 Dejan B. Budimirovic1,2*
Dejan B. Budimirovic1,2* Kelli C. Dominick3,4
Kelli C. Dominick3,4 Lidia V. Gabis5,6
Lidia V. Gabis5,6 Maxwell Adams7
Maxwell Adams7 Mathews Adera7Linda Huang7Pamela Ventola8
Mathews Adera7Linda Huang7Pamela Ventola8 Nicole R. Tartaglia9Elizabeth Berry-Kravis10
Nicole R. Tartaglia9Elizabeth Berry-Kravis10- 1Department of Psychiatry, Kennedy Krieger Institute, Johns Hopkins University, Baltimore, MD, United States
- 2Department of Psychiatry and Behavioral Sciences-Child Psychiatry, Johns Hopkins School of Medicine, Baltimore, MD, United States
- 3Department of Psychiatry, University of Cincinnati College of Medicine, Cincinnati, OH, United States
- 4Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States
- 5Maccabi HMO, Tel Aviv-Yafo, Israel
- 6Sackler School of Medicine, Tel Aviv University, Tel Aviv-Yafo, Israel
- 7Ovid Therapeutics Inc., New York, NY, United States
- 8Child Study Center, Yale University, New Haven, CT, United States
- 9University of Colorado School of Medicine, Children’s Hospital Colorado, Aurora, CO, United States
- 10Department of Pediatrics, Neurological Sciences, Biochemistry, Rush University Medical Center, Chicago, IL, United States
Background: Fragile X syndrome (FXS), the most common single-gene cause of intellectual disability and autism spectrum disorder (ASD), is caused by a >200-trinucleotide repeat expansion in the 5’ untranslated region of the fragile X mental retardation 1 (FMR1) gene. Individuals with FXS can present with a range of neurobehavioral impairments including, but not limited to: cognitive, language, and adaptive deficits; ASD; anxiety; social withdrawal and avoidance; and aggression. Decreased expression of the γ-aminobutyric acid type A (GABAA) receptor δ subunit and deficient GABAergic tonic inhibition could be associated with symptoms of FXS. Gaboxadol (OV101) is a δ-subunit–selective, extrasynaptic GABAA receptor agonist that enhances GABAergic tonic inhibition, providing the rationale for assessment of OV101 as a potential targeted treatment of FXS. No drug is approved in the United States for the treatment of FXS.
Methods: This 12-weeks, randomized (1:1:1), double-blind, parallel-group, phase 2a study was designed to assess the safety, tolerability, efficacy, and optimal daily dose of OV101 5 mg [once (QD), twice (BID), or three-times daily (TID)] when administered for 12 weeks to adolescent and adult men with FXS. Safety was the primary study objective, with key assessments including treatment-emergent adverse events (TEAEs), treatment-related adverse events leading to study discontinuation, and serious adverse events (SAEs). The secondary study objective was to evaluate the effect of OV101 on a variety of problem behaviors.
Results: A total of 23 participants with FXS (13 adolescents, 10 adults) with moderate-to-severe neurobehavioral phenotypes (Full Scale Intelligence Quotient, 41.5 ± 3.29; ASD, 82.6%) were randomized to OV101 5 mg QD (n = 8), 5 mg BID (n = 8), or 5 mg TID (n = 7) for 12 weeks. OV101 was well tolerated across all 3 treatment regimens. The most common TEAEs were upper respiratory tract infection (n = 4), headache (n = 3), diarrhea (n = 2), and irritability (n = 2). No SAEs were reported. Improvements from baseline to end-of-treatment were observed on several efficacy endpoints, and 60% of participants were identified as treatment responders based on Clinical Global Impressions-Improvement.
Conclusions: Overall, OV101 was safe and well tolerated. Efficacy results demonstrate an initial signal for OV101 in individuals with FXS. These results need to be confirmed in a larger, randomized, placebo-controlled study with optimal outcomes and in the most appropriate age group.
Clinical Trial Registration: www.ClinicalTrials.gov, identifier: NCT03697161
Introduction
Fragile X syndrome (FXS) is an inherited neurodevelopmental disorder caused by a full-mutation expansion [>200 trinucleotide (CGG) repeats] in the promoter region of the fragile X mental retardation 1 (FMR1) gene. The resulting epigenetic silencing of FMR1 causes a deficiency in or absence of the gene’s encoded protein, fragile X mental retardation protein (FMRP) (Bagni et al., 2012). With an estimated prevalence of approximately 1 in 4,000 males and 1 in 6,000 females, FXS is the most common single-gene cause of intellectual disability (ID) and autism spectrum disorder (ASD) (Crawford et al., 2001; Tassone et al., 2012). Individuals with FXS can present with a broad range of neurobehavioral abnormalities, including cognitive deficits e.g., ID, 95% of males, 35% of females (Rousseau et al., 1994; Wright-Talamante et al., 1996); language disorders (non-verbal to perseverative); ASD (51% of males); psychiatric and behavioral impairments, such as anxiety, hyperarousal, and other sensory processing difficulties; repetitive behaviors; attentional network deficits; and irritability often accompanied by aggressive and/or self-injurious behaviors (Hagerman et al., 2009; Kaufmann et al., 2017; Raspa et al., 2018; Budimirovic et al., 2020; Reisinger et al., 2020). Patients with FXS also present with neurological abnormalities, such as motor and coordination difficulties (Gabis et al., 2011) and a higher incidence of epilepsy (Hagerman et al., 2009). As an X-linked disorder, males with FXS have a more severe phenotype than females, with evidence suggesting an inverse relationship between FMRP deficiency and severity of FXS-associated neurobehavioral phenotype (Kim et al., 2019; Budimirovic et al., 2020). FXS phenotype severity can also be affected by size mosaicism (premutation), X-chromosome inactivation in females, and variation in the methylation status of full mutations (Nolin et al., 1994; Budimirovic et al., 2020).
Symptom-based, off-label treatments used in the management of patients with FXS include psychostimulants for attention deficit hyperactivity disorder (ADHD) symptoms; α2-adrenergic receptor agonists for sensory overstimulation, hyperarousal, hyperactivity, and sleep disturbances; anticonvulsants for seizures and mood instability; selective serotonin reuptake inhibitors for anxiety; and antipsychotics and antidepressants for aggression, anxiety, and sleep disturbances (Berry-Kravis et al., 2012; Eckert et al., 2019). However, few randomized, controlled studies have been conducted to formally evaluate these symptomatic interventions in FXS (Berry-Kravis et al., 2018). Although safe and effective treatments for FXS are needed, particularly for targeted treatments that surpass symptom-based management, no medication is approved in the United States (Lee et al., 2018).
Evidence suggests that GABAergic dysfunction and the resulting excitatory and inhibitory imbalance can contribute to the pathophysiology of FXS (Berry-Kravis et al., 2018). GABA (γ-aminobutyric acid) is the primary inhibitory neurotransmitter in the brain. Fmr1 knockout (KO) mice exhibit decreased GABA type A (GABAA) receptor δ subunit expression, GABA synthesis, and GABA levels, resulting in reduced tonic inhibition. Reduced tonic inhibition may, in turn, lead to abnormal excitatory signaling in the brain, culminating in a range of symptoms (Olmos-Serrano et al., 2011; Zafarullah and Tassone, 2019). Tonic inhibition, mediated by δ-subunit–containing GABAA receptors, plays an important role in various functions of different regions in the brain (Cope et al., 2005; Brickley and Mody, 2012; Lee and Maguire, 2014; Whissell et al., 2015) and is a potential treatment target for FXS.
Evidence from clinical studies of GABAergic therapies, specifically arbaclofen (GABAB agonist), ganaxolone (GABAA receptor modulator), and acamprosate (GABAB and GABAA receptor modulator), suggests that GABA receptor modulation may hold therapeutic potential for treating the core behavioral problems associated with FXS (Zafarullah and Tassone, 2019). For example, in 2 placebo-controlled, phase 3 studies of patients with FXS aged 5–11 or 12–50 years, arbaclofen did not differ statistically from placebo on the primary outcome measure of social avoidance on the Aberrant Behavior Checklist-Community Edition (ABC-C) refactored for FXS (ABC-CFXS) (Sansone et al., 2012). However, improvements in irritable behavior and parenting stress were observed for children who received the highest drug dose [10 mg three-times daily (TID)] (Berry-Kravis et al., 2017).
Gaboxadol (OV101) is a δ-subunit–selective extrasynaptic GABAA receptor agonist that has been shown to be well tolerated and to confer improvements in sleep induction in 2 double-blind, placebo-controlled studies of adults with insomnia (Roth et al., 2010). Mechanistically, in Fmr1 KO mice, OV101 restored tonic inhibition in the amygdala, reduced sensory hypersensitivity and motor hyperactivity, and improved pre-pulse inhibition (Olmos-Serrano et al., 2011). In other Fmr1 KO mouse studies, OV101 also normalized hyperactivity and repetitive, social, and anxiety-like behavior, which have been associated with decreased expression of the GABAA receptor δ subunit and deficient GABAergic tonic inhibition (Gantois et al., 2006; Olmos-Serrano et al., 2010; Martin et al., 2014). Thus, OV101 may be effective in treating humans with FXS.
Here, we report results from the first interventional clinical study of OV101 in FXS. This 12-weeks, randomized, double-blind, proof-of-concept, phase 2a study evaluated the safety and efficacy profiles of multiple doses of OV101 in adolescent and adult males with FXS.
Materials and Methods
Study Participants
Adolescent and adult males aged 13–22 years inclusive who had a clinically- and molecularly-confirmed full mutation of the FMR1 gene, were moderately-to-severely affected by FXS [score of ≥4 on the Clinical Global Impressions–Severity (CGI-S) scale], and had a Full Scale Intelligence Quotient score <75, were eligible to participate. Antiepileptic and/or psychoactive medication use was permitted if no more than 3 such medications were being used and the dose-regimen for each medication was stable for at least 4 weeks before randomization and then maintained throughout the study.
To ensure that the effects of OV101 were evaluated in a patient population with minimal FMRP expression, females were excluded from the study. Other exclusion criteria included a history of uncontrolled seizure disorder or seizure episodes within 6 months of screening, a change in anticonvulsant pharmacotherapy within 3 months of screening, use of a GABAergic agent on a regular schedule, use of a cannabinoid derivative, a history of suicidal behavior, and any clinically significant medical condition or laboratory finding at screening that could interfere with study conduct/participation or pose an unacceptable risk.
Study Design
This randomized, double-blind, parallel-group, phase 2a study (ClinicalTrials.gov identifier: NCT03697161), conducted at 7 sites in the United States and 1 site in Israel, was designed to assess the safety, tolerability, and efficacy of 3 OV101 dose-regimens administered over 12 weeks to patients with FXS (Figure 1). Independent ethics review boards approved the trial protocol at each trial site. Eligible participants were randomized (1:1:1) to receive OV101 5 mg once daily (QD), twice daily (BID), or TID for 12 weeks. Randomization was stratified by age group (adolescent vs. adult). All patients received study medication TID (morning, afternoon, and evening), with OV101 administered to all participants in the morning and either OV101 or placebo administered in the afternoon and evening throughout the treatment period (including baseline). Dose adjustments were not permitted.
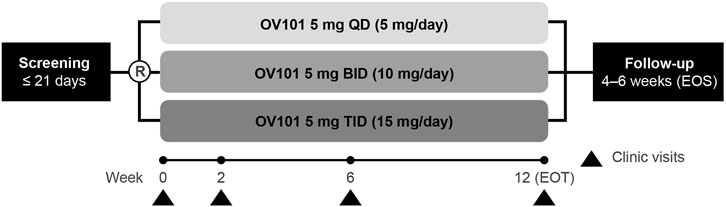
FIGURE 1. Study design. R, randomization; OV101, gaboxadol; QD, once daily; BID, twice daily; TID, 3-times daily; EOT, end of treatment; EOS, end of study.
Clinic visits were scheduled at weeks 2, 6, and 12 [end of treatment (EOT)], with an end-of-study (EOS) follow-up visit at some time during weeks 16 and 18. In addition to the assessments performed during every clinic visit, information on adverse events, concomitant medication use, and suicidality were collected during phone calls at the end of weeks 1, 4, 8, and 10. The caregiver or legally acceptable representative (LAR) completed paper sleep diaries on behalf of participants, and participants wore actigraphs (wrist-worn sleep monitors) (Dueck et al., 2020) for 7 days immediately preceding the baseline, week 2, week 6, EOT, and EOS visits.
The study was designed by the sponsor in collaboration with a consortium of experts in FXS and approved by the internal review board at each participating site. Informed consent was provided by a caregiver or LAR, and, to the extent possible, participants also assented to the protocol.
Objectives and Assessments
The primary objective was to assess the safety and tolerability of OV101 during the 12-weeks treatment period. Safety endpoints were treatment-emergent adverse events (TEAEs), treatment-related adverse events, TEAEs leading to study discontinuation, and serious adverse events (SAEs). Rate of OV101 compliance was calculated as: (actual dosage of OV101/expected dosage of OV101) × 100%; patients achieving a rate of ≥80% based on a medication diary maintained by the LAR was considered compliant.
The secondary objective was to evaluate the efficacy of OV101 during the 12-weeks treatment period. Efficacy endpoints were Clinical Global Impressions-Improvement (CGI-I) scale score and changes from baseline in CGI-S total and subscale scores, ABC-C total and subscale scores (Aman et al., 1985), ABC-CFXS total and subscale scores (Sansone et al., 2012), Anxiety, Depression, and Mood Scales (ADAMS) total and subscale scores (Esbensen et al., 2003), Repetitive Behavior Scale–Revised (RBS-R) total and subscale scores (Lam and Aman, 2007), Short Sensory Profile–2 total and subscale scores, and Conners 3 subscale scores (Conners et al., 1998). The ABC-C is a 58-item questionnaire completed by the LAR/caregiver that assesses a range of behaviors, including irritability, lethargy/social withdrawal, inappropriate speech, hyperactivity, and stereotypic behavior (Aman et al., 1985). Each item is rated on a scale of 0–3 (“not at all a problem” to “the problem is severe in degree”). Compared with the ABC-C, ABC-CFXS includes a new evaluation on social avoidance, modified evaluations of irritability, hyperactivity, lethargy/withdrawal, and stereotypy, and a similar evaluation of inappropriate speech (Sansone et al., 2012). ADAMS, a LAR/caregiver-completed assessment that screens comprehensively for anxiety and depression in persons with ID, is a 28-item questionnaire grouped into 5 subscales that assesses the frequency and severity of manic/hyperactive behavior, depressed mood, social avoidance, general anxiety, and obsessive behavior that are rated on a scale of 0–3 (“not a problem” to “severe problem”) (Esbensen et al., 2003). Also completed by the LAR/caregiver, the RBS-R is a 43-item questionnaire assessing a variety of repetitive behaviors with 6 behavior subscales: stereotyped, ritualistic, self-injurious, compulsive, restricted, and sameness (Lam and Aman, 2007). Behaviors are rated on a scale of 0–3 (“behavior does not occur” to “behavior occurs and is a severe problem”). The Short Sensory Profile-2 (PsychCorp, San Antonio, TX) is a LAR/caregiver-completed, 34-item questionnaire that evaluates sensory processing patterns in the context of home, school, and community-based activities based on a scale of 1–5 (“almost never” to “almost always”) (Simpson et al., 2019). The Conners 3rd Edition (North Tonawanda Multi-Health System, North Tonawanda, NY) was used by a LAR/caregiver to rate ADHD; subscales include Assessment of Validity (positive impression, negative impression, inconsistency index), Content Scales (inattention, hyperactivity/impulsivity, learning problems, executive functioning, defiance/aggression, peer relations), and Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) Symptom Scales (i.e., ADHD predominantly inattention type, ADHD predominantly hyperactive-impulsive) (Conners et al., 1998).
Exploratory efficacy endpoints included changes from baseline in Parent Global Impressions–Severity (PGI–S) and Parent Global Impressions–Improvement (PGI–I) scores and clinician-rated changes from baseline in the top 3 concerns identified by caregivers [per a visual analog scale (VAS)] (Adams et al., 2011). The concerns were identified on a per-patient basis and could have derived from any symptom domain related to FXS. The severity of each concern was scored by caregivers using a 10-cm VAS and was based on the number of centimeters from the left margin, with anchors of “not at all severe” (left side of the line, 0 cm) and “very severe” (right side of the line, 10 cm).
Statistical Methods
The safety analyses were performed on the safety population, which comprised all patients receiving ≥1 dose of study drug. The efficacy analyses were performed on the full-analysis set, which comprised all patients receiving ≥1 dose of study drug and having ≥1 post-baseline efficacy assessment. Participants were analyzed according to the treatment to which they were randomized.
Safety and efficacy outcomes were analyzed using descriptive statistics. To investigate any trends visible in the descriptive analyses, a mixed-effects model for repeated measures (MMRM), with dosing regimen, visit, and age group as fixed effects; age and baseline as covariates (if appropriate); and visit × dosing regimen as an interaction, was used. With an assumed unstructured covariance structure, the least squares mean change from baseline [and 95% confidence interval (CI)] for each dosing regimen, as well as the overall study population, at each post-baseline visit was estimated. The least squares mean difference (and corresponding 95% CI) was estimated for each pairwise comparison among the 3 dosing regimens. In the case of a statistically significant age group main effect (p < 0.05) in the MMRM or age effects in the descriptive analyses, the mean change (and corresponding 95% CI) was estimated for each age group separately, as well as the overall study population, by including an additional interaction term (age × week × dosing regimen) in the MMRM. Post-hoc analyses of the changes or percent changes from baseline for CGI-S total and subscale scores, ABC-CFXS total and subscale scores, and ADAMS total and subscale scores, were performed using the parametric Student’s t-test.
Results
Patients
A total of 23 participants with FXS (13 adolescents, 10 adults) were randomized to OV101 5 mg QD (n = 8), BID (n = 8), or TID (n = 7) (Figure 2). One participant in each dosing group discontinued the study due to withdrawn consent (QD, n = 1; TID, n = 1) or TEAE (agitation; BID, n = 1).
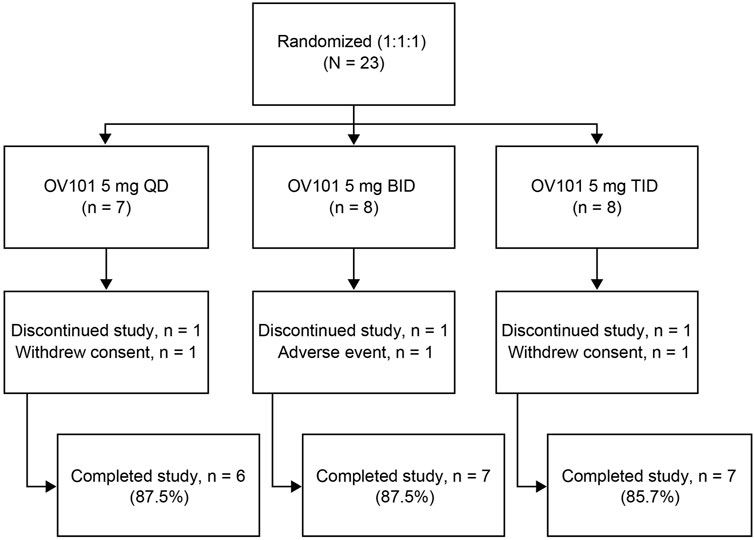
FIGURE 2. Patient disposition. OV101, gaboxadol; QD, once daily; BID, twice daily; TID, 3-times daily.
Baseline demographic and clinical characteristics are summarized in Tables 1, 2, respectively. The overall study population (n = 23) exhibited mostly severe neurobehavioral abnormalities, with a mean ± standard deviation (SD) Stanford Binet-5 IQ score (derived according to standard methodology and not z-deviation method) of 41.5 ± 3.29 and verbal IQ score of 44.5 ± 3.23 (Sansone et al., 2014). Of these 23 participants, 82.6% met the Diagnostic and Statistical Manual of Mental Disorders (5th edition) criteria for ASD. The mean rate of OV101 compliance was 95.4% (n = 22), with 21 patients considered compliant (≥80% compliance rate) with assigned OV101 treatment.
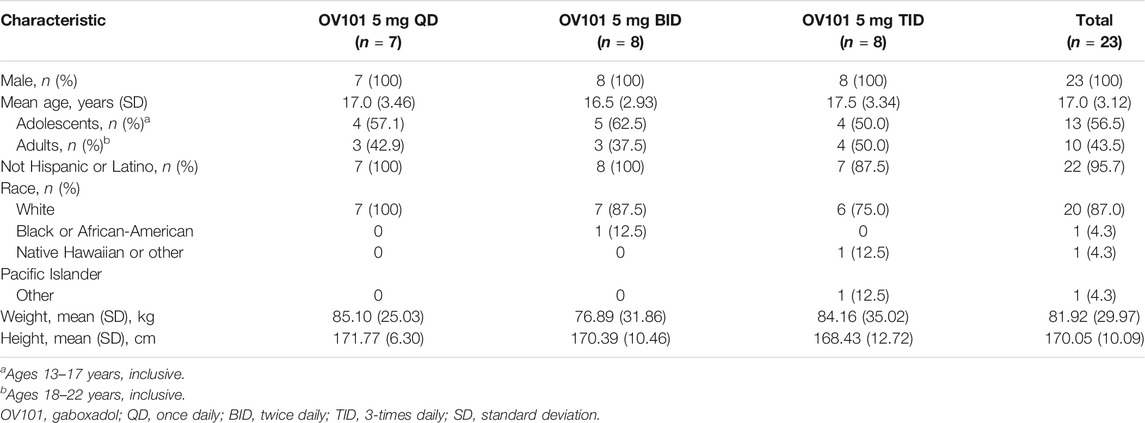
TABLE 1. Baseline demographic characteristics.
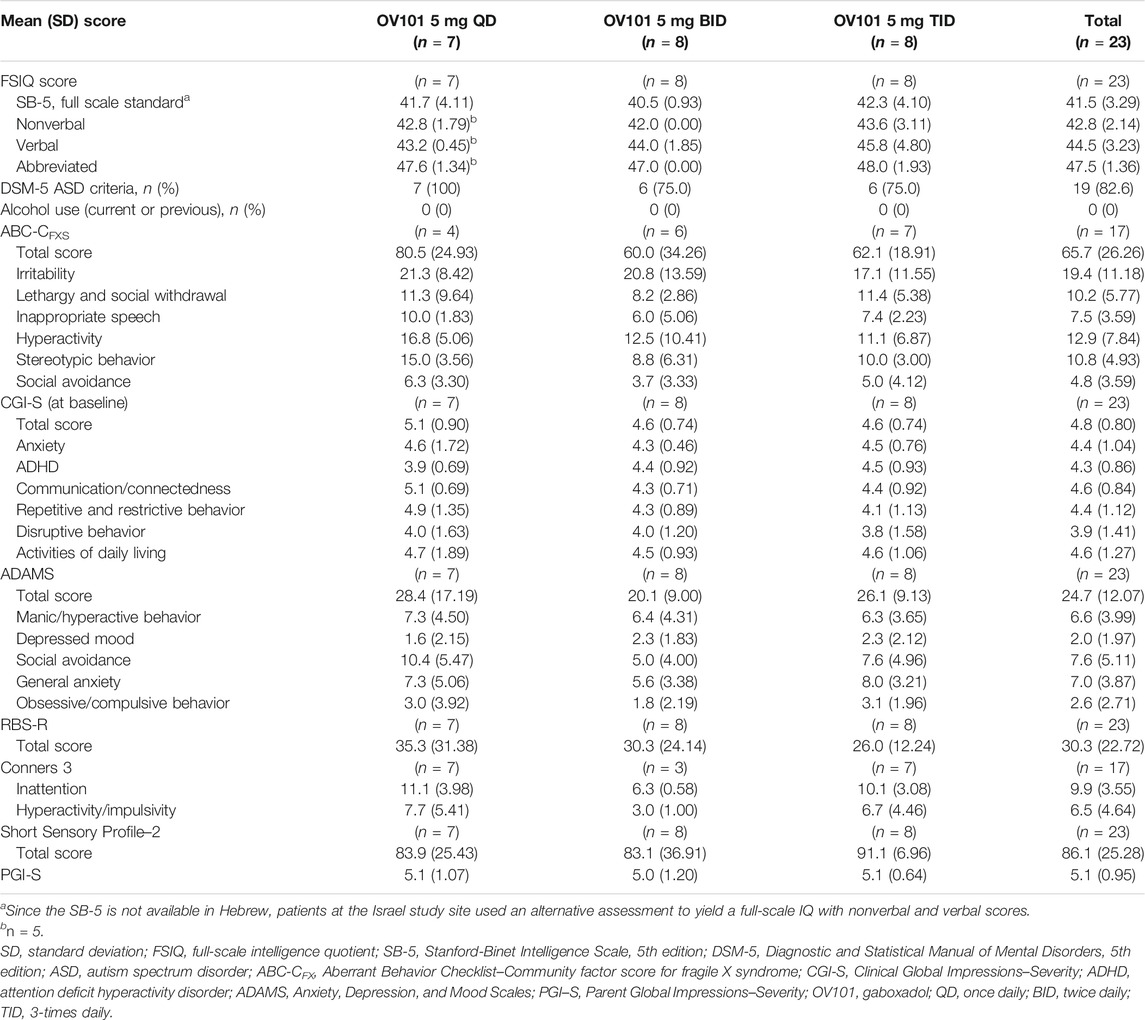
TABLE 2. Baseline clinical characteristics.
Safety and Tolerability
Table 3 summarizes safety and tolerability measures for OV101. The mean ± SD duration of exposure to OV101 was 73.1 ± 31.85, 77.1 ± 19.47, and 75.3 ± 25.64 days for the QD, BID, and TID regimens, respectively. The majority of participants (n = 16, 69.6%) reported ≥1 TEAE (QD, 28.6%; BID, 100.0%; TID, 75.0%). Across all dosing regimens, the only TEAEs occurring in ≥2 patients were upper respiratory tract infection (n = 4, 17.4%), headache (n = 3, 13.0%), diarrhea (n = 2, 8.7%), and irritability (n = 2, 8.7%).
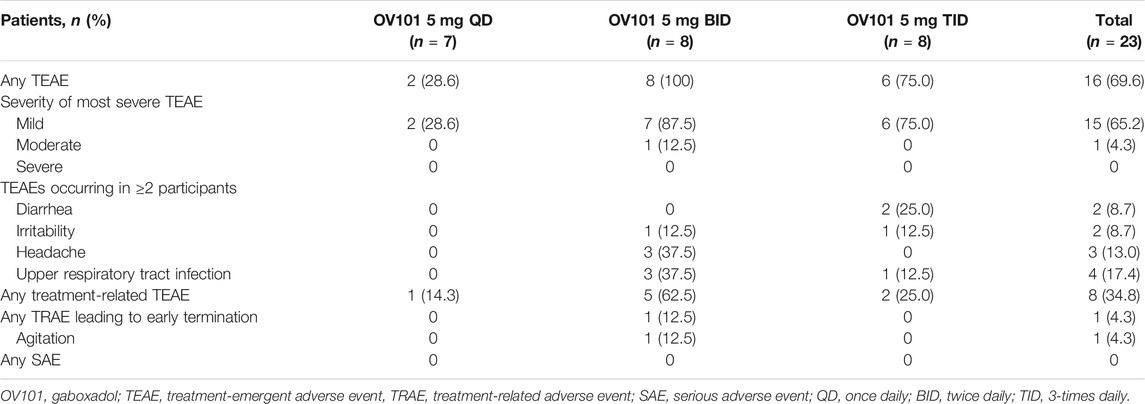
TABLE 3. Summary of OV101 safety and tolerability.
All TEAEs were mild or moderate in severity, with most affected patients experiencing an event of mild severity [overall, 93.8% (15/16); QD, 100% (2/2); BID, 87.5% (7/8); TID, 100% (6/6)]. One-third of all patients (8/23, 34.8%) experienced ≥1 TEAE possibly or probably related to the study medication, with 5/8 (62.5%) of those with any treatment-related TEAE assigned to the OV101 5 mg BID group. No SAE or death was reported. Overall, no trends were observed for hematology, clinical chemistry, and urinalysis laboratory evaluations. There were no notable differences between OV101 treatment groups or discernable trends in vital signs or physical examination changes.
Efficacy
Clinician-Rated Assessments
Figure 3 shows that the majority (60.0%) of all OV101-treated participants were rated by clinicians as being CGI-I responders, defined as a score of ≤3 (minimally improved), at week 12, with 40.0% considered “much improved” and 20.0% considered “minimally improved.” The highest percentage of CGI-I responders (71.5%) was observed in the OV101 5 mg BID group, followed by 66.6% in the QD group and 42.9% in the TID group; in the OV101 5 mg BID group, 42.9% of participants were considered “much improved.” Clinician-rated secondary endpoints, such as CGI-S total score (−0.4, p < 0.01) and subscale scores related to communication and connectedness (−0.60, p < 0.001), anxiety (−0.50, p < 0.01), ADHD (−0.5, p < 0.05), and VABS-III activities of daily living (ADLs; −0.3, p < 0.05), also showed significant improvements from baseline to week 12 (Table 4). No meaningful differences between age groups in clinician-rated assessments were noted (data not shown).
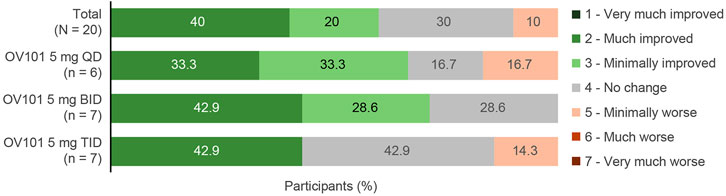
FIGURE 3. CGI-I score at week 12. Among the 3 dosing regimens, the percentage of CGI-I responders was greatest with OV101 5 mg BID. CGI-I, Clinical Global Impressions–Improvement; OV101, gaboxadol; QD, once daily; BID, twice daily; TID, 3-times daily.
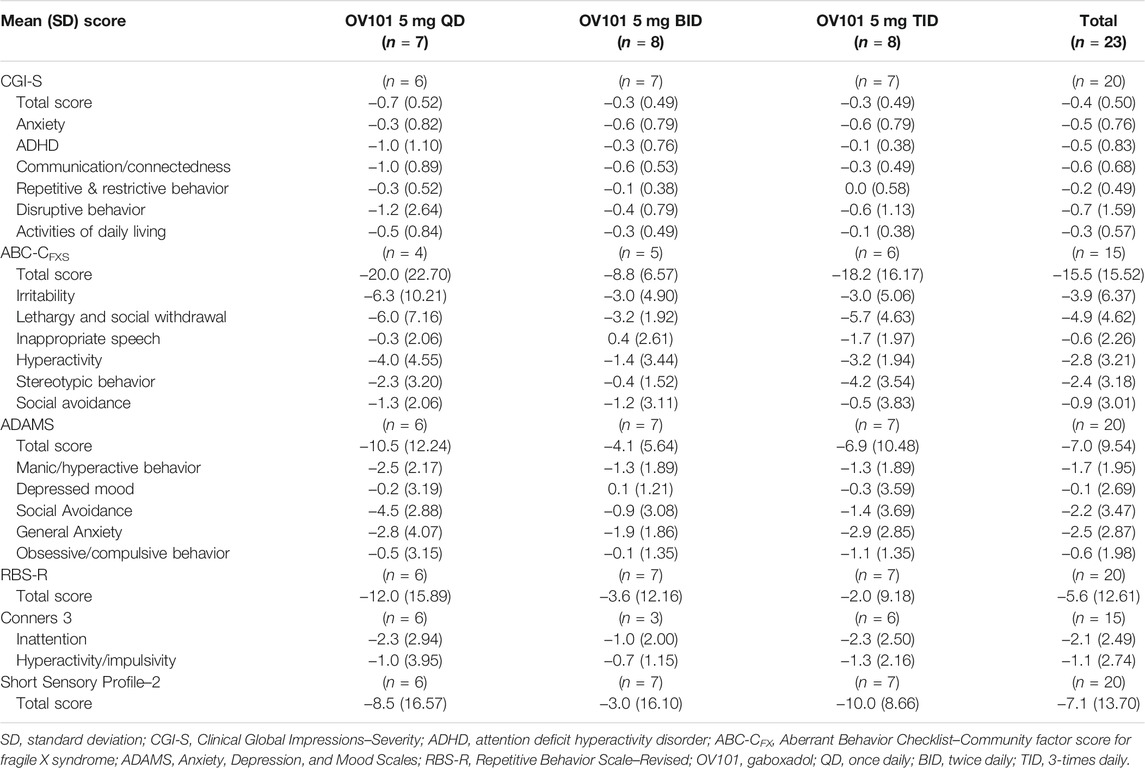
TABLE 4. Change in secondary efficacy measures from baseline to week 12.
Caregiver/LAR Assessments
Changes from baseline to week 12 in secondary efficacy measures are summarized in Table 4. There were significant improvements from baseline to week 12 in caregiver-rated ABC-CFXS total score (26%, p < 0.01), indicating improvements in common behavioral problems associated with FXS, and secondary endpoints [lethargy/social withdrawal (38%, p < 0.01), hyperactivity (29%, p < 0.01), stereotypic behavior (21%, p < 0.05), and irritability (20%, p < 0.05) subdomains]. Using the original scoring method, ABC-C scores were similar to the ABC-CFXS scoring method. There were significant improvements from baseline to week 12 in caregiver-rated ADAMS total score (−0.7, p < 0.01) and secondary endpoints [general anxiety (−2.5, p < 0.01), social avoidance (−2.2, p < 0.05), and manic/hyperactive (−1.7, p < 0.01) subscales]. Other secondary efficacy endpoints, such as RBS-R, Short Sensory Profile-2, and Conners 3, did not show meaningful improvements from baseline. Changes in exploratory measures from baseline to week 12 are reported in Table 5. In an exploratory analysis, an improvement in PGI-I, defined as a PGI-I score of ≤3 (modest improvement) at week 12, was reported by 55.0% of caregivers (QD, 50.0%; BID, 71.4%; TID, 42.9%). The top 3 concerns identified by caregivers via a VAS total score varied, but the most frequently cited were related to anxiety, disruptive behavior, and ADLs. Based on a preliminary analysis, there do not seem to be noteworthy differences in sleep behavior based on the sleep CGI and actigraphy (data not shown). No meaningful differences between age groups in caregiver/LAR assessments were noted (data not shown).
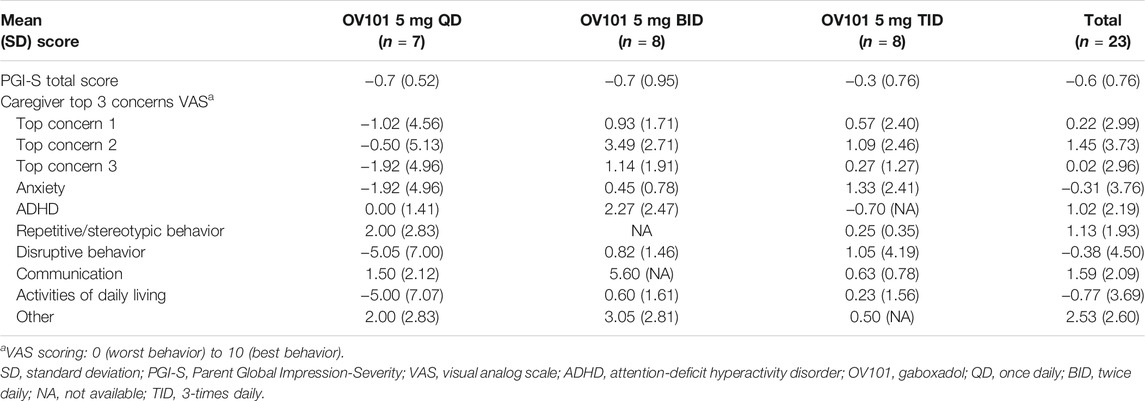
TABLE 5. Change in exploratory efficacy measures from baseline to week 12.
Discussion
We conducted a 12-weeks, double-blind, parallel-group, phase 2a study of 23 adolescent and adult males with FXS with moderate-to-severe neurobehavioral phenotypes to assess the safety of OV101 and its efficacy in targeting core behavioral symptoms in FXS. By showing OV101 5 mg to be generally safe and well tolerated when administered QD, BID, or TID, this first interventional clinical study of OV101 in FXS achieved its primary outcome. Clinical laboratory tests, electrocardiograms, vital signs, and physical examinations showed no trends or distinct safety signals. The majority of participants (16/23, 69.6%) reported ≥1 dose-dependent TEAE of mild severity (15/16, 93.8%). One-third of participants (8/23, 34.8%) experienced ≥1 TEAE possibly or probably related to the study medication, with 62.5% (5/8) of those with any treatment-related adverse event assigned to the OV101 5 mg BID group. This same group also had mild TEAEs [diarrhea and irritability, 2/23 (9%) each; headache, 3/23 (13%); upper respiratory tract infection, 4/23 (17%)]; and 1 participant who terminated treatment early (moderate agitation). No SAEs or deaths were reported, and there was no evidence of withdrawal effects after the study. Importantly, the favorable safety and tolerability profile of OV101 in this FXS study is consistent with that reported in previous OV101 clinical studies of other conditions (Roth et al., 2010; Bird et al., 2021).
This study also demonstrated an initial efficacy signal for OV101 in FXS based on secondary clinician- and caregiver-rated endpoint outcomes, including CGI-I, CGI-S, ABC-CFXS, and ADAMS scores. Using CGI-I as a clinician-rated, syndrome-specific global measure of core features in patients with FXS, the majority of participants treated with OV101 (60%, 12/2) were considered CGI-I responders and 40% (8/20) were rated as “much improved” at week 12. This efficacy signal appeared to be stronger in OV101 5 mg BID and QD groups (71.5 and 66.6% of participants considered CGI-I responders, respectively) versus the TID group (42.9%). From the caregiver perspective, key patient concerns (as captured via VAS) were related to anxiety, disruptive behavior, and ADLs. A high rate of anxiety associated with FXS has been previously reported in the literature (Cordeiro et al., 2011; Weber et al., 2019; Budimirovic et al., 2020), which together with social withdrawal (Budimirovic et al., 2006; Kaufmann et al., 2008), can be present in FXS with and without ASD (Cordeiro et al., 2011; Budimirovic and Subramanian, 2016; Niu et al., 2017; Budimirovic et al., 2020). Results of this study showed that OV101 treatment was associated with converging improvements from baseline in maladaptive behaviors at week 12 as assessed by ABC-CFXS (26.2% improvement in total score and improvements in irritability and social withdrawal subscales) and ADAMS (21.6% improvement in total score and improvements in anxiety and social avoidance subscales). Our results also showed an initial signal of improvements in disruptive behavior/ADHD symptoms and ADLs with OV101 as assessed by CGI-S and VABS-III, respectively. Finally, caregiver-rated exploratory analyses of PGI-I at week 12 showed a modest improvement of 55.0%, which again showed the strongest signal in the 5 mg BID (71.4%) versus QD (50.0%) and TID (42.9%) groups. Overall, the initial signal of improvements across multiple domains of the core phenotype of FXS is promising. However, given this is the first clinical study to assess OV101 in FXS, these findings need to be replicated and confirmed in a larger, placebo-controlled study with optimal outcomes and in the most appropriate age group (Duy and Budimirovic, 2017; Berry-Kravis et al., 2018).
The design on this OV101 study had both strengths and limitations that should be considered for future FXS studies. A strength of this study was that it had a sufficiently long duration (12 weeks) to assess behavioral changes related to any potential symptomatic effects of OV101; this is supported by previous studies of autism and other psychiatric conditions in which treatment-derived improvements in behavioral symptoms could be observed within 4 weeks (Berry-Kravis et al., 2018). It also allowed for preliminary evaluation of safety and efficacy at 3 different doses. There were several limitations in the design of this study, the first being the lack of a placebo-control arm. FXS studies have been shown to have large placebo effect for behavioral measures (Berry-Kravis et al., 2018; Luu et al., 2020), with some reporting a placebo effect size similar to the OV101 effect size observed in the present study (Berry-Kravis et al., 2016; Youssef et al., 2018). Another limitation was the small sample size (n = 23), which may have been insufficient to assess behavioral efficacy, as prior studies that enrolled more than 100 participants have yielded ambiguous results (Berry-Kravis et al., 2016; Berry-Kravis et al., 2018; Youssef et al., 2018; Berry-Kravis et al., 2020). The size of each treatment arm was small making evaluations of individual OV101 regimens difficult within this study; additional evaluations will be needed to confirm the optimal dose of OV101. Patient stratification is another important factor to consider in FXS studies. Relationships between anxiety and FMRP level and ASD status may help to stratify patients with FXS in clinical studies (Budimirovic et al., 2017; Budimirovic et al., 2020), with results of this study showing that participants taking OV101 5 mg QD or BID achieved greater improvements in problem behaviors then those receiving the highest dosage (5 mg TID). Finally, a separate study is needed to assess the safety, tolerability, and effects of OV101 in female adolescents and adults with FXS.
Given the inherent subjective nature and placebo effects associated with caregiver-rated endpoints (Budimirovic et al., 2017), future FXS studies should also include biomarker endpoints since they can objectively evaluate the efficacy of investigational treatments. The identification and evaluation of valid, sensitive-to-treatment biomarkers is increasingly necessary to reliably track treatment changes in the unfolding wave of clinical studies in FXS (Budimirovic et al., 2017; Erickson et al., 2018; Lee et al., 2018) and to substantially mitigate the large placebo effect in FXS studies. Indeed, an effort to differentiate objective from subjective improvements in individuals with FXS is recommended by experts in the field of FXS (Luu et al., 2020). For example, electroencephalography is a potential neural biomarker sensitive to treatment (Erickson et al., 2018; Ethridge et al., 2019), and molecular studies have shown a link between FMR1 expansion, gene methylation, and FMRP deficit and the overall severity of the neurobehavioral phenotype (Berry-Kravis et al., 2018; Budimirovic et al., 2020). Newer performance-rated measures, such as expressive language sampling and the NIH Toolbox, may be able to capture real change and better control the placebo effect. Quantification of key receptor expression in the living human brain of men with FXS is also needed to better understand the results of failed FXS clinical studies and to continue to optimize FXS clinical study designs. This measurement may constitute an effective tool to confirm target engagement, for example of NAMs for mGluR5s, in both FXS and ASD (Brašić et al., 2020; Brašić et al., 2021).
In conclusion, the safety, tolerability, and efficacy data from this phase 2a study demonstrate an initial efficacy signal for OV101 in individuals with FXS. The interpretation of the results is confounded by lack of placebo control and small sample size. These results need to be confirmed in a larger, randomized, placebo-controlled study with optimal outcomes and in the most appropriate age group.
Data Availability Statement
The datasets presented in this article are not readily available because Ovid Therapeutics Inc. is committed to providing qualified scientific researchers appropriate access to anonymized data and clinical study information from the company’s clinical trials for the purpose of conducting legitimate scientific research. Requests for specific data will be considered along with the rationale, description of use need, and clinical value of the proposed analysis. Ovid supports an approach to sharing data that responsibly reflects the interests of all parties involved in clinical trials, including protecting the rights and privacy of trial participants, the innovator’s intellectual property rights, and other incentives for innovation, and as such, will evaluate requests for sharing company clinical trial data with qualified external scientific researchers. Requests to access the data from this clinical trial may be made at Y2xpbmljYWxAb3ZpZHJ4LmNvbQ==. Data will be made available for request after product approval in the United States and European Union, after product development is discontinued, or as otherwise required by law or regulation. There are circumstances that may prevent Ovid from sharing the requested data as the product is investigational at this time. Requests to access the datasets should be directed to Y2xpbmljYWxAb3ZpZHJ4LmNvbQ==.
Ethics Statement
The studies involving human participants were reviewed and approved by Rush University Medical Center Institutional Review Board, The Johns Hopkins Institutional Review Board, Quorum Review IRB, Western IRB, Vanderbilt Human Research Protection Program, Cincinnati Children’s Hospital Medical Center Institutional Review Board, UC Davis Institutional Review Board, and The Chaim Sheba Medical Center Institutional Helsinki Committee. Written informed consent to participate in this study was provided by the participant’s legal guardian/next of kin.
Author Contributions
DB contributed to study concept and design, data interpretation, and was a study investigator. KD contributed to data interpretation and was a study investigator. LG contributed to data interpretation and was a study investigator. MA (4th author) contributed to the study operation and data interpretation. MA (5th author) contributed to the study operation and data interpretation. LH contributed to the study operation and data interpretation. PV contributed to the study design, operation, and data interpretation. NT contributed to the study concept and design, data interpretation and was a study investigator. EB-K contributed to the study concept and design, data interpretation, and was a study investigator. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This study was funded by Ovid Therapeutics, Inc. Medical writing and editorial supported was provided by Ashfield MedCo.
Conflict of Interest
Authors MA (4th author), MA (5th author), and LH were employed by the company Ovid Therapeutics, Inc.
DB has received research funding from Akili, Alcobra, Forest, Lundbeck, Medgenics, Neuren, Ovid Therapeutics, Inc., Pfizer, Purdue, Roche, Seaside Therapeutics, Shire, Sunovion, Supernus, SyneuRX, and Zynerba Pharmaceuticals. All funds have been directed to the Kennedy Krieger Institute and Johns Hopkins medical institutions.
KD has received research funding from the American Academy of Child and Adolescent Psychiatry, Roche, and Ovid Therapeutics Inc. All funds have been directed to Cincinnati Children’s Hospital Medical Center.
NT has received research funding from Alcobra, Neuren, Novartis, Ovid Therapeutics, Inc., Roche, Seaside Therapeutics, and Zynerba. All funds have been directed to University of Colorado or CHCO in support of Fragile X and autism programs.
EB-K has received research funding from Acadia, Alcobra, Anavex, Asuragen Inc., Biogen, BioMarin, Cydan, Fulcrum, GeneTx, GW, Ionis, Lumos, Marinus, Neuren, Neurotrope, Novartis, Orphazyme, Ovid, Roche, Seaside Therapeutics, Tetra Discovery Partners, Ultragenyx, Vtesse/Sucampo/Mallinckrodt Pharmaceuticals, Yamo, and Zynerba. All funds have been directed to Rush University Medical Center in support of rare disease programs.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors wish to thank Jeannie Visootsak for her contributions to the study design, Ying Yan for undertaking the statistical analyses, and the ROCKET Study Sites at Children’s Hospital Colorado, Cincinnati Children’s Hospital Medical Center, Kennedy Krieger Institute/the Johns Hopkins Medical Institutions, Rush University Medical Center, Southwest Autism Research & Resource Center, Suburban Research Associates, University of California Davis MIND Institute, Vanderbilt University Medical Center, The Chaim Sheba Medical Center. Medical writing support was provided, under the direction of the authors, by Ashfield MedComms, an Ashfield Health company.
References
Adams, J. B., Audhya, T., McDonough-Means, S., Rubin, R. A., Quig, D., Geis, E., et al. (2011). Effect of a Vitamin/mineral Supplement on Children and Adults with Autism. BMC Pediatr. 11, 111. doi:10.1186/1471-2431-11-111
Aman, M. G., Singh, N. N., Stewart, A. W., and Field, C. J. (1985). Psychometric Characteristics of the Aberrant Behavior Checklist. Am. J. Ment. Defic. 89, 492–502.
Bagni, C., Tassone, F., Neri, G., and Hagerman, R. (2012). Fragile X Syndrome: Causes, Diagnosis, Mechanisms, and Therapeutics. J. Clin. Invest. 122, 4314–4322. doi:10.1172/JCI63141
Berry-Kravis, E., Des Portes, V., Hagerman, R., Jacquemont, S., Charles, P., Visootsak, J., et al. (2016). Mavoglurant in Fragile X Syndrome: Results of Two Randomized, Double-Blind, Placebo-Controlled Trials. Sci. Transl. Med. 8, 321ra5. doi:10.1126/scitranslmed.aab4109
Berry-Kravis, E., Hagerman, R., Visootsak, J., Budimirovic, D., Kaufmann, W. E., Cherubini, M., et al. (2017). Arbaclofen in Fragile X Syndrome: Results of Phase 3 Trials. J. Neurodev. Disord. 9, 3. doi:10.1186/s11689-016-9181-6
Berry-Kravis, E., Sumis, A., Hervey, C., and Mathur, S. (2012). Clinic-based Retrospective Analysis of Psychopharmacology for Behavior in Fragile X Syndrome. Int. J. Pediatr. 2012, 843016. doi:10.1155/2012/843016
Berry-Kravis, E. M., Erickson, C., Hagerman, R., Tartaglia, N., Cohen, J., Sebree, T., et al. (2020). “ZYN002 Cannabidiol Transdermal Gel in Children and Adolescents with Fragile X Syndrome: Role of Methylation Status as a Correlate to Disease Severity and as a Prognostic Biomarker,” in Poster presented at: The Joint International Child Neurology Congress and Child Neurology Society Meeting, October, 2020. virtual.
Berry-Kravis, E. M., Lindemann, L., Jønch, A. E., Apostol, G., Bear, M. F., Carpenter, R. L., et al. (2018). Drug Development for Neurodevelopmental Disorders: Lessons Learned from Fragile X Syndrome. Nat. Rev. Drug Discov. 17, 280–299. doi:10.1038/nrd.2017.221
Bird, L. M., Ochoa-Lubinoff, C., Tan, W. H., Heimer, G., Melmed, R. D., Rakhit, A., et al. (2021). The STARS Phase 2 Study: a Randomized Controlled Trial of Gaboxadol in Angelman Syndrome. Neurology 96, e1024–e1035. doi:10.1212/WNL.0000000000011409
Brašić, J. R., Nandi, A., Russell, D. S., Jennings, D., Barret, O., Martin, S. D., et al. (2021). Cerebral Expression of Metabotropic Glutamate Receptor Subtype 5 in Idiopathic Autism Spectrum Disorder and Fragile X Syndrome: a Pilot Study. Int. J. Mol. Sci. 22, 2863. doi:10.3390/ijms22062863
Brašić, J. R., Nandi, A., Russell, D. S., Jennings, D., Barret, O., Mathur, A., et al. (2020). Reduced Expression of Cerebral Metabotropic Glutamate Receptor Subtype 5 in Men with Fragile X Syndrome. Brain Sci. 10, 899. doi:10.3390/brainsci10120899
Brickley, S. G., and Mody, I. (2012). Extrasynaptic GABA(A) Receptors: Their Function in the CNS and Implications for Disease. Neuron 73, 23–34. doi:10.1016/j.neuron.2011.12.012
Budimirovic, D. B., Berry-Kravis, E., Erickson, C. A., Hall, S. S., Hessl, D., Reiss, A. L., et al. (2017). Updated Report on Tools to Measure Outcomes of Clinical Trials in Fragile X Syndrome. J. Neurodev. Disord. 9, 14. doi:10.1186/s11689-017-9193-x
Budimirovic, D. B., Bukelis, I., Cox, C., Gray, R. M., Tierney, E., and Kaufmann, W. E. (2006). Autism Spectrum Disorder in Fragile X Syndrome: Differential Contribution of Adaptive Socialization and Social Withdrawal. Am. J. Med. Genet. A. 140A, 1814–1826. doi:10.1002/ajmg.a.31405
Budimirovic, D. B., Schlageter, A., Filipovic-Sadic, S., Protic, D. D., Bram, E., Mahone, E. M., et al. (2020). A Genotype-Phenotype Study of High-Resolution FMR1 Nucleic Acid and Protein Analyses in Fragile X Patients with Neurobehavioral Assessments. Brain Sci. 10, 694. doi:10.3390/brainsci10100694
Budimirovic, D. B., and Subramanian, M. (2016). “Neurobiology of Autism and Intellectual Disability: Fragile X Syndrome,” in Neurobiology of Disease. 2nd ed. (New York, NY, USA: Oxford University Press), 375–384.
Conners, C. K., Sitarenios, G., Parker, J. D., and Epstein, J. N. (1998). Revision and Restandardization of the Conners Teacher Rating Scale (CTRS-R): Factor Structure, Reliability, and Criterion Validity. J. Abnorm. Child. Psychol. 26, 279–291. doi:10.1023/a:1022606501530
Cope, D. W., Hughes, S. W., and Crunelli, V. (2005). GABAA Receptor-Mediated Tonic Inhibition in Thalamic Neurons. J. Neurosci. 25, 11553–11563. doi:10.1523/JNEUROSCI.3362-05.2005
Cordeiro, L., Ballinger, E., Hagerman, R., and Hessl, D. (2011). Clinical Assessment of DSM-IV Anxiety Disorders in Fragile X Syndrome: Prevalence and Characterization. J. Neurodev. Disord. 3, 57–67. doi:10.1007/s11689-010-9067-y
Crawford, D. C., Acuña, J. M., and Sherman, S. L. (2001). FMR1 and the Fragile X Syndrome: Human Genome Epidemiology Review. Genet. Med. 3, 359–371. doi:10.1097/00125817-200109000-00006
Dueck, A., Reis, O., Bastian, M., van Treeck, L., Weirich, S., Haessler, F., et al. (2020). Feasibility of a Complex Setting for Assessing Sleep and Circadian Rhythmicity in a Fragile X Cohort. Front. Psychiatry 11, 361. doi:10.3389/fpsyt.2020.00361
Duy, P. Q., and Budimirovic, D. B. (2017). Fragile X Syndrome: Lessons Learned from the Most Translated Neurodevelopmental Disorder in Clinical Trials. Transl. Neurosci. 8, 7–8. doi:10.1515/tnsci-2017-0002
Eckert, E. M., Dominick, K. C., Pedapati, E. V., Wink, L. K., Shaffer, R. C., Andrews, H., et al. (2019). Pharmacologic Interventions for Irritability, Aggression, Agitation and Self-Injurious Behavior in Fragile X Syndrome: an Initial Cross-Sectional Analysis. J. Autism Dev. Disord. 49, 4595–4602. doi:10.1007/s10803-019-04173-z
Erickson, C. A., Kaufmann, W. E., Budimirovic, D. B., Lachiewicz, A., Haas-Givler, B., Miller, R. M., et al. (2018). Best Practices in Fragile X Syndrome Treatment Development. Brain Sci. 8, 224. doi:10.3390/brainsci8120224
Esbensen, A. J., Rojahn, J., Aman, M. G., and Ruedrich, S. (2003). Reliability and Validity of an Assessment Instrument for Anxiety, Depression, and Mood Among Individuals with Mental Retardation. J. Autism Dev. Disord. 33, 617–629. doi:10.1023/b:jadd.0000005999.27178.55
Ethridge, L. E., De Stefano, L. A., Schmitt, L. M., Woodruff, N. E., Brown, K. L., Tran, M., et al. (2019). Auditory EEG Biomarkers in Fragile X Syndrome: Clinical Relevance. Front. Integr. Neurosci. 13, 60. doi:10.3389/fnint.2019.00060
Gabis, L. V., Baruch, Y. K., Jokel, A., and Raz, R. (2011). Psychiatric and Autistic Comorbidity in Fragile X Syndrome across Ages. J. Child. Neurol. 26, 940–948. doi:10.1177/0883073810395937
Gantois, I., Vandesompele, J., Speleman, F., Reyniers, E., D'Hooge, R., Severijnen, L. A., et al. (2006). Expression Profiling Suggests Underexpression of the GABA(A) Receptor Subunit delta in the Fragile X Knockout Mouse Model. Neurobiol. Dis. 21, 346–357. doi:10.1016/j.nbd.2005.07.017
Hagerman, R. J., Berry-Kravis, E., Kaufmann, W. E., Ono, M. Y., Tartaglia, N., Lachiewicz, A., et al. (2009). Advances in the Treatment of Fragile X Syndrome. Pediatrics 123, 378–390. doi:10.1542/peds.2008-0317
Kaufmann, W. E., Capone, G. T., Clarke, M., and Budimirovic, D. B. (2008). Autism in Genetic Intellectual Disability: Insights into Idiopathic Autism. Totowa, NJ, USA: Humana Press.
Kaufmann, W. E., Kidd, S. A., Andrews, H. F., Budimirovic, D. B., Esler, A., Haas-Givler, B., et al. (2017). Autism Spectrum Disorder in Fragile X Syndrome: Cooccurring Conditions and Current Treatment. Pediatrics 139, S194. doi:10.1542/peds.2016-1159F
Kim, K., Hessl, D., Randol, J. L., Espinal, G. M., Schneider, A., Protic, D., et al. (2019). Association between IQ and FMR1 Protein (FMRP) across the Spectrum of CGG Repeat Expansions. PLoS One 14, e0226811. doi:10.1371/journal.pone.0226811
Lam, K. S., and Aman, M. G. (2007). The Repetitive Behavior Scale-Revised: Independent Validation in Individuals with Autism Spectrum Disorders. J. Autism Dev. Disord. 37, 855–866. doi:10.1007/s10803-006-0213-z
Lee, A. W., Ventola, P., Budimirovic, D., Berry-Kravis, E., and Visootsak, J. (2018). Clinical Development of Targeted Fragile X Syndrome Treatments: an Industry Perspective. Brain Sci. 8, 214. doi:10.3390/brainsci8120214
Lee, V., and Maguire, J. (2014). The Impact of Tonic GABAA Receptor-Mediated Inhibition on Neuronal Excitability Varies across Brain Region and Cell Type. Front. Neural Circuits 8, 3. doi:10.3389/fncir.2014.00003
Luu, S., Province, H., Berry-Kravis, E., Hagerman, R., Hessl, D., Vaidya, D., et al. (2020). Response to Placebo in Fragile X Syndrome Clinical Trials: an Initial Analysis. Brain Sci. 10, 629. doi:10.3390/brainsci10090629
Martin, B. S., Corbin, J. G., and Huntsman, M. M. (2014). Deficient Tonic GABAergic Conductance and Synaptic Balance in the Fragile X Syndrome Amygdala. J. Neurophysiol. 112, 890–902. doi:10.1152/jn.00597.2013
Niu, M., Han, Y., Dy, A. B. C., Du, J., Jin, H., Qin, J., et al. (2017). Autism Symptoms in Fragile X Syndrome. J. Child. Neurol. 32, 903–909. doi:10.1177/0883073817712875
Nolin, S. L., Glicksman, A., Houck, G. E., Brown, W. T., and Dobkin, C. S. (1994). Mosaicism in Fragile X Affected Males. Am. J. Med. Genet. 51, 509–512. doi:10.1002/ajmg.1320510444
Olmos-Serrano, J. L., Corbin, J. G., and Burns, M. P. (2011). The GABA(A) Receptor Agonist THIP Ameliorates Specific Behavioral Deficits in the Mouse Model of Fragile X Syndrome. Dev. Neurosci. 33, 395–403. doi:10.1159/000332884
Olmos-Serrano, J. L., Paluszkiewicz, S. M., Martin, B. S., Kaufmann, W. E., Corbin, J. G., and Huntsman, M. M. (2010). Defective GABAergic Neurotransmission and Pharmacological rescue of Neuronal Hyperexcitability in the Amygdala in a Mouse Model of Fragile X Syndrome. J. Neurosci. 30, 9929–9938. doi:10.1523/JNEUROSCI.1714-10.2010
Raspa, M., Wylie, A., Wheeler, A. C., Kolacz, J., Edwards, A., Heilman, K., et al. (2018). Sensory Difficulties in Children with an FMR1 Premutation. Front. Genet. 9, 351. doi:10.3389/fgene.2018.00351
Reisinger, D. L., Shaffer, R. C., Tartaglia, N., Berry-Kravis, E., and Erickson, C. A. (2020). Delineating Repetitive Behavior Profiles across the Lifespan in Fragile X Syndrome. Brain Sci. 10, 239. doi:10.3390/brainsci10040239
Roth, T., Lines, C., Vandormael, K., Ceesay, P., Anderson, D., and Snavely, D. (2010). Effect of Gaboxadol on Patient-Reported Measures of Sleep and Waking Function in Patients with Primary Insomnia: Results from Two Randomized, Controlled, 3-month Studies. J. Clin. Sleep Med. 6, 30–39. doi:10.5664/jcsm.27707
Rousseau, F., Heitz, D., Tarleton, J., MacPherson, J., Malmgren, H., Dahl, N., et al. (1994). A Multicenter Study on Genotype-Phenotype Correlations in the Fragile X Syndrome, Using Direct Diagnosis with Probe StB12.3: the First 2,253 Cases. Am. J. Hum. Genet. 55, 225–237.
Sansone, S. M., Schneider, A., Bickel, E., Berry-Kravis, E., Prescott, C., and Hessl, D. (2014). Improving IQ Measurement in Intellectual Disabilities Using True Deviation from Population Norms. J. Neurodev. Disord. 6, 16. doi:10.1186/1866-1955-6-16
Sansone, S. M., Widaman, K. F., Hall, S. S., Reiss, A. L., Lightbody, A., Kaufmann, W. E., et al. (2012). Psychometric Study of the Aberrant Behavior Checklist in Fragile X Syndrome and Implications for Targeted Treatment. J. Autism Dev. Disord. 42, 1377–1392. doi:10.1007/s10803-011-1370-2
Simpson, K., Adams, D., Alston-Knox, C., Heussler, H. S., and Keen, D. (2019). Exploring the Sensory Profiles of Children on the Autism Spectrum Using the Short Sensory Profile-2 (SSP-2). J. Autism Dev. Disord. 49, 2069–2079. doi:10.1007/s10803-019-03889-2
Tassone, F., Iong, K. P., Tong, T. H., Lo, J., Gane, L. W., Berry-Kravis, E., et al. (2012). FMR1 CGG Allele Size and Prevalence Ascertained through Newborn Screening in the United States. Genome Med. 4, 100. doi:10.1186/gm401
Weber, J. D., Smith, E., Berry-Kravis, E., Cadavid, D., Hessl, D., and Erickson, C. (2019). Voice of People with Fragile X Syndrome and Their Families: Reports from a Survey on Treatment Priorities. Brain Sci. 9, 18. doi:10.3390/brainsci9020018
Whissell, P. D., Lecker, I., Wang, D. S., Yu, J., and Orser, B. A. (2015). Altered Expression of δGABAA Receptors in Health and Disease. Neuropharmacology 88, 24–35. doi:10.1016/j.neuropharm.2014.08.003
Wright-Talamante, C., Cheema, A., Riddle, J. E., Luckey, D. W., Taylor, A. K., and Hagerman, R. J. (1996). A Controlled Study of Longitudinal IQ Changes in Females and Males with Fragile X Syndrome. Am. J. Med. Genet. 64, 350–355. doi:10.1002/(SICI)1096-8628(19960809)64:2<350:AID-AJMG23>3.0.CO;2-D
Youssef, E. A., Berry-Kravis, E., Czech, C., Hagerman, R. J., Hessl, D., Wong, C. Y., et al. (2018). Effect of the mGluR5-NAM Basimglurant on Behavior in Adolescents and Adults with Fragile X Syndrome in a Randomized, Double-Blind, Placebo-Controlled Trial: FragXis Phase 2 Results. Neuropsychopharmacology 43, 503–512. doi:10.1038/npp.2017.177
Keywords: OV101, gaboxadol, fragile X syndrome, FMR1, GABAA, safety, efficacy, randomized study
Citation: Budimirovic DB, Dominick KC, Gabis LV, Adams M, Adera M, Huang L, Ventola P, Tartaglia NR and Berry-Kravis E (2021) Gaboxadol in Fragile X Syndrome: A 12-Week Randomized, Double-Blind, Parallel-Group, Phase 2a Study. Front. Pharmacol. 12:757825. doi: 10.3389/fphar.2021.757825
Received: 12 August 2021; Accepted: 23 September 2021;
Published: 08 October 2021.
Edited by:
Giovanni Provenzano, University of Trento, ItalyReviewed by:
Clinton Canal, Mercer University, United StatesBjarke Ebert, Lundbeck Pharma A/S, Denmark
Copyright © 2021 Budimirovic, Dominick, Gabis, Adams, Adera, Huang, Ventola, Tartaglia and Berry-Kravis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dejan B. Budimirovic, YnVkaW1pcm92aWNAa2VubmVkeWtyaWVnZXIub3Jn