 Hajar El Hamss1*
Hajar El Hamss1* M. N. Maruthi1
M. N. Maruthi1 Hadija M. Ally2,3Christopher A. Omongo4
Hadija M. Ally2,3Christopher A. Omongo4 Hua-Ling Wang5
Hua-Ling Wang5 Sharon van Brunschot1,6
Sharon van Brunschot1,6 John Colvin1Hélène Delatte2*
John Colvin1Hélène Delatte2*- 1Natural Resources Institute, University of Greenwich, Central Avenue, Chatham Maritime, Kent, United Kingdom
- 2CIRAD, UMR PVBMT, Saint Pierre, France
- 3Tanzania Agricultural Research Institute Ukiriguru, Mwanza, Tanzania
- 4Root Crops Programme, National Crops Resource Research Institute (RCP-NaCRRI), Kampala, Uganda
- 5The Ministry of Agriculture Key Laboratory of Molecular Biology of Crop Pathogens and Insects, Institute of Insect Sciences, Zhejiang University, Hangzhou, China
- 6School of Biological Sciences, The University of Queensland, Brisbane, QLD, Australia
Sap-sucking insects, including whiteflies, are amongst the most devastating and widely distributed organisms on the planet. They are often highly invasive and endosymbiont communities within these insects help them adapt to new or changing environments. Bemisia tabaci (Gennadius; Hemiptera: Aleyrodidae) whitefly species are vectors of more than 500 known plant-viruses and harbour highly diverse endosymbionts communities. To date, however, whitefly–endosymbiont interactions, community structure and their spatio-temporal changes are still poorly understood. In this study, we investigated the spatio-temporal changes in the composition and diversity of bacterial endosymbionts in the agricultural crop pest whitefly species, Bemisia tabaci sub-Saharan Africa 1-subgroup 1 and 2 (SSA1-SG1 and SSA1-SG2). 16S rRNA amplicon sequencing analysis was carried out to characterise endosymbiont compositionsin field-collected SSA1 (SSA1-SG1 and SSA1-SG2) populations infesting cassava in Uganda in 1997 and 2017. We detected Portiera, Arsenophonus, Wolbachia, Hamiltonella and Hemipteriphilus, with Arsenophonus and Wolbachia infections being predominant. Hemipteriphilus and Hamiltonella frequencies were very low and were detected in seven and two samples, respectively. Bacterial diversity based on three independent parameters including Simpson index, number of haplotypes and Bray–Curtis dissimilarity matrix was significantly higher in 1997 than in 2017. This period also coincided with the advent of super-abundant cassava-whitefly populations on cassava crops in Uganda. We discuss how endosymbionts may influence the biology and behaviour of whiteflies leading to population explosions.
Introduction
Whiteflies belong to the Sternorrhyncha suborder, which also includes aphids, psyllids, and mealybugs, all of which feed on plant phloem-sap (Gullan and Martin, 2009). Plant sap is generally lacking amino acid elements required for balanced insect nutrition (Douglas, 2006). As a result, all of these insects have a symbiotic relationship with bacteria called “endosymbionts.” Bacterial endosymbionts are generally localised in vesicles within specialised insect cells (bacteriocytes; Baumann, 2005; Douglas, 2016). In these cells, endosymbionts aggregate into a bacteriome within the body cavity to help them synthesise missing dietary elements and create a balanced diet (Buchner, 1965; Baumann et al., 2006; Douglas, 2016). Early light microscopy studies revealed that each of these insect groups has a morphologically identical endosymbiont (referred to as the primary endosymbiont [P-endosymbiont]) that is found in all members of the group (Buchner, 1965). Despite morphological similarity, primary (and secondary) endosymbionts are comprised of multiple species (across host taxa; Bell-Roberts et al., 2019). Portiera aleyrodidarum, for instance, is the primary endosymbiont of Bemisia tabaci (Santos-Garcia et al., 2012) whilst Buchnera aphidicola is the primary endosymbiont of aphids (Munson et al., 1991). Some members may contain secondary endosymbionts (S-endosymbionts), which are morphologically distinct from the primary endosymbionts (Buchner, 1965; Baumann et al., 2006; Luan et al., 2015). Both endosymbiont types are maternally transmitted to the next generations (Luan et al., 2018).
The whitefly B. tabaci is a highly invasive pest of economically important vegetable and ornamental crops worldwide (Liu et al., 2009). It causes serious damage to crops such as cassava by direct feeding and vectoring plant viruses that cause economically important diseases (Barro et al., 2011; Fiallo-Olivé et al., 2019). B. tabaci is a complex of at least 44 morphologically indistinguishable biological species that are distributed in the tropical and subtropical parts of the world and the protected environments of temperate regions (Tay et al., 2017). Amongst these, B. tabaci sub-Saharan Africa 1 (SSA1) is highly prevalent in sub-Saharan Africa. Based on the partial mitochondrial cytochrome oxidase I gene (mtCOI) marker, this species was divided into five subgroups (Ghosh et al., 2015). However, SSA1-SG3 has recently been considered a separate species based on mating incompatibility studies (Mugerwa et al., 2021) and molecular genetic analysis using specific microsatellite markers (Ally et al., 2019; Mugerwa et al., 2020, 2021).
Endosymbiotic bacteria are also widespread in whiteflies (Gueguen et al., 2010). They are present in the whitefly body cavity, haemolymph or intracellularly in special cells called bacteriocytes (Raina et al., 2015). Portiera aleyrodidarum, a P-endosymbiont present in all whiteflies, supplements the amino acid-deficient diets (Thao and Baumann, 2004). The S-endosymbionts such as Rickettsia, Hamiltonella, Wolbachia, Arsenophonus, Cardinium, and Fritschea are not present in all whiteflies but have a wide variety of roles such as affecting fitness, reproduction (Thierry et al., 2011), sex determination (Wang et al., 2020), insecticide susceptibility or virus transmission capabilities (Ghanim and Kontsedalov, 2009; Himler et al., 2011; Barman et al., 2021).
The infection dynamics of S-endosymbionts in B. tabaci SSA1 showed a high abundance of Arsenophonus and Wolbachia in Uganda (Legg et al., 2014; Ghosh et al., 2015). Ghosh et al. (2015) also found that about 62% of B. tabaci SSA1-SG1 individuals were infected with Wolbachia, Arsenophonus and/or Rickettsia in single or mixed infections in Uganda. Tajebe et al. (2015), however, found Arsenophonus as a single infection or coinfected with Cardinium in Tanzania, indicating the huge differences in the results obtained between the studies. In Nigeria, Arsenophonus, Rickettsia, Wolbachia, Cardinium and Hamiltonella were detected in B. tabaci SSA1 (Akintola et al., 2020). Arsenophonus, Wolbachia, Rickettsia, and Cardinium have been detected in B. tabaci SSA1 from Uganda and Tanzania on cassava (Sseruwagi et al., 2018). In addition, infection of Rickettsia increased in B. tabaci MEAM1 in the United States from 2000 to 2016, indicating a change in the interaction between S-endosymbiont and their host (Bockoven et al., 2019). Hamiltonella infection frequencies were significantly higher in MEAM1 and MED females compared to males, suggesting that the sex of the hosts could also influence S-endosymbionts infections (Pan et al., 2012). In this study, we investigated how the spatio-temporal changes have influenced the endosymbiont composition and diversity in the African cassava whitefly species, B. tabaci SSA1, using samples collected two decades apart (1997 and 2017) and from various different locations in Uganda.
Materials and methods
Whitefly sampling
A total of 65 young cassava leaves having eggs, nymphs and pupae were collected from Uganda in 1997, and stored at −80°C at NRI, University of Greenwich, United Kingdom. Samples were collected in 14 locations (~ 5 Km apart) in three districts of Mityana, Kampala and Masaka. Another sampling was carried out in 2017 in 13 locations in a slightly larger area of seven districts (Mityana, Mpigi, Wakisa, Kalungu, Masaka, Rakai, and Gomba). A total of 48 B. tabaci adults on cassava were collected and preserved in ethanol for subsequent molecular analysis. GPS coordinates collected in 2017 were matched with the names of villages in the 1997 samples using QGIS v.2.18.17 online software1 (Ally et al., 2019; Figure 1; Supplementary Table S1).
Figure 1. Map showing the distribution of whitefly sampling sites used in this study.
DNA extraction and molecular typing of Bemisia tabaci
For this study, the DNA was extracted from 121 whiteflies (Supplementary Table S1). Of these, 65 DNA from single whitefly were extracted from 1997 samples from individual eggs and nymphs of whiteflies using the Chelex method (Walsh et al., 1991; Ghosh et al., 2015). Briefly, each whitefly was ground in a 100 μl TE solution (10 mM Tris–HCl and 1 mM EDTA, pH 8.0) containing 20% Chelex (BIO-RAD, United Kingdom) and 300 μg Proteinase K. Samples were incubated at 58°C for 1.5 h followed by protein denaturation at 96°C for 10 min (Ghosh et al., 2015). Samples were then centrifuged at 13,000 rpm and the supernatant was collected and stored at −20°C. A total of 56 DNA samples collected in 2017 were obtained from a non-destructive method by overnight incubation of whiteflies in buffer solution, as previously described (Delatte et al., 2011; Ally et al., 2019). A total of 121 SSA1 samples (Supplementary Table S1) were selected from the two locations. Their identities were confirmed from their partial mtCOI sequences (Ally et al., 2019). DNA isolated from insects were amplified using the PCR genus-specific primer pair: 2195Bt (5′-TGRTTTTTTGGTCATCCRGAAGT-3′) and C012/Bt-sh2 (5′-TTTACTGCACTTTCTGCC-3′; Mugerwa et al., 2018). Amplification of mtCOI gene was carried out in 25 μl volumes consisting of 2 μl of DNA template, 0.4 μM of each primer, 0.15 mM of dNTPs, 1 × DreamTaq Green buffer and 0.5-unit DreamTaq Green DNA polymerase (Thermo Scientific Ltd., Uniteed Kingdom). The PCR amplification began with a denaturation step at 94°C for 3 min, followed by an amplification consisting of 38 cycles at 94°C for 30 s, an annealing step for 54 s, 72°C for 1.5 min and a final extension for 7 min at 72°C. PCR products were visualised on 1% agarose gels containing RedSafe nucleic acid staining solution (Intron Biotechnology, Korea). PCR products were then visualised under UV light (402 nm) and fragments of the expected size (~ 867 bp) were purified and sent for sequencing (Macrogen, Netherlands).
16S rRNA library preparation
The Hi-Seq Illumina platform with 466 bp paired-end reads was used for sequencing the V4–V5 region of the 16S rRNA gene as previously described (El Hamss et al., 2021). A duplicate PCR strategy was adopted involving two PCR amplifications per sample to decrease PCR selection bias. In every single round of PCR, primers containing the index sequences were used to prepare the 16S rDNA sequencing libraries. Each reaction contained 2 μl of template DNA, 1 U of Dream Taq DNA polymerase, 1 mM dNTPs, and 0.2 μM of each primer in a 25 μl reaction mixture. The PCR conditions started with an initial denaturation step at 95°C for 2 min, followed by an amplification of 38 cycles at 94°C for 30 s, an annealing step for 54 s, 72°C for 1.5 min and a final extension for 7 min at 72°C in a DNA Engine thermocycler (Applied Biosystems, United Kingdom). The dual-index paired-end sequencing approach was adopted for sequencing the 16S rRNA products (Caporaso et al., 2011, 2012). Both the reverse and forward primers consisted of eight nucleotides as index sequences, 4–5 nucleotides as a linker, followed by the gene-specific primer. The index sequences and linkers together formed the barcodes. The barcodes were unique to each sample. Following the separation of products from primers and primer dimers by 1% agarose gel electrophoresis, PCR products of the correct size 400 bp were recovered using an electrophoresis Gel and PCR purification kit (NucleoSpin, Macherey-Nagel, Germany). Samples were pooled into two separate pools (121 amplicons per pool) by mixing duplicate whitefly PCR products with unique indices in equal quantities. Total DNA was quantified on a qPCR machine (Biorad, CFX manager, United Kingdom) using PicoGreen dsDNA Quantification kit (Thermo Fischer Scientific, United Kingdom). A composite pooled sample, having 121 amplified DNA with positive control Echerichia coli to control bias from multiple 16S rRNA gene copies or other sequencing errors was then prepared. Single amplicons were combined in equimolar ratios was purified using the same Gel and PCR purification kit. Quantification of pooled DNA was performed using NanoDrop 2000 (Thermo Scientific, United Kingdom). The pooled samples were sequenced by Fasteris Ltd., Switzerland by Illumina sequencing.
Sequence analysis
Paired-end reads were demultiplexed according to their barcode/primers using the software Metafast_BCsorting version (2.10; Ulyantsev et al., 2016), and the barcode sequences were subsequently removed from the output reads. Standard Illumina adapters and low-quality bases were trimmed using the Trimmomatic version 0.32 (Bolger et al., 2014). The Trimmomatic package was used to remove bases that were below the quality threshold and those that corresponded to the standard Illumina adapters. Trimmomatic parameters were selected so no mismatches were allowed in the barcode sequences, whereas in the primer sequences, the number of mismatches allowed was the number of degenerated bases + 2 mismatches.
Sequences were subjected to a second filtering process using the DADA2 plugin (Callahan et al., 2016) which was implemented in QIIME2 software v2018.6 (Bolyen et al., 2018). DADA2 enables additional read quality filtering and trimming, chimera filtering, denoising, and joining paired reads using the “Denoise and dereplicate paired-end sequences” method of QIIME2. Parameters of forward and reverse read truncation or length filtering were set to 0 while the other parameters were kept as defaut.
DADA2 generated Amplicon Sequence Variants (ASVs) and reported their relative abundance within each sample. VSEARCH consensus taxonomy classifier (Rognes et al., 2016) was used to assign taxonomy to ASVs using the SILVA database. The generated taxonomic groups of sequences were re-analysed to look for unique haplotypes using DNAsp software v6.12.0.3. Unique haplotypes were then generated with a matrix of read abundance using the gplot library in R (Warnes et al., 2009). The tree with unique haplotypes was constructed using maximum-likelihood method in Geneious tree builder, bootstrap was the resampling method and the number of replicates was set at 1,000,000 while other parameters were kept as default.
Statistical analysis
Given the high prevalence of Portiera, it was excluded from the analysis to avoid skewing the results. Therefore, only S-endosymbionts were used in this analysis. Then, Vegan Library of R (Oksanen et al., 2014) and Capscale function (Oksanen et al., 2013) were used to investigate the effect of time and diversity indices including alpha diversity measured by Simpson, the number of haplotypes and Beta diversity measured by Bray–Curtis matrix was calculated on binary matrix of ASV. The non-metric Multi-Dimensional Scaling analysis (NMDS) was used to condense and therefore simplify multidimensional data about the samples into a few important axes to facilitate visualisation and interpretation. As microbiome dataset is compositional by nature with an irrelevant total number of counts within a sample, the centred log-ratio (CLR) transformation on ASV matrix of total reads was also applied to correct for biases (Greenacre et al., 2021). Aitchison distance between transformed samples was subsequently calculated and visualised using Principal Coordinate Analysis (PCoA).
One-way analysis of variance (ANOVA) was used to investigate mean differences between Simpson index and the number of observed haplotypes based on the 2 years of collection.
Analysis of Compositions of Microbiomes with Bias Correction (ANCOM-BC), a robust methodology of differential abundance for microbial absolute abundances, was used to further confirm the date effect on S-endosymbionts composition (Gloor et al., 2017). Permutational Multivariate Analysis of Variance (ADONIS) was used to analyse S-endosymbionts communities with environmental variables (date, space and life stage). Relative abundance of each haplotype in single whitefly sample was also calculated and the ASV matrix of abundance was transformed into a binary matrix before calculating Bray–Curtis matrix.
Results
All individuals in this study were identified as B. tabaci SSA1-SG1 and SSA1-SG2 which are the same species (Mugerwa et al., 2021). Moreover, all of the whitefly individuals sampled in 2017 were adults, while whiteflies sampled in 1997 were at various juvenile life stages. It was, in fact, not possible to standardise the life stages across the 2 years included in the study. Nevertheless, S-endosymbionts are maternally transmitted, and therefore, we are not expecting them to be influenced by life stage. The results of the statistical tests also confirm this hypothesis as the life stage was not influencing the S-endosymbionts at both alpha diversity level (Shannon index: p = 0.2, Haplotype: p = 0.19) and beta diversity level (ADONIS: p = 0.06). The results presented here are, therefore, solely from both the temporal and spatial changes.
The overall diversity of endosymbionts according to the time of the whitefly collection
The diversity of S-endosymbionts changed over time significantly both based on the Shannon index, p = 0.0036 (Figure 2A) and total haplotypes, p = 0.00034 (Figure 2B). Shannon index average value changed from 0.71 in 1997 to 1.09 in 2017 (Figure 2A). The S-endosymbiont diversity and composition did not change across sites based on Shannon index (p = 0.19; Figure 2C), total haplotypes (p = 0.13; Figure 2D) and Bray–Curtis matrix (p = 0.28). Haplotypes average changed from 3 in 1997 to 7 in 2017, while the Bray-Curtis matrix measuring S-endosymbiont structure was significantly different (p = 0.001; Figure 2E). Based on the NMDS, old samples, represented in red dots, are clustered meaning that they have similar compositions (Figure 2E). Furthermore, results of the PCoA on CLR-transformed data showed significant differences in S-endosymbionts between whiteflies collected in 1997 and 2017 (Supplementary Figure S1). These results showed that S-endosymbiont diversity in the B. tabaci SSA1 population had changed over time.
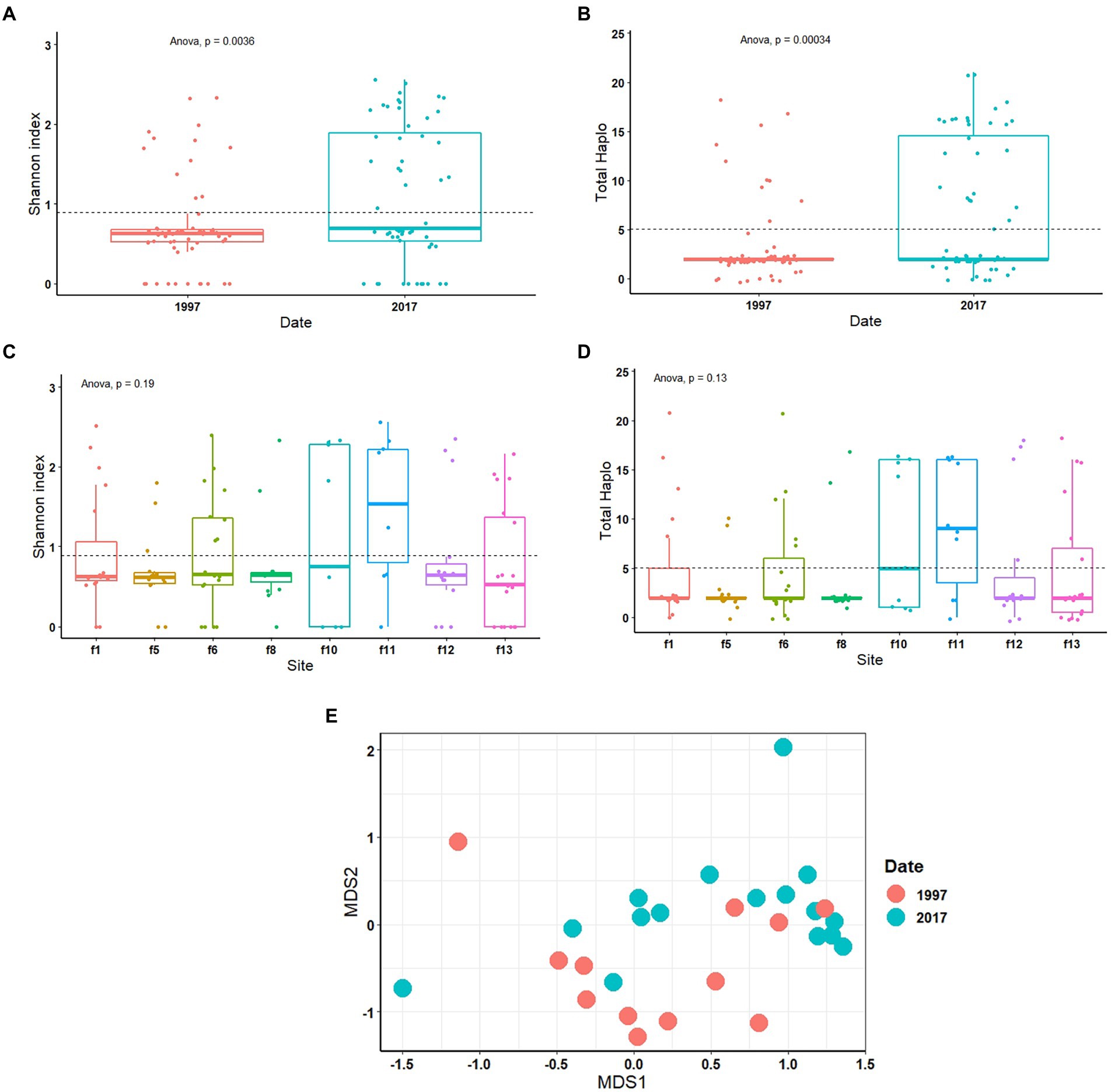
Figure 2. One-way ANOVA of diversity analysis including Shannon index and total haplotype in relation to date (A,B) and site (C,D). Non-metric multi-dimensional scaling analysis (NMDS) test on Bray–Curtis matrix (E) of S-endosymbionts only. Portiera was excluded from the statistical tests.
The temporal prevalence of endosymbionts and their haplotypes
Total reads of 102,493,996 were generated from the 1997 (60,318,306) and 2017 (42,175,690) samples (Supplementary Table S1). Of the 102,493,996 total recovered reads after quality filtering, 96% (92,355,604) were assigned to Portiera whilst the remaining 4% (10,138,3,924) reads were assigned to S-endosymbionts and non-endosymbiotic bacteria (Figure 3). Amongst Portiera sequences, 19 haplotypes were found in this study, with three of them; Por-Hap_3, and Por-Hap_1 detected in one sample collected in 1997, and Por-Hap_2 detected in three SSA1 whiteflies, but with low frequencies (Figure 3), whereas the others with high frequency were prevalent on both dates of collection (Figure 2; Supplementary Table S1). Their accession numbers were deposited in the GENEBANK database (accession numbers from OP160987 to OP160997).
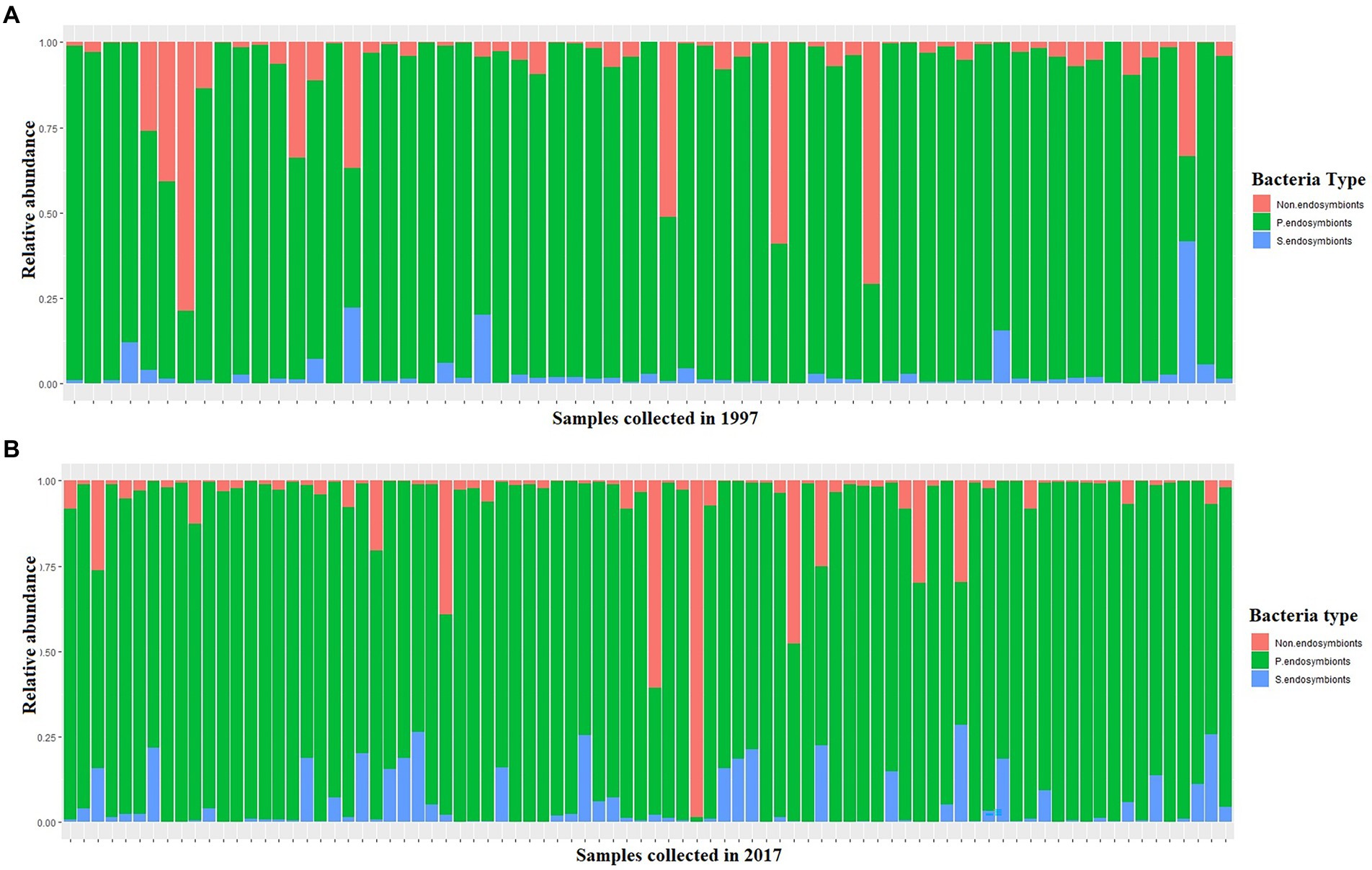
Figure 3. A stacked barplot of the three bacteria types that were detected in this study in each single whitefly showing their relative abundance in relation to date in 1997 (A) and 2017 (B).
Four S-endosymbionts including Arsenophonus Wolbachia, Hemipteriphilus and Hamiltonella were detected in our samples with different prevalences (Figure 4). The analysis of bacterial composition with bias correction (ANCOM-BC) showed that the relative abundances of 23 haplotypes belonging to Arsenophonus and Wolbachia changed significantly between 1997 and 2017 in tested SSA whiteflies (Figure 4; Supplementary Table S2).
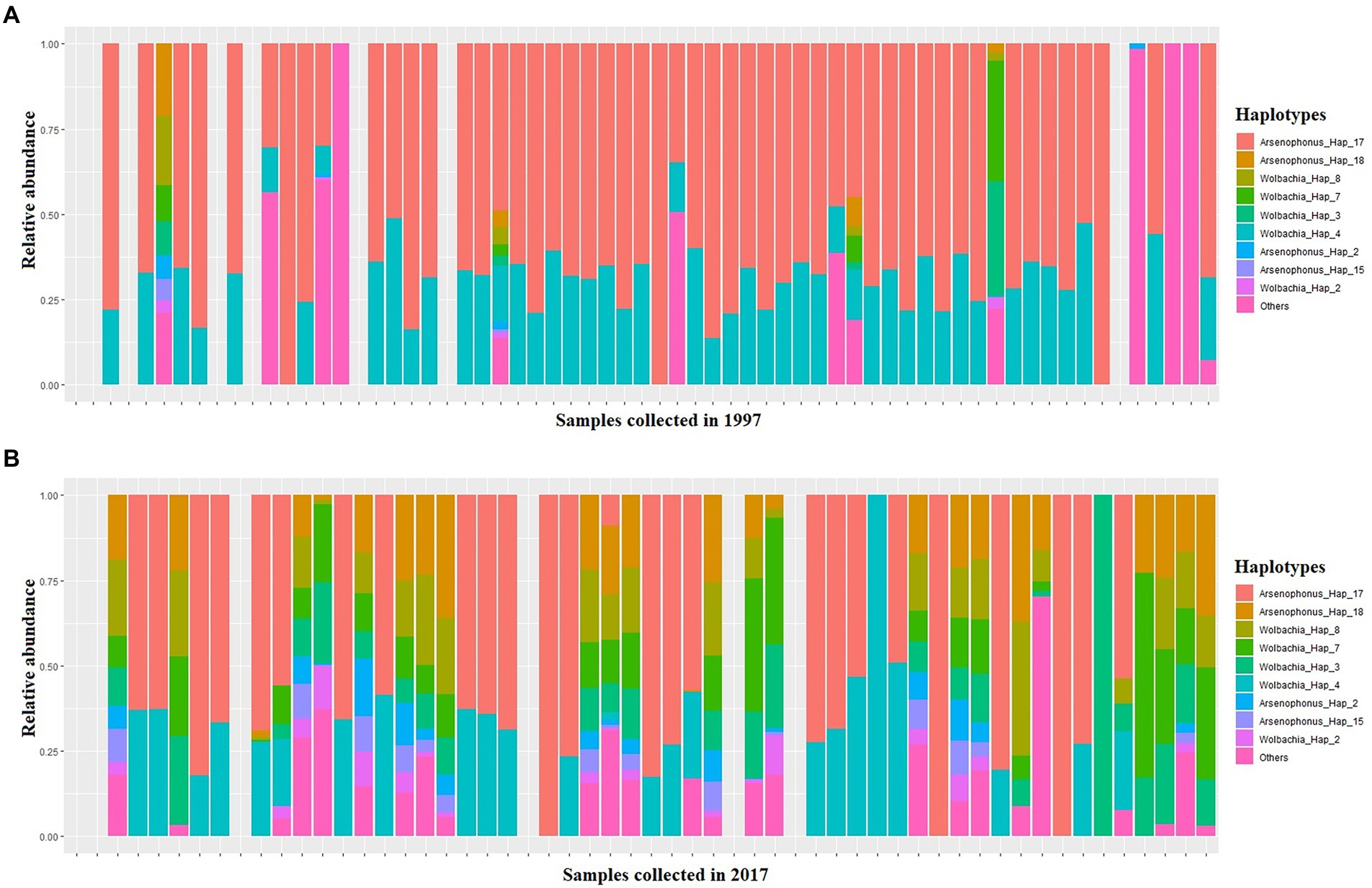
Figure 4. A stacked barplot of S-endosymbiont haplotypes detected in this study in each single whitefly which exhibit significantly different relative abundance in relation to date, as indicated by ANCOM-BC (Supplementary Table S5), in 1997 (A) and 2017 (B).
A total of 69 samples harboured 100% of the relative abundance of Arsenophonus (OP160971 to OP160986), 49 samples in 1997 and 20 in 2017 (Figure 4). In both years, Arsenophonus was the most abundant S-endosymbiont followed by Wolbachia (OP161006–OP161013) which was observed in 5 whitefly samples with relative abundance ranging between 25 to 91% in 1997 and 28 samples in 2017 having a relative abundance ranging from 0.2 to 100% (Figure 4). Hemipteriphilus (OP160998 to OP161005) was least abundant in both years present in four samples in 1997 with relative frequency ranging between 3 and 47% and only present in one sample from 2017 with relative frequency reaching 7% (Figures 4). Hamiltonella (OP160970) was present in one whitefly sample only from the 2017 collections (Figure 4).
Arsenophonus sequences showed the highest haplotype diversity, assigned to 18 haplotypes. Amongst them, only two were found in 1997 collections with both occurring in three samples (Figure 4). The top abundant haplotype was Ars_Hap_7 as the relative frequency assigned to this haplotype was between 0 and 100% in the 1997 samples and 0–100% in the 2017 samples (Figure 4).
Wolbachia and Hemipteriphilus sequences were less diverse, as eight haplotypes were detected for each bacteria (Supplementary Table S1). Five Hemipteriphilus haplotypes were found only in 1997 but not in 2017. Hemi-Hap_39, Hemi-Hap_42 (Hemi-Hap_43 Hemi-Hap_44, Hemi-Hap_45) occurred in less than three samples. Wolbachia prevalence was different between dates but the diversity did not change over time (Figure 2; Supplementary Table S1). Two haplotypes, Ham-Hap_46 (25) and Ham-Hap_47 belonging to Hamiltonella sequences were found only in two samples in 2017, but not in 1997 (Supplementary Table S1). All the sequences generated were deposited in the Genebank database with their accession numbers.
Infection dynamics of S-endosymbionts and their phylogeny at haplotype level
The infection dynamics between the three endosymbionts Arsenophonus, Wolbachia and Hemipteriphilus changed over time (Table 1). Dual infections of Wolbachia and Arsenophonus changed from 9 to 28% from 1997 to 2017 (Table 1). Wolbachia was not found in 1997 (0%) but represented 5% of reads in 2017 (Table 1). Single infection of Arsenophonus changed from 91 to 68% from 1997 to 2017 (Table 1). S-endosymbiont-free whiteflies were found in both years with 12 and 17% in 1997 and 2017, respectively (Table 1).
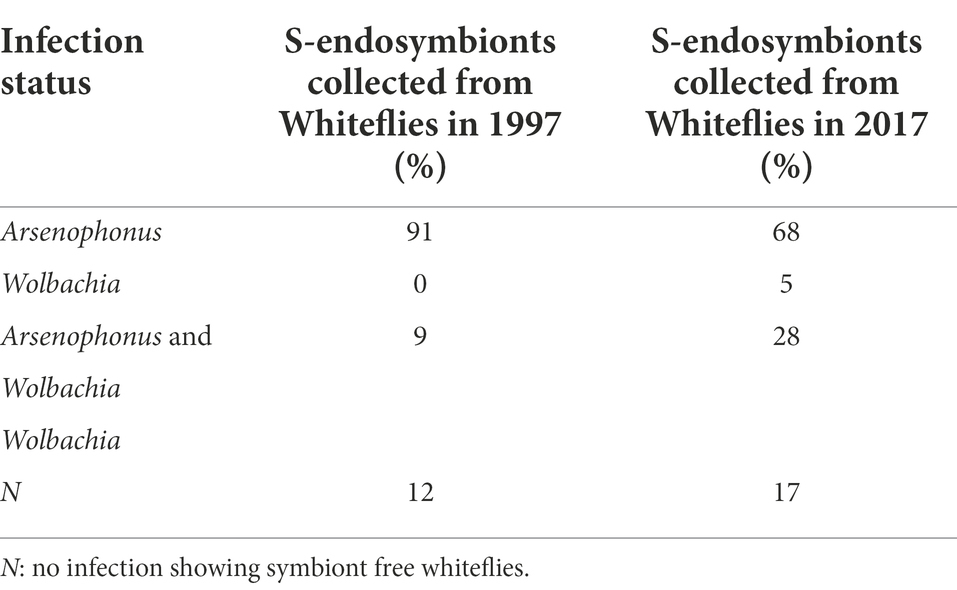
Table 1. Temporal change of S-endosymbiont coinfections.
The phylogenetic relationships between and within S-endosymbionts showed that, apart from Hemipteriphilus, each S-endosymbiont clustered separately and, as expected, with their given reference sequences (downloaded from the GENBANK; Figure 5). Two clusters of Arsenophonus were present in the tree, suggesting the high genetic diversity of this symbiont (Figure 3). Hemipteriphilus was clustered with one Rickettsia sequence, suggesting the need to further investigate other genes of Hemipteriphilus. A heat map of the relative abundance showed the temporal variations of all detected bacteria at the haplotype level (Figure 6).
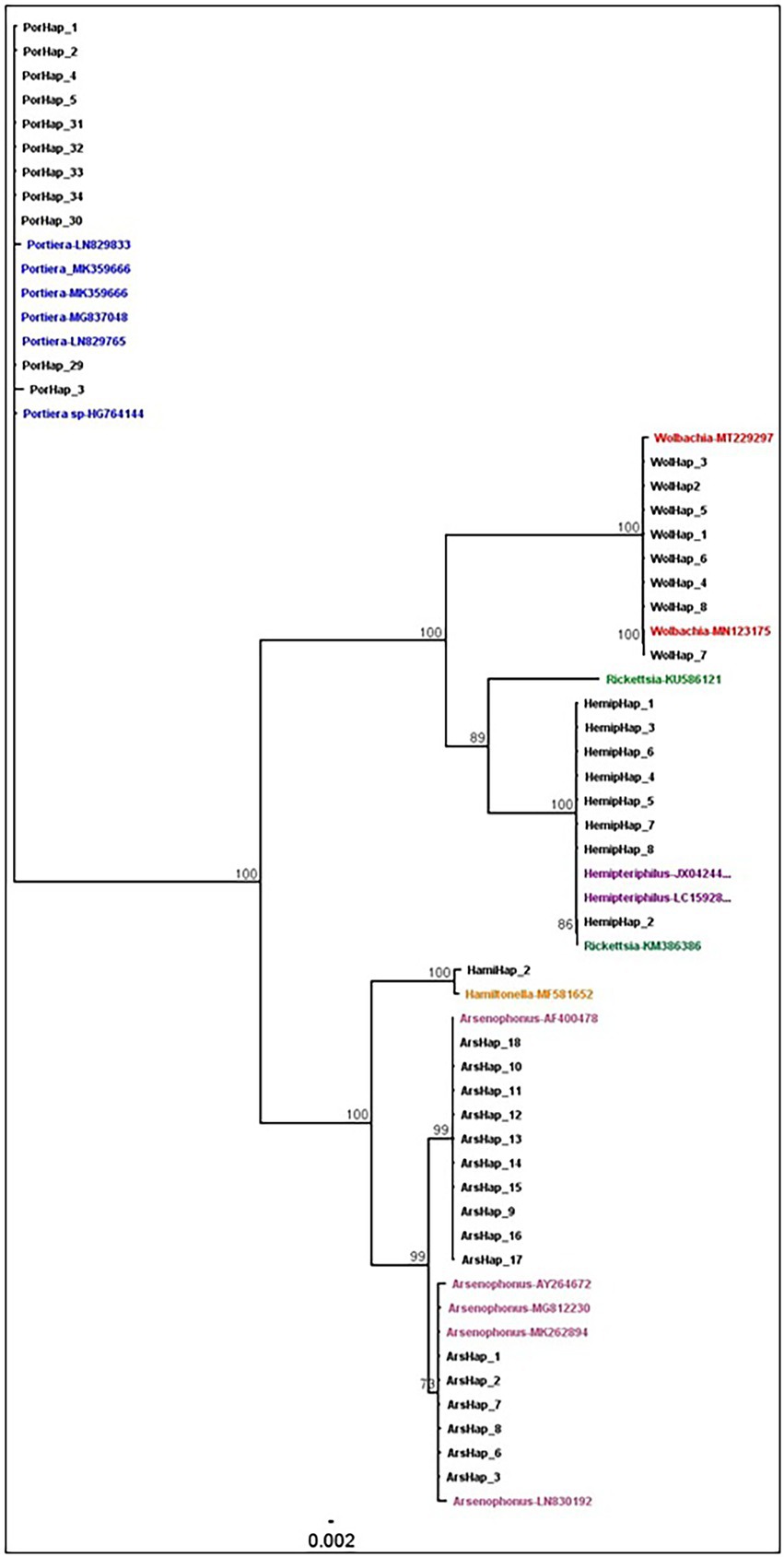
Figure 5. Phylogenetic analysis of the endosymbionts haplotypes based on the region V4–V5 in 16S rRNA sequences using the neighbour-joining method with 1,000,000 bootstraps in Geneious. The coloured names showed the reference sequences with their accession numbers all the black names were generated from this study.
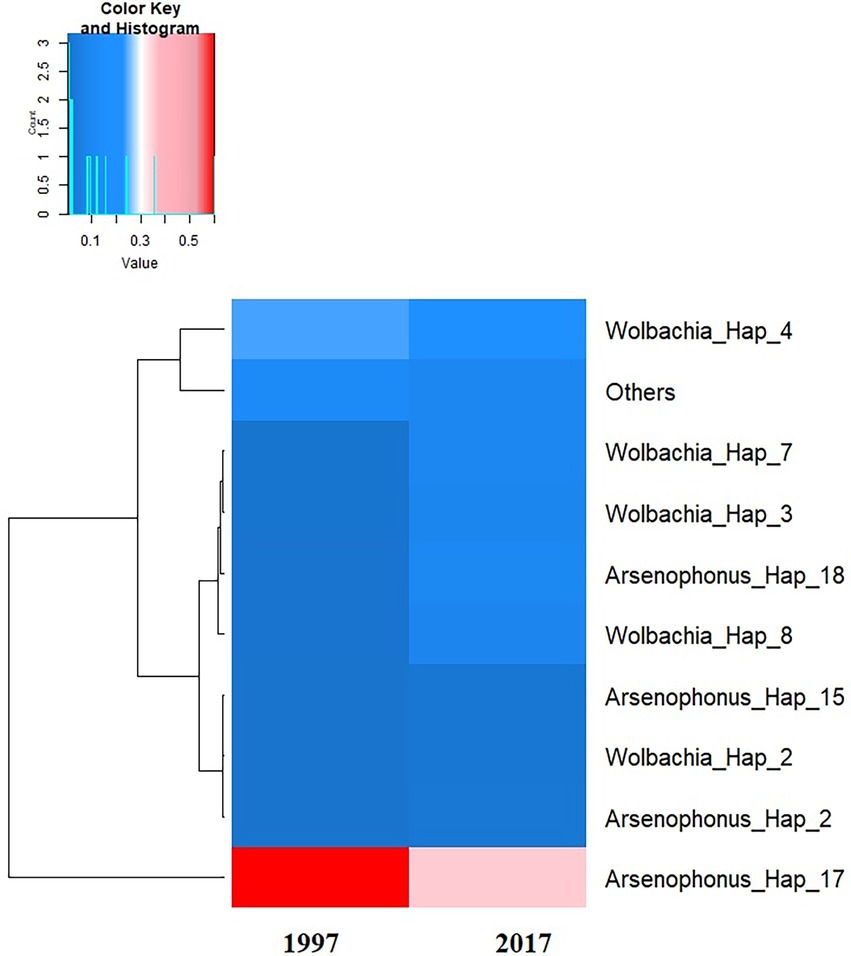
Figure 6. Heat map of the average of the relative abundance of S-endosymbiont haplotypes detected according to the date of the investigations. The dendrogram on the left was constructed based on the overall relative frequency of each haplotype, meaning that the haplotypes which are clustered together have similar overall relative prevalence.
Discussion
Despite their importance to whitefly, relatively little is known about the endosymbionts in African cassava whitefly populations. We present here, for the first time, the full endosymbiont diversity and composition in B. tabaci SSA1 by deep sequencing of 16S rRNA gene, using samples collected at different time points in Uganda. We found that the S-endosymbiont diversity in the B. tabaci SSA1 population has changed since 1997 and that this has happened in parallel with the increased whitefly populations attacking cassava crops.
Before discussing these findings, it is important to note that reads produced by Illumina sequencing create large amounts of data with errors that are difficult to differentiate from real biological variation. To overcome this challenge, DADA2 filtering was adopted to clean Illumina errors following the parametric error model (err). Reads with number of expected errors higher than 2 were discarded. Nevertheless, other errors or misinterpretations can still persist, notably, 16S rRNA gene copies that can occur in some bacteria species, such as polyploidy (Sun et al., 2013; Větrovský and Baldrian, 2013; Espejo and Plaza, 2018; Louca et al., 2018). In this study, we further used Echerichia coli as the mock community in all pools of our samples to further confirm the assignment of reads.
This work is also the first study to investigate intraspecies diversity of Portiera within B. tabaci SSA1. We discovered six prevalent Portiera haplotypes whose relative frequency was stable between the two dates of investigation. Portiera 16S rRNA was also found to contain a highly homologous sequence (Paredes-Montero et al. (2020)). However, we found intraspecies diversity in their variable regions V4-V5 within the 16S rRNA gene sharing 99% identity, which is similar to the 0–0.67% divergence reported by Paredes-Montero et al. (2020), suggesting Portiera evolution is ongoing (Santos-Garcia et al., 2020). The divergence between Portiera variants is concordant with the long period of coevolution between the host and endosymbiont.
The differences found between the study in Paredes-Montero et al., 2020 and our studies could be due to the different methodologies adopted and the depth of sequences obtained. We used amplicon sequence variants (ASV) showing the differences of the V4-V5 region in 16S rRNA in Portiera sequences and obtained deeper sequencing than any previous studies. The differences in diversity metrics of S-endosymbionts observed between Ghosh et al. (2015) and Tajebe et al. (2015) in seemingly similar whitefly populations from East African cassava whitefly populations, highlighted the sensitivity of these types of analyses to the methodology used in detecting endosymbionts. Nevertheless, our approach detected all previously known endosymbionts associated with whiteflies. A standardised set of protocols is therefore needed to detect and identify reliably the various endosymbionts infecting insect species. This conclusion is further emphasised by Ghosh et al. (2015), reporting three S-endosymbionts (Wolbachia, Arsenophonus and Rickettsia) in B. tabaci SSA1 using a simple PCR and sequencing approach, while we found a new and different combination of S-endosymbionts (Arsenophonus Wolbachia, Hemipteriphilus and Hamiltonella). Hemipteriphilus was found for the first time in seven samples and Hamiltonella in a single sample in African whiteflies. This is the first study to detect Hemipteriphilus in African whiteflies. Fritschea and Cardinium, however, were not detected in this study. Previous studies did not find Hemipteriphilus in B. tabaci SSA1, whilst Rickettsia infection was less than 1% (Ghosh et al., 2015), further highlighting high endosymbiont diversity in African cassava whiteflies and differences in the methodologies used.
The high depth of sequences obtained here also allowed us to investigate intraspecies diversity within each S-endosymbiont. This is to show that if there are multiple genetic variants within the population, we might predict that the presence/absence and relative abundance of different haplotypes would show variation among whitefly samples harbouring a particular species of the endosymbiont. Similarly, three strains of Hemipteriphilus were detected in one study on MED Q1 and MED Q3 and ASL whiteflies using three genes including 16S rRNA gene (483 bp), GroEL (269 bp) and GltA (190 bp; Mouton et al., 2022).
The two most prevalent Arsenophonus haplotypes (amongst 18) were present on both tested dates. While the two least prevalent Arsenophonus haplotypes were present only in 1997, they were not detected in 2017 specimens. Three Wolbachia haplotypes were highly prevalent among the eight detected, but they did not change between the two dates. In this study, three Hemipteriphilus haplotypes were also found in only 1997. Little is known about the intraspecies diversity of Hemipteriphilus in African whiteflies, which was first characterised as Candidatus Hamiltonella defensa (Bing et al., 2013) in China 1 whiteflies and subsequently found only in Asian B. tabaci. In that study using both 16S rRNA and gltA genes suggested that Hemipteriphilus is clustered within the Alphaproteobacteria subdivision of Proteobacteria. Here, we found that Hemipteriphilus sequences were clustered with one strain of Rickettsia. Another study in Burkina Faso also identified Hemipteriphilus similar to Rickettsia using 16S rRNA gene in Illumina sequencing technology in MED-Q1, MED-Q3 and ASL genetic groups (Mouton et al., 2022). The sequences of Hemipteriphilus and Rickettsia are so similar that they are difficult to differentiate especially using 16S rRNA primers. This issue could be due to the specificity of the primers used to detect Hemipteriphilus. When a mismatch within V4-V5 sequences occurs, it reduces the thermal stability of the primer-template duplex, thus affecting PCR specificity. Nevertheless in this study, some Hemipteriphilus sequences were not differentiated from Rickettsia, the use of other housekeeping genes (Christensen et al., 2004; Farré et al., 2007) is therefore needed to further study the inter- and intra-species diversity of Hemipteriphilus in African whiteflies. These observed similarities and differences between Hemipteriphilus and Rickettsia imply that the group is not well defined and probably in need of a taxonomic update.
In this study, we found that S-endosymbionts diversity and composition were not significantly different across sites in SSA1 whiteflies. In previous work, it was reported that site-to-site variations subsequently influence the prevalence of endosymbionts in a given whitefly population (Zchori-Fein et al., 2014). Endosymbionts composition of over 2000 studied whiteflies derived from several independent screenings was positively correlated with the distance from the equator (Zchori-Fein et al., 2014). Similarly in China, Wolbachia and Rickettsia were influenced by geography and host plants (Pan et al., 2012). These differences between studies could be linked to the adopted sampling. In our work, the sampling was carried out in Uganda with sites only 5 km apart from each other, compared to other studies where samplings were done at the country level. Rickettsia was also shown to boost its invasive ability within B. tabaci by the increase of infection frequency from 1% in 2000 to 97% in 2006 in Arizona (Himler et al., 2011).
Our results demonstrate the dynamic nature of bacterial endosymbionts. Both single and dual infections of Wolbachia changed in SSA1 over time. Dual infection of Arsenophonus and Wolbachia also changed from 9 to 28%. In contrast, single infections of Arsenophonus changed from 91 to 68%. Wolbachia and Arsenophonus are reported to coexist in other insect species such as brown planthopper (Nilaparvata lugens; Guo et al., 2021) and ants (Wang et al., 2016). Portiera genome is highly reduced and lacks genes involved in the synthesis of certain vitamins, cofactors, and some essential amino-acids, suggesting that there is a metabolic niche that could be filled by secondary endosymbionts (Rao et al., 2015). Similarly, the co-occurrence of both Arsenophonus and Wolbachia has been found in different whitefly species from China MEAM1 and MED (Chu et al., 2011) and East Africa in SSA1-SG2, but their role is not well understood. At the metabolic pathway level, both Arsenophonus and Wolbachia influence whiteflies nutritionally by synthesising vitamin B (Santos-Garcia et al., 2018; Zhu et al., 2022). Their effect on B. tabaci SSA1 should further be investigated.
In this study, a single infection of Wolbachia changed from 0 to 5%. This change may be an indication of the emerging role of Wolbachia in B. tabaci SSA1 or the beginning of a new invasion by these Wolbachia species. Wolbachia is well known to influence the reproduction of their host by reproductive incompatibility within and between insect species including B. tabaci SSA1 and B. tabaci SSA1-SG2. The latter harboured Wolbachia (Mugerwa et al., 2020), suggesting that the incompatibility is species dependent. Nevertheless, the change in Wolbachia infections over time in field-collected whiteflies can have implications on their reproduction that should be investigated further.
A total of 12 and 17% of the samples were S-endosymbionts free in 1997 and 2017, respectively. Similarly, Ghosh et al. (2015) also showed about 38.0% of SSA1 was free from S-endosymbionts. Whiteflies without any S-endosymbionts in them can be found in both field and laboratory whiteflies (El Hamss et al., 2021). Some S-endosymbiont have a negative effect on whitefly development and their removal from whiteflies increased the number of eggs and nymphs laid as well as decreased adult emergence time (Ghosh et al., 2017). Generally, S-endosymbiont’s role is species-specific and therefore may or may not be present depending on the whitefly species. Spatio-temporal change of endosymbionts can be linked to their facultative or vital presence in whiteflies. In this study, S-endosymbionts diversity and composition in B. tabaci SSA1 individuals changed over time, coinciding with the advent of whitefly outbreaks in Uganda. This whitefly upsurge was estimated at more than 1,000 adults per top five leaves (Legg and Raya, 1998; Colvin et al., 2004; Legg et al., 2006). The reasons behind whitefly outbreaks have not been fully understood and one explanation for this high whitefly infestation was related to synergistic interactions between S-endosymbionts providing a high fitness advantage to their whitefly hosts. In one study, infection of B. tabaci MEAM1 by Rickettsia produced more eggs and females with higher survival to adulthood which led to its invasion across the southwestern United States (Himler et al., 2011). The presence of Arsenophonus or other hidden S-endosymbiont in B. tabaci SSA1 whiteflies may be responsible for improved fecundity and survival as suggested in previous studies (Ghosh et al., 2015; Tajebe et al., 2015). Both a high frequency of Arsenophonus and high haplotype diversity were detected from SSA1 in this study, suggesting that this S-endosymbiont can potentially affect whitely behaviour and abundance. In one reciprocal crossing experiment, Arsenophonus, the most prevalent S-endosymbiont in Ugandan B. tabaci SSA1, influenced whitefly reproduction (El Hamss et al., 2021).
Using a population dynamics technique, we looked at endosymbiont diversity and composition. We demonstrated how these discoveries are helping us get closer to our aim to better understand factors changing endosymbiont communities in whiteflies. This research, therefore, adds to our understanding of how S-endosymbiotic communities change over time. Further research is now needed to understand in more depth the role played by the diverse endosymbiont communities in African cassava whitefly outbreaks.
Data availability statement
The data presented in the study are deposited and available in the GenBank repository, accession number from OP160970 to OP161013.
Author contributions
JC, HD, and MM: conceptualisation. HH, CO, and HA: data collection. HH, H-LW, SB, HD, and MM: data analysis and interpretation. HH: writing—original draft. MM and HD: writing, review and editing. JC, MM, and CO: funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This work was supported, in whole or in part, by the Bill & Melinda Gates Foundation [Grant Agreement OPP1058938]. Under the grant conditions of the Foundation, a Creative Commons Attribution 4.0 Generic License has already been assigned to the Author Accepted Manuscript version that might arise from this submission.
Acknowledgments
We acknowledge the advice received from Pierre Lefeuvre that helped us to greatly improve the bioinformatic analysis carried out in this research. We acknowledge the technical help received from Douglas one of the HPC admins in NRI, University of Greenwich that greatly helped us in the analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.986226/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Principal Coordinate Analysis (PCoA) calculated based on Aitchison community dissimilarity distance matrix of CLR-transformed 16s rRNA gene read data generated from Illumina of S-endosymbionts in single whitefly collected in two different time points (1997 and 2017).
SUPPLEMENTARY TABLE S1 | Sample information, overall reads and Amplicon Sequence Variants (ASVs) table of reads of microbiome generated from each whitefly sample using16S rRNA amplicon sequencing.
SUPPLEMENTARY TABLE S2 | ANCOM-BC test results for differential abundance of S-endosymbionts that differed significantly in abundance levels between dates.
Footnotes
References
Akintola, A. A., Hwang, H.-S., Khatun, M. F., Ande, A. T., and Lee, K.-Y. (2020). Genetic diversity of Bemisia tabaci cryptic species in Nigeria and their relationships with endosymbionts and acquired begomoviruses. J. Asia Pac. Entomol. 23, 1003–1009. doi: 10.1016/j.aspen.2020.08.007
Ally, H. M., Hamss, H. E., Simiand, C., Maruthi, M. N., Colvin, J., Omongo, C. A., et al. (2019). What has changed in the outbreaking populations of the severe crop pest whitefly species in cassava in two decades? Sci. Rep. 9:14796. doi: 10.1038/s41598-019-50259-0
Barman, M., Samanta, S., Thakur, H., Chakraborty, S., Samanta, A., Ghosh, A., et al. (2021). Effect of neonicotinoids on bacterial symbionts and insecticide-resistant gene in whitefly, Bemisia tabaci. Insects 12:742. doi: 10.3390/insects12080742
Barro, P. J. D., Liu, S.-S., Boykin, L. M., and Dinsdale, A. B. (2011). Bemisia tabaci: a statement of species status. Annu. Rev. Entomol. 56, 1–19. doi: 10.1146/annurev-ento-112408-085504
Baumann, P. (2005). Biology of bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu. Rev. Microbiol. 59, 155–189. doi: 10.1146/annurev.micro.59.030804.121041
Baumann, P., Moran, N. A., Baumann, L., and Dworkin, M. (2006). Bacteriocyte-associated endosymbionts of insects. PRO 1, 403–438. doi: 10.1007/0-387-30741-9_16
Bell-Roberts, L., Douglas, A. E., and Werner, G. D. (2019). Match and mismatch between dietary switches and microbial partners in plant sap-feeding insects. Proc. R. Soc. B 286:20190065. doi: 10.1098/rspb.2019.0065
Bing, X.-L., Yang, J., Zchori-Fein, E., Wang, X.-W., and Liu, S.-S. (2013). Characterization of a newly discovered symbiont of the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae). Appl. Environ. Microbiol. 79, 569–575. doi: 10.1128/AEM.03030-12
Bockoven, A. A., Bondy, E. C., Flores, M. J., Kelly, S. E., Ravenscraft, A. M., and Hunter, M. S. (2019). What goes up might come down: the spectacular spread of an endosymbiont is followed by its decline a decade later. Microb. Ecol. 79, 1–13. doi: 10.1007/s00248-019-01417-4
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C., Al-Ghalith, G. A., et al. (2018). QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Preprints 6:e27295v27292. doi: 10.7287/peerj.preprints.27295v2
Buchner, P. (1965). Endosymbiosis of Animals With Plant Microorganisms. New York, NY: John Wiley & Sons.
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Caporaso, J. G., Lauber, C., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA. 108 (suppl 1), 4516–4522. doi: 10.1073/pnas.1000080107
Christensen, H., Kuhnert, P., Olsen, J. E., and Bisgaard, M. (2004). Comparative phylogenies of the housekeeping genes atpD, infB and rpoB and the 16S rRNA gene within the Pasteurellaceae. Int. J. Syst. Evol. Microbiol. 54, 1601–1609. doi: 10.1099/ijs.0.03018-0
Chu, D., Gao, C. S., De Barro, P., Zhang, Y. J., Wan, F. H., and Khan, I. A. (2011). Further insights into the strange role of bacterial endosymbionts in whitefly, Bemisia tabaci: comparison of secondary symbionts from biotypes B and Q in China. Bull. Entomol. Res. 101, 477–486. doi: 10.1017/S0007485311000083
Colvin, J., Omongo, C., Maruthi, M., Otim-Nape, G., and Thresh, J. (2004). Dual begomovirus infections and high Bemisia tabaci populations: two factors driving the spread of a cassava mosaic disease pandemic. Plant Pathol. 53, 577–584. doi: 10.1111/j.0032-0862.2004.01062.x
Delatte, H., Holota, H., Warren, B. H., Becker, N., Thierry, M., and Reynaud, B. (2011). Genetic diversity, geographical range and origin of Bemisia tabaci (Hemiptera: Aleyrodidae) Indian Ocean Ms. Bull. Entomol. Res. 101, 487–497. doi: 10.1017/S0007485311000101
Douglas, A. E. (2006). Phloem-sap feeding by animals: problems and solutions. J. Exp. Bot. 57, 747–754. doi: 10.1093/jxb/erj067
Douglas, A. E. (2016). How multi-partner endosymbioses function. Nat. Rev. Microbiol. 14, 731–743. doi: 10.1038/nrmicro.2016.151
El Hamss, H., Ghosh, S., Maruthi, M. N., Delatte, H., and Colvin, J. (2021). Microbiome diversity and reproductive incompatibility induced by the prevalent endosymbiont Arsenophonus in two species of African cassava Bemisia tabaci whiteflies. Ecol. Evol. 11, 18032–18041. doi: 10.1002/ece3.8400
Espejo, R. T., and Plaza, N. (2018). Multiple ribosomal RNA operons in bacteria; their concerted evolution and potential consequences on the rate of evolution of their 16S rRNA. Front. Microbiol. 9:1232. doi: 10.3389/fmicb.2018.01232
Farré, D., Bellora, N., Mularoni, L., Messeguer, X., and Albà, M. M. (2007). Housekeeping genes tend to show reduced upstream sequence conservation. Genome Biol. 8:R140. doi: 10.1186/gb-2007-8-7-r140
Fiallo-Olivé, E., Pan, L.-L., Liu, S.-S., and Navas-Castillo, J. (2019). Transmission of Begomoviruses and other whitefly-borne viruses: dependence on the vector species. Phytopathology 110, 10–17. doi: 10.1094/PHYTO-07-19-0273-FI
Ghanim, M., and Kontsedalov, S. (2009). Susceptibility to insecticides in the Q biotype of Bemisia tabaci is correlated with bacterial symbiont densities. Pest Manag. Sci. 65, 939–942. doi: 10.1002/ps.1795
Ghosh, S., Bouvaine, S., and Maruthi, M. (2015). Prevalence and genetic diversity of endosymbiotic bacteria infecting cassava whiteflies in Africa. BMC Microbiol. 15, 1–17. doi: 10.1186/s12866-015-0425-5
Ghosh, S., Bouvaine, S., Richardson, S. C., Ghanim, M., and Maruthi, M. (2017). Fitness costs associated with infections of secondary endosymbionts in the cassava whitefly species Bemisia tabaci. J. Pest. Sci. 91, 17–28. doi: 10.1007/s10340-017-0910-8
Gloor, G. B., Macklaim, J. M., Pawlowsky-Glahn, V., and Egozcue, J. J. (2017). Microbiome datasets are compositional: and this is not optional. Front. Microbiol. 8:2224. doi: 10.3389/fmicb.2017.02224
Greenacre, M., Martínez-Álvaro, M., and Blasco, A. (2021). Compositional data analysis of microbiome and any-omics datasets: a validation of the additive Logratio transformation. Front. Microbiol. 12:727398. doi: 10.3389/fmicb.2021.727398
Gueguen, G., Vavre, F., Gnankine, O., Peterschmitt, M., Charif, D., Chiel, E., et al. (2010). Endosymbiont metacommunities, mtDNA diversity and the evolution of the Bemisia tabaci (Hemiptera: Aleyrodidae) species complex. Mol. Ecol. 19, 4365–4376. doi: 10.1111/j.1365-294X.2010.04775.x
Gullan, P. J., and Martin, J. H. (2009). “Chapter 244-Sternorrhyncha: (jumping plant–lice, whiteflies, aphids, and scale insects)” in Encyclopedia of Insects (second edition). eds. V. H. Resh and R. T. Cardé (San Diego: Academic Press), 957–967.
Guo, H., Wang, N., Niu, H., Zhao, D., and Zhang, Z. (2021). Interaction of Arsenophonus with Wolbachia in Nilaparvata lugens. BMC Ecol. Evol. 21:31. doi: 10.1186/s12862-021-01766-0
Himler, A. G., Adachi-Hagimori, T., Bergen, J. E., Kozuch, A., Kelly, S. E., Tabashnik, B. E., et al. (2011). Rapid spread of a bacterial symbiont in an invasive whitefly is driven by fitness benefits and female bias. Science 332, 254–256. doi: 10.1126/science.1199410
Legg, J., Owor, B., Sseruwagi, P., and Ndunguru, J. (2006). Cassava mosaic virus disease in east and Central Africa: epidemiology and management of a regional pandemic. Adv. Virus Res. 67, 355–418. doi: 10.1016/S0065-3527(06)67010-3
Legg, J., and Raya, M. (1998). Survey of cassava virus diseases in Tanzania. Int. J. Pest Manag. 44, 17–23. doi: 10.1080/096708798228473
Legg, J. P., Sseruwagi, P., Boniface, S., Okao-Okuja, G., Shirima, R., Bigirimana, S., et al. (2014). Spatio-temporal patterns of genetic change amongst populations of cassava Bemisia tabaci whiteflies driving virus pandemics in east and Central Africa. Virus Res. 186, 61–75. doi: 10.1016/j.virusres.2013.11.018
Liu, J., Zhao, H., Jiang, K., Zhou, X.-P., and Liu, S.-S. (2009). Differential indirect effects of two plant viruses on an invasive and an indigenous whitefly vector: implications for competitive displacement. Ann. Appl. Biol. 155, 439–448. doi: 10.1111/j.1744-7348.2009.00366.x
Louca, S., Doebeli, M., and Parfrey, L. W. (2018). Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 6:41. doi: 10.1186/s40168-018-0420-9
Luan, J.-B., Chen, W., Hasegawa, D. K., Simmons, A. M., Wintermantel, W. M., Ling, K.-S., et al. (2015). Metabolic coevolution in the bacterial Symbiosis of whiteflies and related plant sap-feeding insects. Genome Biol. Evol. 7, 2635–2647. doi: 10.1093/gbe/evv170
Luan, J., Sun, X., Fei, Z., and Douglas, A. E. (2018). Maternal inheritance of a single somatic animal cell displayed by the Bacteriocyte in the whitefly Bemisia tabaci. Curr. Biol. 28, 459–465.e3. doi: 10.1016/j.cub.2017.12.041
Mouton, L., Henri, H., Romba, R., Belgaidi, Z., Gnankiné, O., and Vavre, F. (2022). Analyses of symbiotic bacterial communities in the plant pest Bemisia tabaci reveal high prevalence of Candidatus Hemipteriphilus asiaticus on the African continent. Peer Commun. J. 2, 1–17. doi: 10.24072/pcjournal.103
Mugerwa, H., Seal, S., Wang, H.-L., Patel, M. V., Kabaalu, R., Omongo, C. A., et al. (2018). African ancestry of New World, Bemisia tabaci-whitefly species. Sci. Rep. 8, 1–11. doi: 10.1038/s41598-018-20956-3
Mugerwa, H., Wang, H.-L., Sseruwagi, P., Seal, S., and Colvin, J. (2020). Whole-genome single nucleotide polymorphism and mating compatibility studies reveal the presence of distinct species in sub-Saharan Africa Bemisia tabaci whiteflies. Insect Sci. 28, 1553–1566. doi: 10.1111/1744-7917.12881
Mugerwa, H., Wang, H. L., Sseruwagi, P., Seal, S., and Colvin, J. (2021). Whole-genome single nucleotide polymorphism and mating compatibility studies reveal the presence of distinct species in sub-Saharan Africa Bemisia tabaci whiteflies. Insect Sci. 28, 1553–1566. doi: 10.1111/1744-7917.12881
Munson, M. A., Baumann, P., and Kinsey, M. G. (1991). Buchnera gen. Nov. and Buchnera aphidicola sp. nov., a taxon consisting of the Mycetocyte-associated, primary endosymbionts of aphids. Int. J. Syst. Evol. Microbiol. 41, 566–568. doi: 10.1099/00207713-41-4-566
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’Hara, R., et al. (2013). Package ‘vegan’. Community ecology package, version 2.
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’Hara, R., et al. (2014). Package ‘vegan’. Community ecology package, R package version 2.
Pan, H., Li, X., Ge, D., Wang, S., Wu, Q., Xie, W., et al. (2012). Factors affecting population dynamics of maternally transmitted endosymbionts in Bemisia tabaci. PLoS One 7:e30760. doi: 10.1371/journal.pone.0030760
Paredes-Montero, J. R., Zia-Ur-Rehman, M., Hameed, U., Haider, M. S., Herrmann, H.-W., and Brown, J. K. (2020). Genetic variability, community structure, and horizontal transfer of endosymbionts among three Asia II-Bemisia tabaci mitotypes in Pakistan. Ecol. Evol. 10, 2928–2943. doi: 10.1002/ece3.6107
Raina, H. S., Singh, A., Popli, S., Pandey, N., and Rajagopal, R. (2015). Infection of bacterial endosymbionts in insects: a comparative study of two techniques viz PCR and FISH for detection and localization of symbionts in whitefly, Bemisia tabaci. PLoS One 10:e0136159. doi: 10.1371/journal.pone.0136159
Rao, Q., Rollat-Farnier, P.-A., Zhu, D.-T., Santos-Garcia, D., Silva, F. J., Moya, A., et al. (2015). Genome reduction and potential metabolic complementation of the dual endosymbionts in the whitefly Bemisia tabaci. BMC Genom. 16:226. doi: 10.1186/s12864-015-1379-6
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Santos-Garcia, D., Farnier, P.-A., Beitia, F., Zchori-Fein, E., Vavre, F., Mouton, L., et al. (2012). Complete genome sequence of “Candidatus Portiera aleyrodidarum” BT-QVLC, an obligate symbiont that supplies amino acids and carotenoids to Bemisia tabaci. J. Bacteriol. 194, 6654–6655. doi: 10.1128/JB.01793-12
Santos-Garcia, D., Juravel, K., Freilich, S., Zchori-Fein, E., Latorre, A., Moya, A., et al. (2018). To B or not to B: comparative genomics suggests Arsenophonus as a source of B vitamins in whiteflies. Front. Microbiol. 9:2254. doi: 10.3389/fmicb.2018.02254
Santos-Garcia, D., Mestre-Rincon, N., Ouvrard, D., Zchori-Fein, E., and Morin, S. (2020). Portiera gets wild: genome instability provides insights into the evolution of both whiteflies and their endosymbionts. Genome Biol. Evol. 12, 2107–2124. doi: 10.1093/gbe/evaa216
Sseruwagi, P., Wainaina, J., Ndunguru, J., Tumuhimbise, R., Tairo, F., Guo, J.-Y., et al. (2018). The first transcriptomes from field-collected individual whiteflies (Bemisia tabaci, Hemiptera: Aleyrodidae): a case study of the endosymbiont composition. Gates Open Res. 1:16. doi: 10.12688/gatesopenres.12783.3
Sun, D.-L., Jiang, X., Wu, Q. L., and Zhou, N.-Y. (2013). Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Appl. Environ. Microbiol. 79, 5962–5969. doi: 10.1128/AEM.01282-13
Tajebe, L., Guastella, D., Cavalieri, V., Kelly, S., Hunter, M., Lund, O. S., et al. (2015). Diversity of symbiotic bacteria associated with Bemisia tabaci (Homoptera: Aleyrodidae) in cassava mosaic disease pandemic areas of Tanzania. Ann. Appl. Biol. 166, 297–310. doi: 10.1111/aab.12183
Tay, W., Elfekih, S., Polaszek, A., Evans, G., Gordon, K., and De Barro, P. (2017). Novel molecular approach to define pest species status and tritrophic interactions from historical Bemisia specimens. Sci. Rep. 7, 1–13. doi: 10.1038/s41598-017-00528-7
Thao, M. L., and Baumann, P. (2004). Evolutionary relationships of primary prokaryotic endosymbionts of whiteflies and their hosts. Appl. Environ. Microbiol. 70, 3401–3406. doi: 10.1128/AEM.70.6.3401-3406.2004
Thierry, M., Becker, N., Hajri, A., Reynaud, B., Lett, J.-M., and Delatte, H. (2011). Symbiont diversity and non-random hybridization among indigenous (Ms) and invasive (B) biotypes of Bemisia tabaci. Mol. Ecol. 20, 2172–2187. doi: 10.1111/j.1365-294X.2011.05087.x
Ulyantsev, V. I., Kazakov, S. V., Dubinkina, V. B., Tyakht, A. V., and Alexeev, D. G. (2016). MetaFast: fast reference-free graph-based comparison of shotgun metagenomic data. Bioinformatics 32, 2760–2767. doi: 10.1093/bioinformatics/btw312
Větrovský, T., and Baldrian, P. (2013). The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One 8:e57923. doi: 10.1371/journal.pone.0057923
Walsh, P. S., Metzger, D. A., and Higuchi, R. (1991). Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10, 506–513.
Wang, L., Jiang, J., Xu, Y., Zeng, L., and Lu, Y. (2016). Occurrence of three intracellular symbionts (Wolbachia, Arsenophonus, Cardinium) among ants in southern China. J. Asia Pac. Entomol. 19, 981–988. doi: 10.1016/j.aspen.2016.07.019
Wang, Y.-B., Ren, F.-R., Yao, Y.-L., Sun, X., Walling, L. L., Li, N.-N., et al. (2020). Intracellular symbionts drive sex ratio in the whitefly by facilitating fertilization and provisioning of B vitamins. ISME J. 14, 2923–2935. doi: 10.1038/s41396-020-0717-0
Warnes, G. R., Bolker, B., Bonebakker, L., Gentleman, R., Huber, W., Liaw, A., et al. (2009). Gplots: various R programming tools for plotting data. R Package Version 2:1.
Zchori-Fein, E., Lahav, T., and Freilich, S. (2014). Variations in the identity and complexity of endosymbiont combinations in whitefly hosts. Front. Microbiol. 5:310. doi: 10.3389/fmicb.2014.00310
Keywords: cassava, Illumina HiSeq, temporal change, endosymbionts, whitefly
Citation: El Hamss H, Maruthi MN, Ally HM, Omongo CA, Wang H-L, van Brunschot S, Colvin J and Delatte H (2022) Spatio-temporal changes in endosymbiont diversity and composition in the African cassava whitefly, Bemisia tabaci SSA1. Front. Microbiol. 13:986226. doi: 10.3389/fmicb.2022.986226
Edited by:
Solomon Maina, Elizabeth Macarthur Agricultural Institute, AustraliaReviewed by:
Kilaza Samson Mwaikono, Dar es Salaam Institute of Technology, TanzaniaFrancis Onono Wamonje, National Institute of Agricultural Botany (NIAB), United Kingdom
Katie Louise Robinson, Elizabeth Macarthur Agricultural Institute, Australia
Copyright © 2022 El Hamss, Maruthi, Ally, Omongo, Wang, van Brunschot, Colvin and Delatte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hajar El Hamss, SGFqYXIuZWxoYW1zc0BnbWFpbC5jb20=; Hélène Delatte, aGVsZW5lLmRlbGF0dGVAY2lyYWQuZnI=