
- 1 Genome Stability Laboratory, Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
- 2 Tembusu College, National University of Singapore, Singapore
Silver nanoparticles (Ag-np) have been used in medicine and commercially due to their anti-microbial properties. Therapeutic potentials of these nanoparticles are being explored extensively despite the lack of information on their mechanism of action at molecular and cellular level. Here, we have investigated the DNA damage response and repair following Ag-np treatment in mammalian cells. Studies have shown that Ag-np exerts genotoxicity through double-strand breaks (DSBs). DNA-PKcs, the catalytic subunit of DNA dependent protein kinase, is an important caretaker of the genome which is known to be the main player mediating Non-homologous End-Joining (NHEJ) repair pathway. We hypothesize that DNA-PKcs is responsible for the repair of Ag-np induced DNA damage. In vitro studies have been carried out to investigate both cytotoxicity and genotoxicity induced by Ag-np in normal human cells, DNA-PKcs proficient, and deficient mammalian cells. Chemical inhibition of DNA-PKcs activity with NU7026, an ATP-competitive inhibitor of DNA-PKcs, has been performed to further validate the role of DNA-PKcs in this model. Our results suggest that Ag-np induced more prominent dose-dependent decrease in cell viability in DNA-PKcs deficient or inhibited cells. The deficiency or inhibition of DNA-PKcs renders the cells with higher susceptibility to DNA damage and genome instability which in turn contributed to greater cell cycle arrest/cell death. These findings support the fact that DNA-PKcs is involved in the repair of Ag-np induced genotoxicity and NHEJ repair pathway and DNA-PKcs particularly is activated to safeguard the genome upon Ag-np exposure.
Introduction
Nanotechnology is a rapidly growing industry which promises tremendous scientific and economic potential. Engineered nanomaterial, particularly, nanoparticles have successfully drawn international attention owing to the novel physico-chemical properties they offer. Among the metallic nanomaterial, silver nanoparticles (Ag-np) currently have the highest degree of commercialization due to their anti-microbial potential (Ip et al., 2006), which made them component of wound-dressing, bandages, catheters, and plenty of other medical devices. Ag-np have also been shown to have less chance of drug resistance (Barbu et al., 2009). Ag-np have been the center of attention in nanotechnology development for several other reasons too. Ag-np can be synthesized easily through chemical reaction by reducing the silver salt using a reducing agent to produce small Ag-np and a surfactant to prevent unwanted agglomeration of Ag-np (Wiley et al., 2005). Ag-np display long-term stability at room temperature in aqueous medium with small changes in morphology, shape, and size (Pinto et al., 2010). These properties are particularly important for Ag-np to control their fate, transport, and bioavailability in both in vitro and in vivo models.
Apart from the ease of synthesis and superior optico-chemical properties, Ag-np causes significant cytotoxicity and genotoxicity which are closely associated with resulting oxidative stress and inflammatory responses (Carlson et al., 2008; Asharani et al., 2009a; AshaRani et al., 2009b). The cytotoxicity of Ag-np is an active research topic while genotoxicity is comparatively much less explored. Evidence from published reports has shown that Ag-np induces DNA damage (DNA double-strand breaks, DSBs) and chromosomal aberrations such as acentric and dicentric chromosomes, chromosomal fusions, and fragments (Ahamed et al., 2008; Asharani et al., 2009a; AshaRani et al., 2009b). Up-regulation of DNA damage-signaling proteins, RAD51 and phosphorylated H2AX (γ-H2AX), indicative of signaling events were recorded in Ag-np exposure (Ahamed et al., 2008; Asharani et al., 2012). However, these findings were primarily focused only on the toxicological endpoints. The complex molecular interplay triggering the onset and downstream signaling cascades in response to Ag-np induced DNA damage, in particular, DNA damage repair are largely unexplored.
DNA dependent protein kinase, DNA-PK is a nuclear, serine/threonine protein kinase consisting of a 470-kDa catalytic subunit (DNA-PKcs) and a heterodimeric regulatory complex Ku70/80 (Smith and Jackson, 1999; Doherty et al., 2001). This kinase functions as a notable caretaker of genome (Lees-Miller and Meek, 2003) and mediates non-homologous end-joining (NHEJ) pathway, the main DNA repair pathway responsible for DSB repair in mammalian cells (Hefferin and Tomkinson, 2005; Sonoda et al., 2006). In the current study, we, for the first time, provide mechanistic insights on Ag-np induced DNA damage and repair in mammalian cells with primary interest revolving around the role of DNA-PKcs in response to DNA damage. Chemical inhibition of DNA-PKcs activity with NU7026 was performed to further substantiate the role of DNA-PKcs in our model. It is concluded that DNA-PKcs is responsible for the repair of Ag-np induced DNA damage and may protect the genome from the toxic effects of Ag-np. The absence of functional DNA-PKcs greatly enhanced the genotoxicity of Ag-np in mammalian cells.
Results
Characterization of Ag-np
Characterization of Ag-np was carried out in order to determine the shape, size, state of dispersion, absorbance, and charge which are major determinants of nanoparticle toxicity. Transmission electron microscopy (TEM) analysis revealed that the nanoparticles employed in this study were spherical in shape and mono-dispersed (Figure 1A). UV–vis spectroscopy showed an absorption maximum at around 400 nm indicating a stable homogenous nanoparticle suspension with narrow size distribution (Asharani et al., 2010; Figure 1B). The size distribution analysis showed nanoparticles of size between 12 and 40 nm with an average diameter of 20 nm (Figure 1C). Zeta potential is a measurement of the charge of the synthesized nanoparticles. The Ag-np has zeta potential value of −0.309 ± 0.13 mV (Figure 1D). The elemental composition of the nanoparticles synthesized was further confirmed through elemental analysis. Data indicated that the Ag-np used in our investigations contain an average weight percentage of 15.24% of silver (Figure 1E).
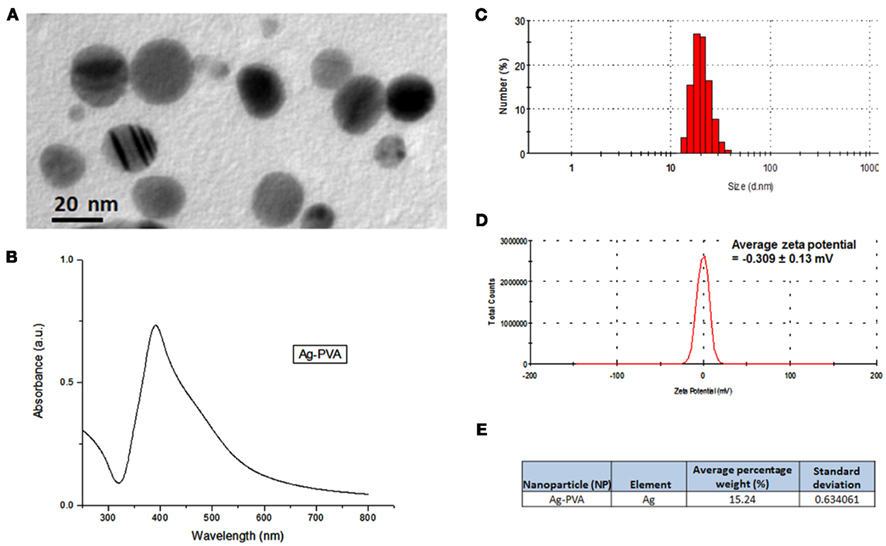
Figure 1. Ag-np characterization. (A) Typical transmission electron microscopy (TEM) image, (B) UV-visible spectrum, (C) size distribution, (D) zeta potential, and (E) elemental analysis of silver nanoparticles (Ag-np) reconstituted after lyophilization. TEM images were captured with JEOL JSM 1220. The size distribution and zeta potential of Ag-np was measured with Zetasizer Nano.
Cells Lacking Functional DNA-PKcs are More Sensitive to Ag-np Induced Reduction in Cell Viability
To understand how DNA-PKcs affects viability of cells under the influence of Ag-np, luminescence based cell viability assay was performed on Ag-np treated DNA-PKcs proficient and deficient cells. To further validate the role(s) of DNA-PKcs, cells were treated with DNA-PKcs inhibitor, NU7026, and the results were compared with DNA-PKcs deficient cells. Normal human cells, IMR-90, showed less susceptibility to Ag-np induced cell damage (Figure 2A). Interestingly, the human DNA-PKcs proficient glioblastoma cells, MO59K, displayed significant dose-dependent decrease in cell viability after treatment with Ag-np. Human DNA-PKcs deficient glioblastoma cells, MO59J, were less sensitive to Ag-np exposure and showed no obvious decrease in cell survival with increasing doses (Figure 2B). DNA-PKcs deficient Chinese hamster cells, CHO V33 cells, showed greater reduction in cell viability than DNA-PKcs proficient CHO AA8 cells at all doses (Figure 2C). In IMR-90, MO59K, and CHO AA8 cells pre-treated with NU7026, there was significant decrease in cell viability compared to their respective proficient cells without NU7026. These observations suggest a potential role played by DNA-PKcs in supporting cell survival in response to Ag-np treatment.
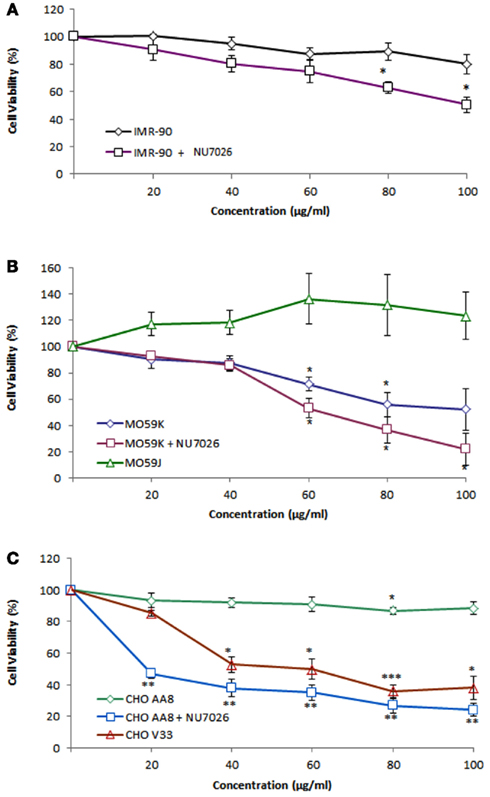
Figure 2. Cell viability in mammalian cells following Ag-np treatment. Cell viability of (A) IMR-90 cells, (B) MO59K and MO59J cells, and (C) CHO AA8 and CHO V33 cells after 48 h treatment with Ag-np at different concentrations is shown. The cell viability percentage was normalized against the untreated controls (0 μg/ml) for Ag-np treatment alone or DMSO control (0 μM) for each cell type pre-treated with NU7026 (10 μM). The values represent the mean ± standard error of three sets of independent experiments; *denotes p < 0.05; **denotes p < 0.01; and ***denotes p < 0.001 as obtained using Student’s t-test, where the statistical significance between controls and Ag-np treated samples was analyzed.
In addition, we have observed that MO59K cells are more susceptible to Ag-np as compared to IMR-90 cells, which are the representatives of cancer and normal cell population, respectively. The difference in survival between the IMR-90 and MO59K cells following Ag-np treatment became notable at 60 μg/ml and above (Figures 2A,B).
Lack of Functional DNA-PKcs Shows Compromised Repair Capacity for Ag-np Induced DNA Damage
Earlier reports have demonstrated a critical role for DNA-PKcs in DNA repair via NHEJ pathway (Gilley et al., 2001; Jackson, 2001, 2002; Uematsu et al., 2007). We examined whether NHEJ pathway is responsible for the repair of the DNA damage induced by Ag-np with regards to the status of DNA-PKcs in mammalian cells. Single-cell gel electrophoresis (also known as Comet assay) under alkaline condition (pH 13) was used to determine the DNA damage induced by Ag-np. DNA damage such as DSBs, single-strand breaks, and alkali labile sites were measured using Comet assay. Representative images of comet analysis are shown in Figures 3Ai,ii Nuclei with undamaged DNA appear round; nuclei with damaged DNA result in DNA fragments which migrate faster during gel electrophoresis and give rise to a “tail.” The tail moment (product of tail length and fraction of DNA) was used as a measure of DNA damage. As presented in Figure 3B, IMR-90 cells are less susceptible to Ag-np as data show minimal increase in DNA damage (1.5 μm) after 48 h treatment. The tail moment in MO59K cells increased from 0.9 to 3.3 μm after treated with Ag-np. To our surprise, we observed that MO59J cells did not show severe DNA damage after Ag-np exposure. Instead, the DNA damage in MO59J cells (3.3 μm, p < 0.05) are comparable to MO59K after Ag-np exposure. This observation is in agreement with our cell viability data for MO59J cells which showed lesser susceptibility to Ag-np. CHO V33 showed much higher total DNA damage induced (21 μm) as compared to CHO AA8 (3.3 μm). Induction of DNA damage by Ag-np in IMR-90 cells was increased significantly after pre-treatment of NU7026 (4 μm, p < 0.01; Figure 3B). MO59K cells pre-treated with NU7026 showed greater DNA damage (5.8 μm, p < 0.05) compared to MO59K cells without NU7026 treatment. Pre-treatment with NU7026 renders higher sensitivity in CHO AA8 cells to Ag-np induced DNA damage (9.3 μm). Collectively, the data imply an essential role played by DNA-PKcs in DNA repair upon Ag-np exposure.
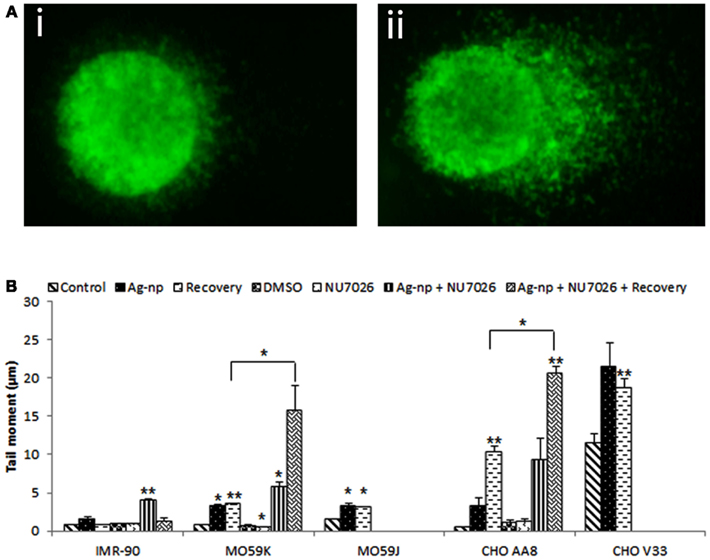
Figure 3. Induction of DNA damage in Ag-np treated cells. DNA damage as measured by the Comet assay in different cell types following 48 h treatment with Ag-np (100 μg/ml). (A) Representative of SYBR Green-stained comets prepared from control (i) and Ag-np treated cells (ii). Magnification = 20×. (B) Mean tail moment (μm) represents the damage distribution in the attached cells. Mean ± standard error of three sets of independent experiments are shown. *Denotes p < 0.05 and **denotes p < 0.01.
To investigate whether the Ag-np induced DNA damage could be recovered, Ag-np containing media were removed 48 h post treatment and following which cells were incubated with fresh media without Ag-np for another 48 h (Figure 3B). Our data showed that the DNA damage was repaired (0.9 μm) in IMR-90 cells following 48 h of recovery. DNA damage was persisted in both MO59K cells (3.6 μm, p < 0.01) and MO59J cells (3.2 μm, p < 0.05). In CHO AA8 cells, the DNA damage was enhanced significantly after recovery (10.4 μm, p < 0.01) meanwhile we observed that the DNA damage remained high in CHO V33 cells (18.8 μm, p < 0.01). Similarly, IMR-90 cells pre-treated with NU7026 showed recovery from DNA damage (1.3 μm) whilst DNA damage was enhanced in both NU7026 pre-treated MO59K cells (15.8 μm) and CHO AA8 cells (20.7 μm, p < 0.01).
DNA-PKcs Deficiency or Inhibition Increases Ag-np Induced Genome Instability
The activities of repair pathways and their coordination with other cell functions are regulated by a DNA damage-signaling network. Dysregulation of this network would significantly impair the coordinated effort to repair DNA damage thus can contribute to genomic instability (Shen, 2011). In the present study, we show that the DNA repair was compromised when functional DNA-PKcs was lacking in the cells. To examine whether Ag-np induce genome instability when the repair network is dysregulated in the absence of functional DNA-PKcs, we used micronuclei induction, a chromosome instability marker, to assess if loss or inhibition of DNA-PKcs renders cells more susceptible to Ag-np induced genomic instability. Micronuclei in the resulting binucleated cells were scored following exposure to Ag-np. Representative images of binucleated cells with and without micronuclei are shown Figures 4Ai,ii respectively.
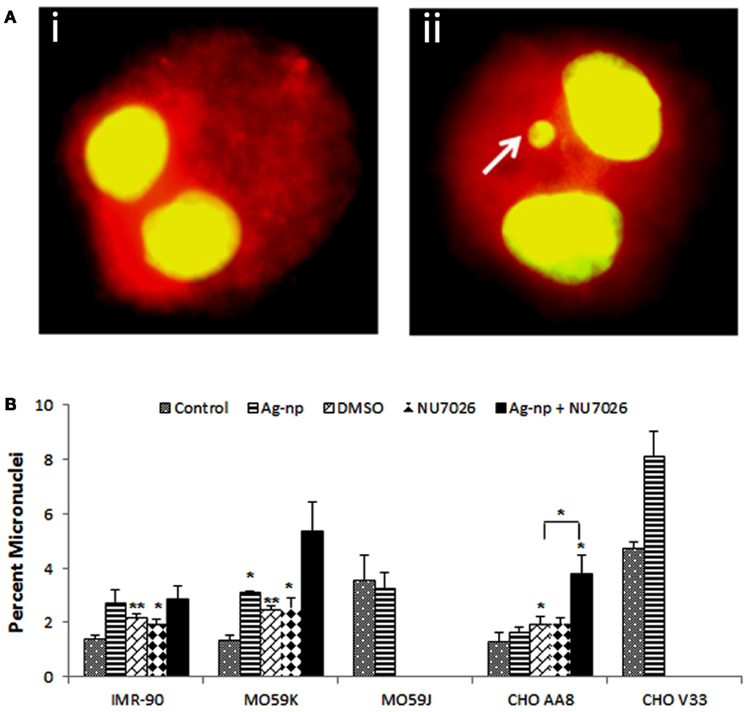
Figure 4. Induction of micronuclei in cells following Ag-np treatment. (A) Representative image of binucleated cells without (Ai) and with micronuclei (Aii). White arrow (Aii) indicates the micronucleus formed among the binucleated cells. Cells and nuclei were stained with acridine orange (30 μg/ml). Magnification = 60×. (B) Data from CBMN show chromosomal aberrations. The data represent 1000 binucleated cells. *Denotes p < 0.05 and **denotes p < 0.01.
As shown in Figure 4B, the percentage of micronuclei in IMR-90 cells was increased from 1.4 to 2.7% after Ag-np treatment. There is no significant increase in the percentage of micronuclei in IMR-90 cells pre-treated with NU7026 followed by Ag-np treatment (2.9%) indicating the fact that normal cells were able to safeguard the stability of genome even without functional DNA-PKcs. In Ag-np treated MO59K cells, there was 2.4-fold increase in percentage of micronuclei from 1.3 to 3.1%. MO59K cells pre-treated with NU7026 showed 4.2-fold after Ag-np exposure, compared with that of the control group, which is a higher increase in micronuclei percentage (5.4%) compared to Ag-np treatment alone. In agreement with comet data, there was no increase in micronuclei percentage in MO59J cells without (3.5%) and with Ag-np treatment (3.2%). The loss of DNA-PKcs sensitized CHO V33 cells to Ag-np and resulted in higher increase in micronuclei percentage from 4.7 to 8.1% compared to CHO AA8 cells from 1.3 to 1.6% micronuclei. CHO AA8 cells pre-treated with NU7026 are more susceptible to genome instability with higher percentage of micronuclei (3.8%) after Ag-np exposure compared to Ag-np treatment alone. The results observed in CHO V33 cells, NU7026 pre-treated MO59K cells and CHO AA8 cells following Ag-np treatment imply that both Chinese hamster cells and human brain cancer cells are sensitized to Ag-np induced genome instability when they lack functional DNA-PKcs.
Ag-np Induce DNA DSBs in Mammalian Cells
Earlier studies have shown that metallic nanoparticles can induce DNA damage either directly through physical assault (Ahamed et al., 2008; Asharani et al., 2009a; AshaRani et al., 2009b) or indirectly by triggering the production of oxidative stress (Asharani et al., 2009a) or both. Studies have also indicated that DNA damage and repair proteins responsible for DSBs were shown to be up-regulated in response to Ag-np treatment (Sun and Ding, 2009). Our results reveal that total DNA damage is greater in Ag-np-treated DNA-PKcs deficient or inhibited cells than DNA-PKcs proficient cells or normal mammalian cells. Therefore, we looked at DSBs in the cells since DNA-PKcs plays pivotal role in DSB repair in mammalian cells. As previously reported, DSBs can induce phosphorylation of histone H2AX at serine 139 (Rogakou et al., 1998, 1999). The phosphorylated form of histone H2AX is termed γ-H2AX and it is considered as an excellent marker for DSBs since γ-H2AX is one of the first proteins recruited to the site of DSBs upon induction (Rogakou et al., 1998, 1999). Immunofluorescence based γ-H2AX foci assay and western blot analysis had been carried out to assay DSBs. The results showed that multiple γ-H2AX foci were induced indicating the presence of DSBs. Our data indicate that there was an increase in the number of γ-H2AX foci in the nuclei of treated cells (Figure 5Aii) compared to untreated control cells (Figure 5Ai). In IMR-90 cells, number of γ-H2AX foci increased after 48 h treatment but decreased following 48 h of recovery in fresh medium. Similarly, in IMR-90 cells pre-treated with NU7026, highest number of foci were observed at 24 h post treatment and decreased thereafter. This implies that normal human cells are capable of repairing DSBs even when DNA-PKcs activity was chemically inhibited (Figure 5B). In contrast, there was time dependent increase in γ-H2AX foci in MO59K cells and CHO AA8 cells. The number of foci has persisted or further increased significantly after 48 h of recovery indicating persistence of DSBs. In addition, higher number of foci detected in MO59K cells and CHO AA8 cells infers higher susceptibility of these cells to Ag-np induced DSBs compared with IMR-90 cells. Interestingly, significantly low number of γ-H2AX foci was observed in MO59J cells and CHO V33 cells which is not line with our hypothesis that more DSBs are expected to be produced in cells lacking functional DNA-PKcs (Figure 5B). γ-H2AX protein expression data as measured by Western blotting are in agreement with observations deduced from the foci assay (Figure 5C). The protein expression level of γ-H2AX in IMR-90 cells was highest at 48 h post treatment and decreased after 48 h of recovery. Similar observation was made for in NU7026 pre-treated IMR-90 cells (Figure 5C). Both MO59K cells and CHO AA8 cells showed time dependent increase in γ-H2AX protein expression and the expression was greatly enhanced after 48 h of recovery. γ-H2AX expression was not detected in MO59J cells. The expression level of γ-H2AX in CHO V33 was relatively low compared with CHO AA8 cells (Figure 5C).
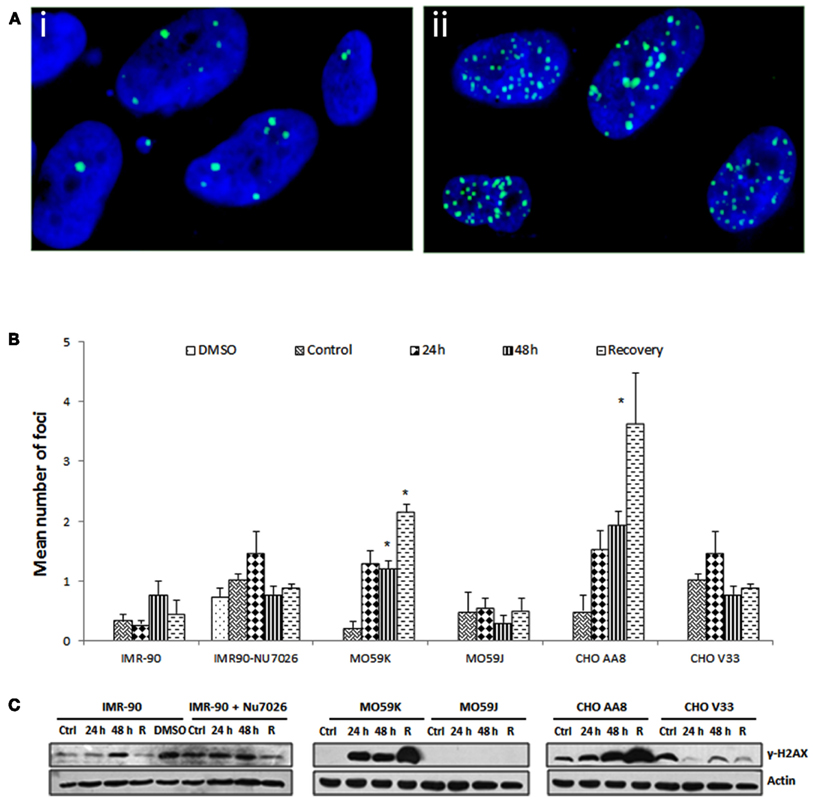
Figure 5. Induction of DNA double strand breaks in response to Ag-np treatment. (A) Representative of immunofluorescence images of γ-H2AX foci without (Ai) and with (Aii) 100 μg/ml Ag-np treatment, detected with anti-γ-H2AX antibodies. Nuclei were stained with DAPI (blue) and γ-H2AX was stained green with FITC. Magnification = 60×. (B) Represents mean no. of γ-H2AX induced in various cell types. *Denotes p < 0.05. (C) Western blots analysis of γ-H2AX following Ag-np treatment. Whole cells were trypsinized and lyzed. Equal amounts of proteins were loaded and separated using 12% SDS-PAGE, transferred to nitrocellulose membrane and immunoreacted with antibodies against proteins of interest. Actin was used as loading control. R denotes 48 h recovery.
Ag-np Induced DSBs Trigger the Activation of DNA-PKcs and ATM
As explained above, Ag-np induce DSBs in treated mammalian cells. Since DNA-PKcs mediates NHEJ repair pathway, which is the primary DSB repair pathway in mammalian cells, we examined whether DNA-PKcs expression and activation (in term of phosphorylation) will be triggered under the influence of Ag-np. We have also included investigation of Ataxia-telangiectasia mutated (ATM), a key signal transducer in the DNA damage response in order to understand further the DNA damage potential of Ag-np in our model. Western blot analysis revealed that IMR-90 cells treated with Ag-np show neither up-regulation nor increase in phosphorylation of DNA-PKcs. There was no significant difference in the expression level of this protein in IMR-90 cells pre-treated with NU7026 followed by Ag-np treatment as verified by densitometry. There was 2.2-fold increase in phosphorylated ATM in Ag-np treated IMR-90 cells and 2.3-fold increase in NU7026 pre-treated IMR-90 cells after Ag-np exposure (Figures 6A,D). As shown in Figures 6B,E, Ag-np treated MO59K cells showed activation and up-regulation of DNA-PKcs and ATM. There was 1.7-fold increment in phosphorylated DNA-PKcs and 4.3-fold increase in phosphorylated ATM. DNA-PKcs was not detected in MO59J cells. Similar results were obtained for CHO AA8 cells following treatment with Ag-np; phosphorylated DNA-PKcs increased by 3.3-fold and phosphorylated ATM by 4.4-fold. DNA-PKcs remained undetected in CHO V33 (Figures 6C,F). These data suggest the induction of DNA damage in all the tested mammalian cell types. It is possible that similar repair mechanisms are initiated in human glioblastoma cells and Chinese hamster cells in response to Ag-np exposure. Higher levels of activated DNA-PKcs and ATM were detected in MO59K and CHO AA8 cells compared to normal human cells. This is in agreement with our above observations in which more DSBs were shown to be induced in MO59K and CHO AA8 cells and therefore higher amount of the DNA repair proteins were activated to repair the DSBs induced compared to IMR-90 cells.
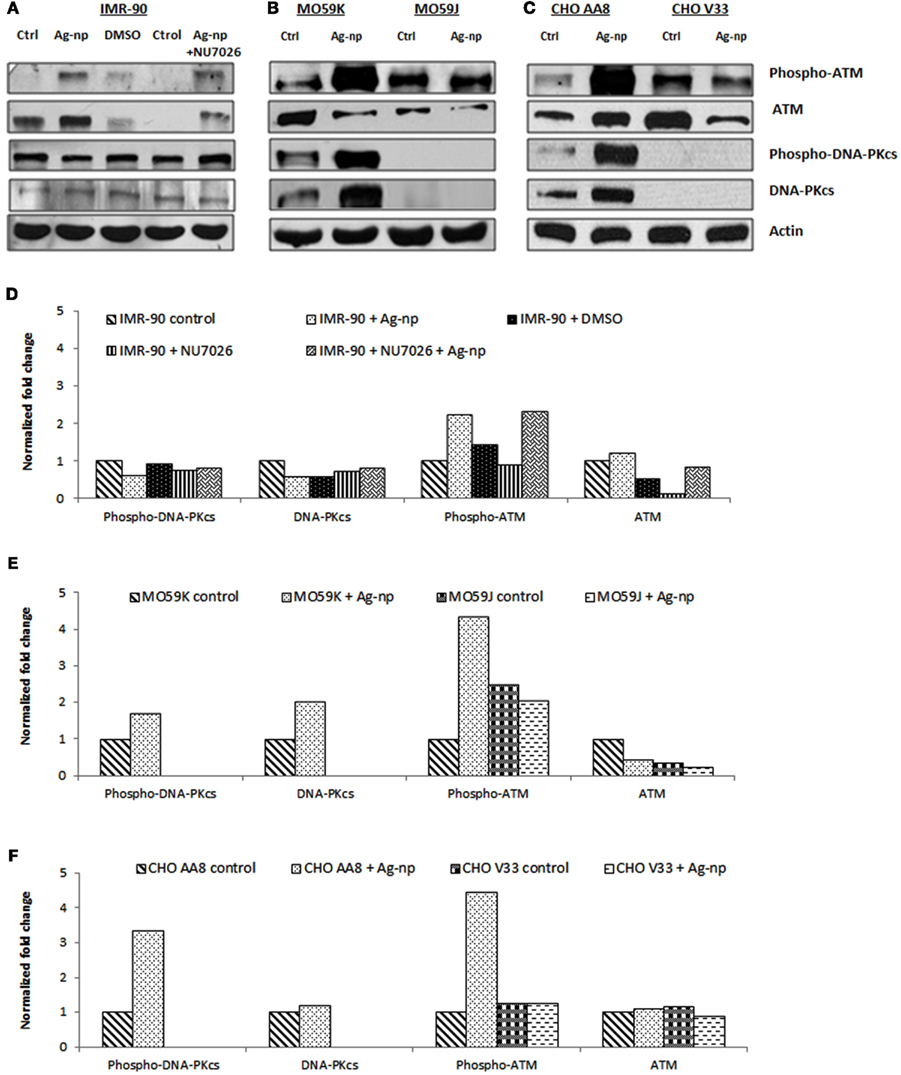
Figure 6. Ag-np upregulated and activated DNA-PKcs and ATM in mammalian cells. Western blot analysis of DNA-PKcs and ATM following 48 h Ag-np treatment (100 μg/ml) and NU7026 (10 μM) in (A) IMR-90 cells, (B) MO59K cells and MO59J cells, and (C) CHO AA8 cells and CHO V33 cells. (D–F) Fold change for protein levels were obtained by densitometry analysis of X-ray films using Kodak molecular imaging software and normalized against actin levels to represent accurate values for proteins which deviated from the control.
We analyzed the levels of phosphorylated ATM in MO59J cells and CHO V33 cells in comparison with MO59K cells and CHO AA8 cells, respectively. Results show that phosphorylated ATM level remained low in both treated and untreated MO59J cells and CHO V33 cells (Figures 6B,C,E,F). The low level of ATM in MO59J cells and CHO V33 cells can be correlated to the mutated status of p53 in these cells.
DNA-PKcs Deficiency or Inhibition Induces Greater G2/M Arrest and Cell Death in Ag-np Treated Cells
DNA damage to cells causes cell cycle arrest. Cells with irreversible damage might undergo apoptosis, giving rise to accumulation of cells in sub-G1 phase (Canman et al., 1994; Boonstra and Post, 2004). We show here that Ag-np induce more significant DNA damage and genome instability in cells lacking functional DNA-PKcs (Figures 3 and 4). To examine whether the extensive DNA damage induced in DNA-PKcs deficient or inhibited cells will perturb the cell cycle progression, cell cycle analysis was performed. We have previously shown that Ag-np induced G2/M arrest in cells (AshaRani et al., 2009b). Cell cycle profiles of DNA-PKcs deficient or DNA-PKcs inhibited cells were compared with respective DNA-PKcs proficient cells.
As shown in Figure 7A, there was no phase shift in IMR-90 cells with and without NU7026 pre-treatment at increasing doses. MO59K cells exhibited a dose-dependent increase in S and G2/M phase up to 60 μg/ml (1.7-fold, p < 0.05), above which the value remained constant, with increase in sub-G1 populations at 60 μg/ml (8.5%) and 100 μg/ml (10.8%) compared to untreated control cells. Data showed no marked changes in cell cycle profile of MO59J cells of all doses. Cell populations arrested at S and G2/M in MO59K cells pre-treated with NU7026 were significant only at 100 μg/ml (6.6-fold, p < 0.01). It is also noteworthy that NU7026 pre-treated MO59K cells showed dose-dependent increase in sub-G1 population and were significant at 60 μg/ml (∼30%, p < 0.01) and 100 μg/ml (∼33%, p < 0.01) as compared to Ag-np treated MO59K cells alone (Figure 7B). There was 1.5-fold increase in G2/M in CHO AA8 cells at 20 μg/ml and 60 μg/ml and only 1.3-fold at 100 μg/ml compared to untreated cells. Percentage of cells in sub-G1 population increased slightly with increasing dose which is not statistically significant. There is 2.2- and 2.4-fold increase in G2/M in CHO V33 cells at 60 and 100 μg/ml (p < 0.05), respectively compared with untreated control. Minimal increase in sub-G1 populations detected in CHO V33 cells. In CHO AA8 cells pre-treated with NU7026, there was 1.5-fold increase in cell population at G2/M phase at 60 μg/ml and this fold increase remained at 100 μg/ml of Ag-np treatment (Figure 7C).
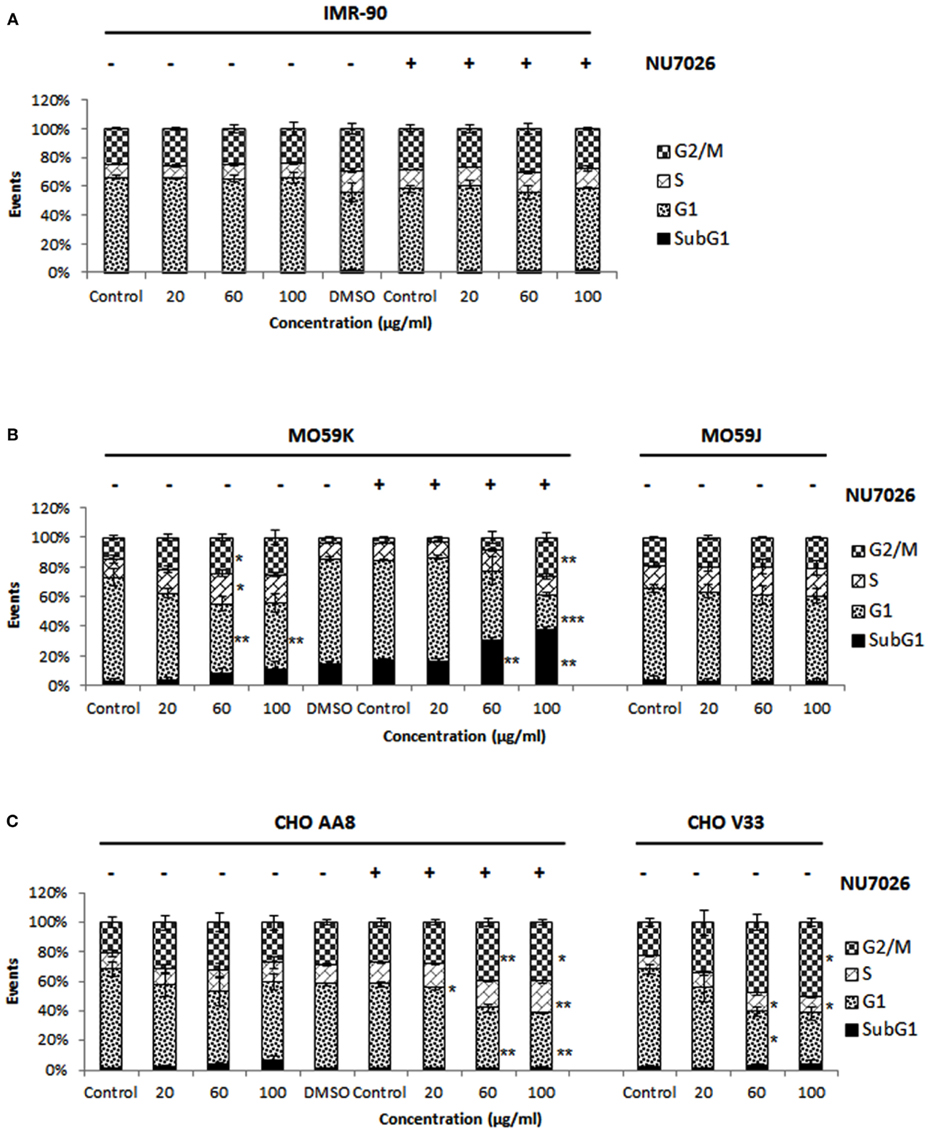
Figure 7. Cell cycle analysis by flow-cytometry. Cell cycle histograms for (A) IMR-90 cells, (B) MO59K cells and MO59J cells, and (C) CHO AA8 cells and CHO V33 cells after 48 h exposure to Ag-np and NU7026 (10 μM) as measured by propidium iodide staining. Markers were set at regions of interest (subG1, G1, S, and G2/M), and the percentage of cells (events) under each area was generated using Microsoft excel.
The data obtained in this study demonstrate two major scenarios following nanoparticle treatment in mammalian cells when DNA-PKcs is absent or inhibited. The efficacy of DNA repair was compromised and more cells were either arrested at S or G2/M phase to enable the repair of Ag-np induced DNA damage as shown in CHO V33 cells and in NU7026 treated CHO AA8 cells. Lack of functional DNA-PKcs following NU7026 in MO59K cells might have caused elevated DNA damage which was beyond repair thereby resulting in cell death. The cell cycle analysis has indirectly implied the importance of DNA-PKcs in the repair of Ag-np induced DNA damage.
Discussion
DNA damage and repair dynamics following Ag-np treatment are not well understood. Thorough investigation of these mechanisms could shed light on the mechanisms of toxicity as well as potential therapeutic applications of nanoparticles. Here, we demonstrate for the first time that Ag-np with average size of 20 nm and negative zeta potential (−0.309 ± 0.13 mV) are able to trigger DNA repair pathway particularly NHEJ repair pathway. Deficiency or inhibition of NHEJ repair pathway sensitized cells to Ag-np. The combined effect of small size and negative surface charges on Ag-np are believed to be the main contributors to our observed results. Ag-np of 20 nm can easily penetrate cell (Asharani et al., 2009a). Toxicity of nanoparticles could have been enhanced with their negative zeta potential as previously shown in our laboratory that Ag-BSA and Ag-PVA bearing negative surface charges show a high affinity for the cell membrane and, as a consequence, are captured by cells with higher efficiency (Wilhelm et al., 2003; Patil et al., 2007; Asharani et al., 2010). Possibly, through electrostatic interaction with positively charged cell surface receptors or nuclear import receptors, negatively charged Ag-np further entered the nucleus generating stress to the genomic material.
Deficiency or pharmacological inhibition of DNA-PKcs has increased sensitivity to Ag-np induced genotoxicity and cytotoxicity. DNA-PKcs plays a direct role in DSBs repair by acting as a key component of NHEJ and defects in kinase activity result in radio- and chemosensitivity (Allalunis-Turner et al., 1993; Bentle et al., 2007). The NHEJ repair mechanism was assumingly impaired in the absence or functional loss of DNA-PKcs (Burma and Chen, 2004). We speculate that the repair capacity for Ag-np induced DNA damage was compromised which resulted in extensive DNA damage and contributed to genomic instability of cells. Since one implication of DNA damage is cell cycle arrest (Pellegata et al., 1996), the transient delay at G2/M phase of cell cycle and decrease in cell viability of DNA-PKcs deficient or inhibited cells could have been due to extensive DNA damage.
In this study, differential susceptibility to Ag-np was observed in MO59J cells and CHO V33 cells despite the fact that both cell types are deficient in DNA-PKcs. CHO V33 cells are highly susceptible to Ag-np as demonstrated by the data obtained in cell viability assay, Comet assay, CBMN assay and cell cycle analysis. In contrast, MO59J cells displayed significant resistance to Ag-np as a deviation from our hypothesis that DNA-PKcs deficiency rendered cells more susceptible to Ag-np. Prolonged absence of DNA-PKcs in MO59J cells might have caused inadequate regulation of p53. The cell death stimulus imbalance, i.e., increased levels of anti-apoptotic Bcl-2 and decreased expression of pro-apoptotic Bcl-2 members (Bak and Bax), could have contributed to the higher resistance in MO59J cells (Chen et al., 2005). Another possibility for remarkable resistance of MO59J cells against Ag-np cytotoxicity may be that these cells are unable to quickly liberate toxic Ag+ from the accumulated Ag-np but rather store Ag-np in non-toxic form, as such accumulation of Ag-np was observed in a recent study (Luther et al., 2011). In addition, this observation can also be considered as a form of cancer cell adaptation (Di Nicolantonio et al., 2005). We speculate that resistance genes are up-regulated under unfavorable condition to facilitate cancer cell survival by conferring protection to MO59J cells against Ag-np in both cellular and macromolecular aspect, causing M059J cells to become less susceptible to Ag-np. Since CHO V33 cells are stable and immortalized hamster cells deficient in DNA-PKcs, the adaptive mechanism in cancer cells is unlikely to be triggered in the CHO V33 cells and hence it is plausible that these cells are more susceptible to Ag-np.
Persistence or enhancement of DNA damage was observed in DNA-PKcs deficient or inhibited cells. On the contrary, efficient recovery of DNA damage was evident in normal human cells. Human cancer cells and immortalized rodent cells may express receptors, ligands, or functional group such as sulfate on cells surface which are not expressed in normal human cells (Moghimi et al., 2005). These surface moieties can either facilitate the uptake of Ag-np into intracellular region or they may have higher affinity for Ag-np which allows intake of greater amount of Ag-np into the cells. Moreover, Ag-np are metallic nanomaterials. Longer time frame is needed to expel these nanoparticles from cells via exocytosis (Asharani et al., 2009a) which suggests that short-term recovery (48 h) is insufficient to remove Ag-np from cells. This could have resulted in the persistence or enhancement of DNA damage in human glioblastoma cells and Chinese hamster cells. The lack of certain surface molecules in normal human cells may render them less susceptible for Ag-np uptake and binding (Moghimi et al., 2005). In addition, alternative repair mechanisms such as homologous recombination (HR) can be triggered at the same time (Takata et al., 1998) to safeguard the genome when NHEJ is inhibited in normal cells to faithfully maintain the genome to counteract potentially mutagenic and cytotoxic insults (Lindahl and Wood, 1999). This could be another reason for efficient recovery in normal cells.
DSBs activate an array of responses which lead to damage repair and continuation of cellular life (Hopfner et al., 2002). In mammalian cells, DSBs initiate the generation γ-H2AX foci, which function as essential indicators for recognition and repair of DSBs (Kinner et al., 2008). We show in this study that Ag-np induce DSBs as indicated by the presence of γ-H2AX foci. DNA damage response is presumably initiated (Sanchez-Perez, 2006) to trigger NHEJ repair pathway as evidenced by DNA-PKcs phosphorylation (serine 2056). Activation of NHEJ repair pathway is a crucial defence mechanism evolved to combat the potential deleterious consequences of DNA damage for maintaining genomic integrity. However, lack of proper NHEJ repair in DNA-PKcs deficient or inhibited cells results in cell death or survival of cells with mis-repaired DNA, which may then lead to tumorigenesis. Under these circumstances it may be advantageous for cells with severely damaged DNA to convert into lethal events, promoting cell death instead of transformation (Sanchez-Perez, 2006).
We have observed a lower level of γ-H2AX protein in MO59J cells and CHO V33 cells compared to MO59K cells and CHO AA8 cells respectively, despite higher amount of DNA damage observed in particular in Ag-np treated CHO V33 cells. On the contrary, γ-H2AX expression increased in both IMR-90 cells treated with Ag-np alone and with NU7026 and Ag-np. It was previously reported that DNA-PKcs and ATM can complement each other in H2AX phosphorylation (Quanz et al., 2009; Shrivastav et al., 2009). When DNA-PKcs activity was inhibited by NU7026 in IMR-90 cells, ATM was presumably activated to phosphorylate H2AX as an effort to trigger DNA repair machinery. These opposing results in human glioblastoma cells and Chinese hamster cells could be explained due to the dominant role played by DNA-PKcs in regulating H2AX phosphorylation in response to DNA damage (Reitsema et al., 2005; Mukherjee et al., 2006; An et al., 2010) due to the reduced level of ATM in both MO59J cells and CHO V33 cells (Kuhne et al., 2003; Peng et al., 2005), which has made H2AX phosphorylation unattainable. Based on this observation, we speculate that the DSBs are induced albeit under detected due to the fact that H2AX is not phosphorylated efficiently when DNA-PKcs is lacking in MO59J cells and CHO V33 cells (An et al., 2010).
ATM activation as evidenced by phosphorylation was also noted in DNA-PKcs proficient cells after Ag-np treatment. Although some studies have shown that ATM might be involved in single-strand breaks repair (Nur et al., 2003) and HR (Morrison et al., 2000), it also plays a vital role as transducer in general damage-signaling and DSB repair (Riballo et al., 1999; Zhou and Elledge, 2000). This explains the activation of ATM in normal human cells, DNA-PKcs proficient human glioblastoma cells and Chinese hamster cells in response to DNA damage induced by Ag-np. Interestingly, we found no change in phospho-ATM level in Ag-np treated DNA-PKcs deficient cells though greater total DNA damage observed. Previous studies correlated the reduced ATM level in these cells to the truncating mutations in both ATM alleles (Tsuchida et al., 2002) and p53 (Anderson and Allalunis-Turner, 2000). Peng et al. (2005) also reported that DNA-PKcs deficiencies could have consequences in processes connected to the network of damage response pathways controlled by ATM that have not been recognized previously which causes down-regulation of ATM.
Conclusion
Based on the data obtained in the present study, it can be concluded that DNA-PKcs deficiency or inhibition impairs NHEJ repair pathway, renders cells to higher susceptibility to Ag-np induced DNA damage and genome instability. DNA-PKcs is an essential component of DNA DSB repair that serves as defence mechanism in cells in response to Ag-np and lack of functional DNA-PKcs sensitize cells to Ag-np induced genotoxicity and cytotoxicity. Normal human cells are less susceptible to Ag-np compared to human glioblastoma cells and Chinese hamster cells. Clearly, further studies are required to understand the molecular interplay and physiological response by which DNA-PK protects cells against Ag-np. Pharmacological inhibition of DNA-PKcs can act as a potent target in cancer cells to enhance the efficacy of Ag-np. The combination of Ag-np treatment and DNA-PKcs inhibition may provide new avenues for the development of novel anticancer therapeutics.
Materials and Methods
Synthesis of Polyvinyl Alcohol (PVA) Capped Ag-np
PVA (150 mg, MW: 16,000 g/mol, Sigma-Aldrich, USA) was dissolved in 15 ml of boiling ultrapure water. Ag-np capped with PVA were synthesized by reducing AgNO3 solution (1.5 mM) with 0.02 g of NaBH4 (Sigma-Aldrich, USA), followed by the addition of PVA solution, under constant stirring at 500 rpm at room temperature. A color change of the solution to light brown color indicated Ag-np formation. Stirring was continued for 2 h. The nanoparticle solution was centrifuged at 25,000 rpm for 1 h and resuspended in ultrapure water to remove traces of reducing agent and unbound PVA. The washing was done twice. The final pellet was lyophilized overnight (Labconco, USA).
Characterization of Ag-np
The lyophilized Ag-np pellet was resuspended in ultrapure water and sonicated to prepare a homogenous nanoparticle suspension. The shape of the nanoparticles was determined by transmission electron microscope (JEOL JSM 1220, JAPAN) analysis. Presence of Ag-np was confirmed by performing Ultraviolet (UV) visible spectroscopy (Agilent, USA). Additionally, a size and charge distribution of the pure nanoparticle suspension was recorded on Zetasizer Nano ZS (Malvern, UK) and analyzed with Dispersion Technology Software (DTS, Malvern, UK). The elemental composition of nanoparticles synthesized was determined using Inductively Coupled Plasma-Optical Emission Spectroscopy (ICP-OES, Perkin Elmer, USA).
Cells, Cell Culture, and Treatment with Nanoparticles and DNA-PKcs Inhibitor
Five commercially available cell types were used in this study, apparently normal human fibroblasts, IMR-90 (Coriell Cell Repositories, USA); human glioblastoma cells, M059K (CRL-2365) and M059J (CRL-2366; American Type Culture Collection, USA); and Chinese hamster ovarian cells, CHO AA8 and CHO V33 (kindly provided by Dr. Paul Wilson, Lawrence Livermore National Laboratory, Livermore, CA, USA). MO59K and CHO AA8 cells express normal level of DNA-PKcs. Both MO59J and CHO V33 cells are deficient in DNA-PKcs expression and activity (Lees-Miller et al., 1995; Natarajan et al., 2008). IMR-90 cells at passage 18 ± 2 were maintained in modified eagles medium with glutamine (MEM, Gibco, Invitrogen, USA) supplemented with 15% fetal bovine serum (FBS), 2% essential amino acids, 1% each of non-essential amino acids, vitamins, and penicillin streptomycin (Gibco, Invitrogen, USA). MO59K and MO59J cells were maintained in Dulbecco’s modified eagles medium (DMEM, Sigma-Aldrich, USA) supplemented with 10% FBS and 1% penicillin streptomycin. CHO AA8 and CHO V33 cells were maintained in MEM alpha medium with glutamine, ribonucleosides, and deoxyribonucleosides (MEM α, Gibco, Invitrogen, USA) supplemented with 10% FBS and 1% penicillin streptomycin. All cells were maintained in a humidified 5% CO2 incubator at 37°C. Stock solution of an organic based DNA-PKcs inhibitor NU7026 (2-(morpholin-4-yl)-benzo[h]chroomen-4-one, Calbiochem, USA) were prepared in dimethyl sulfoxide (DMSO, AppliChem, Germany) and working concentration (10 μM) were made from the stock solution using complete cell culture medium. Stock solutions of nanoparticles (5 mg/ml) were prepared in autoclaved distilled water and diluted to the required concentrations using the complete cell culture medium. Appropriate concentrations of Ag-np stock solution were added to the cultures and incubated for 48 h.
Cell Viability Assay
Following NU7026 and/or Ag-np treatment, the viability of the treated cells was measured using CellTiter-Glo® luminescent cell viability assay (Promega, USA) as described in our previous publication (AshaRani et al., 2009b). For cell viability assay, 5 × 103 cells per well were plated in transparent 96-well plates (Corning, Costar, USA) and treated with various concentration of Ag-np (20, 40, 60, 80, and 100 μg/ml) for 48 h. The CellTiter-Glo® reagent and cell culture mixture were then transferred to white opaque walled 96-well plates (Corning, Costar, USA) for measurement of luminescence using plate reader (Tecan, Switzerland). Following which 100 μg/ml dose was selected for subsequent studies.
Cell Cycle Analysis
Approximately 5 × 105 cells were seeded in 100 mm tissue culture dish (Falcon, USA). Following treatment with NU7026 and/or Ag-np, cells were harvested, washed in 1 × phosphate-buffered saline (PBS, 1st Base, Singapore) and fixed in 3:1 70% ethanol:PBS and stored at −20°C. Fixed cells were then washed with 1× PBS and stained with propidium iodide (PI, Sigma, USA): RNase A (Roche, USA) solution (2 mg propidium iodide and 2 mg RNaseA/100 ml in 1× PBS). Samples were analyzed by flow cytometry (FACS Calibur™, USA) at 488 nm excitation λ and 610 nm emission λ. A total of 10,000 cells were collected and data obtained was analyzed using Summit 4.3 software.
Western Blot Analysis
Cells were seeded as described above and harvested 48 h after Ag-np treatment. Total cellular protein was extracted by lyzing cells in 50–200 μl lysis buffer (10 mM Tris-HCl [pH 7.4], 1% SDS, 1 mM sodium-ortho-vanadate in distilled H2O) for 60 min at 4°C. The whole cell lysate was recovered by centrifugation at 13,200 rpm for 60 min at 4°C. The supernatants were collected. Protein concentrations were determined using Quick Start™ Bradford Protein Assay (Bio-Rad, USA) with bovine serum albumin (BSA, Sinopharm Chemical Reagent, China) as a standard. One hundred micrograms protein from each sample was resolved on a 7.5% SDS-Polyacrylamide gel and transferred onto a nitrocellulose membrane (Bio-Rad, USA). Membranes were blocked for 60 min with 5% non-fat milk in TBS-T (0.1% Tween-20) (Sigma, USA) in 1 × TBS (0.1 M NaCl, 0.1 M Tris pH 7.4 in ddH2O; All purchased from NUMI, National University of Singapore). Subsequently, membranes were immunoreacted with primary antibodies against proteins of interest overnight at 4°C. The membrane was then incubated in secondary antibody conjugated to HRP for 60 min at room temperature. The protein bands were visualized using Chemiluminescence substrate (chemiluminescence Reagent Plus, Perkin Elmer, USA) followed by exposure to X-ray film (Pierce, USA).
Alkaline Single-Cell Gel Electrophoresis (Comet Assay)
Ag-np treated cells were harvested and Ag-np induced DNA damage was detected using alkaline single-cell gel electrophoresis (Comet assay) as described in our previous publication (Poonepalli et al., 2005). Approximately 2 × 105 cells were seeded in 6-well plate (NUNC, Thermo Fisher Scientific, Singapore). Following 48 h treatment, cells were harvested and alkaline electrophoresis was performed to detect DNA damage induced. A total of 50 nuclei per sample were randomly selected and scored with Comet Analysis Software (Metasystems, Germany). The images were captured using Zeiss Axioplan 2 imaging fluorescence microscope (Carl Zeiss, Germany) equipped with triple band filter. The tail moments of the nuclei which corresponded to the fraction of the DNA in the tail of the comet, were measured as a function of DNA damage.
Cytokinesis-Blocked Micronucleus Analysis (CBMN)
Cytokinesis-blocked micronucleus analysis (CBMN), an established biomarker for genomic instability was carried out to evaluate the susceptibility of cells to Ag-np induced chromosomal damage endpoint (micronuclei). Approximately 5 × 105 cells were seeded in 100 mm tissue culture dish. The cells were treated with Ag-np for 48 h and further incubated with cytochalasin B (Sigma, USA, 4 μg/ml) for 22 h. Cells were harvested and the assay was performed as described in our previous publication (AshaRani et al., 2009b). Micronuclei originate from either lagging chromosomes or chromosome fragments which excluded from daughter nuclei following nuclear division prior to cytokinesis. Cytochalasin B was added to stop dividing cells from performing cytokinesis, thus allowing cells that have completed one nuclear division to be recognized by their binucleate appearance. Fixed cells were stained with acridine orange that allows differential staining of nuclear material and cytoplasm. One thousand binucleated cells were scored under Zeiss Axioplan 2 imaging fluorescence microscope equipped under FITC-TRITC filter, and the number of micronuclei was recorded. The analysis was performed according to a reported procedure (Hande et al., 1996).
γ-H2AX foci Assay
Approximately 2 × 104 cells were seeded in 6-well plates (Greiner Bio-One, USA) and treated with Ag-np for 48 h. Cells were washed once with 1× PBS, fixed with 4% formaldehyde for 15 min and permeabilized with 0.2% Triton-X for 5 min. Cells were blocked in 3% BSA for 60 min at room temperature. Cells were incubated in monoclonal γ-H2AX antibody (Millipore, USA, 1:300 3% BSA) for 60 min at room temperature. Unbound antibody was removed by washing three times with 1× PBS and further incubated in secondary antibody conjugated with Fluorescein isothiocyanate (FITC, Santa Cruz Biotechnology, USA, 1:600 FITC/3% BSA) for 60 min. Cells were washed three times with 1× PBS and mounted with slow fade 4’, 6-diamidino-2-phenylindole (DAPI, Vector Laboratories, USA). Fifty cells per sample were captured randomly with Zeiss Axioplan 2 imaging fluorescence microscope equipped with appropriate filters. Number of foci on each cell was recorded.
Authors’ Contributions
Hui Kheng Lim, P. V. Asharani, and M. Prakash Hande designed the experiments. Hui Kheng Lim performed nanoparticle synthesis, cell viability, Comet, CBMN, γ-H2AX foci assay, cell cycle analysis, and western blots. Hui Kheng Lim, P. V. Asharani, and M. Prakash Hande wrote and edited the manuscript. All the authors approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Paul Wilson (Lawrence Livermore National Laboratory, Livermore, CA, USA) for providing CHO AA8 and CHO V33 cells. This study was supported by grants from the Singapore Delft Water Alliance (SDWA). Dr. Veena Hande is thanked for critically reading the manuscript.
References
Ahamed, M., Karns, M., Goodson, M., Rowe, J., Hussain, S. M., Schlager, J. J., and Hong, Y. (2008). DNA damage response to different surface chemistry of silver nanoparticles in mammalian cells. Toxicol. Appl. Pharmacol. 233, 404–410.
Allalunis-Turner, M. J., Barron, G. M., Day, R. S. III, Dobler, K. D., and Mirzayans, R. (1993). Isolation of two cell lines from a human malignant glioma specimen differing in sensitivity to radiation and chemotherapeutic drugs. Radiat. Res. 134, 349–354.
An, J., Huang, Y. C., Xu, Q. Z., Zhou, L. J., Shang, Z. F., Huang, B., Wang, Y., Liu, X. D., Wu, D. C., and Zhou, P. K. (2010). DNA-PKcs plays a dominant role in the regulation of H2AX phosphorylation in response to DNA damage and cell cycle progression. BMC Mol. Biol. 11, 18. doi:10.1186/1471-2199-11-18
Anderson, C. W., and Allalunis-Turner, M. J. (2000). Human TP53 from the malignant glioma-derived cell lines M059J and M059K has a cancer-associated mutation in exon 8. Radiat. Res. 154, 473–476.
Asharani, P., Sethu, S., Lim, H. K., Balaji, G., Valiyaveettil, S., and Hande, M. P. (2012). Differential regulation of intracellular factors mediating cell cycle, DNA repair and inflammation following exposure to silver nanoparticles in human cells. Genome Integr. 3, 2.
Asharani, P. V., Hande, M. P., and Valiyaveettil, S. (2009a). Anti-proliferative activity of silver nanoparticles. BMC Cell Biol. 10, 65. doi:10.1186/1471-2121-10-65
AshaRani, P. V., Low Kah Mun, G., Hande, M. P., and Valiyaveettil, S. (2009b). Cytotoxicity and genotoxicity of silver nanoparticles in human cells. ACS Nano 3, 279–290.
Asharani, P. V., Sethu, S., Vadukumpully, S., Zhong, S. P., Lim, C. T., Hande, M. P., and Valiyaveettil, S. (2010). Investigations on the structural damage in human erythrocytes exposed to silver, gold, and platinum nanoparticles. Adv. Funct. Mater. 20, 1233–1242.
Barbu, E., Molnar, E., Tsibouklis, J., and Gorecki, D. C. (2009). The potential for nanoparticle-based drug delivery to the brain: overcoming the blood-brain barrier. Expert Opin. Drug Deliv. 6, 553–565.
Bentle, M. S., Reinicke, K. E., Dong, Y., Bey, E. A., and Boothman, D. A. (2007). Nonhomologous end joining is essential for cellular resistance to the novel antitumor agent, beta-lapachone. Cancer Res. 67, 6936–6945.
Boonstra, J., and Post, J. A. (2004). Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 337, 1–13.
Burma, S., and Chen, D. J. (2004). Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst.) 3, 909–918.
Canman, C. E., Chen, C. Y., Lee, M. H., and Kastan, M. B. (1994). DNA damage responses: p53 induction, cell cycle perturbations, and apoptosis. Cold Spring Harb. Symp. Quant. Biol. 59, 277–286.
Carlson, C., Hussain, S. M., Schrand, A. M., Braydich-Stolle, L. K., Hess, K. L., Jones, R. L., and Schlager, J. J. (2008). Unique cellular interaction of silver nanoparticles: size-dependent generation of reactive oxygen species. J. Phys. Chem. B 112, 13608–13619.
Chen, G. G., Sin, F. L., Leung, B. C., Ng, H. K., and Poon, W. S. (2005). Glioblastoma cells deficient in DNA-dependent protein kinase are resistant to cell death. J. Cell. Physiol. 203, 127–132.
Di Nicolantonio, F., Mercer, S. J., Knight, L. A., Gabriel, F. G., Whitehouse, P. A., Sharma, S., Fernando, A., Glaysher, S., Di Palma, S., Johnson, P., Somers, S. S., Toh, S., Higgins, B., Lamont, A., Gulliford, T., Hurren, J., Yiangou, C., and Cree, I. A. (2005). Cancer cell adaptation to chemotherapy. BMC Cancer 5, 78. doi:10.1186/1471-2407-5-78
Doherty, A. J., Jackson, S. P., and Weller, G. R. (2001). Identification of bacterial homologues of the Ku DNA repair proteins. FEBS Lett. 500, 186–188.
Gilley, D., Tanaka, H., Hande, M. P., Kurimasa, A., Li, G. C., Oshimura, M., and Chen, D. J. (2001). DNA-PKcs is critical for telomere capping. Proc. Natl. Acad. Sci. U.S.A. 98, 15084–15088.
Hande, M. P., Boei, J., and Natarajan, A. T. (1996). Induction and persistence of cytogenetic damage in mouse splenocytes following whole-body X-irradiation analysed by fluorescence in situ hybridization.2. Micronuclei. Int. J. Radiat. Biol. 70, 375–383.
Hefferin, M. L., and Tomkinson, A. E. (2005). Mechanism of DNA double-strand break repair by non-homologous end joining. DNA Repair (Amst.) 4, 639–648.
Hopfner, K. P., Putnam, C. D., and Tainer, J. A. (2002). DNA double-strand break repair from head to tail. Curr. Opin. Struct. Biol. 12, 115–122.
Ip, M., Lui, S. L., Poon, V. K., Lung, I., and Burd, A. (2006). Antimicrobial activities of silver dressings: an in vitro comparison. J. Med. Microbiol. 55, 59–63.
Jackson, S. P. (2001). Detecting, signalling and repairing DNA double-strand breaks. Biochem. Soc. Trans. 29, 655–661.
Kinner, A., Wu, W., Staudt, C., and Iliakis, G. (2008). Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 36, 5678–5694.
Kuhne, C., Tjornhammar, M. L., Pongor, S., Banks, L., and Simoncsits, A. (2003). Repair of a minimal DNA double-strand break by NHEJ requires DNA-PKcs and is controlled by the ATM/ATR checkpoint. Nucleic Acids Res. 31, 7227–7237.
Lees-Miller, S. P., Godbout, R., Chan, D. W., Weinfeld, M., Day, R. S. III, Barron, G. M., and Allalunis-Turner, J. (1995). Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science 267, 1183–1185.
Lees-Miller, S. P., and Meek, K. (2003). Repair of DNA double strand breaks by non-homologous end joining. Biochimie 85, 1161–1173.
Luther, E. M., Koehler, Y., Diendorf, J., Epple, M., and Dringen, R. (2011). Accumulation of silver nanoparticles by cultured primary brain astrocytes. Nanotechnology 22, 375101.
Moghimi, S. M., Hunter, A. C., and Murray, J. C. (2005). Nanomedicine: current status and future prospects. FASEB J. 19, 311–330.
Morrison, C., Sonoda, E., Takao, N., Shinohara, A., Yamamoto, K., and Takeda, S. (2000). The contro role of ATM in homologous recombinational repair of DNA damage. EMBO J. 19, 463–471.
Mukherjee, B., Kessinger, C., Kobayashi, J., Chen, B. P., Chen, D. J., Chatterjee, A., and Burma, S. (2006). DNA-PK phosphorylates histone H2AX during apoptotic DNA fragmentation in mammalian cells. DNA Repair (Amst.) 5, 575–590.
Natarajan, A. T., Berni, A., Marimuthu, K. M., and Palitti, F. (2008). The type and yield of ionising radiation induced chromosomal aberrations depend on the efficiency of different DSB repair pathways in mammalian cells. Mutat. Res. 642, 80–85.
Nur, E. K. A., Li, T. K., Zhang, A., Qi, H., Hars, E. S., and Liu, L. F. (2003). Single-stranded DNA induces ataxia telangiectasia mutant (ATM)/p53-dependent DNA damage and apoptotic signals. J. Biol. Chem. 278, 12475–12481.
Patil, S., Sandberg, A., Heckert, E., Self, W., and Seal, S. (2007). Protein adsorption and cellular uptake of cerium oxide nanoparticles as a function of zeta potential. Biomaterials 28, 4600–4607.
Pellegata, N. S., Antoniono, R. J., Redpath, J. L., and Stanbridge, E. J. (1996). DNA damage and p53-mediated cell cycle arrest: a reevaluation. Proc. Natl. Acad. Sci. U.S.A. 93, 15209–15214.
Peng, Y., Woods, R. G., Beamish, H., Ye, R., Lees-Miller, S. P., Lavin, M. F., and Bedford, J. S. (2005). Deficiency in the catalytic subunit of DNA-dependent protein kinase causes down-regulation of ATM. Cancer Res. 65, 1670–1677.
Pinto, V. V., Ferreira, M. J., Silva, R., Santos, H. A., Silva, F., and Pereira, C. M. (2010). Long time effect on the stability of silver nanoparticles in aqueous medium: effect of the synthesis and storage conditions. Colloids Surf. A Physicochem. Eng. Asp. 364, 19–25.
Poonepalli, A., Balakrishnan, L., Khaw, A. K., Low, G. K. M., Jayapal, M., Bhattacharjee, R. N., Akira, S., Balajee, A. S., and Hande, M. P. (2005). Lack of poly(ADP-ribose) polymerase-1 gene product enhances cellular sensitivity to arsenite. Cancer Res. 65, 10977–10983.
Quanz, M., Chassoux, D., Berthault, N., Agrario, C., Sun, J. S., and Dutreix, M. (2009). Hyperactivation of DNA-PK by double-strand break mimicking molecules disorganizes DNA damage response. PLoS ONE 4, e6298. doi:10.1371/journal.pone.0006298
Reitsema, T., Klokov, D., Banath, J. P., and Olive, P. L. (2005). DNA-PK is responsible for enhanced phosphorylation of histone H2AX under hypertonic conditions. DNA Repair (Amst.) 4, 1172–1181.
Riballo, E., Critchlow, S. E., Teo, S. H., Doherty, A. J., Priestley, A., Broughton, B., Kysela, B., Beamish, H., Plowman, N., Arlett, C. F., Lehmann, A. R., Jackson, S. P., and Jeggo, P. A. (1999). Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr. Biol. 9, 699–702.
Rogakou, E. P., Boon, C., Redon, C., and Bonner, W. M. (1999). Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146, 905–916.
Rogakou, E. P., Pilch, D. R., Orr, A. H., Ivanova, V. S., and Bonner, W. M. (1998). DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868.
Sanchez-Perez, I. (2006). DNA repair inhibitors in cancer treatment. Clin. Transl. Oncol. 8, 642–646.
Shrivastav, M., Miller, C. A., De Haro, L. P., Durant, S. T., Chen, B. P., Chen, D. J., and Nickoloff, J. A. (2009). DNA-PKcs and ATM co-regulate DNA double-strand break repair. DNA Repair (Amst.) 8, 920–929.
Sonoda, E., Hochegger, H., Saberi, A., Taniguchi, Y., and Takeda, S. (2006). Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair (Amst.) 5, 1021–1029.
Sun, J., and Ding, T. (2009). Differences in DNA damage pathways induced by two ceramic nanoparticles. IEEE Trans. Nanobioscience 8, 78–82.
Takata, M., Sasaki, M. S., Sonoda, E., Morrison, C., Hashimoto, M., Utsumi, H., Yamaguchi-Iwai, Y., Shinohara, A., and Takeda, S. (1998). Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 17, 5497–5508.
Tsuchida, R., Yamada, T., Takagi, M., Shimada, A., Ishioka, C., Katsuki, Y., Igarashi, T., Chessa, L., Delia, D., Teraoka, H., and Mizutani, S. (2002). Detection of ATM gene mutation in human glioma cell line M059J by a rapid frameshift/stop codon assay in yeast. Radiat. Res. 158, 195–201.
Uematsu, N., Weterings, E., Yano, K., Morotomi-Yano, K., Jakob, B., Taucher-Scholz, G., Mari, P. O., Van Gent, D. C., Chen, B. P., and Chen, D. J. (2007). Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J. Cell Biol. 177, 219–229.
Wiley, B., Sun, Y., and Xia, Y. (2005). Polyol synthesis of silver nanostructures: control of product morphology with Fe(II) or Fe(III) species. Langmuir 21, 8077–8080.
Wilhelm, C., Billotey, C., Roger, J., Pons, J. N., Bacri, J. C., and Gazeau, F. (2003). Intracellular uptake of anionic superparamagnetic nanoparticles as a function of their surface coating. Biomaterials 24, 1001–1011.
Keywords: silver nanoparticles, DNA damage and repair, genotoxicity, DNA-PKcs
Citation: Lim HK, Asharani PV and Hande MP (2012) Enhanced genotoxicity of silver nanoparticles in DNA repair deficient mammalian cells. Front. Gene. 3:104. doi: 10.3389/fgene.2012.00104
Received: 04 March 2012; Accepted: 21 May 2012;
Published online: 13 June 2012.
Edited by:
Takeki Uehara, Shionogi & Co., Ltd., JapanReviewed by:
Xueping Chen, Vitargent (International) Biotechnology Ltd., ChinaRyou Fukushima, Shionogi & Co., Ltd., Japan
Copyright: © 2012 Lim, Asharani and Hande. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: M. Prakash Hande, Genome Stability Laboratory, Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Block MD9, 2 Medical Drive, Singapore 117597. e-mail: phsmph@nus.edu.sg