell-mediated immune response is tightly regulated via both the repertoire of immunosuppressive cells in the microenvironment and cell-intrinsic regulation of anergy and exhaustion (47). T cell anergy is the state of T cells in which they are hyporesponsive to triggers of naïve T cell differentiation (47). And T cell exhaustion describes a process by which effector T cells become resistant to persistent reactivation (47). Under physiological conditions, T cell activation upon MHC engagement is balanced via co-regulation of both stimulatory and inhibitory signals, referred to as immune checkpoints. The balance between stimulatory and inhibitory signals is crucial to generate self-tolerance and to maintain the ability to fight with non-self. However, tumor cells shift this balance toward their benefit by abrogating co-activatory signals and augmenting co-inhibitory signals ultimately heightening anergy and exhaustion (48).
Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4 or CD152) and programmed cell death protein 1 (PD-1 or CD279) are the most studied co-inhibitory receptors of T cell receptor (TCR) signaling (40). The first antibody against CTLA-4, ipilimumab, was approved in 2011 (19), while pembrolizumab and nivolumab, antibodies that both target PD-1, were approved in 2014 for the treatment of melanoma (20, 21, 38). The clinical success of antibodies targeting CTLA-4 and PD-1 marks a breakthrough as these agents established immunotherapy as a new pillar of cancer treatment strategies next to surgery, chemotherapy, and radiation therapy (49).
After TCR engagement with cognate peptide presented by a MHC molecule, costimulatory receptor CD28 binding with CD80 (B7.1) or CD86 (B7.2) amplifies TCR signaling (50). CTLA-4, on the other hand, has higher affinity for CD80 and CD86, outcompeting CD28 binding (50, 51), and subsequently sequestering CD80 and CD86 from the APC surface (52). Initial TCR activation with CD28 co-activation increases IL-2 release, which induces metabolism, proliferation, and survival in a paracrine manner. However, gradual CTLA-4 accumulation on the T cell membrane replaces the activation signal of CD28, blocking IL-2 accumulation (53). Since B7 proteins are expressed on APCs but not on solid tumor cells, the action of CTLA-4 inhibition is thought to take place in secondary lymphoid organs where early T cell activation occurs. CTLA-4 action on CD8+ CTLs is inhibitory, as shown in several studies (54, 55). Still, the overall inhibitory action of CTLA-4 is thought to mainly show itself through its action on CD4+ Foxp3+ Tregs, indirectly modulating CD8+ CTL action (48). Tregs produce CTLA-4 constitutively through the action of their subset defining transcription factor Foxp3 (56–58). Deletion of CTLA-4 in Tregs reduces their activity, blocking their immune-suppressive action (59, 60). Still, use of CTLA4 antibodies in preclinical mouse models of PDAC did not affect Treg infiltration in tumors while enhancing total CD4+ T cell presence (61). Tregs might also mediate effector T cell activation through APCs, impairing their B7 ligand expression, and thereby decreasing the CD28 co-activation signal on effector T cells (52). Overall, CTLA-4 engagement downregulates effector T cell activity, while enhancing Treg immunosuppressive activity (59, 62). Inhibiting CTLA-4 action might enhance immunosurveillance through both its action on effector and Tregs.
Programmed cell death protein 1 belongs to the family of CD28 proteins, initiating co-inhibitory signaling upon TCR engagement (63, 64). Ligands of PD-1 receptor PD-L1 (B7-H1 or CD274) and PD-L2 (B7-DC or CD273) belong to the B7 family of proteins (64–67). PD-1 is expressed mostly on late effector phase CD4+ helper T cells and CD8+ cytotoxic T cells in peripheral tissues (63, 68). Especially chronically activated, then exhausted CD8+ cytotoxic T cells show constitutive PD-1 production (69–72). Therefore, PD-1 action is mostly associated with the late phase of immune response, which counterbalances cytotoxic T cell activity. PD-1 is also expressed on Tregs and PD-1 blockage leads Treg apoptosis (73). Also, PD-L1 stimulation of naïve T cells can skew differentiation toward the Treg subset (74). Therefore, anti-PD-1 treatment might show an indirect effect on antitumor T cells through its inhibitory actions on Tregs (75).
Programmed cell death protein 1 knock out mice show reduced peripheral tolerance and display autoimmunity (76, 77), with a milder phenotype compared with CTLA-4 knock out mice (78, 79). There is a prominent difference between CTLA-4 and PD-1 effects. Anti-CTLA-4 action mostly results in changes in secondary lymphoid organs during the initial phase of naïve T cell activation, while anti-PD-1 treatment targets the effector phase of T cell activation in the periphery where the activated T cells attack the target (40, 48, 80). In addition, CTLA-4 is mobilized to the cell membrane upon TCR engagement in naïve T cells directly from the protein stores, implicating its importance for initial T cell activation (81). By contrast, PD-1 transport requires an initial transcriptional production causing a 6–12 h delay in response upon TCR engagement (48). Considering the differences in mode of action between CTLA-4 and PD-1, PD-1 blockage is thought to be effective in TME (80). Tumor-infiltrating lymphocytes (TILs) in the TME are frequently exhausted due to chronic exposure to the tumor antigens and PD-L1 directly produced by the tumor cells or anti-inflammatory cells of the TME (82). Anti-PD-1 or anti-PD-L1 therapy aims to reduce this exhausted state of TILs in the TME. Of note, PD-1 blockage (e.g., nivolumab) shows milder autoimmunity-related side effects than anti-CTLA-4 treatment (e.g., ipilimumab) in melanoma patients (19, 83). Considering anti-CTLA-4 and PD-1 therapy has implications in different phases of immune response, combination therapy with nivolumab and ipilimumab showed prolonged progression free survival and a higher objective response rate than ipilimumab alone, albeit with concomitant higher toxicity (84).
The Immune-Privileged Nature of PDAC: Immunosurveillance and Immunoediting
The immunosurveillance hypothesis was proposed by Paul Ehrlich (85) in the early 1900s and later developed further by Thomas and Burnet (86, 87). As a very important concept for cancer immunotherapy, immunosurveillance states that immune cells continually survey somatic cells for any malignant transformation to then destroy them (88). The concept of cancer immunoediting is a byproduct of the immunosurveillance process, in which cancer cells undergo a Darwinian-like selection for their capacity to evade an attack by the immune system (88). The concept of tumor immunoediting proposed by Schreiber and colleagues in 2002 states three different phases of tumor immunoediting: elimination, equilibrium, and escape (i.e., triple E hypothesis) (Figure 1) (88, 89). As being more comprehensive than immunosurveillance, immunoediting proposes that not only innate immunity but also adaptive immunity is involved in the elimination process of tumor cells. During the equilibrium phase, tumor cell variants surviving the dynamic but relentless pressure of adaptive and innate immunity undergo a Darwinian-like selection. At the end of the equilibrium phase, many of the tumor cells are dead, whereas new clones generated, likely through genetic instability with better resistance to the immune response, remain. In the escape phase, survivors of the equilibrium phase start to expand in numbers, maintaining an immune-privileged state (89).
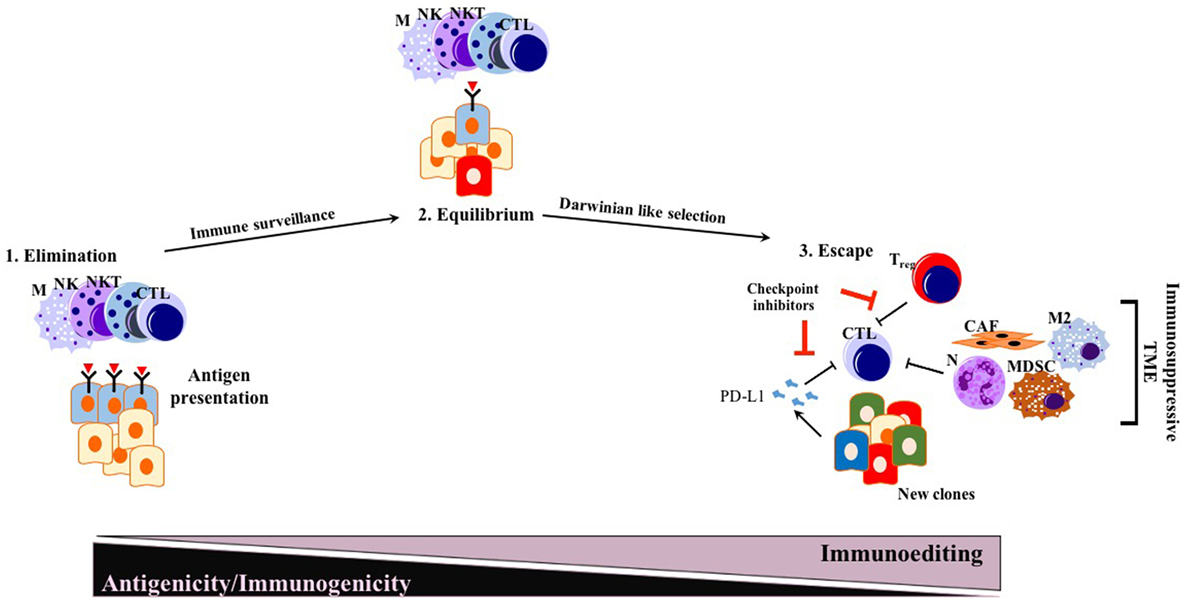
Figure 1. Conventional triple E hypothesis: elimination, equilibrium, and escape. While many solid tumors responding to ICI therapy follow triple E of immunoediting, PDAC is mostly an exception. Generated new clones due to Darwinian-like selection reduce their antigenicity and immunogenicity, escaping from immunosurveillance. Abbreviations: M, macrophages; NK, natural killer; NKT, natural killer T cells; CTLs, cytotoxic T lymphocytes; N, neutrophils; M2, M2 phenotype macrophages; MDSCs, myeloid-derived suppressor cells; Treg, regulatory T cells; CAFs, cancer-associated fibroblasts; ICI, immune checkpoint inhibition.
Before the wide use of genetically engineered mouse models (GEMMs) of PDAC, human or mouse tumor transplantation into mice had been the main model for preclinical studies of therapeutic response (90). To eliminate simple tissue rejection of tumor xenografts, mostly immune-incompetent mouse models had been utilized. However, these models are unsuitable for studies of the immune response toward tumors. Furthermore, syngeneic murine transplantation models do not provide information regarding the tumorigenesis process. GEMMs for PDAC harboring pancreas-specific expression of mutant Kras recapitulate carcinogenesis of human PDAC, as pre-neoplastic lesions (PanIN) reliably progress to invasive and metastatic cancer (91). In this mouse model, CD45+ leukocytes were shown to accumulate in time as the disease progresses. However, CD4+ T cells observed in PanIN lesions were mostly of the Foxp3+ Treg subtype, accompanied by an abundance of myeloid-derived suppressor cells (MDSCs) and M2 macrophages (92). Strikingly, infiltration by CD8+ antitumor cytotoxic T cells was very scarce in early PanIN lesions, and only a small portion of advanced tumors actually showed presence of active CD8+ CTLs (92). This spontaneous carcinogenesis model of PDAC highlights the immune-privileged status of PDAC even in the early neoplastic state (92). Unlike for many other solid tumors, the elimination phase of the triple E hypothesis is almost absent or substantially impaired during murine carcinogenesis due to the scarcity of cytotoxic immune cells and the abundant presence of immunosuppressive cells (92). Thus, ablation of T cells did not affect the spontaneous formation of cancer in KPC models (LSL—KrasG12D/+; LSL—Trp53R172H/+; Pdx—1Cre) (93). However, ectopic expression of a strong neoantigen (e.g., ovalbumin) in cancer cells boosted T cell-mediated immunity, rescuing the elimination phase of the immunoediting sequence. Expression of a single, yet strong, neoantigen thus allowed tumor control via CTL infiltration and “Triple E” (immune active) immunoediting. This implies that the scarcity of neoantigens in PDAC is not a result of the elimination step of immunoediting, but rather due to an alternative mechanism more like immune quiescence (Figure 2). Because of immune quiescence in tumors with low basal adaptive immune activation, CTLs cannot invade into the TME to initiate conventional immunoediting during carcinogenesis, which is true for the KPC model (93). This model represents human PDAC fairly well, showing an “immune quiescence like” phenotype rather than an “immune active” one (94). Reduced CD8+ CTL and increased CD4+ Foxp3+ Treg infiltration in progressive PDAC has also been validated in human patient samples (95).
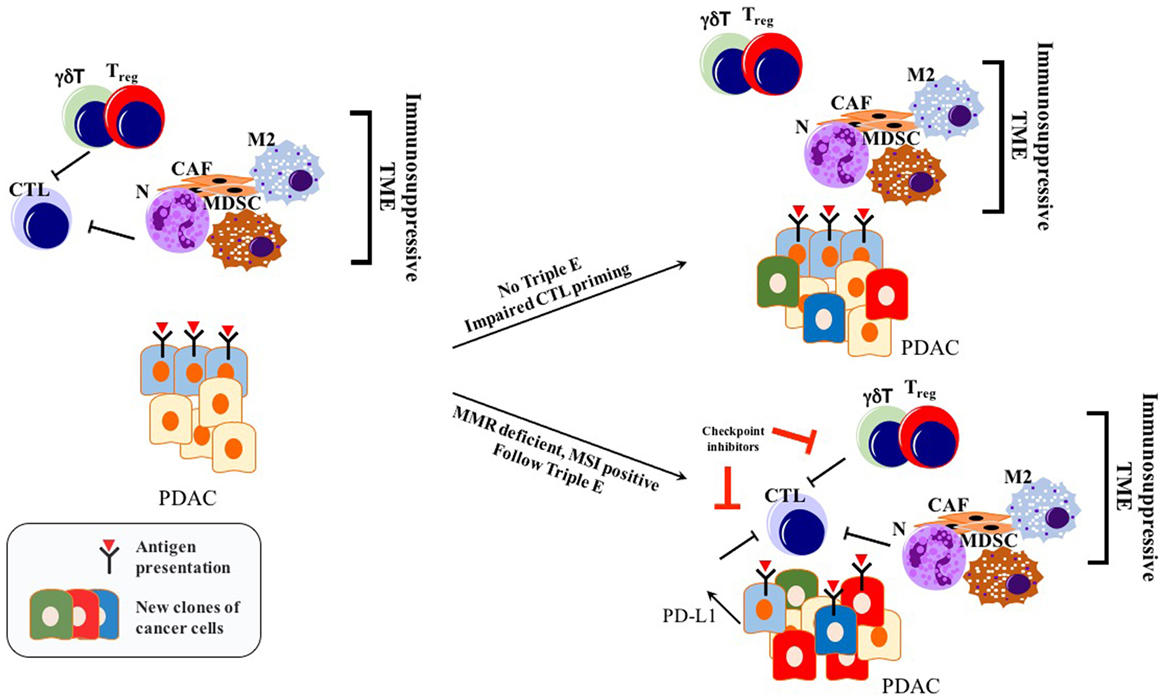
Figure 2. Immunoediting in PDAC: only tumors with genetic instability follow Triple E, while others cannot. Immunosuppressive TME blocks initial CTL priming. Therefore, cancer cells are not forced to undergo Darwinian-like selection. PDAC can still retain its antigenic capacity while impairing immunogenicity making it unresponsive to checkpoint inhibitors. Abbreviations: CTLs, cytotoxic T lymphocytes; N, neutrophils; M2, M2 phenotype macrophages; MDSCs, myeloid-derived suppressor cells; Tregs, regulatory T cells; CAFs, cancer-associated fibroblasts; γδT, γδT cells; MMR, mismatch repair; MSI, microsatellite instability; TME, tumor microenvironment.
In summary, PDAC frequently does not undergo a Darwinian-like selection with respect to the adaptive immune response. Thus, it retains vulnerability toward the natural T cell repertoire. Thus, strategies boosting T cell priming, activation levels, and attraction are promising for the treatment of this cancer (96).
Factors Determining the Efficacy of ICI and Failure in PDAC
Two important factors determine the prospects of immunotherapy of cancer in general, and checkpoint inhibition, in particular, antigenicity and immunogenicity, the latter being modulated by both intrinsic properties of tumor cells and TME (97) (Figure 3). Considering the, to date, low efficacy of immunotherapy, and especially checkpoint inhibition in PDAC, a better understanding of the immune escape mechanisms present in PDAC will pave the way for combination factors of checkpoint inhibition for the treatment of this generally intractable disease. A list of selected preclinical mouse model studies focusing on ICI combination therapies in PDAC can be found in Table 1.
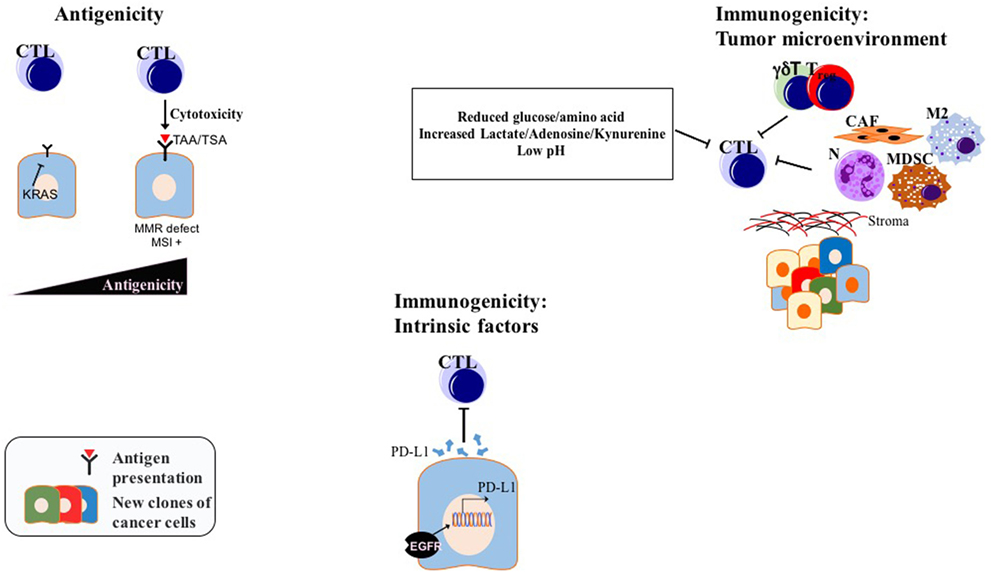
Figure 3. Factors determining ICI efficiency in PDAC: while modulation of antigenicity, intrinsic immunogenicity, and extrinsic immunogenicity via TME might be valid for many tumors, drawn examples above are experimentally shown for PDAC. Abbreviations: TAAs, tumor-associated antigens; TSAs, tumor-specific antigens; CTLs, cytotoxic T lymphocytes; N, neutrophils; M2, M2 phenotype macrophages; MDSCs, myeloid-derived suppressor cells; Tregs, regulatory T cells; CAFs, cancer-associated fibroblasts; γδT, γδT cells; MMR, mismatch repair; MSI, microsatellite instability; TME, tumor microenvironment; ICI, immune checkpoint inhibition.
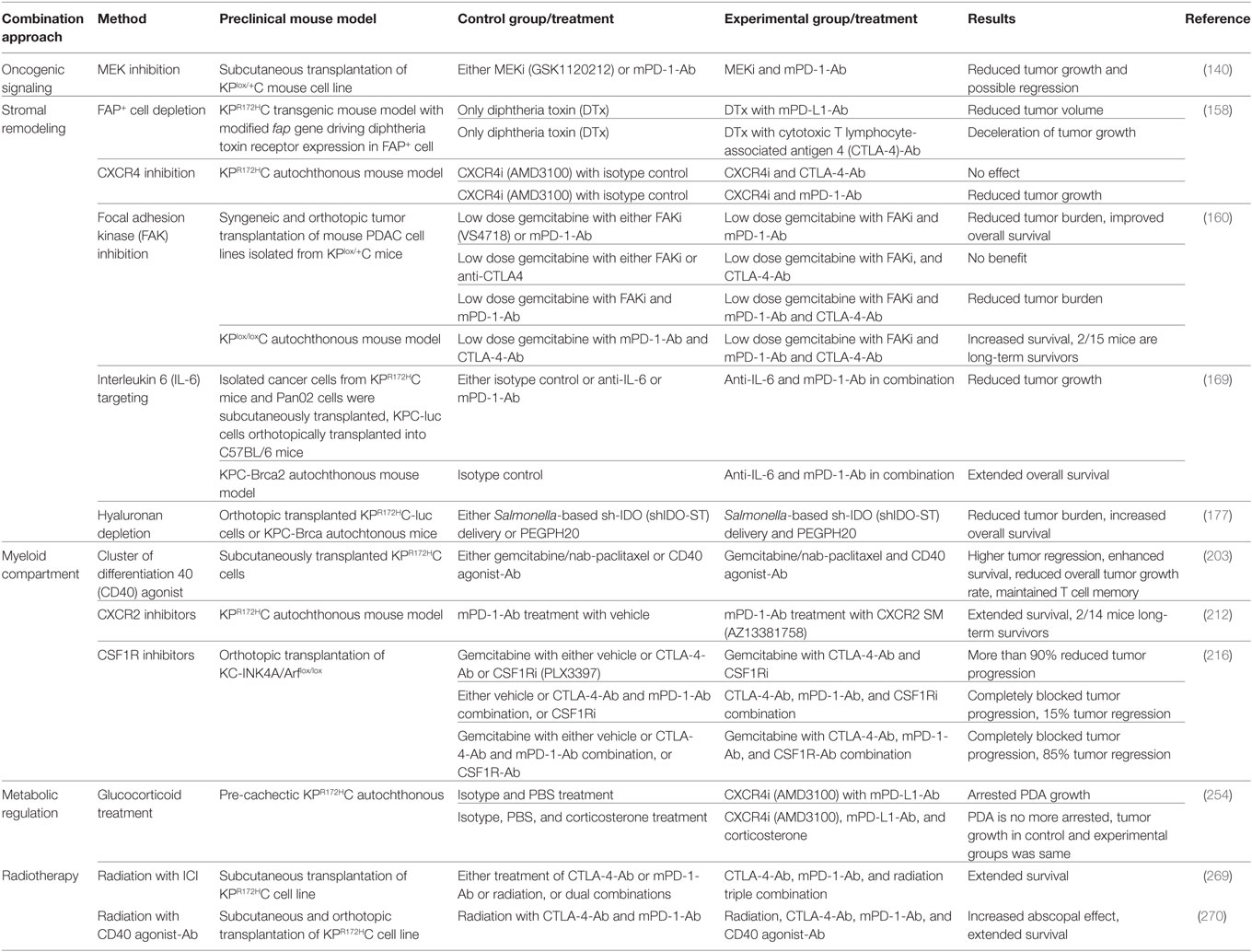
Table 1. Selection of studies focusing on immune checkpoint inhibition (ICI) combination therapies in preclinical mouse PDAC model.
Antigenicity
Antigenicity refers to the ability of tumor cells to produce and present tumor-specific antigens (TSA) and tumor-associated antigens (TAAs) to the adaptive immune system (97). The bottlenecks of antigenicity include the range of TAA and TSA production, and their ability to be presented to the immune system through MHC complexes (human leukocyte antigen—HLA—in humans) (97, 98). TAAs are overexpressed in cancer cells while their expression is low in normal cells, whereas TSA subtype neoantigens are produced de novo upon mutational changes of tumor cells (98). These mutations can favor neoantigen tethering to MHCs, produce a new residue on neoantigens increasing TCR recognition, or generate a proteolytic cleavage site, providing better processing for antigen presentation (99). Since TSAs are expressed only in malignant cells, they provide great specificity for T cell cytotoxicity (98). Epigenetic regulation of TAAs in tumor cells can also represent an important target for T cell action (99). In melanoma patients, even tumors with a low mutational burden, but with a high expression of TAAs, which is likely mediated by epigenetic mechanisms, showed considerable response to immunotherapy (100).
Cancers with high mutation rates such as melanoma, bladder cancer, and lung cancer show better response to check point inhibition compared with other types with a lower mutational burden, for instance, PDAC (101–105). Especially tumors with mismatch repair (MMR) deficiency or with more microsatellite instability (MSI) are shown to respond better to immunotherapy (106). As a matter of fact, impairing MMR through genetic inactivation of mutL homolog 1 gene (MLH1) in PDAC mouse models provoked hypermutation, triggering more neoantigen production. This, in turn, prolonged immunosurveillance with better therapeutic response to immune check point inhibitors (Figure 2) (107). Humphris et al. reported that among the 385 resected patient samples only 1% of showed MSI with inactivation of MLH1 and MSH2 (mutS protein homolog 2). This may provide a possible explanation for low response rate to immunotherapy in PDAC (108). Pembrolizumab, a PD-1 antibody, was approved by the Food and Drug Administration in 2017 for solid tumors with MMR defects or MSI, including PDAC (106). Use of DNA damage response (DDR) inhibitors may also enhance the genetic instability of the cancer cells upon exposure to DNA damaging agents, increasing the production of neoantigens. On the other hand, DDR inhibition may show a tumorigenic effect by acting on antitumor immune cells (109). Still, mutational load is not a reliable biomarker for the prediction of response to immunotherapy, considering the patients who were not responding to immunotherapy even if they had a high mutation burden. Likewise, tumors with a low mutational load, such as renal cell carcinomas, responded well to immunotherapy (28, 110).
Recently, Balachandran et al. described a neoantigen quality fitness model identifying long-term survivors of PDAC via selecting neoantigens with great resemblance to disease derived peptides (111). On the other hand, a neoantigen quantity model showing more immunosurveillance in response to increasing neoantigen numbers revealed no long-term survivors by itself. Only tumors showing both, high neoantigen numbers and abundant CD8+ cytotoxic T cell infiltration, were associated with a significant survival benefit for the patients. Supporting the immune quiescence-like phenotype of PDAC, a modest decrease in high-quality neoantigen transcript levels was seen. More strikingly, they identified a loss of high-quality neoantigen expression in metastatic tumors compared with their primary counterparts. In conclusion, identification of hotspot neoantigens and methods to exploit or target them may increase the response to checkpoint inhibition, not only regarding the primary tumor but also regarding metastatic lesions.
Another mode of reduced antigenicity is the loss of antigen presentation, which can reduce immunosurveillance either by blocking priming of naïve T cells, or by making cancer cells invisible to effector T cell function (97, 112). In other cancer types, reduced antigen presentation was achieved by downregulation of MHC class proteins or impaired antigen processing and shuttling (113–116). Oncogenic RAS signaling was shown to reduce antigen presentation in different cancer types, including PDAC (113, 117, 118). Also, HLA-1 and transporter for antigen presentation production was demonstrated to be reduced in human PDAC specimens (113, 117–119). Manipulating cancer cells for enhanced antigen presentation can reinforce the checkpoint inhibition response.
As a matter of fact, Pommier et al. recently showed that disseminated cancer cells (DCCs, metastatic, quiescent single cancer cells) are undergoing a Darwinian-like selection during immune surveillance of metastasis (120). These investigators elegantly showed that only the metastatic cancer cells, negative for MHC-I and CK-19 expression on the surface, could form DCCs, avoiding T cell-mediated killing in pre-immunized mice. ER stress was the barrier for DCCs to maintain a quiescent state, and also to escape from T-cell-mediated immunity. Therefore, to form macrometastasis, in addition to ER stress relieve, a systemic immunity depletion was required. All these results show how important it is for cancer cells (both primary and metastatic tumor cells) to have good quality neoantigens, and a competency to present neoantigens through MHC complexes to immune cells.
The correlation between total neoantigen load and checkpoint inhibition response is absent in PDAC unlike in other, immunogenic tumors, such as melanoma or lung cancer (104, 121–125). This implies that other factors, determined by the immunogenic properties of PDAC, play an important role in the response to immunotherapy of this malignancy.
Immunogenicity
Immunogenicity of cancer refers to its ability to induce an adaptive immune response. Based on comprehensive integrated genomic analysis, PDAC was classified into different subgroups by several studies (126–130). RNA expression analysis identified an immunogenic subtype of PDAC in 25 among 96 PDAC patient specimens. This subtype is associated with an increased immune cell infiltration, and enriched signatures such as CD4+ and CD8+ T cell signaling, antigen presentation, B cell signaling, and most notably CTLA-4 and PD-1 signaling. Signatures enriched in immunogenic subtype might represent predictive biomarkers for immunotherapeutic response in PDAC (130).
Cytolytic activity is determined by the transcription levels of granzyme A (GZMA) and perforin (PRF1), which are known cytotoxicity markers of CD8+ T cells (131). Interestingly, genetic amplification of MYC and/or deletion of CDKN2A/B were associated with reduced cytolytic activity in TCGA PDAC datasets (125). Mutant Kras-mediated immunosuppression via GM-CSF or IL17R production might be another reason for impaired cytolytic activity in PDAC (132–134). Other than oncogenic drivers, stromal composition may have an impact: PDAC with so-called “normal” stroma (127) (i.e., a good version of stroma, characterized by high ACTA2, VIM, and DES pancreatic stellate cell-PSC markers) was associated with a higher cytolytic activity (125). Considering various factors determine cytolytic activity other than neoantigenic capacity, it is important to adapt individualized precision immunotherapy covering different determinants of immunogenicity in PDAC (125, 135). For an ease of understanding, the determinants of immunogenicity can be divided into two: intrinsic and extrinsic factors.
Intrinsic Determinants of Immunogenicity
Antigenic tumors can still evade ICI therapy via downregulation of tumor cell-intrinsic immunogenicity (97). In various cancers, stimulation of oncogenic pathways such as PI3K (136, 137), MYC (138), TAZ (139), and JAK-STAT (35) through either excessive ligand production or their mutations induces constitutive PD-L1 production (Figure 2). Myeloid cell induction of EGFR and MAPK signaling in PDAC cells enhanced PD-L1 production inhibiting CD8+ T cell infiltration (140). The expression of PD-L1 in various tumors was associated with higher immune cell infiltration and the presence of lymphoid aggregates, and tumors with naturally high levels of PD-L1 in these showed comparably high response rates to anti-PD-1 or anti-PD-L1 (38, 83, 141). Regulation of PD-L1 and other checkpoint inhibitors or oncogenic signaling cascades in cancer cells also constitute an important place for the regulation of tumor immunogenicity (142–144).
Although initial IFNγ production is favorable for CTL activity, chronic exposure may lead to immunoediting in tumor cells. As a result of this, tumors develop genetic or epigenetic modifications in IFNγ signaling components such as IFNγ receptors (IFNGR1 and IFNGR2), JAK-STAT pathway components and IRF1 transcription factors (145, 146). A loss of function mutation on Apelin receptor has recently been identified impairing IFNγ induced JAK-STAT signaling cascade in melanoma (147). Although IFNγ is considered to be antitumorigenic, its induction of PD-L1 transcription in cancer cells might positively correlate to anti-PD-1 or PD-L1 therapy response in established tumors (148). Since IFNγ exposure of cancer cells induces PD-L1 production, mutations in IFNγ signaling components JAK1 and JAK2 would lead to clonal evolution of PD-L1-negative tumor cells, which are not responsive to anti-PD-1 treatment (149). Although no such mutations have been identified in PDAC, personalized medicine can favor the prediction of checkpoint inhibition response through identification of these type of mutations.
Several solid tumors including PDAC showed anti-PD-1 resistance signatures (IPRES) such as enhanced mesenchymal transformation, cell adhesion, extracellular matrix modeling, angiogenesis, hypoxia, and wound healing in TCGA datasets (123). Overall, the differential mutational and transcriptional landscape of tumors does not only determine neoantigen quality and quantity but also regulates several signaling pathways responsible for intrinsic and extrinsic properties of immunogenicity in cancer.
Extrinsic Determinants of Immunogenicity: Modulation of TME
Cytotoxic T lymphocyte infiltration into the TME is essential for ICI therapy (150). Even if the anti-tumor CTL infiltration is seen in many tumor types, PDAC represents an outlier in this manner (92). Starting from the premalignant lesions, its microenvironment restricts the cytotoxic T cell infiltration. The cytotoxic T cell function is limited through the actions of immunosuppressive cells in the TME such as cancer-associated fibroblasts (CAFs), myeloid cells, and inhibitory actions of some T cell subsets, albeit they infiltrate in the TME (97, 151). In support of this, strategies eliminating immunosuppressive populations in the TME enhanced CTL infiltration in various cancers (152, 153). To shift the immunosuppressive environment to a non-immune-privileged status, it is important to be aware of the individual components of the TME and to know how to modulate them.
Stromal Remodeling
The characteristic abundant desmoplastic stroma of PDAC can be both beneficial and harmful in terms of carcinogenesis. Studies showed that transplantation of PDAC cancer cells with pancreatic stellate cells increased tumorigenic potential and metastasis (154). However, depletion of stroma in preclinical mouse models also revealed further accumulation of Tregs in the TME showing the dual nature of stromal compartment (155). In a study performed on human PDAC tissues, the fibrotic reaction did not impair TIL infiltration, rather fibrosis associated collagen-I amount positively correlated with effector T cell presence (156). However, previous studies showed the inhibitory actions of αSMA+ CAFs on CD8+ CTLs in PDAC TME (157, 158). These results indicated the presence of (?) tumor heterogeneity not only in terms of cancer cells but also stromal compartments of PDAC (159).
One study revealed that depletion of CAFs could actually be employed to increase the immunotherapy response of PDAC: fibroblast activation protein (FAP+) CAFs were shown to induce chemokine (C-X-C motif) ligand 12 (CXCL-12) mediating immunosuppression through limiting effector T cell infiltration (158). Targeted inhibitors of FAP+ CAFs or CXCL-12 chemokine (C-X-C motif) receptor (CXCR-4) inhibition via AMD3100 increased CD3+ T cell accumulation and revealed a synergistic effect with anti-PD-L1 therapy in mouse models (158).
Further studies focusing on focal adhesion kinase (FAK) showed FAK inhibition in cancer cells can remodel stroma, inhibiting immunosuppressive TME cells (160). Combination of FAK inhibitor with gemcitabine and anti-PD-1 increased CD8+ CTL infiltration, reducing tumor burden and prolonging overall survival (160). Even though single agents targeting FAK inhibition in PDAC showed no objective response in clinic (161–163), trials combining iFAK (vs.-4718) with gemcitabine and anti-PD-1 are ongoing (NCT02758587).
The importance of interleukin 6 (IL-6) signaling in PDAC has been shown by several groups revealing its importance on both carcinogenesis and persistency (164–166). CAFs are also responsible for the production of pro-inflammatory cytokines other than myeloid cells such as IL-6 (167). Unfortunately, clinical trials targeting IL-6 alone demonstrated no benefit (168). However, preclinical studies targeting IL-6 in combination with PD-L1 showed decreased αSMA+ stromal cells and increased CD3+ lymphocyte infiltration in KPC and Panc02 subcutaneous and orthotopic transplantation models and a survival benefit in the KPC-Brca2 autochthonous mouse model (169).
Hyaluronan, an extracellular matrix component, is a linear glycosaminoglycan in PDAC, associated with multiple markers of aggressiveness of cancer for instance increased cell proliferation, invasion, and metastasis (170). High hyaluronan expression correlates with worse prognosis in PDAC patients (171). Several drugs have been developed to deplete stromal hyaluronan, such as PEGPH20. In preclinical models, hyaluronan depletion via PEGPH20 remodeled stroma, decreased interstitial fluid pressure, and increased drug delivery by enhancing micro-vessel permeability (172–174). As PEGPH20 increased delivery of chemotherapeutic agents in PDAC preclinical models, the same was seen for monoclonal antibodies (trastuzumab) in breast cancer (175). With the use of transplanted and autochthonous PDAC mouse models, Salmonella-based IDO-1 depletion (176) was also enhanced with (by means of gibi mi?) PEGPH20 treatment (177). Vitamin D receptor (VDR) was identified as a PSC master regulator for dynamic regulation of stromal composition. Treatment with VDR ligand reduced inflammation and enhanced gemcitabine delivery and efficacy in a mode of action similar to hyaluronan depletion (178). Based on these results, hyaluronan depletion or VDR activation appear as promising combination partners of checkpoint inhibitor monoclonal antibodies in clinical trials.
Modulation of Immunosuppressive Myeloid Cells
Tumor-associated macrophages (TAMs) differentiate from resident macrophages or mobile inflammatory monocytes (179). TAM polarization can be both beneficial and harmful in terms of carcinogenesis. M1 differentiation of TAMs is known to be antitumorigenic due to their tumoricidal nature via releasing pro-inflammatory cytokines. By contrast, the M2 subtype is pro-tumorigenic, since it suppresses immunosurveillance by secreting anti-inflammatory cytokines, e.g., TGFβ and IL-10 or by remodeling tumor stroma (180). Consistent with this, expression of M2-related markers such as CD204 and CD163 negatively correlate with patient survival (181, 182). Derived from immature cells of myeloid origin, MDSCs are known for their neoangiogenic and immune-suppressive activities in TME. MDSCs have been shown to inhibit CTL activity by recruiting Treg subset, modulating amino acid reserves in TME, and pushing T cells toward apoptosis via ROS production (183). Also, the presence of immunosuppressive cells such as M2 macrophages, Tregs, and MDSCs in PDAC negatively correlates with overall survival (155, 184–188). Both pro- and antitumorigenic properties of neutrophils in cancer are reported, and their inhibitory action on CTL activity is known to be mediated by various mechanisms (189). Considering the complexity of immune cells in TME and their crosstalk with T cell activity, it is challenging but important to modulate these mechanisms to boost ICI response in cancer.
Cluster of differentiation 40 (CD40) is a member of the tumor necrosis factor receptor superfamily and is expressed on APCs including monocyte subsets, DCs, macrophages, and B cells (190). CD40 signaling is important for licensing APCs (to maximize their capacity to present antigens) followed by cross-priming of CD8+ CTL in lymph nodes (191–193). CD40 agonists mediated an enhancement of adaptive antitumor immunity in preclinical mouse models in various cancer types (194–196). By contrast, treatment of KPC mice with CD40 agonist (FGK45) and gemcitabine transiently blocked PDAC development through re-education of tumor-infiltrating macrophages and stromal remodeling, but was not able to invoke an adaptive antitumor immune response (197). On the other hand, subcutaneous transplantation of KPC cancer cells into syngeneic mice revealed that the same treatment strategy induced an adaptive immune response with CD4+ and CD8+ T cell infiltration. Consequently, the authors used another “two tumor” model, in which intact KPC tumors (cancer cells with intact TME) were transplanted into endogenous tumor-bearing KPC mice. Here, gemcitabine with FGK45 treatment induced a CD4+ and CD8+ T cell infiltration into subcutaneous tumor but only CD4+ infiltration into endogenous tumor. The barrier for CD8+ repletion in spontaneous tumors was exceeded through systemic macrophage depletion. Upon deeper analysis, Ly6Clow F4/80+ macrophages residing in vicinity of PDAC TME were identified as the responsible physical barrier for CTL infiltration (198). In a similar manner, CTL-mediated antitumor immune responses were not seen in clinical trials with CD40 agonists in various cancers even with the addition of gemcitabine to increase tumor immunogenicity (199–202). CD40 agonist treatment finally acted as a checkpoint co-activator through its action on APCs inducing T cell priming upon gemcitabine/nab-paclitaxel dual treatment (203). In conclusion, PDAC retains its antigenic properties to induce both innate and adaptive immune response. This antigenicity might be increased through the use of chemotherapeutics or targeted therapy. Yet, since PDAC is immunologically cold (i.e., very scarce resident CTL infiltration) to respond to increased antigenicity, mechanisms to enhance CTL infiltration must be elucidated. Combination of gemcitabine with nab-paclitaxel remodels TME to permissive conditions for CTL infiltration, but not with gemcitabine alone (203). Furthermore, once T cell priming barrier is exceeded through CD40 agonist treatment, CTL activity might be more expedited with checkpoint inhibitor usage.
C-X-C motif chemokine receptor 2 (interleukin 8 receptor beta, CXCR-2) is a G-protein-coupled receptor for various CXCL ligands including IL-8. CXCR-2 in a cell type-specific manner can act both as a tumor suppressor where it induces senescence in premalignant lesions of PDAC (204, 205) and as tumor promoting via enhancing neutrophil and MDSC recruitment to TME (152, 206–208). Through inhibition of CXCR-2 either genetically or pharmacologically with CXCR-2 pepducin (209, 210) or AZ13381758 (211) inhibitors, Steele et al. showed an enhanced response to anti-PD-1 therapy and decreased metastasis in PDAC (212). The enhanced therapy response is reasoned by reduced infiltration of monocytes and MDSCs, which augments T cell infiltration in TME. Also, they propose that stromal remodeling through T cell recruitment might enhance gemcitabine efficacy in tumors (212, 213).
Colony-stimulating factor 1 receptor (CSF1R) is an important regulator of TAMs’ differentiation and sustenance in microenvironment (214, 215). Therefore, inhibition of CSF1R is considered to have potential for cancer therapeutics. Yet, single-agent use targeting CSF1R did not yield clinical benefits in various tumor types (215). In mouse models, treatment of PDAC with CSF1R inhibitors enhanced antitumor immune response; however, this effect was diminished due to the production of checkpoint proteins such as PD-L1 and CTLA-4 (216). Combination of checkpoint inhibitors with CSF1R blockage showed regression of tumors in mouse models (216). CSF1R inhibition was shown to have an effect not only on TAMs but also on CAFs in various subcutaneously transplanted mouse models (217). Recently, CSF1R blockage was shown to enhance the production of granulocyte-specific chemokine expression such as CXCL-1 by CAFs increasing polymorphonuclear MDSC (PMN-MDSC) recruitment as a resistance mechanism (217). PMN-MDSC cells are known for their pro-tumorigenic and anti-immunogenic properties (218). Therefore, combination treatment of CSF1R and CXCR2 inhibitors (see above) targeting, respectively, both TAMs and MDSCs enhanced anti-PD-1 therapy response in transplanted tumor models (217). Considering response enhancement by usage of either CXCR2 or CSF1R inhibitor in combination with immune checkpoint inhibitors, simultaneous use of the three might exploit a broader benefit for therapy response also in PDAC (208, 212).
B Cells
Bruton’s tyrosine kinase (BTK) is an enzyme expressed in B cells, macrophages, and mast cells, and targeting BTW in combination with ibrutinib was shown to be effective in chronic lymphocytic leukemia, Mantle cell lymphoma, and Waldenstrom’s macroglobulinemia (219–221). Besides targeting BTK, ibrutinib also inhibits interleukin-2-inducible T-cell kinase in T cells, skewing Th differentiation toward Th1 (222). Because of this effect, dual combination of ibrutinib with anti-PD-L1 inhibitor was shown to have a synergistic effect in a T cell-dependent manner, but not MDSC dependently in studies with mouse transplantation models of lymphoma, breast, and colon cancer (219). In various PDAC preclinical mouse models, ibrutinib demonstrated its antitumorigenic effect via depletion of macrophage deposition and fibrosis (220). In another study, on the other hand, ibrutinib enhanced macrophage production of Th1 differentiation cytokines, while inhibiting Th2, and augmenting the CD8+ cytotoxic T cell deposition in tumors. The effect on macrophage activity was also dependent on B cells, and B cell-specific BTK signaling, still there was no change in fibrosis (221). Based on these results, checkpoint inhibition in combination with BTK inhibitor ibrutinib might enhance the therapeutic benefit of single use of each in PDAC, accordingly clinical trials are ongoing.
γδT Cells (γδT)
T cells are broadly divided into two subtypes based on the antigen receptor types they express: αβT and γδT (223). While 95% of the CD3+ T cells in blood express αβTCR (includes CD4+ and CD8+ T cells) recognizing MHC class I–II, 5% have γδTCR which does not require MHC engagement for activation: γδT are cytolytic through the release of inflammatory cytokines (224, 225). There are conflicting data about the function of γδT in PDAC, with both pro- and antitumorigenic potential. Isolated γδT were shown to be tumoricidal to PDAC cell lines in vitro (226). By contrast, in mouse models, pre-neoplastic lesions with KRASG12D were shown to recruit IL-17-expressing immune cells including γδT, which accelerated carcinogenesis through IL-17 receptor oncogenic signaling (133). In support of this, genetic and therapeutic depletion of γδT in mouse models prolonged survival. Other than the IL-17-mediated oncogenic effect on PanIN lesions, γδT directed checkpoint receptor inhibitory action (through galectin-9 and PD-L1 expression) on αβT cells, accelerating carcinogenesis. While ablation of CD4+ and CD8+ T cells had no impact on PDAC generation and persistency, this was different upon δTCR knock out: γδT cell deletion increased CD8+ CTL and CD4+ Th1 tumor infiltration, and skewed CD4+ differentiation toward the Th1 type. More importantly, the immunosuppressive action of γδT cell was not due to an effect on MDSCs or TAMs; instead, it was directly dependent on checkpoint co-inhibitory receptor engagement with antitumor T cells. PD-L1 and Galectin-9 checkpoint inhibition was effective in tumors with γδT cell present, but not in their absence. This implies the importance of personalized medicine, through which the γδT cell presence may be characterized in patients, to predict checkpoint inhibition therapy response (227).
Metabolic Regulation
Enhancing checkpoint inhibition efficiency may also be achieved through regulation of metabolic properties of T cells. For cytotoxic and effector T cell activity, a metabolic switch from a catabolic to anabolic state is important (228–231). While naïve T cells rely mostly on oxidative phosphorylation, activated T cells prefer to switch aerobic glycolysis for faster ATP production (230, 231). In support of this, T cells in anergic state even with TCR engagement and costimulator checkpoint activation can retain their hyporesponsive state in a nutrient poor environment (232). The nutrient poor microenvironment with low glucose and amino acid reservoir is regulated by both cancer cells and the TME (233). Cancer cells, for example, outcompete T cells for glucose uptake having implications for intrinsic immunogenicity regulation (234). Furthermore, glutamine usage by cancer cells also limits its presence in TME, limiting its activator function on T cells (235, 236). ARG-1 (Arginase 1) produced by TAMs and MDSCs degrades arginine (237, 238), while indoleamine 2,3-dioxygenase (IDO-1) produced by cancer cells, TAMs, and MDSCs converts tryptophane to an immunosuppressive metabolite kynurenine reducing T cell activity (239–244). Other than limiting nutrient availability, production of immunosuppressive intermediary metabolic products can also impair T cell activation. Cancer cell production of lactate as a result Warburg effect can impair T cell immunity by both decreasing TME pH and lactate shuttling into T cell (245–247). In addition, adenosine produced by cancer and Tregs (248–250), and prostaglandin E2 produced by TAMs and MDSCs are known to inhibit T cell signaling (251). Even though PDAC with its cancer cell and TME components shows similarities in metabolic properties as discussed above, how these metabolic properties effect T cell immunity specifically in PDAC has not been well studied (252). Overall, other than modulation of cytokine–chemokine–receptor axis, nutrient availability and production of immunosuppressive metabolites might also affect the extent of T cell immunity.
The impact of metabolism on checkpoint inhibition efficacy may not be only relevant on a micro environmental but also on a more systemic level. Cachexia is a systemic disorder with an excessive weight loss through the consumption of muscles and adipose tissues (253). Many diseases are associated with cachexia, including cancer in general and PDAC in particular (253). An increase in serum IL-6 levels was shown to impair hepatic ketogenesis inducing cachexia in C26 colon cancer and autochthonous KPC-PDAC mouse models (254–256). Physiologically, the body responded to cachexia with an upregulation of glucocorticoids like (?) corticosterone, which inhibits T cell infiltration into tumors of C26 cells. In support of this, transcriptomics analysis of pre-cachectic and cachectic C26 transplanted mice revealed an impaired immunological phenotype. However, this signature was not seen in the KPC model of PDAC, again implying its innate immunocompromised nature (254). With the use of the CXCR-4 inhibitor AMD3100, this barrier was overcome, increasing T cell infiltration and PD-L1 checkpoint inhibitor efficiency (158, 254). However, with the addition of corticosterone to the AMD3100-PD-L1 combination, the therapeutic effect was diminished (254). These results have multiple implications for PDAC therapeutics: the checkpoint inhibition resistance might be tackled with glucocorticoid synthesis inhibition, though this might first require a prior consideration for CTL infiltration. Second, serum glucocorticoid levels might be important markers for checkpoint inhibition response in PDAC patients. And finally, serum IL-6 depletion might provide further opportunities to increase the checkpoint inhibition efficacy, not only because of its direct effect in the TME but also due to its physiological role in cancer-related cachexia (254).
Other Combination Strategies Exploiting Antigenicity/Immunogenicity of Tumors to Enhance Checkpoint Inhibition Therapy
Approaches including specific inhibitors (small molecules and antibodies) of various signaling pathways are described thus far and listed in Table 2. Apparently, combining the classical, untargeted treatment strategies, chemotherapy and RT, with checkpoint inhibition in clinical trials appears reasonable. Other targeted immunotherapeutic options, e.g., oncolytic viruses, vaccines, and chimeric antigen receptor-T cell (CAR-T) therapies aim to treat cancer in a more specific manner with minimal side effects. Selected clinical trials combining immune checkpoint inhibitors with untargeted and other targeted immunotherapeutic options are listed in Table 3. Combination therapies can modulate both antigenic and immunogenic landscape of tumors (Figure 4). Likely more important than just developing novel combination partners, exact understanding of the mode of action of combination partners, their tolerability and toxicity, a determination of dosing and appropriate sequencing of the combinations are required (257).
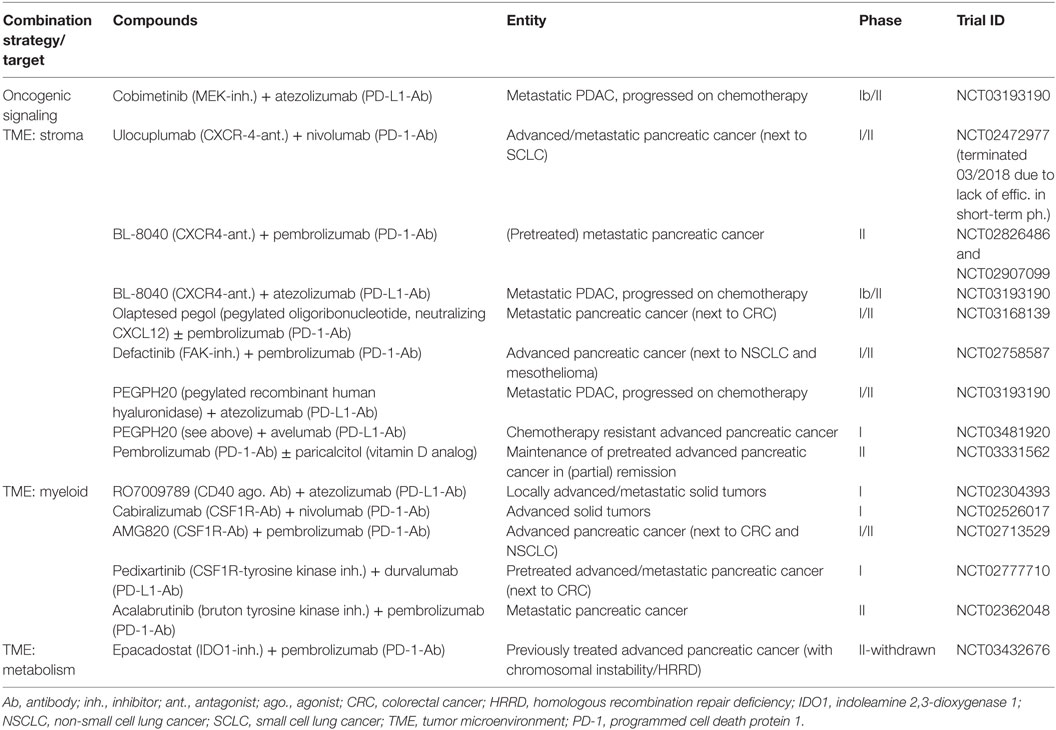
Table 2. Selection of currently ongoing clinical trials evaluating CTLA4 or/and PD1/PD-L1 checkpoint blockade in combination with targeted therapy approaches for pancreatic cancer as indicated.
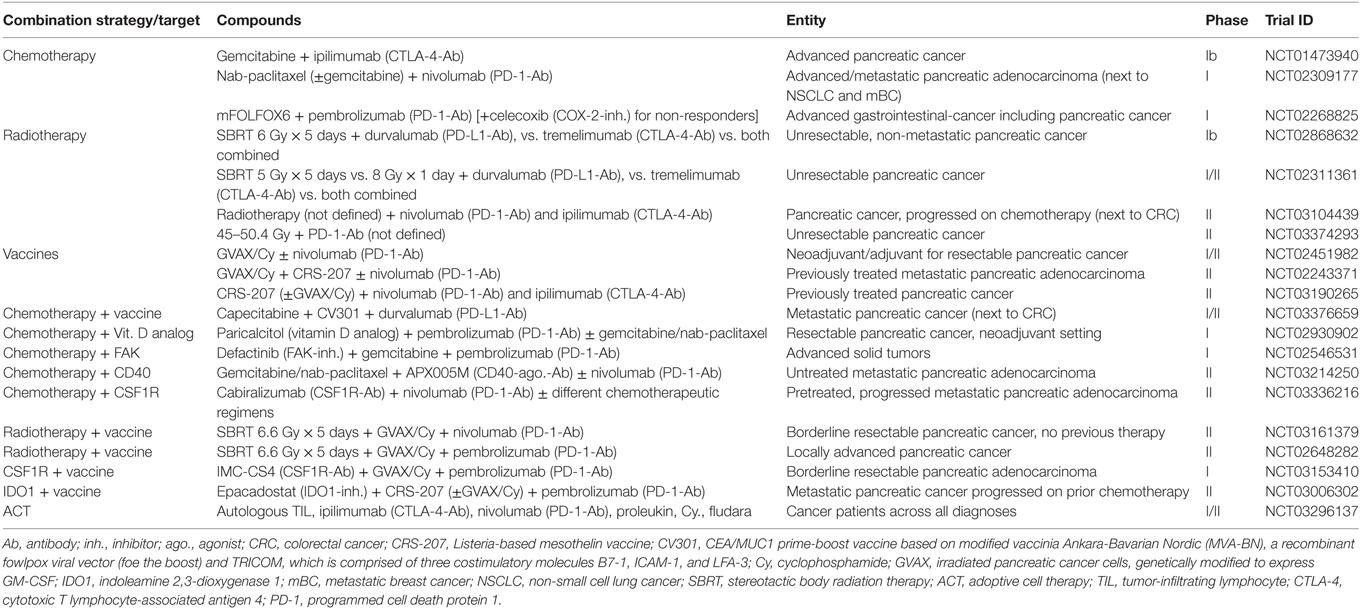
Table 3. Selection of currently ongoing clinical trials evaluating CTLA4 or/and PD1/PD-L1 checkpoint blockade in combination with untargeted and targeted options including other immunotherapeutic approaches for pancreatic cancer as indicated.
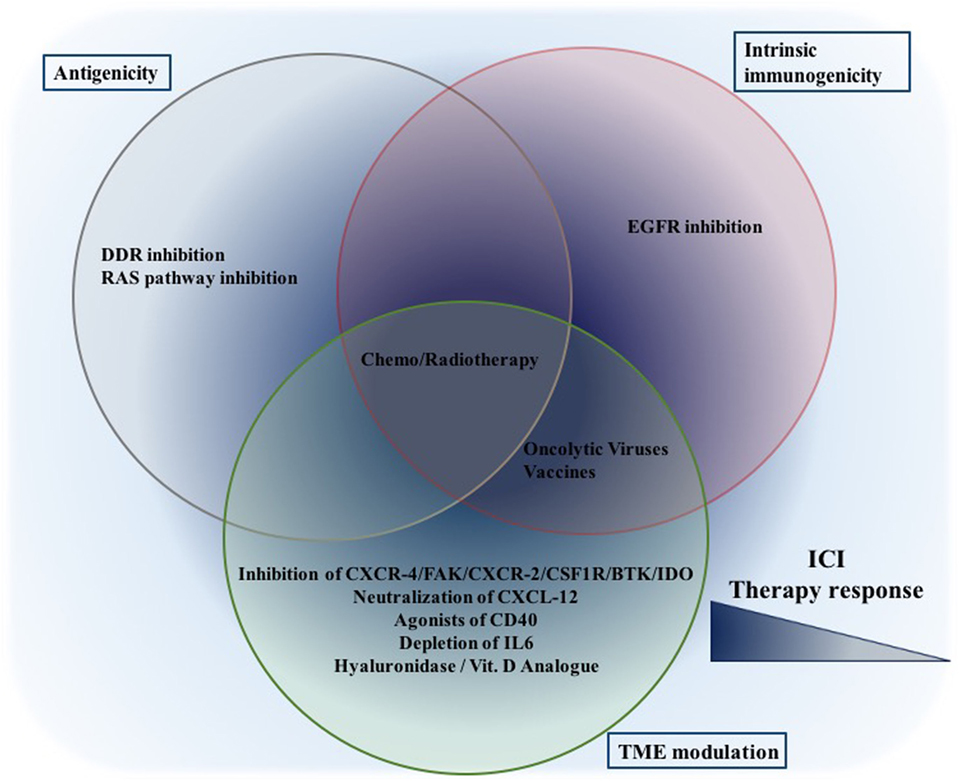
Figure 4. Combination therapeutic options to increase ICI efficiency: while given therapeutic options are placed in the corresponding cluster, only published data thus far are taken into consideration. This still does not eliminate their potential to affect other aspects. While treatments focusing on a single aspect (either one of antigenicity/intrinsic immunogenicity/TME modulation) might be effective, the best synergism will probably be achieved through combinations focusing on all aspects. Abbreviations: ICI, immune checkpoint inhibition, DDR, DNA damage response.
Combination of Immune Checkpoint Inhibitors With Untargeted Therapeutic Options
Chemotherapy
The first-line PDAC therapeutics used in clinic are chemotherapeutic agents such as gemcitabine with/without nab-paclitaxel, and FOLFIRINOX (folinic acid, fluorouracil, irinotecan, and oxaliplatin) (258). These agents are known for their ability to induce cytotoxicity due to impaired cell division. The mutagenic effect of chemotherapy (or RT) may enhance neoantigen production and MHC class I antigen presentation on cancer cells, increasing tumor antigenicity (109). Still, even if sub-clones with reactive neoantigenic properties might evolve, they might not be substantial enough to result in a broad clonal response in response to checkpoint inhibition (109). Furthermore, considering that PDAC already retains its antigenic capacity but its immunosuppressive microenvironment is the main barrier to pass as explained above, chemotherapy might exert its effect rather by altering immunogenicity. Immunogenic cell death (ICD) upon chemotherapy releases danger signals and cytokines for the generation of a more immunogenic TME (109). As also seen in the gemcitabine with nab-paclitaxel example above, remodeling of the immunosuppressive TME can bolster up T cell cytotoxicity due to enhanced immunogenicity. Chemotherapy can increase immunogenicity by also its direct action on immunosuppressive cells of TME. For example, fluorouracil and paclitaxel were shown to induce MDSC apoptosis in various tumor models, while low dose gemcitabine was shown to deplete Tregs in panc02 orthotopic mouse model (259). Furthermore, it will be important to select chemotherapeutic agents, their dosing and time and sequence of administration with regard to their ability to induce ICD and remodel the microenvironment.
Radiotherapy
Although the use of RT for the treatment of PDAC has been controversially discussed due to rather disappointing results in clinical trials (260), radiation treatment in combination with ICI might be a promising strategy for pancreatic cancer patients. In a phenomenon known as the abscopal response, RT was shown to induce immune responses that mediate regression of metastatic lesions lying outside the field of radiation (261). RT could activate the immune system, increase trafficking of T cells to the tumor, and elicit antitumor immune responses following ICD (262). Several preclinical and clinical studies in different cancer types showed synergistic effects in cohorts treated with RT and immune checkpoint blockade (263–266). Although not many studies have been published thus far, evidence for synergism can also be seen in PDAC and has been related to increased immunogenicity (95, 267, 268). In the PDAC mouse model used by Twyman-Saint Victor et al., any combination of immune checkpoint inhibitor with RT substantially increased overall survival, compared with immune checkpoint blockade with either CTLA-4 antibody or PD-1 antibody alone. The highest response rate and longest overall survival was seen in the triple combination therapy (two checkpoint inhibitors + RT) group (269).
Recently, CD40 agonist treatment was demonstrated to be beneficial upon a RT + ICI regimen in murine pancreatic cancer models (270). While RT alone or in combination with ICI resulted in reduction of irradiated tumor growth, only the triple therapy, RT + αCD40 + ICI (RCP4), affected the growth of both irradiated and unirradiated tumors. These observations were also reflected in the long-term survival. Furthermore, CD4 and CD8 T cells, as well as short-lived myeloid cells were shown to be necessary for optimal response to RCP4 and that RCP4 antitumor immunity. This immunity was dependent on host CD40, Batf3, and IFNγ but not on B cells and canonical innate immune activation pathways. The three therapies all showed non-redundant impact on the antitumor immune response. While RT triggered an early pro-inflammatory stimulus, αCD40 caused systemic myeloid compartment reorganization and ICI increases intratumoral T cell infiltration, thus improving the CD8/Treg cell ratio.
In conclusion, RT can enhance the “visibility” of tumor antigens and make the tumor more immunogenic. While the combination of RT and ICI shows promise in preclinical and clinical trials in various cancer entities, challenges still exist for the safe and efficacious application of the combination. Tumor-type and immune therapy-specific optimization of radiation dose and timing and the identification of potential biomarkers is likely to further enhance the effectiveness (271). Also, the addition of αCD40 agonists appears to be a promising avenue to pursue in clinical PDAC trials.
Combination of Immune Checkpoint Inhibitors With Other Immunotherapeutic Approaches
Oncolytic Viruses
Tumor-targeted oncolytic viruses (TOVs) are viruses that selectively infect, replicate in, and lyse tumor cells, while leaving healthy, normal tissues unharmed. TOVs can have intrinsic tumor-selectivity, making them naturally nonpathogenic to humans and sensitive to antiviral signaling (272) or depend on oncogenic signaling pathways, e.g., constitutively activated RAS (273, 274). Viral tumor specificity can also be genetically engineered by deleting genes required for replication in normal tissues (273) or by placing viral replication under the control of a tumor-specific promoter (274–276), TOVs can also be designed to express tumor-specific cell surface receptors (277, 278). TOVs can thus be engineered to increase safety, efficacy, and tissue tropism.
The advantages of TOVs are their specificity, modest toxicity, low probability for resistance, and most importantly, their induction of an inflammatory cascade and engagement of the adaptive immune system (273). In contrast to any other drug, the therapeutic dose of TOVs increases over time, as the virus replicates and spreads to neighboring cells (273). Although TOVs directly lyse infected malignant cells, causing acute tumor debulking, it is the ability of the virus to spread from cell to cell and potentiate an inflammatory response through ICD that make oncolytic viruses such promising new therapies (279–281). However, oncolytic virus therapy faces challenges in solid tumors and especially PDAC. These challenges, i.e., overcoming the TME, avoiding neutralization by the host immune system, and acquired resistance in tumor cells culminate in the main problem, i.e., the systemic delivery of TOVs for the targeting of metastatic cancer cells (282). Thus, it is not surprising that thus far, no studies investigating ICI and oncolytic viral therapy in pancreatic cancer have been published. However, Mahalingam et al. (283) conducted a phase II study of pelareorep, a proprietary replication-competent isolate of reovirus type 3 dearing in combination with gemcitabine in advanced PDAC and observed the upregulation of PD-L1 in following treatment. They suggested to investigate the combination of oncolytic virus therapy with anti-PD-L1 inhibitors in PDAC.
Congruent with this finding, recent research in other cancer entities revealed that antiviral immunological events induced by the administration of oncolytic viruses can turn tumors “hot” (284, 285) and establish a TME that is conducive for enhancing the efficacy of checkpoint inhibitors (286–288). Using intravenous infusion of oncolytic human orthoreovirus, Samson et al. (288) found that TOV treatment increases cytotoxic T cell tumor infiltration, upregulates IFN-regulated gene expression, and the PD-1/PD-L1 axis in tumors, via an IFN-mediated mechanism. And finally, addition of PD-1 blockade to reovirus treatment enhanced systemic therapy in a preclinical glioma model. In their simultaneously published triple-negative breast cancer (TNBC) study, Bourgeois-Daigneault et al. (286) reported that TOV therapy sensitizes otherwise refractory TNBC to immune checkpoint blockade, preventing relapse in most of the treated animals.
In conclusion, once the problem of systemic delivery is solved, oncolytic viruses are not only valuable therapies in terms of tumor debulking but are also useful in a “prime and boost” approach in combination with ICIs.
Vaccines
Another promising approach to enhance the immunogenicity of pancreatic cancer cells and boost the antitumor T cell response is the use of cancer vaccines. Vaccines have been designed to generate a humoral/cellular immune response with the aim of stimulating the host immune system to recognize and eliminate tumor cells with specific effector and memory T cells. There are two major categories of tumor vaccines: whole cell vaccines and antigen-specific vaccines (289). A brief review on the different vaccines currently investigated for pancreatic cancer can be found in the publication by Skelton et al. (290). Although early studies using single-agent tumor vaccines against PDAC showed improved immune profiles, they were largely unable to produce a positive clinical response (291). This can be explained by the upregulation of immunosuppressive signaling, as well as other immune modulating mechanisms, which negate the positive effects of the vaccine (267, 292).
The induction of T cell infiltration and PD-L1 expression in the TME by vaccine treatment was hypothesized to prime PDACs for anti-PD-1/PD-L1 therapies. Indeed, the whole cell vaccine GVAX, consisting of two allogeneic irradiated PDAC cell lines engineered to secrete GM-CSF, converted a non-immunogenic or “cold” neoplasm into an immunogenic or “hot” neoplasm by inducing infiltration of T cells and development of tertiary lymphoid structures (267, 285). In a subsequent phase Ib study, Le et al. (293) were able to show that the combination of GVAX with ipilimumab induced objective responses in patients with metastatic PDAC that were not observed with either single therapy alone.
Preclinical data suggested beneficial effects when two vaccination treatments were co-implemented, e.g., GVAX and CRS-207, a live-attenuated Listeria monocytogenes vaccine expressing the TAA mesothelin, in a sequential combination—a so-called prime/boost approach. The first vaccine was given to initiate or “prime” the immune system, and this immune response was then “boosted” following re-administration of antigen resulting in the induction of a synergistic enhancement of T cell induction and antitumor effect (289). Based on the preclinical data, a phase II trial (NCT01417000) was conducted resulting in the conclusion that heterologous prime/boost with Cy/GVAX and CRS-207 extended the survival of patients with pancreatic cancer, with minimal toxicity (294). Unfortunately, a subsequent phase IIb trial of CRS-207 and GVAX (NCT02004262) did not show a significant difference in overall survival between the groups treated with either CRS-207/GVAX or CRS207 alone and the group treated with chemotherapy, i.e., physicians’ choice of therapies including: gemcitabine, capecitabine, fluorouracil, leucovorin, irinotecan, and erlotinib.
Although the last mentioned study was quite a set-back, the strategy of combining different immunotherapy options with each other still holds a merit, especially for “prime and boost” approach.
Chimeric Antigen Receptor (CAR)—T Cell Therapy
Chimeric antigen receptors are fusion proteins that can be comprised of three major domains. These are the antigen-specific ectodomain, commonly derived from a single-chain variable antibody-fragment (scFv); a transmembrane domain fused to a spacer that links to the ectodomain; and an endodomain consisting of different cytoplasmic proteins responsible for T cell activation (295). Unlike endogenous TCRs, CARs recognize their target antigen in an MHC (or HLA)-independent manner, due to their engineered antibody fragment. Upon antigen recognition, CAR-T cells are activated, leading to cytokine secretion, T cell proliferation, and antigen-specific cytotoxicity (296). The production of CAR-T cells for adoptive T cell transfer requires the isolation, stimulation, expansion, transduction, i.e., viral vector-mediated insertion of specific CAR genes, and ultimately reinfusion of autologous or allogeneic T cells (297–299).
Although impressive clinical activities of CAR-T cells in hematological malignancies were reported, CAR T-cell trials in solid tumors have yet to yield the same level of success (300, 301). The most prominent obstacles standing in the way of successful CAR-T cell therapy are (1) lack of ideal TSAs, (2) inefficient trafficking of CAR-T cells to tumor sites, (3) the immune-suppressive TME, and (4) the risk of developing on-target/off-tumor toxicities, i.e., the attack of normal cells expressing the targeted tumor antigen (296).
While the investigation of CAR-T cells in pancreatic cancer is still in early stages, it is fair to say that the first above mentioned obstacle does not apply. PDAC exhibits a number of TSAs and, conceptually, is a promising candidate tumor for investigating CAR T-cell therapy. Thus far, there have been preclinical studies on various pancreatic cancer cell surface antigens, namely, MSLN, CEA, MUC1, PSCA, CD24, HER2, and natural killer receptors (302).
The main obstacles in pancreatic cancer are most likely the strong immunosuppressive TME, already discussed in this review, and improper homing and inefficient infiltration of CAR T-cells to the tumor bed. Especially challenging is the high number of infiltrating Tregs and MDSCs, which can deactivate CAR-T cells through cytokines inhibitory cytokines such as TGFβ and IL-10, and the upregulation of inhibitory receptors, e.g., PD-1 on adoptively transferred CAR-T cells after homing to the tumor (95, 302–304). T cell hypofunction was reversed when the cells were isolated from the tumor, or after treatment with a blocking PD-1 antibody (304–306), and there are promising preclinical studies on CAR-T cells engineered to secrete PD-1 checkpoint inhibitors (307, 308) or PD-1 dominant negative receptor (304).
These results provide rationale for combination therapies, with CAR-T cells and checkpoint blockade, as a new strategy to overcome the tumor escape and to further strengthen CAR-T cells, especially in patients with PDAC shown to express high levels of PD-L1.
Adoptive Cell Therapy (ACT) With Endogenous TILs
Adoptive cell therapy using endogenous TILs taken from surgically resected tumors, expanded in vitro, and re-infused back into the patient, is a promising approach for otherwise untreatable cancer types (309). In metastatic melanoma patients, for example, TIL-ACT was associated with a 20% complete response lasting beyond 3 years (310). Gastrointestinal tumor patients with CD3+ T cell infiltration showed a higher rate of progression-free survival (311), and pancreatic adenocarcinomas containing both CD4+ and CD8+ T cells correlated with an improved prognosis and significantly greater 5-year survival (181, 312, 313). This evidence of a host T cell immune response in patients with pancreatic adenocarcinoma drove both Hall et al. (309) and Poschke et al. (314) to expand and analyze the T cell repertoire in resected primary PDAC specimen. Contrary to the common description of PDAC as an immunologically “cold” tumor, they found that most resectable PDA tumors actually contained significant numbers of T-cells and, along with that, tertiary lymphoid structures in which clonal T-cell expansion takes place and were able to expand them in vitro using high levels of IL-2 (309, 314). The majority of these TILs were CD4+ T cells and were highly activated and resembled those extracted from melanoma samples. Media supplemented with anti-4-1BB significantly increased the TIL yield per fragment and shifted the T cell population to predominantly CD8+ cells compared with control cultures. The population of 4-1BB positive CD8+ lymphocytes represented the population of tumor-resident TILs specific for expressed tumor antigens on the surface of pancreatic adenocarcinoma cells (309, 314).
Thus far, there has been no studies investigating the combination of TIL ACT and ICI in pancreatic cancer. However, pretreating PDAC patients with immune checkpoint inhibitors and thus enriching the population of tumor-specific lymphocytes prior to surgical resection might be a worthwhile strategy. This way the yield of tumor reactive cells could be increased, the expansion time and the time between surgery and infusion shortened, and thus the risk of recurrent growth during the expansion period decreased.
This is exactly the approach taken by Mullinax et al. when analyzing the combination of TIL ACT and Ipilimumab in a clinical pilot study (NCT01701674) for metastatic melanoma, and in clinical trials for metastatic ovarian cancer (NCT03287674). The pretreatment with Ipilimumab followed by ACT in metastatic melanoma patients was reported as feasible, well tolerated, and associated with a low rate of attrition due to progression during cell expansion (315). The investigators are currently recruiting patients for a similar trial in metastatic melanoma, now including 4-1BB (NCT02652455), and another study with a similar design is currently recruiting patients with locally advanced or metastatic cancers of various types (NCT03296137).
Considering the positive results in metastatic melanoma, as well as the similarity in the population of extractable T cells from melanoma and PDAC, the investigation of ICI + TIL ACT in PDAC is recommendable.
Concluding Remarks
The revolution of immunotherapy is changing our perspective in cancer therapeutics. For some solid tumors, immunotherapy has already entered into clinical practice. While PDAC is unresponsive and refractory to many of the conventional therapies, immunotherapy holds a promise for future improvement. However, single-agent ICI has largely failed. Based on the findings thus far, the decisive drawback for ICI efficiency in PDAC is the initial T cell priming. Only less than 1% of human PDAC samples are projected to show aberrant genomic instability, enabling T cell priming despite the immunosuppressive microenvironment. However, this does not mean the other 99% are not antigenic, rather its antigenic strength likely cannot beat reduced immunogenicity. Each patient, each tumor, and each cancer cell are distinct. T cells might provide the best repertoire for the recognition of each single difference, yet to overcome immunogenic obstacles, combination strategies are required. Development of the best combinations comes along with better characterization of the patient samples. Characterization of these samples might help us to better classify the individual distinctions that patients, tumors, and cancer cells have, and to find the best combination partners with checkpoint inhibition. Even though complete regression of the primary tumor might not be achieved, reduction and control of metastasis can still provide a considerable prognostic value in PDAC patients. While we know that metastatic lesions evade the expression of high quality neoantigens of their cognate primary tumor and antigen presenting machinery, they might still retain their unique antigenic and immunogenic master regulators to be targeted. Most importantly, T cell memory provides the best tool to minimize disease recurrence, therefore strategies exploiting T cell memory may provide long-term disease control. Before achieving T cell memory, to make PDAC responsive to first time checkpoint inhibition, we have to elucidate and exploit the mechanisms discussed above: (1) increasing initial T cell priming, (2) exceeding immunosuppressive TME, and (3) inhibiting compensatory mechanisms of T cell anergy and exhaustion.
Author Contributions
DK wrote the manuscript, prepared the figures and tables. KC wrote and edited the manuscript. DR provided the tables and edited the manuscript. HA edited and supervised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank to Munich International Max Planck Research Schools-Life Sciences Program (IMPRS-LS) for their financial support of DK. We thank to Dr. Med. Vet. Joerg Steiner and M. A. Zeynep Yueksel for their help during the revision of the manuscript.
Funding
This work is supported by the Mildred-Scheel-Professur der Deutschen Krebshilfe 111464 and Deutsches Forschungsgemeinschaft (SFB 1321/ P04) to HA.
References
1. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res (2014) 74:2913–21. doi:10.1158/0008-5472.CAN-14-0155
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin (2018) 68:7–30. doi:10.3322/caac.21442
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin (2017) 67:7–30. doi:10.3322/caac.21387
4. Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, et al. Pancreatic cancer. Nat Rev Dis Prim (2016) 2:1–22. doi:10.1038/nrdp.2016.22
5. Neesse A, Algül H, Tuveson DA, Gress TM. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut (2015) 64:1476–84. doi:10.1136/gutjnl-2015-309304
7. Ko AH, Bekaii-Saab T, Van Ziffle J, Mirzoeva OM, Joseph NM, Talasaz AA, et al. A multicenter, open-label phase II clinical trial of combined MEK plus EGFR inhibition for chemotherapy-refractory advanced pancreatic adenocarcinoma. Clin Cancer Res (2016) 22:61–8. doi:10.1158/1078-0432.CCR-15-0979
8. Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz-Ares L, Vansteenkiste J, et al. A Phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res (2015) 21:730–8. doi:10.1158/1078-0432.CCR-14-1814
9. Tolcher AW, Bendell JC, Papadopoulos KP, Burris HA, Patnaik A, Jones SF, et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann Oncol (2015) 26:58–64. doi:10.1093/annonc/mdu482
10. Wolpin BM, Hezel AF, Abrams T, Blaszkowsky LS, Meyerhardt JA, Chan JA, et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol (2009) 27:193–8. doi:10.1200/JCO.2008.18.9514
11. Infante JR, Somer BG, Park JO, Li C-P, Scheulen ME, Kasubhai SM, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer (2014) 50:2072–81. doi:10.1016/j.ejca.2014.04.024
12. Bodoky G, Timcheva C, Spigel DR, La Stella PJ, Ciuleanu TE, Pover G, et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest New Drugs (2012) 30:1216–23. doi:10.1007/s10637-011-9687-4
13. Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med (2011) 364:1817–25. doi:10.1056/NEJMoa1011923
14. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med (2013) 369:1691–703. doi:10.1056/NEJMoa1304369
15. Goldstein D, El-Maraghi RH, Hammel P, Heinemann V, Kunzmann V, Sastre J, et al. Nab-paclitaxel plus gemcitabine for metastatic pancreatic cancer: long-term survival from a phase III trial. J Natl Cancer Inst (2015) 107. doi:10.1093/jnci/dju413
16. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer (2015) 136:E359–86. doi:10.1002/ijc.29210
17. Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, et al. Drug resistance in cancer: an overview. Cancers (2014) 6:1769–92. doi:10.3390/cancers6031769
18. Groenendijk FH, Bernards R. Drug resistance to targeted therapies: Déjà vu all over again. Mol Oncol (2014) 8:1067–83. doi:10.1016/j.molonc.2014.05.004
19. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi:10.1056/NEJMoa1003466
20. Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol (2015) 16:908–18. doi:10.1016/S1470-2045(15)00083-2
21. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med (2015) 372:2521–32. doi:10.1056/NEJMoa1503093
22. Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol (2015) 16:375–84. doi:10.1016/S1470-2045(15)70076-8
23. Rizvi NA, Mazières J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol (2015) 16:257–65. doi:10.1016/S1470-2045(15)70054-9
24. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med (2015) 372:2018–28. doi:10.1056/NEJMoa1501824
25. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med (2016) 375:1823–33. doi:10.1056/NEJMoa1606774
26. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med (2015) 373:1627–39. doi:10.1056/NEJMoa1507643
27. Taylor M, Antonia S, Bendell J, Calvo E, Jager D, Braud F, et al. Phase I/II study of nivolumab with or without ipilimumab for treatment of recurrent small cell lung cancer (SCLC): CA209-032. J Immunother Cancer (2015) 3:P376. doi:10.1186/2051-1426-3-S2-P376
28. Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, et al. Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J Clin Oncol (2015) 33:1430–7. doi:10.1200/JCO.2014.59.0703
29. Weinstock M, McDermott D. Targeting PD-1/PD-L1 in the treatment of metastatic renal cell carcinoma. Ther Adv Urol (2015) 7:365–77. doi:10.1177/1756287215597647
30. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature (2014) 515:558–62. doi:10.1038/nature13904
31. Powles T, O’Donnell PH, Massard C, Arkenau H-T, Friedlander TW, Hoimes CJ, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma. JAMA Oncol (2017) 3:e172411. doi:10.1001/jamaoncol.2017.2411
32. Massard C, Gordon MS, Sharma S, Rafii S, Wainberg ZA, Luke J, et al. Safety and efficacy of durvalumab (MEDI4736), an anti-programmed cell death ligand-1 immune checkpoint inhibitor, in patients with advanced urothelial bladder cancer. J Clin Oncol (2016) 34:3119–25. doi:10.1200/JCO.2016.67.9761
33. Seiwert TY, Haddad RI, Gupta S, Mehra R, Tahara M, Berger R, et al. Antitumor activity and safety of pembrolizumab in patients (pts) with advanced squamous cell carcinoma of the head and neck (SCCHN): preliminary results from KEYNOTE-012 expansion cohort. J Clin Oncol (2015) 33:LBA6008. doi:10.1200/jco.2015.33.18_suppl.lba6008
34. Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol (2016) 17:956–65. doi:10.1016/S1470-2045(16)30066-3
35. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med (2015) 372:311–9. doi:10.1056/NEJMoa1411087
36. Armand P, Shipp MA, Ribrag V, Michot JM, Zinzani PL, Kuruvilla J, et al. Programmed death-1 blockade with pembrolizumab in patients with classical Hodgkin lymphoma after brentuximab vedotin failure. J Clin Oncol (2016) 34:3733–9. doi:10.1200/JCO.2016.67.3467
37. Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib study. J Clin Oncol (2016) 34:2698–704. doi:10.1200/JCO.2015.65.9789
38. Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366:2455–65. doi:10.1056/NEJMoa1200694
39. Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother (2010) 33:828–33. doi:10.1097/CJI.0b013e3181eec14c
40. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi:10.1038/nrc3239
41. Gao GF, Rao Z, Bell JI. Molecular coordination of αβ T-cell receptors and coreceptors CD8 and CD4 in their recognition of peptide-MHC ligands. Trends Immunol (2002) 23:408–13. doi:10.1016/S1471-4906(02)02282-2
42. Steimle V, Siegrist C, Mottet A, Lisowska-Grospierre B, Mach B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science (1994) 265:106–9. doi:10.1126/science.8016643
43. Stout RD, Bottomly K. Antigen-specific activation of effector macrophages by IFN-gamma producing (TH1) T cell clones. Failure of IL-4-producing (TH2) T cell clones to activate effector function in macrophages. J Immunol (1989) 142:760–5.
44. Dobrzanski MJ. Expanding roles for CD4 T cells and their subpopulations in tumor immunity and therapy. Front Oncol (2013) 3:63. doi:10.3389/fonc.2013.00063
45. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4+T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell (2009) 16:91–102. doi:10.1016/j.ccr.2009.06.018
46. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res (2017) 27:109–18. doi:10.1038/cr.2016.151
47. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol (2013) 25:214–21. doi:10.1016/j.coi.2012.12.003
48. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27:451–61. doi:10.1016/j.ccell.2015.03.001
49. Iwai Y, Hamanishi J, Chamoto K, Honjo T. Cancer immunotherapies targeting the PD-1 signaling pathway. J Biomed Sci (2017) 24:26. doi:10.1186/s12929-017-0329-9
50. Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell (1992) 71:1065–8. doi:10.1016/S0092-8674(05)80055-8
51. Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity (1994) 1:793–801. doi:10.1016/S1074-7613(94)80021-9
52. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332:600–3. doi:10.1126/science.1202947
53. Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med (1996) 183:2533–40. doi:10.1084/jem.183.6.2533
54. Fallarino F, Fields PE, Gajewski TF. B7-1 engagement of cytotoxic T lymphocyte antigen 4 inhibits T cell activation in the absence of CD28. J Exp Med (1998) 188:205–10. doi:10.1084/jem.188.1.205
55. Chambers CA, Sullivan TJ, Truong T, Allison JP. Secondary but not primary T cell responses are enhanced in CTLA-4-deficient CD8+ T cells. Eur J Immunol (1998) 28:3137–43. doi:10.1002/(SICI)1521-4141(199810)28:10<3137::AID-IMMU3137>3.0.CO;2-X
56. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by Cd25+ Cd4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med (2000) 192:303–10. doi:10.1084/jem.192.2.303
57. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi:10.1126/science.1079490
58. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi:10.1038/ni904
59. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322:271–5. doi:10.1126/science.1160062
60. Klocke K, Holmdahl R, Wing K. CTLA-4 expressed by FOXP3+ regulatory T cells prevents inflammatory tissue attack and not T-cell priming in arthritis. Immunology (2017) 152:125–37. doi:10.1111/imm.12754
61. Bengsch F, Knoblock DM, Liu A, McAllister F, Beatty GL. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol Immunother (2017) 66:1609–17. doi:10.1007/s00262-017-2053-4
62. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti–CTLA-4 antibodies. J Exp Med (2009) 206:1717–25. doi:10.1084/jem.20082492
63. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11:3887–95.
64. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the Pd-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192:1027–34. doi:10.1084/jem.192.7.1027
65. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med (1999) 5:1365–9. doi:10.1038/70932
66. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol (2001) 2:261–8. doi:10.1038/85330
67. Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med (2001) 193:839–46. doi:10.1084/jem.193.7.839
68. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol (1996) 8:765–72. doi:10.1093/intimm/8.5.765
69. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8+ T cells. Immunity (2011) 35:400–12. doi:10.1016/j.immuni.2011.06.015
70. Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T. PD-1 inhibits antiviral immunity at the effector phase in the liver. J Exp Med (2003) 198:39–50. doi:10.1084/jem.20022235
71. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2006) 439:682–7. doi:10.1038/nature04444
72. Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8+ T cells during chronic infection. Immunity (2014) 41:802–14. doi:10.1016/j.immuni.2014.10.013
73. Asano T, Meguri Y, Yoshioka T, Kishi Y, Iwamoto M, Nakamura M, et al. PD-1 modulates regulatory T-cell homeostasis during low-dose interleukin-2 therapy. Blood (2017) 129:2186–97. doi:10.1182/blood-2016-09-741629
74. Wang L, Pino-Lagos K, De Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+ CD4+ regulatory T cells. Proc Natl Acad Sci U S A (2008) 105:9331–6. doi:10.1073/pnas.0710441105
75. Wang C, Thudium KB, Han M, Wang X-T, Huang H, Feingersh D, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res (2014) 2:846–56. doi:10.1158/2326-6066.CIR-14-0040
76. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11:141–51. doi:10.1016/S1074-7613(00)80089-8
77. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science (2001) 291:319–22. doi:10.1126/science.291.5502.319
78. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3:541–7. doi:10.1016/1074-7613(95)90125-6
79. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science (1995) 270:985–8. doi:10.1126/science.270.5238.985
80. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi:10.1146/annurev.immunol.26.021607.090331
81. Linsley PS, Bradshaw J, Greene JA, Peach R, Bennett KL, Mittler RS. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity (1996) 4:535–43. doi:10.1016/S1074-7613(00)80480-X
82. Davoodzadeh Gholami M, Kardar GA, Saeedi Y, Heydari S, Garssen J, Falak R. Exhaustion of T lymphocytes in the tumor microenvironment: significance and effective mechanisms. Cell Immunol (2017) 322:1–14. doi:10.1016/j.cellimm.2017.10.002
83. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366:2443–54. doi:10.1056/NEJMoa1200690
84. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373:23–34. doi:10.1056/NEJMoa1504030
85. Ehrlich P. Vol. II. Immunology and cancer research. In: Himmelweit F, Marquardt M, Dale H, editors. Collected Papers of Paul Ehrlich: In Four Volumes Including a Complete Bibliography. London: Pergamon Press (1960).
86. Burnet FM. Immunological surveillance in neoplasia. Immunol Rev (1971) 7:3–25. doi:10.1111/j.1600-065X.1971.tb00461.x
88. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol (2002) 3:991–8. doi:10.1038/ni1102-991
89. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol (2004) 22:329–60. doi:10.1146/annurev.immunol.22.012703.104803
90. Gopinathan A, Morton JP, Jodrell DI, Sansom OJ. GEMMs as preclinical models for testing pancreatic cancer therapies. Dis Model Mech (2015) 8:1185–200. doi:10.1242/dmm.021055
91. Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell (2003) 4:437–50. doi:10.1016/S1535-6108(03)00309-X
92. Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res (2007) 67:9518–27. doi:10.1158/0008-5472.CAN-07-0175
93. Evans RA, Diamond MS, Rech AJ, Chao T, Richardson MW, Lin JH, et al. Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight (2016) 1. doi:10.1172/jci.insight.88328
94. Foley K, Kim V, Jaffee E, Zheng L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett (2016) 381:244–51. doi:10.1016/j.canlet.2015.12.020
95. Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res (2006) 12:5423–34. doi:10.1158/1078-0432.CCR-06-0369
96. Vonderheide RH. The immune revolution: a case for priming, not checkpoint. Cancer Cell (2018) 33:563–9. doi:10.1016/j.ccell.2018.03.008
97. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res (2015) 21:687–92. doi:10.1158/1078-0432.CCR-14-1860
98. Yarchoan M, Johnson BA, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer (2017) 17:209–22. doi:10.1038/nrc.2016.154
99. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer (2016) 16:275–87. doi:10.1038/nrc.2016.36
100. Coulie PG, Van Den Eynde BJ, Van Der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer (2014) 14:135–46. doi:10.1038/nrc3670
101. Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, et al. Comprehensive analysis of hypermutation in human cancer. Cell (2017) 171:1042.e–56.e. doi:10.1016/j.cell.2017.09.048
102. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science (2013) 339:1546–58. doi:10.1126/science.1235122
103. Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther (2017) 16(11):2598–608. doi:10.1158/1535-7163.MCT-17-0386
104. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348:124–8. doi:10.1126/science.aaa1348
105. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med (2017) 377:2500–1. doi:10.1056/NEJMc1713444
106. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (2017) 357:409–13. doi:10.1126/science.aan6733
107. Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature (2017) 552:1–5. doi:10.1038/nature24673
108. Humphris JL, Patch AM, Nones K, Bailey PJ, Johns AL, McKay S, et al. Hypermutation in pancreatic cancer. Gastroenterology (2017) 152:68.e–74.e. doi:10.1053/j.gastro.2016.09.060
109. Brown JS, Sundar R, Lopez J. Combining DNA damaging therapeutics with immunotherapy: more haste, less speed. Br J Cancer (2018) 118:312–24. doi:10.1038/bjc.2017.376
110. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature (2013) 499:214–8. doi:10.1038/nature12213
111. Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature (2017) 551:S12–6. doi:10.1038/nature24462
112. de Charette M, Marabelle A, Houot R. Turning tumour cells into antigen presenting cells: the next step to improve cancer immunotherapy? Eur J Cancer (2016) 68:134–47. doi:10.1016/j.ejca.2016.09.010
113. Delp K, Momburg F, Hilmes C, Huber C, Seliger B. Functional deficiencies of components of the MHC class I antigen pathway in human tumors of epithelial origin. Bone Marrow Transplant (2000) 25:S88–95. doi:10.1038/sj.bmt.1702363
114. Herrmann F, Lehr HA, Drexler I, Sutter G, Hengstler J, Wollscheid U, et al. HER-2/neu-mediated regulation of components of the MHC class I antigen-processing pathway. Cancer Res (2004) 64:215–20. doi:10.1158/0008-5472.CAN-2522-2
115. Paulson KG, Tegeder A, Willmes C, Iyer JG, Afanasiev OK, Schrama D, et al. Downregulation of MHC-I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol Res (2014) 2:1071–9. doi:10.1158/2326-6066.CIR-14-0005
116. Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst (2013) 105:1172–87. doi:10.1093/jnci/djt184
117. Lohmann S, Wollscheid U, Huber C, Seliger B. Multiple levels of MHC class I down-regulation by ras oncogenes. Scand J Immunol (1996) 43:537–44. doi:10.1046/j.1365-3083.1996.d01-73.x
118. Seliger B, Harders C, Wollscheid U, Staege MS, Reske-Kunz AB, Huber C. Suppression of MHC class I antigens in oncogenic transformants: association with decreased recognition by cytotoxic T lymphocytes. Exp Hematol (1996) 24:1275–9.
119. Mimura K, Shiraishi K, Mueller A, Izawa S, Kua L-F, So J, et al. The MAPK pathway is a predominant regulator of HLA-A expression in esophageal and gastric cancer. J Immunol (2013) 191:6261–72. doi:10.4049/jimmunol.1301597
120. Pommier A, Anaparthy N, Memos N, Kelley ZL, Gouronnec A, Yan R, et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science (2018) 360:eaao4908. doi:10.1126/science.aao4908
121. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371:2189–99. doi:10.1056/NEJMoa1406498
122. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science (2015) 350:207–11. doi:10.1126/science.aad0095
123. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell (2016) 165:35–44. doi:10.1016/j.cell.2016.02.065
124. Bailey P, Chang DK, Forget MA, Lucas FAS, Alvarez HA, Haymaker C, et al. Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Sci Rep (2016) 6:1–8. doi:10.1038/srep35848
125. Balli D, Rech AJ, Stanger BZ, Vonderheide RH. Immune cytolytic activity stratifies molecular subsets of human pancreatic cancer. Clin Cancer Res (2017) 23:3129–38. doi:10.1158/1078-0432.CCR-16-2128
126. Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med (2011) 17:500–3. doi:10.1038/nm.2344
127. Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet (2015) 47:1168–78. doi:10.1038/ng.3398
128. Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature (2012) 491:399–405. doi:10.1038/nature11547
129. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature (2015) 518:495–501. doi:10.1038/nature14169
130. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature (2016) 531:47–52. doi:10.1038/nature16965
131. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell (2015) 160:48–61. doi:10.1016/j.cell.2014.12.033
132. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell (2012) 21:836–47. doi:10.1016/j.ccr.2012.04.024
133. McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, et al. Oncogenic kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell (2014) 25:621–37. doi:10.1016/j.ccr.2014.03.014
134. Vonderheide RH. Tumor-promoting inflammatory networks in pancreatic neoplasia: another reason to loathe kras. Cancer Cell (2014) 25:553–4. doi:10.1016/j.ccr.2014.04.020
135. Zhang J, Wolfgang C, Zheng L. Precision immuno-oncology: prospects of individualized immunotherapy for pancreatic cancer. Cancers (Basel) (2018) 10:39. doi:10.3390/cancers10020039
136. Lastwika KJ, Wilson W, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res (2016) 76:227–38. doi:10.1158/0008-5472.CAN-14-3362
137. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med (2007) 13:84–8. doi:10.1038/nm1517
138. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science (2016) 352:227–31. doi:10.1126/science.aac9935
139. Feng J, Yang H, Zhang Y, Wei H, Zhu Z, Zhu B, et al. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene (2017) 36:5829–39. doi:10.1038/onc.2017.188
140. Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut (2017) 66:124–36. doi:10.1136/gutjnl-2016-312078
141. Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med (2013) 369:134–44. doi:10.1056/NEJMoa1305133
142. Khandelwal N, Breinig M, Speck T, Michels T, Kreutzer C, Sorrentino A, et al. A high-throughput RNAi screen for detection of immune-checkpoint molecules that mediate tumor resistance to cytotoxic T lymphocytes. EMBO Mol Med (2015) 7:450–63. doi:10.15252/emmm.201404414
143. Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell (2016) 30:925–39. doi:10.1016/j.ccell.2016.10.010
144. Lu C, Paschall AV, Shi H, Savage N, Waller JL, Sabbatini ME, et al. The MLL1-H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion. J Natl Cancer Inst (2017) 109. doi:10.1093/jnci/djw283
145. Dunn GP, Bruce AT, Sheehan KCF, Shankaran V, Uppaluri R, Bui JD, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol (2005) 6:722–9. doi:10.1038/ni1213
146. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell (2016) 167:397.e–404.e. doi:10.1016/j.cell.2016.08.069
147. Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature (2017) 548:537–42. doi:10.1038/nature23477
148. Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I. Dual faces of ifnγ in cancer progression: a role of pd-l1 induction in the determination of pro and antitumor immunity. Clin Cancer Res (2016) 22:2329–34. doi:10.1158/1078-0432.CCR-16-0224
149. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov (2017) 7:188–201. doi:10.1158/2159-8290.CD-16-1223
150. Cogdill AP, Andrews MC, Wargo JA. Hallmarks of response to immune checkpoint blockade. Br J Cancer (2017) 117:1–7. doi:10.1038/bjc.2017.136
151. Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol (2008) 8:74–80. doi:10.1038/nri2264
152. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med (2014) 6:237ra67. doi:10.1126/scitranslmed.3007974
153. Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, et al. Monocytic CCR2+ myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res (2012) 72:876–86. doi:10.1158/0008-5472.CAN-11-1792
154. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res (2008) 68:918–26. doi:10.1158/0008-5472.CAN-07-5714
155. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2014) 25:719–34. doi:10.1016/j.ccr.2014.04.005
156. Carstens JL, De Sampaio PC, Yang D, Barua S, Wang H, Rao A, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun (2017) 8:15095. doi:10.1038/ncomms15095
157. Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology (2013) 145:1121–32. doi:10.1053/j.gastro.2013.07.025
158. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A (2013) 110:20212–7. doi:10.1073/pnas.1320318110
159. Johnson BA, Yarchoan M, Lee V, Laheru DA, Jaffee EM. Strategies for increasing pancreatic tumor immunogenicity. Clin Cancer Res (2017) 23:1656–69. doi:10.1158/1078-0432.CCR-16-2318
160. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med (2016) 22:851–60. doi:10.1038/nm.4123
161. Infante JR, Camidge DR, Mileshkin LR, Chen EX, Hicks RJ, Rischin D, et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J Clin Oncol (2012) 30:1527–33. doi:10.1200/JCO.2011.38.9346
162. Jones SF, Siu LL, Bendell JC, Cleary JM, Razak ARA, Infante JR, et al. A phase i study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Invest New Drugs (2015) 33:1100–7. doi:10.1007/s10637-015-0282-y
163. Soria JC, Gan HK, Blagden SP, Plummer R, Arkenau HT, Ranson M, et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Ann Oncol (2016) 27:2268–74. doi:10.1093/annonc/mdw427
164. Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Klöppel G, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell (2011) 19:456–69. doi:10.1016/j.ccr.2011.03.009
165. Fukuda A, Wang SC, Morris JP, Folias AE, Liou A, Kim GE, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell (2011) 19:441–55. doi:10.1016/j.ccr.2011.03.002
166. Kang R, Loux T, Tang D, Schapiro NE, Vernon P, Livesey KM, et al. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc Natl Acad Sci U S A (2012) 109:7031–6. doi:10.1073/pnas.1113865109
167. Zhang Y, Yan W, Collins MA, Bednar F, Rakshit S, Zetter BR, et al. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res (2013) 73:6359–74. doi:10.1158/0008-5472.CAN-13-1558-T
168. Angevin E, Tabernero J, Elez E, Cohen SJ, Bahleda R, van Laethem J-L, et al. A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res (2014) 20:2192–204. doi:10.1158/1078-0432.CCR-13-2200
169. Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut (2018) 67:320–32. doi:10.1136/gutjnl-2016-311585
170. Sato N, Kohi S, Hirata K, Goggins M. Role of hyaluronan in pancreatic cancer biology and therapy: once again in the spotlight. Cancer Sci (2016) 107:569–75. doi:10.1111/cas.12913
171. Cheng XB, Sato N, Kohi S, Yamaguchi K. Prognostic impact of hyaluronan and its regulators in pancreatic ductal adenocarcinoma. PLoS One (2013) 8:e80765. doi:10.1371/journal.pone.0080765
172. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut (2013) 62:112–20. doi:10.1136/gutjnl-2012-302529
173. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell (2012) 21:418–29. doi:10.1016/j.ccr.2012.01.007
174. Thompson CB, Shepard HM, O’Connor PM, Kadhim S, Jiang P, Osgood RJ, et al. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol Cancer Ther (2010) 9:3052–64. doi:10.1158/1535-7163.MCT-10-0470
175. Singha NC, Nekoroski T, Zhao C, Symons R, Jiang P, Frost GI, et al. Tumor-associated hyaluronan limits efficacy of monoclonal antibody therapy. Mol Cancer Ther (2015) 14:523–32. doi:10.1158/1535-7163.MCT-14-0580
176. Blache CA, Manuel ER, Kaltcheva TI, Wong AN, Ellenhorn JDI, Blazar BR, et al. Systemic delivery of Salmonella typhimurium transformed with IDO shRNA enhances intratumoral vector colonization and suppresses tumor growth. Cancer Res (2012) 72:6447–56. doi:10.1158/0008-5472.CAN-12-0193
177. Manuel ER, Chen J, D’Apuzzo M, Lampa MG, Kaltcheva TI, Thompson CB, et al. Salmonella-based therapy targeting indoleamine 2,3-dioxygenase coupled with enzymatic depletion of tumor hyaluronan induces complete regression of aggressive pancreatic tumors. Cancer Immunol Res (2015) 3:1096–107. doi:10.1158/2326-6066.CIR-14-0214
178. Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell (2014) 159:80–93. doi:10.1016/j.cell.2014.08.007
179. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37:208–20. doi:10.1016/j.it.2016.01.004
180. Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, et al. Macrophage polarization in tumour progression. Semin Cancer Biol (2008) 18:349–55. doi:10.1016/j.semcancer.2008.03.004
181. Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer (2013) 108:914–23. doi:10.1038/bjc.2013.32
182. Protti MP, De Monte L. Immune infiltrates as predictive markers of survival in pancreatic cancer patients. Front Physiol (2013) 4:210. doi:10.3389/fphys.2013.00210
183. Monu NR, Frey AB. Myeloid-derived suppressor cells and anti-tumor T cells: a complex relationship. Immunol Invest (2012) 41:595–613. doi:10.3109/08820139.2012.673191
184. Shibuya KC, Goel VK, Xiong W, Sham JG, Pollack SM, Leahy AM, et al. Pancreatic ductal adenocarcinoma contains an effector and regulatory immune cell infiltrate that is altered by multimodal neoadjuvant treatment. PLoS One (2014) 9:e96565. doi:10.1371/journal.pone.0096565
185. Tassi E, Gavazzi F, Albarello L, Senyukov V, Longhi R, Dellabona P, et al. Carcinoembryonic antigen-specific but not antiviral CD4+ T cell immunity is impaired in pancreatic carcinoma patients. J Immunol (2008) 181:6595–603. doi:10.4049/jimmunol.181.9.6595
186. De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med (2011) 208:469–78. doi:10.1084/jem.20101876
187. Soares KC, Rucki AA, Kim V, Foley K, Solt S, Wolfgang CL, et al. TGF-β blockade depletes T regulatory cells from metastatic pancreatic tumors in a vaccine dependent manner. Oncotarget (2015) 6:43005–15. doi:10.18632/oncotarget.5656
188. Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother (2015) 38:1–11. doi:10.1097/CJI.0000000000000062
189. Leliefeld PHC, Koenderman L, Pillay J. How neutrophils shape adaptive immune responses. Front Immunol (2015) 6:471. doi:10.3389/fimmu.2015.00471
190. Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol (2004) 22:307–28. doi:10.1146/annurev.immunol.22.012703.104533
191. Schoenberger SP, Toes REM, Van der Voort EIH, Offringa R, Melief CJM. T-cell help for cytotoxic T-lymphocytes is mediated by CD40-CD40L interactions. Nature (1998) 393:480–3. doi:10.1038/31002
192. Bennett SRM, Carbone FR, Karamalis F, Flavell RA, Miller JFAP, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature (1998) 393:478–80. doi:10.1038/30996
193. Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature (1998) 393:474–8. doi:10.1038/30989
194. French RR, Chan HTC, Tutt AL, Glennie MJ. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med (1999) 5:548–53. doi:10.1038/8426
195. Diehl L, Den Boer AT, Schoenberger SP, Van Der Voort EIH, Schumacher TNM, Melief CJM, et al. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat Med (1999) 5:774–9. doi:10.1038/10495
196. Sotomayor EM, Borrello I, Tubb E, Rattis FM, Bien H, Lu Z, et al. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of cd40. Nat Med (1999) 5:780–7. doi:10.1038/10503
197. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (2011) 331:1612–6. doi:10.1126/science.1198443
198. Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, et al. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6Clow F4/80+ extratumoral macrophages. Gastroenterology (2015) 149:201–10. doi:10.1053/j.gastro.2015.04.010
199. Vonderheide RH, Burg JM, Mick R, Trosko JA, Li D, Shaik MN, et al. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology (2013) 2:e23033. doi:10.4161/onci.23033
200. Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol (2007) 25:876–83. doi:10.1200/JCO.2006.08.3311
201. Nowak AK, Cook AM, McDonnell AM, Millward MJ, Creaney J, Francis RJ, et al. A phase 1b clinical trial of the CD40-activating antibody CP-870,893 in combination with cisplatin and pemetrexed in malignant pleural mesothelioma. Ann Oncol (2015) 26:2483–90. doi:10.1093/annonc/mdv387
202. Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res (2013) 19:6286–95. doi:10.1158/1078-0432.CCR-13-1320
203. Byrne KT, Vonderheide RH. CD40 stimulation obviates innate sensors and drives T cell immunity in cancer. Cell Rep (2016) 15:2719–32. doi:10.1016/j.celrep.2016.05.058
204. Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell (2008) 133:1006–18. doi:10.1016/j.cell.2008.03.038
205. Lesina M, Wörmann SM, Morton J, Diakopoulos KN, Korneeva O, Wimmer M, et al. RelA regulates CXCL1/CXCR2-dependent oncogene-induced senescence in murine Kras-driven pancreatic carcinogenesis. J Clin Invest (2016) 126:2919–32. doi:10.1172/JCI86477
206. Cacalano G, Lee J, Kikly K, Ryan AM, Pitts-Meek S, Hultgren B, et al. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science (1994) 265:682–4. doi:10.1126/science.8036519
207. Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest (2010) 120:2423–31. doi:10.1172/JCI41649
208. Chao T, Furth EE, Vonderheide RH. CXCR2-dependent accumulation of tumor-associated neutrophils regulates T-cell immunity in pancreatic ductal adenocarcinoma. Cancer Immunol Res (2016) 4:968–82. doi:10.1158/2326-6066.CIR-16-0188
209. Kaneider NC, Agarwal A, Leger AJ, Kuliopulos A. Reversing systemic inflammatory response syndrome with chemokine receptor pepducins. Nat Med (2005) 11:661–5. doi:10.1038/nm1245
210. Jamieson T, Clarke M, Steele CW, Samuel MS, Neumann J, Jung A, et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J Clin Invest (2012) 122:3127–44. doi:10.1172/JCI61067
211. Nicholls DJ, Wiley K, Dainty I, MacIntosh F, Phillips C, Gaw A, et al. Pharmacological characterization of AZD5069, a slowly reversible CXC chemokine receptor 2 antagonist. J Pharmacol Exp Ther (2015) 353:340–50. doi:10.1124/jpet.114.221358
212. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell (2016) 29:832–45. doi:10.1016/j.ccell.2016.04.014
213. Stromnes IM, Schmitt TM, Hulbert A, Brockenbrough JS, Nguyen HN, Cuevas C, et al. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell (2015) 28:638–52. doi:10.1016/j.ccell.2015.09.022
214. Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med (2001) 193:727–40. doi:10.1084/jem.193.6.727
215. Cannarile MA, Weisser M, Jacob W, Jegg A-M, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer (2017) 5:53. doi:10.1186/s40425-017-0257-y
216. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res (2014) 74:5057–69. doi:10.1158/0008-5472.CAN-13-3723
217. Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell (2017) 32:654–68.e5. doi:10.1016/j.ccell.2017.10.005
218. Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN-MDSC: their biological role and interaction with stromal cells. Semin Immunol (2018) 35:19–28. doi:10.1016/j.smim.2017.12.004
219. Sagiv-Barfi I, Kohrt HEK, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci U S A (2015) 112:E966–72. doi:10.1073/pnas.1500712112
220. Masso-Valles D, Jauset T, Serrano E, Sodir NM, Pedersen K, Affara NI, et al. Ibrutinib exerts potent antifibrotic and antitumor activities in mouse models of pancreatic adenocarcinoma. Cancer Res (2015) 75:1675–81. doi:10.1158/0008-5472.CAN-14-2852
221. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discov (2016) 6:270–85. doi:10.1158/2159-8290.CD-15-0827
222. Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood (2013) 122:2539–49. doi:10.1182/blood-2013-06-507947
223. Zou C, Zhao P, Xiao Z, Han X, Fu F, Fu L. γδ T cells in cancer immunotherapy. Oncotarget (2017) 8(5):8900–9. doi:10.18632/oncotarget.13051
224. Tanaka Y, Morita CT, Tanaka Y, Nieves E, Brenner MB, Bloom BR. Natural and synthetic non-peptide antigens recognized by human γδ T cells. Nature (1995) 375:155–8. doi:10.1038/375155a0
225. Lawand M, Déchanet-Merville J, Dieu-Nosjean MC. Key features of gamma-delta T-cell subsets in human diseases and their immunotherapeutic implications. Front Immunol (2017) 8:761. doi:10.3389/fimmu.2017.00761
226. Oberg HH, Peipp M, Kellner C, Sebens S, Krause S, Petrick D, et al. Novel bispecific antibodies increase GD T-cell cytotoxicity against pancreatic cancer cells. Cancer Res (2014) 74:1349–60. doi:10.1158/0008-5472.CAN-13-0675
227. Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, et al. γδ T cells support pancreatic oncogenesis by restraining αβ T cell activation. Cell (2016) 166:1485.e–99.e. doi:10.1016/j.cell.2016.07.046
228. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186:3299–303. doi:10.4049/jimmunol.1003613
229. van der Windt GJW, Pearce EL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev (2012) 249:27–42. doi:10.1111/j.1600-065X.2012.01150.x
230. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi:10.1016/j.immuni.2011.09.021
231. Chang C-H, Curtis JD, Maggi LB, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153:1239–51. doi:10.1016/j.cell.2013.05.016
232. Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol (2009) 183:6095–101. doi:10.4049/jimmunol.0803510
233. Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, et al. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front Immunol (2017) 8:248. doi:10.3389/fimmu.2017.00248
234. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162:1229–41. doi:10.1016/j.cell.2015.08.016
235. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol (2010) 185:1037–44. doi:10.4049/jimmunol.0903586
236. Speiser DE, Ho P-C, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol (2016) 16:599–611. doi:10.1038/nri.2016.80
237. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med (2005) 202:931–9. doi:10.1084/jem.20050715
238. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res (2009) 69:1553–60. doi:10.1158/0008-5472.CAN-08-1921
239. Löb S, Königsrainer A, Zieker D, Brücher BLDM, Rammensee HG, Opelz G, et al. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol Immunother (2009) 58:153–7. doi:10.1007/s00262-008-0513-6
240. Braun D, Longman RS, Albert ML. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood (2005) 106:2375–81. doi:10.1182/blood-2005-03-0979
241. Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9:1269–74. doi:10.1038/nm934
242. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of t cell proliferation by macrophage tryptophan catabolism. J Exp Med (1999) 189:1363–72. doi:10.1084/jem.189.9.1363
243. Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ (2002) 9:1069–77. doi:10.1038/sj.cdd.4401073
244. Platten M, Wick W, Van Den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res (2012) 72:5435–40. doi:10.1158/0008-5472.CAN-12-0569
245. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109:3812–9. doi:10.1182/blood-2006-07-035972
246. Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E. Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation. Int J Cancer (2012) 131:633–40. doi:10.1002/ijc.26410
247. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res (2012) 72:2746–56. doi:10.1158/0008-5472.CAN-11-1272
248. Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood (1997) 90:1600–10.
249. Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol (2014) 5. doi:10.3389/fimmu.2014.00304
250. Ohta A. A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol (2016) 7:1–11. doi:10.3389/fimmu.2016.00109
251. Sreeramkumar V, Fresno M, Cuesta N. Prostaglandin E2 and T cells: friends or foes? Immunol Cell Biol (2012) 90:579–86. doi:10.1038/icb.2011.75
252. Halbrook CJ, Lyssiotis CA. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell (2017) 31:5–19. doi:10.1016/j.ccell.2016.12.006
253. Mueller TC, Burmeister MA, Bachmann J, Martignoni ME. Cachexia and pancreatic cancer: are there treatment options? World J Gastroenterol (2014) 20:9361–73. doi:10.3748/wjg.v20.i28.9361
254. Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, Coll AP, et al. Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab (2016) 24:672–84. doi:10.1016/j.cmet.2016.10.010
255. Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS One (2011) 6:e22538. doi:10.1371/journal.pone.0022538
256. Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab (2012) 303:E410–21. doi:10.1152/ajpendo.00039.2012
257. Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer (2017) 17:286–301. doi:10.1038/nrc.2017.17
258. Ruess DA, Görgülü K, Wörmann SM, Algül H. Pharmacotherapeutic management of pancreatic ductal adenocarcinoma: current and emerging concepts. Drugs Aging (2017) 34:331–57. doi:10.1007/s40266-017-0453-y
259. Zheng Y, Dou Y, Duan L, Cong C, Gao A, Lai Q, et al. Using chemo-drugs or irradiation to break immune tolerance and facilitate immunotherapy in solid cancer. Cell Immunol (2015) 294:54–9. doi:10.1016/j.cellimm.2015.02.003
260. Kim SK, Wu CC, Horowitz DP. Stereotactic body radiotherapy for the pancreas: a critical review for the medical oncologist. J Gastrointest Oncol (2016) 7:479–86. doi:10.21037/jgo.2015.10.01
261. Kalina JL, Neilson DS, Comber AP, Rauw JM, Alexander AS, Vergidis J, et al. Immune modulation by androgen deprivation and radiation therapy: implications for prostate cancer immunotherapy. Cancers (2017) 9:13. doi:10.3390/cancers9020013
262. Burnette B, Weichselbaum RR. Radiation as an immune modulator. Semin Radiat Oncol (2013) 23:273–80. doi:10.1016/j.semradonc.2013.05.009
263. Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med (2012) 366:925–31. doi:10.1056/NEJMoa1112824
264. Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst (2013) 105:256–65. doi:10.1093/jnci/djs629
265. Golden EB, Demaria S, Schiff PB, Chachoua A, Formenti SC. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol Res (2013) 1:365–72. doi:10.1158/2326-6066.CIR-13-0115
266. Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest (2014) 124:687–95. doi:10.1172/JCI67313
267. Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res (2014) 2:616–31. doi:10.1158/2326-6066.CIR-14-0027
268. Azad A, Yin Lim S, D’Costa Z, Jones K, Diana A, Sansom OJ, et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol Med (2017) 9:167–80. doi:10.15252/emmm.201606674
269. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature (2015) 520:373–7. doi:10.1038/nature14292
270. Rech AJ, Dada H, Kotzin JJ, Henao-Mejia J, Minn AJ, Twyman-Saint Victor C, et al. Radiotherapy and CD40 activation separately augment immunity to checkpoint blockade in cancer. Cancer Res (2018) 78:4282–91. doi:10.1158/0008-5472.CAN-17-3821
271. Wang Y, Deng W, Li N, Neri S, Sharma A, Jiang W, et al. Combining immunotherapy and radiotherapy for cancer treatment: current challenges and future directions. Front Pharmacol (2018) 9:185. doi:10.3389/fphar.2018.00185
272. Naik S, Russell SJ. Engineering oncolytic viruses to exploit tumor specific defects in innate immune signaling pathways. Expert Opin Biol Ther (2009) 9:1163–76. doi:10.1517/14712590903170653
273. Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res (2014) 2:295–300. doi:10.1158/2326-6066.CIR-14-0015
274. Wong HH, Lemoine NR, Wang Y. Oncolytic viruses for cancer therapy: overcoming the obstacles. Viruses (2010) 2:78–106. doi:10.3390/v2010078
275. Pan W, Bodempudi V, Esfandyari T, Farassati F. Utilizing Ras signaling pathway to direct selective replication of herpes simplex virus-1. PLoS One (2009) 4:e6514. doi:10.1371/journal.pone.0006514
276. Doloff JC, Waxman DJ, Jounaidi Y. Human telomerase reverse transcriptase promoter-driven oncolytic adenovirus with E1B-19 kDa and E1B-55 kDa gene deletions. Hum Gene Ther (2008) 19:1383–99. doi:10.1089/hum.2008.056
277. Nishimoto T, Yoshida K, Miura Y, Kobayashi A, Hara H, Ohnami S, et al. Oncolytic virus therapy for pancreatic cancer using the adenovirus library displaying random peptides on the fiber knob. Gene Ther (2009) 16:669–80. doi:10.1038/gt.2009.1
278. Conner J, Braidwood L, Brown SM. A strategy for systemic delivery of the oncolytic herpes virus HSV1716: redirected tropism by antibody-binding sites incorporated on the virion surface as a glycoprotein D fusion protein. Gene Ther (2008) 15:1579–92. doi:10.1038/gt.2008.121
279. Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer (2014) 14:559–67. doi:10.1038/nrc3770
280. Sobol PT, Boudreau JE, Stephenson K, Wan Y, Lichty BD, Mossman KL. Adaptive antiviral immunity is a determinant of the therapeutic success of oncolytic virotherapy. Mol Ther (2011) 19:335–44. doi:10.1038/mt.2010.264
281. Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol (2002) 20:197–216. doi:10.1146/annurev.immunol.20.083001.084359
282. Rahal A, Musher B. Oncolytic viral therapy for pancreatic cancer. J Surg Oncol (2017) 116:94–103. doi:10.1002/jso.24626
283. Mahalingam D, Goel S, Aparo S, Patel Arora S, Noronha N, Tran H, et al. A phase II study of pelareorep (REOLYSIN®) in combination with gemcitabine for patients with advanced pancreatic adenocarcinoma. Cancers (2018) 10:160. doi:10.3390/cancers10060160
284. Gujar S, Pol JG, Kim Y, Lee PW, Kroemer G. Antitumor benefits of antiviral immunity: an underappreciated aspect of oncolytic virotherapies. Trends Immunol (2018) 39(3):209–21. doi:10.1016/j.it.2017.11.006
285. Lutz ER, Kinkead H, Jaffee EM, Zheng L. Priming the pancreatic cancer tumor microenvironment for checkpoint-inhibitor immunotherapy. Oncoimmunology (2014) 3:e962401. doi:10.4161/21624011.2014.962401
286. Bourgeois-Daigneault M-C, Roy D, Aitken A, El Sayes N, Martin N, Varrette O, et al. Neo-adjuvant oncolytic virotherapy prior to surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Transl Med (2018) 10:1–12. doi:10.1126/scitranslmed.aao1641
287. Lee P, Gujar S. Potentiating prostate cancer immunotherapy with oncolytic viruses. Nat Rev Urol (2018) 15:235–50. doi:10.1038/nrurol.2018.10
288. Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med (2018) 10. doi:10.1126/scitranslmed.aam7577
289. Blair AB, Zheng L. Rational combinations of immunotherapy for pancreatic ductal adenocarcinoma. Chin Clin Oncol (2017) 6:31–31. doi:10.21037/cco.2017.06.04
290. Skelton RA, Javed A, Zheng L, He J. Overcoming the resistance of pancreatic cancer to immune checkpoint inhibitors. J Surg Oncol (2017) 116:55–62. doi:10.1002/jso.24642
291. Jaffee EM, Hruban RH, Canto M, Kern SE. Focus on pancreas cancer. Cancer Cell (2002) 2:25–8. doi:10.1016/S1535-6108(02)00093-4
292. Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol (2014) 11:509–24. doi:10.1038/nrclinonc.2014.111
293. Le DT, Lutz E, Uram JN, Sugar EA, Onners B, Solt S, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother (2013) 36:382–9. doi:10.1097/CJI.0b013e31829fb7a2
294. Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, Springett G, et al. Safety and survival with GVAX pancreas prime and Listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol (2015) 33:1325–33. doi:10.1200/JCO.2014.57.4244
295. Abate-Daga D, Davila ML. CAR models: next-generation CAR modifications for enhanced T-cell function. Mol Ther Oncolytics (2016) 3:16014. doi:10.1038/mto.2016.14
296. Marin-Acevedo JA, Soyano AE, Dholaria B, Knutson KL, Lou Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J Hematol Oncol (2018) 11:8. doi:10.1186/s13045-017-0552-6
297. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. Biomark Res (2017) 5:22. doi:10.1186/s40364-017-0102-y
298. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev (2014) 257:107–26. doi:10.1111/imr.12131
299. Kenderian SS, Ruella M, Gill S, Kalos M. Chimeric antigen receptor T-cell therapy to target hematologic malignancies. Cancer Res (2014) 74:6383–9. doi:10.1158/0008-5472.CAN-14-1530
300. Brown CE, Adusumilli PS. Next frontiers in CAR T-cell therapy. Mol Ther oncolytics (2016) 3:16028. doi:10.1038/mto.2016.28
301. Yu S, Li A, Liu Q, Li T, Yuan X, Han X, et al. Chimeric antigen receptor T cells: a novel therapy for solid tumors. J Hematol Oncol (2017) 10:78. doi:10.1186/s13045-017-0444-9
302. DeSelm CJ, Tano ZE, Varghese AM, Adusumilli PS. CAR T-cell therapy for pancreatic cancer. J Surg Oncol (2017) 116:63–74. doi:10.1002/jso.24627
303. Bauer C, Kühnemuth B, Duewell P, Ormanns S, Gress T, Schnurr M. Prevailing over T cell exhaustion: new developments in the immunotherapy of pancreatic cancer. Cancer Lett (2016) 381:259–68. doi:10.1016/j.canlet.2016.02.057
304. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest (2016) 126:3130–44. doi:10.1172/JCI83092
305. Moon EK, Wang L-CS, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T cell hypofunction that is reversible can limit the efficacy of chimeric antibody receptor-transduced human T cells in solid tumors. Clin Cancer Res (2014) 20:4262–73. doi:10.1158/1078-0432.CCR-13-2627
306. John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res (2013) 19:5636–46. doi:10.1158/1078-0432.CCR-13-0458
307. Suarez ER, Chang D-K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget (2016) 7:34341–55. doi:10.18632/oncotarget.9114
308. Li S, Siriwon N, Zhang X, Yang S, Jin T, He F, et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin Cancer Res (2017) 23:6982–92. doi:10.1158/1078-0432.CCR-17-0867
309. Hall ML, Liu H, Malafa M, Centeno B, Hodul PJ, Pimiento J, et al. Expansion of tumor-infiltrating lymphocytes (TIL) from human pancreatic tumors. J Immunother Cancer (2016) 4:61. doi:10.1186/s40425-016-0164-7
310. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17:4550–7. doi:10.1158/1078-0432.CCR-11-0116
311. Rusakiewicz S, Semeraro M, Sarabi M, Desbois M, Locher C, Mendez R, et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res (2013) 73:3499–510. doi:10.1158/0008-5472.CAN-13-0371
312. Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y, Oshikiri T, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas (2004) 28:e26–31. doi:10.1097/00006676-200401000-00023
313. Sideras K, Braat H, Kwekkeboom J, van Eijck CH, Peppelenbosch MP, Sleijfer S, et al. Role of the immune system in pancreatic cancer progression and immune modulating treatment strategies. Cancer Treat Rev (2014) 40:513–22. doi:10.1016/j.ctrv.2013.11.005
314. Poschke I, Faryna M, Bergmann F, Flossdorf M, Lauenstein C, Hermes J, et al. Identification of a tumor-reactive T-cell repertoire in the immune infiltrate of patients with resectable pancreatic ductal adenocarcinoma. Oncoimmunology (2016) 5:e1240859. doi:10.1080/2162402X.2016.1240859
Keywords: pancreatic ductal adenocarcinoma, immune checkpoint inhibitors, triple E, antigenicity, immunogenicity, tumor microenvironment
Citation: Kabacaoglu D, Ciecielski KJ, Ruess DA and Algül H (2018) Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Current Limitations and Future Options. Front. Immunol. 9:1878. doi: 10.3389/fimmu.2018.01878
Received: 14 May 2018; Accepted: 30 July 2018;
Published: 15 August 2018
Edited by:
Alexandr Bazhin, Klinikum der Universität München, GermanyReviewed by:
Carmen Guerra, Centro Nacional de Investigaciones Oncológicas CNIO—Fundación Cáncer FUCA, SpainCara Haymaker, University of Texas MD Anderson Cancer Center, United States
Copyright: © 2018 Kabacaoglu, Ciecielski, Ruess and Algül. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hana Algül, aGFuYS5hbGd1ZWxAbXJpLnR1bS5kZQ==
†These authors have contributed equally to this work.