 Huang Huang
Huang Huang Zhe Sun
Zhe Sun Michael A. Hill
Michael A. Hill Gerald A. Meininger
Gerald A. Meininger- Dalton Cardiovascular Research Center, Department of Medical Pharmacology and Physiology, University of Missouri, Columbia, MO, United States
Efficient mechanotransduction in vascular smooth muscle cells (VSMCs) is intimately coupled to physical coupling of the cell to extracellular matrix proteins (ECM) by integrins. Integrin adhesion receptors are essential for normal vascular function and defective integrin signaling is associated with cardiovascular disease. However, less is known about the mechanism of integrin activation in VSMCs in relation to vasoregulation. Our laboratory previously demonstrated that the vasoconstrictor Angiotensin II increases VSMC stiffness in concert with enhanced adhesion to fibronectin (FN), indicating an important role for adhesion in contraction. However, the mechanism of this coordination remains to be clarified. In this study, intracellular Ca2+ ([Ca2+]i) was hypothesized to link integrin activation through inside-out signaling pathways leading to enhanced adhesion in response to AII. By using atomic force microscopy (AFM) with an anti-α5 antibody coated AFM probe, we confirmed that cell stiffness was increased by AII, while we observed no change in adhesion to an α5 integrin antibody. This indicated that increases in cell adhesion to FN induced by AII were occurring through an integrin activation process, as increased membrane integrin expression/receptor density would have been accompanied by increased adhesion to the anti-α5 antibody. Further studies were performed using either KCl or BAPTA-AM to modulate the level of [Ca2+]i. After KCl, VSMCs showed a rapid transient increase in cell stiffness as well as cell adhesion to FN, and these two events were synchronized with superimposed transient increases in the level of [Ca2+]i, which was measured using the Ca2+ indicator, fluo-4. These relationships were unaffected in VSMCs pretreated with the myosin light chain kinase inhibitor, ML-7. In contrast, unstimulated VSMCs incubated with an intracellular calcium chelator, BAPTA-AM, showed reduced cell adhesion to FN as well the expected decrease in [Ca2+]i. These data suggest that in VSMCs, integrin activation is linked to signaling events tied to levels of [Ca2+]i while being less dependent on events at the level of contractile protein activation. These findings provide additional evidence to support a role for adhesion in VSMC contraction and suggest that following cell contractile activation, that adhesion may be regulated in tandem with the contractile event.
Introduction
The biomechanical properties of blood vessels are important determinants of cardiovascular function, and changes in these properties are characteristic of many cardiovascular diseases including hypertension, stroke, aneurysms, atherosclerosis and heart disease (Evans et al., 2016). The mechanical properties of the blood vessel wall are a function of the various wall components and their organization. Therefore, the stiffness of vascular tissue is resulted from both the composition and organization of the extracellular matrix (ECM) as well as the cells in the vascular wall which are interconnected by cell-matrix and cell-cell contacts (Saphirstein et al., 2013). It is commonly accepted that changes in the ECM are the dominate influence on vascular stiffness and that changes in collagen and elastin content and organization are of major importance (Dinardo et al., 2014; Xu and Shi, 2014). In addition to the ECM, it has been recently established that vascular smooth muscle cells (VSMCs), as the main cell type within the vascular wall, can exhibit increased stiffness that impacts the stiffness of the blood vessel wall. For example, Sehgel et al. (2013) found increased elastic stiffness of VSMCs in spontaneously hypertensive rats, and Zhu et al. (2012) have demonstrated higher stiffness in VSMCs from old monkeys compared to young monkeys. Cytoskeletal proteins and the contractile state have been associated with cell stiffness in VSMCs (Hong et al., 2012, 2014; Lim et al., 2012). In these studies, VSMC adhesion to ECM proteins was also shown to be enhanced in stiffer VSMCs. Fibronectin (FN) is a major ECM protein that interacts with integrin receptors on VSMCs and it is reported to accumulate in diseased artery walls (Pickering et al., 2000). Of importance, the α5β1 integrin is recognized as a major FN receptor and plays an important role in VSMCs mechanoreception linking it to vascular regulation (Francis et al., 2002; Sun et al., 2005).
Integrins, as heterodimeric membrane receptors, composed of two non-covalent associated transmembrane subunits –α and -β, provide an important physical connection between the ECM and the cytoskeleton, maintaining a dynamic adhesion between the cell and its microenvironment. This unique feature positioning of integrins makes them ideally situated for bi-directionally transmitting and conducting mechanical signals through both inside-out and outside-in mechanisms. Integrins are known to regulate cell differentiation, proliferation, and survival through controlling critical intracellular signals (Lee et al., 2010; Paszek et al., 2014; Sarrazy et al., 2014). Importantly, evidence exists to support an important role for integrins in regulating vascular function (Mogford et al., 1996; D’Angelo et al., 1997; Platts et al., 1998). Among the multitude of integrin receptors identified on the surface of VSMCs, integrin α5β1 acts as the dominant FN receptor (Pickering et al., 2000; Sun et al., 2005; Trache et al., 2005). In previous work from our laboratory, Hong et al. demonstrated the existence of coordination between cell stiffness and cell adhesion of VSMCs to FN as they respond to the contractile vasoactive agonist Angiotensin II (AII) or to the vasodilators adenosine and nitric oxide (Hong et al., 2012, 2015). This coordination of adhesion with the contractile state was suggested to be regulated by changes in integrin behavior (Hong et al., 2012). The function of integrin can be controlled by both ‘outside-in’ signaling and “inside-out signaling” (Banno and Ginsberg, 2008). In the case of VSMC activation by soluble vasoactive agonists, it is likely that changes in the integrin activation state are occurring through an ‘inside-out activation’ process from within the cell. Such changes in integrin activation occur through conformational changes in the extracellular integrin head domains in which they can switch reversibly from a low-affinity conformation to a high-affinity active conformation (Calderwood, 2004). Integrin α5β1 has been observed to undergo conformational changes in focal adhesions (Askari et al., 2010), and these changes have been demonstrated to occur with alterations in cytoskeletal tension (Friedland et al., 2009; Kong et al., 2009). However, the mechanism underlying the changes in integrin activation in VSMCs in response to vasoactive-agonists requires further study.
In this study, we hypothesized that the coordination of cell stiffness and cell adhesion to FN of VSMCs is linked to changes in intracellular calcium ([Ca2+]i) which is known to activate the contractile process. Therefore, the enhanced adhesion in response to AII may result from integrin activation through an ‘inside-out’ signaling pathway linked to an increase in [Ca2+]i. To address this hypothesis, we utilized atomic force microscopy (AFM) combined with confocal microscopy. Using AFM it is possible to detect and apply mechanical forces at the nano- and pico-newton ranges, providing a powerful tool for investigating biomechanical properties of VSMCs (Smith et al., 2005) and for simultaneously measuring changes in cell adhesion molecules (Baumgartner et al., 2000; Kong et al., 2009; Carvalho and Santos, 2012). In this study, AFM was used for real-time dynamic observations of cell stiffness and adhesion and correlate them with [Ca2+]i, measured using the calcium indicator fluo-4. We show that cell stiffness and cell adhesion to FN change directionally with [Ca2+]i. We also observed that increased cell stiffness and adhesion to FN was not abolished by treatment with a myosin light chain kinase (MLCK) inhibitor. Thus, the changes in cell stiffness and adhesion appear to occur by a signaling pathway parallel to but not downstream of contraction.
Materials and Methods
Reagents and Materials Applied
For all studies described, human plasma fibronectin (FN), 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (acetoxymethyl ester) (BAPTA-AM), and fluo 4-AM were purchased from Invitrogen (Carlsbad, CA, United States), Bovine Serum Albumin (BSA), KCl, AII were purchased from Sigma (St. Louis, MO, United States). Antibodies to integrin α5 (CD49e, cat: 553350), phalloidin 488 were purchased from Abcam (Cambridge, United Kingdom). For all cell isolations, dithioerythritol (Dith), papain, collagenase, soybean trypsin inhibitor, and elastase were purchased from Sigma (St. Louis, MO, United States). For studies using atomic force microscopy (AFM), a Bioscope II AFM system (Bruker, Santa Barbara, CA, United States) was mounted on a Fluoview confocal microscope (Olympus, Thornwood, NY, United States). AFM data were collected and analyzed using Nanoscope (Bruker, Santa Barbara, CA, United States) and Matlab (MathWorks, Natick, MA, United States) software. Confocal images were analyzed by using Image-Pro Plus software (Media Cybernetics, Carlsbad, CA, United States).
Cell Preparation
Animal and Tissue Preparation
Male Sprague-Dawley rats (200–300 g) were used in this study and were maintained in accordance with the protocol of the Guide for the Care and Use of Laboratory Animals (NIH 83-23, revised 1996). The animal protocol was reviewed and approved by the Laboratory Animal Use Committee (ACUC) of the University of Missouri. Animals were housed individually in a 12:12 light-dark cycle with free access to food and water. Arterioles were isolated as described in previous work with minor modification (Hong et al., 2012). Briefly, rats were anesthetized by intraperitoneal injection of pentobarbital sodium (Lundbeck, Inc., Deerfield, IL, United States) at 0.1 g/kg. After the anesthesia was verified by lack of spinal reflex, the cremaster muscle was removed through a scrotal surgical incision while rats remained under anesthesia. The tissue was immediately placed in cold (4°C), Ringer’s buffer (composed of 145 μM NaCl, 4.7 μM KCl, 2.0 μM CaCl2-2H2O, 1.0 μM MgSO4-7H20, 1.2 μM NaH2PO4-H2O, 0.02 μM EDTA-2H2O, 3.0 MOPS, 5 μM Glucose, 2.0 μM Pyruvic Acid). Vessels and cell isolations were performed in solution based on Ringer’s buffer. Anesthetized rats were euthanized by intracardiac injection of saturated KCl solution (3 ml). The cremaster muscles were then pinned flat onto a silicone rubber pad in a bath of cold Ringer’s buffer. With the aid of a stereomicroscope (Olympus SZX10, Thornwood, NY, United States), long segments of the first and second order cremasteric feed arterioles vessel were dissected and isolated from surrounding skeletal muscle tissue. The isolated arteriole segments were removed and cut into small pieces (∼2–3 μm) and were placed into culture dishes (35 μm) with Ringer’s buffer in room temperature, and subsequently processed to isolate vascular smooth muscle cells (VSMCs).
VSMCs Isolation and Culture
Vascular smooth muscle cells were enzymatically isolated using previously described methods (Jackson et al., 1997). Briefly, arteriolar segments were transferred to a glass culture tube (10 μm × 75 μm) that contained 1 ml of the first cell dissociation solution that Ringer’s buffer included 27 U/ml papain and 1 mg/ml dithioerythritol. The segments were incubated at 37°C for 30 min without agitation. Following this incubation, the first cell dissociation solution was carefully decanted and replaced with 1 ml of second cell dissociation solution consisting of Ringer’s buffer and 0.5 U/ml collagenase, 75 U/ml elastase, and 1 mg/ml soybean trypsin inhibitor. The segments were further incubated for 12–15 min at 37°C. At the end of the incubation period, the dissociation solution was carefully decanted with that not disturb the arteriole segment. Arteriole segments were then very carefully and gently washed twice by adding 1 ml Ringer’s buffer, followed by 1 ml serum-free supplemented Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Invitrogen, Carlsbad, CA, United States). The vessels segments were pipetted with a glass pipette (1 ml) for about 20–30 times to disperse individual cells from the tissue. The solution containing the dispersed VSMCs was then decanted into 60 μm tissue culture dishes (Corning incorporated, Corning, NY, United States), and maintained under culture conditions in a humidified incubator with 5% CO2, at 37°C, in DMEM supplemented with 20% fetal bovine serum (FBS, ATLANTA Biologicals, Lawrenceville, GA, United States), 10 mM HEPES (Sigma, St. Louis, MO, United States), 1 mM sodium pyruvate (Sigma, St. Louis, MO, United States), 2 μM L-glutamine (Sigma, St. Louis, MO, United States), 100 U/ml penicillin (Sigma, St. Louis, MO, United States), and 100 g/ml streptomycin (Sigma, St. Louis, MO, United States). After 48 h incubation, the culture medium was changed to DMEM with 10% FBS. The cells used in all experiments were maintained under these conditions for 5–10 days without passage. Prior to an experiment, the VSMCs were serum starved overnight.
Force Spectroscopy Measurement
Bio-Functionalized AFM Probes
The AFM probe tip was modified by gluing a 5 μm diameter glass bead (Structure Probe Inc., West Chester, PA, United States) to the tip of cantilever (MLCT-O10, Bruker Corp., Santa Barbara, CA, United States). The bead tip was then coated with FN or anti-α5 antibody using the protocol previously used in our laboratory (Sun et al., 2005). Briefly, the bead was first glued to a clean AFM probe and then FN was attached to bead by a cross-linker. Before gluing the glass bead to the cantilever tip, each cantilever was calibrated before a given experiment using thermal noise amplitude analysis. Cantilevers were then cleaned first by rinsing in 100% ethanol twice followed by rinsing in acetone for three times. The cantilever was dipped in glue (Progressive Epoxy Polymers Inc., United States) and then brought into contact with a glass bead mixture in solution that had been smeared on a glass slide. After bead attachment, AFM probes were stored for 48 h to allow bead glue to cure. The AFM probe with attached bead was then processed to bio-coat the bead tip. The tip was immersed for 5 min in 10 mM Polyethylene glycol (PEG, Sigma, St. Louis, MO, United States), which acted as a cross-linker used for attachment of proteins of interest to the bead. This was followed by washing with distilled water four times, and incubating in a solution of FN (0.25 mg/ml) or anti-α5 antibody (1 mg/ml) for 5 min, followed by cleaning four times with Phosphate-Buffered Saline (PBS) (1.05 μM KH2P04, 155.17 μM NaCl, 2.96 μM Na2HPO4-7H2O). For all experiments, the AFM spring constants were assumed to be unchanged after protein labeling (Lehenkari and Horton, 1999).
Nano-Indentation Protocol of AFM
The biomechanical properties of the VSMCs were measured using a Bioscope II AFM system (Bruker, Santa Barbara, CA, United States) that was mounted on an IX81 OLYMPUS inverted microscope (Olympus Inc., NY, United States). AFM measurements were carried out at room temperature in a 1% HEPES supplemented DMEM without antibiotics. For each experiment, cells were randomly selected and the functionalized AFM probe was repeatedly brought into contact with and retract from the cell surface. This cyclic approach/retraction cycle was performed at a site midway between the nucleus and cell margin for frequency of 0.1 Hz and ramp size of 1000 nm. 60 force curves were collected over 10 min for a control (pre-drug) period followed by 240 force curves over 40 min for the post-drug condition. To minimalize the machine drift, after the probe was initially submerged in the cell bath, the AFM-microscope system was thermally equilibrated for 1 h. The analysis of force curves was processed by a custom analysis program written in MATLAB.
Measurement of Biomechanical Properties With AFM
For the nano-indentation protocol, the stiffness measured is an estimation of Young’s modulus of elasticity (E-modulus) for the cell cortex, reflecting the cell’s viscoelastic properties. A length of approximately 100–300 nm of the AFM indentation curve, following the initial point of contact, was fitted to a modified Hertz Model, which was used to assess the VSMC elasticity.
Here, F is the force applied by the AFM probe on the cell surface; E is the E-modulus; v is the Poisson ratio (assumed as 0.5); r is the radius of spherical AFM tip; δ is the indentation depth into the cell membrane. Rupture force, also referred to here as adhesion force between FN and integrin adhesion complexes, was the product of rupture height and cantilever spring constant, measured from the retraction curve (Hong et al., 2012).
AFM Contact Mode Imaging
To obtain a topographical cell image, the AFM tip was placed on the cell surface and then using scanning mode was moved horizontally along the cell surface while applying a constant force (500 – 800 pN) to the cell surface. Scanned images were 100 μm × 100 μm at the digital density of 512 pixels × 512 pixels. A stylus-type AFM probe (model MLCT-C, k = 15 pN nm-1, Bruker, Santa Barbara, CA, United States) was used to perform the cell surface scanning at 0.15 Hz frequency at room temperature. Height and deflection images were collected with Bioscope software and analyzed using Nanoscope software.
Measurement of Intracellular Calcium
Fluo-4 AM Loading of VSMCs
Intracellular calcium was measured by imaging fluo-4 AM. Cells cultured on glass-bottomed tissue culture dishes (Corning incorporated, Corning, NY, United States) were rinsed by loading buffer (150 μM NaCl, 5 mM KCl, 1 μM MgCl, 10 μM glucose, and 20 μM HEPES, pH 7.4) twice and then bathed with fluo 4-AM solution (2.5 μM, Invitrogen Corp., Carlsbad, CA, United States), which is dissolved in loading buffer supplemented with 2% BSA and 0.01 % Pluronic F-127 (BASF) protected for light for 25 min at room temperature on a rolling plate. Cells were then washed twice using loading buffer and incubated in serum-free DMEM at 30°C for 20 min to allow the de-esterification.
BAPTA-AM Loading of VSMCs
Cells were cultured on glass-bottomed tissue culture dishes. Cells were rinsed twice with loading buffer followed by a 25-min incubation with 20 μM BAPTA-AM (Invitrogen, Carlsbad, CA, United States) dissolved in loading buffer supplemented with 2% BSA and 0.01 % Pluronic F-127 (BASF, Sigma, St Louis, MO, United States) on a rolling plate agitator at room temperature. After loading, cells were then washed twice with loading buffer and incubated in serum-free DMEM for 20 min at 30°C to allow the de-esterification.
Fluorescence Imaging
To assess the correlation between cell stiffness and adhesion with intracellular calcium levels, we performed calcium imaging and made AFM measurements simultaneously. Calcium fluorescence images were continuously recorded 20 s before pulling the AFM until 20 s after pulling. A confocal microscope (FV1000) was used to imagine Fluo-4-loaded VSMCs with laser of 488 nm. Fluorescence used for all imaging experiments was collected at 510∼550 nm ×60 oil immersion objective. Fluo-4 images were analyzed using Fluoview software.
Immunocytochemistry and Confocal Microscopy
Prior to immunocytochemistry, VSMCs were serum-starved overnight. After treatment under different conditions (AII treated at 10-6M for 30 min versus PBS exposure for 30 min, as a control), cells were washed three times with PBS followed by fixation with 2% paraformaldehyde for 30 min at room temperature. Cells were then quenched with glycine buffer (0.1 μM glycine) three times for 10 min. After quenching, cells were bathed with primary antibody overnight at 4°C, 1:100 diluted in labeling buffer (150 μM NaCl, 15 μM Na3C6H5O7, 0.05% Triton X-100, 2% BSA). Cells were then rinsed (six times) with cold washing buffer (150 μM NaCl, 15 μM Na3C6H5O7, 0.05% Triton X-100), followed by secondary antibody incubation (Cy5-labeled or Alex 488-conjugated phalloidin), 1:200 diluted in labeling buffer, for 2 h in a dark environment on a rolling plate at room temperature. The cells were washed again with cold washing buffer and visualized with a confocal microscope. Laser of 647 and 488 nm were used, respectively, for Cy5 label and phalloidin. A through-focus image set was collected for each cell with a z step interval of 0.2 μm.
Statistical Analysis
All data are reported as mean ± SEM. Statistical differences for comparisons of pre-drug controls and post-drug treatments were analyzed with paired t-test. ANOVA was used for between-group analysis. All statistical analyses were performed using open source software SciDAVis and StatPlus (AnalystSoft Inc., CA, United States). Differences were considered significant at p ≤ 0.05.
Results
Membrane Expression of Integrin α5 Following Angiotensin II (AII) Treatment
Previous work in our laboratory has shown that AII increases VSMCs stiffness as well as adhesion to FN (Hong et al., 2012, 2014). Specificity of FN adhesion to α5β1-integrin on VSMCs was confirmed by demonstrating that the adhesion was inhibited with function-blocking antibodies to α5 or β1 integrins (Sun et al., 2005). To determine whether the increased cell adhesion during AII treatment was due to enhanced membrane expression of integrin α5β1, we used AFM probes coated with anti-α5 antibody, since β1 integrin would also be present in other integrin dimers. We hypothesized that if AII was increasing the number of α5 integrins on the cell surface, then adhesion to the antibody-coated probes would increase. In these experiments, 8 cells were treated with AII and of these five responded with the characteristic increase in cell stiffness whereas 3 of the cells did not respond. Only responsive cells were analyzed. The increase in cell stiffness (blue circle) was gradual after AII exposure and reached a plateau after approximately 15 min, which is consistent with previous reports from our laboratory (Figure 1A). Before treatment, average cell stiffness was 6.04 ± 0.26 kPa, and which increased to 7.47 ± 0.21 kPa after AII representing a statistically significant increase by 24% (Figure 1B). As a control, VSMCs were treated with PBS which showed no effect on cell stiffness (Figures 1E,F).
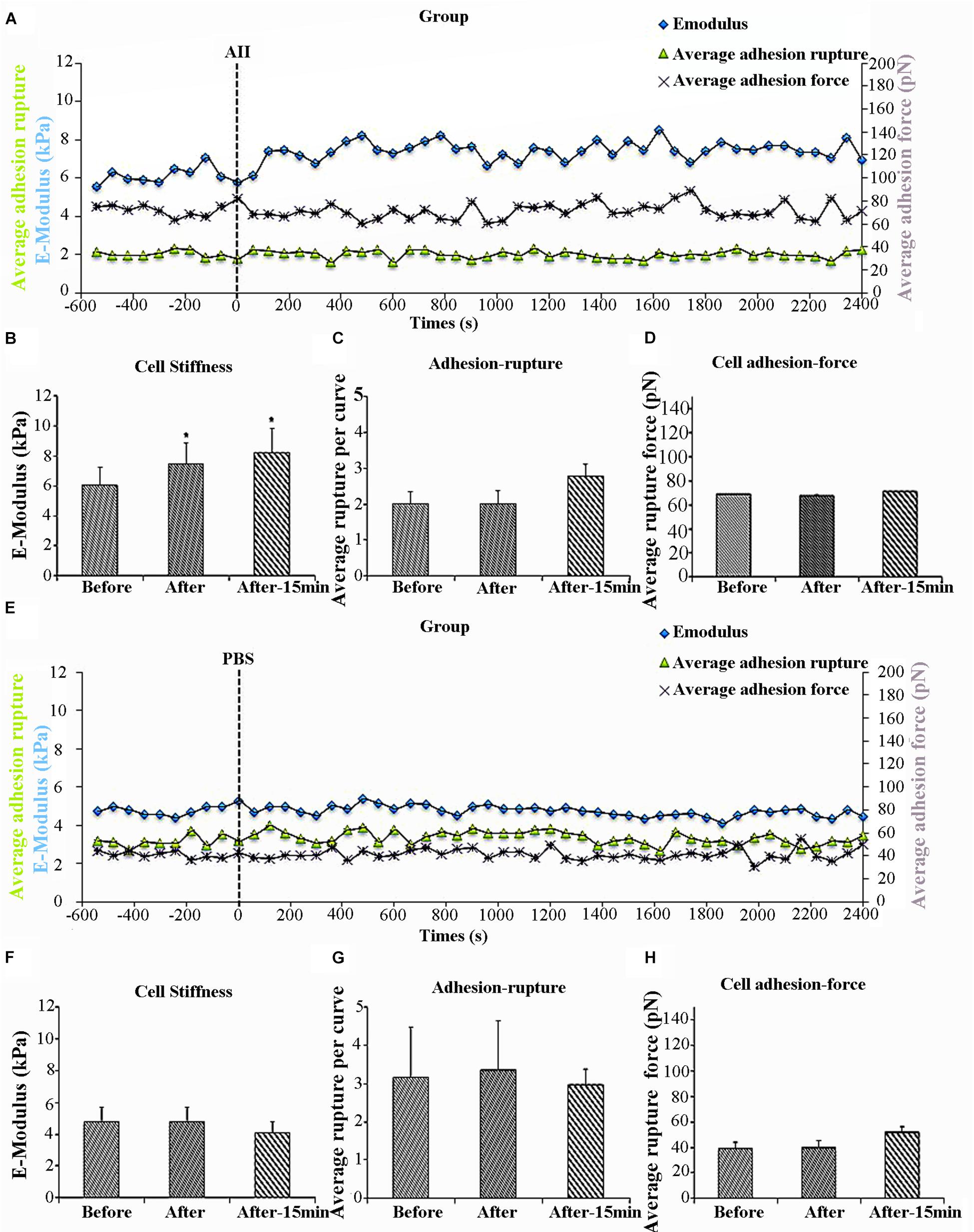
Figure 1. Angiotensin II (AII) increased vascular smooth muscle cells (VSMCs) stiffness but had no effect on cell adhesion to anti-α5 antibody. Group average real-time recordings of cell stiffness (blue square), rupture events (green triangle), and rupture force (purple cross) for AII (n = 5t) treatment (A) and the addition of PBS (n = 10) (E). Average elastic modulus summed across all or 15 min of time points for the group of VSMCs before and after AII (10-6 M) treatment (B) (n = 5, ∗p < 0.05) and PBS addition (F) (n = 10, ∗p < 0.05). Average rupture numbers per curve summed across all or 15 min of time points for the group of VSMCs before and after addition of AII (C) (n = 5, ∗p < 0.05) and PBS (G) (n = 10, ∗p < 0.05). (D) Average rupture force summed across all or 15 min of time points for the group of VSMCs before and after addition of AII (D) (n = 5, ∗p < 0.05) and PBS (H) (n = 10, ∗p < 0.05). Data were collected by integrin α5 antibody coated AFM tip at 0.1 Hz of indentation frequency and are presented as mean ± SEM. tAs detailed in the text while eight cells were treated with AII analysis was limited to the five cells responding with the characteristic increase in cell stiffness as observed in previous studies.
Cell adhesion was analyzed in the AII responsive cells and was quantified as the number of ruptures per curve (Figure 1A, green triangles) and the total rupture force (Figure 1A, purple crosses). After AII treatment, cell adhesion to the anti-α5 antibody, measured as rupture number per curve (Figure 1C), was not significantly increased. In addition, rupture force showed no observable increase with AII (Figure 1D). PBS-treated cells also showed no effect on average rupture number per curve or total rupture force (Figures 1E,G,H).
As a further test to confirm a lack of change in expression of integrin α5, we also performed immunostaining of α5 on VSMCs. Cells were co-labeled with phalloidin to visualize cell stress fibers. Distribution of α5 integrin was diffusely present on the apical surface of VSMCs observed by immunofluorescence confocal microscopy (Figure 2A). After treatment with AII, there were no observable changes of α5 distribution on the cell surface or intensity of labeling (Figure 2B). As the control group, PBS treatment also had no effect on α5 distribution or labeling intensity (Figure 2C). Collectively, these data suggest that AII-induced increases in VSMCs adhesion to FN does not result from enhanced membrane expression of integrin α5. Rather, these data are consistent with an effect of AII to increase integrin activation. As caveat to the experiments shown in Figure 2 it cannot be excluded that some of these cells were non-responders (as in Figure 1) as these cells were not subjected to AFM functional studies for technical reasons.
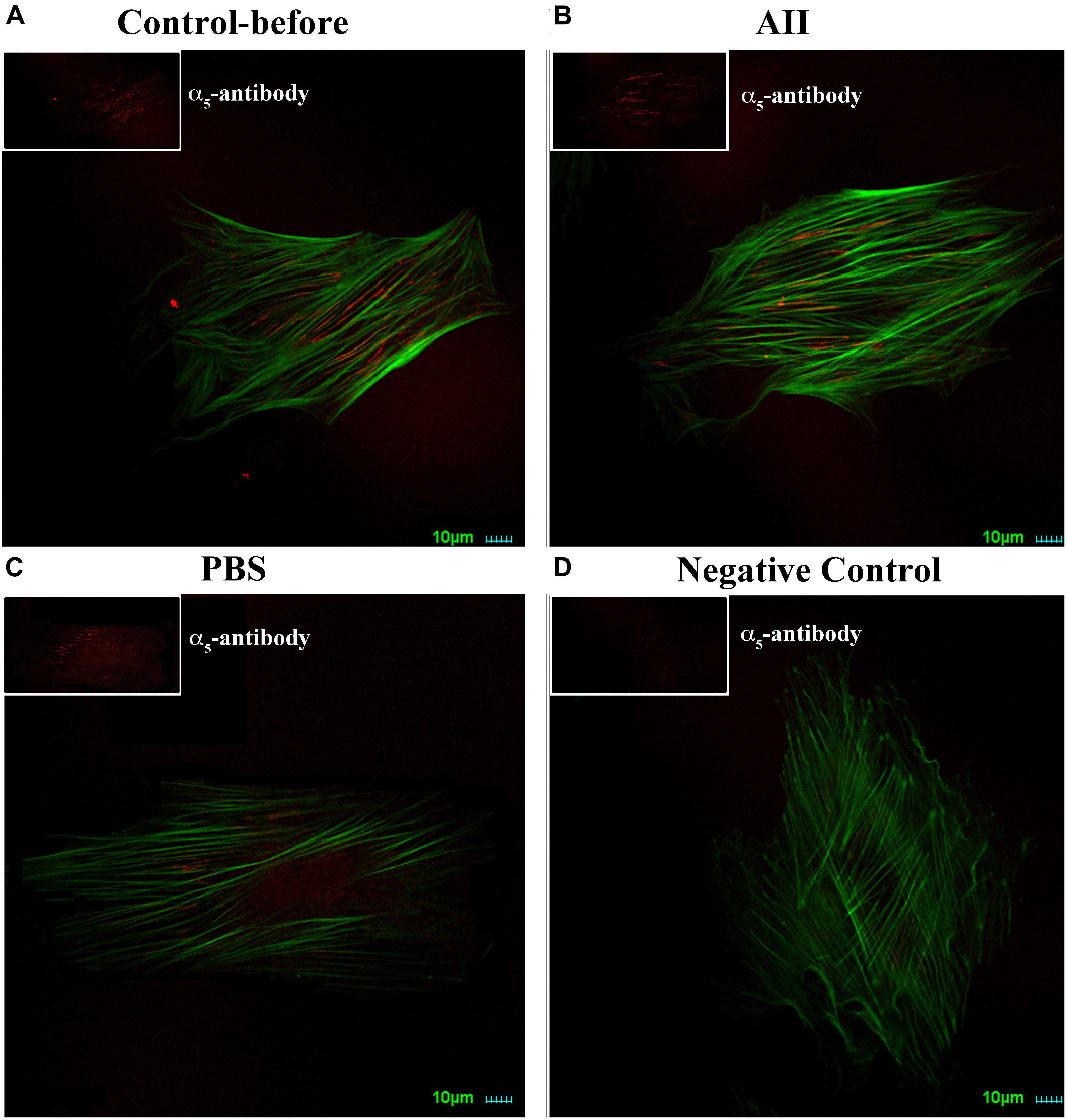
Figure 2. AII has no effect on expression of integrin α5 in VSMCs. Representative images for six cells studied under each condition. Note should be made that it cannot be excluded that some of the cells were non-responders as these cells were not subjected to AFM functional studies for technical reasons. (A–D) Immunostaining of integrin α5 (red) and labeling with phalloidin (green) on VSMCs before treatment (A), after addition of AII (10-6 M) for 30 min (B) and PBS (C). (D) Is negative control without anti-integrin α5 as the primary antibody. Scale bars represent 10 μm.
Effect of KCl Treatment on VSMC Stiffness and Adhesion to FN
AII is known to act on the AT1R and to cause increases in intracellular Ca2+ ([Ca2+]i) in VSMCs (Touyz and Schiffrin, 2000; Simo-Cheyou et al., 2017). To test the hypothesis that [Ca2+]i was playing a role in the enhanced adhesion to FN and increase in cell stiffness we utilized KCl (60 μM). KCl is a non-receptor agonist which increases levels of [Ca2+]i secondary to cell depolarization. In these experiments, FN (0.25 mg/ml) coated AFM probes were used to measure cell stiffness and adhesion response to KCl. In this protocol, all 10 cells studied showed a response to KCl treatment. Before treatment, average cell stiffness was 7.64 ± 1.65 kPa, and application of KCl induced a rapid transient and significant increase in cell stiffness to 18.87 ± 4.39 kPa (Figure 3A, blue circle) representing a 246% increase (Figure 3B, blue square) (Figure 3C). During the period when cell stiffness was transiently elevated we observed that cell adhesion to FN was also transiently increased. The increase in adhesion was apparent as a significant increase (248%) in the numbers of ruptures per curve (Figures 3A,D, green triangle) and increase in total adhesion force (284%) (Figures 3A,E, purple cross).
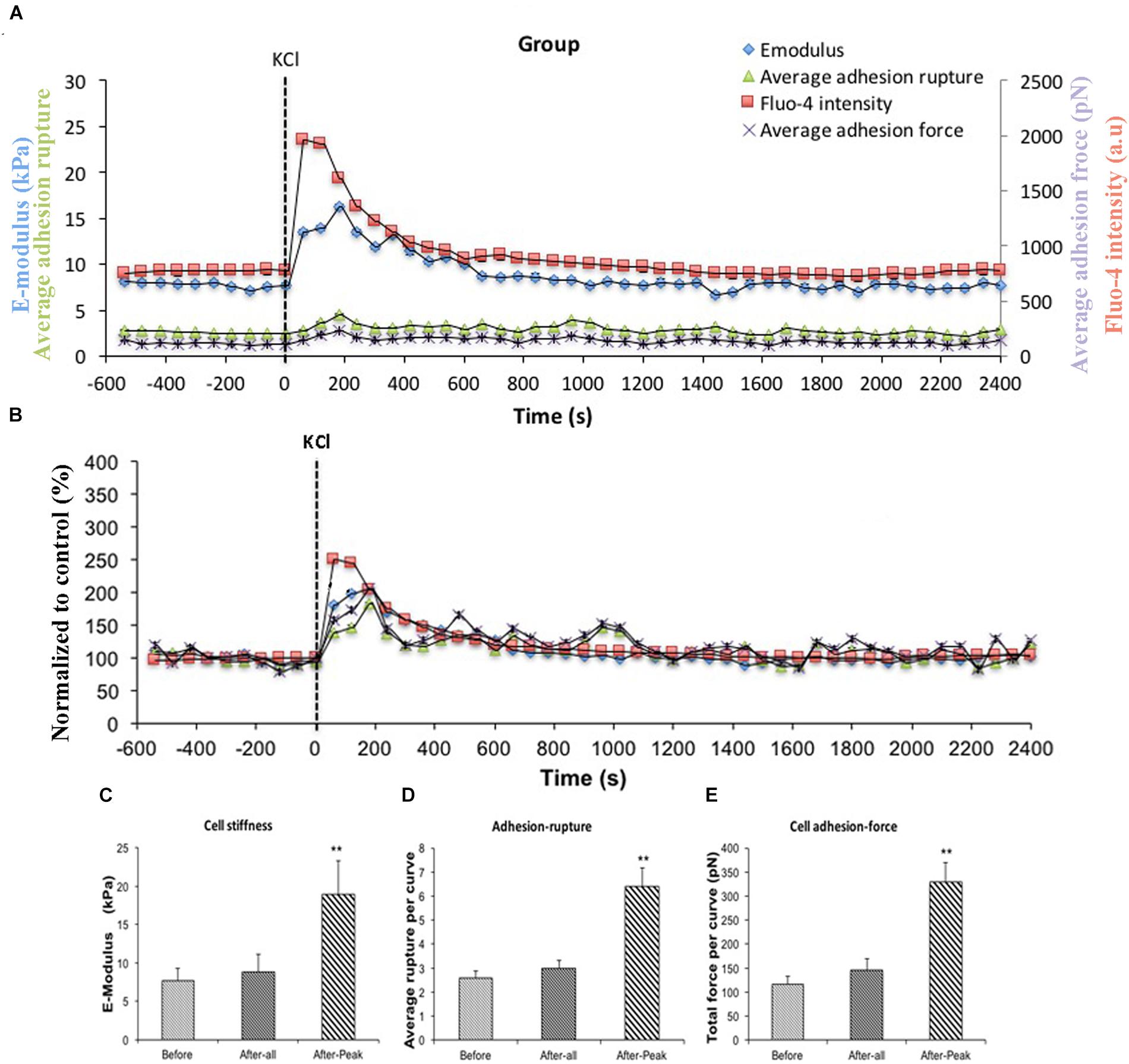
Figure 3. KCl transiently enhanced cell stiffness and cell adhesion to Fibronectin (FN) as well as intracellular calcium level ([Ca2+]i). All cells responded to KCl treatment. (A) Group average real-time recordings of [Ca2+]i (red), cell stiffness (blue), rupture events (green), and rupture force (purple) before and after addition of KCl (n = 10). (B) Group recordings of [Ca2+]i (red), cell stiffness (blue), rupture events (green), and rupture force (purple) normalized to average summed all time points of before-treatment (n = 10). (C) Average elastic modulus summed across all of time points and peak response for the group of VSMCs before and after addition of KCl (60 μM) (n = 10, ∗∗p < 0.01). (D) Average rupture numbers per curve summed across all of the time points and peak response for the group of VSMCs before and after addition of KCl (n = 10, ∗∗p < 0.01). (E) Average rupture force summed across all of the time points and peak response for the group of VSMCs before and after addition of KCl (n = 10, ∗∗p < 0.01). Data were collected by FN-coated AFM tip at 0.1 Hz of indentation frequency and are presented as mean ± SEM.
Involvement of [Ca2+]i as a Mediator of Changes in Cell Stiffness and Adhesion
To confirm Ca2+ was changing following KCl treatment, VSMCs were loaded with the fluorescent calcium indicator fluo-4. [Ca2+]i measurements were obtained while simultaneously performing AFM to measure cell stiffness and adhesion to FN. A transient increase in levels of [Ca2+]i induced by KCl that correlated with the increased cell stiffness, as well as the increases in adhesion ruptures and total adhesion force, which is cumulative ‘pull-off’ force measured for each curve (Figures 3A,B, red square). It was noted that [Ca2+]i levels reached maximum levels immediately preceding the changes in cell stiffness and cell adhesion. These data suggest that calcium-related signaling is linked to the observed changes in cell stiffness and adhesion. As a further test of the involvement of [Ca2+]i as a mediator of the observed changes in stiffness and adhesion, resting VSMCs were treated with the calcium chelator, BAPTA to remove Ca2+ from the intracellular compartment. BAPTA was applied under basal or resting conditions (i.e., in the absence of AII or KCl) as our earlier studies have shown that cells have a baseline level of stiffness that can be decreased by treatment with agents that cause relaxation (for example adenosine and sodium nitroprusside) (Hong et al., 2014, 2015). As shown in Figure 4A, BAPTA-treated groups of cells showed slightly decreased stiffness, however, this reduction was not statistically significant compared to cells of PBS-treated groups. However, average adhesion ruptures significantly declined 24% (p < 0.01) in BAPTA-loaded cells (Figure 4B) compared to the control group. BAPTA treated cells also showed a significant reduction in total adhesion force, which decreased 25% (Figure 4C). Fluo-4 was loaded VSMCs to measure [Ca2+]i in BAPTA treatment, BAPTA-loaded cells showed significantly decreasing [Ca2+]i compared to the PBS-treated control group at the p level of 0.01 (Figure 4D). Thus, these data further suggest [Ca2+]i plays a role in altering integrin adhesion.

Figure 4. BAPTA treatment decreased cell adhesion to FN but has no effect on cell stiffness. (A) The average elastic modulus for the group of VSMCs with and without BAPTA-treatment (20 μM). (B) Average rupture events per curve for the group of VSMCs with and without BAPTA treatment (n = 10, ∗∗p < 0.01). (C) Average rupture for the group of VSMCs with and without addition of BAPTA (n = 10, ∗∗p < 0.01). (D) Level of [Ca2+]i for the group of VSMCs with and without addition of BAPTA (n = 10, ∗∗p < 0.01). Data were collected by FN-coated AFM on randomly selected five cells, each cell was recorded 2 min at 0.1 Hz of indentation frequency. Data are presented as mean ± SEM.
Effects of Inhibition of Myosin Light Chain Kinase
[Ca2+]i elevation leads to VSMC contraction through activation of myosin light chain kinase (MLCK) via calcium–calmodulin complex (Misárková et al., 2016). To further investigate the role of [Ca2+]i in cell stiffness and adhesion to FN, VSMCs were treated with ML-7, a myosin light chain kinase inhibitor prior to treatment with KCl. [Ca2+]i was also measured using fluo-4. We have previously shown that ML-7 inhibits depolarization-induced induced smooth muscle contraction while not significantly altering the associated intracellular Ca2+ signal increases (Zou et al., 1995). Following treatment with ML-7, VSMCs E-modulus was decreased to 35% (Figure 5B, blue diamond) consistent with a loss of contractile tone in the cells. These data were normalized to the pre-drug period and compared to the last 10 min of ML-7 treatment (Figure 5C) In contrast there were no significant effects on adhesion rupture (Figure 5D) or adhesion force (Figure 5E). [Ca2+]i by ML-7 while KCl continued to cause a similar acute increase in Ca2+i (compare Figure 5B with Figure 3B, Supplementary Figure S1). Compared to the pre-drug condition (Figure 6A), ML-7 treatment was associated with a loss of stress fibers or the large bands of actin cytoskeletal filaments underlying the cell membrane (Figure 6B). Topographic images of the VSMCs also revealed cell height decreased following ML-7 (Figures 6C,D); maximal cell height 3.2 ± 0.2 μm before and 2.6 ± 0.2 μm after ML-7 (n = 9; p < 0.01). Following application of KCl to the ML-7-treated cells (Figure 5) there were no observable changes in the effect of KCl on stiffness or adhesion as either rupture numbers or adhesion force. Overall, these results are consistent with an involvement of [Ca2+]i in cortical cell stiffness and adhesion to FN. Furthermore these data suggest that increased cell stiffness and adhesion were not dependent on actin-myosin interaction.
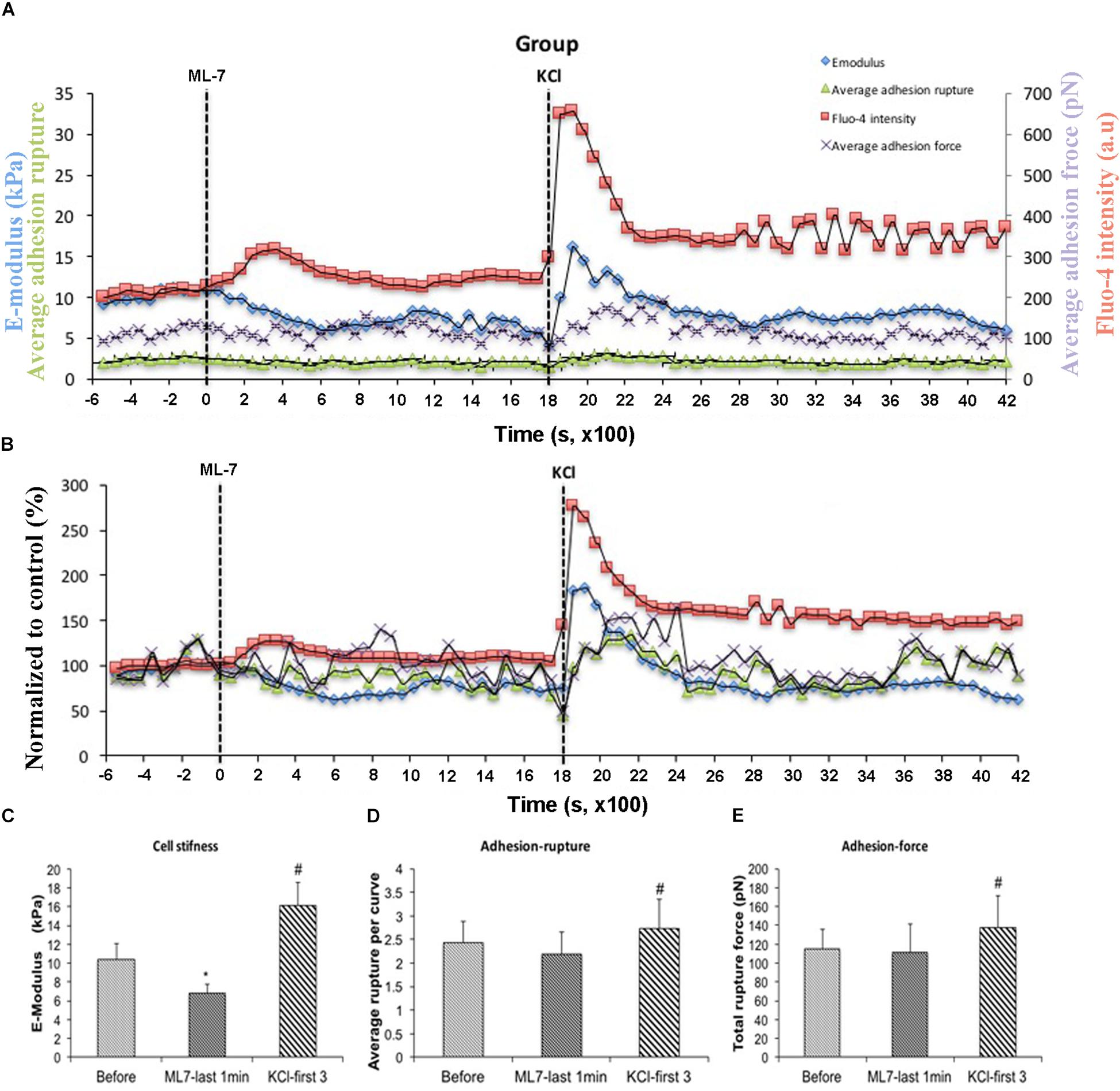
Figure 5. With pre-treatment of ML-7, KCl elevated cell stiffness, cell adhesion to FN as well as the level of [Ca2+]i. All cells responded to KCl treatment. (A) Group average real-time recordings of [Ca2+]i (red), cell stiffness (blue), rupture events (green), and rupture force (purple) before and after addition of ML-7 and KCl (n = 10). (B) Group recordings of [Ca2+]i (red), cell stiffness (blue), rupture events (green), and rupture force (purple) normalized to average summed all time points without treatment (n = 10). (C) Average elastic modulus summed across all time points for the group of VSMCs before treatment, time points of 15 min of ML-7 (15 μM) and first three time points after addition of KCl (60 μM) (n = 10, ∗p < 0.05 compared to no-treatment, #p < 0.05 compared to ML-7). (D) Average rupture numbers per curve summed across all time points for the group of VSMCs before treatment, time points of 15 min of ML-7 (15 μM) and first three time points of after addition of KCl (60 μM) (n = 10, ∗p < 0.05 compared to no-treatment, #p < 0.05 compared to ML-7). (E) Average rupture force summed across all of time points for the group of VSMCs before treatment, following 15 min pre-incubation with ML-7 (15 μM) and first three time points of after addition of KCl (60 μM) (n = 10, ∗p < 0.05 compared to no-treatment, #p < 0.05 compared to ML-7). Data were collected by FN-coated AFM at 0.1 Hz of indentation frequency and are presented as mean ± SEM.

Figure 6. Representative contact-mode images of VSMCs treated with ML-7. (A,B) Deflection error images of single cultured VSMC before treatment (A) and after ML-7 (15 μM, 45 min) treatment (B). (C,D) Representative height images of single cultured VSMC of before treatment (C) and after addition of ML-7 (15 μM) (D). Group data for maximal cell height were 3.2 ± 0.2 μm before and 2.6 ± 0.2 μm after ML-7 (n = 9; p < 0.01).
Discussion
Previously, Hong et al. (2012, 2015) have shown in VSMCs that vasoactive agonists induce coordinated changes in cell elasticity and integrin-mediated cell adhesion. That specifically, vasoconstrictors increase VSMC elastic modulus and simultaneously enhance cell adhesion whereas vasodilators have the opposite effect (Hong et al., 2012). In this study, we performed a series of experiments to examine the hypothesis that intracellular Ca2+ ([Ca2+]i) is a signaling link that plays a role in coordinating changes in cell adhesion that occur when contraction is activated. We first examined, whether contractile activation with AII would alter integrin expression or activation on the cell surface. Our results supported an integrin activation process which led to additional experiments designed to specifically probe the role intracellular Ca2+. KCl was used as a contractile agonist to elevate intracellular Ca2+, BAPTA was used to lower intracellular Ca2+ and ML-7 to explore the role of a Ca2+ dependent actin-myosin interaction. The ensuing discussion is organized in support of the concept that the regulation of ECM adhesion is linked to the contractile activation state of vascular smooth muscle. The results suggest this coordinated link is synchronized with [Ca2+]i. We tested the hypothesis that adhesion to the ECM protein FN would be enhanced with elevated [Ca2+]i in VSMCs, and that the change in adhesion would occur along with increases in VSMC cortical stiffness. The increase in cortical stiffness was assumed to act as an indication of changes in the state of VSMC contractile activation. As a corollary to this, we postulated that lower [Ca2+]i would produce opposite results characterized by decreased adhesion to FN and a parallel reduction in VSMC cortical stiffness.
Real-time fluo-4 confocal image combined with AFM was used to record cell stiffness and adhesion to FN along with [Ca2+]i. We confirmed that increases in cell stiffness and adhesion to FN occurred in the presence of a rapid but transiently increase [Ca2+]i that was induced by KCl. The results indicated a high degree of correlation between the increase in [Ca2+]i and cell stiffness and adhesion. As the contraction of VSMC follows from increased [Ca2+]i, we inhibited the contractile interaction of actin and myosin with ML-7. By presumably preventing contractile filament interaction, we sought to determine whether the changes in cortical stiffness and cell adhesion were linked as a downstream event of the contractile process or occurred as a set parallel event coordinated with contraction through changes in [Ca2+]i. The results of these experiments indicated that the changes in cell stiffness and adhesion persisted in the presence of the inhibition of MLCK supporting the interpretation that that coordination with contraction is likely regulated by a pathway parallel to but not downstream of contraction. We then sought to lower [Ca2+]i by incubation with the calcium chelator BAPTA. We reasoned that lowering [Ca2+]i would not only inhibit contractile filament interaction but would also inhibit any parallel pathway dependent on [Ca2+]i. The results confirmed that lowering [Ca2+]i reduced cellular adhesion to FN providing additional support for the conclusion that there is a parallel pathway activated upstream of the contractile pathway. It should be noted that BAPTA was applied under basal conditions (i.e., in the absence of added contractile agents) as our earlier studies have shown that cells have a basal level of stiffness which can be decreased by the addition of agents known to cause relaxation (e.g., Adenosine and nitroprusside) (Hong et al., 2014, 2015). While this approach was taken to examine the effects of Ca2+ depletion on inherent VSMC mechanical properties future studies could repeat this approach after contractile activation in response to exogenous agents.
The cytoskeleton of VSMC provides a structural framework that determines cell shape/morphology and mechanical properties. These characteristics are determined by both dynamic actomyosin interactions as well as by the capacity of the actin cytoskeleton to rapidly depolymerize and repolymerize (Gerthoffer and Gunst, 2001; Yamin and Morgan, 2012). In this study, VSMC stiffness was measured by using the force required to indent the cell surface to a depth of ∼100–300 nm. In this case, the cell elasticity should be regarded as mainly representing the cortical stiffness of the cell’s stress fibers underlying the cell membrane. It is well established that the cortical cytoskeleton network physically connects to deeper cytoskeletal elements within the cytosol as well as to adhesion zones that regulate adhesion to the ECM (Fabry et al., 2011; Field and Lenart, 2011; Murrel and Gardel, 2012). Cortical stiffness is mechanically determined by cellular cortex elasticity and cortex tension, which is believed to be mostly governed by myosin-generated contractility (Reymann et al., 2012; Munjal and Lecuit, 2014). The changes in VSMC stiffness we observed in the presence of inhibition of MLCK are consistent with interpreting the alterations in stiffness as changes in the polymerization state of the cortical cytoskeleton.
Phenylephrine (PE) is another vasoconstrictor that has also been reported to induce cortical stiffening in VSMC without affecting focal adhesion size via FAK–ERK signaling. These data support a correlation between cellular cortical stiffness and cellular contractile state (Saphirstein et al., 2015). Together with the present result that pretreatment of ML-7 did not blunt KCl effects on cell stiffness and adhesion (Figure 5), these data further indicate calcium exerts its effect via calcium-related signaling pathway other than pathways downstream of actin-myosin interactions. Given previous findings from Hong et al. that cytoskeletal remodeling occurs in VMSCs following AII treatment (Hong et al., 2014), calcium appears to exert its effect on cell stiffness through changes in actin polymerization. Calcium-calmodulin-dependent protein kinase II (CaMKII) has been reported to regulate actin cytoskeletal assembly (Hoffman et al., 2013), which might suggest a possible mechanism of calcium effect. However, more experiments will be needed to demonstrate underlying mechanisms of involving cytoskeletal remodeling and calcium.
The responsiveness of VSMCs used in this study to AII confirmed the retention of characteristics expected of a contractile phenotype. This is important because cultured VSMCs carried in culture over many passages exhibit a transition from a contractile phenotype to a synthetic phenotype (Huber and Badylak, 2012; Chang et al., 2014). In our studies, only primary cell cultures were used without passage to minimize these phenotypic changes. Despite these precautions, it is not possible to completely prevent phenotypic shifting and this may account for why only 80% of the cells we studied were AII responsive (data are not shown).
Vascular smooth muscle cells cellular adhesion to FN was assessed by measuring the force required to produce rupture events between the FN-coated AFM probe adhering to the VSMC. This was quantified as the number of rupture events per force curve and the force that break adhesion bonds formed between VSMCs and FN on the AFM probe. An increase in detectable rupture events and/or force was interpreted as a change in integrin-mediated cell adhesion. Another possibility is that an increase in the number of rupture events induced by an agonist might be related to decrease in the cell surface area that resulted from mechanical crinkling or ruffling of the cell membrane, such that density of integrin per unit membrane area would be augmented and the number of detected adhesions would be also affected. However, pretreatment of VSMC with ML-7 to prevent any cell contraction did not change KCl-induced adhesion to FN (Figure 5). This evidence argues against a membrane contraction-induced change in integrin density. Also, in previous studies by Hong et al. (2012) increases in adhesion force were detected following AII suggested that the increase in adhesion was accompanied by changes in the molecular connections of the integrin to cytoskeletal components. This also argues against the argument of the change in membrane area.
The adhesion events and corresponding rupture forces measured by FN-coated AFM probes are due to interactions with α5β1 integrins (Sun et al., 2005; Trache et al., 2005). Thus, in this study, changes in adhesion are also likely α5β1 integrin-dependent. It has been demonstrated in migratory cells that integrin trafficking to and from the membrane can regulate focal adhesion size through assembly and/or disassembly resulting in spatial integrin redistribution of membrane integrins (Caswell et al., 2009). In our study, to assess whether there was a possible change of integrin expression on the membrane that could contribute to an increase in adhesion via the number of detectable rupture events, we coated spherical AFM probe with an anti-α5 antibody. The anti-α5 antibody that was selected shows the affinity for both active and inactive forms of the α5 integrin. We reasoned that if the AII was increasing the amount of integrin on the membrane then the anti-α5 antibody would show an increase in adhesion events following AII. Our results demonstrated that there was no change in adhesion to the anti-α5 antibody coated AFM probe following AII treatment (Figure 1). These data support the interpretation that AII-induced changes in adhesion to FN was occurring through a process of integrin activation rather than integrin turnover.
In summary, our studies show coordination of VSMC cortical stiffness and cell adhesion following contractile activation that is synchronized with changes in [Ca2+]i. ML-7 results further confirmed cell stiffness was related to contractile state since inhibition of MLCK reduced resting cell stiffness, but ML-7 did not inhibit KCl induced coordination of enhanced cortical stiffness and adhesion to FN. These observations suggest that calcium-induced cell stiffness and adhesion is parallel to the contractile activation pathway and not downstream of actin and myosin interaction. This information is an important advance in our understanding of the mechanisms underlying coordination of cell stiffness and adhesion to ECM. We propose this coordination is essential to provide the mechanical efficiency and ability of the VSMCs to appropriately adapt itself to mechanical changes brought about through intracellular contractile activation and likely extracellular changes in applied force. Increased vascular smooth muscle cell stiffness is a significant feature of aging and occurs in cardiovascular diseases, such as hypertension (Zhu et al., 2012; Sehgel et al., 2013). Further investigations into the underlying mechanisms governing both the cellular mechanics and adhesion of VSMC may lead to possible therapeutic targets that through the link between these pathways and vascular disease.
Author Contributions
GM and MH conceived and designed the study, and analyzed and interpreted the data. ZS performed the experiments, analyzed the data, and interpreted the data. HH performed the experiments, analyzed the data, made figures and wrote the manuscript. GM edited the manuscript. All authors approved the final version of the manuscript.
Funding
This study was supported by NIH grant NIH 1PO1HL095486 to GM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewers XP and XW and handling Editor declared their shared affiliation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2018.01810/full#supplementary-material
FIGURE S1 | Calcium level for ML-7 in Figure 5. Level of [Ca2+]i summed across all of time points for the group of VSMCs before treatment, time points of 15 min of ML-7 (15 μM) and first three time points of after addition of KCl (60 μM) (n = 10, #p < 0.05 compared to ML-7). Data were collected by FN-coated AFM at 0.1 Hz of indentation frequency and are presented as mean ± SEM.
References
Askari, J. A., Tynan, C. J., Webb, S. E., Martin-Fernandez, M. L., Ballestrem, C., and Humphries, M. J. (2010). Focal adhesions are sites of integrin extension. J. Cell Biol. 188, 891–903. doi: 10.1083/jcb.200907174
Banno, A., and Ginsberg, M. H. (2008). Integrin activation. Biochem. Soc. Trans. 36(Pt 2), 229–234. doi: 10.1042/BST0360229
Baumgartner, W., Hinterdorfer, P., Ness, W., Raab, A., Vestweber, D., Schindler, H., et al. (2000). Cadherin interaction probed by atomic force microscopy. Proc. Natl. Acad. Sci. U.S.A. 97, 4005–4010. doi: 10.1073/pnas.070052697
Calderwood, D. A. (2004). Integrin activation. J. Cell Sci. 117(Pt 5), 657–666. doi: 10.1242/jcs.01014
Carvalho, F. A., and Santos, N. C. (2012). Atomic force microscopy-based force spectroscopy–biological and biomedical applications. IUBMB Life 64, 465–472. doi: 10.1002/iub.1037
Caswell, P. T., Vadrevu, S., and Norman, J. C. (2009). Integrins: masters and slaves of endocytic transport. Nat. Rev. Mol. Cell Biol. 10, 843–853. doi: 10.1038/nrm2799
Chang, S., Song, S., Lee, J., Yoon, J., Park, J., Choi, S., et al. (2014). Phenotypic modulation of primary vascular smooth muscle cells by short-term culture on micropatterned substrate. PLoS One 9:e88089. doi: 10.1371/journal.pone.0088089
D’Angelo, G., Mogford, J. E., Davis, G. E., Davis, M. H., and Meininger, G. A. (1997). Integrin-mediated reduction in vascular smooth muscel calcium induced by RGD-containing peptide. Am. J. Physiol. 272(4 Pt 2),H2065–H2070.
Dinardo, C. L., Venturini, G., Zhou, E. H., Watanabe, I. S., Campos, L. C., Dariolli, R., et al. (2014). Variation of mechanical properties and quantitative proteomics of VSMC along the arterial tree. Am. J. Physiol. Heart Circ. Physiol. 306, H505–H516. doi: 10.1152/ajpheart.00655.2013
Evans, P. C., Gijsen, F. J., Wentzel, J. J., and van der Heiden, K. (2016). Biomechanics in vascular biology and cardiovascular disease. Thromb. Haemost. 115, 465–466. doi: 10.1160/th16-01-0075
Fabry, B., Klemm, A. H., Kienle, S., Schaffer, T. E., and Goldmann, W. H. (2011). Focal adhesion kinase stabilizes the cytoskeleton. Biophys. J. 101, 2131–2138. doi: 10.1016/j.bpj.2011.09.043
Field, C. M., and Lenart, P. (2011). Bulk cytoplasmic actin and its functions in meiosis and mitosis. Curr. Biol. 21, R825–R830. doi: 10.1016/j.cub.2011.07.043
Francis, S. E., Goh, K. L., Hodivala-Dilke, K., Bader, B. L., Stark, M., Davidson, D., et al. (2002). Central roles of alpha5beta1 integrin and fibronectin in vascular development in mouse embryos and embryoid bodies. Arterioscler. Thromb. Vasc. Biol. 22, 927–933. doi: 10.1161/01.ATV.0000016045.93313.F2
Friedland, J. C., Lee, M. H., and Boettiger, D. (2009). Mechanically activated integrin switch controls a5b1 function. Sience 323, 642–644. doi: 10.1126/science.1168441
Gerthoffer, W. T., and Gunst, S. J. (2001). Invited review: focal adhesion and small heat shock proteins in the regulation of actin remodeling and contractility in smooth muscle. J. Appl. Physiol. 91, 963–972. doi: 10.1152/jappl.2001.91.2.963
Hoffman, L., Farley, M. M., and Waxham, M. N. (2013). Calcium-calmodulin-dependent protein kinase II isoforms differentially impact the dynamics and structure of the actin cytoskeleton. Biochemistry 52, 1198–1207. doi: 10.1021/bi3016586
Hong, Z., Reeves, K. J., Sun, Z., Li, Z., Brown, N. J., and Meininger, G. A. (2015). Vascular smooth muscle cell stiffness and adhesion to collagen I modified by vasoactive agonists. PLoS One 10:e0119533. doi: 10.1371/journal.pone.0119533
Hong, Z., Sun, Z., Li, M., Li, Z., Bunyak, F., Ersoy, I., et al. (2014). Vasoactive agonists exert dynamic and coordinated effects on vascular smooth muscle cell elasticity, cytoskeletal remodelling and adhesion. J. Physiol. 592, 1249–1266. doi: 10.1113/jphysiol.2013.264929
Hong, Z., Sun, Z., Li, Z., Mesquitta, W. T., Trzeciakowski, J. P., and Meininger, G. A. (2012). Coordination of fibronectin adhesion with contraction and relaxation in microvascular smooth muscle. Cardiovasc. Res. 96, 73–80. doi: 10.1093/cvr/cvs239
Huber, A., and Badylak, S. F. (2012). Phenotypic changes in cultured smooth muscle cells: limitation or opportunity for tissue engineering of hollow organs? J. Tissue Eng. Regen. Med. 6, 505–511. doi: 10.1002/term.451
Jackson, W. F., Huebner, J. M., and Rusch, N. J. (1997). Enzymatic isolation and characterization of single vascular smooth muscle cells from creamsteric arterioles. Microcirculation 4, 35–50. doi: 10.3109/10739689709148316
Kong, F., Garcia, A. J., Mould, A. P., Humphries, M. J., and Zhu, C. (2009). Demonstration of catch bonds between an integrin and its ligand. J. Cell Biol. 185, 1275–1284. doi: 10.1083/jcb.200810002
Lee, D. Y., Li, Y. S., Chang, S. F., Zhou, J., Ho, H. M., Chiu, J. J., et al. (2010). Oscillatory flow-induced proliferation of osteoblast-like cells is mediated by alphavbeta3 and beta1 integrins through synergistic interactions of focal adhesion kinase and Shc with phosphatidylinositol 3-kinase and the Akt/mTOR/p70S6K pathway. J. Biol. Chem. 285, 30–42. doi: 10.1074/jbc.M109.010512
Lehenkari, P. P., and Horton, M. A. (1999). Single integrin molecule adhesion forces in intact cells measured by atomic force microscopy. Biochem. Biophys. Res. Commun. 259, 645–650. doi: 10.1006/bbrc.1999.0827
Lim, S. M., Trzeciakowski, J. P., Sreenivasappa, H., Dangott, L. J., and Trache, A. (2012). RhoA-induced cytoskeletal tension controls adaptive cellular remodeling to mechanical signaling. Integr. Biol. 4, 615–627. doi: 10.1039/c2ib20008b
Misárková, E., Behuliak, M., Bencze, M., and Zicha, J. (2016). Excitation-contraction coupling and excitation-transcription coupling in blood vessels: their possible interactions in hypertensive vascular remodeling. Physiol. Res. 65, 173–191.
Mogford, J. E., Davis, G. E., Platts, S. H., and Meininger, G. A. (1996). Vascular smooth muscle alpha v beta 3 integrin mediates arteriolar vasodilation in response to RGD peptides. Circ. Res. 79, 821–826. doi: 10.1161/01.RES.79.4.821
Munjal, A., and Lecuit, T. (2014). Actomyosin networks and tissue morphogenesis. Development 141, 1789–1793. doi: 10.1242/dev.091645
Murrel, M. P., and Gardel, M. L. (2012). F-actin buckling coordinates contractility and servering in a biomimetic actomyosin cortex. Proc. Natl. Acad. Sci. U.S.A. 109, 20820–20825. doi: 10.1073/pnas.1214753109
Paszek, M. J., DuFort, C. C., Rossier, O., Bainer, R., Mouw, J. K., Godula, K., et al. (2014). The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature 511, 319–325. doi: 10.1038/nature13535
Pickering, J. G., Chow, L. H., Li, S., Rogers, K. A., Rocnik, E. F., Zhong, R., et al. (2000). a5b1 integrin expression and luminal edge fibronectin matrix assembly by smooth muscle cells after arterial injury. Am. J. Pathol. 156, 453–465. doi: 10.1016/S0002-9440(10)64750-5
Platts, S. H., Mogford, J. E., Davis, M. J., and Meininger, G. A. (1998). Role of K+ channels in arteriolar vasodilation mediated by integrin interaction with RGD-containing peptide. Am. J. Physiol. 275(4 Pt 2), H1449–H1454. doi: 10.1152/ajpheart.1998.275.4.H1449
Reymann, A. C., Boujemaa-Paterski, R., Martiel, J. L., Guerin, C., Cao, W., Chin, H. F., et al. (2012). Actin network architecture can determine myosin motor activity. Science 336, 1310–1314. doi: 10.1126/science.1221708
Saphirstein, R. J., Gao, Y. Z., Jensen, M. H., Gallant, C. M., Vetterkind, S., Moore, J. R., et al. (2013). The focal adhesion: a regulated component of aortic stiffness. PLoS One 8:e62461. doi: 10.1371/journal.pone.0062461
Saphirstein, R. J., Gao, Y. Z., Lin, Q. Q., and Morgan, K. G. (2015). Cortical actin regulation modulates vascular contractility and compliance in veins. J. Physiol. 593, 3929–3941. doi: 10.1113/JP270845
Sarrazy, V., Koehler, A., Chow, M. L., Zimina, E., Li, C. X., Kato, H., et al. (2014). Integrins alphavbeta5 and alphavbeta3 promote latent TGF-beta1 activation by human cardiac fibroblast contraction. Cardiovasc. Res. 102, 407–417. doi: 10.1093/cvr/cvu053
Sehgel, N. L., Zhu, Y., Sun, Z., Trzeciakowski, J. P., Hong, Z., Hunter, W. C., et al. (2013). Increased vascular smooth muscle cell stiffness: a novel mechanism for aortic stiffness in hypertension. Am. J. Physiol. Heart Circ. Physiol. 305, H1281–H1287. doi: 10.1152/ajpheart.00232.2013
Simo-Cheyou, E. R., Tan, J. J., Grygorczyk, R., and Srivastava, A. K. (2017). STIM-1 and ORAI-1 channel mediate angiotensin-II-induced expression of Egr-1 in vascular smooth muscle cells. J. Cell. Physiol. 232, 3496–3509. doi: 10.1002/jcp.25810
Smith, B. A., Tolloczko, B., Martin, J. G., and Grutter, P. (2005). Probing the viscoelastic behavior of cultured airway smooth muscle cells with atomic force microscopy: stiffening induced by contractile agonist. Biophys. J. 88, 2994–3007. doi: 10.1529/biophysj.104.046649
Sun, Z., Martinez-Lemus, L. A., Trache, A., Trzeciakowski, J. P., Davis, G. E., Pohl, U., et al. (2005). Mechanical properties of the interaction between fibronectin and alpha5beta1-integrin on vascular smooth muscle cells studied using atomic force microscopy. Am. J. Physiol. Heart Circ. Physiol. 289, H2526–H2535. doi: 10.1152/ajpheart.00658.2004
Touyz, R. M., and Schiffrin, E. L. (2000). Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol. Rev. 52, 639–672.
Trache, A., Trzeciakowski, J. P., Gardiner, L., Sun, Z., Muthuchamy, M., Guo, M., et al. (2005). Histamine effects on endothelial cell fibronectin interaction studied by atomic force microscopy. Biophys. J. 89, 2888–2898. doi: 10.1529/biophysj.104.057026
Xu, J., and Shi, G. P. (2014). Vascular wall extracellular matrix proteins and vascular diseases. Biochim. Biophys. Acta 1842, 2106–2119. doi: 10.1016/j.bbadis.2014.07.008
Yamin, R., and Morgan, K. G. (2012). Deciphering actin cytoskeletal function in the contractile vascular smooth muscle cell. J. Physiol. 590, 4145–4154. doi: 10.1113/jphysiol.2012.232306
Zhu, Y., Qiu, H., Trzeciakowski, J. P., Sun, Z., Li, Z., Hong, Z., et al. (2012). Temporal analysis of vascular smooth muscle cell elasticity and adhesion reveals oscillation waveforms that differ with aging. Aging Cell 11, 741–750. doi: 10.1111/j.1474-9726.2012.00840.x
Keywords: integrins, fibronectin, calcium, vascular smooth muscle, cell adhesion, cell stiffness, angiotensin II, atomic force microscopy
Citation: Huang H, Sun Z, Hill MA and Meininger GA (2018) A Calcium Mediated Mechanism Coordinating Vascular Smooth Muscle Cell Adhesion During KCl Activation. Front. Physiol. 9:1810. doi: 10.3389/fphys.2018.01810
Received: 08 February 2018; Accepted: 04 December 2018;
Published: 18 December 2018.
Edited by:
Mariappan Muthuchamy, Texas A&M University, United StatesReviewed by:
Xu Peng, Texas A&M University, United StatesXin Wu, Texas A&M University, United States
Uthayashanker Ezekiel, Saint Louis University, United States
Copyright © 2018 Huang, Sun, Hill and Meininger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerald A. Meininger, bWVpbmluZ2VyZ0BtaXNzb3VyaS5lZHU=