 Rezvan Zabihi1
Rezvan Zabihi1 Mina Zamani1,2Majid Aminzadeh3Niloofar Chamanrou1,2,4Fatemeh Zahra Kiani1,2Tahere Seifi1,2Jawaher Zeighami2Tahere Yadegari2Alireza Sedaghat2,3Alihossein Saberi5,6
Mina Zamani1,2Majid Aminzadeh3Niloofar Chamanrou1,2,4Fatemeh Zahra Kiani1,2Tahere Seifi1,2Jawaher Zeighami2Tahere Yadegari2Alireza Sedaghat2,3Alihossein Saberi5,6 Mohammad Hamid7Gholamreza Shariati2,5*
Mohammad Hamid7Gholamreza Shariati2,5* Hamid Galehdari1*
Hamid Galehdari1*- 1Department of Biology, Faculty of Science, Shahid Chamran University of Ahvaz, Ahvaz, Iran
- 2Narges Medical Genetics and Prenatal Diagnosis Laboratory, Kianpars, Ahvaz, Iran
- 3Diabetes Research Center, Ahvaz Jundishapour University of Medical Sciences, Ahvaz, Iran
- 4Department of Genetics, Faculty of Science, Shahrekord University, Shahrekord, Iran
- 5Department of Medical Genetics, Faculty of Medicine, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran
- 6Cellular and Molecular Research Center, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran
- 7Department of Molecular Medicine, Biotechnology Research Center, Pasteur Institute of Iran, Tehran, Iran
Introduction: Mucopolysaccharidoses are a group of lysosomal storage disorders that include seven types that are classified based on the enzymes that are disrupted. Malfunction of these enzymes leads to the accumulation of glycosaminoglycans (GAGs) in various tissues. Due to genetic and clinical heterogeneity, diagnosing and distinguishing the different types is challenging. Genetic methods such as whole exome sequencing (WES) and Sanger sequencing are accurate methods for detecting pathogenic variants in patients.
Methods: Thirty-two cases of mucopolysaccharidosis, predominantly from families with consanguineous marriages, were genetically examined. Out of these, fourteen cases underwent targeted sequencing, while the rest underwent WES. The results of WES were analyzed and the pathogenicity of the variants was examined using bioinformatics tools. In addition, a segregation analysis within families was carried out.
Results: In most cases, a pathogenic or likely pathogenic variant was detected. Sixteen previously reported variants and six new variants were detected in the known IDS (c.458G>C, c.701del, c.920T>G), GNS (c.1430A>T), GALNS (c.1218_1221dup), and SGSH (c.149T>C) genes. Furthermore, we discovered a c.259G>C substitution in the NAGLU gene for the first time in three homozygous patients. This substitution was previously reported as heterozygous. Except for the variants related to the IDS gene, which were hemizygous, all the other variants were homozygous.
Discussion: It appears that the high rate of consanguineous marriages in the families being studied has had a significant impact on the occurrence of this disease. Overall, these findings could expand the spectrum of pathogenic variants in mucopolysaccharidoses. Genetic methods, especially WES, are very accurate and can be used alone or in conjunction with other diagnostic methods for a more precise and rapid diagnosis of mucopolysaccharidoses. Additionally, they could be beneficial for family screening and disease prevention.
1 Introduction
Mucopolysaccharidoses refer to a group of metabolic disorders resulting from a deficiency of lysosomal enzymes involved in the breakdown of glycosaminoglycans (GAGs) and generally classified into seven types. The main cause of mucopolysaccharidosis type I (Hurler syndrome OMIM #607014, Scheie syndrome OMIM #607016, Hurler-Scheie syndrome OMIM #607015) is a mutation in the α-L-iduronidase (IDUA) gene, which results in a deficiency or relative deficiency of the α-L-iduronidase enzyme. This enzyme is responsible for the breakdown of dermatan sulfate and heparan sulfate (Hampe et al., 2020). In contrast to other MPS types with autosomal recessive inheritance, mucopolysaccharidosis type II (Hunter syndrome OMIM #309900) is an X-linked disease caused by a mutation in the iduronate-2-sulfatase (IDS) gene. The IDS protein triggers the initial cleavage of dermatan sulfate and heparan sulfate in the lysosome. Therefore, deficiency of this enzyme leads to the formation of these GAGs in various tissues (Verma et al., 2021). Mucopolysaccharidosis type III (Sanfilippo syndrome) is caused by a defect in one of four genes: N-sulfoglucosamine sulfohydrolase (SGSH) (MPS IIIA OMIM#252900), N-acetyl-alpha-glucosaminidase (NAGLU) (MPS IIIB OMIM#252920), heparan alpha-glucosaminide N-acetyltransferase (HGSNAT) (MPS IIIC OMIM#252930), and N-acetylglucosamine–Sulfohydrolase (GNS) (MPS IIID OMIM#252940). The protein products of these genes are involved in the lysosomal degradation of heparan sulfate (Yogalingam and Hopwood, 2001). Mucopolysaccharidosis type IV (Morquio syndrome) is determined by impairment of the enzymes galactosamine-6-sulfate sulfatase (GALNS) (MPS IVA OMIM#253000) or beta-galactosidase 1 (GLB1) (MPS IVB OMIM#253010), which are involved in the breakdown of keratan sulfate and chondroitin sulfate. Mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome OMIM#253200) results from a deficiency of the enzyme arylsulfatase B (ARSB). This type begins with the accumulation of dermatan sulfate and chondroitin sulfate in the lysosomes. Finally, mucopolysaccharidosis type VII and type IX are caused by mutations in genes encoding the enzymes beta-glucuronidase (GUSB) and hyaluronoglucosaminidase-1 (HYAL1), respectively. These types are characterized by the accumulation of chondroitin sulfate, dermatan sulfate and heparan sulfate in MPS VII and hyaluronan in MPS IX (Celik et al., 2021; McBride and Flanigan, 2021; Nagral et al., 2022).
Overall, reduced activity of these enzymes leads to the accumulation of GAGs that affect various tissues, including the brain, eyes, ears, upper and lower respiratory tract, liver, spleen, heart, bones, cartilage, and joints, resulting in a variety of clinical manifestations (Münzer, 2011; McBride and Flanigan, 2021). The prevalence of each mucopolysaccharidosis subtype depends on geographic region and/or ethnic background. However, demographic data show that MPS is most common in Saudi Arabia, possibly due to consanguineous marriage or a founder effect. This is followed by Portugal, Brazil, the Netherlands and Australia (Celik et al., 2021). There is currently no curative treatment for mucopolysaccharidoses and treatment includes surgical, supportive and disease-specific treatments. Additionally, current standard treatments such as enzyme replacement therapy (ERT) and hematopoietic stem cell transplantation (HSCT) cannot prevent or reverse the abnormalities in the cornea, bones, central nervous system, and heart valves. In addition, ERT can trigger the formation of antibodies against the recombinant enzyme (Sawamoto et al., 2019). Therefore, early diagnosis of MPS is crucial to prevent numerous clinical symptoms. Since MPS syndromes are an extremely heterogeneous group of inherited diseases, the identification of new pathogenic variants could be of great benefit for family screening and prediction of the risk of recurrence. In addition, it can help with early diagnosis and provide therapeutic options for potential new cases. The aim of this study was to evaluate and validate potential novel variants involved in the pathogenesis of MPS using whole-exome sequencing and Sanger sequencing. We have reported several new variants in different types of genes associated with mucopolysaccharidosis and confirmed their association with the disease in the affected families.
2 Material and method
2.1 Patients
We enrolled 32 unrelated cases, including one neonatal death, who were referred to the Narges Medical Genetics and Prenatal Diagnosis laboratory in the Southwest of Iran from 2014 to 2022. These patients were given a preliminary diagnosis of Mucopolysaccharidosis by a metabolism specialist based on clinical symptoms and biochemical tests, such as cellular enzymatic activities and urine GAG levels. To accurately diagnose the type of disease and screen it in the patient’s families, we conducted genetic examinations. Genetic counseling was provided to all patients by genetic specialists from the laboratory. The study was approved by the Institutional Review Board of Ahvaz Jundishapur University of Medical Sciences, and informed consent was obtained from all enrolled patients and their families.
2.2 DNA extraction
Ten milliliters of peripheral blood were isolated from each case and their available respective parents. Genomic DNA was extracted using the standard salting-out protocol (Miler et al., 1988). The quality and quantity of the extracted DNA were evaluated using gel electrophoresis and a nanodrop, respectively.
2.3 Exome sequencing
Exome enrichment was performed on 18 samples using the SureSelect Human All Exon kit V6 (Agilent Technologies, Santa Clara, CA, United States), followed by sequencing using the Illumina HiSeq 2000 genome analyzer platform (Illumina, San Diego, CA, United States). The short reads were aligned to the human genome reference version B37 using BWA, and duplicate reads were marked using Picard v2.6.0 (https://broadinstitute.github.io/picard). GATK and ANNOVAR were used for variant detection and annotation, respectively.
2.4 Assessment of variant pathogenicity
To identify disease-causing variants, filtering was performed for each WES data file based on allele frequency. Variants with allele frequencies lower than 0.01 in the 1,000 Genomes (https://www.internationalgenome.org), ExAC (http://exac.broadinstitute.org), and gnomAD (https://gnomad.broadinstitute.org/) databases were selected for the next step. Then, the data files were filtered based on the variant consequence, selecting exonic, exonic-splicing, and splicing locations. Afterward, we focused on zygosity and the inheritance of diseases in families. Mucopolysaccharidosis-related genes were then filtered based on the OMIM and GeneCards databases. In the next step, the pathogenicity of the remaining variants was evaluated using in silico tools such as Mutation-Taster (Steinhaus et al., 2021), Polyphen2 (Adzhubei et al., 2010), PredictSNP2 (Bendl et al., 2016), VarSome (Kopanos et al., 2019), and CADD (Kircher et al., 2014). Finally, the selected variants were searched in our in-house database (which includes more than 2,500 WES files), and variants with low allele frequency were chosen for additional segregation analysis in the families.
2.5 Sanger sequencing
Direct Sanger sequencing was performed on DNA samples from 14 cases. Specific primers were designed using OLIGO 7 to target the entire coding exons and flanking intronic sequences of the IDS, NAGLU, SGSH, ARSB, and GALNS genes in order to identify any existing variants. The selected regions were amplified using PCR. The PCR products were then sequenced and analyzed using the ABI Prism 3700 automated genetic analyzer (Applied Biosystems, ThermoFisher, United States). The results were interpreted using Chromas 2.6.6 and compared with reference gene sequences using the BLASTN and ClustalW programs. Furthermore, the presence of all detected variants was confirmed through Sanger sequencing of cases that underwent whole exome sequencing, as well as available relatives of all the studied cases.
3 Result
The initial diagnosis of mucopolysaccharidosis in patients was based on clinical symptoms and biochemical tests. However, the specific type of disease was not clear in some patients. After sequencing, the exact type of disease in each person was determined by identifying the presence of pathogenic or likely pathogenic variants in different known genes that encode enzymes responsible for mucopolysaccharidosis. The general information of the probands is provided in Table 1. Based on Table 1, the majority of patients were of Arab or Lor ethnicity, and 71.8% (23/32) of them were born to consanguineous parents. Furthermore, MPS III has the highest prevalence among all types of the disease, accounting for 50% (16/32) of cases. We identified various disease-causing variants in known genes associated with different types of Mucopolysaccharidosis in all 32 cases. The genetic findings and in silico analysis of these variants are summarized in Table 2. The distribution of genes containing these variants is shown in Figure 1, depicted as a pie chart. The NAGLU, IDS, SGSH, GNS, GALNS, ARSB, HGSNAT, and IDUA genes each contained one or more variants. Based on the data presented in Figure 1, it is evident that the NAGLU and IDS genes had the highest number of variants among the patients, respectively. Among all the variants, we detected 6 novel variants in the IDS, GNS, GALNS, and SGSH genes. All of these new variants occur in the protein-coding regions of the genes, with 66.6% (4/6) being missense mutations and 33.3% (2/6) being frameshift mutations. The novel variants are as follows, based on the gene in which the mutation occurred. Additionally, the pedigree of the proband’s families with new variants is shown in Figure 3.
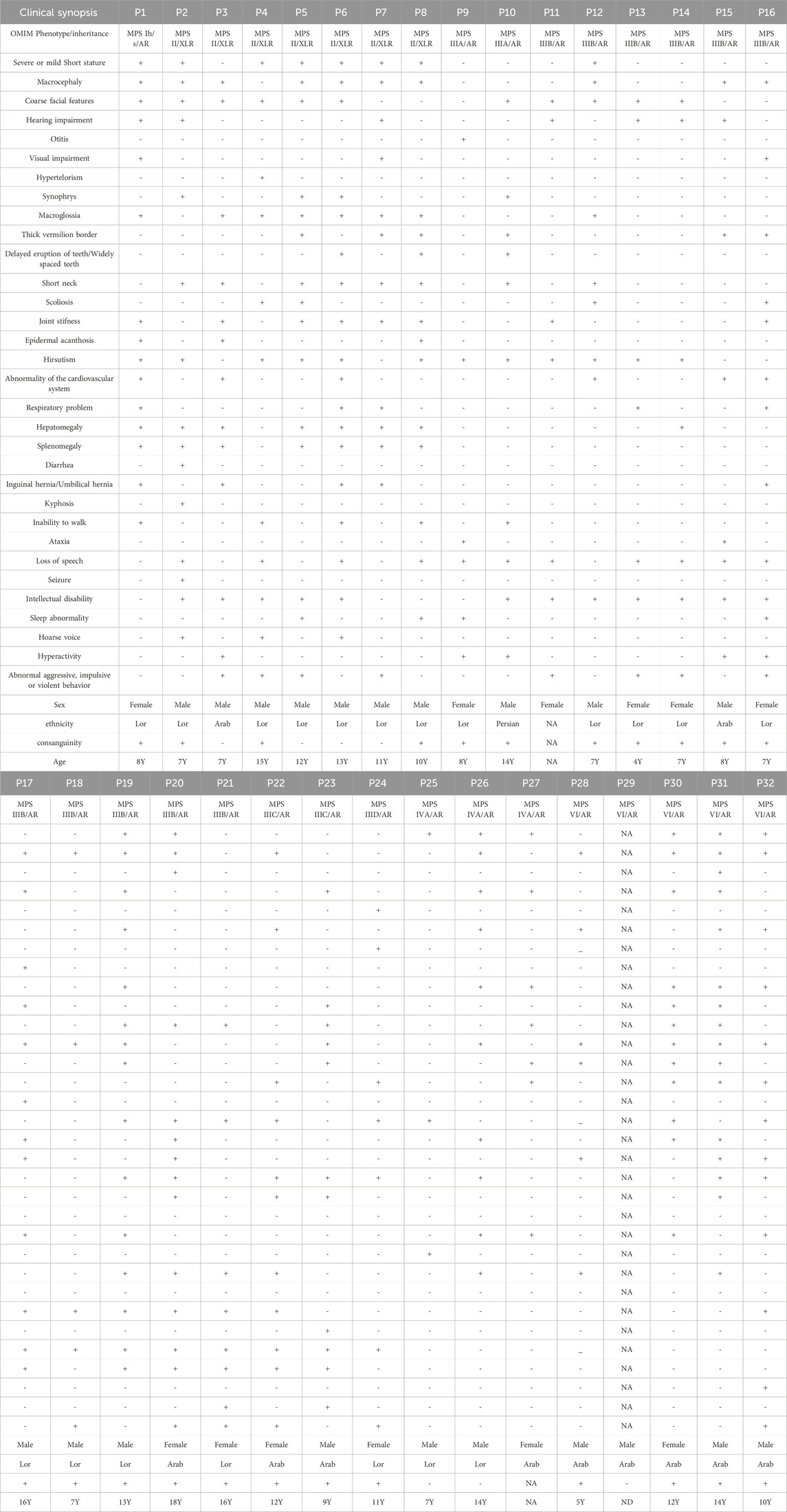
TABLE 1. A brief description of individuals. P29 clinical data were not available due to neonatal death. P: proband, AR: autosomal recessive, NA: not available, Y:year, ND: neonatal death.
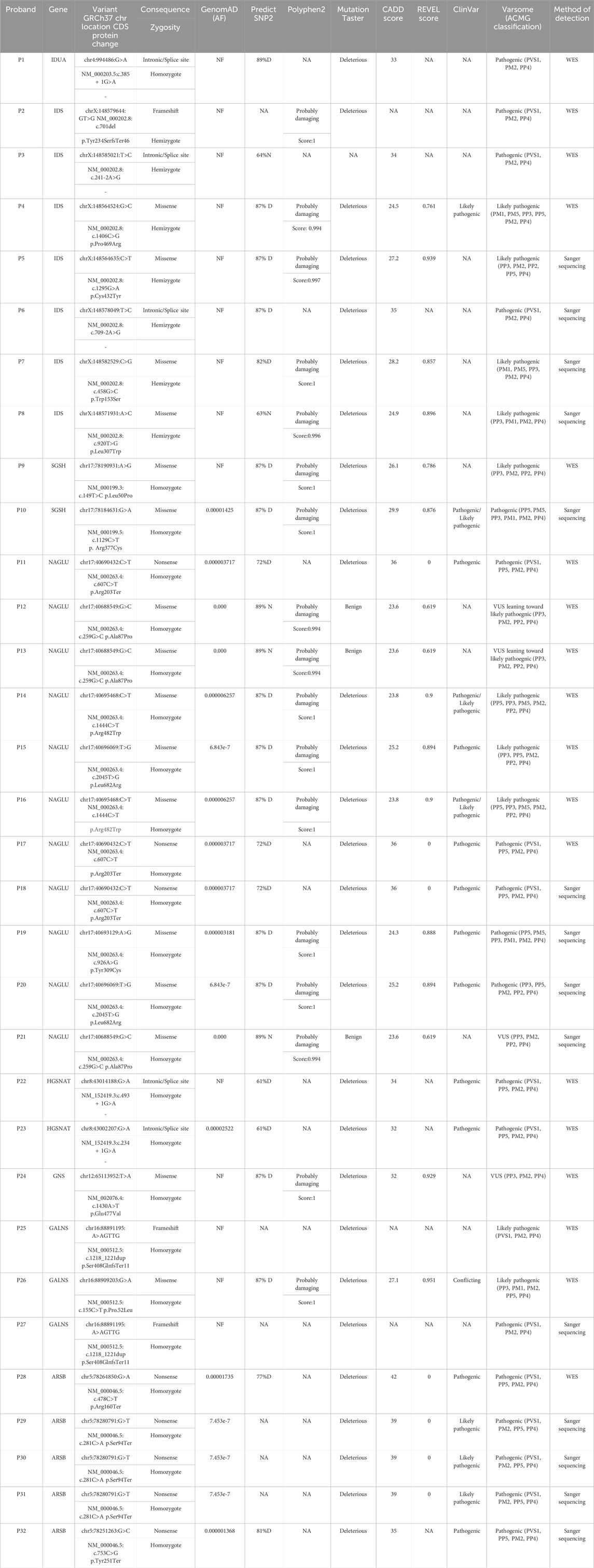
TABLE 2. In silico pathogenicity analysis of the detected variants. WES: Whole Exome Sequencing, NF: not found, D: deleterious, N: neutral, NA: not available.
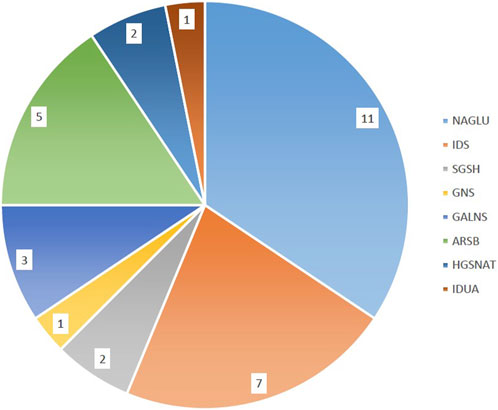
FIGURE 1. Distribution of variants in MPS genes. The majority of patients had disease-causing variants in the NAGLU and IDS genes respectively.
3.1 IDS
Three different variants were detected in the Iduronate-2-sulfatase (IDS) gene in P2, P7, and P8. As shown in Figure 2, all of the variants were found within the sulfatase domain of the protein, which is crucial for the enzyme’s activity. In the case of P2, genetic screening identified a single nucleotide deletion (NM_000202.8, c.701del, p.Tyr234Ser) in exon 5. Additionally, two novel missense variants, (NM_000202.8, c.458G>C, p.Trp153Ser (exon 4)) and (NM_000202.8, c.920T>G, p.Leu307Trp (exon 7)), were detected in subjects P7 and P8, respectively. All three variants were found in a hemizygous state in the probands, while being heterozygous in their mother and wild type in their father. According to the pedigrees of the families (Figure 3), the other patients in these three families were also males, confirming the type of disease as mucopolysaccharidosis type II.
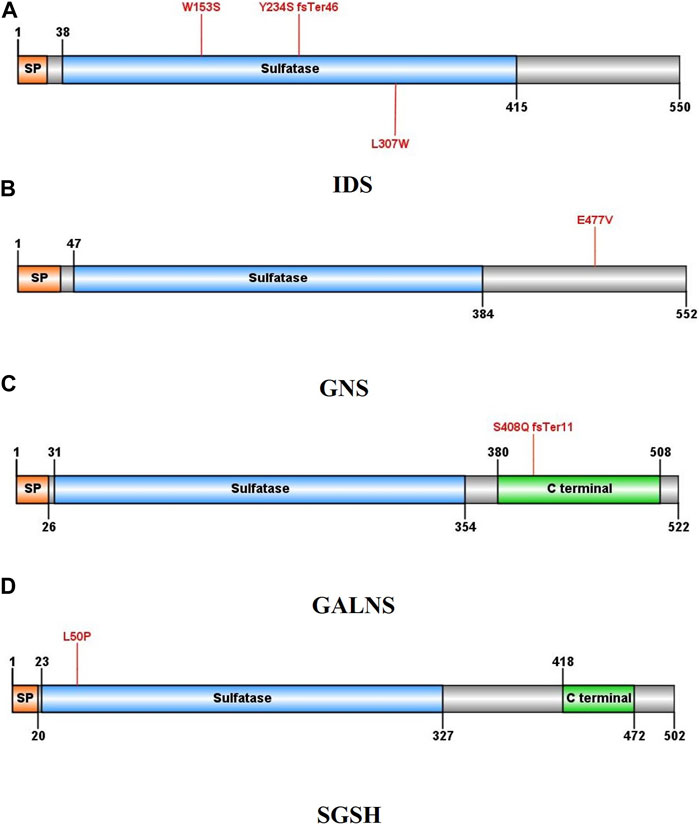
FIGURE 2. Schematic illustration of the protein structures in which at least one new variant was identified: (A) Iduronate-2-sulfatase, (B) N-acetylglucosamine-6-sulfatase, (C) galactosamine-6-sulfate sulfatase, (D) N-sulfoglucosamine sulfohydrolase. Mutated residues are indicated by red lines.
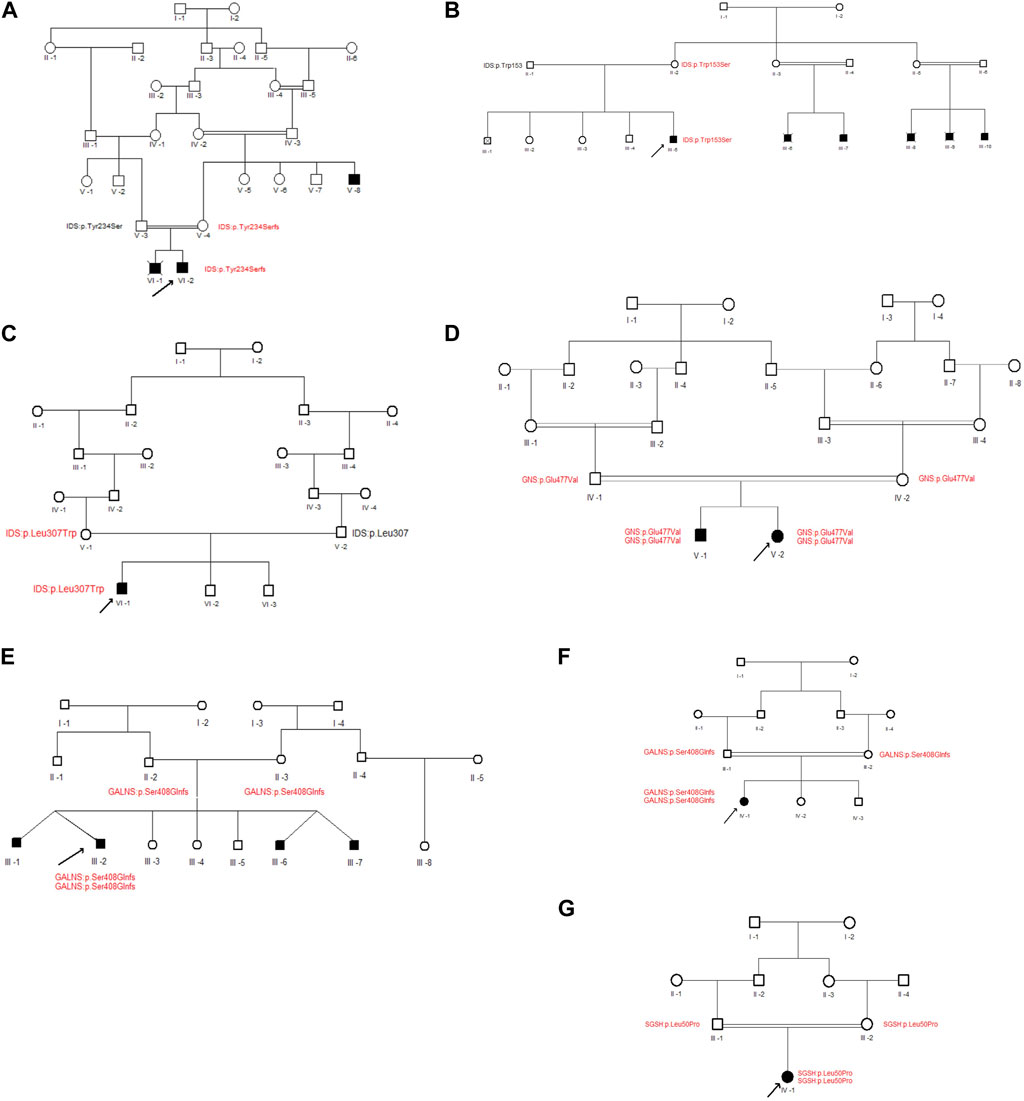
FIGURE 3. Pedigrees of families with new variants. (A–G) represent the family pedigrees of P2, P7, P8, P9, P24, P25 and P27 cases, respectively. Squares and circles denote males and females, respectively. Filled symbols represent affected individuals, and consanguinity is indicated by double marriage lines. The normal and mutated protein locations of participating individuals have been written beneath each individual in black and red, respectively. The proband is indicated by an arrow.
3.2 GNS
In case P24, a missense variant was detected in the N-acetylglucosamine-6-sulfatase (GNS) gene (NM_002076.4, c.1430A>T, p.Glu477Val). This variant was located in exon 13 and similar to the variants in the IDS gene, it affects the sulfatase domain of the protein. As shown in Figure 3, The proband was homozygous for the mentioned variant, and both of her parents were heterozygous. After performing Sanger sequencing on the proband’s brother, who was also suspected of having mucopolysaccharidosis, it was revealed that he also has this variant in a homozygous form.
3.3 GALNS
In patients P25 and P27, a novel duplication variant was identified in exon 11 of the galactosamine-6-sulfate sulfatase (GALNS) gene (NM_000512.4, c.1218_1221dup, p.Ser408GlnfsTer11). In both cases, the probands inherited the variant homozygously from their heterozygous parents. Interestingly, in case P25, the patient also had an identical twin with him, along with two other identical twin brothers who also had mucopolysaccharidosis type IVA.
3.4 SGSH
A missense variant was found in exon 2 of the N-sulfoglucosamine sulfohydrolase (SGSH) gene in case P9 (NM_000199.3, c.149T>C, p.Leu50Pro). This variant also affects the sulfatase domain of the protein, potentially interfering with its activity. Here, the proband was the only affected individual in the family and had received the variant in a homozygous form from her heterozygous parents.
4 Discussion
In the present study, a total of 32 cases with a primary diagnosis of mucopolysaccharidosis from separate families underwent genetic analysis. In some families, there was only one patient, while in others, there were additional patients besides the proband, clearly indicating the hereditary nature of the disease. Among all the families, MPS I was reported in only one family, and the affected individual was characterized as having the MPS Ih/s subtype. Genetic analysis revealed a homozygous splice site variant located in intron 3 of the IDUA gene (c.385 + 1G>A) in the proband. This variant had previously been detected in the European population and was shown to cause the loss of a neutral donor splice site that is conserved among vertebrate species (Bertola et al., 2011).
Probands in seven families were classified as MPS II, and we identified four previously reported and three novel hemizygous variants in the IDS gene. X-linked inheritance was apparent in the pedigrees and all of the patients were male. Among these variants, an intronic splice variant (c.241-2A>G) was detected, which can lead to alternative splicing and the creation of two mutant transcripts (Alves et al., 2006). Another splice site variant (c.709-2A>G) was detected, in which a nucleotide transition leads to a 3′ splice site alteration in intron 5 of the IDS gene (Lissens et al., 1997). This alteration may result in the production of an incorrect transcript or a premature stop codon, leading to the absence of a functional protein. Further investigation and functional assays are needed to determine the exact mechanism of this vatiant. Other previously reported alterations in MPS II patients were two missense variants, including c.1406C>G and c.1295G>A (Karsten et al., 1998; Pollard et al., 2013). Additionally, two previously unreported missense variants were identified in this study. In one of these variants (c.458G>C, p.Trp153Ser), a hydrophobic, nonpolar, aromatic, and large amino acid is replaced with a neutral, polar, and small amino acid. Hence, factors such as hydrophobicity, size and polarity exchange may affect the final protein folding. Two other pathogenic variants in the same amino acid have been previously reported, in which tryptophan is converted to arginine and leucine, respectively (Froissart et al., 2007; Zhang et al., 2019). In another missense variant (c.920T>G, p.Leu307Trp), an aliphatic amino acid is changed to an aromatic amino acid. This change allows the amino acid to donate hydrogene, unlike its previous form. In addition to the missense variants, one frameshift variant was also found in the IDS gene (c.701del, Tyr234Ser), which likely results in the production of incomplete and non-functional protein.
Sanfilippo syndrome, the most frequent and heterogeneous type of mucopolysaccharidoses (van de Kamp et al., 1981), was observed in the families with different subtypes ranging from MPS IIIA to MPS IIID. In two of the families, we identified two distinct homozygous missense variants in the SGSH gene. These variants included one previously reported variant (c.1129C>T) and one newly discovered variant (c.149T>C), both of which were found to be responsible for causing MPS ⅢA. The c.1129C>T variant, which was initially identified by (Di Natale et al., 1998) leads to a severe form of the disease because position 377 at the C-terminal is crucial for sulfamidase function (Di Natale et al., 1998). Our studied patient had a severe form of the disease, consistent with the research conducted by Di Natale et al. The c.149T>C variant, p.Leu50Pro, involves the substitution of a hydrophobic and large amino acid with a neutral and small amino acid. This change in the size and nature of amino acids at position 50 may affect the protein’s interactions with other molecules and residues, leading to improper protein folding. Among the individuals we studied, the highest frequency of Sanfilippo patients was associated with the subtype of MPS ⅢB. In this subtype, we observed five different variants in the NAGLU gene. Among these variants, we found a previously reported nonsense variant (c.607C>T, p.Arg203ter) and a missense variant (c.2045T>G, p.Leu682Arg) in three and two families, respectively. Schmidtchen et al. (1998) first reported these variants and demonstrated the presence of these variants along with 8 other variants in the fibroblast cell line of patients with MPS ⅢB. They also validated the reduction of enzyme activity in Chinese hamster ovary cells transfected with a mutagenized vector containing the NAGLU missense variants. Furthermore, they found excessive molecular heterogeneity in the NAGLU gene and suggested a role for the amino or carboxyl end of α-N-acetylglucosaminidase in the transport or function of the enzyme (Schmidtchen et al., 1998). In three families, we identified one homozygous missense variant (c.259G>C, p.Ala87Pro) in the NAGLU gene. This variant is similar to the aforementioned variants in terms of its location in the N-terminal domain of the protein. The substitution of alanine with proline converts a hydrophobic amino acid to a neutral one. As a result, the hydrophobic intra and intermolecular interactions that are dependent on this substitution lead to the complete loss of protein activity. Pollard et al. (2013) reported the c.259G>C substitution in a compound heterozygous patient, in combination with another variant (c.1949G>A, p.G650E) (Pollard et al., 2013). However, our patients showed novel homozygous alleles. In two of the families we observed a substitution (c.1444C>T) in the NAGLU gene, where a 5-methylcytosine in a highly mutable CpG dinucleotide position is converted to thymine as a result of deamination (Bunge et al., 1999). The last observed missense variant in the NAGLU gene in our study was a substitution (c.926A>G, p.Tyr309Cys) in a highly conserved amino acid. This variant does not affect the amount or stability of RNA, but it only influences the activity of the enzyme (Lee-Chen et al., 2002). We identified MPS ⅢC in two families, where we observed two different previously described homozygous splice site variants (c.493 + 1G>A, c.234 + 1G>A) in the HGSNAT gene (Fan et al., 2006; Hřebíček et al., 2006). In a comprehensive study, (Martins et al., 2019), described the evolutionary history of MPS ⅡIC by analyzing the clinical presentation, molecular defects, and haplotype context of patients from 22 countries (Martins et al., 2019). Remarkably, both patients in our study were from the Arab ethnic group, suggesting an African origin of these variants. In one of the families, we found the c.1430A>T missense variant in the GNS gene, which is responsible for MPS IIID. This variant was found for the first time in the current study. At the protein level, this variant results in the conversion of a hydrophilic, negatively charged and polar amino acid to a hydrophobic, uncharged and nonpolar amino acid. This change has the potential to generate misfolded proteins. In two families with individuals clinically diagnosed with MPS ⅣA, a novel homozygous frameshift variant was observed in the GALNS gene. This variant is caused by a duplication of CAAC at position 1,218 to 1,221 of the coding sequence (c.1218_1221dup); resulting in the introduction of a premature termination codon (PTC) 11 codons downstream of the duplication. Another MPS ⅣA patient was found to be homozygous for a missense variant (c.155C>T) that had been previously identified in the Indian population. This variant is likely to prevent the formation of homodimers of the GALNS protein, as reported by (Bidchol et al., 2014). Additionally, two different nonsense variants were identified in the ARSB gene, which is mutated in MPS VI. These variants are c.478C>T and c.281C>A, resulting in the conversion of Arg160 and Ser94 to premature stop codons and the creation of a non-functional truncated protein (Voskoboeva et al., 1994). The c.478C>T variant is located in a CpG dinucleotide, which is considered a mutation hotspot in the ARSB gene, as noted by (Zapała et al., 2020). It is also one of the most frequently reported pathogenic and likely pathogenic variants in ARSB, as stated by (Tomanin et al., 2018). The c.281C>A variant was found to have a high frequency in Arab patients, as reported by (Aminzadeh et al., 2019). Its presence in 4 out of 5 families of Arab ethnicity suggests that this variant may be the most common variant among Iranian Arab patients with MPS VI. Furthermore, among the four families with the same variant, one family experienced neonatal death, while the individuals in the other cases were over 10 years old. This may demonstrate the clinical heterogeneity of MPS VI.
In mucopolysaccharidoses, as was the case in the current study, there can be clinical symptom overlap between different types. In some cases, relying solely on clinical symptoms and biochemical tests for an accurate diagnosis of Mucopolysaccharidosis type can lead to errors. However, with the assistance of genetic counseling and genetic tests, an accurate diagnosis of Mucopolysaccharidosis type in patients can be achieved. For instance, in the case of MPS type 2, which has X-linked inheritance, a definitive diagnosis can be made by combining genetic counseling, identifying the disease more frequently in males than females, and detecting mutations in the IDS gene.
This research is unique due to the prevalence of specific ethnic groups and the practice of consanguineous marriage in them. Considering that all types of mucopolysaccharidosis, except type II, have autosomal recessive inheritance, the high rate of inbreeding leads to the emergence and manifestation of potentially pathogenic variants in populations with high levels of inbreeding. Therefore, further genetic investigations and studies, particularly those utilizing exome or genome sequencing, are strongly recommended in inbred populations.
There are several caveats that should be noted regarding the present study. Firstly, in most cases, the other patients in the family, apart from the proband, were not sequenced. This was either due to their unavailability or their refusal to participate in the research. Secondly, the current study does not include functional assays related to the novel detected variants, and our final criteria for determining the pathogenicity of variants were based on ACMG classification. Therefore, future research is needed to validate the pathogenicity and better understand the mechanism and biological effects of the identified new variants. These studies should include more in silico investigations, such as 3D protein structure analysis, as well as in-vitro and in-vivo functional assays.
5 Conclusion
Genetic screenings can provide significant benefits in managing and preventing diseases within families, as well as alleviating clinical symptoms in new cases (Kubaski et al., 2020). In this study, we focused on whole exome sequencing and Sanger sequencing to examine genetic variants in patients with mucopolysaccharidosis from the Khuzestan province in Southwest Iran. We detected six novel variants and confirmed these findings through segregation analysis in family members. Our in silico analysis suggests that these novel variants may be disease-causing variants. The present findings contribute to expanding the spectrum of pathogenic variants associated with mucopolysaccharidosis and can facilitate rapid diagnosis.
Data availability statement
The datasets presented in this article cannot be publicly shared due to privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Ahvaz Jundishapur University of medical Sciences Institutional Review Board. Informed consent was obtained from all enrolled families. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
RZ: Conceptualization, Methodology, Visualization, Writing–original draft, Writing–review and editing. MZ: Formal Analysis, Investigation, Project administration, Supervision, Validation, Visualization, Writing–review and editing. MA: Resources, Writing–review and editing. NC: Methodology, Writing–review and editing. FK: Methodology, Writing–review and editing. TSe: Formal Analysis, Methodology, Software, Visualization, Writing–review and editing. JZ: Data curation, Writing–review and editing. TY: Data curation, Writing–review and editing. ASe: Funding acquisition, Supervision, Writing–review and editing. ASa: Resources, Supervision, Writing–review and editing. MH: Formal Analysis, Visualization, Writing–review and editing. GS: Funding acquisition, Resources, Supervision, Writing–review and editing. HG: Investigation, Supervision, Validation, Visualization, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We grateful to the staff of Narges Genetic Laboratory and all family members of the patients and their relatives for their cooperation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods. 7, 248–249. doi:10.1038/nmeth0410-248
Alves, S., Mangas, M., Prata, M. J., Ribeiro, G., Lopes, L., Ribeiro, H., et al. (2006). Molecular characterization of Portuguese patients with mucopolysaccharidosis type II shows evidence that the IDS gene is prone to splicing mutations. J. Inherit. Metab. Dis. 29 (6), 743–754. doi:10.1007/s10545-006-0403-z
Aminzadeh, M., Malekpour, N., and Ghandil, P. (2019). Identification of arylsulfatase B gene mutations and clinical presentations of Iranian patients with Mucopolysaccharidosis VI. Gene 706, 1–5. doi:10.1016/j.gene.2019.04.050
Bendl, J., Musil, M., Štourač, J., Zendulka, J., Damborský, J., and Brezovský, J. (2016). PredictSNP2: a unified platform for accurately evaluating SNP effects by exploiting the different characteristics of variants in distinct genomic regions. PLoS Comput. Biol. 12 (5), e1004962. doi:10.1371/journal.pcbi.1004962
Bertola, F., Filocamo, M., Casati, G., Mort, M., Rosano, C., Tylki-Szymanska, A., et al. (2011). IDUA mutational profiling of a cohort of 102 European patients with mucopolysaccharidosis type I: identification and characterization of 35 novel α-L-iduronidase (IDUA) alleles. Hum. Mutat. 32 (6), E2189–E2210. doi:10.1002/humu.21479
Bidchol, A. M., Dalal, A., Shah, H., Suryanarayana, S., Nampoothiri, S., Kabra, M., et al. (2014). GALNS mutations in Indian patients with mucopolysaccharidosis IVA. Am. J. Med. Genet. A 164A (11), 2793–2801. doi:10.1002/ajmg.a.36735
Bunge, S., Knigge, A., Steglich, C., Kleijer, W. J., van Diggelen, O. P., Beck, M., et al. (1999). Mucopolysaccharidosis type IIIB (Sanfilippo B): identification of 18 novel alpha-N-acetylglucosaminidase gene mutations. Med. Genet. 36 (1), 28–31.
Çelik, B., Tomatsu, S. C., Tomatsu, S., and Khan, S. A. (2021). Epidemiology of mucopolysacchari doses update. Diagn. (Basel) 11 (2), 273. doi:10.3390/diagnostics11020273
Di Natale, P., Balzano, N., Esposito, S., and Villani, G. R. (1998). Identification of molecular defects in Italian Sanfilippo A patients including 13 novel mutations. Hum. Mutat. 11 (4), 313–320. doi:10.1002/(SICI)1098-1004(1998)11:4<313::AID-HUMU9>3.0.CO;2-P
Fan, X., Zhang, H., Zhang, S., Bagshaw, R. D., Tropak, M. B., Callahan, J. W., et al. (2006). Identification of the gene encoding the enzyme deficient in mucopolysaccharidosis IIIC (Sanfilippo disease type C). Am. J. Hum. Genet. 79 (4), 738–744. doi:10.1086/508068
Froissart, R., Da Silva, I. M., and Maire, I. (2007). Mucopolysaccharidosis type II: an update on mutation spectrum. Acta Paediatr. 96 (455), 71–77. doi:10.1111/j.1651-2227.2007.00213.x
Hampe, C. S., Eisengart, J. B., Lund, T. C., Orchard, P. J., Swietlicka, M., Wesley, J., et al. (2020). Mucopolysaccharidosis type I: a review of the natural history and molecular pathology. Cells 9 (8), 1838. doi:10.3390/cells9081838
Hrebícek, M., Mrázová, L., Seyrantepe, V., Durand, S., Roslin, N. M., Nosková, L., et al. (2006). Mutations in TMEM76* cause mucopolysaccharidosis IIIC (Sanfilippo C syndrome). Am. J. Hum. Genet. 79 (5), 807–819. doi:10.1086/508294
Karsten, S., Voskoboeva, E., Tishkanina, S., Pettersson, U., Krasnopolskaja, X., and Bondeson, M. L. (1998). Mutational spectrum of the iduronate-2-sulfatase (IDS) gene in 36 unrelated Russian MPS II patients. Hum. Genet. 103 (6), 732–735. doi:10.1007/s004390050901
Kircher, M., Witten, D. M., Jain, P., O'roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi:10.1038/ng.2892
Kopanos, C., Tsiolkas, V., Kouris, A., Chapple, C. E., Albarca Aguilera, M., Meyer, R., et al. (2019). VarSome: the human genomic variant search engine. Bioinformatics 35 (11), 1978–1980. doi:10.1093/bioinformatics/bty897
Kubaski, F., de Oliveira Poswar, F., Michelin-Tirelli, K., Burin, M. G., Rojas-Málaga, D., Brusius-Facchin, A. C., et al. (2020). Diagnosis of mucopolysaccharidoses. Diagnostics 10 (3), 172. doi:10.3390/diagnostics10030172
Lee-Chen, G. J., Lin, S. P., Lin, S. Z., Chuang, C. K., Hsiao, K. T., Huang, C. F., et al. (2002). Identification and characterisation of mutations underlying Sanfilippo syndrome type B (mucopolysaccharidosis type IIIB). J. Med. Genet. 39 (2), E3. doi:10.1136/jmg.39.2.e3
Lissens, W., Seneca, S., and Liebaers, I. (1997). Molecular analysis in 23 Hunter disease families. J. Inherit. Metab. Dis. 20 (3), 453–456. doi:10.1023/a:1005335624386
Martins, C., de Medeiros, P. F. V., Leistner-Segal, S., Dridi, L., Elcioglu, N., Wood, J., et al. (2019). Molecular characterization of a large group of Mucopolysaccharidosis type IIIC patients reveals the evolutionary history of the disease. Hum. Mutat. 40 (8), 1084–1100. doi:10.1002/humu.23752
McBride, K. L., and Flanigan, K. M. (2021). Update in the mucopolysaccharidoses. Semin. Pediatr. Neurol. 37, 100874. doi:10.1016/j.spen.2021.100874
Miller, S. A., Dykes, D. D., and Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16 (3), 1215. doi:10.1093/nar/16.3.1215
Muenzer, J. (2011). Overview of the mucopolysaccharidoses. Rheumatol. Oxf. 50 (5), 4–12. doi:10.1093/rheumatology/ker394
Nagral, R., Goyal, R. B., Priyadarshini, K., Kashyap, S., Sharma, M., Sinha, R., et al. (2022). Mucopolysaccharidosis: a broad review. Indian J. Ophthalmol. 70 (7), 2249–2261. doi:10.4103/ijo.IJO_425_22
Pollard, L. M., Jones, J. R., and Wood, T. C. (2013). Molecular characterization of 355 mucopolysaccharidosis patients reveals 104 novel mutations. J. Inherit. Metab. Dis. 36 (2), 179–187. doi:10.1007/s10545-012-9533-7
Sawamoto, K., Stapleton, M., Alméciga-Díaz, C. J., Espejo-Mojica, A. J., Losada, J. C., Suarez, D. A., et al. (2019). Therapeutic options for mucopolysaccharidoses: current and emerging treatments. Drugs 79 (10), 1103–1134. doi:10.1007/s40265-019-01147-4
Schmidtchen, A., Greenberg, D., Zhao, H. G., Li, H. H., Huang, Y., Tieu, P., et al. (1998). NAGLU mutations underlying Sanfilippo syndrome type B. Am. J. Hum. Genet. 62 (1), 64–69. doi:10.1086/301685
Steinhaus, R., Proft, S., Schuelke, M., Cooper, D. N., Schwarz, J. M., and Seelow, D. (2021). MutationTaster2021. Nucleic Acids Res. 49, W446–W451. doi:10.1093/nar/gkab266
Tomanin, R., Karageorgos, L., Zanetti, A., Al-Sayed, M., Bailey, M., Miller, N., et al. (2018). Mucopolysaccharidosis type VI (MPS VI) and molecular analysis: review and classification of published variants in the ARSB gene. Hum. Mutat. 39 (12), 1788–1802. doi:10.1002/humu.23613
van de Kamp, J. J., Niermeijer, M. F., von Figura, K., and Giesberts, M. A. (1981). Genetic heterogeneity and clinical variability in the Sanfilippo syndrome (types A, B, and C). Clin. Genet. 20 (2), 152–160. doi:10.1111/j.1399-0004.1981.tb01821.x
Verma, S., Pantoom, S., Petters, J., Pandey, A. K., Hermann, A., and Lukas, J. (2021). A molecular genetics view on Mucopolysaccharidosis Type II. Mutat. Res. Rev. Mutat. Res. 788, 108392. doi:10.1016/j.mrrev.2021.108392
Voskoboeva, E., Isbrandt, D., von Figura, K., Krasnopolskaya, X., and Peters, C. (1994). Four novel mutant alleles of the arylsulfatase B gene in two patients with intermediate form of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). Hum. Genet. 93 (3), 259–264. doi:10.1007/BF00212019
Yogalingam, G., and Hopwood, J. J. (2001). Molecular genetics of mucopolysaccharidosis type IIIA and IIIB: diagnostic, clinical, and biological implications. Hum. Mutat. 18 (4), 264–281. doi:10.1002/humu.1189
Zapała, B., Chmura, O., Ciałowicz, U., Solnica, B., Krajewska-Włodarczyk, M., and Żuber, Z. (2020). A case of mucopolysaccharidosis type VI in a polish family. Importance of genetic testing and genotype-phenotype relationship in the diagnosis of mucopolysaccharidosis. Mol. Genet. Metab. Rep. 25, 100658. doi:10.1016/j.ymgmr.2020.100658
Keywords: mucopolysaccharidosis (MPS), whole exome sequencing (WES), pathogenic variant, Sanger sequencing, genetic diagnosis
Citation: Zabihi R, Zamani M, Aminzadeh M, Chamanrou N, Kiani FZ, Seifi T, Zeighami J, Yadegari T, Sedaghat A, Saberi A, Hamid M, Shariati G and Galehdari H (2024) Identification of new variants in patients with mucopolysaccharidosis in consanguineous Iranian families. Front. Genet. 15:1343094. doi: 10.3389/fgene.2024.1343094
Received: 22 November 2023; Accepted: 25 January 2024;
Published: 15 February 2024.
Edited by:
Xiaonan Zhao, Baylor College of Medicine, United StatesReviewed by:
Xueyang Pan, Baylor College of Medicine, United StatesEric Kao, Baylor College of Medicine, United States
Casey Thornton, Baylor College of Medicine, United States
Copyright © 2024 Zabihi, Zamani, Aminzadeh, Chamanrou, Kiani, Seifi, Zeighami, Yadegari, Sedaghat, Saberi, Hamid, Shariati and Galehdari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gholamreza Shariati, shariatig@yahoo.com; Hamid Galehdari, Galehdari187@yahoo.com