 Chen Li1†
Chen Li1† Xingwang Wang1†
Xingwang Wang1† Fake Li1Hongke Ding1Ling Liu1Ying Xiong1
Fake Li1Hongke Ding1Ling Liu1Ying Xiong1 Chaoxiang Yang2
Chaoxiang Yang2 Yan Zhang1
Yan Zhang1 Jing Wu1*
Jing Wu1* Aihua Yin1*
Aihua Yin1*- 1Medical Genetic Center, Guangdong Women and Children Hospital, Guangzhou, China
- 2Medical Imaging Department, Guangdong Women and Children Hospital, Guangzhou, China
Background: Joubert syndrome (JBS) is a rare neurodevelopmental disorder associated with progressive renal, liver, and retinal involvement that exhibits heterogeneity in both clinical manifestations and genetic etiology. Therefore, it is difficult to make a definite prenatal diagnosis.
Methods: Whole-exome sequencing and Sanger sequencing were performed to screen the causative gene variants in a suspected JBS family. RNA-seq and protein model prediction were performed to clarify the potential pathogenic mechanism. A more comprehensive review of previously reported cases with OFD1 variants is presented and may help to establish a genotype–phenotype.
Results: We identified a novel non-sense variant in the OFD1 gene, OFD1 (NM_003611.3): c.2848A>T (p.Lys950Ter). Sanger sequencing confirmed cosegregation among this family. RNA-seq confirmed that partial degradation of mutant transcripts, which was predicted to be caused by the non-sense-mediated mRNA decay (NMD) mechanism, may explain the reduction in the proportion of mutant transcripts. Protein structure prediction of the non-sense variant transcript revealed that this variant may lead to a change in the OFD1 protein structure.
Conclusion: The genetic variation spectrum of JBS10 caused by OFD1 was broadened. The novel variants further deepened our insight into the molecular mechanism of the disease.
Introduction
Joubert syndrome (JBS) (MIM213300) is a rare neurodevelopmental disorder that is characterized by distinctive mid-hindbrain malformation, cerebellar ataxia, oculomotor apraxia, breathing difficulties, intellectual impairment, and varying degrees of developmental delay. The presence of a specific cerebellar and brainstem malformation known as the “molar tooth sign (MTS)” (Romani et al., 2013; Poretti et al., 2014) on brain magnetic resonance imaging (MRI) is one of the main diagnostic characteristics of JBS. The MTS comprises a deep interpeduncular fossa; prominent, straight, and thickened superior cerebellar peduncles; and hypoplasia of the cerebellar vermis (Maria et al., 1999). Apart from neurological involvement, JBS is also associated with gradual damage to the retina, kidneys, and liver. In addition, polydactyly has also been observed in some patients (Parisi 2019).
JBS is a clinically and genetically heterogeneous ciliated disease. To date, at least 35 subtypes of JBS have been classified according to different causative genes. Most of them are autosomal recessive, while only JBS10 (MIM 300804) is an X-linked recessive (XLR) disorder. In JBS10, hemizygous male patients are affected, and heterozygous females are asymptomatic (Coene et al., 2009).
JBS10 is caused by OFD1 mutations. The OFD1 protein is essential for the formation of primary cilia and the establishment of left–right asymmetry. Moreover, it has been suggested that this multitasking protein has additional functions, such as controlling cell cycle progression (Alfieri et al., 2020), participating in chromatin remodeling and DNA repair (Abramowicz et al., 2017), acting as a novel autophagy receptor that is independent from the mTOR pathway (Franco and Morleo 2021), interacting with the translation machinery and modulating the translation of specific mRNA targets in the kidney (Iaconis et al., 2017). Pleiotropy is a common characteristic of ciliopathies (Giorgio et al., 2007). Mutations in the OFD1 gene with different degrees of severity may cause oro-facio-digital-syndrome 1 (OFD1 syndrome, MIM 311200) (Macca and Franco 2009), Joubert syndrome (JBS10), retinitis pigmentosa (RP23, MIM 300424) (Webb et al., 2012) and primary ciliary dyskinesia (PCD, MIM 244400) (Coene et al., 2009; Bukowy-Bieryllo et al., 2019). Since the first JBTS10 case was reported in 2009 (Coene et al., 2009), only 23 families (26 patients) with male patients have been reported for JBTS10, and there were a few fetal diagnoses. Thus, it remains difficult to obtain a comprehensive understanding of this rare disease.
In this study, we describe a non-consanguineous healthy Chinese couple with two fetuses who both had unexplained hypoplasia of the cerebellar vermis and polydactyly. Employing whole-exome sequencing (WES) technology, we identified a novel variant in the OFD1 gene. Bioinformatic validations were performed to confirm the pathogenicity of the variant at the transcription and protein levels. Furthermore, we summarized a more comprehensive review of previously reported JBS10 cases with OFD1 variants, which may help to understand how the phenotype emerges from the genotype.
Materials and methods
Ethical approval
The present study was approved by the institutional review board and clinical research ethics committee of Guangdong Women and Children Hospital. Informed consent was provided by all the participants or their statutory guardian in this study.
Subjects
A 28-year-old mother at 30 weeks of gestation was transferred to our Prenatal Diagnosis Center due to brain abnormalities and polydactyly in the fetus. Prenatal ultrasound (US) in our hospital showed thickening of the superior cerebellar feet, cerebellar vermis agenesis, enlarged posterior cranial fossa and bilateral postaxial polydactyly on the hands and right foot (Figures 1AⅠ–Ⅳ). Fetal brain MRI further displayed typical “MTS”, posterior meningocele involving the inferior portion of the occipital bone and superior cervical vertebrae, thin brain stem and polymicrogyria (Figures 1AⅤ–Ⅶ), which signified a poor prognosis. Therefore, the parents decided to terminate the pregnancy due to the fetal deformity. Umbilical cord blood sampling was performed on the fetus, which revealed normal chromosomal microarray (CMA) results. Later, trio-WES identified a novel OFD1 variant (XLR) inherited from the mother.
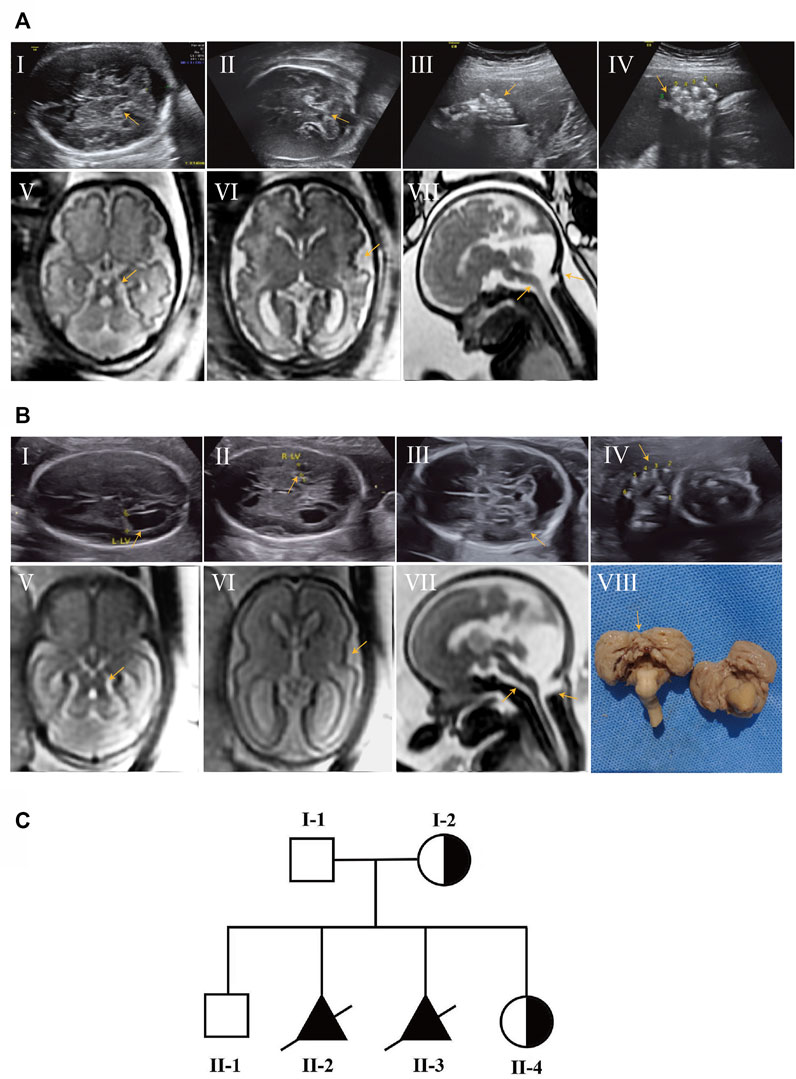
FIGURE 1. Prenatal ultrasound and fetal brain MRI findings of affected subjects and pedigree chart of the family with JBTS10. (AⅠ–Ⅳ) Prenatal ultrasound of II-2 showed thickening of the superior cerebellar feet, cerebellar vermis agenesis, enlarged posterior cranial fossa (AⅠ,Ⅱ) and bilateral postaxial polydactyly on the hands and right foot (AⅢ,Ⅳ) at 30 weeks of gestation. (AⅤ–Ⅶ) Brain MRI of II-2 showed MTS (AⅤ), polymicrogyria (AⅥ), posterior meningocele and thin brain stem (AⅦ). (BⅠ–Ⅳ) Prenatal ultrasound of II-3 showed a similar anomaly to II-2 at 25 weeks of gestation. (BⅤ–Ⅶ) Brain MRI of II-3; polymicrogyria was not obvious compared with II-2. (BⅧ) Autopsy result of II-3 showed absent cerebellar vermis. (C) Pedigrees of the family with JBS10 profiled in this study.
This was the second child of this healthy, unrelated Chinese couple. They already had a healthy son without this OFD1 variant. After 1 year, the mother came to us at 25 weeks of gestation for a similar abnormality (Figures 1BⅠ–Ⅶ) in the fetus. Gene detection confirmed the same OFD1 variant in this fetus (II-3). The couple chose induced abortion and agreed to undergo further autopsy (Figure 1BⅧ). When the mother conceived for the fourth time, she came to us in the first trimester. Through chorionic villus sampling at the 13th week of gestation and normal prenatal US and brain MRI, they finally had a girl who was a carrier (Figure 1C) (Table 1).
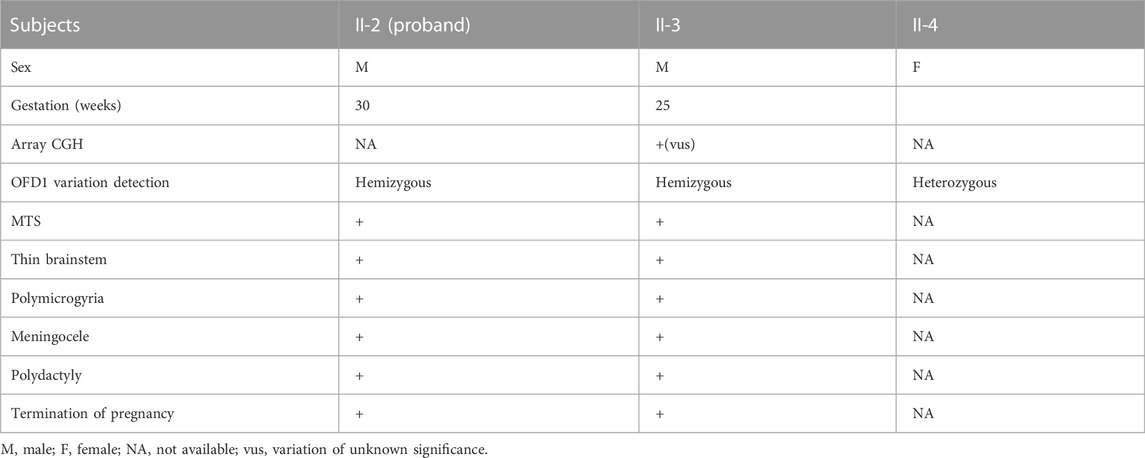
TABLE 1. Clinical data of three fetuses in this family.
Umbilical cord blood sampling, chorionic villus sampling and DNA extraction
Fetal samples of the proband (II-2) were collected by umbilical cord blood sampling. The procedure was performed using the transabdominal approach. Under aseptic conditions, a 22-gauge spinal needle was inserted into the umbilical artery under continuous ultrasound guidance, and umbilical cord blood was collected in an EDTA vacuum tube. Samples of the fetuses (II-3, II-4) were collected by amniocentesis. The procedure was performed in much the same way as umbilical cord blood sampling, except that the spinal needle was 18–20 gauge and inserted into the placenta to obtain chorionic villus samples.
Genomic DNA was extracted from the umbilical cord blood samples (II-2, II-3), chorionic villus samples (II-4), and peripheral blood samples of the parents (I-1, I-2) and their unaffected son (II-1) by using the QIAamp DNA Mini Kit (Qiagen, Germany).
Microarray analysis
Microarray analysis was performed using a high-resolution genotyping single nucleotide polymorphism microarray, Affymetrix CytoScan 750 K Array (Affymetrix, Santa Clara, CA, United States). Copy number variations (CNVs) were identified based on associated records of the human reference genome 37 (NCBI37hg19) of the National Center for Biotechnology Information. Data were analyzed in accordance with American College of Medical Genetics guidelines.
Whole-exome sequencing (WES) and sanger sequencing
Briefly, the sequencing library preparation and target enrichment experiments were performed following the manufacturer’s procedures for the SureSelectXT Clinical Research Exome kit (Agilent Technologies, Santa Clara, CA). Then, paired-end reads were sequenced on a NextSeq 500 platform (Illumina, San Diego, CA) with 150-bp paired-end reads, yielding an average coverage above 110×, with 97.6% of target bases covering at least 10×. Sequence quality analysis and filtering of mapped target sequences were conducted under the “varbank” exome and genome analysis pipeline v.2.1 (Vetro et al., 2020). Standards and guidelines recommended by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology were used to interpret the sequence variants (Richards et al., 2015). The genomic variation database (http://dgv.tcag.ca/dgv/app/home), DECIPHER database (https://decipher.sanger.ac.uk/), and OMIM database (http://www.ncbi.nlm.nih.gov/omim) were employed. Furthermore, the allele frequency and pLI score (representing the tolerance for truncating variants) from gnomAD databases (http://gnomad.broadinstitute.org), REVEL score (a combined method of pathogenicity prediction) (Ioannidis et al., 2016) and searching egnine for variants (Mastermind, https://mastermind.genomenon.com/) were also referenced to assist in the interpretation of variant pathogenicity. VarSome (https://varsome.com/), a widely used online pathogenicity grading tool, was employed to assess the pathogenicity of variation sites (Supplementary Figure S1).
Sanger sequencing was performed to validate the findings of trio WES in the fetuses and parents. The PCR primers for sequencing (targeting OFD1 (NM_003611.3) c.2848A>T) were as follows: forward 5′-CAGGCTGTACTTGAAGGAT-3′, reverse 5′-GTCCCAAGAAGTTAAGCCA-3′.
RNA sequencing and data analysis
RNA sequencing was performed by Aegicare, which has been described in our previous literature (Qi et al., 2022). Total RNA was extracted from peripheral blood samples of one of the carriers (I-2) and two normal females using a Qiagen blood RNA extraction kit I (QIAGEN, United States) according to the manual. After complete property control and quality control of RNA, 1 μg of RNA was aspirated for mRNA library construction by using the TIANSeq Fast RNA Library Kit (Illumina, United States). Finally, the library was amplified by using a high-fidelity enzyme. After quality control, the libraries were sequenced on an Illumina HiSeq 4,000 platform.
The raw reads were subjected to quality control before mapping to the reference genome through the fastp program. Adapter sequences and low-quality reads were filtered out, and high-quality reads were mapped onto the reference genome (Homo sapiens, hg19) by using STAR (Dobin et al. 2013), ultrafast universal RNA-seq aligner STAR_2.3.059 with default parameters. Finally, the read count tables were generated by HTSeq (Anders et al., 2015) software, while the fragment per kilobase of transcript per million reads (FPKM) of each gene was calculated based on the length of the gene and read count tables.
The expression ratio of the OFD1 biallelic gene is estimated by the reads containing specific SNP sites.
Conservational and protein structural analysis
The evolutionary conservation of the affected amino acid (AA) residue by the non-sense variant was analyzed using the MEGA7 online tool (http://www.megasoftware.net/previousVersions.php) with default parameters. Protein structure was predicted by AlphaFold. RoseTTAFold online software (https://robetta.bakerlab.org/) was also used to predict the protein structure (Supplementary Figure S1).
Results
Chromosomal microarray (CMA) analysis
CMA was normal in the proband (II-2) but revealed a 696 kb heterozygous deletion at 5p13.33 (2091630-2787829) in the fetus (II-3), which was inherited from his mother. No similar deletion has been reported to cause disease, nor was a similar deletion found in the DGV database of the general population. The deleted region encompasses four genes. The four genes are LOC100506858, LSINCT5, IRX2, and C5orf38. LOC100506858 is a pseudogene. Although the last three genes are OMIM genes, there is no OMIM causative gene among them. Therefore, it is identified as a variants of unknown significance (VUS) according to the CNV classification guidelines of ACMG.
Gene findings
Trio WES identified a hemizygous variant c.2848A>T (p.Lys950Ter) in the OFD1 (NM_003611.3) gene in the proband (II-2). The OFD1 gene is located in the area between the pseudoautosomal boundary and the Duchenne muscular dystrophy (DMD) gene on the short arm of chromosome X. OFD1 encodes a component of the centrioles controlling mother and daughter centriole length (Singla et al., 2010). The variant c.2848A>T (p.Lys950Ter) resulted in truncation after the first 950 residues. This non-sense mutation was absent in the population databases (GnomAD v2.1.1) and is predicted to undergo non-sense-mediated mRNA decay (NMD). The variant of the proband (II-2) was inherited from the mother, who was a heterozygous carrier. The variant cosegregated with the disease in the family, as shown by direct Sanger sequencing in all family members (Figure 2). This variant has not been reported in public databases, and few papers have examined its impact on the expression or activity of the OFD1 protein. In silico tools predicted that this variant was disease causing (Supplementary Figure S2). Taking all of these findings into consideration, this mutation can be classified as “likely pathogenic” because of the evidence chain (PVS1 + PM2) based on the ACMG guidelines (Richards et al., 2015) (Supplementary Table S1).

FIGURE 2. Genetic findings. (A). WES detected a hemizygous variant c.2848A>T (p.Lys950Ter) of the OFD1 gene in the index fetus, which was heterozygous in the mother and absent in the father. (B). Sanger DNA sequencing chromatogram showed the cosegregation of this variant in all family members.
Evaluation of the effect of the gene mutation on gene function
To further investigate the influence of this variant on gene function, we performed RNA-seq on the mother’s blood sample to assess the effect of the variation on gene expression or transcript length. The results showed that OFD1 had biallelic gene expression in all samples, which was consistent with a previous study showing that OFD1 escaped inactivation (Figures 3C,D). Further analysis found that when the expression of OFD1 was standardized to FPKM, its expression in the mother’s sample was lower than that in the female control samples (Figure 3B). Subsequently, we used the reads containing specific SNPs to calculate the ratio (mutant-type reads/wild-type reads) of OFD1 biallelic gene expression and found that the ratio in the mother’s sample was lower than that in the control female sample (mutant reads/wild-type reads) (Figure 3E). This suggests that the lack of mRNA in the mother sample is due to the lack of mRNA of the mutant allele, which may be caused by RNA degradation caused by non-sense mutations. However, there was still a certain amount of mutant transcripts. This suggests that these RNAs may escape this mechanism and may be translated into proteins. The function of proteins is closely related to their structure. Therefore, we used the AlphaFold model to predict the protein spatial structure of the wild type and mutant type and found that the protein structure of the mutant type changed greatly (Figures 3F,G), which may have affected its function.
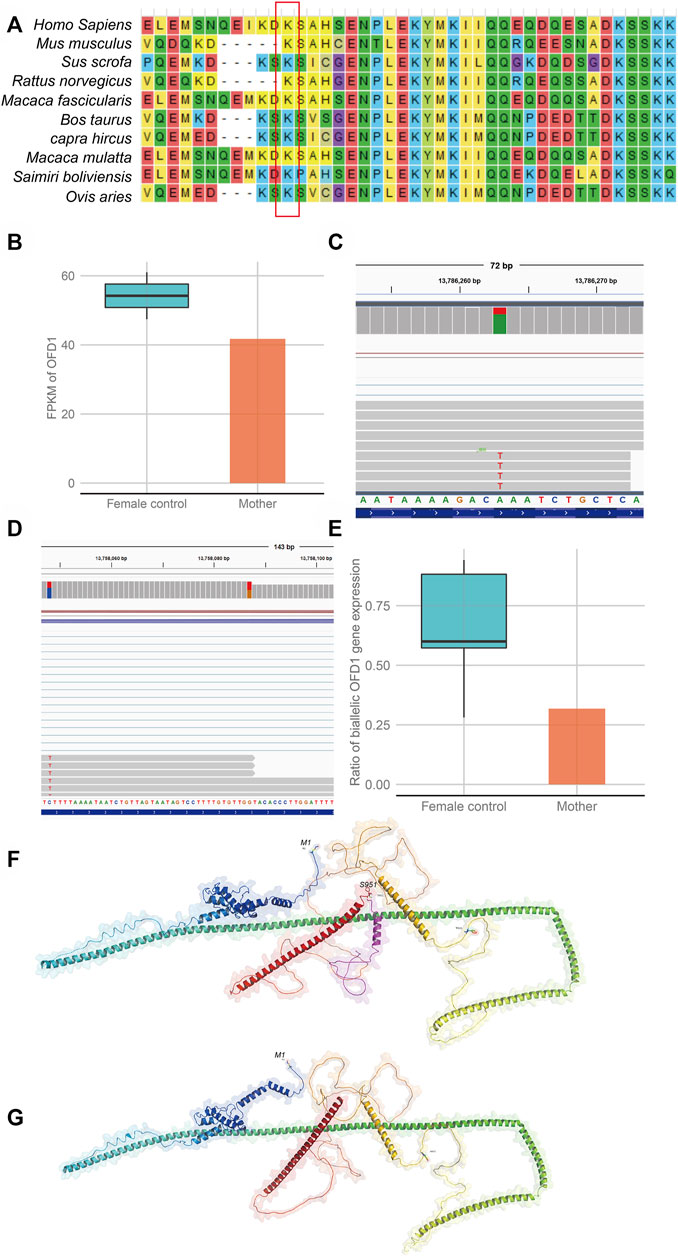
FIGURE 3. Evaluation of the effect of the gene variation on gene function (A). Alignment of multiple OFD1 protein sequences across species. Letters in the red rectangle show that the regions of the variant p.Lys950Ter were highly conserved across different species. (B) Comparison of OFD1 expression in blood between a heterozygous mutant individual (mother) and wild-type female individuals (female controls). (C) Biallelic expression bias of OFD1 occurs in the mother. (D) Biallelic expression bias of OFD1 occurs in wild-type female individuals (female control). (E) The expression bias is more serious in the mutant individual (mother). (F,G) The predicted structure of the mutant protein and wild-type protein by AlphaFold software. (F) (wild-type protein), the amino acid in the red box is missing from the mutant protein. (G) (mutant protein).
Discussion
Herein, we report a non-consanguineous healthy Chinese couple with two male fetuses, both of whom were suspected to suffer from unexplained hypoplasia of the cerebellar vermis, enlarged posterior fossa and polydactyly detected by prenatal US. Further fetal brain MRI presented more severe anomalies, resulting in the couple deciding to terminate the pregnancies. This shows the significance of fetal brain MRI in prognosis evaluation.
Since the embryologic development of the cerebellum extends over a long time, it is more vulnerable to a wide range of destructions and disruptions (Andescavage et al., 2017). Undoubtedly, sonography is the initial imaging tool for screening of the fetal brain. However, prenatal sonography has some difficulties in subdividing multiple similar abnormalities in the cerebellum and posterior fossa due to technical limitations (Wald et al., 2004). Fetal brain MRI, which has higher-contrast resolution than prenatal sonography, has been an adjunct to prenatal ultrasound since the 1980s (Thickman et al., 1984). The use of fetal MRI has increased the diagnostic accuracy of fetal brain abnormalities (Wilson et al., 2020). Griffiths et al. (2017) demonstrated that in the United Kingdom, the overall diagnostic accuracy for detecting an isolated posterior fossa abnormality was only 65.4% for US compared to 87.7% for fetal MRI, and the prognosis was altered after MRI in 44% of cases. In 2021, another large single-center study found that fetal MRI resulted in a change in fetal prognosis in 70% of cases (Schlatterer et al., 2021). In conclusion, it is necessary to use MRI as a secondary technique to confirm, correct, or complement questionable US findings in cases of fetal brain abnormalities.
In our study, CMA revealed no clinically relevant deletions or duplications in the proband (II-2). The fetus (II-3) revealed a 696 kb deletion of 5p15.33 Supplementary Figure S3, which was inherited from his mother and identified as a VUS according to the CNV classification guidelines of ACMG. Although the 696 kb deletion encompasses four genes, none of those genes is related to fetal phenotypes. In addition, the mother did not exhibit the disease phenotypes corresponding to these genes, further indicating that these genes do not seem to be dose-sensitive and that normal expression of an allele can maintain normal biological function. To further explore the pathogenic factors, trio-WES was adopted for the proband (II-2) and the parents. A novel hemizygous non-sense variant c.2848A>T (p.Lys950Ter) of the OFD1 gene was detected in the proband (II-2), and his mother carried this variant. Sanger sequencing confirmed that this variant was in genotype-phenotype segregation among the family members. To our knowledge, this mutation has not been reported in any public databases. OFD1 (NM_003611.3) c.2848A>T (p.Lys950Ter), which occurred in the 21st exon, is predicted to encode a non-sense mutation leading to a premature stop codon, p.Lys950Ter. The conservation of the affected residue was relatively maintained across mammal species (Figure 3A).
To test whether this mutation caused non-sense-mediated mRNA decay and thereby loss of OFD1 function, we measured OFD1 mRNA levels by RNA-seq in the mother (I-2), who was a female carrier. Since the OFD1 gene escapes X inactivation in humans according to previous reports (de Conciliis et al., 1998; Carrel et al., 1999; Carrel and Willard 2005), X inactivation should not be skewed in carrier females. Indeed, in carrier female I-2 and female control samples, there was no preferential expression of either X chromosome. Second, after calculating the statistics of reads with specific SNP sites in the control female sample, the proportion of escaped allele OFD1 expression exceeded more than half of the normal active OFD1 allele expression. This implied that this escape was partial and incomplete. Third, the results also showed that OFD1 gene expression and the ratio of mutant reads to wild-type reads in the mother sample were lower than those in female control samples. These results indicated that the mutant transcripts were partially degraded. Suggesting that this truncation mutation could trigger the NMD pathway. The NMD mechanism may be one of the reasons for the reduction in the proportion of mutant transcripts. As a truncation mutation, it may result in the synthesis of abnormal protein, which can be toxic to cells through dominant negative effects. To our knowledge, carriers I-2 and II-4 in this family are both without obvious disease-associated phenotypes, and we can conclude that this abnormal protein would not cause a dominant negative effect and would only retain residual wild-type activity. Alternatively, even if it can cause this effect, the NMD mechanism would reduce the amount of abnormal protein, resulting in a loss-of-function effect (Khajavi et al., 2006).
Moreover, as predicted by AlphaFold software, the non-sense variant may lead to a change in the OFD1 protein structure (Figure 3F). It is tempting to speculate that the function of the protein may be affected. The OFD1 gene encodes a 1011-amino acid protein that does not have similarities to previously known proteins. There is a Lis1 homology (LisH) motif in the N-terminal region and five coiled-coil α-helical domains among almost the entire length of the molecule (de Conciliis et al., 1998). As reported, the C-terminal OFD1 region containing the last two predicted coiled-coil motifs (aa 615-1012) was found to bind to the C-terminal region of SDCCAG8 (aa 533-713), which also carried two predicted coiled-coil motifs (Otto et al., 2010). At the protein level, which is predicted by AlphaFold, the substitution (Lys950Ter) would give rise to a truncated protein missing an alpha helix compared to the wild-type protein. Since the coiled-coil motifs are built by two or more alpha-helices that wind around each other to form a supercoil (UniProtKB), the lack of an alpha-helix may influence the formation of coiled-coil motifs and affect the binding of the OFD1 protein to other proteins. Thus, if a male fetus inherits this variant allele, the amount of OFD1 mRNA will be reduced, and the protein produced by the remaining mRNA will be altered.
Mutations in OFD1 can cause four diseases, OFD1 syndrome, JBS10, RP23, and PCD. OFD1 syndrome is caused by dominant mutations of OFD1, and the last three diseases are caused by recessive OFD1 mutations. RP23 is limited to ocular symptoms, with no extraocular manifestations (Wang et al., 2017; Chen et al., 2018). PCD is a motile ciliopathy that mainly causes respiratory phenotypes (Bukowy-Bieryllo et al., 2019). OFD1 syndrome and JBS10 are primary cilia dysfunction diseases that affect multiple organs. They have both overlapping and unique phenotypes. The overlapping phenotypes presented in JBS10 and OFD1 syndrome still have some notable differences. As Gangaram et al. (2022) reported, the degree of ID is more severe and the structural CNS malformations are more extensive in JBS10 than in OFD1 syndrome. CNS malformations reported in JBS10, which are summarized in Table 2, include cerebellar vermis hypoplasia (with or without MTS), Dandy-Walker malformations, ventriculomegaly, cephalocele, meningocele, brain stem hypoplasia, polymicrogyria, microgyria, hypothalamic hamartoma, corpus callosum hypoplasia, and pituitary hypoplasia. Corpus callosum anomalies are the only commonly reported CNS malformations in OFD1 syndrome. Polydactyly is often seen in JBS10 but is uncommon in OFD1 syndrome, while polycystic kidney is much more prevalent in OFD1 syndrome.
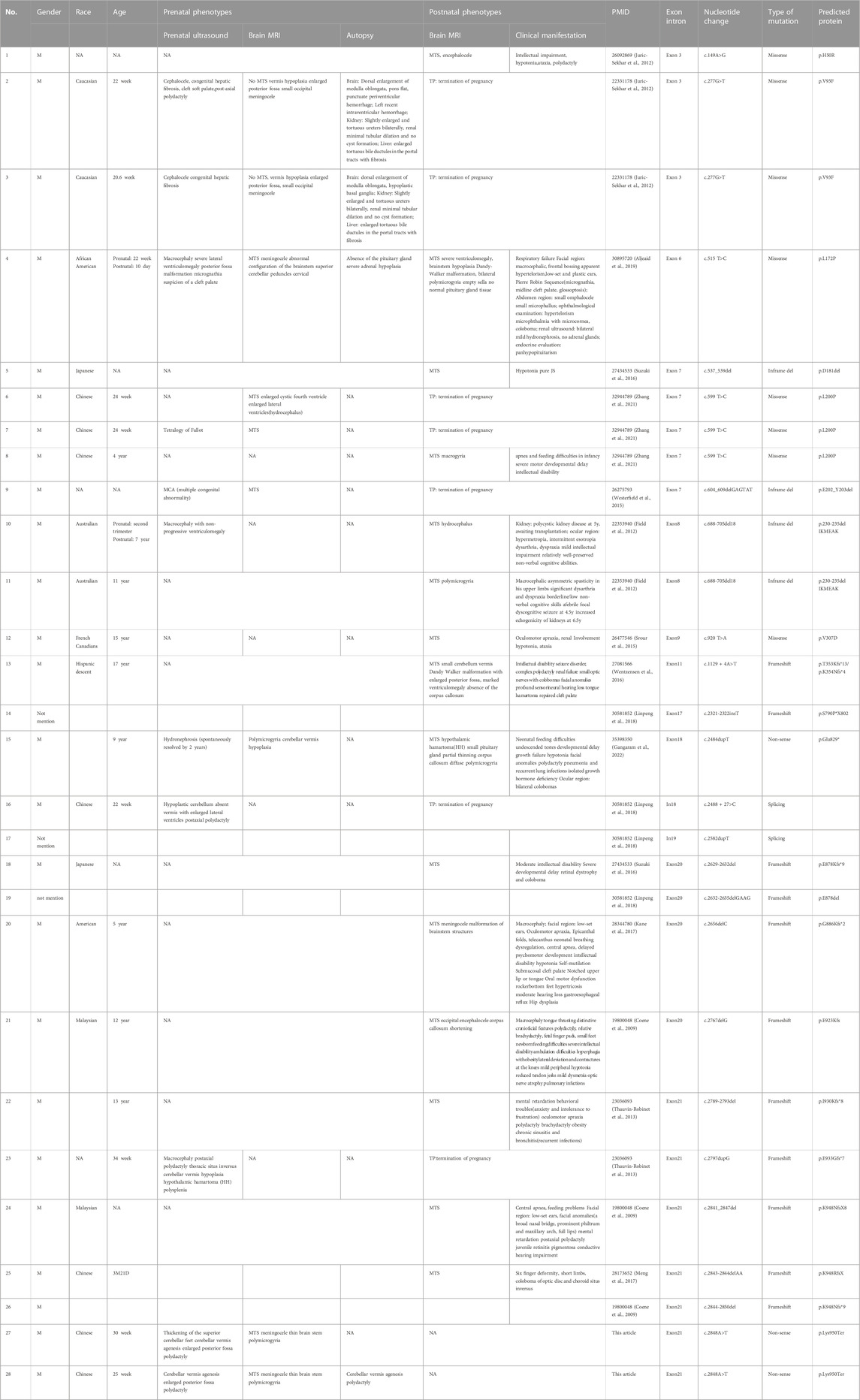
TABLE 2. Prenatal and postnatal features of individuals with previously described OFD1 variants in JBS10 (including the individuals in this study).
To date, only 23 variants of OFD1 have been reported to cause JBS10 (Figure 4), including 10 deletion/insertion mutations, four in-frame deletions, five missense mutations, two splicing mutations and two non-sense mutations. Mutations, especially frameshift mutations in the first 16 exons of the OFD1 gene, mostly lead to OFD1 syndrome. OFD1 syndrome is an X-linked dominant disease that shows typical dysmorphic features, malformations of the oral cavity, and skeletal and CNS abnormalities in female patients (Franco and Thauvin-Robinet 2016; Pezzella et al., 2022). However, it is often lethal in males. Notably, the five missense mutations (OFD1 c.149A>G, c.277G>T, c.515 T>C, c.599 T>C, c.920 T>A) and the three in-frame deletions (OFD1 c.537_539del, c.604_609del, c.688-705del) shown in Table 2, which were located in the first 16 exons but caused JBS10, not OFD1 syndrome, may have occurred because these mild mutations may not lead to OFD1 protein truncation and likely do not abolish its binding affinity to functionally interacting proteins, such as lebercilin (Coene et al., 2009; Bachmann-Gagescu et al., 2015; Kroes et al., 2016). Other mutations are located up to exon 17 of the protein, mainly in exon 21, generating proteins retaining the fifth coiled-coil domain and predicting reduced binding of the protein to its partners without affecting pericentriolar localization (Coene et al., 2009). In addition, as shown in Table 2, there is a frameshift mutation (OFD1 c.1129 + 4A>T) in exon 11 of Patient 13 presenting as JBS10 rather than OFD1 syndrome. Another frameshift mutation (OFD1 c.2789-2793del) in exon 21 presented as JBS10 in one patient but PCD in another. The mechanism accounting for this phenomenon requires further study.
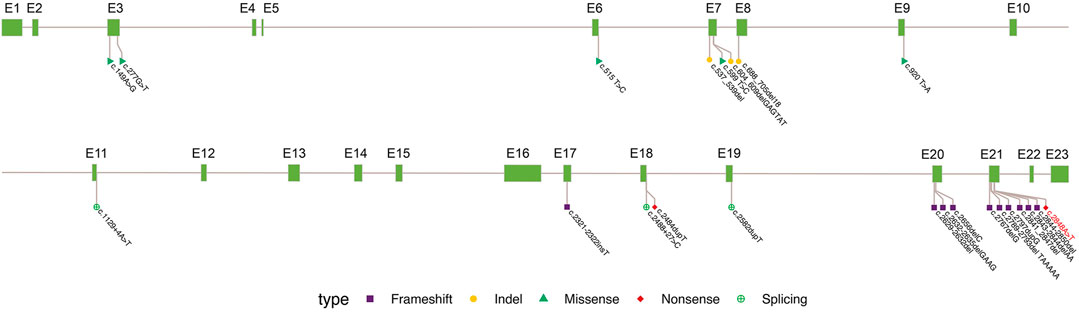
FIGURE 4. Summary of OFD1 variations in JBS10. Exon (grass–green square); variation types: frameshift (purple square), indel not causing frameshift (yellow circle), missense (green triangle), non-sense (red diamond), and synonymous (green cross circle). The site marked in red is reported in the paper.
The phenotypic spectrum associated with OFD1 mutations resulting in JBS10 has been extended over the years. To date, 28 patients from 23 families with JBS10 have been identified (Table 2), four of whom had no phenotypic description in the articles (Patients 14, 17, 19, and 26). The fetal phenotypes were obtained in 16 fetuses, and there were mainly brain abnormalities, such as MTS, meningocele, polymicrogyria, thin brainstem, ventriculomegaly, and macrocephaly. Some fetuses (31%, 5/16) had polydactyly, and other anomalies were also found in the individual fetuses. Overall, 12% (2/16) of fetuses had congenital hepatic fibrosis; 6% (1/16) had tetralogy of Fallot; 6% (1/16) presented with hypothalamic hamartoma (HH), thoracic situs inversus and polysplenia; 6% (1/16) had cleft palate; and 6% (1/16) displayed hydronephrosis. Postnatal phenotypes were obtained in 15 male patients. Common clinical manifestations among these patients included facial anomalies, intellectual impairment, motor developmental delay, hypotonia, ataxia, seizure, polydactyly, renal involvement, retinal dystrophy and colobomas, hearing impairment, cleft palate, pulmonary infections, dyspnea and newborn feeding difficulties. Panhypopituitarism was seen in Patient 4, and HH was present in Patient 15. When we encounter a fetus with the dominant prenatal phenotypes described above, we should consider the possibility of genetic disease, and chromosome detection alone is not sufficient. Trio-WES should be performed on these fetuses to identify etiological factors in genetics. In addition, when gestation was less than 22 weeks (Patients 2 and 3), the MTS was not obvious, and we should be vigilant about the suspected brain signs. When a fetus has been diagnosed with JBS10, possible postnatal phenotypes should be provided to the couple to evaluate prognosis.
Conclusion
In this study, we first identified a novel variant of the OFD1 gene employing WES technology and bioinformatics strategies, broadening the spectrum of OFD1 mutations. Our results provide data for genetic counseling and prenatal diagnosis in this family and contribute to comprehending the possible pathogenic mechanisms of JBS10, which is beneficial to genetic counseling and timely perinatal management.
Data availability statement
The data presented in the study are deposited in the NCBI BioProject repository, accession number PRJNA914088.
Ethics statement
The studies involving human participants were reviewed and approved by institutional review board and clinical research ethics committee of Guangdong Women and Children Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author contributions
All authors participated in the study and manuscript preparation. CL participated in the design of the work and wrote the draft; XW carried out bioinformatics analysis; FL and YZ performed genetic analysis; HD and LL participated in genetic testing; YX and CY collected and analyzed clinical data; and AY and JW designed the work and revised the manuscript. All authors critically reviewed the manuscript and provided approval for submission.
Funding
The study was financially supported by the Science and Technology Planning Project of Guangzhou (Grant No. 202102080263) and Guangzhou Basic and Applied Basic Research Funding Project (Grant No. 202102020847).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1064762/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Assessment of the pathogenicity of variation OFD1 (NM_003611.3): c.2848A>T (p.Lys950Ter) by VarSome (https://varsome.com/).
SUPPLEMENTARY FIGURE S2 | The predicted structure of the mutant protein and wild-type protein by RoseTTAFold online software. (A) (wild-type protein), (B) (mutant protein).
SUPPLEMENTARY FIGURE S3 | The CMA analysis of the fetus (II-3), which shows a 696 kb heterozygous deletion at 5p13.33 (2091630-2787829).
References
Abramowicz, I., Carpenter, G., Alfieri, M., Colnaghi, R., Outwin, E., Parent, P., et al. (2017). "Oral-facial-digital syndrome type I cells exhibit impaired DNA repair; unanticipated consequences of defective OFD1 outside of the cilia network.". Hum. Mol. Genet. 26 (1), 19–32. doi:10.1093/hmg/ddw364
Alfieri, M., Iaconis, D., Tammaro, R., Perone, L., Calì, G., Nitsch, L., et al. (2020). The centrosomal/basal body protein OFD1 is required for microtubule organization and cell cycle progression.". Tissue Cell 64, 101369. doi:10.1016/j.tice.2020.101369
Aljeaid, D., Lombardo, R. C., Witte, D. P., and Hopkin, R. J. (2019). A novel pathogenic variant in OFD1 results in X-linked Joubert syndrome with orofaciodigital features and pituitary aplasia.". Am. J. Med. Genet. A 179 (6), 1010–1014. doi:10.1002/ajmg.a.61018
Anders, S., Pyl, P. T., and Huber, W. (2015). "HTSeq–a Python framework to work with high-throughput sequencing data.". Bioinformatics 31 (2), 166–169. doi:10.1093/bioinformatics/btu638
Andescavage, N. N., du Plessis, A., McCarter, R., Serag, A., Evangelou, I., Vezina, G., et al. (2017). "Complex trajectories of brain development in the healthy human fetus.". Cereb. Cortex 27 (11), 5274–5283. doi:10.1093/cercor/bhw306
Bachmann-Gagescu, R., Dempsey, J. C., Phelps, I. G., O'Roak, B. J., Knutzen, D. M., Rue, T. C., et al. (2015). Joubert syndrome: A model for untangling recessive disorders with extreme genetic heterogeneity. J. Med. Genet. 52 (8), 514–522. doi:10.1136/jmedgenet-2015-103087
Bukowy-Bieryllo, Z., Rabiasz, A., Dabrowski, M., Pogorzelski, A., Wojda, A., Dmenska, H., et al. (2019). "Truncating mutations in exons 20 and 21 of OFD1 can cause primary ciliary dyskinesia without associated syndromic symptoms.". J. Med. Genet. 56 (11), 769–777. doi:10.1136/jmedgenet-2018-105918
Carrel, L., Cottle, A. A., Goglin, K. C., and Willard, H. F. (1999). A first-generation X-inactivation profile of the human X chromosome.". Proc. Natl. Acad. Sci. U. S. A. 96 (25), 14440–14444. doi:10.1073/pnas.96.25.14440
Carrel, L., and Willard, H. F. (2005). "X-inactivation profile reveals extensive variability in X-linked gene expression in females.". Nature 434 (7031), 400–404. doi:10.1038/nature03479
Chen, X., Sheng, X., Liu, Y., Li, Z., Sun, X., Jiang, C., et al. (2018). "Distinct mutations with different inheritance mode caused similar retinal dystrophies in one family: A demonstration of the importance of genetic annotations in complicated pedigrees.". J. Transl. Med. 16 (1), 145. doi:10.1186/s12967-018-1522-7
Coene, K., Roepman, R., Dan, D., Afroze, B., Kroes, H. Y., Letteboer, S., et al. (2009). "OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin.". Am. J. Hum. Genet. 85 (4), 465–481. doi:10.1016/j.ajhg.2009.09.002
de Conciliis, L., Marchitiello, A., Wapenaar, M. C., Borsani, G., Giglio, S., Mariani, M., et al. (1998). "Characterization of Cxorf5 (71-7A), a novel human cDNA mapping to Xp22 and encoding a protein containing coiled-coil alpha-helical domains.". Genomics 51 (2), 243–250. doi:10.1006/geno.1998.5348
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). Star: Ultrafast universal RNA-seq aligner. Bioinformatics 29 (1), 15–21. doi:10.1093/bioinformatics/bts635
Field, M., Scheffer, I. E., Gill, D., Wilson, M., Christie, L., Shaw, M., et al. (2012). "Expanding the molecular basis and phenotypic spectrum of X-linked Joubert syndrome associated with OFD1 mutations.". Eur. J. Hum. Genet. 20 (7), 806–809. doi:10.1038/ejhg.2012.9
Franco, B., and Morleo, M. (2021). The role of OFD1 in selective autophagy.". Mol. Cell Oncol. 8 (3), 1903291. doi:10.1080/23723556.2021.1903291
Franco, B., and Thauvin-Robinet, C. (2016). "Update on oral-facial-digital syndromes (OFDS).". Cilia 5, 12. doi:10.1186/s13630-016-0034-4
Gangaram, B., Devine, W. P., and Slavotinek, A. (2022). "Expanding the phenotype of males with OFD1 pathogenic variants-a case report and literature review.". Eur. J. Med. Genet. 65 (6), 104496. doi:10.1016/j.ejmg.2022.104496
Giorgio, G., Alfieri, M., Prattichizzo, C., Zullo, A., Cairo, S., and Franco, B. (2007). Functional characterization of the OFD1 protein reveals a nuclear localization and physical interaction with subunits of a chromatin remodeling complex. Mol. Biol. Cell 18 (11), 4397–4404. doi:10.1091/mbc.e07-03-0198
Griffiths, P. D., Brackley, K., Bradburn, M., Connolly, D. J. A., Gawne-Cain, M. L., Kilby, M. D., et al. (2017). Anatomical subgroup analysis of the MERIDIAN cohort: Posterior fossa abnormalities. Ultrasound Obstet. Gynecol. 50 (6), 745–752. doi:10.1002/uog.17485
Iaconis, D., Monti, M., Renda, M., van Koppen, A., Tammaro, R., Chiaravalli, M., et al. (2017). The centrosomal OFD1 protein interacts with the translation machinery and regulates the synthesis of specific targets.". Sci. Rep. 7 (1), 1224. doi:10.1038/s41598-017-01156-x
Ioannidis, N. M., Rothstein, J. H., Pejaver, V., Middha, S., McDonnell, S. K., Baheti, S., et al. (2016). Revel: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99 (4), 877–885. doi:10.1016/j.ajhg.2016.08.016
Juric-Sekhar, G., Adkins, J., Doherty, D., and Hevner, R. F. (2012). Joubert syndrome: Brain and spinal cord malformations in genotyped cases and implications for neurodevelopmental functions of primary cilia. Acta Neuropathol. 123 (5), 695–709. doi:10.1007/s00401-012-0951-2
Kane, M. S., Davids, M., Bond, M. R., Adams, C. J., Grout, M. E., Phelps, I. G., et al. (2017). "Abnormal glycosylation in Joubert syndrome type 10.". Cilia 6, 2. doi:10.1186/s13630-017-0048-6
Khajavi, M., Inoue, K., and Lupski, J. R. (2006). "Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease.". Eur. J. Hum. Genet. 14 (10), 1074–1081. doi:10.1038/sj.ejhg.5201649
Kroes, H. Y., Monroe, G. R., van der Zwaag, B., Duran, K. J., de Kovel, C. G., van Roosmalen, M. J., et al. (2016). Joubert syndrome: Genotyping a northern European patient cohort. Eur. J. Hum. Genet. 24 (2), 214–220. doi:10.1038/ejhg.2015.84
Linpeng, S., Liu, J., Pan, J., Cao, Y., Teng, Y., Liang, D., et al. (2018). Diagnosis of joubert syndrome 10 in a fetus with suspected dandy-walker variant by wes: A novel splicing mutation in OFD1.". Biomed. Res. Int. 2018, 4032543. doi:10.1155/2018/4032543
Macca, M., and Franco, B. (2009). The molecular basis of oral-facialヾ igital syndrome, type 1. American Journal of Medical Genetics Part C: Seminars in Medical Genetics. pathologic significance. J. Child. Neurol. 14 (6), 368–376.
Meng, C., Zhang, K. H., Ma, J., Gao, X., Yu, K., Zhang, H. Y., et al. (2017). Clinical and genetic analysis of a family with Joubert syndrome type 10 caused by OFD1 gene mutation. Zhonghua Er Ke Za Zhi 55 (2), 131–134. doi:10.3760/cma.j.issn.0578-1310.2017.02.015
Otto, E. A., Hurd, T. W., Airik, R., Chaki, M., Zhou, W., Stoetzel, C., et al. (2010). "Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy.". Nat. Genet. 42 (10), 840–850. doi:10.1038/ng.662
Parisi, M. A. (2019). The molecular genetics of Joubert syndrome and related ciliopathies: The challenges of genetic and phenotypic heterogeneity.". Transl. Sci. Rare Dis. 4 (1-2), 25–49. doi:10.3233/TRD-190041
Pezzella, N., Bove, G., Tammaro, R., and Franco, B. (2022). OFD1: One gene, several disorders. Am. J. Med. Genet. C Semin. Med. Genet. 190 (1), 57–71. doi:10.1002/ajmg.c.31962
Poretti, A., Boltshauser, E., and Valente, E. M. (2014). The molar tooth sign is pathognomonic for Joubert syndrome!". Pediatr. Neurol. 50 (6), e15–e16. doi:10.1016/j.pediatrneurol.2013.11.003
Qi, Y., Ji, X., Ding, H., Liu, L., Zhang, Y., and Yin, A. (2022). "Novel biallelic variant in the BRAT1 gene caused nonprogressive cerebellar ataxia syndrome.". Front. Genet. 13, 821587. doi:10.3389/fgene.2022.821587
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). "Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology.". Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Romani, M., Micalizzi, A., and Valente, E. M. (2013). Joubert syndrome: Congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 12 (9), 894–905. doi:10.1016/S1474-4422(13)70136-4
Schlatterer, S. D., Sanapo, L., du Plessis, A. J., Whitehead, M. T., and Mulkey, S. B. (2021). The role of fetal MRI for suspected anomalies of the posterior fossa.". Pediatr. Neurol. 117, 10–18. doi:10.1016/j.pediatrneurol.2021.01.002
Singla, V., Romaguera-Ros, M., Garcia-Verdugo, J. M., and Reiter, J. F. (2010). Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev. Cell 18 (3), 410–424. doi:10.1016/j.devcel.2009.12.022
Srour, M., Hamdan, Fadi F., McKnight, D., Davis, E., Mandel, H., Schwartzentruber, J., et al. (2015). "Joubert syndrome in French Canadians and identification of mutations in CEP104.". Am. J. Hum. Genet. 97 (5), 744–753. doi:10.1016/j.ajhg.2015.09.009
Suzuki, T., Miyake, N., Tsurusaki, Y., Okamoto, N., Alkindy, A., Inaba, A., et al. (2016). "Molecular genetic analysis of 30 families with Joubert syndrome.". Clin. Genet. 90 (6), 526–535. doi:10.1111/cge.12836
Thauvin-Robinet, C., Thomas, S., Sinico, M., Aral, B., Burglen, L., Gigot, N., et al. (2013). "OFD1 mutations in males: Phenotypic spectrum and ciliary basal body docking impairment.". Clin. Genet. 84 (1), 86–90. doi:10.1111/cge.12013
Thickman, D., Mintz, M., Mennuti, M., and Kressel, H. Y. (1984). "MR imaging of cerebral abnormalities in utero.". J. Comput. Assist. Tomogr. 8 (6), 1058–1061. doi:10.1097/00004728-198412000-00003
Vetro, A., Pisano, T., Chiaro, S., Procopio, E., Guerrini, R., Parrini, E., et al. (2020). "Early infantile epileptic-dyskinetic encephalopathy due to biallelic PIGP mutations.". Neurol. Genet. 6 (1), e387. doi:10.1212/NXG.0000000000000387
Wald, M., Lawrenz, K., Deutinger, J., and Weninger, M. (2004). "Verification of anomalies of the central nervous system detected by prenatal ultrasound.". Ultraschall Med. 25 (3), 214–217. doi:10.1055/s-2004-813180
Wang, X., Zheng, C., Liu, W., and Yang, H. (2017). "Retinitis pigmentosa and bilateral idiopathic demyelinating optic neuritis in a 6-year-old boy with OFD1 gene mutation.". Case Rep. Ophthalmol. Med. 2017, 5310924. doi:10.1155/2017/5310924
Webb, T. R., Parfitt, D. A., Gardner, J. C., Martinez, A., Bevilacqua, D., Davidson, A. E., et al. (2012). "Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23).". Hum. Mol. Genet. 21 (16), 3647–3654. doi:10.1093/hmg/dds194
Wentzensen, I. M., Johnston, J. J., Patton, J. H., Graham, J. M., Sapp, J. C., and Biesecker, L. G. (2016). "Exome sequencing identifies a mutation in OFD1 in a male with Joubert syndrome, orofaciodigital spectrum anomalies and complex polydactyly.". Hum. Genome Var. 3, 15069. doi:10.1038/hgv.2015.69
Westerfield, L. E., Stover, S. R., Mathur, V. S., Nassef, S. A., Carter, T. G., Yang, Y., et al. (2015). "Reproductive genetic counseling challenges associated with diagnostic exome sequencing in a large academic private reproductive genetic counseling practice.". Prenat. Diagn 35 (10), 1022–1029. doi:10.1002/pd.4674
Wilson, M., Muir, K., Reddy, D., Webster, R., Kapoor, C., and Miller, E. (2020). "Prognostic accuracy of fetal MRI in predicting postnatal neurodevelopmental outcome.". AJNR Am. J. Neuroradiol. 41 (11), 2146–2154. doi:10.3174/ajnr.A6770
Zhang, Y. W., Qu, H. B., Long, N., Leng, X. Y., Liu, Y. Q., and Yang, Y. (2021). A rare mutant of OFD1 gene responsible for Joubert syndrome with significant phenotype variation.". Mol. Genet. Genomics 296 (1), 33–40. doi:10.1007/s00438-020-01726-1
Keywords: Joubert syndrome, OFD1, novel non-sense variant, MRI, whole exome sequencing (WES), RNA-seq
Citation: Li C, Wang X, Li F, Ding H, Liu L, Xiong Y, Yang C, Zhang Y, Wu J and Yin A (2023) A novel non-sense variant in the OFD1 gene caused Joubert syndrome. Front. Genet. 13:1064762. doi: 10.3389/fgene.2022.1064762
Received: 08 October 2022; Accepted: 13 December 2022;
Published: 10 January 2023.
Edited by:
Victor Martinez-Glez, Parc Taulí Hospital Universitari, SpainReviewed by:
Joshi Stephen, Human Genome Sequencing Center, Baylor College of Medicine, United StatesSteven L. C. Pei, Yale University, United States
Rebecca Marie Dewhurst, Newcastle University, United Kingdom
Copyright © 2023 Li, Wang, Li, Ding, Liu, Xiong, Yang, Zhang, Wu and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Wu, wujing020@126.com; Aihua Yin, yinaihua0131@163.com
†These authors have contributed equally to this work