 Chae-Ok Yun
Chae-Ok Yun JinWoo Hong
JinWoo Hong A-Rum Yoon
A-Rum Yoon- 1Department of Bioengineering, College of Engineering, Hanyang University, Seoul, South Korea
- 2Institute of Nano Science and Technology (INST), Hanyang University, Seoul, South Korea
- 3Hanyang Institute of Bioscience and Biotechnology (HY-IBB), Hanyang University, Seoul, South Korea
- 4GeneMedicine CO., Ltd., Seoul, South Korea
Oncolytic viruses (OVs) have been gaining attention in the pharmaceutical industry as a novel immunotherapeutic and therapeutic adjuvant due to their ability to induce and boost antitumor immunity through multiple mechanisms. First, intrinsic mechanisms of OVs that enable exploitation of the host immune system (e.g., evading immune detection) can nullify the immune escape mechanism of tumors. Second, many types of OVs have been shown to cause direct lysis of tumor cells, resulting in an induction of tumor-specific T cell response mediated by release of tumor-associated antigens and danger signal molecules. Third, armed OV-expressing immune stimulatory therapeutic genes could be highly expressed in tumor tissues to further improve antitumor immunity. Last, these OVs can inflame cold tumors and their microenvironment to be more immunologically favorable for other immunotherapeutics. Due to these unique characteristics, OVs have been tested as an adjuvant of choice in a variety of therapeutics. In light of these promising attributes of OVs in the immune-oncology field, the present review will examine OVs in clinical development and discuss various strategies that are being explored in preclinical stages for the next generation of OVs that are optimized for immunotherapy applications.
1 Introduction
The last decade has seen considerable success of immune checkpoint inhibitors (ICI) and chimeric antigen receptor (CAR)-T cells that has highlighted immuno-oncology (IO) (1–4). Although both ICI and CAR-T cell therapy led to complete tumor regression and durable remission in a small subset of cancer patients, a larger fraction of the patients did not respond or showed limited response to these immunotherapeutics (5). Detailed examination of these poor responders to immunotherapy led to characterization of immunologically ‘cold’ tumors that possess low density of tumor-infiltrating lymphocytes and a highly immunosuppressive microenvironment (6, 7). Global pharmaceutical companies have been exploring various strategies to overcome the limited efficacy of immunotherapeutics against such poorly responding tumors.
To this end, oncolytic viruses (OVs) have garnered the attention of biopharmaceutical industries since the US Food and Drug Administration (FDA)- and European Medicines Agency (EMA)-approved the first OVs, Imlygic, in 2015. Both preclinical and clinical data of Imlygic, as well as numerous other OVs, have shown that OVs can warm immunologically cold tumors to improve overall antitumor immune response of various immunotherapeutics drugs (8). OVs possess several unique features that are beneficial for cancer immunotherapy applications, and these attributes cannot be mimicked by other conventional cancer therapeutics. In particular, OVs selectively propagate in and eradicate cancer cells through a domino-like cascading infection and subsequent lysis of tumor cells (9, 10), leading to generation of tumor lysates, pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), and tumor-associated antigens (TAA) as well as increasing production of various cytokines and chemokines, such as type I interferons (IFNs) (11). These byproducts of the oncolytic process can augment various aspects of the antitumor immune (both innate and adaptive) response, such as TAA presentation by antigen-presenting cells (APCs), subsequent induction of tumor-specific T cell response, and immune activation of the tumor microenvironment (12, 13). Other noteworthy attributes of OVs are their strong abscopal effect leading to regression of distant metastatic tumors and establishment of tumor-specific immune memory that can confer protection against tumor recurrence/relapse (14). Furthermore, arming OVs with immune stimulatory genes (e.g., cytokines, chemokines, co-stimulators, and modalities that can nullify negative immune regulators like immune checkpoints) can further improve the induction of tumor-specific immune response and restore immune surveillance function in the tumor microenvironment (15–19).
These immune boosting properties of OVs are being actively explored both alone for therapy and in combination with other immunotherapeutics in clinical landscape. The therapeutic strategies with OV range from monotherapies to combination of other cancer therapies, including traditional cancer treatments and also other immunotherapies. Since 2013 the majority of clinical trials developing an OV were combination trials (183 out of 289), whereby an OV was administered in conjunction with another therapy (Figure 1). Among all the combination trials, the most common modality administered in combination with an OV is ICI, as more than 105 trials have been conducted. The trial start year distribution of these trials and it has grown over the past 8 years, and this growth is expected to continue. Further, much of the preliminary data forecast that OVs are likely to be an integral part of cancer immunotherapy in the near future.
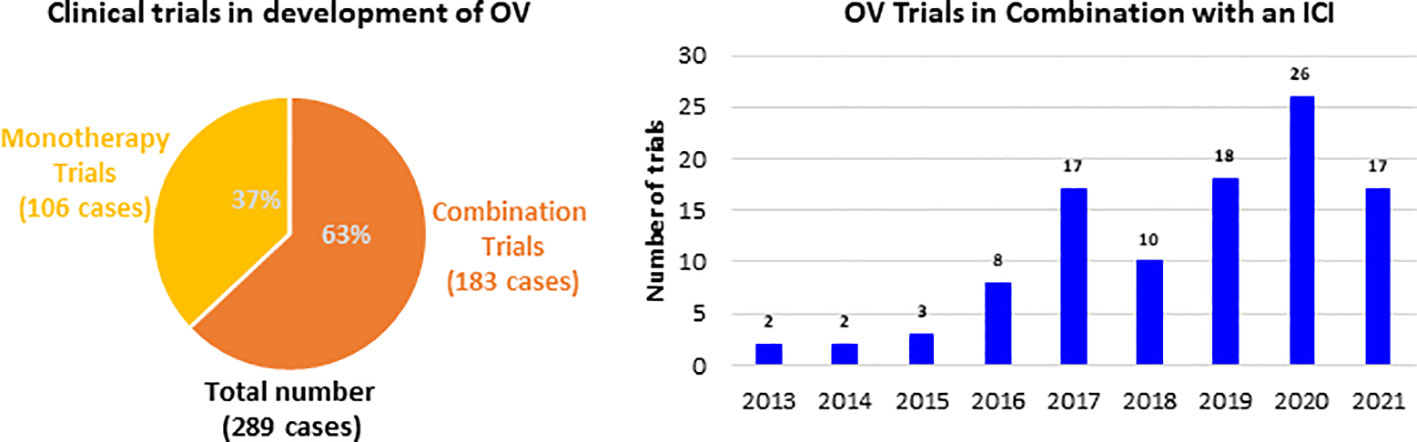
Figure 1 Clinical trials investigating OV and number of clinical trials combining OV with ICI.
There are many different types of OVs that had been investigated in clinical trials, but three viruses, oncolytic adenovirus (oAd), oncolytic herpes simplex virus (oHSV), oncolytic vaccinia virus (oVV), have the longest history and highest number of clinical trials conducted to date. This review will focus on preclinical and clinical development of these three OVs. Further, some of the other types of OVs, like oncolytic reovirus, oncolytic measles virus (oMV) and oncolytic picornaviruses, which are under active clinical development and where detailed and recent clinical studies are available will also be highlighted in the review. We explore the emergence and evolution of these OVs in preclinical and clinical landscapes and their role in advancement and understanding of IO. The review also discusses groundbreaking innovations and breakthroughs in OV application as both stand-alone and combination regimens to improve antitumor immunity, demonstrating that OVs could be widely adopted across different standard care options as promising adjuvant.
2 Current clinical trial landscape
2.1 Oncolytic adenovirus
Adenovirus was one of the earliest gene therapy vectors to be investigated in clinical trial (first human trial dates back to 1993) (20), and its clinical safety has been evaluated and documented extensively. Oncorine (which is similar to ONYX-015) was the first OV to be approved for clinical use in China in 2005, predating the US FDA and EMA approval of oHSV and Imlygic by a decade. Oncorine often is noted as a good testament to the extensive and historical development of oncolytic adenovirus (oAd) in the clinic (21). Major strengths and advantages of oAds rely on their ability to induce strong antitumor immune response (16, 22–24), anti-angiogenic effects (25, 26), high transgene expression, and synergistic anticancer effects in conjunction with conventional cancer therapies (9, 10, 27–29). Additionally, facile viral production at high titer makes the oAd production process economically advantageous (21). In lieu of these attributes, oAds are the most frequently used OV in clinical trials, accounting for ~42% of all trials (40 of 96 clinical trials, as presented in Table 1).
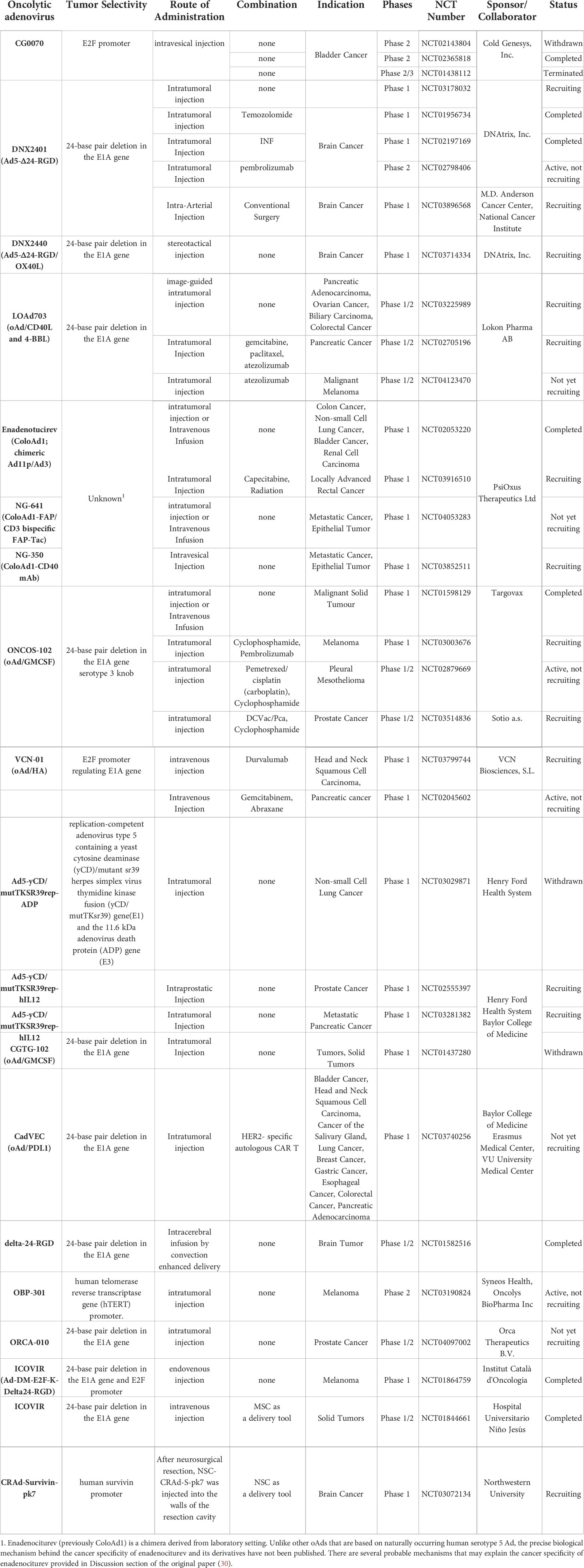
Table 1 Oncolytic adenoviruses tested in current clinical trials.
Despite of commercialization of Oncorine and its yearly growth in total usage in China, it has failed to demonstrate sufficient therapeutic benefit as a single agent for refractory solid tumors (21, 31). One of the likely explanations of the limited efficacy of Oncorine is the deletion of the adenoviral E1B 55 kDa gene, which endows the oAd with cancer specificity but restricts its overall viral replication capacity (32). Since the preliminary clinical trials with ONYX-015 in the 1990s, significant technological advancements have been witnessed in vector design and construction. In detail, majority of the newer human serotype 5 oAd constructs, like ONCOS-102, LoAd703, TILT-123, ORCA-010, CG0070, under active clinical development contain intact E1B 55 kDa gene to circumvent the attenuation in viral replication capacity, and rather employ different genetic engineering strategies like cancer-specific promoter driven Ad E1A expression or deletion of the Rb binding site in Ad E1A gene to achieve preferential replication in cancer cells (33–38). Most of these oAds also harbor a genetically engineered fiber on the viral capsid to enhance their cellular uptake into tumor cells in a coxsackie and adenovirus receptor (CAR)-independent manner: this is important in a clinical environment where heterogeneity of tumor can lead to variable or abrogated CAR expression levels that can lead to suboptimal infection by Ad with wild-type fiber (39, 40).
Indeed, phase I results of ICOVIR-5, an oAd that is being investigated in several ongoing clinical trials (NCT03714334, NCT03178032, NCT02798406), contains a functional copy of the E1B 55 kDa gene, and its cancer specificity is acquired through deletion in the Rb binding domain of the Ad E1A region and insertion of a tumor-targeting RGD motif at the fiber region of the viral capsid to improve its cancer specificity. Despite significant advancements made to the viral constructs, poor systemic administrability of the virus remain a major challenge within the field as an ideal cancer therapeutic should be systemically administrable to effectively treat noninjectable or metastatic lesions in advanced stages of cancer. Patients treated with ICOVIR-5 (single intravenous (IV) infusion of 1 × 1011 to 1 × 1013 viral particles (VP)) demonstrated that only a small portion of systemically administered virus could accumulate in melanoma metastases but ultimately failed to induce objective response (41). A phase I trial results of IV infused enadenotucirev also failed to induce clinically beneficial response in patients with epithelial solid tumors or those that underwent tumor resection (29). Progressive disease was observed in ~56% of patients treated with systemically administered enadenotucirev. Similarly, another phase I trial results of IV administered enadenotucirev in combination with paclitaxel in platinum-resistant ovarian cancer patients only led to overall response rate of 10% at the highest dose of the virus of 1 × 1012 VP, which was lower than those achieved by paclitaxel monotherapy in similar patient demographic (42). Collectively, these clinical findings demonstrate that the intravenous administration of oAd remain suboptimal in current state and majority of the ongoing clinical trials evaluating locoregional administration of oAds.
Although systemic delivery of oAds remain a major challenge, strategies to maximize the induction of systemic antitumor immune response mediated by oAds could be a more practical approach to maximize the antitumor effect of oAd in the noninjected lesions. Indeed, many of the ongoing or recruiting phase I trials (NCT01437280, NCT02143804, and NCT02365818) are evaluating oAds expressing pro-inflammatory cytokines (granulocyte-macrophage colony stimulating factor (GM-CSF), interleukin (IL)-2, IL-12) or co-stimulators (4-1BB or CD40 ligand (4-1BBL and CD40L) to enhance the induction of antitumor immune response mediated by these viruses (43, 44). Although clinical trial results for many of these oAd-expressing antitumor immune transgenes are not available, their increasing prevalence in the current clinical landscape strongly indicates that identifying the correct combination of therapeutic transgenes will be integral for maximizing oAd application in immmuno-oncology.
The therapeutic transgenes, which was initially and mainly applied to clinical studies, are IL-12 and GM-CSF. For example, Ad5-yCD/mutTKSR39rep-hIL12, an oAd expressing the human IL-12 gene and two suicide genes (yeast cytosine deaminase (yCD) and HSV thymidine kinase (TK), yielded promising antitumor immune response and tumor growth inhibition in pre-clinical and clinical studies (45–48). Both suicide genes, yCD (cytosine deaminase) and mutTKSR39 (HSV thymidine kinase), expressed by Ad5-yCD/mutTKSR39rep-hIL12 successfully converted respective prodrugs 5-fluorocytosine and ganciclovir to induce irreversible inhibition of DNA synthesis and yielded potent anti-tumor effects (49, 50). Further, Ad5-yCD/mutTKSR39rep-hIL12 treatment improved induction of antitumor immune response through expression of hIL12, as evidenced by activation of NKs and secretion of IFN-γ by cytotoxic T lymphocytes (CTL) against tumor cells (51). Based on these findings from the preclinical study, two phase-1 clinical trials (NCT02555397 and NCT03281382) have been initiated to evaluate Ad5-yCD/mutTKSR39rep-hIL12 for the treatment of patients with either prostate or metastatic pancreatic cancer, respectively (47).
There are two clinical trials ongoing with oAds-expressing GM-CSF (ONCOS-102 and CG0070). ONCOS-102, developed by Targovax, possesses a 24 bp deletion in the Rb binding site of E1A to improve its cancer specificity. ONCOS-102 has shown encouraging phase I results for patients heavily pretreated for solid tumors (38, 52). The patients treated with ONCOS-102 intratumoral injection of a dose range at 3 × 1010 VP, 1 × 1011 VP, or 3 × 1011 VP/injection on days 1, 4, 8, 15, 29, 57, 85, 113 and 141 showed stable disease in 40% of evaluable cases (of the 12 patients in this study, two passed away before the first clinical assessment). ONCOS-102 treatment elevated number of tumor-infiltrating cytotoxic CD8+ T cells and cancer-specific CD8+ T cells in blood, indicating systemic activation of the immune system. Importantly, activation of antitumor immune system correlated with overall survival. Furthermore, upregulation of programmed death ligand 1 (PD-L1) after treatment with ONCOS-102 suggested that the combination of ONCOS-102 with immune checkpoint inhibitors (ICI), including PD-1/PD-L1 axis inhibitors, offers a promising strategy to treat refractory tumors. In support, a clinical trial (NCT03003676) combining ONCOS-102 and Keytruda (pembrolizumab; an anti-PD-1 antibody) is under investigation.
Another GM-CSF-expressing oAd, CG0070, was assigned cancer specificity by transcribing Ad E1A through E2F-1 promoter. In a phase I trial, 1012 VP of CG0070 induced complete response (CR) in bladder cancer patients who did not respond to standard care (bacillus calmette-guerin (BCG) treatment). Recently, CG0070 showed success in a phase II study against BCG-unresponsive high-grade non-muscle invasive bladder cancer (NMIBC; NCT02365818). Recently, CG0070 completed phase II study in a successful manner against BCG-unresponsive high-grade non-muscle invasive bladder cancer (NMIBC; NCT02365818). In specific, it was reported in American Society of Clinical Oncology (ASCO) meeting that CR rate for CG0070-phase II trials in the single dose cohort was 23% (3/13) (53). Their findings showed that CR response rate was greatly improved in patients who received multiple injections of CG0070, reaching CR rate of 64% (14/22). Six patients from multiple dose cohorts remained in remission (duration ranging from 3.3 to 38.2 months) as of the last follow-up. Currently, phase III study of CG0700 monotherapy is ongoing (NCT04452591), while the combination of CG0070 with Keytruda is in phase II clinical trials for treatment of BCG-unresponsive NMIBC patients (NCT04387461). The clinical results of the combination therapy trial reported in April of 2022 reported that 89% of patients evaluable for efficacy (16/18) had CR at 3-month time point and 75% (8/18) maintained CR at the 12-month assessment (https://www.cgoncology.com/news/press-releases/041322/). Together, these reports demonstrate that arming oAds with immune stimulatory cytokines could improve overall patient response rate and clinical benefit.
LOAd703, which is under phase 1/2a clinical trial (NCT02705196 and NCT03225989), expresses trimerized CD40L and 4-1BBL as immune activators to stimulate the CD40 and 4-1BB pathways, respectively (34). Many cells in the tumor microenvironment, including stromal cells and the infiltrating immune cells, express CD40 and 4-1BB; thus, expression of complementary activating ligands via LOAd703 could activate many types of cells in the tumor milieu to induce antitumor immune response. For example, dendritic cells (DC) were stimulated by LOAd703 to upregulate co-stimulators, cytokines, and chemokines, ultimately leading to increased antigen-specific T cells and NK cell population to mount potent antitumor immune response. Interim phase I/II trial results reported in 2020 revealed that intratumoral administration of LOAd703 with nab-paclitaxel/gemcitabine chemotherapy was well-tolerated in pancreatic ductal adenocarcinoma patients (13 patients were evaluable); most adverse events were transient grade 1-2 with only a single patient at the highest dose (5 × 1011 VP) exhibiting dose-limiting grade 3 transaminase elevation. The decreased number of immunosuppressive myeloid-derived suppressor cells in circulation was reported (8/13), suggesting alleviation of immunosuppression. Further, elevated effector memory T cell (10/13) and tumor antigen-specific T cell counts were reported, ultimately leading to 6/10 patients at higher virus doses exhibiting partial response.
Collectively, there has been significant advancements in the genetic constructs of oAds that are currently being evaluated in clinical environment to enhance their safety profile and efficacy. Notably, there are increasing number of trials evaluating the oAds armed with pro-inflammatory immune transgenes to maximize the viruses’ potential to induce robust systemic antitumor immune response, which is an essential parameter to control the growth of noninjectable and metastatic lesions in patients with advanced stages of cancer.
2.2 Oncolytic herpes simplex virus
oHSV, like several other types of OVs, can directly kill tumor cells and promote antitumor immune response. The pathogenicity and function of viral proteins of HSV have been well-characterized, and most oHSVs in development have deletion of several viral genes to prevent potential neurotoxicity and confer cancer specificity (54). Furthermore, clinicians have proper knowledge, training, and means to treat HSV infection in an efficient manner, as HSV is one of the few viruses with well-established antiviral drugs. These attributes endow oHSV another extra layer of safety in the clinic since uncontrolled viremia and other virus-related adverse events can be managed by clinicians in an efficient manner. Additionally, nearly all types of cancer can be infected with oHSV, which is beneficial in clinical scenarios where heterogeneity of tumors and resulting phenotypic variations necessitate flexibility and wide target coverage to induce optimal therapeutic effect. oHSV also possesses a large genome size (55) and a relatively large transgene insertion capacity.
Talimogene laherparepvec, an oHSV expressing GM-CSF also known as Imlygic, was the first oncolytic virus to be approved by FDA and EMA. It was shown to possess promising antitumor activity in melanoma patients. This landmark approval led to significant improvement in understanding of OV mechanisms in patients, such as shedding, biodistribution, induction of antitumor immune response, and transmissibility. Imlygic usage and number of clinical reports have been on an upward trajectory, and its prevalence has improved clinicians’ and government regulators’ understanding and handling of oncolytic virotherapy in medical settings. The approval of Imlygic has accelerated the development of other OVs, and the promising clinical outcomes achieved via its combination with other clinically approved immunotherapeutics forecast their critical role in advancing cancer immunotherapy paradigm.
While Imlygic remains the only oncolytic HSV to be approved by US FDA and EMA to date, several other oHSVs are under clinical investigation (Table 2). Some of the earliest oHSV clinical trial results were published as early as 2002 (58), using HSV1716 that lacks γ134.5. These findings showed that 1 × 105 plaque-forming unit (PFU) of HSV1716 can be administered intratumorally in a safe manner without dose-limiting toxicity in both HSV-seropositive and -negative patients. HSV1716 replicated actively in brain tumors (in two of 12 patients, HSV genome copies were detected at a higher level than the input dose at 9 days after inoculation), and infectious particles were recovered from the tumor biopsies of these two patients. In another phase I trial, HSV1716 was injected into the normal brain tissues surrounding the resection cavity following surgical resection of glioma (59). Injection of HSV1716 into normal brain tissues did not induce any observable HSV1716-related toxicity. Three of the 12 enrolled patients remained alive and clinically stable at 15-22 months post-surgical resection and HSV1716 injection into normal brain tissues surrounding the resection cavity. Remarkably, one of the surviving patients who had extensive recurrent disease at the time of trial enrollment demonstrated reduction in residual tumor volume over the 22-month period after HSV1716 administration, despite not receiving any adjuvant treatment. The patient at this period remained in complete clinical and radiological remission. In 2017, a phase I trial of intratumorally-administered HSV1716 (a single dose of 105 to 107 PFU) in young cancer patients revealed that HSV1716 is safe and well-tolerated in young patients (60). However, there was no tumor shrinkage (either in injected or uninjected lesions) in any of these patients, suggesting further optimization of the clinical protocol will be necessary for future HSV1716 trials.
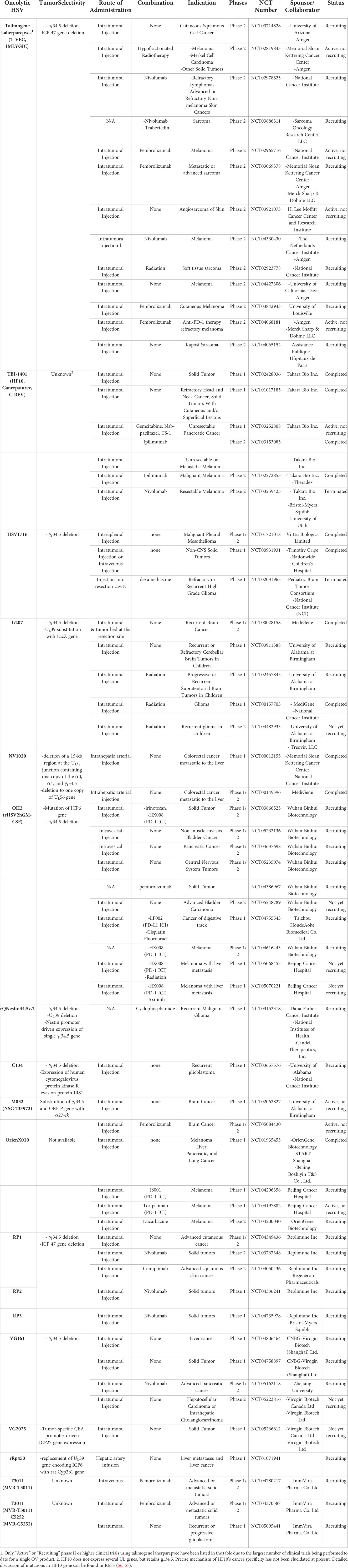
Table 2 Oncolytic HSVs tested in current clinical trials.
In a phase I glioma clinical trial using G207 (with deletion of γ134.5 and inactivated UL39), the virus was intratumorally administered pre-resection than subsequently into the normal brain tissues surrounding the resection cavity post-resection of recurrent glioblastoma multiforme (GBM) (61). In detail, six patients were treated initially with 1.5 × 108 PFU via stereotactic injection into the GBM tumor (pre-resection), followed by tumor resection at 2 to 5 days after the final virus administration. Immediately after surgical resection, second dose of G207 was administered into resected tumor bed using multiple injections. The viral replication was noted in resected tumor tissues in 50% of the patients. Although no determination regarding efficacy could be made due to the small cohort, the injected tumor lesions showed elevated T cell, monocyte, and macrophage infiltration in comparison to those observed prior to G207 administration, which would be integral to induction of OV-mediated antitumor immune response. G47Δ, a third-generation oHSV based on G207, harbors addition deletion of the α47 gene and has been under extensive clinical investigation in Japan (62). A phase I-IIa clinical trial of G47Δ in patients with recurrent glioblastoma was completed in 2014 (UMIN000002661), and a subsequent phase II trial examining (UMIN000015995) multiple stereotactic administration at 1 × 109 PFU (a maximum of six times) revealed that locally administered G47Δ was well tolerated. In February 2016, G47Δ was designated as a breakthrough therapy drug by the Ministry of Health, Labor, and Welfare of Japan (63) and it has been given conditional approval for the treatment of patients with malignant glioma or any primary brain cancer in 2021 (64).
Imlygic was the only antitumor cytokine-expressing oncolytic HSV being evaluated in phase II/III clinical trials as of 2019, with other oHSVs in phase II/III trials not expressing any therapeutic transgenes, thus failing to fully exploit the large transgene capacity of oHSV. With growing number of preclinical data demonstrating that oHSVs expressing any antitumor immune transgene exert more potent tumor growth inhibition than do cognate controls lacking any transgenes (65–68), majority of the recruiting or ongoing clinical trials (14 out of 17) listed on http://clinicaltrials.gov as of July 2022 utilizes an oHSV expressing at least one antitumor immune transgene. Currently, combinations of various immune stimulatory transgenes are being actively explored in either preclinical stage or early phase of clinical trial to improve antitumor immunogenicity of oHSVs. Notably, in view of the reports suggesting that the effect of potent antitumor cytokines like IL-12 supersedes the tumor growth inhibiting effect of GM-CSF, the transgene payload of oHSV is diverging away from GM-CSF of Imlygic (69). In lieu of these trends, there are increasing number of oHSV pipelines harboring IL-12 as a transgene in clinical trials: two phase I clinical trial utilizing an oHSV expressing IL-12 (M032) against recurrent malignant glioma (NCT02062827 and NCT02062827) (70), three phase I or phase I/II trials examining either intratumorally or IV administered C5252 or MVR-T3011 (both are oHSV co-expressing IL-12 and PD-1 antibody in NCT04370587, NCT04780217, and NCT05095441), three phase I or phase I/II trials evaluating VG161 (oHSV co-expressing IL-12, IL-15 with its receptor α unit, and Fc-fused PD-L1 blocking peptide in NCT04806464, NCT05162118, and NCT04758897), and a single phase I study of ONCR-117 (an oHSV expressing IL-12, extracellular domain of FLT3LG, CCL4, anti-CTLA-4 ICI ipilimumab, and anti-PD-1 single variable heavy chain domain fused with Fc region NCT04348916) is ongoing.
2.3 Oncolytic vaccinia virus
Vaccinia virus (VV) is a membrane-coated virus with a linear double-strand DNA virus and was shown to efficiently infect, replicate, and kill a wide-range of cancer cells (71). Further, large viral genome size of VV allows insertion of large transgenes (~40 kb) with minimal change in viral production (72). VV also possesses attributes that ensure good safety profile: (1) VV replication cycle occurs in cytoplasm (73), thus there is no risk of genome integration (74) and (2) there is no associated-human disease (72) reported so far. However, one major shortcoming is that 50% of VV genes have unknown functions, which can lead to unforeseeable side-effects when interacting with other cancer therapeutics (72). Despite incomplete understanding of VV viral proteins, development of various oncolytic VV (oVV)s has been pursued since the 1990s (75–77).
The most extensively tested oVV in clinical trials is Pexa-Vec (pexastimogene devacirepvec, also known as JX-594) that expresses human GM-CSF (78). Pexa-Vec was well-tolerated in patients with refractory solid tumors, showing a good safety profile (NCT01169584) (79). Importantly, a phase II clinical trial of Pexa-Vec in combination with sorafenib was shown to improve long-term survival rate (~35% and 11% at 18 months for respectively high-dose and low-dose groups of virus injection) of liver cancer patients (NCT00554372) (80). Other promising clinical results demonstrated that Pexa-Vec treatment elevated IFN-γ and tumor necrosis factor (TNF)-α in tumor sites to lead to activation and/or recruitment of neutrophils, eosinophils, and lymphocytes to the tumor tissues, ultimately suggesting immune activation at injected tissues (79–82). Furthermore, tumor vascular disruption was also observed (80), suggesting an anti-vascular effect of Pexa-Vec. Despite these results from phase I/II clinical trials, Independent Data Monitoring Committee recently concluded that phase III trial results of Pexa-Vec in combination with sorafenib for liver cancer (NCT02562755) failed to improve the clinical outcome of patients in respect to the standard care.
Although phase III trial results of Pexa-Vec have been disappointing, there are several other oVVs in clinical trials targeting wide range of tumor types that might yield promising results (Table 3). For example, phase I trial results of oVV (vvDD) derived from Western Reserve strain, which is the most virulent strain of VV, has shown some promising outcomes (83). Two viral genes (TK and vaccinia growth factor (VGF) genes) have been deleted in vvDD to endow tumor specificity and decrease viral replication in resting cells (84). Subsequently, vvDD was engineered to co-express somatostatin receptor (SR) to track the virus easily in an in vivo setting (85) and cytosine deaminase (CD) as a suicide gene (86), generating a vvDD-CDSR (also known as JX-929) that entered phase I clinical trial. In 2015, phase I trial results of intratumorally administered JX-929 demonstrated good safety profile and tumor specificity (83). In specific, infectious JX-929 particle was detected in the injected lesions in 4 of 5 biopsied patients in a high-dose cohort (1 x 108 to 3 x 109 PFU), whereas 3/3 biopsy samples from injected lesions in a low-dose cohort (3 x 107 PFU) were negative for infectious particles at 8-day post administration. Notably, 4 patients exhibited infectious viral persistence in the injected lesion, and 50% of these patients tested positive for infectious JX-929 in the non-injected lesions, suggesting distal viral spread from the injected site. Antitumor activity and tumor regression were observed in injected lesions for 2 of 3 patients with active viral replication in higher dose cohorts, but numerous other non-injected nodules failed to show any sign of infection or respond to treatment. CD4+ and CD8+ T cell populations among peripheral blood mononuclear cells (PBMC)s of 1 ×109 and 3 × 109 PFU dose cohorts showed dose-dependent increase in the levels of pERK, pS6, and Ki67, suggesting T cell proliferation. JX-929 did not induce any significant elevation in serum chemokine or cytokine levels. Interestingly, one patient with a large tumor burden who had received two injections of JX-929 under a compassionate use protocol showed complete resolution of both injected tumors, thus demonstrating a potentially promising antitumor effect of JX-929.
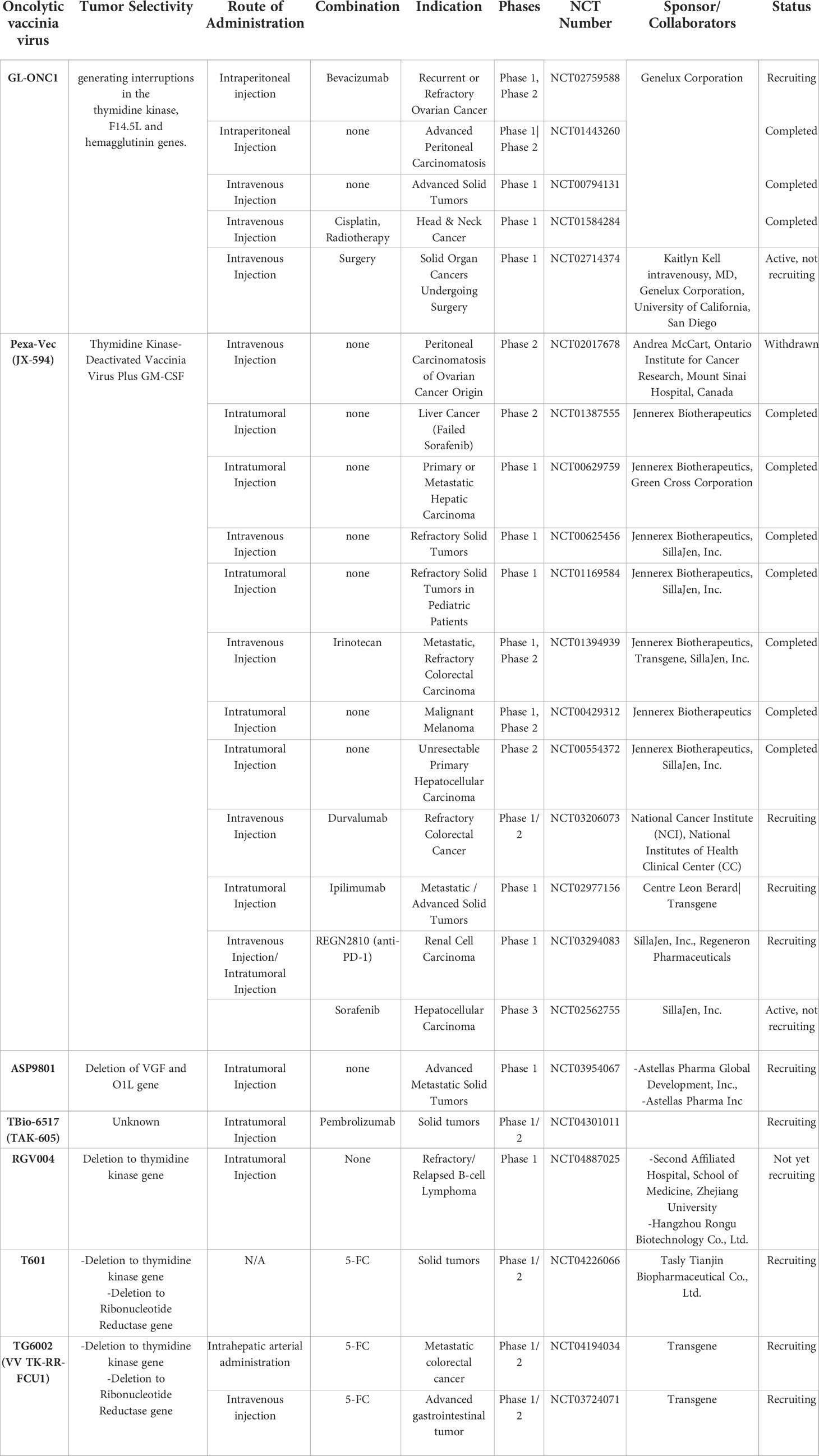
Table 3 Oncolytic vaccinia viruses tested in current clinical trials.
Another phase I trial of JX-929 where IV administration was employed against solid tumors (NCT00574977) demonstrated a good safety profile with no dose-limiting toxicity or serious adverse events in cohorts ranging from 3 × 108 to 3 × 109 PFU (87). At 4 h post systemic administration, significant elevation of Th1 or Th1-related cytokines (IL-2, IFN-γ, IL-7, and GM-CSF) was observed, while the expression level of Th2 cytokines remained unaffected after the treatment, suggesting acute Th1 immune activation likely due to antiviral immune response. Most patients were cleared of the virus quickly after IV infusion, and the viral genome was only detected in tumor biopsies of 2 patients on days 8 and 22 after treatment, demonstrating that systemic administration of JX-929 leads to insufficient viral accumulation in tumor tissues. Poor viral accumulation in tumor tissues likely resulted in the poor antitumor activity of IV-administered JX-929, as evidenced by failure to show any sign of necrosis or change in PET signal intensity on PET-CT scan results at 3 weeks post-administration.
There are other clinical cases using different oVVs clearly illustrating that systemic administration of oVV should be avoided in future clinical trials. Two phase I trials evaluating two systemic administration routes (IV and intraperitoneal injection - NCT01584284 and NCT01443260)) for Lister strain oVV (also known as GL-ONC1 or GLV-1h68) failed to elicit an antitumor effect (88, 89). In detail, a phase I trial evaluating IV-administered GL-ONC1 in combination with chemoradiotherapy to treat head and neck cancer stage IV patients showed that IV infusion was safe with no grade 4 toxicity observed and 18 of 19 patients completed the injection course. Of the 14 patients bearing p16-negative tumors, 7 deaths and 7 treatments failures were observed by 30 months. Despite the lack of virus accumulation in tumor tissues and the absence of significant improvement, the combination therapy (NCT01584284) is safe and that the viral MTD was not reached (88). Similarly, intraperitoneal infusion of GL-ONC1 in late stage carcinomatosis patients failed to exert a meaningful antitumor effect. Despite the increase in lymphocyte count in the peritoneal cavity and even though 8 of 9 patients showed efficient infection in ascitic fluids, only 4 of them had virus-infected cells in peritoneal fluids. As a consequence, of the 4 patients who completed the 4-cycle-treatment, only 2 had stable disease. Furthermore, grade 4 adverse events were not observed at any dosage. However, no correlation could be made with the virus dosage levels.
Collectively, these clinical trial results of oVV therapy clearly demonstrated that intratumoral injection of oVV should be the preferred route of administration in future clinical trials as systemic administration cannot sufficiently deliver oVV to tumor tissues to induce notable therapeutic effect. Furthermore, the limitation of the oVV therapy might be due to the highly advanced stage of patients used in clinical trials, but absence of critical side-effects is a big advantage of the vaccinia virus. The clinical benefits of oVV may be enhanced further with combination therapy such as radio-, chemo- or immunotherapy, as discussed in Section 3 of the review.
2.4 Other OVs
Although oAd, oHSV, and oVV have been most extensively evaluated in clinical environment (90) and primary scope of the review, there are other types of viruses, like reovirus, measles virus (MV), and picornaviruses, that are currently being evaluated in the clinical environment. Among these OVs, reovirus has been most extensively evaluated in clinical environment across multiple phase I and II clinical trials across multiple types of tumors, and thus will be discussed in-depth. Some of the other OVs with recently completed clinical studies and ongoing clinical trials will also be highlighted in this section of the review (Table 4).

Table 4 Characteristics of oncolytic viruses.
2.4.1 Oncolytic reovirus
A mammalian orthoreovirus type three Dearing strain, previously known as Reolysin and now manufactured as pelareorep, is one of the most extensively evaluated OV in clinical trials. Pelareorep is a non-enveloped and double-stranded RNA virus that is known to be relatively nonpathogenic in adults. The first-in-man phase I study of pelareorep, REO-001, enrolled 19 patients with accessible and advanced solid tumors that were intratumorally injected with the virus (94). No dose limiting toxicities were observed and majority of the treatment-related adverse effect being grade two or below, and tumor responses were observed in 37% of the patients. Subsequent phase I clinical trials investigating systemically administered pelareorep demonstrated that IV administered virus was well-tolerated in patients (36, 95, 96). Despite its safety, IV administered pelareorep as monotherapy only elicited modest therapeutic benefit across multiple trials (97).
Due to inconsistent and insufficient therapeutic benefit of pelareorep in multiple clinical trials as monotherapy, a series of phase II trials were launched to evaluate IV administered pelareorep in combination with standard of care chemotherapy across different types of cancer (1: pancreatic adenocarcinoma, 2: recurrent ovarian, tubal, or peritoneal cancer, 3: metastatic non-small cell lung cancer, 4: metastatic colon cancer, 5: advanced melanoma, and 6: metastatic breast cancer) and the results from these trials were published during 2016 to 2018 (98–102). Unfortunately, majority of these trials (4 out of 6) demonstrated that pelareorep in combination with standard of care chemotherapy failed to improve progression-free survival period compared with chemotherapy alone (98–101); 3 out of 4 trials also reported increased risk of severe adverse events (grade 3 or 4) in the pelareorep combination arm versus chemotherapy arm. Still, two of these phase II trials yielded potentially promising results when pelareorep was used in combination with standard of care chemotherapy for the treatment of patients with advanced melanoma or metastatic breast cancer (98, 103). In detail, pelareorep in combination with carboplatin and paclitaxel in patients with advanced melanoma met the efficacy goal for the first stage of the trial design with partial responses being observed in 3 out of 14 patients (ORR of 21%), stable disease in 9 out of 14 patients, median PFS of 5.2 months, and OS of 10.9 months (98); median PFS and OS showed minor improvement compared with historical controls (5.2 vs. 3 months & 10.9 vs. 9 months, respectively). Despite meeting the efficacy goal in the first stage of the trial, the second stage was terminated due to success of novel targeted therapies and immunotherapy for the treatment of melanoma during the course of first stage of this trial. The multicenter and randomized phase II trial that enrolled 74 patients with previously treated metastatic breast cancer demonstrated that combination of pelareorep with paclitaxel significantly improved the median OS versus paclitaxel alone (17.4 vs. 10 months, respectively), despite no differences being observed in median PFS and disease response rate between the two arms. Despite this substantial difference in OS, the result should be interpreted with caution as the study was not powered to detect a difference in OS and the study cohort favored pelareorep combination arm as the OS for the control chemotherapy arm was lower than expected.
As the combination of pelareorep with standard of care chemotherapy was largely unsuccessful across multiple types of cancer in several clinical trials, more recent clinical development using pelareorep has focused on the immune stimulatory aspect of pelareorep and are being conducted in combination with ICIs (NCT04102618, NCT04215146, NCT04445844, NCT03723915, NCT03605719, and Eudra-CT Number: 2020-003996-16) or GM-CSF (NCT02444546) with only two of the ongoing trials being evaluated in absence of other cancer immunotherapeutics for the treatment of patients with relapsed or refractory myeloma (NCT02101944 and NCT02514382). Two recent clinical studies demonstrated that this strategy of combining pelareorep with other cancer immunotherapy may yield promising results (104). In detail, PBMC isolated from metastatic colorectal cancer patients treated with pelareorep and chemotherapy in a phase I trial revealed that several pro-inflammatory cytokines IL-12p40, IL-12p70, GM-CSF, and IFN-γ were upregulated at day 8 or 15 after IV administration of pelareorep compared to the baseline, and reduction of pro-tumoral chemokines associated with angiogenesis or immunosuppression, like IL-8, VEGF, and RANTES/CCL5, was observed. Ultimately, pelareorep infusion induced APC stimulation and activation of T cells, suggesting that pelareorep could initiate antitumor and pro-inflammatory immune response (105).
Similarly, a phase Ib study evaluating the combination of pelareorep, PD-1-targeted ICI pembrolizumab, and standard chemotherapy for the treatment of patients with advanced pancreatic adenocarcinoma revealed that the combination therapy increased number of CD8+ T cells in tumor tissues in 2 out of 7 evaluable patients. Pelareorep infusion prior to pembrolizumab administration was shown to elevate the expression level of CTL attracting cytokines CXCL10 and CXCL11 in the peripheral blood of patients as well as promoting clonal expansion of T cells; the effects were further augmented upon additional treatment with pembrolizumab in patients. The study demonstrated that increased clonal expansion of T cells in patients positively correlated with higher OS, as patient who achieved partial response for 17.4 months and two patients who achieved stable disease for 4 and 9 months all exhibited higher peripheral T cell clonality, which is indicative of increased generation of tumor-associated neoantigens (106), as well as elevated expression level of antitumor cytokines, suggesting that pelareorep may inflame the tumor microenvironment and improve the efficacy of concomitantly administered ICI treatment. Based on these preliminary findings, a phase II trial evaluating the combination of pelareorep with pembrolizumab for the treatment of pancreatic cancer patients was initiated in 2018 (NCT03723915). Unfortunately, the combination therapy failed to meet the stage 1 evaluation criteria, which was to reach two or more PR or CR in patients from stage 1, thus ultimately leading to termination of the trial.
Despite the early termination of phase II trial exploring pelareorep and pembrolizumab, the interim results from phase I/II trial evaluating the combination of pelareorep and anti-PD-L1 ICI atezolizumab (Eudra-CT Number: 2020-003996-16) for the treatment of advanced gastrointestinal cancers revealed that the combination therapy administered to 3 out of 3 patients with locally advanced/metastatic unresectable pancreatic ductal adenocarcinoma led to partial response at week 16 after the treatment with no safety signals. Similarly, interim results from the phase I trial evaluating the combination of pelareorep and atezolizumab in patients with early breast cancer (NCT04102618) that are hormone receptor-positive and HER2-negative were promising (107). The study evaluated CelTIL score, a metric that quantitates changes in tumor cellularity and TIL with higher score correlating to favorable therapeutic responses, and met the primary endpoint of the trial when greater than 30% increase in CelTIL score was achieved in 40% of the patients receiving pelareorep in absence of atezolizumab and 60% in the combination therapy arm. Their findings demonstrated that increased CelTIL score was associated with (1) upregulation of PD-L1 expression level and (2) higher infiltration of CD8+ or memory T cells in the tumor tissues, as well as higher CD8+ T cell to Treg ratio, which are all indicative of polarization toward pro-inflammatory response and amelioration of tumor-induced immunosuppression. A gene panel analysis comparing the biopsy samples from pre-treatment and day 21 after the treatment revealed that aggressive luminal B breast cancer subtype was converted to luminal A subtype (with 100% conversion being achieved in the combination therapy arm) and panel of risk factors associated with tumor recurrence was markedly decreased in both pelareorep alone and pelareorep plus atezolizumab arms.
Collectively, IV infusion of pelareorep has been shown to be well-tolerated and induce pro-inflammatory changes to the tumor microenvironment across multiple types of cancers in different trials, but the therapeutic efficacy of the agent as monotherapy or in combination with standard of care chemotherapy were largely underwhelming. Although more recent clinical development strategy centered on immune stimulatory property of pelareorep seems to be yielding promising results as demonstrated by the interim results of two trials examining pelareorep in combination with anti-PD-L1 ICI atezolizumab, these initial findings should be taken with caution as the promising results of phase I trial exploring the combination of pelareorep with pembrolizumab in pancreatic cancer patients did not translate to successful phase II trial.
2.4.2 Oncolytic measles virus
Measles virus (MV) is an enveloped RNA virus with a long history of antitumor activity in lymphoma patients, as there were many case studies from the 1970~80s reporting tumor regression or “spontaneous” remission following infection with MV (108). Due to this historical background, first in-human clinical trial of Edmonston vaccine strain of MV was conducted in patients with cutaneous T-cell lymphomas (109). The live-attenuated Edmonston vaccine strain of MV has a long history of excellent safety record, as it has been administered to vaccinate countless children, and it predominantly internalizes into the cells via CD46, which is known to be overexpressed in many cases of human tumors (109–112). In support, the CD46 was either shown to be expressed in tumor tissues or at a higher level in malignant tissues than the normal counterpart across multiple clinical studies evaluating oncolytic MV: the tumor biopsies from 5 out of 5 patients with cutaneous T-cell lymphomas tested positive for CD46 (109), 13 out of 15 patients with ovarian cancer showed high expression level of CD46 (110), and CD138+ myeloma cells from patients were shown to express higher level of CD46 than CD138- normal counterpart (111).
In terms of safety and efficacy, first phase I study reported for an oncolytic MV (oMV) in 5 patients with cutaneous T-cell lymphomas demonstrated that five of the six injected lesions exhibited tumor regression and partial regression of the distant noninjected lesions in 2 patients with no adverse events higher than grade 1 being observed even with the highest dose of 1,000 TCID50 (109). The regression of both injected and noninjected lesions suggest that oncolytic effect by the virus and potential induction of systemic antitumor immunity. Other evidence like elevated serum IL-2, IL-12, and IFN-γ expression level and elevated intratumoral infiltration of CD8+ T cells in the injected lesions also suggest induction of pro-inflammatory changes in the patients following oMV administration. Still, these findings should be interpreted with caution as patients were treated with systemic INF-α therapy (113), which could also induce pro-inflammatory changes, to minimize oMV activity in normal tissues of immune-compromised lymphoma patients prior to oMV administration.
More recently published clinical studies utilized oMV expressing either soluble extracellular domain of human carcinoembryonic antigen (CEA; MV-CEA) or human thyroidal sodium-iodide symporter (NIS; MV-NIS) to monitor real-time viral gene expression in vivo (110–112). In phase I trial evaluating intraperitoneally administered MV-CEA (103 to 109 TCID50) was shown to be well-tolerated in platinum- and paclitaxel-resistant ovarian cancer patients with only one grade 3 arthralgia being observed in one patient (NCT00408590). Viral kinetics could be monitored by increased CEA level in peritoneal fluid in high dose cohort (one patient from 108 and two patients at 109 TCID50) and dose-dependent objective response was observed with best objective response of stable disease being observed in 9 out of 9 patients at the dose level of 107 – 109 TCID50 while only 5 out of 12 patients achieved stable disease at dose level of 103 – 106 TCID50. The median overall survival of the patients receiving MV-CEA was 12.15 months, which is greater than expected median survival of 6 months in similar historical patient cohort. Although immune stimulatory aspect of MV-CEA was not examined in detail in this study, there was no changes in CD4 and CD8 levels following MV-CEA administration and further evaluation of the product in the scope of IO will be needed. Unfortunately, only one other phase I clinical trial utilizing MV-CEA has been completed in patients with recurrent glioblastoma (NCT00390299) and there is no ongoing studies utilizing MV-CEA, thus its immune regulatory properties will likely remain unknown.
Currently, majority of the ongoing clinical trials are utilizing MV-NIS construct rather than MV-CEA, possibly due to NIS having greater clinical applicability as MV-NIS could enhance the accumulation of therapeutic radioisotopes at the tumor lesions and induce additional antitumor effect in preclinical models (114). A phase I/II trial evaluating IV administered MV-NIS either in combination with or without cyclophosphamide (the drug was included to attenuate antiviral immune response) in patients with advanced multiple myeloma (NCT00450814) demonstrated that MV-NIS monotherapy was well-tolerated up to the dose of 1011 TCID50 with no dose limiting toxicities being observed (111, 112). In terms of efficacy, one patient who received 1011 TCID50 achieved durable and long-lasting complete response and >25% reduction in serum free light chain levels (a biomarker of plasma cell malignancy like multiple myeloma) being observed in four other patients out of 32 patients. Unfortunately, the uptake of 123I was positive in the tumor deposits of only four patients with modest uptake being observed in fraction of the lesions, suggesting virus-induced NIS expression at the current level would not be sufficient to induce radioisotope-mediated antitumor effect. A more in-depth immune profiling of 10 patients who were treated with 1011 TCID50 revealed 8 out of the 10 patients who did not clinically respond to MV-NIS therapy also exhibited negligible increase in cytotoxic T cell response from the baseline observed prior to virotherapy (112). In general, the patients showed elevated CD8+ T cell count in the PBMC and increased proportion of both effector memory and central memory CD8+ T cell population following MV-NIS treatment, providing preliminary evidence of pro-inflammatory reaction following systemic virus administration. Increased PD-1 expression level was also observed in CD8+ T cell following virus administration, which suggests that MV-NIS in conjunction with ICI may enhance clinical response in patients. Unfortunately, only trial registered to evaluate MV-NIS with ICI has been terminated due to low recruitment (NCT02919449).
Although several phase I or II clinical trials are either active or recruiting for evaluation of MV-NIS in wide-range of cancer types (NCT02364713, NCT01846091, NCT02962167, NCT02700230, and NCT03171493), a clinical trials that focuses on IO property of oMV are needed in the future. Currently, there is only a single ongoing clinical trial that utilizes oMV that expresses pro-inflammatory transgene, Helicobacter pylori Neutrophil-activating Protein (NAP; MV-s-NAP), for the treatment of patients with invasive metastatic breast cancer (NCT04521764). Although preclinical models have demonstrated MV-s-NAP to induce pro-inflammatory response (115), its immune regulatory properties in patients has not been reported to date.
2.4.3 Oncolytic picornaviruses
Currently, two different oncolytic picornaviruses, lerapolturev (previously known as PVSRIPO) and V937 (previously known as CAVATAK and CVA21) are under active clinical development. Another oncolytic picornavirus NTX-010, a Seneca Valley virus, has conducted one phase I trial and II trial reported to date (NCT01048892 & NCT01017601, respectively), but will not be discussed in this section (116). This is due to phase II trial in patients with small cell lung cancer leading to early termination of the trial due to NTX-010 treatment failing to improve overall survival or progression free survival rate compared to the placebo group and early termination of the trial, and no subsequent clinical trial being conducted since the failure (116). Both lerapolturev and V937 natively have a tropism that may be beneficial for cancer therapy application and demonstrated promising therapeutic efficacy in early phases of clinical trials with good safety record, thus these two viruses will be reviewed in greater detail.
Lerapolturev is a genetically modified attenuated version of the poliovirus type 1 Sabin that had its internal ribosome entry site (IRES) replaced with IRES of human rhinovirus type 2 to ablate neurovirulence (117, 118), internalizes into cells via CD155, which is upregulated in solid tumors and APCs. Importantly, the infection of APC with lerapolturev has been shown to be nonlethal and reported to induce sustained proinflammatory response and activation of APC (118, 119), which could be beneficial for the instigation of tumor-specific immune response. In support, a phase I clinical trial result evaluating intratumorally administered lerapolturev in patients with unresectable and PD-1 ICI treatment-refractory melanoma revealed that one patient (Patient #11) who was negative for CD155 in pretreatment tumor biopsy (biopsy had small area of viable tumor on the slide, but rather contained abundant CD155+ abundant pigment-laden macrophages) showed partial response to treatment per immune-related response criteria (irRC), suggesting that antitumor response may have been achieved via infection of immune cells in the tumor microenvironment (120). Overall, the intratumoral administration of lerapolturev led to objective response in 33% of the patients (4 out of 12) who were administered with three doses of lerapolturev in the lesions with tumor regression being observed 10 days after the virus administration. Two patients showed pathological complete responses in both the injected and non-injected lesions with post treatment biopsy samples at the injection site showing abundant macrophage accumulation. Notably, 6 out of 12 patients resuming ICI therapy after lerapolturev treatment had durable disease control and remained progression free at a median follow-up period of 18 months, which suggests potential resensitization of PD-1 ICI refractory tumors to PD-1 blockade. Building on this promising results, multicenter phase II trial evaluating lerapolturev in patients with confirmed PD-1 ICI refractory melanoma with or without pembrolizumab is now ongoing (NCT04577807).
Another phase I study evaluating the convection-enhanced infusion of lerapolturev directly into the tumor tissues in 61 patients with recurrent World Health Organization grade IV glioma also yielded promising results without any sign of neurovirulence symptoms (encephalomyelitis, poliomyelitis, and meningitis) typically associated with wild-type polio infection (118). The overall survival rate was 21% in lerapolturev-treated patients at 24 and 34 months after virus administration and this was higher compared with 14% and 4% survival rate expected in the historical control group at the same timepoint. Eight patients had a durable radiographic response in the lerapolturev-treated tumor with two patients having complete response and surviving for 15.1 and 70.4 months at the time of last follow-up prior to publication of the study and three patients achieving stable to partial radiographic response for 26 to 60 months. A transcriptomic analysis of lerapolturev-treated patient biopsies revealed that very low tumor mutation burden is associated with longer survival after lerapolturev treatment in recurrent glioblastoma patients, likely due to recurrent glioblastoma with lower tumor mutation burden exhibiting enrichment of inflammatory gene signature. Further, anti-PD-1 ICI in recurrent glioblastoma patients also achieved better survival rate in patients with lower tumor mutation burden, suggesting that lerapolturev in combination with PD-1 ICI could be beneficial in similar subset of patients.
Collectively, the results from phase I trials of lerapolturev as monotherapy have provided preliminary clinical evidence that lerapolturev in combination with ICI could be synergistic as lerapolturev may either resensitize the PD-1 ICI refractory tumors or be beneficial in recurrent glioblastoma patients with low tumor burden. Currently, several phase I/II or II trials evaluating lerapolturev in combination with ICIs are ongoing; phase II trials in combination with anti-PD-1 ICI in patients with recurrent glioblastoma (NCT04479241) or PD-1 refractory melanoma (NCT04577807) and phase I/II trial in combination with Anti-PD-1 or PD-L1 ICI in patients with advanced solid tumors (NCT04690699). The interim results from these trials are awaited.
V937, a wild-type coxsackievirus A21, is another oncolytic picornavirus that is under active clinical development and intrinsically possesses tropism to cells expressing intracellular adhesion molecule-(ICAM)-1 and decay-accelerating factor (DAF) (120, 121). This native tropism is beneficial for cancer therapy, as (1) both ICAM-1 and DAF are known to be overexpressed in several cancer types and (2) increased expression of ICAM-1 correlates with metastatic progression of multiple cancers (122–127). Due to this native tropism favoring infection of tumor cells by V937, no additional genetic engineering was performed to attenuate the virulence of the virus or enable cancer-specific replication of the virus.
Lack of additional safety measure other than ICAM-1- and DAF-targeted tropism of the virus could be a safety concern, as ICAM-1 and DAF are both expressed in normal tissues, which could lead to off-target cytolytic effect and adverse events (121, 128, 129). Still, two recently published phase I and phase II study results demonstrated that locoregional (intravesical or intratumoral) administration of the virus in patients with non-muscle-invasive bladder cancer (NMIBC; NCT02316171) or unresectable melanoma (NCT01227551 & NCT01636882), respectively, was well-tolerated with no grade 2 or higher virus-related adverse events observed (120, 127). Further, IV administration of V937 up to 1 × 109 median tissue culture infectious dose (TCID50) in patients with advanced cancer was reported to be safe with no grade 3 or 4 product-related adverse events (NCT02043665) (130). These findings suggest that the tropism-mediated cancer specificity of V937 was sufficient to ensure safe locoregional and systemic delivery of the virus to patients, despite normal tissues also expressing its entry molecules ICAM-1 and DAF.
In terms of efficacy, intravesical administration of V937 led to increased surface hemorrhage and inflammation of the tumors and one case of complete tumor regression from 15 NMIBC patients enrolled in the phase I trial (NCT02316171). ICAM-1 expression level in the tumors was shown to correlate with higher virus infectivity and no virus was detectable by IHC in adjacent stromal areas; another entry molecule DAF was also expressed at a high level across all tumor biopsy of patients, showing that V937 infection/replication was dependent on high level of ICAM-1 and DAF expression in NMIBC tumors. V937 treatment led to higher level of high mobility group box 1 (HMGB1) in the urine samples and cytosolic localization of the HMGB1 in the tumor tissues than the paired untreated NMIBC patient samples, suggesting V937-mediated induction of immunogenic cell death. Further, V937-treated tumors exhibited high level of perforin (127, 131), which is indicative of immune cell activation, likely due to elevation of CXCL9 and CXCL10 expression level following virus administration. V937 treatment led to upregulation of immune checkpoint or immunosuppression-related molecules (PD-L1 and LAG3), suggesting that the combination with ICIs targeting these immune checkpoint axes could be beneficial to boost the antitumor immunity of the V937. Phase II clinical trial of V937 in 57 patients with unresectable melanoma also yielded promising therapeutic outcome with 12-month PFS of 32.9% and durable response rate of 21.1%, ultimately resulting in 75.4% of overall survival at 12-month follow-up (NCT01227551 & NCT01636882). Notably, more than 30% reduction in tumor volume at the noninjected lesions at distal metastases sites (lung or liver) were observed in 4 out of the 13 visceral lesions from eight patients, demonstrating that V937 induced systemic antitumor immune response.
In lieu of these immune stimulatory properties of V937 and elevation of immune checkpoint molecules following virus administration, several phase I, I/II, or II trials evaluating the combination of V937 with pembrolizumab or ipilimumab have been either completed (pembrolizumab: NCT02043665, NCT02565992 and ipilimumab: NCT03408587, NCT02307149) or ongoing (pembrolizumab: NCT02824965, NCT04152863, NCT04152863, NCT04303169). Although detailed or final results of the completed or ongoing studies have not yet been published, the interim results reported from some of these trials seem promising: (1) phase 1b trial of V937 in combination with pembrolizumab reported objective response rate of 100% 5 out of 5 evaluable patients with stage IVM1c melanoma and overall objective response rate of 73% out of the 11 patients in 2017 (132) and (2) phase 1b trial in combination with ipilimumab yielding median overall survival of 45.1 months with objective response rate of 30% and median duration of response of 8.8 months in patients with advanced melanoma (133) with manageable serious adverse events in both trials.
In sum, those oncolytic picornaviruses (lerapolturev and V937) have shown promising efficacy in clinical trials both as a monotherapy and as a combination therapy with ICI, showing strong indications of robust antitumor immune response activation by both viruses. Although detailed and finalized study results from the combination therapy trials are not yet available, the interim results are promising and continued clinical development seems warranted.
3 OV in combination therapy regimen
Ideally, a new therapeutic modality is expected to improve the therapeutic outcome when used in conjunction with standard care, and at the least, the combination therapy should not be antagonistic. Based on these premises, the next part of this review will explore how OVs can improve the therapeutic potential of standard treatments such as radio-, chemo-, and immunotherapy in both preclinical and clinical studies.
3.1 OV in combination with radiotherapy
Radiotherapy, along with surgery, remains the preferred treatment for locoregional tumors, especially in early stages of cancer (134). Radiation regimens have improved and matured over time, leading to improved disease management and patient outcome. Despite these improvements, a locoregional anticancer effect exerted by radiotherapy limits its efficacy in advanced and metastatic stages of the disease (135). Additionally, locoregional tumor recurrence remains a major challenge for efficient disease management by localized cancer therapeutics (136, 137). To this end, OVs that exert the most potent anticancer effect via intratumoral administration could be a promising addition to address these limitations of conventional locoregional therapies. In support, several combination strategies of OV with radiotherapy have demonstrated promising therapeutic outcome.
Synergism of the combination of oAd with radiotherapy has been investigated during the last two decades (138, 139), and several oAds in combination with radiotherapy are being evaluated in phase I and II clinical trials (140, 141). One of the main mechanisms of synergism of the combination therapy involves upregulation of transgene expression by radiation through increase in oAd replication (142, 143). Particularly, radiation has been shown to increase cellular internalization of Ad (142), likely due to radiation-induced CAR, integrin, and dynamin 2 expression levels that are integral to endocytosis of Ads (142, 144–148). Alternatively, through preclinical studies, several oAds in combination with radiation have been shown to promote a pro-apoptotic effect in tumor cells over individual therapies (149–153). Additionally, the Ad E1A gene has been shown to sensitize cancer cells to DNA-damaging agents like radiation (149, 154), and deletion of the E1B 19 kDa gene, a homolog of anti-apoptotic Bcl-2-related protein, enhanced the induction of apoptosis in tumor cells in combination with radiation (153).
In support of these preclinical results, a phase I clinical trial examining the combined therapeutic effect of oAd expressing dual suicide genes (Ad5-yCD/mutTKSR39rep-ADP) in combination with intensity-modulated radiotherapy (IMRT) against newly diagnosed intermediate- to high-risk prostate cancer yielded promising outcomes (141). In detail, patients received intraprostatic injections of Ad5-yCD/mutTKSR39rep-ADP (1011 VP and 1012 VP on Days 1 and 22, respectively), each followed by a 2.6-week cycle of 5-fluorocytosine + ganciclovir prodrug therapy and concomitant 74 Gy IMRT. The combination therapy led to lower tumor positivity in biopsies performed during follow-up (at 6, 12, and 24 months) with respect to historically matched patients who underwent only radiotherapy. Specifically, more than 40% of intermediate- to high-risk patients in the historical cohort tested positive for adenocarcinoma during post-treatment biopsy when treated with radiotherapy alone. On the other hand, only 22% of the evaluable patients receiving combination therapy were positive for adenocarcinoma. More notable therapeutic benefit was achieved by combination therapy in the intermediate-risk group: ≥30% positivity in biopsy was expected in the historical cohort following radiation monotherapy, but none of the 12 intermediate-risk patients were positive for tumor during the last biopsy following combination therapy. None of these 12 intermediate-risk patients (0%) exhibited prostate specific antigen (PSA) relapse during the follow-up period (12 - 48 months). In contrast, frequency of positive biopsy in high-risk patients following combination therapy (45%) did not differ statistically from the expected result (56%) for this prognostic risk group. In terms of safety, the combined treatment did not increase any adverse effects compared with side-effects induced by either monotherapy examined in separate trials or historically. However, no dose-limiting toxicities or treatment-related serious adverse events have been recorded. Overall, this clinical study showed that the combined treatment of oAd and radiation can be beneficial toward improving therapeutic outcomes of prostate cancer patients with no additional safety hazard.
One clinical study examining replication-incompetent Ad in combination with radiation provided some evidence that this combination strategy induces a favorable antitumor immune response. A phase I trial combining replication-incompetent Ad in combination with radiation has been shown to elevate HLA DR+ CD8+ and CD4+ T cell levels in combination therapy compared to radiation monotherapy, suggesting development of a Th1 immune response favorable for IO application (155). Another preclinical study provided further evidence that the combination of oAd and radiation could exert synergistic antitumor effect via robust activation of immune cell infiltration (156). Specifically, oAd co-expressing GM-CSF and IL-12 in combination with radiation was shown to inhibit primary tumor growth and its lung metastasis. Importantly, CD4+, CD8+, and CD11c+ immune cell infiltration into tumor tissues was significantly improved in combination therapy with respect to radiotherapy alone. These clinical and preclinical data support the combination of oAd and radiation by exerting a potent antitumor immune response in future clinical trials.
The oHSVs in combination with radiation have been shown to elicit more potent anticancer effect than either treatment administered alone (40, 157, 158). Several mechanisms behind additive or synergistic tumor growth control via combination of oHSV and irradiation have been proposed. For example, Mehzir et al. demonstrated that oHSV with γ134.5 gene deletion in combination with ionizing radiation (IR) elicited a more potent anticancer effect than the respective monotherapies due to irradiation-mediated improvement in viral production (159). Their findings demonstrated that γ134.5-deleted oHSV exhibited poorly sustained synthesis of viral DNA at late stages of the infection cycle compared to wild-type HSV. This restricted viral replication could be overcome by combination with IR; the radiation restored late viral gene expression and replication through activation of the p38 pathway, leading to improved viral replication of γ134.5-deleted oHSV (96). Collectively, their findings showed that p38 activation by irradiation enhanced late viral gene expression that subsequently improved viral replication of γ134.5-deleted oHSVs. In another report, G207 (oHSV deficient in viral ribonucleotide reductase (RR) and γ134.5 neurovirulence protein) in combination with IR resulted in better anticancer effects compared with mono therapy via upregulation of cellular RR (160). G207 in combination with radiation elicited dose-dependent and synergistic cytotoxic effects against colorectal cancer cells through radiation-mediated enhancement in viral replication of G207. Similar trends were observed in vivo, where G207 in combination with IR induced a more potent tumor-growth-inhibiting effect than did the respective monotherapies. Interestingly, the parental strain of G207 named R3616 that only harbors γ134.5 deletion while the RR encoding gene remains intact failed to induce synergistic killing effect in combination with the same irradiation condition as used with G207. These findings are in disagreement with those observed by Mehzir et al. (161), where irradiation improved the viral replication of γ134.5 gene-deleted oHSV. One plausible explanation is that this could be due to (i) different doses (in vitro radiation of 250 rad versus 5 Gy (= 500 rad), or (ii) different cancer cell lines used in the two studies. Nonetheless, these discrepancies indicate that more thorough comparative evaluation be explored in the future to better elucidate how irradiation can improve the efficacy of oHSVs.
Although the main mechanism of synergism during combination therapy using oHSVs and radiation remains elusive, this approach has been evaluated in phase I/II clinical trials (162, 163). A phase I trial of G207 in combination with IR against recurrent and progressive glioma showed that the combination was well-tolerated, and no patients developed HSV encephalitis (163). The patients enrolled in the study did not respond to standard therapy, yet six of nine patients achieved stable disease or partial response, at least, at one time point. Importantly, two patients who underwent retreatment under a compassionate use protocol showed significant radiographic response, showing increase in necrotic tumor region and decrease in tumor mass. Notably, the two patients with most significant radiographic response were HSV-1 seronegative at enrollment, suggesting that the pre-existing neutralizing antibody impedes the potency of locally administered oHSVs. In another phase I/II clinical trial, dose-escalating Imlygic (dose range: 106 to 108 PFU) in combination with chemoradiotherapy (70 Gy/35 fractions with concomitant cisplatin 100 mg/m2) for treatment of patients with untreated stage III/IV squamous cell cancer of the head and neck (SCCHN) has been evaluated (163). Their findings revealed that Imlygic in combination with chemoradiotherapy was well-tolerated as no dose-limiting toxicity was observed even with multiple administrations (four administrations over 64 day period). HSV was detected in injected and adjacent un-injected tumor lesions at levels higher than the administered dose, showing efficient replication of Imlygic. Importantly, 82.3% of the treated patients showed tumor response by Response Evaluation Criteria in Solid Tumors (RECIST), and 93% of the patients achieved complete remission at the time of neck dissection, performed 6-10 weeks after completion of combination therapy. Further, no patients developed locoregional recurrence, and disease-specific survival was 82.4% at a median follow up of 29 months, a remarkable achievement compared to the 35-55% of SCCHN patients who develop locoregional or metastatic recurrence within two years of conventional therapy. Together, these results clearly illustrate that the combination of oHSV and IR exerts promising therapeutic effects where locoregional tumor control was critical for patient outcome.
Like other OVs, the precise mechanism of synergism between oVV and radiation remains elusive. In one instance, IR has been shown to upregulate viral genes essential for viral replication, improving overall viral production (88). In marked contrast, others have shown that the synergy behind combination therapy of oVV and IR does not rely on increased viral replication, since IR inhibited JNK signaling and subsequently attenuated viral replication (164). In another report, radiotherapy failed to improve oVV replication. External beam radiation therapy (EBRT) used at clinical dose neither affected GL-ONC1 viability nor accelerated the virus replication. Rather, the combination therapy of EBRT and GL-ONC1 showed a synergistic killing effect due to activation of the apoptosis pathway resulting in delayed tumor growth in an orthotopic sarcoma model. While mice showed survival of 16 and 18 days for EBRT and GL-ONC1, respectively, compared to 12 days for the control group, the combination therapy-treated group showed survival up to 27 days with no toxicity (165). Activation of apoptosis in the combination (GL-ONC1 and X-radiation) group was confirmed in a mouse model of head & neck xenograft tumors. The results showed that X-radiation at clinical dose failed to inhibit virus replication, and the combination was most effective to stop tumor growth (166). Similar results with combination of GL-ONC1 and radiation have been obtained in melanoma, glioma, and sarcoma models (164, 165, 167), leading to a phase I trial (NCT01584284) combining IV-administered GL-ONC1 with standard chemoradiotherapy in head & neck carcinoma patients. Results of this trial also showed that the therapy outcome depends on p16 status. Indeed, after 30 months of follow-up in 19 patients, 7 showed treatment failure and 7 deaths were recorded among p16-negative tumors. In contrast, the five patients with p16-positive tumors were alive and disease-free after 36 months (168). Collectively, these reports suggest that further optimization in dosing regimen for combined treatment of OVs and radiation is necessary to translate promising preclinical outcomes into clinical benefits.
3.2 OV in combination with chemotherapy
Unlike surgical resection or radiotherapy, chemotherapy exerts its therapeutic effect in a systemic manner and remains integral in treating cancer patients with disseminated disease. The systemic chemotherapy used as an adjuvant therapy for localized surgical resection has been shown to achieve similar therapeutic outcomes to those achieved by radical resection, as early as 1981 (169, 170). Recent studies revealed that chemotherapeutics are also capable of inducing immunogenic cell death (ICD) of cancer cells (171, 172). There are many factors involved in chemotherapeutics-mediated ICD, such as exposure of calreticulin (CRT) (173, 174), adenosine triphosphate (ATP) (175, 176) and release of high mobility group box 1 (HMGB1) (175, 177). Due chemotherapy is used commonly in conjunction with OVs that require localized delivery to induce a notable antitumor effect. In general, systemically administered OVs in several clinical trials (discussed in greater detail in Section 2) induced suboptimal therapeutic benefit, and delivery of OVs to metastatic sites remains a major challenge. For these reasons, chemotherapy as a systemic adjuvant to localized OV therapy is a topic of clinical interest and under active clinical investigation.
oAds in combination with chemotherapy can induce synergistic anticancer effects through several distinct mechanisms, in which both oAd and chemotherapeutics can function as a potent adjuvant to one another. For example, Ad E1A protein can force the cell cycle into S-phase to sensitize cancer cells to DNA-damaging agents (149, 154, 178). On the other hand, several chemotherapeutic drugs have been reported to enhance the cellular internalization of viruses, their replication inside the cells, and expression of transgenes (179–184). Indeed, oAd that contains E1A but has a double deletion of E1B 19- and E1B 55-genes, in combination with cisplatin exerted enhanced cytolytic and apoptotic activities against a wide range of cancer cell types (32). In clinical trial, patients who received intratumoral Oncorine in combination with platinum-based chemotherapy showed 79% response rate compared to 40% observed in the control arm that lacked virus treatment (185). Based on a phase III clinical trial, in 2006, Oncorine was approved by China’s State Food and Drug Administration for treatment of head & neck cancer in combination with chemotherapy. GM-CSF-expressing oAd (ONCOS-102) in combination with pemetrexed, cisplatin, or carboplatin has been shown to induce synergistic antitumor effects in a malignant mesothelioma model (186). Whereas combination chemotherapy (Pemetrexed + Cisplatin or Pemetrexed + Carboplatin) alone or ONCOS-102 monotherapy showed either no or inadequate tumor suppression in an immune-competent mesothelioma model, the combination of these drugs and ONCOS-102 resulted in a synergistic antitumor effect. Based on these preclinical results, a phase I trial (NCT02879669) to examine ONCOS-102 in combination with first-line chemotherapy in patients suffering from malignant mesothelioma has been initiated.
Despite significant improvements in survival of patients with pancreatic cancers by combination of interferon-alpha (IFN) and chemoradiation in clinical trials (16-36% increase in 2-year survival and 35% increase in 5-year survival), it demonstrated limited overall efficacy due to systemic toxicity of IFN and low intratumoral level of the cytokine (187, 188). To overcome these limitations in therapeutic efficacy and safety issues, oAd expressing hamster IFN (OAd-hamIFN) was tested in combination with chemotherapy and/or radiation in regimens mimicking the IFN-based therapies in a preclinical setting (189). oAd-hamIFN potentiated the cytotoxicity of chemotherapeutic drugs (5-FU, gemcitabine, and cisplatin) to yield enhanced pancreatic cancer cell death in both in vitro and in vivo experimental settings in a hamster model of pancreatic cancer. Particularly, combining OV therapy with 5-FU showed significant tumor growth inhibition in an in vivo immunocompetent hamster model.
In line with these preclinical findings, phase I clinical study evaluating intratumorally administered Ad5-yCD/mutTKSR39rep-hIL12 in combination with chemotherapeutics for the treatment of patients with metastatic pancreatic cancer has demonstrated promising results (NCT03281382). For chemotherapy component of the trial, 5-fluorocytosine (5-FC), which is a prodrug that can be converted to 5-FU by yCD/mutTKSR39rep transgene, in combination with one of the standard of care chemotherapy options for the treatment of pancreatic cancer (FOLFIRINOX or gemcitabine plus nab-paclitaxel) were utilized. The treatment was well-tolerated with only one serious adverse event being observed in one patient from the highest dose level (1 x 1012 VP). There were strong evidences of immune activation as the combination therapy elevated the serum levels of pro-inflammatory cytokines and chemokines, like IL-12, IFN-γ, and CXCL10, in a virus dose-dependent manner. Further, Ad5-yCD/mutTKSR39rep-hIL12 treatment elevated the number of proliferating NK and T cells (both CD4+ and CD8+ subsets) in the PBMC of patients. Interestingly, in the low dose cohort (1 x 1011 VP) exhibited elevated Tim3, a T cell exhaustion marker, expression level in NK and CD8+ T cell population, whereas the expression level was maintained at a similar level to the baseline observed pretreatment in patients receiving higher doses (3 x 1011 or 1 x 1012 VP). This finding suggests that prevention of T cell exhaustion requires high level of viral doses and pro-inflammatory cytokine expression level. Additionally, 2 out of 6 patients at the highest dose cohort achieved stable disease in both the virus-treated tumors and metastatic lesions, which indicates induction of systemic antitumor immune response. The highest dose cohort also had median progression free survival period of 10.6 months in comparison to 6.4 or 3.3 months expected in patients who receive FOLFIRINOX or gemcitabine alone. Although no conclusive comparison can be made due to small patient number in early phase clinical trial, the preliminary findings seem encouraging.
Several studies in the last two decades have explored the anticancer activity of numerous combinations of oHSVs and chemotherapeutics (190–194), and many potent combination regimens have been identified. One of the strongest merits of oHSV is that its anticancer effect is not inhibited by genotypic alterations commonly observed in tumors (e.g., p53 and Rb pathways), whereas many of the conventional therapies are nullified by these oncogenic mutations (195). Mechanisms underlying synergistic interactions among different oHSV and chemotherapeutics have been described (196). In general, the synergistic outcome of combination therapy requires that chemotherapy not interfere with replication of oHSV in infected tumor cells. Consistently, enhancement of viral replication has been reported as a common mechanism behind the synergistic anticancer effect (197–200).
Based on these strong lines of evidence supporting synergy between a diverse range of chemotherapeutic drugs and different oHSVs, phase I and I/II trials have been conducted to evaluate the safety profile of these combination therapies (201, 202). In a phase I trial, endoscopic ultrasound (EUS)-guided administration of HF10 (a spontaneously mutated oHSV lacking expression of multiple viral genes, γ134.5, UL43, UL49.5, UL55, and UL56) into localized and unresectable pancreatic tumors in combination with chemotherapeutics (erlotinib and gemcitabine) was explored (202). In this study, patients underwent one cycle of erlotinib and gemcitabine treatment followed by intratumoral injections (four repeated administrations of 1 × 106 – 1 × 107 PFU of HF10) via EUS guidance on the first day of the second chemotherapy cycle. This combination treatment did not yield any grade III or IV adverse effect, suggesting that addition of HF10 to standard chemotherapy was well-tolerated. Of the nine enrolled patients, three showed partial response, four showed stable disease, and two exhibited progressive disease. Notably, tumor shrinkage in two patients was observed, and the tumors were resected. In both cases, surgically resected patients achieved long-term survival, and significant infiltration of CD4+ and CD8+ T cells was detected either in fibrosis near the residual cancer cells or in the resected tumor specimen. In one case, invasion to the plexus of the superior mesenteric artery decreased following HF10 administration, and the resected specimen showed 90% reduction in cancer cells with fibrosis. Of note, tumors were considered unresectable at the time of enrollment. These results demonstrate that the combination of chemotherapy and oHSV could exert antitumor immune responses critical in IO applications. Although most reports examining the combination therapeutic index of oHSV and chemotherapeutic drug reported additive or synergistic effects, some studies have reported an antagonistic effect on viral replication (203). Thus, the combination of chemotherapy with oHSV warrants careful optimization of parameters such as sequence and timing of administration as well as transgene and drug selection (196).
Several chemotherapies in combination with oVV have been explored. However, in some cases, the combination has been shown to restrict the therapeutic efficacy of oVV and demands careful selection of drug candidates and dosing regimen. For instance, 5-FU and irinotecan (a topoisomerase inhibitor) have been shown to impede viral replication (204). Despite the attenuation in viral replication, the combination therapy with irinotecan resulted in a significant synergy, with a median survival of 87 days in mice with a colorectal tumor treated with combination vvDD and irinotecan compared to 57 and 48 days for the groups receiving vvDD or chemotherapy alone, respectively. Indeed, the authors showed that the virus-infected cells were arrested in S-phase, where they were more sensitive to the irinotecan. The mechanism of synergy was assigned to the direct killing effect of the vaccinia virus and the early recruitment of macrophages (205). A phase I clinical trial (NCT01469611) using Pexa-Vec showed that administration of multiple doses of oVV was safe with no adverse events other than flu-like symptoms in colorectal cancer patients. Furthermore, IV infusion (3.1 × 109 pfu/kg injected biweekly over 8 weeks) resulted in stable disease in 8 of 9 patients (89%) (206). In view of these pre-clinical and clinical data indicating the synergistic effect of oVV and irinotecan combination, a clinical trial on Pexa-Vec and irinotecan combination on refractory colorectal carcinoma patients (NCT01394939) has been initiated. Despite some oVV and chemotherapy combination therapy regimens achieving promising therapeutic outcome, a recent phase III clinical trial evaluating the combined therapeutic effect of Pexa-Vec with the kinase inhibitor sorafenib failed to show any benefits over sorafenib monotherapy in patients with advanced liver cancer (207). Those results illustrate that there are many obstacles to overcome prior to successful clinical adaptation of various OV plus chemotherapeutic combination therapy regimens.
Paclitaxel, targeting tubulin, also has been investigated as a possible combination therapy component. Although paclitaxel caused reduction in viral replication, G2/M phase cell cycle arrest yielding a 2-fold increase in infectivity of the vaccinia virus was reported. The combination of paclitaxel and IV vvDD led to 50% complete and durable response in mice bearing colorectal tumors. The data were in contrast to a 10% response in the vvDD-alone group and 0% in the paclitaxel-only group (208). Compared to paclitaxel, the effects of other drugs in combination therapy are unclear. Gemcitabine, a pro-drug that acts like a nucleoside analog, failed to show any synergistic effect when combined with GL-ONC1 in pancreatic cancer cell lines (209), while its combination with oVV-Smac (an oncolytic vaccinia virus encoding a caspase activator) effectively reduced tumor volume and enhanced the survival rate of pancreatic-cancer bearing mice (210).
Despite synergistic effects observed in the cases described above, some combinations did not show any enhancement in cancer cell killing. A triple therapy with GL-ONC1, nab-paclitaxel, and gemcitabine did not show any significant increase in cytotoxicity in pancreatic cell lines (209). However, this finding was of special interest for clinical application because the combination of nab-paclitaxel and gemcitabine has been approved. GL-ONC1 possessing 3 transgenes (Ruc-GFP, β-glucuronidase and β-galactosidase) provided the potential to non-invasively monitor tumors (89). In this case, despite not bringing a therapeutic benefit to the actual treatment, it could be used safely to monitor tumors.
Taken together, these results suggest that further optimization regarding detailed mechanism of chemotherapeutics how they improve therapeutic outcome of OV is needed to translate promising preclinical results into clinical benefits.
3.3 OV in combination with immunotherapy
For a new treatment modality, such as OVs, to enter the clinical landscape, the product should not impede the therapeutic effect of standard care. Ideally, a new therapeutic modality is expected to improve the therapeutic outcome when used in conjunction with standard care, and at the least, the combination therapy should not be antagonistic. Based on these premises, the next part of this review will explore how OVs can improve the therapeutic potential of immunotherapy.
Despite remarkable success in cancer immunotherapeutics in recent times, immunologically ‘cold’ tumors remain a major obstacle for these innovative drugs. Specifically, a wide range of clinically approved immunotherapeutics, including the chimeric antigen receptor (CAR)-T cells and ICIs, are beneficial in only a small subset of cancer patients, as a large subset of patients with immunologically cold tumor respond poorly to the treatment (14, 211–213). In this regard, OVs with their unique inflammatory properties are investigated as promising adjuvants to inflame ‘cold’ tumors and convert them into ‘hot’ tumors that are more responsive to immunotherapy-induced antitumor immune reaction. This OV-induced immunological conversion of the tumor milieu has been shown to be highly favorable toward maximizing the antitumor immune response of several clinically approved cancer immunotherapies. Additionally, cancer vaccines and OVs have been found to exert lower side-effects in cancer patients than do other systemic immunotherapies (214, 215). Since the increased risk of safety hazard in mono- or combination-therapies of clinically approved immunotherapies is a major concern in IO, the low toxicity profile of OV-combined therapies is highly favorable and is being explored actively in preclinical and clinical studies.
Even though immune checkpoint inhibitors (ICI) have changed the treatment paradigm for many cancers, there is medical unmet need in 70-80% of patients who did not response to ICI treatment (216). Certain cancers have a unique tumor microenvironment that has a relative paucity of infiltrating immune effector cells, creating a “cold tumor” (216). Therefore, strategies to utilize OV for warming cold tumor microenvironments are attractive to increase the effectiveness of ICIs, and thus there are multiple ongoing clinical trials evaluating the combination of oAd with ICI. Several preclinical studies involving oAd-ICIs have revealed promising candidates (217). oAd expressing IL-2 and TNF-α has been reported to yield marked increase in intratumoral CD8+ T cells when each of these was combined with an anti-PD-1 antibody compared to virus alone. Furthermore, combination therapy with the anti-PD-1 antibody and viral therapy resulted in statistically significant tumor growth suppression and increase in survival compared to virus monotherapy. A clinical trial employing an oAd encoding TNF-α and IL-2 (TILT-123) in combination with an anti-PD-1 antibody is warranted. Recently, two phase I clinical trials evaluating TILT-123 in combination with ICI (pembrolizumab NCT05271318 or avelumab NCT05222932) have been posted (https://clinicaltrials.gov/).
Penetration of ICI into tumor tissues is limited due to abnormal vasculature, tumor interstitial pressure, and excessive extracellular matrix (ECM) accumulation (218). To overcome these challenges, strategies combine the therapeutic efficacy of oAd (oAd/IL12/GM-RLX), which induces antitumor immune response and ECM degradation in tumor microenvironments, and PD-1 immune checkpoint blockade (αPD-1). The combination of oAd/IL12/GM-RLX and αPD-1 induced effective degradation of the tumor ECM. Further, it enhanced intratumoral infiltration of αPD-1 and activated antitumor immune cells. This strategy elicited a potent and durable antitumor immune response against cold tumors. This is the first study showing that expression of four genes (three immune stimulatory genes and another gene specializing in ECM degradation) by a single oAd can overcome the major limitations of ICI therapies, which has emerged in recent clinical trials, by promoting favorable remodeling of both physical and immunological aspects of the tumor.
Combination of oHSV, HSV1716, and PD-1 blockade significantly prolonged survival compared with PD-1 blockade alone or HSV1716 monotherapy in murine rhabdomyosarcoma models (219). This therapeutic outcome was due to increased tumor infiltration of CD4+ and CD8+ T cells, but not Tregs. Furthermore, this combination strategy was effective to treat glioblastoma (GBM), which is lethal, highly immunosuppressive, and posited to contain GBM stem-like cells (GSCs) (220). Triple combination of oHSV expressing IL-12 with anti-PD-1 and anti-CTLA-4 cure most mice with GSC-derived orthotopic GBM. This curative therapy is associated with large increases in M1-like macrophages and T effector cells (CD4+ and CD8+ T cells) and decreases in regulatory T cells. These data suggest the combination of ICI and oHSV as an effective treatment strategy for tumor. Similar trends have been observed in early phase clinical trials evaluating oHSV with ICI. For instance, preliminary phase I/II trial results of RP-1 (an oHSV armed with GM-CSF and a truncated highly fusogenic form of the envelope glycoprotein of gibbon ape leukemia virus (GALV-GP-R) to enhance immunogenic cell death) in combination with nivolumab (NCT03767348) have been demonstrating promising therapeutic efficacy in melanoma and non-melanoma skin cancer patients refractory to ICI therapy. In detail, 13 out of 36 melanoma patients showed therapeutic response and 8 out of 13 patients with non-melanoma skin cancer achieved therapeutic response with 5 of these 8 patients achieving complete response (221). Although many of the phase I or II clinical trials evaluating different oHSVs in combination with ICI has seemed to yield promising results (212, 215), these results should be interpreted with caution due to small sample size of patients in early phases of clinical trials. For instance, T-VEC in combination with pembrolizumab, which yielded promising CR rate of 33% in a phase 1b trial (NCT02263508) (222), failed to demonstrate superior PFS or OR over pembrolizumab plus placebo in a phase III trial. Similarly, phase Ib/III trial of same combination therapy regimen in patients with metastatic head and neck squamous cell carcinoma (HNSCC) also failed to demonstrate superior efficacy over historical HNSCC cohort treated with pembrolizumab monotherapy in phase 1b portion of the trial and phase III was not further pursued (NCT02626000) (35).
Combination treatment of anti-CTLA-4 ICI with an oVV was effective in eradicating tumors and provided extended survival compared to monotherapies (223). Building on these preclinical data, clinical studies of Pexa-Vec in combination with ICIs have been initiated, particularly in view of the termination of phase III trial of Pexa-Vec in combination with sorafenib for liver cancer in 2019. Phase I/II clinical trial of Pexa-Vec in combination with anti-CTLA-4 ICI ipilimumab in patients with metastatic or advanced solid tumors is ongoing (NCT02977156). Another phase I/II study of Pexa-Vec in combination with either anti-PD1 ICI durvalumab alone or in triple combination with anti-CTLA-4 ICI tremelimumab has been ongoing for the treatment of patients with advanced colorectal cancer (NCT3206073). Majority of the newly registered and “recruiting” clinical trials listed in http://clinicaltrials.gov as of July of 2022 aim to maximize the immune stimulatory aspects of oVV; 5 out of 7 trials plan to evaluate oVV in combination with ICI (NCT04301011, NCT05061537, NCT04725331, NCT03954067, and NCT03294083) and 5 out of 7 trials utilize oVV harboring either a single or combination of immune stimulatory transgenes (GM-CSF: NCT05376527 & NCT03294083, anti-CTLA4 antibody + GM-CSF: NCT04725331 (224), Fit3 Ligand + anti-CTLA4 antibody+ IL-12: NCT04301011, and IL-7 + IL-12: NCT03954067 (225)).
The potential benefit of combining cytokine-expressing OV with DCs, efficient and specialized APCs that can stimulate naive and memory T cells, was explored for treatment of established tumors (226). Ad-ΔB7/IL-12/4-1BBL, an oAd co-expressing IL-12 and 4-1BBL, exhibited significantly enhanced IFN-γ expression and antitumor efficacy in vivo, suggesting that the antitumor type 1 immune response was significantly activated by co-expression of these transgenes. Moreover, Ad-ΔB7/IL-12/4-1BBL in combination with DCs further enhanced antitumor and anti-metastatic effects by enhancing antitumoral type 1 immune response and suppression of type 2 immune response. Furthermore, oAd co-expressing IL-12 and GM-CSF in combination with DC resulted in strong and synergistic antitumor effects compared to the single treatments (227). Of note, combination of this virus with DC caused upregulation of CCL21+ lymphatic vessels in tumor tissues and led to considerable increase in endogenous and exogenous DC in DLN, indicating that oAd expressing the proper cytokine could increase the function of DC by stimulating it to differentiate.
Despite several advantages in using oAd and DC in combination therapy, their limited bioavailability and short half-life in solid tumor are critical drawbacks (228). Development of injectable and biodegradable gelatin-based hydrogel as a matrix to protect oAd and DCs in the hostile tumor microenvironment has been attempted. This matrix was shown to protect therapeutic components in the solid tumor and offered sustained release while preserving biological activity (229). oAd- and DC-loaded hydrogel (oAd+DC/gel) showed significantly greater expression of IL-12, GM-CSF, and IFN-γ than either the single treatment (oAd or DC) or oAd in combination with DC (oAd+DC). Furthermore, efficient activation of both endogenous and exogenous DCs, migration of DCs to draining lymph nodes, and tumor infiltration of CD4+ and CD8+ T cells was observed. oAd+DC/gel significantly increased tumor-specific IFN-γ-secreting immune cells and attenuated tumor-mediated thymic atrophy, associated with immunosuppression in the tumor microenvironment, compared with oAd+DC. These findings suggested that the gel-mediated co-delivery of oAd and DCs offer potent and prolonged antitumor effects and hence warrant future cancer clinical trials.
As innate immune cells, natural killer (NK) cells are unique and play pivotal functions in cancer immune surveillance. These can eliminate a variety of abnormal or stressed cells without prior sensitization and can preferentially kill stem-like cells or cancer stem cells (230). However, the antitumor response of NK cells faces many limitations, including (i) poor ability of NK cells to reach tumor tissues (231), (ii) changes in NK cell-activating receptors and their ligands in tumors (232, 233), and (iii) tumor microenvironment infiltrating suppressive or tolerogenic macrophages and regulatory T (Treg) cells (234) that inhibit their action. A recent study demonstrated that the combination of NK with oAd enhanced its tumor suppression activity (235, 236). The telomerase reverse transcriptase (TERT)-positive tumor treated with CCL20/IL15-armed oAd that replicated under control of the TERT promoter plus NK showed significantly higher antitumor efficacy than either of the treatments alone and induced tumor-specific cytotoxicity of CTLs. These results demonstrated that immunomodulation of the tumor milieu by cytokine-expressing oAd could induce synergistic antitumor immunity in combination with cell-based immunotherapeutics.
Adoptive or CAR-T cell therapy is a promising candidate for cancer immunotherapy. Clinical trials against B cell malignancies with anti-CD19 CAR-T cells demonstrated remarkable therapeutic efficacy, leading to durable complete remission (237–240). However, CAR-T therapy elicits low therapeutic efficacy against solid tumors, possibly due to T cell hypofunction that hinders T cell infiltration and activity (241, 242). Based on these backgrounds, there has been increasing number of reports investigating various OVs to improve the activity of CAR-T cell therapies (243, 244). For example, oAd armed with the chemokine RANTES and IL-15 has been shown to facilitate migration and survival of CAR-T cells and enhance its cytolytic effect in preclinical setting (245). Further, the oAd expressing an EGFR-targeting bispecific T-cell engager (OAd-BiTE) improved the outcome of CAR-T cell therapy in solid tumors (246). CAR-T cells targeting folate receptor alpha (FR-α) successfully infiltrated FR-α-positive and EGFR-positive SK-OV3 xenograft tumors but failed to induce complete responses. However, BiTEs secreted from infected cells redirected CAR-T cells toward EGFR in the absence of FR-α in tumor, indicating increased BiTE-mediated T-cell activation in tumors. As a result, combination of a BiTE-expressing oAd with adoptive CAR-T therapy improved antitumor efficacy and prolonged survival compared with the monotherapies. In lieu of these trends, there is one phase 1 clinical trial evaluating the combination of oAd with HER2-targeted CAR-T cell is ongoing for the treatment of patients with advanced HER2-positive solid tumors (NCT03740256).
Although there are strong scientific rationales and preclinical evidence that support the combination of OV with CAR-T cells to induce synergistic antitumor efficacy, there are growing evidence that contrarily demonstrate that OV-induced type I IFN response can be detrimental to the functionality of T cells, including CAR-T cells, and induce their apoptotic cell death (48, 247, 248). One promising strategy to circumvent this issue is to infect the CAR-T cells directly with OVs which enables systemic delivery of both CAR-T cell and OV to solid tumors (249). In detail, dual-specific CAR-T cells that recognizes viral antigens of the OVs and EGFR of tumors infected with oncolytic reovirus or vesicular stomatitis virus (VSV) exhibited augmented proliferation and biological function due to stimulation of native T cell receptor by viral epitopes. Notably, systemic treatment with OVs-loaded CAR-T cell led to potent antitumor activity, which could be further enhanced by subsequent IV administration of the OV for immune boosting application. This research demonstrated that CAR-T therapy can be combined with OV therapy as a systemic delivery regimen to activate the TCR and bypass the requirement for lymphodepletion.
Collectively, these studies have demonstrated that OV with immune-modulating therapeutic genes and their combination with immunotherapeutics such as DC, NK, CAR-T, and ICI offer a viable strategy to elicit potent induction of antitumor immune response by improving activation, recruitment, and infiltration of immune cells to tumor tissues.
4 Future perspective
Both the preclinical and clinical development of OVs are growing at an exponential rate, as these viruses could potentially enhance the poor efficacy of conventional immunotherapy options against immunologically cold tumors in an increasingly immuno-oncology dominant clinical landscape. Further, OVs with more traditional cancer treatment options, like chemo- and radiotherapy, have also yielded promising results, demonstrating the wide applicability of OVs in combination with standard of care. Still, there are many obstacles and unknowns that require further investigation in both preclinical and clinical setting as not all of the clinical trial results have been positive, as evidenced by some landmark phase III trials (Pexa-Vec in combination with sorafenib and T-VEC in combination with pembrolizumab) failing to meet the primary endpoint of trial design despite promising results from early phases of respective clinical trials. In general, there is great variability in patient outcome following OV treatment and insufficient information regarding biological markers that can predict which patient demographic responds to particular OV therapy. More in-depth profiling of responders and nonresponders to OV therapies will be needed to characterize the limitations of OVs more precisely and strategically overcome these issues. Suboptimal systemic administrability of OVs is another hurdle that must be addressed to effectively treat advanced stages of cancer where metastatic or noninjectable lesions are present. To this end, cell-based carriers (CAR-T and mesenchymal stem cells) or synthetic carriers (nanomaterial, polymers, and liposomes) with tumor homing properties have been shown promising preliminary results in preclinical and clinical setting to enhance tumor accumulation of OVs and warrant further investigation.
5 Conclusion
This review highlighted various clinical applications of OVs in both monotherapy and combination therapy applications. Since the first clinical trials using OVs in the 1990s, there have been significant advancements in understanding of OV genetics and biology for development of more potent and safer OVs for clinical application. Currently, numerous OVs are under clinical development by multiple pharmaceutical powerhouses as these viruses have shown strong potential to improve the therapeutic outcome of standard cancer treatment options like radiotherapy, chemotherapy, and immunotherapy. Although most clinical data of OV pipelines investigated in this review originated from phase I/II trials and the interpretation of the efficacy results should be taken with caution due to the small sample sizes, the initial reports are highly promising and strongly suggest that OVs could become a common cancer treatment option in the near future.
Many of clinical trials has been developed OVs in conjunction with another therapy and the numbers in combination trials of OVs with ICI have grown over the past 8 years.
Author contributions
C-OY and A-RY substantially contributed to the conception and design of the article and interpreting the relevant literature. JH revised it critically for important intellectual content and all authors approved the final version of the manuscript.
Funding
This research was supported by grants from the National Research Foundation of Korea (2016M3A9B5942352 and 2021R1A2C301016611, C-OY; 2021M2E8A1049151 and 2022R1I1A1A01071162, A-RY).
Conflict of interest
Authors C-OY and JH were employed by company GeneMedicine Co., Ltd.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl J Med (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
2. Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. New Engl J Med (2011) 364:2517–26. doi: 10.1056/NEJMoa1104621
3. Sehgal A, Whiteside TL, Boyiadzis M. Programmed death-1 checkpoint blockade in acute myeloid leukemia. Expert Opin Biol Ther (2015) 15:1191–203. doi: 10.1517/14712598.2015.1051028
4. Grosso JF, Jure-Kunkel MN. CTLA-4 blockade in tumor models: An overview of preclinical and translational research. Cancer Immun (2013) 13:5. doi: 10.3390/cancers11050685
5. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. New Engl J Med (2017) 377:1345–56. doi: 10.1056/NEJMoa1709684
6. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res (2015) 75:2139–45. doi: 10.1158/0008-5472.CAN-15-0255
7. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer (2016) 16:275–87. doi: 10.1038/nrc.2016.36
8. Pol J, Kroemer G, Galluzzi L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology (2016) 5:e1115641. doi: 10.1080/2162402X.2015.1115641
9. Kirn D. Oncolytic virotherapy for cancer with the adenovirus dl1520 (Onyx-015): Results of phase I and II trials. Expert Opin Biol Ther (2001) 1:525–38. doi: 10.1517/14712598.1.3.525
10. Ganly I, Kirn D, Eckhardt G, Rodriguez GI, Soutar DS, Otto R, et al. A phase I study of onyx-015, an E1B attenuated adenovirus, administered intratumorally to patients with recurrent head and neck cancer. Clin Cancer Res Off J Am Assoc Cancer Res (2000) 6:798–806.
11. Brown MC, Holl EK, Boczkowski D, Dobrikova E, Mosaheb M, Chandramohan V, et al. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen–specific CTLs. Sci Trans Med (2017) 9:eaan4220. doi: 10.1126/scitranslmed.aan4220
12. Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol (2018) 18:498–513. doi: 10.1038/s41577-018-0014-6
13. Felt SA, Grdzelishvili VZ. Recent advances in vesicular stomatitis virus-based oncolytic virotherapy: A 5-year update. J Gen Virol (2017) 98:2895–911. doi: 10.1099/jgv.0.000980
14. Ferrucci PF, Pala L, Conforti F, Cocorocchio E. Talimogene laherparepvec (T-VEC): An intralesional cancer immunotherapy for advanced melanoma. Cancers (Basel) (2021) 13:1383. doi: 10.3390/cancers13061383
15. Oh E, Hong J, Yun CO. Regulatory T cells induce metastasis by increasing tgf-β and enhancing the epithelial–mesenchymal transition. Cells (2019) 8:1387. doi: 10.3390/cells8111387
16. Oh E, Choi IK, Hong J, Yun CO. Oncolytic adenovirus coexpressing interleukin-12 and decorin overcomes treg-mediated immunosuppression inducing potent antitumor effects in a weakly immunogenic tumor model. Oncotarget (2017) 8:4730–46. doi: 10.18632/oncotarget.13972
17. Kim JH, Lee YS, Kim H, Huang JH, Yoon AR, Yun CO. Relaxin expression from tumor-targeting adenoviruses and its intratumoral spread, apoptosis induction, and efficacy. J Natl Cancer Inst (2006) 98:1482–93. doi: 10.1093/jnci/djj397
18. Hermiston T. Gene delivery from replication-selective viruses: Arming guided missiles in the war against cancer. J Clin Invest (2000) 105:1169–72. doi: 10.1172/JCI9973
19. Bonadio J. Tissue engineering via local gene delivery: Update and future prospects for enhancing the technology. Adv Drug Delivery Rev (2000) 44:185–94. doi: 10.1016/S0169-409X(00)00094-6
20. Crystal RG. Adenovirus: the first effective in vivo gene delivery vector. Hum Gene Ther (2014) 25:3–11. doi: 10.1089/hum.2013.2527
21. Liang M. Oncorine, the world first oncolytic virus medicine and its update in China. Curr Cancer Drug Targets (2018) 18:171–6. doi: 10.2174/1568009618666171129221503
22. El-Shemi AG, Ashshi AM, Na Y, Li Y, Basalamah M, Al-Allaf FA, et al. Combined therapy with oncolytic adenoviruses encoding TRAIL and IL-12 genes markedly suppressed human hepatocellular carcinoma both in vitro and in an orthotopic transplanted mouse model. J Exp Clin Cancer Res CR (2016) 35:74. doi: 10.1186/s13046-016-0353-8
23. Choi IK, Li Y, Oh E, Kim J, Yun CO. Oncolytic adenovirus expressing IL-23 and p35 elicits IFN-gamma- and TNF-alpha-co-producing T cell-mediated antitumor immunity. PloS One (2013) 8:e67512. doi: 10.1371/journal.pone.0067512
24. Marelli G, Howells A, Lemoine NR, Wang Y. Oncolytic viral therapy and the immune system: A double-edged sword against cancer. Front Immunol (2018) 9:866. doi: 10.3389/fimmu.2018.00866
25. Saito Y, Sunamura M, Motoi F, Abe H, Egawa S, Duda DG, et al. Oncolytic replication-competent adenovirus suppresses tumor angiogenesis through preserved E1A region. Cancer Gene Ther (2006) 13:242–52. doi: 10.1038/sj.cgt.7700902
26. Yoo JY, Kim JH, Kwon YG, Kim EC, Kim NK, Choi HJ, et al. VEGF-specific short hairpin RNA-expressing oncolytic adenovirus elicits potent inhibition of angiogenesis and tumor growth. Mol Ther J Am Soc Gene Ther (2007) 15:295–302. doi: 10.1038/sj.mt.6300023
27. Jung KH, Choi IK, Lee HS, Yan HH, Son MK, Ahn HM, et al. Oncolytic adenovirus expressing relaxin (YDC002) enhances therapeutic efficacy of gemcitabine against pancreatic cancer. Cancer Lett (2017) 396:155–66. doi: 10.1016/j.canlet.2017.03.009
28. Koom WS, Park SY, Kim W, Kim M, Kim JS, Kim H, et al. Combination of radiotherapy and adenovirus-mediated p53 gene therapy for MDM2-overexpressing hepatocellular carcinoma. J Radiat Res (2012) 53:202–10. doi: 10.1269/jrr.11110
29. Garcia-Carbonero R, Salazar R, Duran I, Osman-Garcia I, Paz-Ares L, Bozada JM, et al. Phase 1 study of intravenous administration of the chimeric adenovirus enadenotucirev in patients undergoing primary tumor resection. J immunother Cancer (2017) 5:71. doi: 10.1186/s40425-017-0277-7
30. Kuhn I, Harden P, Bauzon M, Chartier C, Nye J, Thorne S, et al. Directed evolution generates a novel oncolytic virus for the treatment of colon cancer. PloS One (2008) 3:e2409. doi: 10.1371/journal.pone.0002409
31. Kirn D. Clinical research results with dl1520 (Onyx-015), a replication-selective adenovirus for the treatment of cancer: What have we learned? Gene Ther (2001) 8:89–98. doi: 10.1038/sj.gt.3301377
32. Yoon AR, Kim JH, Lee YS, Kim H, Yoo JY, Sohn JH, et al. Markedly enhanced cytolysis by E1B-19kD-deleted oncolytic adenovirus in combination with cisplatin. Hum Gene Ther (2006) 17:379–90. doi: 10.1089/hum.2006.17.379
33. Dong W, van Ginkel JW, Au KY, Alemany R, Meulenberg JJ, van Beusechem VW. ORCA-010, a novel potency-enhanced oncolytic adenovirus, exerts strong antitumor activity in preclinical models. Hum Gene Ther (2014) 25:897–904. doi: 10.1089/hum.2013.229
34. Eriksson E, Milenova I, Wenthe J, Ståhle M, Leja-Jarblad J, Ullenhag G, et al. Shaping the tumor stroma and sparking immune activation by CD40 and 4-1BB signaling induced by an armed oncolytic virus. Clin Cancer Res (2017) 23:5846–57. doi: 10.1158/1078-0432.CCR-17-0285
35. Harrington KJ, Kong A, Mach N, Chesney JA, Fernandez BC, Rischin D, et al. Talimogene laherparepvec and pembrolizumab in recurrent or metastatic squamous cell carcinoma of the head and neck (MASTERKEY-232): A multicenter, phase 1b study. Clin Cancer Res (2020) 26:5153–61. doi: 10.1158/1078-0432.CCR-20-1170
36. Havunen R, Siurala M, Sorsa S, Grönberg-Vähä-Koskela S, Behr M, Tähtinen S, et al. Oncolytic adenoviruses armed with tumor necrosis factor alpha and interleukin-2 enable successful adoptive cell therapy. Mol Ther Oncol (2017) 4:77–86. doi: 10.1016/j.omto.2016.12.004
37. Packiam VT, Lamm DL, Barocas DA, Trainer A, Fand B, Davis RL 3rd, et al. An open label, single-arm, phase II multicenter study of the safety and efficacy of CG0070 oncolytic vector regimen in patients with BCG-unresponsive non-muscle-invasive bladder cancer: Interim results. Urol Oncol (2018) 36:440–7. doi: 10.1016/j.urolonc.2017.07.005
38. Ranki T, Pesonen S, Hemminki A, Partanen K, Kairemo K, Alanko T, et al. Phase I study with ONCOS-102 for the treatment of solid tumors - an evaluation of clinical response and exploratory analyses of immune markers. J Immunother Cancer (2016) 4:17. doi: 10.1186/s40425-016-0121-5
39. Reeh M, Bockhorn M, Görgens D, Vieth M, Hoffmann T, Simon R, et al. Presence of the coxsackievirus and adenovirus receptor (CAR) in human neoplasms: A multitumour array analysis. Br J Cancer (2013) 109:1848–58. doi: 10.1038/bjc.2013.509
40. Giaginis CT, Zarros AC, Papaefthymiou MA, Papadopouli AE, Sfiniadakis IK, Theocharis SE. Coxsackievirus and adenovirus receptor expression in human endometrial adenocarcinoma: Possible clinical implications. World J Surg Oncol (2008) 6:59. doi: 10.1186/1477-7819-6-59
41. Garcia M, Moreno R, Gil-Martin M, Cascallo M, de Olza MO, Cuadra C, et al. A phase 1 trial of oncolytic adenovirus ICOVIR-5 administered intravenously to cutaneous and uveal melanoma patients. Hum Gene Ther (2019) 30:352–64. doi: 10.1089/hum.2018.107
42. Moreno V, Barretina-Ginesta MP, García-Donas J, Jayson GC, Roxburgh P, Vázquez RM, et al. Safety and efficacy of the tumor-selective adenovirus enadenotucirev with or without paclitaxel in platinum-resistant ovarian cancer: A phase 1 clinical trial. J Immunother Cancer (2021) 9:e003645. doi: 10.1136/jitc-2021-003645
43. Ramesh N, Ge Y, Ennist DL, Zhu M, Mina M, Ganesh S, et al. CG0070, a conditionally replicating granulocyte macrophage colony-stimulating factor–armed oncolytic adenovirus for the treatment of bladder cancer. Clin Cancer Res Off J Am Assoc Cancer Res (2006) 12:305–13. doi: 10.1158/1078-0432.CCR-05-1059
44. Du T, Shi G, Li YM, Zhang JF, Tian HW, Wei YQ, et al. Tumor-specific oncolytic adenoviruses expressing granulocyte macrophage colony-stimulating factor or anti-CTLA4 antibody for the treatment of cancers. Cancer Gene Ther (2014) 21:340–8. doi: 10.1038/cgt.2014.34
45. Freytag SO, Rogulski KR, Paielli DL, Gilbert JD, Kim JH. A novel three-pronged approach to kill cancer cells selectively: concomitant viral, double suicide gene, and radiotherapy. Hum Gene Ther (1998) 9:1323–33. doi: 10.1089/hum.1998.9.9-1323
46. Barton KN, Paielli D, Zhang Y, Koul S, Brown SL, Lu M, et al. Second-generation replication-competent oncolytic adenovirus armed with improved suicide genes and ADP gene demonstrates greater efficacy without increased toxicity. Mol Ther J Am Soc Gene Ther (2006) 13:347–56. doi: 10.1016/j.ymthe.2005.10.005
47. Freytag SO, Khil M, Stricker H, Peabody J, Menon M, DePeralta-Venturina M, et al. Phase I study of replication-competent adenovirus-mediated double suicide gene therapy for the treatment of locally recurrent prostate cancer. Cancer Res (2002) 62:4968–76.
48. Barton KN, Siddiqui F, Pompa R, Freytag SO, Khan G, Dobrosotskaya I, et al. Phase I trial of oncolytic adenovirus-mediated cytotoxic and interleukin-12 gene therapy for the treatment of metastatic pancreatic cancer. Mol Ther Oncol (2021) 20:94–104. doi: 10.1016/j.omto.2020.11.006
49. Freytag SO, Paielli D, Wing M, Rogulski K, Brown S, Kolozsvary A, et al. Efficacy and toxicity of replication-competent adenovirus-mediated double suicide gene therapy in combination with radiation therapy in an orthotopic mouse prostate cancer model. Int J Radiat Oncol Biol Phys (2002) 54:873–85. doi: 10.1016/S0360-3016(02)03005-5
50. Rogulski KR, Wing MS, Paielli DL, Gilbert JD, Kim JH, Freytag SO. Double suicide gene therapy augments the antitumor activity of a replication-competent lytic adenovirus through enhanced cytotoxicity and radiosensitization. Hum Gene Ther (2000) 11:67–76. doi: 10.1089/10430340050016166
51. Freytag SO, Barton KN, Zhang Y. Efficacy of oncolytic adenovirus expressing suicide genes and interleukin-12 in preclinical model of prostate cancer. Gene Ther (2013) 20:1131–9. doi: 10.1038/gt.2013.40
52. Ranki T, Joensuu T, Jager E, Karbach J, Wahle C, Kairemo K, et al. Local treatment of a pleural mesothelioma tumor with ONCOS-102 induces a systemic antitumor CD8(+) T-cell response, prominent infiltration of CD8(+) lymphocytes and Th1 type polarization. Oncoimmunology (2014) 3:e958937. doi: 10.4161/21624011.2014.958937
53. Burke JM, Lamm DL, Meng MV, Nemunaitis JJ, Stephenson JJ, Arseneau JC, et al. A first in human phase 1 study of CG0070, a GM-CSF expressing oncolytic adenovirus, for the treatment of nonmuscle invasive bladder cancer. J Urol (2012) 188:2391–7. doi: 10.1016/j.juro.2012.07.097
54. Sanchala DS, Bhatt LK, Prabhavalkar KS. Oncolytic herpes simplex viral therapy: A stride toward selective targeting of cancer cells. Front Pharmacol (2017) 8:270. doi: 10.3389/fphar.2017.00270
55. Ma W, He H, Wang H. Oncolytic herpes simplex virus and immunotherapy. BMC Immunol (2018) 19:40. doi: 10.1186/s12865-018-0281-9
56. Nawa A, Luo C, Zhang L, Ushjima Y, Ishida D, Kamakura M, et al. Non-engineered, naturally oncolytic herpes simplex virus HSV1 HF-10: applications for cancer gene therapy. Curr Gene Ther (2008) 8:208–21. doi: 10.2174/156652308784746422
57. Eissa IR, Naoe Y, Bustos-Villalobos I, Ichinose T, Tanaka M, Zhiwen W, et al. Genomic signature of the natural oncolytic herpes simplex virus HF10 and its therapeutic role in preclinical and clinical trials. Front Oncol (2017) 7:149. doi: 10.3389/fonc.2017.00149
58. Papanastassiou V, Rampling R, Fraser M, Petty R, Hadley D, Nicoll J, et al. The potential for efficacy of the modified (ICP 34.5(-)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther (2002) 9:398–406. doi: 10.1038/sj.gt.3301664
59. Harrow S, Papanastassiou V, Harland J, Mabbs R, Petty R, Fraser M, et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther (2004) 11:1648–58. doi: 10.1038/sj.gt.3302289
60. Streby KA, Geller JI, Currier MA, Warren PS, Racadio JM, Towbin AJ, et al. Intratumoral injection of HSV1716, an oncolytic herpes virus, is safe and shows evidence of immune response and viral replication in young cancer patients. Clin Cancer Res Off J Am Assoc Cancer Res (2017) 23:3566–74. doi: 10.1158/1078-0432.CCR-16-2900
61. Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, et al. Phase ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther J Am Soc Gene Ther (2009) 17:199–207. doi: 10.1038/mt.2008.228
62. Taguchi S, Fukuhara H, Todo T. Oncolytic virus therapy in Japan: progress in clinical trials and future perspectives. Jpn J Clin Oncol (2019) 49:201–9. doi: 10.1093/jjco/hyy170
63. Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci (2016) 107:1373–9. doi: 10.1111/cas.13027
64. Otani Y, Yoo JY, Shimizu T, Kurozumi K, Date I, Kaur B. Implications of immune cells in oncolytic herpes simplex virotherapy for glioma. Brain Tumor Pathol (2022) 39:57–64. doi: 10.1007/s10014-022-00431-8
65. Parker JN, Meleth S, Hughes KB, Gillespie GY, Whitley RJ, Markert JM. Enhanced inhibition of syngeneic murine tumors by combinatorial therapy with genetically engineered HSV-1 expressing CCL2 and IL-12. Cancer Gene Ther (2005) 12:359–68. doi: 10.1038/sj.cgt.7700784
66. Pearl TM, Markert JM, Cassady KA, Ghonime MG. Oncolytic virus-based cytokine expression to improve immune activity in brain and solid tumors. Mol Ther Oncol (2019) 13:14–21. doi: 10.1016/j.omto.2019.03.001
67. Ino Y, Saeki Y, Fukuhara H, Todo T. Triple combination of oncolytic herpes simplex virus-1 vectors armed with interleukin-12, interleukin-18, or soluble B7-1 results in enhanced antitumor efficacy. Clin Cancer Res Off J Am Assoc Cancer Res (2006) 12:643–52. doi: 10.1158/1078-0432.CCR-05-1494
68. Cheema TA, Wakimoto H, Fecci PE, Ning J, Kuroda T, Jeyaretna DS, et al. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci United States America (2013) 110:12006–11. doi: 10.1073/pnas.1307935110
69. Varghese S, Rabkin SD, Liu R, Nielsen PG, Ipe T, Martuza RL. Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther (2006) 13:253–65. doi: 10.1038/sj.cgt.7700900
70. Patel DM, Foreman PM, Nabors LB, Riley KO, Gillespie GY, Markert JM. Design of a phase I clinical trial to evaluate M032, a genetically engineered HSV-1 expressing IL-12, in patients with Recurrent/Progressive glioblastoma multiforme, anaplastic astrocytoma, or gliosarcoma. Hum Gene Ther Clin Dev (2016) 27:69–78. doi: 10.1089/humc.2016.031
71. Lee SS, Eisenlohr LC, McCue PA, Mastrangelo MJ, Lattime EC. Intravesical gene therapy: in vivo gene transfer using recombinant vaccinia virus vectors. Cancer Res (1994) 54:3325–8.
72. Guo ZS, Lu B, Guo Z, Giehl E, Feist M, Dai E, et al. Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J immunother Cancer (2019) 7:6. doi: 10.1186/s40425-018-0495-7
73. Tolonen N, Doglio L, Schleich S, Krijnse Locker J. Vaccinia virus DNA replication occurs in endoplasmic reticulum-enclosed cytoplasmic mini-nuclei. Mol Biol Cell (2001) 12:2031–46. doi: 10.1091/mbc.12.7.2031
74. Smith GL, Vanderplasschen A, Law M. The formation and function of extracellular enveloped vaccinia virus. J Gen Virol (2002) 83:2915–31. doi: 10.1099/0022-1317-83-12-2915
75. Parato KA, Breitbach CJ, Le Boeuf F, Wang J, Storbeck C, Ilkow C, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther J Am Soc Gene Ther (2012) 20:749–58. doi: 10.1038/mt.2011.276
76. Mastrangelo MJ, Maguire HC Jr., Eisenlohr LC, Laughlin CE, Monken CE, McCue PA, et al. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther (1999) 6:409–22. doi: 10.1038/sj.cgt.7700066
77. Puhlmann M, Brown CK, Gnant M, Huang J, Libutti SK, Alexander HR, et al. Vaccinia as a vector for tumor-directed gene therapy: Biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther (2000) 7:66–73. doi: 10.1038/sj.cgt.7700075
78. Kim JH, Oh JY, Park BH, Lee DE, Kim JS, Park HE, et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol Ther J Am Soc Gene Ther (2006) 14:361–70. doi: 10.1016/j.ymthe.2006.05.008
79. Cripe TP, Ngo MC, Geller JI, Louis CU, Currier MA, Racadio JM, et al. Phase 1 study of intratumoral pexa-vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol Ther J Am Soc Gene Ther (2015) 23:602–8. doi: 10.1038/mt.2014.243
80. Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med (2013) 19:329–36. doi: 10.1038/nm.3089
81. Hwang TH, Moon A, Burke J, Ribas A, Stephenson J, Breitbach CJ, et al. A mechanistic proof-of-concept clinical trial with JX-594, a targeted multi-mechanistic oncolytic poxvirus, in patients with metastatic melanoma. Mol Ther J Am Soc Gene Ther (2011) 19:1913–22. doi: 10.1038/mt.2011.132
82. Kim MK, Breitbach CJ, Moon A, Heo J, Lee YK, Cho M, et al. Oncolytic and immunotherapeutic vaccinia induces antibody-mediated complement-dependent cancer cell lysis in humans. Sci Trans Med (2013) 5:185ra63. doi: 10.1126/scitranslmed.3005361
83. Zeh HJ, Downs-Canner S, McCart JA, Guo ZS, Rao UN, Ramalingam L, et al. First-in-man study of western reserve strain oncolytic vaccinia virus: safety, systemic spread, and antitumor activity. Mol Ther J Am Soc Gene Ther (2015) 23:202–14. doi: 10.1038/mt.2014.194
84. McCart JA, Puhlmann M, Lee J, Hu Y, Libutti SK, Alexander HR, et al. Complex interactions between the replicating oncolytic effect and the enzyme/prodrug effect of vaccinia-mediated tumor regression. Gene Ther (2000) 7:1217–23. doi: 10.1038/sj.gt.3301237
85. McCart JA, Mehta N, Scollard D, Reilly RM, Carrasquillo JA, Tang N, et al. Oncolytic vaccinia virus expressing the human somatostatin receptor SSTR2: molecular imaging after systemic delivery using 111In-pentetreotide. Mol Ther J Am Soc Gene Ther (2004) 10:553–61. doi: 10.1016/j.ymthe.2004.06.158
86. Chalikonda S, Kivlen MH, O'Malley ME, Eric Dong XD, McCart JA, Gorry MC, et al. Oncolytic virotherapy for ovarian carcinomatosis using a replication-selective vaccinia virus armed with a yeast cytosine deaminase gene. Cancer Gene Ther (2008) 15:115–25. doi: 10.1038/sj.cgt.7701110
87. Downs-Canner S, Guo ZS, Ravindranathan R, Breitbach CJ, O'Malley ME, Jones HL, et al. Phase 1 study of intravenous oncolytic poxvirus (vvDD) in patients with advanced solid cancers. Mol Ther J Am Soc Gene Ther (2016) 24:1492–501. doi: 10.1038/mt.2016.101
88. Mell LK, Brumund KT, Daniels GA, Advani SJ, Zakeri K, Wright ME, et al. Phase I trial of intravenous oncolytic vaccinia virus (GL-ONC1) with cisplatin and radiotherapy in patients with locoregionally advanced head and neck carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res (2017) 23:5696–702. doi: 10.1158/1078-0432.CCR-16-3232
89. Lauer UM, Schell M, Beil J, Berchtold S, Koppenhöfer U, Glatzle J, et al. Phase I study of oncolytic vaccinia virus GL-ONC1 in patients with peritoneal carcinomatosis. Clin Cancer Res Off J Am Assoc Cancer Res (2018) 24:4388–98. doi: 10.1158/1078-0432.CCR-18-0244
90. Macedo N, Miller DM, Haq R, Kaufman HL. Clinical landscape of oncolytic virus research in 2020. J Immunother Cancer (2020) 8:e001486. doi: 10.1136/jitc-2020-001486
91. McCarthy C, Jayawardena N, Burga LN, Bostina M. Developing picornaviruses for cancer therapy. Cancers (Basel) (2019) 11:685. doi: 10.3390/cancers11050685
92. Yang L, Gu X, Yu J, Ge S, Fan X. Oncolytic virotherapy: From bench to bedside. Front Cell Dev Biol (2021) 9:790150. doi: 10.3389/fcell.2021.790150
93. Melero I, Castanon E, Alvarez M, Champiat S, Marabelle A. Intratumoural administration and tumour tissue targeting of cancer immunotherapies. Nat Rev Clin Oncol (2021) 18:558–76. doi: 10.1038/s41571-021-00507-y
94. Morris DG, Feng X, DiFrancesco LM, Fonseca K, Forsyth PA, Paterson AH, et al. REO-001: A phase I trial of percutaneous intralesional administration of reovirus type 3 dearing (Reolysin®) in patients with advanced solid tumors. Invest New Drugs (2013) 31:696–706. doi: 10.1007/s10637-012-9865-z
95. Gollamudi R, Ghalib MH, Desai KK, Chaudhary I, Wong B, Einstein M, et al. Intravenous administration of reolysin, a live replication competent RNA virus is safe in patients with advanced solid tumors. Invest New Drugs (2010) 28:641–9. doi: 10.1007/s10637-009-9279-8
96. Karaca G, Hargett D, McLean TI, Aguilar JS, Ghazal P, Wagner EK, et al. Inhibition of the stress-activated kinase, p38, does not affect the virus transcriptional program of herpes simplex virus type 1. Virology (2004) 329:142–56. doi: 10.1016/j.virol.2004.08.020
97. Müller L, Berkeley R, Barr T, Ilett E, Errington-Mais F. Past, present and future of oncolytic reovirus. Cancers (Basel) (2020) 12:3219. doi: 10.3390/cancers12113219
98. Mahalingam D, Fountzilas C, Moseley J, Noronha N, Tran H, Chakrabarty R, et al. A phase II study of REOLYSIN(®) (pelareorep) in combination with carboplatin and paclitaxel for patients with advanced malignant melanoma. Cancer Chemother Pharmacol (2017) 79:697–703. doi: 10.1007/s00280-017-3260-6
99. Jonker DJ, Tang PA, Kennecke H, Welch SA, Cripps MC, Asmis T, et al. A randomized phase II study of FOLFOX6/Bevacizumab with or without pelareorep in patients with metastatic colorectal cancer: IND.210, a Canadian cancer trials group trial. Clin Colorectal Cancer (2018) 17:231–239.e7. doi: 10.1016/j.clcc.2018.03.001
100. Bradbury PA, Morris DG, Nicholas G, Tu D, Tehfe M, Goffin JR, et al. Canadian Cancer trials group (CCTG) IND211: A randomized trial of pelareorep (Reolysin) in patients with previously treated advanced or metastatic non-small cell lung cancer receiving standard salvage therapy. Lung Cancer (2018) 120:142–8. doi: 10.1016/j.lungcan.2018.03.005
101. Cohn DE, Sill MW, Walker JL, O'Malley D, Nagel CI, Rutledge TL, et al. Randomized phase IIB evaluation of weekly paclitaxel versus weekly paclitaxel with oncolytic reovirus (Reolysin®) in recurrent ovarian, tubal, or peritoneal cancer: An NRG Oncology/Gynecologic oncology group study. Gynecol Oncol (2017) 146:477–83. doi: 10.1016/j.ygyno.2017.07.135
102. Noonan AM, Farren MR, Geyer SM, Huang Y, Tahiri S, Ahn D, et al. Randomized phase 2 trial of the oncolytic virus pelareorep (Reolysin) in upfront treatment of metastatic pancreatic adenocarcinoma. Mol Ther (2016) 24:1150–8. doi: 10.1038/mt.2016.66
103. Bernstein V, Ellard SL, Dent SF, Tu D, Mates M, Dhesy-Thind SK, et al. A randomized phase II study of weekly paclitaxel with or without pelareorep in patients with metastatic breast cancer: final analysis of Canadian cancer trials group IND.213. Breast Cancer Res Treat (2018) 167:485–93. doi: 10.1007/s10549-017-4538-4
104. Mahalingam D, Wilkinson GA, Eng KH, Fields P, Raber P, Moseley JL, et al. Pembrolizumab in combination with the oncolytic virus pelareorep and chemotherapy in patients with advanced pancreatic adenocarcinoma: A phase ib study. Clin Cancer Res (2020) 26:71–81. doi: 10.1158/1078-0432.CCR-19-2078
105. Mohammad NS, Nazli R, Zafar H, Fatima S. Effects of lipid based multiple micronutrients supplement on the birth outcome of underweight pre-eclamptic women: A randomized clinical trial. Pak J Med Sci (2022) 38:219–26. doi: 10.12669/pjms.38.1.4396
106. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
107. Manso L, Salvador F, Villagrasa P, Chic N, Bermejo B, Cejalvo JM, et al. Abstract CT191: A window-of-opportunity study with atezolizumab and the oncolytic virus pelareorep in early breast cancer (AWARE-1). Cancer Res (2021) 81:CT191–1. doi: 10.1158/1538-7445.AM2021-CT191
108. Bluming AZ, Ziegler JL. Regression of burkitt's lymphoma in association with measles infection. Lancet (1971) 2:105–6. doi: 10.1016/S0140-6736(71)92086-1
109. Heinzerling L, Künzi V, Oberholzer PA, Kündig T, Naim H, Dummer R. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon-resistant tumor cells. Blood (2005) 106:2287–94. doi: 10.1182/blood-2004-11-4558
110. Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res (2010) 70:875–82. doi: 10.1158/0008-5472.CAN-09-2762
111. Dispenzieri A, Tong C, LaPlant B, Lacy MQ, Laumann K, Dingli D, et al. Phase I trial of systemic administration of edmonston strain of measles virus genetically engineered to express the sodium iodide symporter in patients with recurrent or refractory multiple myeloma. Leukemia (2017) 31:2791–8. doi: 10.1038/leu.2017.120
112. Packiriswamy N, Upreti D, Zhou Y, Khan R, Miller A, Diaz RM, et al. Oncolytic measles virus therapy enhances tumor antigen-specific T-cell responses in patients with multiple myeloma. Leukemia (2020) 34:3310–22. doi: 10.1038/s41375-020-0828-7
113. Guo J, Xiao Y, Iyer R, Lu X, Lake M, Ladror U, et al. Empowering therapeutic antibodies with IFN-α for cancer immunotherapy. PloS One (2019) 14:e0219829. doi: 10.1371/journal.pone.0219829
114. Opyrchal M, Allen C, Iankov I, Aderca I, Schroeder M, Sarkaria J, et al. Effective radiovirotherapy for malignant gliomas by using oncolytic measles virus strains encoding the sodium iodide symporter (MV-NIS). Hum Gene Ther (2012) 23:419–27. doi: 10.1089/hum.2011.158
115. Panagioti E, Kurokawa C, Viker K, Ammayappan A, Anderson SK, Sotiriou S, et al. Immunostimulatory bacterial antigen-armed oncolytic measles virotherapy significantly increases the potency of anti-PD1 checkpoint therapy. J Clin Invest (2021) 131:e141614. doi: 10.1172/JCI141614
116. Kelly CM, Antonescu CR, Bowler T, Munhoz R, Chi P, Dickson MA, et al. Objective response rate among patients with locally advanced or metastatic sarcoma treated with talimogene laherparepvec in combination with pembrolizumab: A phase 2 clinical trial. JAMA Oncol (2020) 6:402–8. doi: 10.1001/jamaoncol.2019.6152
117. Dobrikova EY, Goetz C, Walters RW, Lawson SK, Peggins JO, Muszynski K, et al. Attenuation of neurovirulence, biodistribution, and shedding of a poliovirus:rhinovirus chimera after intrathalamic inoculation in macaca fascicularis. J Virol (2012) 86:2750–9. doi: 10.1128/JVI.06427-11
118. Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med (2018) 379:150–61. doi: 10.1056/NEJMoa1716435
119. Beasley GM, Nair SK, Farrow NE, Landa K, Selim MA, Wiggs CA, et al. Phase I trial of intratumoral PVSRIPO in patients with unresectable, treatment-refractory melanoma. J Immunother Cancer (2021) 9:e002203. doi: 10.1136/jitc-2020-002203
120. Annels NE, Mansfield D, Arif M, Ballesteros-Merino C, Simpson GR, Denyer M, et al. Phase I trial of an ICAM-1-Targeted immunotherapeutic-coxsackievirus A21 (CVA21) as an oncolytic agent against non muscle-invasive bladder cancer. Clin Cancer Res (2019) 25:5818–31. doi: 10.1158/1078-0432.CCR-18-4022
121. Vainer ED, Meir K, Furman M, Semenenko I, Konikoff F, Vainer GW. Characterization of novel CD55 isoforms expression in normal and neoplastic tissues. Tissue Antigens (2013) 82:26–34. doi: 10.1111/tan.12138
122. Kotteas EA, Boulas P, Gkiozos I, Tsagkouli S, Tsoukalas G, Syrigos KN. The intercellular cell adhesion molecule-1 (icam-1) in lung cancer: implications for disease progression and prognosis. Anticancer Res (2014) 34:4665–72.
123. Rosette C, Roth RB, Oeth P, Braun A, Kammerer S, Ekblom J, et al. Role of ICAM1 in invasion of human breast cancer cells. Carcinogenesis (2005) 26:943–50. doi: 10.1093/carcin/bgi070
124. Lim EJ, Kang JH, Kim YJ, Kim S, Lee SJ. ICAM-1 promotes cancer progression by regulating SRC activity as an adapter protein in colorectal cancer. Cell Death Dis (2022) 13:417. doi: 10.1038/s41419-022-04862-1
125. Benedicto A, Romayor I, Arteta B. Role of liver ICAM-1 in metastasis. Oncol Lett (2017) 14:3883–92. doi: 10.3892/ol.2017.6700
126. Kageshita T, Yoshii A, Kimura T, Kuriya N, Ono T, Tsujisaki M, et al. Clinical relevance of ICAM-1 expression in primary lesions and serum of patients with malignant melanoma. Cancer Res (1993) 53:4927–32. doi: 10.1016/0923-1811(93)90825-A
127. Andtbacka RHI, Curti B, Daniels GA, Hallmeyer S, Whitman ED, Lutzky J, et al. Clinical responses of oncolytic coxsackievirus A21 (V937) in patients with unresectable melanoma. J Clin Oncol (2021) 39:3829–38. doi: 10.1200/JCO.20.03246
128. Pluskota E, D'Souza SE. Fibrinogen interactions with ICAM-1 (CD54) regulate endothelial cell survival. Eur J Biochem (2000) 267:4693–704. doi: 10.1046/j.1432-1327.2000.01520.x
129. van Den Engel NK, Heidenthal E, Vinke A, Kolb H, Martin S. Circulating forms of intercellular adhesion molecule (ICAM)-1 in mice lacking membranous ICAM-1. Blood (2000) 95:1350–5. doi: 10.1182/blood.V95.4.1350.004k07_1350_1355
130. Läubli H, Balmelli C, Bossard M, Pfister O, Glatz K, Zippelius A. Acute heart failure due to autoimmune myocarditis under pembrolizumab treatment for metastatic melanoma. J Immunother Cancer (2015) 3:11. doi: 10.1186/s40425-015-0057-1
131. Osińska I, Popko K, Demkow U. Perforin: an important player in immune response. Cent Eur J Immunol (2014) 39:109–15. doi: 10.5114/ceji.2014.42135
132. Silk AW, Kaufman H, Gabrail N, Mehnert J, Bryan J, Norrell J, et al. Abstract CT026: Phase 1b study of intratumoral coxsackievirus A21 (CVA21) and systemic pembrolizumab in advanced melanoma patients: Interim results of the CAPRA clinical trial. Cancer Res (2017) 77:CT026–6. doi: 10.1158/1538-7445.AM2017-CT026
133. Curti B, Richards J, Hyngstrom J, Daniels G, Faries M, Feun L, et al. 381 intratumoral oncolytic virus V937 plus ipilimumab in patients with advanced melanoma: the phase 1b MITCI study. J ImmunoTher Cancer (2021) 9:A415. doi: 10.1136/jitc-2021-SITC2021.381
134. Cihoric N, Tsikkinis A, Filipovic N, Jeremic B. Treatment options for isolated locoregional recurrences of nonsmall cell lung cancer after surgery: yes, radiation therapy too! Eur Respir J (2016) 48:276–8. doi: 10.1183/13993003.00388-2016
135. Chandra RA, Kachnic LA, Thomas CR Jr. Contemporary topics in radiation medicine, part II: Disease sites. Hematol Oncol Clinics (2020) 34. doi: 10.1016/S0889-8588(19)30142-X
136. Brizel DM, Albers ME, Fisher SR, Scher RL, Richtsmeier WJ, Hars V, et al. Hyperfractionated irradiation with or without concurrent chemotherapy for locally advanced head and neck cancer. New Engl J Med (1998) 338:1798–804. doi: 10.1056/NEJM199806183382503
137. Isles MG, McConkey C, Mehanna HM. A systematic review and meta-analysis of the role of positron emission tomography in the follow up of head and neck squamous cell carcinoma following radiotherapy or chemoradiotherapy. Clin Otolaryngol Off J ENT-UK ; Off J Netherlands Soc Oto-Rhino-Laryngol Cervico-Facial Surg (2008) 33:210–22. doi: 10.1111/j.1749-4486.2008.01688.x
138. Taccioli GE, Amatucci AG, Beamish HJ, Gell D, Xiang XH, Torres Arzayus MI, et al. Targeted disruption of the catalytic subunit of the DNA-PK gene in mice confers severe combined immunodeficiency and radiosensitivity. Immunity (1998) 9:355–66. doi: 10.1016/S1074-7613(00)80618-4
139. Collis SJ, DeWeese TL, Jeggo PA, Parker AR. The life and death of DNA-PK. Oncogene (2005) 24:949–61. doi: 10.1038/sj.onc.1208332
140. Lupold SE, Rodriguez R. Adenoviral gene therapy, radiation, and prostate cancer. Rev Urol (2005) 7:193–202.
141. Freytag SO, Movsas B, Aref I, Stricker H, Peabody J, Pegg J, et al. Phase I trial of replication-competent adenovirus-mediated suicide gene therapy combined with IMRT for prostate cancer. Mol Ther J Am Soc Gene Ther (2007) 15:1016–23. doi: 10.1038/mt.sj.6300120
142. Zhang M, Li S, Li J, Ensminger WD, Lawrence TS. Ionizing radiation increases adenovirus uptake and improves transgene expression in intrahepatic colon cancer xenografts. Mol Ther J Am Soc Gene Ther (2003) 8:21–8. doi: 10.1016/S1525-0016(03)00143-6
143. Chen Y, DeWeese T, Dilley J, Zhang Y, Li Y, Ramesh N, et al. CV706, a prostate cancer-specific adenovirus variant, in combination with radiotherapy produces synergistic antitumor efficacy without increasing toxicity. Cancer Res (2001) 61:5453–60.
144. Bieler A, Mantwill K, Holzmuller R, Jurchott K, Kaszubiak A, Stark S, et al. Impact of radiation therapy on the oncolytic adenovirus dl520: Implications on the treatment of glioblastoma. Radiother Oncol J Eur Soc Ther Radiol Oncol (2008) 86:419–27. doi: 10.1016/j.radonc.2007.10.009
145. Lamfers ML, Idema S, Bosscher L, Heukelom S, Moeniralm S, van der Meulen-Muileman IH, et al. Differential effects of combined Ad5- delta 24RGD and radiation therapy in in vitro versus in vivo models of malignant glioma. Clin Cancer Res Off J Am Assoc Cancer Res (2007) 13:7451–8. doi: 10.1158/1078-0432.CCR-07-1265
146. Ottolino-Perry K, Diallo JS, Lichty BD, Bell JC, McCart JA. Intelligent design: combination therapy with oncolytic viruses. Mol Ther J Am Soc Gene Ther (2010) 18:251–63. doi: 10.1038/mt.2009.283
147. Hingorani M, White CL, Zaidi S, Merron A, Peerlinck I, Gore ME, et al. Radiation-mediated up-regulation of gene expression from replication-defective adenoviral vectors: Implications for sodium iodide symporter gene therapy. Clin Cancer Res Off J Am Assoc Cancer Res (2008) 14:4915–24. doi: 10.1158/1078-0432.CCR-07-4049
148. Qian J, Yang J, Dragovic AF, Abu-Isa E, Lawrence TS, Zhang M. Ionizing radiation-induced adenovirus infection is mediated by dynamin 2. Cancer Res (2005) 65:5493–7. doi: 10.1158/0008-5472.CAN-04-4526
149. Martin-Duque P, Sanchez-Prieto R, Romero J, Martinez-Lamparero A, Cebrian-Sagarriga S, Guinea-Viniegra J, et al. In vivo radiosensitizing effect of the adenovirus E1A gene in murine and human malignant tumors. Int J Oncol (1999) 15:1163–8. doi: 10.3892/ijo.15.6.1163
150. Lamfers ML, Grill J, Dirven CM, Van Beusechem VW, Geoerger B, Van Den Berg J, et al. Potential of the conditionally replicative adenovirus Ad5-Delta24RGD in the treatment of malignant gliomas and its enhanced effect with radiotherapy. Cancer Res (2002) 62:5736–42.
151. Idema S, Lamfers ML, van Beusechem VW, Noske DP, Heukelom S, Moeniralm S, et al. AdDelta24 and the p53-expressing variant AdDelta24-p53 achieve potent anti-tumor activity in glioma when combined with radiotherapy. J Gene Med (2007) 9:1046–56. doi: 10.1002/jgm.1113
152. Geoerger B, Grill J, Opolon P, Morizet J, Aubert G, Lecluse Y, et al. Potentiation of radiation therapy by the oncolytic adenovirus dl1520 (ONYX-015) in human malignant glioma xenografts. Br J Cancer (2003) 89:577–84. doi: 10.1038/sj.bjc.6601102
153. Kim J, Kim PH, Yoo JY, Yoon AR, Choi HJ, Seong J, et al. Double E1B 19 kDa- and E1B 55 kDa-deleted oncolytic adenovirus in combination with radiotherapy elicits an enhanced anti-tumor effect. Gene Ther (2009) 16:1111–21. doi: 10.1038/gt.2009.72
154. Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature (2002) 418:348–52. doi: 10.1038/nature00863
155. Fujita T, Teh BS, Timme TL, Mai WY, Satoh T, Kusaka N, et al. Sustained long-term immune responses after in situ gene therapy combined with radiotherapy and hormonal therapy in prostate cancer patients. Int J Radiat Oncol Biol Phys (2006) 65:84–90. doi: 10.1016/j.ijrobp.2005.11.009
156. Kim W, Seong J, Oh HJ, Koom WS, Choi KJ, Yun CO. A novel combination treatment of armed oncolytic adenovirus expressing IL-12 and GM-CSF with radiotherapy in murine hepatocarcinoma. J Radiat Res (2011) 52:646–54. doi: 10.1269/jrr.10185
157. Evgin L, Vile RG. Parking CAR T cells in tumours: Oncolytic viruses as valets or vandals? Cancers (Basel) (2021) 13:1106. doi: 10.3390/cancers13051106
158. Gillory LA, Megison ML, Stewart JE, Mroczek-Musulman E, Nabers HC, Waters AM, et al. Preclinical evaluation of engineered oncolytic herpes simplex virus for the treatment of neuroblastoma. PloS One (2013) 8:e77753. doi: 10.1371/journal.pone.0077753
159. Mezhir JJ, Advani SJ, Smith KD, Darga TE, Poon AP, Schmidt H, et al. Ionizing radiation activates late herpes simplex virus 1 promoters via the p38 pathway in tumors treated with oncolytic viruses. Cancer Res (2005) 65:9479–84. doi: 10.1158/0008-5472.CAN-05-1927
160. Stanziale SF, Petrowsky H, Joe JK, Roberts GD, Zager JS, Gusani NJ, et al. Ionizing radiation potentiates the antitumor efficacy of oncolytic herpes simplex virus G207 by upregulating ribonucleotide reductase. Surgery (2002) 132:353–9. doi: 10.1067/msy.2002.125715
161. Franco-Lie I, Iversen T, Tretli S, Kringlen E, Berg K, Abdelnoor M. Malignant melanoma of the skin: Risk, tumour characteristics and mortality in adult twins born in Norway between 1905 and 1945 - a cohort study. Melanoma Res (2005) 15:461–6. doi: 10.1097/00008390-200510000-00016
162. Harrington KJ, Hingorani M, Tanay MA, Hickey J, Bhide SA, Clarke PM, et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin Cancer Res Off J Am Assoc Cancer Res (2010) 16:4005–15. doi: 10.1158/1078-0432.CCR-10-0196
163. Markert JM, Razdan SN, Kuo HC, Cantor A, Knoll A, Karrasch M, et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther J Am Soc Gene Ther (2014) 22:1048–55. doi: 10.1038/mt.2014.22
164. Kyula JN, Khan AA, Mansfield D, Karapanagiotou EM, McLaughlin M, Roulstone V, et al. Synergistic cytotoxicity of radiation and oncolytic Lister strain vaccinia in (V600D/E)BRAF mutant melanoma depends on JNK and TNF-α signaling. Oncogene (2014) 33:1700–12. doi: 10.1038/onc.2013.112
165. Wilkinson MJ, Smith HG, McEntee G, Kyula-Currie J, Pencavel TD, Mansfield DC, et al. Oncolytic vaccinia virus combined with radiotherapy induces apoptotic cell death in sarcoma cells by down-regulating the inhibitors of apoptosis. Oncotarget (2016) 7:81208–22. doi: 10.18632/oncotarget.12820
166. Mansfield D, Pencavel T, Kyula JN, Zaidi S, Roulstone V, Thway K, et al. Oncolytic vaccinia virus and radiotherapy in head and neck cancer. Oral Oncol (2013) 49:108–18. doi: 10.1016/j.oraloncology.2012.07.019
167. Advani SJ, Buckel L, Chen NG, Scanderbeg DJ, Geissinger U, Zhang Q, et al. Preferential replication of systemically delivered oncolytic vaccinia virus in focally irradiated glioma xenografts. Clin Cancer Res Off J Am Assoc Cancer Res (2012) 18:2579–90. doi: 10.1158/1078-0432.CCR-11-2394
168. Guo ZS, Liu Z, Sathaiah M, Wang J, Ravindranathan R, Kim E, et al. Rapid generation of multiple loci-engineered marker-free poxvirus and characterization of a clinical-grade oncolytic vaccinia virus. molecular therapy. Methods Clin Dev (2017) 7:112–22. doi: 10.1016/j.omtm.2017.09.007
169. Schirrmacher V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int J Oncol (2019) 54:407–19. doi: 10.3892/ijo.2018.4661
170. Fisher B, Wolmark N. The current status of systemic adjuvant therapy in the management of primary breast cancer. Surg Clinics North America (1981) 61:1347–60. doi: 10.1016/S0039-6109(16)42589-2
171. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer (2012) 12:860–75. doi: 10.1038/nrc3380
172. Wang Q, Ju X, Wang J, Fan Y, Ren M, Zhang H. Immunogenic cell death in anticancer chemotherapy and its impact on clinical studies. Cancer Lett (2018) 438:17–23. doi: 10.1016/j.canlet.2018.08.028
173. Liu P, Zhao L, Kepp O, Kroemer G. Quantitation of calreticulin exposure associated with immunogenic cell death. Methods Enzymol (2020) 632:1–13. doi: 10.1016/bs.mie.2019.05.011
174. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13:54–61. doi: 10.1038/nm1523
175. Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ (2014) 21:79–91. doi: 10.1038/cdd.2013.75
176. Solari JIG, Filippi-Chiela E, Pilar ES, Nunes V, Gonzalez EA, Figueiró F, et al. Damage-associated molecular patterns (DAMPs) related to immunogenic cell death are differentially triggered by clinically relevant chemotherapeutics in lung adenocarcinoma cells. BMC Cancer (2020) 20:474. doi: 10.1186/s12885-020-06964-5
177. Krysko O, Løve Aaes T, Bachert C, Vandenabeele P, Krysko DV. Many faces of DAMPs in cancer therapy. Cell Death Dis (2013) 4:e631. doi: 10.1038/cddis.2013.156
178. Cherubini G, Petouchoff T, Grossi M, Piersanti S, Cundari E, Saggio I. E1B55K-deleted adenovirus (ONYX-015) overrides G1/S and G2/M checkpoints and causes mitotic catastrophe and endoreduplication in p53-proficient normal cells. Cell Cycle (Georgetown Tex.) (2006) 5:2244–52. doi: 10.4161/cc.5.19.3263
179. Zhu H, Zhang L, Huang X, Davis JJ, Jacob DA, Teraishi F, et al. Overcoming acquired resistance to TRAIL by chemotherapeutic agents and calpain inhibitor I through distinct mechanisms. Mol Ther J Am Soc Gene Ther (2004) 9:666–73. doi: 10.1016/j.ymthe.2004.02.007
180. Bernt KM, Steinwaerder DS, Ni S, Li ZY, Roffler SR, Lieber A. Enzyme-activated prodrug therapy enhances tumor-specific replication of adenovirus vectors. Cancer Res (2002) 62:6089–98.
181. Wong ML, Hsu MT. Involvement of topoisomerases in replication, transcription, and packaging of the linear adenovirus genome. J Virol (1990) 64:691–9. doi: 10.1128/jvi.64.2.691-699.1990
182. Khuri FR, Nemunaitis J, Ganly I, Arseneau J, Tannock IF, Romel L, et al. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat Med (2000) 6:879–85. doi: 10.1038/78638
183. Hassan MAIA, Braam SR, Kruyt FAE. Paclitaxel and vincristine potentiate adenoviral oncolysis that is associated with cell cycle and apoptosis modulation, whereas they differentially affect the viral life cycle in non-small-cell lung cancer cells. Cancer Gene Ther (2006) 13:1105–14. doi: 10.1038/sj.cgt.7700984
184. Seidman MA, Hogan SM, Wendland RL, Worgall S, Crystal RG, Leopold PL. Variation in adenovirus receptor expression and adenovirus vector-mediated transgene expression at defined stages of the cell cycle. Mol Ther (2001) 4:13–21. doi: 10.1006/mthe.2001.0414
185. Xia ZJ, Chang JH, Zhang L, Jiang WQ, Guan ZZ, Liu JW, et al. [Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus]. Ai Zheng = Aizheng = Chin J Cancer (2004) 23:1666–70.
186. Kuryk L, Haavisto E, Garofalo M, Capasso C, Hirvinen M, Pesonen S, et al. Synergistic anti-tumor efficacy of immunogenic adenovirus ONCOS-102 (Ad5/3-D24-GM-CSF) and standard of care chemotherapy in preclinical mesothelioma model. Int J Cancer (2016) 139:1883–93. doi: 10.1002/ijc.30228
187. Knaebel HP, Marten A, Schmidt J, Hoffmann K, Seiler C, Lindel K, et al. Phase III trial of postoperative cisplatin, interferon alpha-2b, and 5-FU combined with external radiation treatment versus 5-FU alone for patients with resected pancreatic adenocarcinoma – CapRI: study protocol [ISRCTN62866759]. BMC Cancer (2005) 5:37. doi: 10.1186/1471-2407-5-37
188. Picozzi VJ, Abrams RA, Decker PA, Traverso W, O'Reilly EM, Greeno E, et al. Multicenter phase II trial of adjuvant therapy for resected pancreatic cancer using cisplatin, 5-fluorouracil, and interferon-alfa-2b-based chemoradiation: ACOSOG trial Z05031. Ann Oncol Off J Eur Soc Med Oncol (2011) 22:348–54. doi: 10.1093/annonc/mdq384
189. Salzwedel AO, Han J, LaRocca CJ, Shanley R, Yamamoto M, Davydova J. Combination of interferon-expressing oncolytic adenovirus with chemotherapy and radiation is highly synergistic in hamster model of pancreatic cancer. Oncotarget (2018) 9:18041–52. doi: 10.18632/oncotarget.24710
190. Chahlavi A, Todo T, Martuza RL, Rabkin SD. Replication-competent herpes simplex virus vector G207 and cisplatin combination therapy for head and neck squamous cell carcinoma. Neoplasia (1999) 1:162–9. doi: 10.1038/sj.neo.7900016
191. Toyoizumi T, Mick R, Abbas AE, Kang EH, Kaiser LR, Molnar-Kimber KL. Combined therapy with chemotherapeutic agents and herpes simplex virus type 1 ICP34.5 mutant (HSV-1716) in human non-small cell lung cancer. Hum Gene Ther (1999) 10:3013–29. doi: 10.1089/10430349950016410
192. Cheema TA, Kanai R, Kim GW, Wakimoto H, Passer B, Rabkin SD, et al. Enhanced antitumor efficacy of low-dose etoposide with oncolytic herpes simplex virus in human glioblastoma stem cell xenografts. Clin Cancer Res Off J Am Assoc Cancer Res (2011) 17:7383–93. doi: 10.1158/1078-0432.CCR-11-1762
193. Aghi M, Rabkin S, Martuza RL. Effect of chemotherapy-induced DNA repair on oncolytic herpes simplex viral replication. J Natl Cancer Inst (2006) 98:38–50. doi: 10.1093/jnci/djj003
194. Ning J, Wakimoto H. Oncolytic herpes simplex virus-based strategies: toward a breakthrough in glioblastoma therapy. Front Microbiol (2014) 5:303. doi: 10.3389/fmicb.2014.00303
195. Coukos G, Makrigiannakis A, Kang EH, Rubin SC, Albelda SM, Molnar-Kimber KL. Oncolytic herpes simplex virus-1 lacking ICP34.5 induces p53-independent death and is efficacious against chemotherapy-resistant ovarian cancer. Clin Cancer Res Off J Am Assoc Cancer Res (2000) 6:3342–53.
196. Kanai R, Wakimoto H, Cheema T, Rabkin SD. Oncolytic herpes simplex virus vectors and chemotherapy: Are combinatorial strategies more effective for cancer? Future Oncol (2010) 6:619–34. doi: 10.2217/fon.10.18
197. Adusumilli PS, Chan MK, Chun YS, Hezel M, Chou TC, Rusch VW, et al. Cisplatin-induced GADD34 upregulation potentiates oncolytic viral therapy in the treatment of malignant pleural mesothelioma. Cancer Biol Ther (2006) 5:48–53. doi: 10.4161/cbt.5.1.2237
198. Nakano K, Todo T, Zhao G, Yamaguchi K, Kuroki S, Cohen JB, et al. Enhanced efficacy of conditionally replicating herpes simplex virus (G207) combined with 5-fluorouracil and surgical resection in peritoneal cancer dissemination models. J Gene Med (2005) 7:638–48. doi: 10.1002/jgm.700
199. Petrowsky H, Roberts GD, Kooby DA, Burt BM, Bennett JJ, Delman KA, et al. Functional interaction between fluorodeoxyuridine-induced cellular alterations and replication of a ribonucleotide reductase-negative herpes simplex virus. J Virol (2001) 75:7050–8. doi: 10.1128/JVI.75.15.7050-7058.2001
200. Bennett JJ, Adusumilli P, Petrowsky H, Burt BM, Roberts G, Delman KA, et al. Up-regulation of GADD34 mediates the synergistic anticancer activity of mitomycin c and a gamma134.5 deleted oncolytic herpes virus (G207). FASEB J Off Publ Fed Am Soc Exp Biol (2004) 18:1001–3. doi: 10.1096/fj.03-0610com
201. Geevarghese SK, Geller DA, de Haan HA, Hörer M, Knoll AE, Mescheder A, et al. Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum Gene Ther (2010) 21:1119–28. doi: 10.1089/hum.2010.020
202. Hirooka Y, Kasuya H, Ishikawa T, Kawashima H, Ohno E, Villalobos IB, et al. A phase I clinical trial of EUS-guided intratumoral injection of the oncolytic virus, HF10 for unresectable locally advanced pancreatic cancer. BMC Cancer (2018) 18:596. doi: 10.1186/s12885-018-4453-z
203. Watanabe I, Kasuya H, Nomura N, Shikano T, Shirota T, Kanazumi N, et al. Effects of tumor selective replication-competent herpes viruses in combination with gemcitabine on pancreatic cancer. Cancer Chemother Pharmacol (2008) 61:875–82. doi: 10.1007/s00280-007-0567-8
204. Kulu Y, Kawasaki H, Donahue JM, Kasuya H, Cusack JC, Choi EW, et al. Concurrent chemotherapy inhibits herpes simplex virus-1 replication and oncolysis. Cancer Gene Ther (2013) 20:133–40. doi: 10.1038/cgt.2012.97
205. Ottolino-Perry K, Acuna SA, Angarita FA, Sellers C, Zerhouni S, Tang N, et al. Oncolytic vaccinia virus synergizes with irinotecan in colorectal cancer. Mol Oncol (2015) 9:1539–52. doi: 10.1016/j.molonc.2015.04.009
206. Park SH, Breitbach CJ, Lee J, Park JO, Lim HY, Kang WK, et al. Phase 1b trial of biweekly intravenous pexa-vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus in colorectal cancer. Mol Ther J Am Soc Gene Ther (2015) 23:1532–40. doi: 10.1038/mt.2015.109
207. Sacks D, Baxter B, Campbell BCV, Carpenter JS, Cognard C, Dippel D, et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int J Stroke Off J Int Stroke Soc (2018) 13:612–32. doi: 10.1016/j.jvir.2017.11.026
208. Huang B, Sikorski R, Kirn DH, Thorne SH. Synergistic anti-tumor effects between oncolytic vaccinia virus and paclitaxel are mediated by the IFN response and HMGB1. Gene Ther (2011) 18:164–72. doi: 10.1038/gt.2010.121
209. Binz E, Berchtold S, Beil J, Schell M, Geisler C, Smirnow I, et al. Chemovirotherapy of pancreatic adenocarcinoma by combining oncolytic vaccinia virus GLV-1h68 with nab-paclitaxel plus gemcitabine. Mol Ther Oncol (2017) 6:10–21. doi: 10.1016/j.omto.2017.04.001
210. Chen W, Fan W, Ru G, Huang F, Lu X, Zhang X, et al. Gemcitabine combined with an engineered oncolytic vaccinia virus exhibits a synergistic suppressive effect on the tumor growth of pancreatic cancer. Oncol Rep (2019) 41:67–76. doi: 10.3892/or.2018.6817
211. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J (2021) 11:69. doi: 10.1038/s41408-021-00459-7
212. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell (2017) 170:1109–1119.e10. doi: 10.1016/j.cell.2017.08.027
213. Liu YT, Sun ZJ. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics (2021) 11:5365–86. doi: 10.7150/thno.58390
214. Schirrmacher V. Cancer vaccines and oncolytic viruses exert profoundly lower side effects in cancer patients than other systemic therapies: A comparative analysis. Biomedicines (2020) 8:61. doi: 10.3390/biomedicines8030061
215. Chesney J, Puzanov I, Collichio F, Singh P, Milhem MM, Glaspy J, et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol (2018) 36:1658–67. doi: 10.1200/JCO.2017.73.7379
216. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. New Engl J Med (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
217. Cervera-Carrascon V, Siurala M, Santos JM, Havunen R, Tahtinen S, Karell P, et al. TNFa and IL-2 armed adenoviruses enable complete responses by anti-PD-1 checkpoint blockade. Oncoimmunology (2018) 7:e1412902. doi: 10.1080/2162402X.2017.1412902
218. Jung BK, Ko HY, Kang H, Hong J, Ahn HM, Na Y, et al. Relaxin-expressing oncolytic adenovirus induces remodeling of physical and immunological aspects of cold tumor to potentiate PD-1 blockade. J Immunother Cancer (2020) 8:e000763. doi: 10.1136/jitc-2020-000763
219. Chen CY, Wang PY, Hutzen B, Sprague L, Swain HM, Love JK, et al. Cooperation of oncolytic herpes virotherapy and PD-1 blockade in murine rhabdomyosarcoma models. Sci Rep (2017) 7:2396. doi: 10.1038/s41598-017-02503-8
220. Saha D, Martuza RL, Rabkin SD. Oncolytic herpes simplex virus immunovirotherapy in combination with immune checkpoint blockade to treat glioblastoma. Immunotherapy (2018) 10:779–86. doi: 10.2217/imt-2018-0009
221. Middleton M, Aroldi F, Sacco J, Milhem M, Curti B, Mbioeth AV, et al. 422 an open-label, multicenter, phase 1/2 clinical trial of RP1, an enhanced potency oncolytic HSV, combined with nivolumab: Updated results from the skin cancer cohorts. J ImmunoTher Cancer (2020) 8:A257. doi: 10.1136/jitc-2020-SITC2020.0422
222. Long G, Dummer R, Johnson D, Michielin O, Martin-Algarra S, Treichel S, et al. 429 long-term analysis of MASTERKEY-265 phase 1b trial of talimogene laherparepvec (T-VEC) plus pembrolizumab in patients with unresectable stage IIIB-IVM1c melanoma. J ImmunoTher Cancer (2020) 8:A261. doi: 10.1136/jitc-2020-SITC2020.0429
223. Rojas JJ, Sampath P, Hou W, Thorne SH. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin Cancer Res Off J Am Assoc Cancer Res (2015) 21:5543–51. doi: 10.1158/1078-0432.CCR-14-2009
224. Semmrich M, Marchand JB, Fend L, Rehn M, Remy C, Holmkvist P, et al. Vectorized treg-depleting αCTLA-4 elicits antigen cross-presentation and CD8(+) T cell immunity to reject 'cold' tumors. J Immunother Cancer (2022) 10:e003488. doi: 10.1136/jitc-2021-003488
225. Nakao S, Arai Y, Tasaki M, Yamashita M, Murakami R, Kawase T, et al. Intratumoral expression of IL-7 and IL-12 using an oncolytic virus increases systemic sensitivity to immune checkpoint blockade. Sci Transl Med (2020) 12:eaax7992. doi: 10.1126/scitranslmed.aax7992
226. Huang JH, Zhang SN, Choi KJ, Choi IK, Kim JH, Lee MG, et al. Therapeutic and tumor-specific immunity induced by combination of dendritic cells and oncolytic adenovirus expressing IL-12 and 4-1BBL. Mol Ther J Am Soc Gene Ther (2010) 18:264–74. doi: 10.1038/mt.2009.205
227. Zhang SN, Choi IK, Huang JH, Yoo JY, Choi KJ, Yun CO. Optimizing DC vaccination by combination with oncolytic adenovirus coexpressing IL-12 and GM-CSF. Mol Ther J Am Soc Gene Ther (2011) 19:1558–68. doi: 10.1038/mt.2011.29
228. Hori Y, Winans AM, Huang CC, Horrigan EM, Irvine DJ. Injectable dendritic cell-carrying alginate gels for immunization and immunotherapy. Biomaterials (2008) 29:3671–82. doi: 10.1016/j.biomaterials.2008.05.033
229. Oh E, Oh JE, Hong J, Chung Y, Lee Y, Park KD, et al. Optimized biodegradable polymeric reservoir-mediated local and sustained co-delivery of dendritic cells and oncolytic adenovirus co-expressing IL-12 and GM-CSF for cancer immunotherapy. J Controlled Release Off J Controlled Release Soc (2017) 259:115–27. doi: 10.1016/j.jconrel.2017.03.028
230. Grossenbacher SK, Canter RJ, Murphy WJ. Natural killer cell immunotherapy to target stem-like tumor cells. J immunother Cancer (2016) 4:19. doi: 10.1186/s40425-016-0124-2
231. Melero I, Rouzaut A, Motz GT, Coukos G. T-Cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discovery (2014) 4:522–6. doi: 10.1158/2159-8290.CD-13-0985
232. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol (2016) 17:1025–36. doi: 10.1038/ni.3518
233. McGilvray RW, Eagle RA, Watson NF, Al-Attar A, Ball G, Jafferji I, et al. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin Cancer Res Off J Am Assoc Cancer Res (2009) 15:6993–7002. doi: 10.1158/1078-0432.CCR-09-0991
234. Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol (2014) 44:1582–92. doi: 10.1002/eji.201344272
235. Ye JF, Qi WX, Liu MY, Li Y. The combination of NK and CD8+T cells with CCL20/IL15-armed oncolytic adenoviruses enhances the growth suppression of TERT-positive tumor cells. Cell Immunol (2017) 318:35–41. doi: 10.1016/j.cellimm.2017.06.002
236. Morini M, Albini A, Lorusso G, Moelling K, Lu B, Cilli M, et al. Prevention of angiogenesis by naked DNA IL-12 gene transfer: angioprevention by immunogene therapy. Gene Ther (2004) 11:284–91. doi: 10.1038/sj.gt.3302175
237. Gill S, June CH. Going viral: Chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev (2015) 263:68–89. doi: 10.1111/imr.12243
238. Gross G, Eshhar Z. Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: Counteracting off-tumor toxicities for safe CAR T cell therapy. Annu Rev Pharmacol Toxicol (2016) 56:59–83. doi: 10.1146/annurev-pharmtox-010814-124844
239. Samineni D, Huang W, Gibiansky L, Ding H, Zhang R, Li C, et al. Population pharmacokinetics and exposure-response analyses for venetoclax in combination with r-CHOP in Relapsed/Refractory and previously untreated patients with diffuse Large b cell lymphoma. Adv Ther (2022) 39:598–618. doi: 10.1007/s12325-021-01919-z
240. Hawkes EA, Phillips T, Budde LE, Santoro A, Saba NS, Roncolato F, et al. Avelumab in combination regimens for Relapsed/Refractory DLBCL: Results from the phase ib JAVELIN DLBCL study. Target Oncol (2021) 16:761–71. doi: 10.1007/s11523-021-00849-8
241. Kunert A, Straetemans T, Govers C, Lamers C, Mathijssen R, Sleijfer S, et al. TCR-engineered T cells meet new challenges to treat solid tumors: Choice of antigen, T cell fitness, and sensitization of tumor milieu. Front Immunol (2013) 4:363. doi: 10.3389/fimmu.2013.00363
242. Gilham DE, Debets R, Pule M, Hawkins RE, Abken H. CAR-T cells and solid tumors: tuning T cells to challenge an inveterate foe. Trends Mol Med (2012) 18:377–84. doi: 10.1016/j.molmed.2012.04.009
243. Zaafouri H, Jouini R, Khedhiri N, Khanchel F, Cherif M, Mesbahi M, et al. Comparison between signet-ring cell carcinoma and non-signet-ring cell carcinoma of the stomach: Clinicopathological parameters, epidemiological data, outcome, and prognosis-a cohort study of 123 patients from a non-endemic country. World J Surg Oncol (2022) 20:238. doi: 10.1186/s12957-022-02699-8
244. Rezaei R, Esmaeili Gouvarchin Ghaleh H, Farzanehpour M, Dorostkar R, Ranjbar R, Bolandian M, et al. Combination therapy with CAR T cells and oncolytic viruses: A new era in cancer immunotherapy. Cancer Gene Ther (2022) 29:647–60. doi: 10.1038/s41417-021-00359-9
245. Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res (2014) 74:5195–205. doi: 10.1158/0008-5472.CAN-14-0697
246. Wing A, Fajardo CA, Posey AD Jr., Shaw C, Da T, Young RM, et al. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol Res (2018) 6:605–16. doi: 10.1158/2326-6066.CIR-17-0314
247. Evgin L, Huff AL, Wongthida P, Thompson J, Kottke T, Tonne J, et al. Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat Commun (2020) 11:3187. doi: 10.1038/s41467-020-17011-z
248. Bahl K, Kim SK, Calcagno C, Ghersi D, Puzone R, Celada F, et al. IFN-induced attrition of CD8 T cells in the presence or absence of cognate antigen during the early stages of viral infections. J Immunol (2006) 176:4284–95. doi: 10.4049/jimmunol.176.7.4284
Keywords: oncolytic virus, clinical research, combination, radiation, chemotherapy, immunotherapy
Citation: Yun C-O, Hong J and Yoon A-R (2022) Current clinical landscape of oncolytic viruses as novel cancer immunotherapeutic and recent preclinical advancements. Front. Immunol. 13:953410. doi: 10.3389/fimmu.2022.953410
Received: 26 May 2022; Accepted: 03 August 2022;
Published: 25 August 2022.
Edited by:
Yaohe Wang, Queen Mary University of London, United KingdomReviewed by:
Zong Sheng Guo, University at Buffalo, United StatesJohn Cameron Bell, Ottawa Hospital Research Institute (OHRI), Canada
Copyright © 2022 Yun, Hong and Yoon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: A-Rum Yoon, YXlvb25AaGFueWFuZy5hYy5rcg==