 Emma M. J. Passchier1,2
Emma M. J. Passchier1,2 Quinty Bisseling1,2
Quinty Bisseling1,2 Guy Helman3Rosalina M. L. van Spaendonk4Cas Simons3,5René C. L. Olsthoorn6Hieke van der Veen1,7
Guy Helman3Rosalina M. L. van Spaendonk4Cas Simons3,5René C. L. Olsthoorn6Hieke van der Veen1,7 Truus E. M. Abbink1
Truus E. M. Abbink1 Marjo S. van der Knaap1,2*†
Marjo S. van der Knaap1,2*† Rogier Min1,2*†
Rogier Min1,2*†- 1Department of Child Neurology, Amsterdam Leukodystrophy Center, Emma Children’s Hospital, Amsterdam University Medical Center, Amsterdam Neuroscience, Amsterdam, Netherlands
- 2Department of Integrative Neurophysiology, Center for Neurogenomics and Cognitive Research, Vrije Universiteit Amsterdam, Amsterdam Neuroscience, Amsterdam, Netherlands
- 3Translational Bioinformatics, Murdoch Children’s Research Institute, The Royal Children’s Hospital, Parkville, VIC, Australia
- 4Department of Human Genetics, Amsterdam University Medical Center, Amsterdam, Netherlands
- 5Centre for Population Genomics, Garvan Institute of Medical Research, Sydney, NSW, Australia
- 6Leiden Institute of Chemistry, Leiden University, Leiden, Netherlands
- 7Department of Complex Trait Genetics, Center for Neurogenomics and Cognitive Research, Vrije Universiteit Amsterdam, Amsterdam Neuroscience, Amsterdam, Netherlands
The leukodystrophy megalencephalic leukoencephalopathy with subcortical cysts (MLC) is characterized by infantile-onset macrocephaly and chronic edema of the brain white matter. With delayed onset, patients typically experience motor problems, epilepsy and slow cognitive decline. No treatment is available. Classic MLC is caused by bi-allelic recessive pathogenic variants in MLC1 or GLIALCAM (also called HEPACAM). Heterozygous dominant pathogenic variants in GLIALCAM lead to remitting MLC, where patients show a similar phenotype in early life, followed by normalization of white matter edema and no clinical regression. Rare patients with heterozygous dominant variants in GPRC5B and classic MLC were recently described. In addition, two siblings with bi-allelic recessive variants in AQP4 and remitting MLC have been identified. The last systematic overview of variants linked to MLC dates back to 2006. We provide an updated overview of published and novel variants. We report on genetic variants from 508 patients with MLC as confirmed by MRI diagnosis (258 from our database and 250 extracted from 64 published reports). We describe 151 unique MLC1 variants, 29 GLIALCAM variants, 2 GPRC5B variants and 1 AQP4 variant observed in these MLC patients. We include experiments confirming pathogenicity for some variants, discuss particularly notable variants, and provide an overview of recent scientific and clinical insight in the pathophysiology of MLC.
1 Introduction
Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is a genetic brain white matter disease with onset in infancy (van der Knaap et al., 1995a; Singhal et al., 1996). Compared to many other leukodystrophies, it has a mild clinical course. Almost all patients with MLC present with macrocephaly, which is obvious already in the first year of life (van der Knaap et al., 1995b). Brain MRI is characterized by diffuse signal abnormality and swelling of the cerebral white matter and the presence of cysts in subcortical areas, almost invariably in the anterior temporal lobe (Figures 1A–D) (van der Knaap et al., 1995a; van der Knaap et al., 1995b). Patients typically develop neurologic signs after a few years. Motor development is initially normal or slightly delayed, and later shows slow deterioration with ataxia and spasticity. Half of the patients lose the ability to walk without support and become wheelchair bound in their teens (Hamilton et al., 2018). Most MLC patients experience one or more seizures in their lifetime, and 63% of patients with classic MLC meet the criteria for clinical epilepsy (Hamilton et al., 2018). Seizures can typically be controlled with antiepileptic medication (Yalcinkaya et al., 2003; Dubey et al., 2018). Mild head trauma is often a trigger for seizures, and status epilepticus is more frequent in MLC than expected based on the mild epilepsy (Dubey et al., 2018). Behavioral and cognitive problems are common. The diagnosis of MLC is based on clinical and MRI criteria (van der Knaap et al., 2012; van der Knaap et al., 2018).
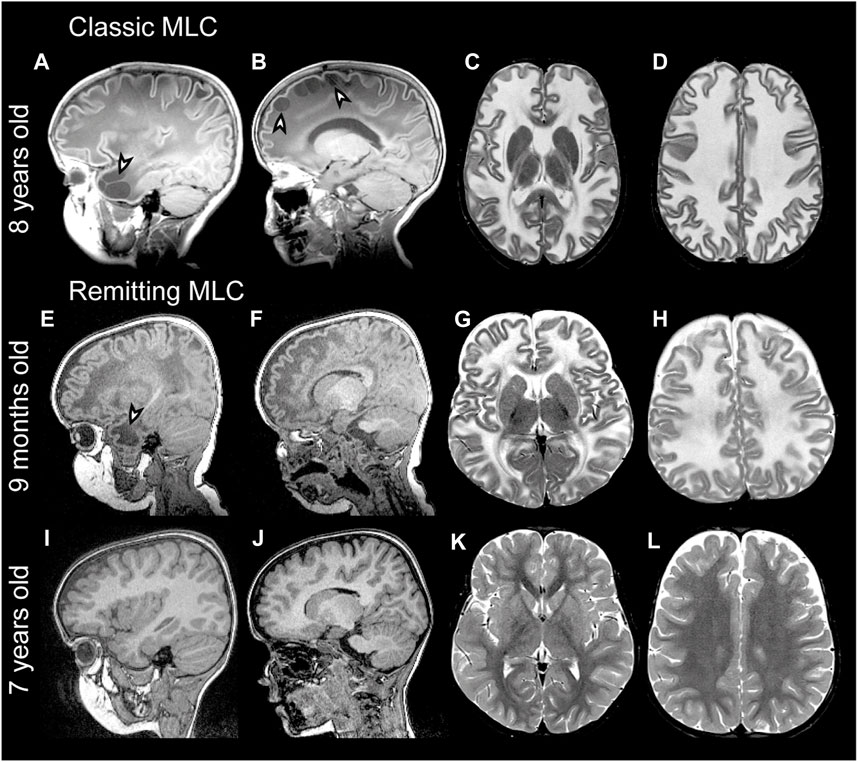
FIGURE 1. MRI findings in classic and remitting MLC patients. (A–D) MRI from an 11-year-old patient depicting an example of classic MLC. Anterior temporal and frontal subcortical cysts are visible in the sagittal T1-weighted MRIs in panels A and B (arrowheads). T2-weighted images in panel C and D show diffuse hyper intensity and swelling of the cerebral white matter with broadening of gyri (compare width of gyri in C and D to K and L). (E–H) MRI in a 9-month-old MLC patient and (I–L) images of the same patient at 7 years, showing the remitting phenotype. The anterior temporal cyst visible in panel E at 9 months (arrowhead) is no longer visible in panel I at 7 years. (F, J) No frontal subcortical cysts are present. Panels G, H, K, and L show that the cerebral white matter is initially T2-hyperintense and slightly swollen and that this T2-hyperintensity and swelling disappear over the years.
In 2001, the first gene linked to MLC was discovered and named MLC1 (Leegwater et al., 2001). Biallelic recessive MLC1 variants were found in many MLC patients. The associated disease is known as MLC1 (OMIM#604004). A remaining group of patients without MLC1 variants could be divided into patients with a classic clinical and MRI MLC phenotype and patients with initial signs of MLC followed by normalization of MRI and absence of motor and cognitive decline (van der Knaap et al., 2010) (Figures 1E-L). In 2011, a second MLC gene was discovered (Lopez-Hernandez et al., 2011a). This gene was initially called HEPACAM, but the name GLIALCAM is preferable because of its prominent expression in glial cells in the brain. Both patients with biallelic recessive GLIALCAM variants and patients with heterozygous dominant GLIALCAM variants were found. The small patient group with biallelic recessive variants in GLIALCAM has classic MLC and the associated disease is also known as MLC2A (OMIM# 613925). The larger group of patients heterozygous for a dominant GLIALCAM variant shows a remitting MLC phenotype, also known as MLC2B (OMIM# 613926). Macrocephaly and MRI properties are similar to classic MLC in the first year of life, but MRI greatly improves or normalizes in the following years and neurological regression does not occur (Lopez-Hernandez et al., 2011a). All patients with remitting MLC remain ambulatory, although some clumsiness can be present. In some patients head circumference also normalizes. Seizures and cognitive problems are less common in patients with remitting MLC, but autism is more common in these patients as compared to classic MLC patients (Lopez-Hernandez et al., 2011a; Hamilton et al., 2018).
Recently two new genes were linked to MLC in the small group of patients that lack variants in MLC1 or GLIALCAM (Passchier et al., 2023). Heterozygous dominant variants in GPRC5B were found in patients with an MRI pattern and clinical course characteristic of classic MLC patients, and the associated disease is known as MLC3 (OMIM# 620447). A homozygous recessive variant in AQP4 was identified in two siblings with MLC typical of the remitting form of the disease, and this disease is known as MLC4 (OMIM# 620448).
The last comprehensive overview of genetic variants linked to MLC dates from 2006 (Boor et al., 2006). This was before the discovery of GLIALCAM, GPRC5B and AQP4 as additional genes linked to MLC. Since then, many new variants in all four MLC genes have been described in literature and new variants were discovered in the Amsterdam Leukodystrophy Center (ALC). In this study we provide an overview of all known variants in MLC1, GLIALCAM, GPRC5B and AQP4 that have been linked to MLC to date. We discuss particularly notable variants. We briefly highlight the link of MLC1 and GLIALCAM variants with psychiatric diseases, recapitulate what is known about MLC disease mechanisms from cellular, molecular and animal studies and provide an outlook for future research.
2 Materials and methods
We made a list of known variants in MLC genes by performing an extensive literature search, supplemented with variants taken from the patient database of the ALC. We used the following accession numbers: NT_011526.7 and NM_015166.3 for MLC1, NT_033899.8 and NM_152722.4 for GLIALCAM (HEPACAM) NM_016235.3 for GPRC5B and NM_001650.7 for AQP4. All found variants were checked against the reference sequence, and nomenclature was updated, if necessary, making use of Alamut Visual version 2.9 (Interactive Biosoftware, Rouen, France). Interpretations of pathogenicity following ACMG guidelines were done for all variants (Richards et al., 2015).
2.1 Literature search
To identify MLC1 and GLIALCAM variants described in the literature, we performed a PUBMED search using the search words ‘MLC1’, ‘GLIALCAM’, ‘HEPACAM’ ‘AQP4 MLC’ and ‘GPRC5B’. Articles published until July 2022 were included. We included papers that were written in English, Dutch or French. Only papers discussing patient data were included (e.g., descriptions of cloned plasmid variants without patient relevance were excluded). Variants were reported only when the coding sequence position was reported and when a conclusive MLC diagnosis (based on MRI) was reported in the study.
2.2 ALC diagnostic workflow and database inclusion
Patients from the database of the ALC were included in this study upon conclusive MLC diagnosis by clinical features, MRI and genetic confirmation of variants in MLC1, GLIALCAM, GPRC5B or AQP4. Written informed consent was obtained from families for phenotyping.
The diagnostic workflow for MLC patients in the ALC is as follows: First, the diagnosis of MLC is established based on the presence of macrocephaly and characteristic MRI abnormalities now or in the past (Figure 1). Genetic testing starts with Sanger sequencing of MLC1. If no potentially pathogenic variants are found, this is followed by Sanger sequencing of GLIALCAM. When both are negative, and the MRI diagnosis is unambiguous, multiplex ligation-dependent probe amplification (MLPA) and cDNA analysis using lymphoblasts are performed for MLC1. If these do not uncover potentially pathogenic variants, next-generation sequencing (NGS, preferably whole genome sequencing (WGS)) is performed to identify potential rare (non-coding) variants.
2.3 Validation of variants impacting on MLC1 expression
Three reporter constructs were generated with the pNL1.1 vector (Promega), in which MLC1-expression regulating DNA sequences c.-2,645 to c.-1 (wild-type or with the c.-190A>G or c.-195T>C variant) were cloned directly upstream of the nanoluciferase open reading frame similarly as previously described (Hamilton et al., 2017). This DNA sequence includes the MLC1 core promoter and encodes the full 5′ untranslated region (5′UTR). Sanger sequencing was performed to confirm the MLC1 sequence with or without either of the two variants in the three p.NL1.1-MLC1 plasmids. Subsequently, U373 cells were cultured in DMEMF12 + 10% FBS. 24 h before transfection 3,000 cells were plated in white half area 96 well plates. The next day cells were co-transfected with a wild-type or mutant pNL1.1-MLC1 plasmid and the pGL3 plasmid (Promega) as internal standard using Fugene6 according to manufacturer’s instructions. The pGL3 plasmid encodes the firefly luciferase open reading frame under regulation of the SV40 promoter. Approximately 40 h after transfection, nanoluciferase and firefly luciferase activities were measured with a plate reader (Victor2; Perkin-Elmer Life Sciences, Waltham, MA), as described (Hamilton et al., 2017). The nanoluciferase signal was normalized to the firefly luciferase signal to obtain the relative expression driven by the wildtype and mutant MLC1 sequences. Statistical analysis was performed with Brown-Forsythe ANOVA followed by Dunnett’s T3 multiple comparisons test using GraphPad Prism 9 (GraphPad, USA). Statistically significant differences were defined as p ≤ 0.05. Data are represented as mean ± SEM.
2.4 Database submission
All variants described in this study have been submitted to the LOVD database (www.lovd.nl): MLC1: https://databases.lovd.nl/shared/transcripts/00013671; HEPACAM: https://databases.lovd.nl/shared/transcripts/00009260; AQP4: https://databases.lovd.nl/shared/transcripts/00002726; GPRC5B: https://databases.lovd.nl/shared/transcripts/00008881.
2.5 UK biobank
To estimate allele frequency for some variants, data was obtained from approximately 500,000 participants from the UK Biobank (Bycroft et al., 2018), a population-based sample of adults in the UK with self-report surveys, linked electronic health records, and genotypic data. The National Research Ethics Service Committee North West–Haydock ethically approved this initiative (reference 11/NW/0382) and participants provided informed written consent. Data were accessed under application #16406.
3 Variants
3.1 MLC1 variants
The MLC1 gene is located on chromosome 22q13. The gene contains 12 exons and 11 introns (Figure 2A). The 5′UTR consists of exon 1 and part of exon 2. Predictions and experimental studies show that the MLC1 protein has 8 transmembrane regions with both the amino- and the carboxy-terminus residing in the cytoplasm (Figure 2B). MLC1 most likely forms a trimeric structure in the membrane (Hwang et al., 2021). The protein has very low homology with other proteins, with highest similarity (less than 20%) with the shaker-related voltage gated potassium channel Kv1.1 α-subunit (Teijido et al., 2004; Brignone et al., 2015). The exact function of MLC1 is not known. However, experimental studies have implicated an indirect role for MLC1 in cell ion and water homeostasis.
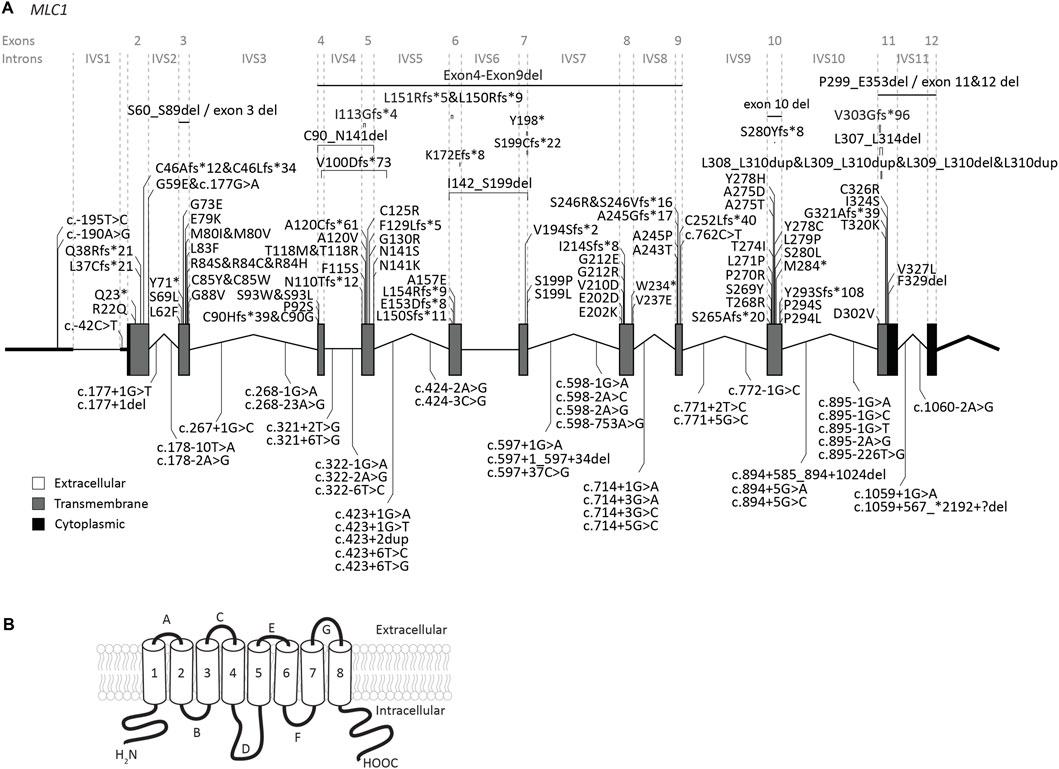
FIGURE 2. An overview of MLC1 variants found in MLC patients. (A) MLC1 is depicted. Exonic regions are indicated by blocks; intronic regions by lines. Exonic regions and intronic regions depicted with a horizontal line are drawn to scale. All variants are indicated above or below the gene schematic. For exonic variants the resulting peptide alterations are indicated, for intronic variants coding DNA alterations are indicated. Exonic variants are depicted in their relative positions. Intronic variants are depicted in their relative position roughly in the first or second half of the respective intronic region. (B) Schematic image of MLC1 in the cell membrane.
Our search yielded a total of 151 unique MLC1 variants in patients with a confirmed MLC diagnosis (Figure 2; Supplementary Table S1). Biochemical studies have been performed for some variants (see Supplementary Table S1) (Teijido et al., 2004; Duarri et al., 2008; Lopez-Hernandez et al., 2011b; Lanciotti et al., 2012; Capdevila-Nortes et al., 2013a; Sirisi et al., 2014; Xu et al., 2021). For most tested variants these studies reveal reduced plasma membrane levels, with retention of the protein in intracellular compartments (possibly the endoplasmic reticulum). In addition, protein stability is reduced for several variants. This might be a consequence of misfolding or defective oligomerization, leading to disrupted protein structure.
Several (possible) founder variants in MLC1 have been described. c.135dup; p.(Cys46Leufs*34) is a founder variant in East Indian individuals from the Agrawal community (Leegwater et al., 2002; Singhal et al., 2003; Gorospe et al., 2004). c.176G>A; p.(Gly59Glu) is a possible founder variant in Libyan Jews (Ben-Zeev et al., 2002). c.278C>T; p.(Ser93Leu) is common in Japanese individuals (Shimada et al., 2014), while c.824C>A; p.(Ala275Asp) is a founder variant accounting for the majority of MLC patients of Korean ancestry (Choi et al., 2017). c.908_918delinsGCA; p.(Val303Glyfs*96) is a founder variant in individuals with Egyptian ancestry.
3.1.1 Genotype-phenotype correlation for MLC1
All MLC1 variants are recessive and cause classic MLC when present in homozygous or compound heterozygous form. It is known that clinical disease severity greatly varies between patients, and can even greatly differ for patients with the same MLC1 variants. For example, two siblings homozygous for the c.736A>C; p.(Ser246Arg) variant show a particularly mild phenotype, but still with considerable differences between them. No clear genotype-phenotype correlation has been established (Gorospe et al., 2004; Hamilton et al., 2018). Most patients with bi-allelic variants in MLC1 have slowly progressing disease. However, a low number of patients with bi-allelic MLC1 variants display radiological improvement in the course of years. Recently such a patient with radiological improvement was described (Mayayo-Vallverdú et al., 2023). The two variants in this patient (c.597+37C>G; p.? and c.895–1G>T; p.?) cause splicing defects and the researchers could detect a small amount of wild-type MLC1 transcript as well as wild-type MLC1 protein in peripheral blood leukocytes taken from the patient. Incomplete penetrance of the splice site variant could explain the residual MLC1 and might underlie the radiological improvement. Similarly, we observed radiological improvement for patients in the ALC database with variants upstream of the MLC1 open reading frame. These variants reduce protein expression (c.-195T>C; p.? and c.-190A>G; p.?; see description below in section 3.1.2). In these cases, a low level of residual wild-type MLC1 is also expected, which may explain the improvement. However, given the broad phenotypic spectrum of MLC patients and the rarity of patients affected by these specific variants, further studies are required to confirm whether low levels of residual wild-type MLC1 are indeed at the basis of radiological improvement.
3.1.2 Two variants upstream of the MLC1 open reading frame reduce expression
Our database contains three MLC patients, in whom Sanger sequencing revealed two variants of unknown significance in exon 1 of MLC1. One of these patients is heterozygous for the c.-190A>G; p.? variant, with the other MLC1 allele affected by another, known pathogenic variant. The second patient is heterozygous for the c.-195T>C; p.? variant, with the other MLC1 allele affected by a different known, likely pathogenic variant. The third patient is homozygous for the c.-195T>C; p.? variant. To assess if and how the c.-195T>C; p.? and the c.-190A>G; p.? variants affect MLC1 expression, a set of three reporter constructs was created, in which nanoluciferase expression was regulated by the wild-type or mutant sequences upstream of the MLC1 open reading frame. Both variants significantly reduce the expression of the downstream reporter by more than 50%, indicating that they are likely to reduce but not fully abrogate MLC1 expression in patients (Figure 3). In combination with the patients’ clinical and MRI phenotypes, we classified these variants as UV4, likely pathogenic.
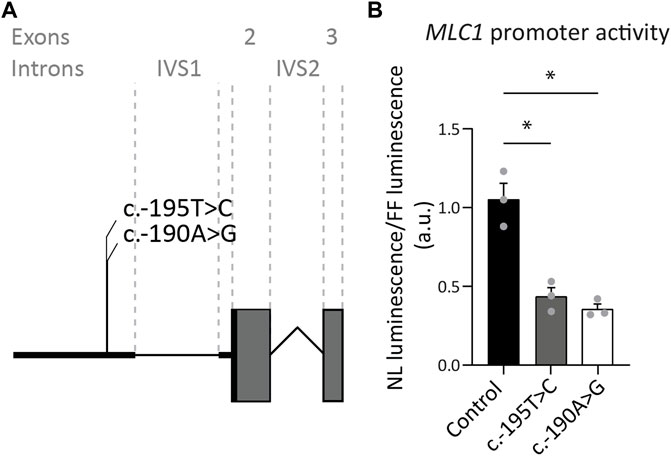
FIGURE 3. Exon 1 variants affecting expression of the downstream reporter. (A) Miniature schematic of 2 variants in the promoter region of MLC1. (B) Reporter gene assay depicting MLC1 promoter and 5′UTR activity in U373 cells. Readout was the ratio of Nano luciferase luminescence over Firefly luciferase luminescence in arbitrary units (a.u.). Experiments were performed in triplicate (n = 3), and each data point represents the average of 4 technical replicates. Graph shows means and individual data points of one experiment. Both the c.-190A>G and c.-195T>C variants significantly reduced expression of the downstream open reading frame of the reporter, reflected in a decrease in luminescence over fluorescence ratio. Brown-Forsythe ANOVA; F = 30.63 (2.000, 3.543) p = 0.0058. Dunnett’s T3 multiple comparisons test Control vs. c.-190A>G t = 6.576 df = 2.392 p = 0.0364. Control vs c.-195T>C t = 5.363 df = 3.085 p = 0.0222. *,p < 0.05.
3.1.3 A large intronic deletion in MLC1
Our database contained a patient homozygous for the c.894+585_894+1024del; p.? variant in MLC1, detected by WGS, with clinical and MRI features of classic MLC and no variants found in GLIALCAM. Both parents were unaffected carriers. The variant causes a deletion of 440 nucleotides from intron 10 of MLC1. The variant is not listed in the 1,000 genomes database (www.1000genomes.org), and not found in the UK Biobank. This makes it unlikely that it represents a common polymorphism. The deletion reduces the number of GGGGGAUGGAGUCACUG repeats present in wild-type MLC1 RNA from 17 to 3. These repeats share similarity with previously described G-rich intronic splicing enhancers (Kralovicova and Vorechovsky, 2007). One possibility therefore is that the variant reduces intron 10 splicing. Alternatively, the deletion could lead to a reduction in RNA stability and thereby reduce MLC1 protein expression. Therefore, we classify the c.894+585_894+1024del; p.? variant as UV3 (variant of unknown significance).
3.1.4 MLC1 variants that affect splicing
41 variants in MLC1 affect splicing. Most of these are in canonical splice sites. For variants outside of canonical splice sites, including some deep intronic variants (Mancini et al., 2012) a splicing defect was confirmed using cDNA analysis. The c.597+37C>G; p.? variant discussed in section 3.1.1 (Mayayo-Vallverdú et al., 2023), creates a splice acceptor site in intron 7. cDNA analysis shows that the variant affects RNA splicing and leads to skipping of exon 7 and partial retention of intron 7.
3.1.5 (Likely) benign variants in MLC1 and variants with an uncertain link to MLC
Several likely benign MLC1 variants have been reported (Leegwater et al., 2002; Shukla et al., 2011; Wang et al., 2011), and one was found in the ALC database. These are listed in Table 1, together with their allele frequency in the UK Biobank. For some variants in MLC patients reported in literature it is not clear whether they cause disease. These variants are listed in Table 1. The variant c.858C>G; p.(Ile286Met) was observed on the paternal allele in only one individual that carried another known pathogenic variant on the same allele (Cao et al., 2016). The authors describe that this patient has classic MLC. No other patients with this variant have been found. The variant is not found in the gnomAD database or in the UK Biobank, a large UK cohort (Table 1). Based on this information it is not possible to conclude whether the variant is disease causing. An individual with MLC with a heterozygous c.95C>T; p.(Ala32Val) variant on the maternal allele was described by Wang and others (Wang et al., 2011). No MLC1 variant was found on the paternal allele. At the time of this study MLC1 was the only known MLC gene. It is therefore possible that this individual had variants in another MLC gene, or that additional hard to detect MLC1 variants were missed. In a follow-up study from the same team, Cao and others (Cao et al., 2016) describe this variant in a patient who also has a dominant variant in GLIALCAM. This could be the same patient as described in the earlier study. A follow-up MRI for this patient was not described, making it impossible to assess whether this patient had a remitting phenotype. The c.95C>T; p.(Ala32Val) MLC1 variant has an allele frequency of 2.02*10^-5 and an allele count of 19 in the UK Biobank (Table 1).
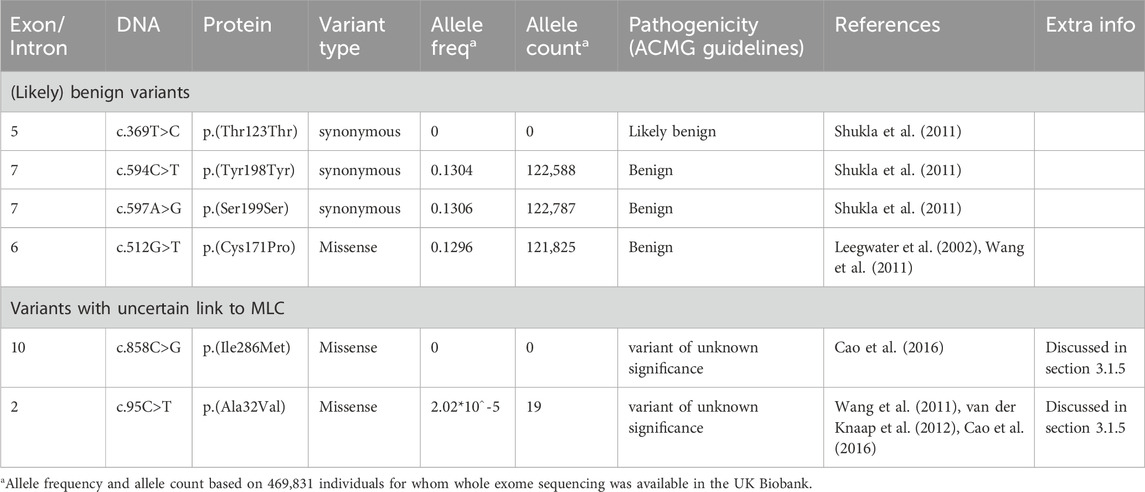
TABLE 1. (Likely) benign variants in MLC1 and variants with an uncertain link to MLC.
3.2 GLIALCAM variants
The GLIALCAM gene is located on chromosome 11q24. It contains 7 exons and 6 introns (Figure 4A). The protein GlialCAM, encoded by GLIALCAM, is an immunoglobulin-like transmembrane adhesion protein of 417 amino acids. It contains two N-terminal immunoglobulin domains (IgV and IgC2), a transmembrane domain and an intracellular C-terminal domain (Capdevila-Nortes et al., 2015) (Figure 4B). GlialCAM tightly interacts with MLC1 (Capdevila-Nortes et al., 2013b), acts as an auxiliary subunit for ClC-2 chloride channels (Jeworutzki et al., 2012) and regulates Connexin-43 mediated gap-junctional coupling (Wu et al., 2016). Before GLIALCAM was linked to MLC, it was mainly known as a cancer gene (Moh et al., 2008; He et al., 2010; Zhang et al., 2011). A total of 29 unique variants in GLIALCAM were identified in patients with either classic or remitting MLC (Figure 4; Table 2).
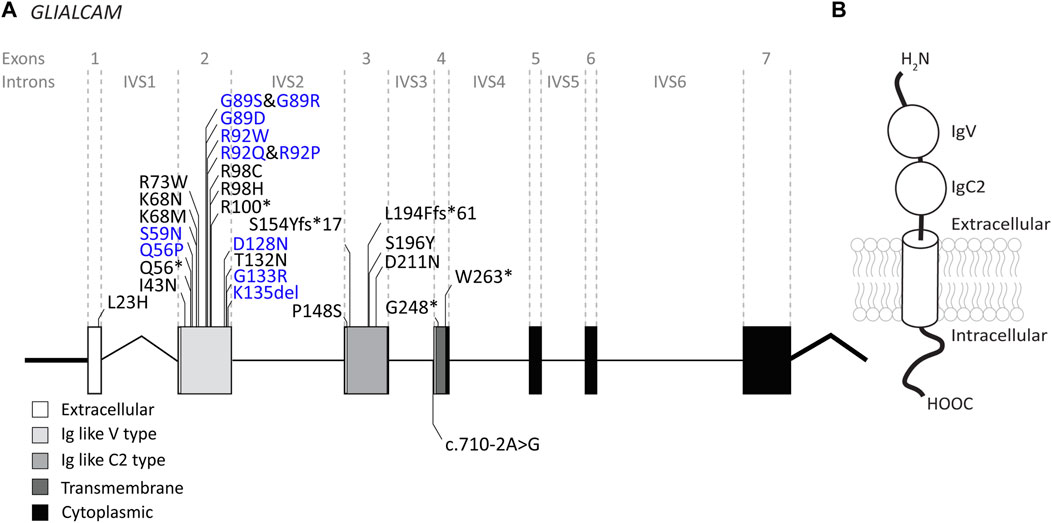
FIGURE 4. An overview of GLIALCAM variants found in MLC patients. (A) The GLIALCAM gene is depicted. Exonic regions are indicated by blocks; intronic regions by lines. Exonic regions and intronic regions depicted with a horizontal line are drawn to scale. All variants are indicated above or below the gene schematic. Resulting peptide alterations are indicated for exonic variants, and coding DNA alteration for an intronic variant. Variants are depicted in their relative positions. Dominant GLIALCAM variants are depicted in blue; recessive variants are depicted in black. (B) Schematic representation of GlialCAM in a cell membrane.
TABLE 2. GLIALCAM variants found in MLC patients.
3.2.1 Genotype-phenotype correlation: dominant GLIALCAM variants cause remitting MLC
For GLIALCAM variants, there is a clear genotype-phenotype correlation: Dominant variants lead to remitting MLC when present in heterozygous form. Variants classified as recessive lead to classic MLC when present in homozygous or compound heterozygous form. Some patients from the ALC database have a dominant variant on one allele and a recessive variant on the second allele. These patients have classic MLC.
Of the 29 GLIALCAM variants found in MLC patients, 11 have been reported to have a dominant effect (Figure 4; depicted in blue; bold in Table 2). All of these variants are located in exon 2. In addition, with the exception of one amino acid deletion, all dominant variants are missense. Heterozygous presence of these variants leads to remitting MLC. For some variants (c.167A>C; p.(Gln56Pro), c.274C>T; p.(Arg92Trp) and c.275G>C; p.(Arg92Pro)) it was reported that family members carried these variants but were not diagnosed with MLC in their youth. This indicates either reduced penetrance of the variants (meaning that some individuals with a dominant variant do not have a disease), or it means that the remitting MLC phenotype can be so mild in some individuals that it remains undiagnosed.
Functional experiments on recessive and dominant GLIALCAM variants show that most variants disrupt localization of GlialCAM to cell-cell junctions (Lopez-Hernandez et al., 2011a; Lopez-Hernandez et al., 2011b; Arnedo et al., 2014a). Structurally, all dominant variants affect the first extracellular immunoglobulin domain of the GlialCAM protein, while recessive variants are found in parts of the gene encoding the extracellular or transmembrane parts of the protein. Biochemical experiments suggest that dominant variants disrupt homophilic GlialCAM-GlialCAM interactions; possibly more specifically the interactions in trans with GlialCAM on neighbouring cells (Elorza-Vidal et al., 2020).
We report the c.217C>T; p.(Arg73Trp) and c.166C>T; p.(Gln56*) variants as recessive, because they have only been found in compound heterozygous form in one patient (Arnedo et al., 2014b). However, there is marked improvement of brain MRI abnormalities and mild clinical symptoms, more consistent with remitting MLC. c.166C>T; p.(Gln56*) is predicted to produce no functional protein. No clear effect of the c.217C>T; p.(Arg73Trp) variant was observed in cellular and biochemical assays. One possibility is that the full loss of function caused by the c.166C>T; p.(Gln56*) variant by itself is sufficient to cause a remitting MLC phenotype (see also the discussion on GLIALCAM hemizygosity below). Alternatively, the c.217C>T; p.(Arg73Trp) variant might act in a dominant fashion. To fully understand the consequence of these two variants therefore requires either a better mechanistic understanding of GlialCAM function, or the observation of novel patients with only one of these variants.
3.2.2 GLIALCAM hemizygosity in Jacobsen syndrome
Partial deletion of the terminal part of chromosome 11q leads to Jacobsen syndrome (Mattina et al., 2009). MRI abnormalities resembling MLC have been described in Jacobsen syndrome patients already long ago, which led to the speculation that a leukodystrophy gene could be located in this region (Wardinsky et al., 1990; Gutmann, 1991). Indeed, deletion of 11q24 including GLIALCAM was later linked to MLC-like white matter abnormalities with diffuse signal abnormality and swelling of the cerebral white matter in several patients (Yamamoto et al., 2015; Patel et al., 2019; Wolf and van der Knaap, 2020). Importantly, the MRI phenotype in Jacobsen syndrome is remitting (similar to what is seen in remitting MLC; see Figure 5 for an example from the ALC database). Since MRI abnormalities are not observed in all Jacobsen patients, either penetrance of such abnormalities upon GLIALCAM hemizygosity is not complete, or white matter abnormalities were missed because they resolved before the first MRI (Ono et al., 1996). This suggests that hemizygosity for GLIALCAM may lead to a clinical phenotype similar to remitting MLC.
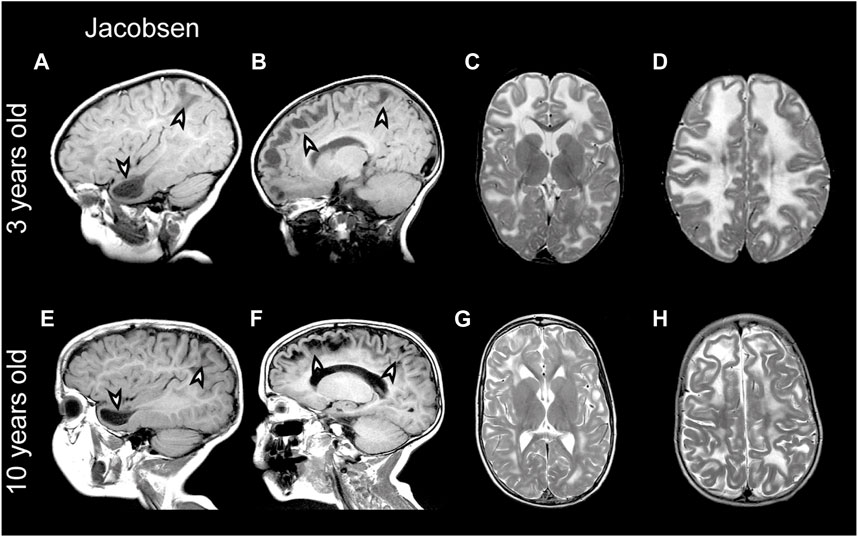
FIGURE 5. MRI findings in a Jacobsen syndrome patient. (A–D) MRIs from a 3-year-old Jacobsen syndrome patient with a chromosomal deletion that includes GLIALCAM. Numerous anterior temporal and frontal subcortical cysts are visible in the sagittal T1-weighted MRIs in panels A and B (arrowheads). T2-weighted images in (C and D) reveal extensive signal abnormality and swelling of the cerebral white matter. (E–H) Follow-up of the same patient at 10 years of age. Subcortical cysts persist [arrowheads in (E,F)], but there is clear improvement in signal abnormality and swelling of the cerebral white matter.
3.2.3 (Likely) benign variants in GLIALCAM and variants with an uncertain link to MLC
For some variants it is not possible to establish their link to MLC. c.461_462del; p.(Ser154Tyrfs*17) (Table 2) and c.789G>A; p.(Trp263*) (Table 2) were found in one patient on the same allele. It is likely that the first variant, which causes a premature stop codon, already disrupts the expression of the full-length GlialCAM protein. Still, because the second variant also causes a premature stop codon, we have classified both variants as pathogenic.
c.862C>T; p.(Arg288Cys) was found in an individual with remitting MLC. This individual had the c.382G>A; p.(Asp128Asn), a known dominant variant, on the same allele (Lopez-Hernandez et al., 2011a). The allele count of c.862C>T; p.(Arg288Cys) in the UK Biobank is 122 (heterozygous only) with an allele frequency of 1.29*10−4 and in the gnomAD database the allele count is 156 with one occurrence in homozygous state (Table 3). Based on this information it is not possible to say whether the variant is pathogenic. We classify this variant as a variant of unknown significance.

TABLE 3. (Likely) benign variants in GLIALCAM and variants with an uncertain link to MLC.
Certain dominant GLIALCAM variants have been observed in compound heterozygous state with an intronic variant of unknown consequence (c.877+101G>T; p.?). This variant was also found in two patients with additional biallelic GLIALCAM variants. The allele frequency of this variant in the UK biobank, gnomAD and 1000genomes (3.43%) database is high (Table 3). We conclude that this variant is benign. c.582C>T; p.(Leu194Leu) was found on one allele together with a known pathogenic variant. The variant is synonymous and not predicted to affect splicing. Allele frequency in the UK Biobank is low. We conclude that this variant is likely benign.
3.3 AQP4 variants
The AQP4 gene is located on chromosome 18q11.2 and contains 5 exons and 4 introns (Figure 6A). It encodes the water channel aquaporin-4 (AQP4), the most abundant aquaporin in the brain (King et al., 2004). AQP4 has two main isoforms due to the use of two different translation initiation sites (Neely et al., 1999; Verkman et al., 2011): the longer M1 isoform and a shorter M23 isoform. Both isoforms, among other isoforms, are expressed in astrocytes and assemble as heterotetramers (Jorgacevski et al., 2020). AQP4 has 6 transmembrane domains and 2 highly conserved NPA domains that form the pore in the membrane (Ho et al., 2009) (Figure 6B). Both the C- and the N-terminus reside intracellularly. AQP4 is of vital importance for brain ion and water homeostasis (Nagelhus and Ottersen, 2013). It shares its location in astrocyte endfeet with MLC1 and GlialCAM (Nagelhus et al., 2004).
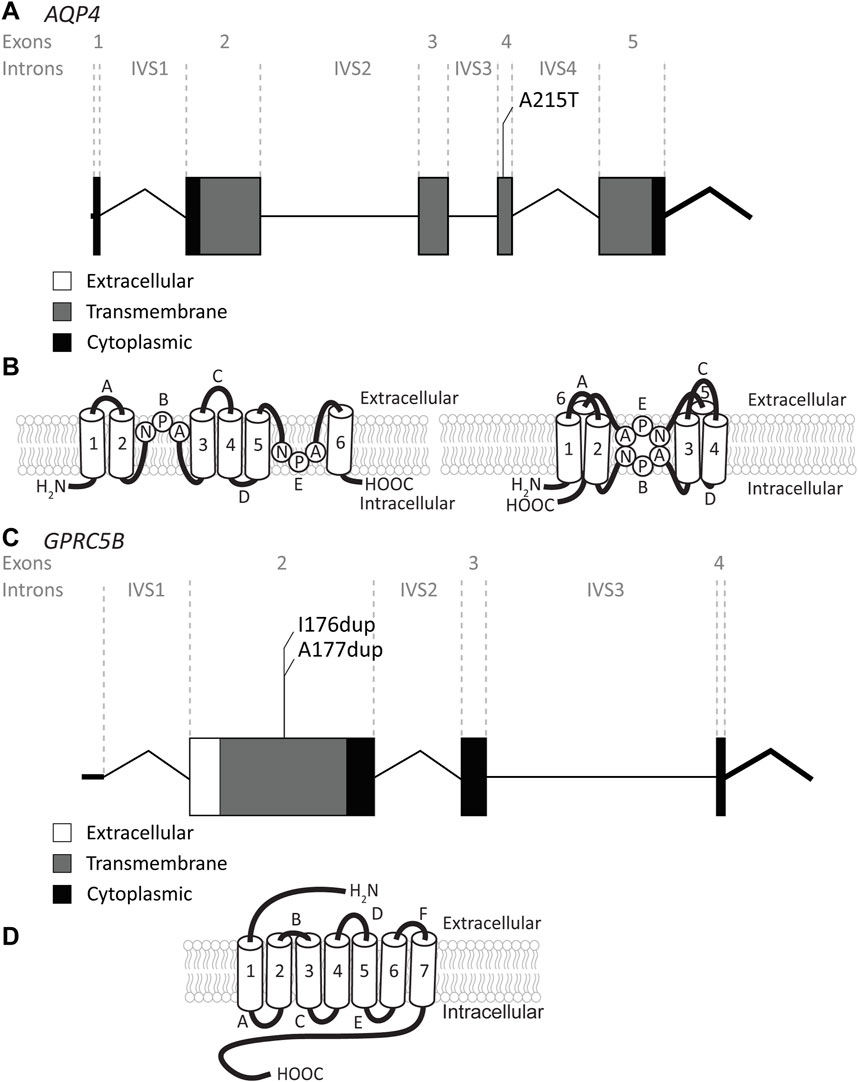
FIGURE 6. An overview of AQP4 and GPRC5B variants found in MLC patients. (A) AQP4 is depicted. Exonic regions are indicated by blocks; intronic regions by lines. Exonic regions and intronic regions depicted with a horizontal line are drawn to scale. All variants are indicated above the gene schematic. The resulting peptide alteration is in its relative position. (B) A schematic representation of AQP4 in the membrane. Amino acids of the NPA motif are depicted. (C) As in (A) however here, GPRC5B is depicted with its two peptide alterations. (D) A schematic representation of GPRC5B in the membrane.
Recently, a recessive variant in AQP4 was found to lead to remitting MLC (Passchier et al., 2023). This is the first variant found in AQP4 to be disease linked and offers new insight into MLC disease mechanisms. The variant affects the key pore forming NPA motif in the AQP4 protein, exchanging an alanine, which is a hydrophobic amino acid, for a threonine, a hydrophilic amino acid (c.643G>A; p.(Ala215Thr) Figure 6A; Table 4). This leads to a loss of AQP4 at the cell membrane and therefore, a loss of function of the protein. Why an AQP4 defect would lead to remitting MLC is unknown. Passchier et al. describe that under certain conditions (e.g., massive overexpression) mutant AQP4 retains some function (Passchier et al., 2023). The physiological relevance of this is unclear, but this might form a basis for the observed radiological improvement. An alternative explanation could be redundancy in brain ion and water homeostasis, potentially with other aquaporins taking over the role of AQP4.
TABLE 4. AQP4 and GPRC5B variants found in MLC patients.
3.4 GPRC5B variants
The GPRC5B gene is located on chromosome 16p12.3 and contains 4 exons and 3 introns. Exon 1 encodes the 5′UTR (Figure 6C). GPRC5B encodes the orphan G protein-coupled receptor (GPCR) GPRC5B. It is a class C GPCR and has 7 transmembrane regions and a relatively short extracellular N terminus compared to other GPCRs of the same class (Figure 6D). Multiple GPRC5B isoforms are known, including a long and a short isoform, as well as a brain specific isoform (Cool et al., 2010). GPRC5B is expressed in various tissues and cell types, including adipocytes, pancreatic islets, kidney podocytes and vascular smooth muscle cells. The receptor modulates inflammatory signalling through the NF-κB pathway, and has been linked to various disease states including diabetes (Soni et al., 2013), kidney disease (Zambrano et al., 2019) and artherosclerosis (Carvalho et al., 2020). Few studies have looked at GPRC5B function in the brain (Kurabayashi et al., 2013; Sano et al., 2018), but recently GPRC5B was identified as an MLC1/GlialCAM interacting protein that is expressed in astrocyte endfeet (Alonso-Gardon et al., 2021). Two dominant de novo variants in the fourth transmembrane region of GPRC5B were recently described in patients with classic MLC that had no variants in MLC1, GLIALCAM or AQP4 (Passchier et al., 2023). These two variants are specific amino acid duplications of two neighbouring amino acids in the fourth transmembrane domain of the GPCR (c.526_528dup; p.(Ile176dup) and c. 528_530dup; p.(Ala177dup); Figure 6C; Table 4). How these variants affect the function of GPRC5B is not yet understood.
3.5 Psychiatric and neurodevelopmental disorders linked with MLC1 and GLIALCAM variants
Abnormalities of the brain white matter have been consistently associated with psychiatric disorders (Fields, 2008). Psychiatric symptoms are therefore often seen in leukodystrophies, even sometimes as presenting symptoms (Costei et al., 2021). A clear example is the frequent occurrence of psychosis in Metachromatic Leukodystrophy (MLD OMIM# 250100) (Hyde et al., 1992).
Regarding neurodevelopmental disorders, autistic features are common within the population of MLC patients (Lopez-Hernandez et al., 2011a). Surprisingly, autistic features are seen in ∼25% of patients with remitting MLC caused by dominant GLIALCAM variants, while this is only 9% for patients with classic MLC (Hamilton et al., 2018). Therefore, while motor symptoms are invariably milder in remitting MLC, the occurrence of autistic features is higher. The basis for this difference is not understood. It suggests that dominant GLIALCAM variants disrupt a function of GlialCAM that is separate from its role in astrocyte endfeet. For example, a recent study showed that astroglial release of GlialCAM from exosomes regulates neuronal axon outgrowth and dendritic spine formation (Jin et al., 2023). Both of these processes have been implicated in the etiology of autism (Gilbert and Man, 2017). While speculative, such a specialized function of GlialCAM might explain why dominant GLIALCAM variants more often lead to autistic features.
Genetic screens in cohorts of patients with autism spectrum disorder (ASD) uncovered additional heterozygous missense variants in GLIALCAM. The c.274C>T; p.(Arg92Trp) variant, which was previously found in a remitting MLC patient (Lopez-Hernandez et al., 2011a), was independently discovered in a study on ASD patients (Iossifov et al., 2014). The c.437C>T; p.(Ser146Leu) variant was discovered in another study on ASD patients (Li et al., 2017). Another new GLIALCAM variant that has not been observed in MLC patients, c.505T>C; p.(Ser169Pro), was found in a study on patients with intellectual disability (Lelieveld et al., 2016). Finally, a splice site variant in GLIALCAM, c.803+1G>A; p.? was found in an ASD cohort. This variant was classified as variant of unknown significance (Zhou et al., 2019). Together, these studies further substantiate the link between specific dominant GLIALCAM variants, ASD and intellectual disability.
For MLC1 it has been suggested that specific variants in heterozygous state are linked to schizophrenia or bipolar affective disorder, two psychiatric disorders thought to share a common etiology. A rare missense variant in MLC1 (c.1121C>A; p.(Leu309Met)) was associated with periodic catatonia, a familial subtype of catatonic schizophrenia (#OMIM605419) in a large pedigree (Meyer et al., 2001), although this variant was not found in other cohorts of schizophrenia or bipolar affective disorder patients (Meyer et al., 2001). Additional MLC1 variants have been significantly associated with schizophrenia and bipolar affective disorder in an Indian cohort (Devaney et al., 2002; Ewald and Lundorf, 2002; Jorgensen et al., 2002; McQuillin et al., 2002; Rubie et al., 2003; Kaganovich et al., 2004), with confirmation of two intronic MLC1 variants in an independent study, where they were specifically associated with periodic catatonia and not with other types of schizophrenia (Selch et al., 2007). Interestingly, variants linked to bipolar disorder or schizophrenia have not been observed in MLC patients. In addition, the c.1121C>A; p.(Leu309Met) variant has no influence on MLC1 protein expression levels in cellular studies (Teijido et al., 2004). Therefore, while these findings suggest that being a heterozygous carrier of specific MLC1 variants is associated with schizophrenia and bipolar disorder, the mechanistic link between MLC1 and psychiatry requires further research.
Finally, gene expression levels for MLC1 show potential association with depression and suicidal behavior. MLC1 transcript levels were significantly downregulated in post-mortem brain tissue of suicide victims (Thalmeier et al., 2008). A later study found reduced transcript levels in blood of patients with major depressive disorder (Spijker et al., 2010). Future studies confirming and extending the link between MLC1 transcript levels and suicidal behavior are required.
4 Disease mechanisms
4.1 Expression pattern and interaction partners of MLC1, GlialCAM, GPRC5B and AQP4
All MLC linked proteins are membrane proteins that are mainly expressed by glial cells in the brain (Zhang et al., 2016). MLC1 and AQP4 expression in the brain is restricted to astrocytes. GLIALCAM and GPRC5B are predominantly expressed by astrocytes, but both can also be detected in oligodendrocytes (Figure 7). Immunohistochemical studies on human brain tissue show that MLC1 and GlialCAM are highly enriched in astrocyte endfeet contacting the vasculature. In addition, both are found in other glial membranes at brain-cerebrospinal fluid interfaces, such as in subpial astrocyte endfeet and in ependymal cells (Boor et al., 2007; Lopez-Hernandez et al., 2011a). This location is shared with AQP4 (Nagelhus and Ottersen, 2013) and numerous other proteins involved in brain ion and water homeostasis, such as members of the dystrophin-associated glycoprotein complex (Boor et al., 2007), cation channels such as Kir4.1 (Higashi et al., 2001) and TRPV4 (Benfenati et al., 2011; Jo et al., 2015), chloride channels such as the essential volume regulated anion channel (VRAC) subunit LRRC8A (Bugiani et al., 2017; Formaggio et al., 2019), the chloride channel ClC-2 (Depienne et al., 2013) and the gap-junction protein Connexin-43 (Wu et al., 2016).
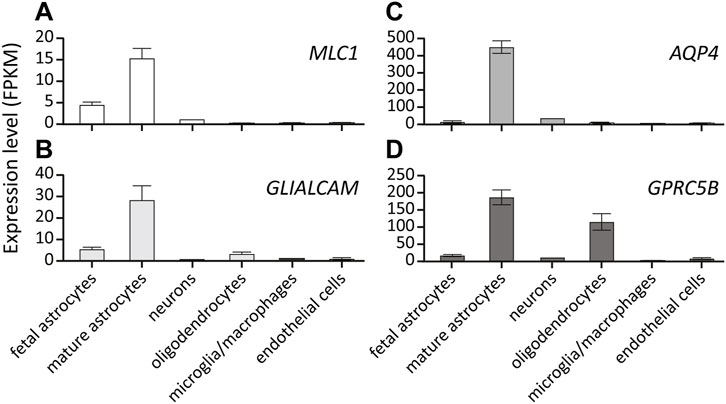
FIGURE 7. Expression of MLC-linked genes in different brain cell types. Expression level of MLC1 (A), GLIALCAM (B), AQP4 (C) and GPRC5B (D) in different cell types in the human brain. Values are depicted as RNA Fragments Per Kilobase Million (FPKM). All genes show predominant expression in mature astrocytes. Data from https://brainrnaseq.org/.
Direct or indirect interactions of many proteins that are enriched in astrocyte endfeet with MLC1 and/or GlialCAM have been described (for review see (Brignone et al., 2015)). Of particular importance is the early finding that a direct interaction between GlialCAM and MLC1 is required for the correct localization of both proteins to astrocyte-astrocyte junctions (Lopez-Hernandez et al., 2011a; Capdevila-Nortes et al., 2013b; Sirisi et al., 2014). A pathogenic variant in either of these proteins can disrupt the junctional localization of both. In addition, GlialCAM forms both cis and trans interactions with other GlialCAM proteins. Understanding the structural nature of these interactions likely holds the key to unravelling why certain GLIALCAM variants act in a dominant fashion while others are recessive (Elorza-Vidal et al., 2020). GlialCAM also directly interacts with, and acts as an auxiliary subunit for the chloride channel ClC-2. It is involved in both correct localization of the channel and in changing its gating kinetics (Jeworutzki et al., 2012). Recently, additional interaction partners of GlialCAM and MLC1 were identified, including the GPCRs, GPRC5B and GPR37L1 (Alonso-Gardon et al., 2021). Together, this information suggests that MLC1, GlialCAM and GPRC5B form an astrocyte endfoot complex interacting with ion channels, AQP4 water channels, gap junction proteins and other players involved in ion and water homeostasis.
Studying healthy human brain tissue has provided insight into the normal distribution of MLC related proteins and their interaction partners in the brain. However, the scarcity of post-mortem MLC patient brain material limits knowledge on how protein levels and protein distribution are altered in the disease context. Rare biopsy tissue has confirmed the linked localization of MLC1, GlialCAM and ClC-2 (Sirisi et al., 2014). Qualitative studies in animal models confirm this, but show that many other endfoot proteins have normal localization in MLC (Dubey et al., 2015; Bugiani et al., 2017). A quantitative study of the endfoot proteome in the context of MLC, in animal models or ideally in patient tissue, would be valuable to confirm and expand on these findings. In addition, how other cell types alter their protein make-up in the MLC disease context has been largely unstudied.
4.2 The functional role of MLC1, GlialCAM, GPRC5B and AQP4 in MLC
Pathological examination of a brain biopsy of an MLC patient revealed the existence of numerous vacuoles in the white matter (van der Knaap et al., 1996). These vacuoles are located in between the myelin lamellae, suggesting that they are the consequence of myelin splitting due to intramyelinic fluid accumulation. In the same tissue, perivascular astrocyte endfeet appear swollen and show intracellular vacuolization (Duarri et al., 2011). Together with the obvious swelling of the white matter observed on MRI, this has led to the hypothesis that MLC is characterized by a disruption of brain ion and water homeostasis (van der Knaap et al., 2012), leading to chronic white matter edema. The unraveling of the network of proteins interacting with MLC1, GlialCAM, GPRC5B and AQP4 described in section 4.1 supports this hypothesis.
Important mechanistic insight into the function of MLC1 and GlialCAM came from the discovery that lymphoblasts from MLC patients and astrocytes from MLC mouse models show a defect in volume regulation. Upon hypotonicity induced cell swelling most mammalian cells, including astrocytes, recover from swelling through a process known as regulatory volume decrease (RVD) (Jentsch, 2016). Lymphoblasts from patients with bi-allelic pathogenic MLC1 variants, as well as astrocytes isolated from Mlc1-null mice show reduced RVD when compared to healthy cells (Ridder et al., 2011; Dubey et al., 2015). A similar disruption of RVD was recently observed in lymphoblasts from patients with either of the dominant GPRC5B variants (Passchier et al., 2023). In addition, loss of AQP4 in astrocytes also disrupts the RVD process (Benfenati et al., 2011).
A key question is how MLC proteins are involved in the process of astrocyte volume regulation. The RVD process involves concerted activity of channels and transporters to orchestrate the net efflux of osmolytes from the cells, leading to associated water efflux through osmosis. A key channel involved in RVD is VRAC, a channel permeable to anions and small osmolytes which opens upon cell swelling (Jentsch, 2016). Loss of MLC1 or GlialCAM from cells reduces VRAC activity (Ridder et al., 2011; Capdevila-Nortes et al., 2013b; Dubey et al., 2015), and overexpression of MLC1 in various cells increases VRAC activity (Ridder et al., 2011). GPRC5B was also recently shown to regulate VRAC activity. Overexpression of GPRC5B, either mutant or wild-type, leads to increased VRAC activity in astrocytoma cells (Passchier et al., 2023), while GPRC5B knockdown in primary astrocytes reduces VRAC activity (Alonso-Gardon et al., 2021).
The observation that MLC1 shows minor homology to ion channels initially led to the speculation that MLC1 might be the long sought protein that forms the VRAC channel. Subsequent identification of LRRC8A-E subunits as the subunits of VRAC (Qiu et al., 2014; Voss et al., 2014) put this hypothesis to rest, and allowed investigation of molecular interactions between VRAC subunits, MLC1 and GlialCAM. This revealed that a direct molecular interaction between these proteins is lacking (Elorza-Vidal et al., 2018; Alonso-Gardon et al., 2021). Instead, the interaction between MLC1, GlialCAM, GPRC5B and VRAC activity likely involves an intermediate cellular process that is not fully resolved. Many signaling cascades and cellular properties are known to modulate VRAC functioning in different cell types (for review see (Jentsch, 2016)). A recent study has suggested that intracellular calcium might be an intermediate (Brignone et al., 2022), through CaMKII-mediated phosphorylation of MLC1. AQP4 forms a complex with TRPV4 and together these proteins affect RVD through a functional interaction with VRAC (Benfenati et al., 2011). Upon osmotic changes, water entry or exit from astrocytes is accelerated by the presence of AQP4. Rapid volume changes greatly enhance the activation of VRAC channels (Benfenati et al., 2007). Additionally, hypo-osmotic challenges increase expression of AQP4 at the plasma membrane (Kitchen et al., 2015). The membrane expression of LRRC8A proteins and AQP4 show mutual dependence in astrocytes (Liu et al., 2023). Therefore, MLC linked proteins converge on regulation of VRAC activity in RVD. Further mechanistic insight into how VRAC function is disrupted in MLC is a crucial step in our understanding of the disease and may aid in directing therapy development.
In addition to the effect on VRAC, MLC1 and GlialCAM interact with various other ion channels and pumps involved in brain ion and water homeostasis. As mentioned earlier, a direct interaction of GlialCAM with the chloride channel ClC-2 was described, with GlialCAM acting as an auxiliary channel subunit, potentiating channel function and changing desensitization kinetics (Jeworutzki et al., 2012). However, since neurological disease caused by loss of ClC-2 function differs substantially from MLC, both clinically and on MRI (Depienne et al., 2013), the role of ClC-2 in the pathogenesis of MLC is not well understood. Furthermore, MLC1, as well as AQP4, directly interacts with the mechanosensitive cation channel TRPV4 (Benfenati et al., 2011; Lanciotti et al., 2012; Jo et al., 2015). The nature of this interaction and its relevance in MLC pathogenesis is not well understood. Alonso-Gardon et al. (2021) described direct interaction between MLC1 and GPRC5B and between GLIALCAM and GPRC5B. They also showed interaction of GlialCAM with another GPCR, GPR37L1. Research into these GPCRs could give new insight into signaling pathways that are dysregulated in MLC.
As mentioned, there is large variability in disease severity between patients and a lack of a clear phenotype/genotype correlation. Because of the many interactions of MLC proteins with other proteins involved in ion and water homeostasis, an important open question is whether there are genetic modifiers of disease severity. Variants in genes involved in brain ion and water homeostasis, which by themselves do not lead to disease, might modulate disease severity in MLC patients. To illustrate this, the presence of several common single-nucleotide polymorphisms in AQP4, which are linked to lower levels of AQP4, have been associated with altered EEG activity during sleep in the healthy population (Ulv Larsen et al., 2020). It will be interesting to see whether the presence of such common or less common haplotypes, which constitute a polygenic risk for brain ion and water homeostasis, correlates with disease severity in MLC patients.
In conclusion, MLC1, GlialCAM, GPRC5B and AQP4 appear to be central in the organization of an osmoregulatory complex in astrocyte endfeet. The disruption of this complex in MLC hampers astrocytes in keeping ion and water homeostasis, thereby causing fluid accumulation in myelin and astrocytes.
4.3 MLC animal models
Several animal models for MLC have been generated. Mlc1-null mice and Glialcam-null mice both recapitulate key disease features: increased brain water content and progressive myelin vacuolization (Hoegg-Beiler et al., 2014; Dubey et al., 2015; Bugiani et al., 2017). However, while in patients vacuolization and swelling is most apparent in the cerebral white matter outside the corpus callosum and to a lesser degree in the cerebellar white matter (van der Knaap et al., 1995b), mice have very little cerebral white matter beyond the corpus callosum and white matter vacuolization in MLC mice is more readily observed in cerebellar white matter and corpus callosum (Dubey et al., 2015; Bugiani et al., 2017; Hoegg-Beiler et al., 2014). Patients develop white matter swelling with megalencephaly in the first year of life, replicated in MLC mice. MLC patients typically develop motor and cognitive decline with a delay of 4–6 years. The short life span of mice probably explains why MLC mice show no obvious behavioral phenotype (Dubey et al., 2015), although detailed investigation does reveal hind limb clasping when the animals are lifted by the tail (Dubey et al., 2018). While the mice show no overt seizure phenotype, recording electrical brain activity in freely moving mice uncovered increased interictal spike occurrence. Furthermore, the threshold for kainate-induced seizure induction is lowered (Dubey et al., 2018). Therefore, similar to MLC patients the mice have a seizure phenotype.
Thus, Mlc1-null mice and Glialcam-null mice replicate early stages of classic MLC (Bugiani et al., 2017). A mouse model based on a dominant Glialcam variant has also been generated (Hoegg-Beiler et al., 2014). As expected, the vacuolization phenotype observed in mice heterozygous for this dominant variant was mild.
A mouse overexpressing wild-type MLC1 has also been generated. Interestingly, this overexpression mouse shows white matter vacuolization similar to classic MLC mouse models, but with a more rapid onset (Sugio et al., 2017). As outlined in section 4.2, MLC linked proteins are part of an osmoregulatory complex in astrocyte endfeet. It is likely that ion channels, pumps and transporters involved in these processes need to be carefully tuned, with deviation in their activity in either direction leading to disease. The fact that both a decrease and an increase in expression levels of MLC1 can lead to a similar phenotype is in line with this idea.
Two zebrafish models for MLC have been generated and characterized. Knockout of the zebrafish orthologs of MLC1 (mlc1) or GLIALCAM (glialcama) led to macrocephaly, but vacuoles were not observed in the brain of adult MLC zebrafish (Sirisi et al., 2014; Perez-Rius et al., 2019).
Long before the clinical discovery that loss of AQP4 function leads to remitting MLC (Passchier et al., 2023), Aqp4-null mice had been generated and extensively characterized (Ma et al., 1997). Similar to Mlc1-null and Glialcam-null mice, Aqp4-null mice show increased brain water content (Vindedal et al., 2016) and an alteration in seizure threshold (Binder et al., 2004; Binder et al., 2006). The alteration in seizure threshold described for Aqp4-null mice differs from what is seen in other MLC mouse models and in patients: Aqp4-null mice have an increased threshold for evoked seizures, but when seizures occur they are longer in duration. Whether this is due to slight differences in experimental conditions remains to be explored.
In conclusion, multiple animal models for MLC have been generated and characterized. These models do not show an exact phenocopy of the human disease, mainly with respect to most prominently affected white matter structures and a reduced severity of overt neurological phenotypes. However, central MLC related phenotypes can be observed in these models, and they enable researchers to study MLC pathomechanisms in the intact brain. This has allowed confirmation of the hypothesis that disturbed astrocyte volume regulation leads to disrupted extracellular potassium regulation in MLC: extracellular potassium dynamics are altered in Mlc1-null and Glialcam-null mice (Dubey et al., 2018). Disrupted potassium homeostasis is also observed in Aqp4-null mice (Binder et al., 2006; Haj-Yasein et al., 2015). In addition, Mlc1-null mice were used to uncover delayed maturation of perivascular astrocyte processes, altered astrocyte morphology and polarity, reduced vascular smooth muscle cell contractility and disturbed neurovascular coupling and parenchymal flow in MLC (Gilbert et al., 2021). MLC mouse models also allow for preclinical testing of therapeutic interventions. To date, this has been only done in the context of gene therapy. Viral transduction with wild-type MLC1 targeted to cerebellum could revert cerebellar white matter vacuolization in Mlc1-null mice (Sanchez et al., 2020). The use of MLC mouse models for preclinical testing of other interventions holds great promise for the future search for an MLC therapy.
4.4 Therapy outlook for MLC
Curative therapy for MLC is still lacking. The fact that white matter abnormalities in some MLC patients show normalization holds promise for treatment, since it indicates that myelin vacuolization in MLC is in principle reversible.
For monogenic diseases like MLC restoring the causative genetic defect through gene editing or overexpression of a healthy gene copy could be considered. A first proof-of-concept gene therapy study showed that cerebellar white matter vacuolization can be prevented or reversed when MLC1 levels are virally restored in the cerebellum of Mlc1-null mice (Sanchez et al., 2020). However, while technological advances in gene therapy are fast, it will take time before the risks associated with gene therapy (immune reactions, cancers, tissue damage upon intracranial delivery) can properly be addressed. Since clinically MLC is a relatively mild disease, it is important to carefully weigh these risks with therapeutic benefit. Furthermore, for effective gene therapy it is likely necessary to target astrocytes throughout the brain, which is not feasible currently. Finally, overexpression of Mlc1 in an animal model causes an MLC-like phenotype, indicating that expression levels of MLC related proteins likely need to be carefully tuned. These factors pose an additional challenge for gene therapy approaches. However, the observation that low residual levels of MLC1 might lead to mild or remitting disease offer new leads for tuning expression levels.
Alternatively, traditional pharmacological targeting of disrupted cellular processes in the MLC brain might hold promise. This relies on detailed mechanistic understanding of the disease and identification of suitable therapeutic targets. Ion and water channels involved in brain fluid homeostasis (VRAC, TRPV4, AQP4) can be considered as targets. The recent identification of two GPCRs that interact with MLC1 and GlialCAM raises additional exciting possibilities (Alonso-Gardon et al., 2021; Passchier et al., 2023), especially since modulation of GPCRs is the mechanism of action for ∼1/3 of current FDA approved drugs (Chan et al., 2019). Unravelling the underlying signalling pathways, how they are disrupted in MLC, and whether pharmaceutical modulators can be identified should be the focus point of future mechanistic studies. Advances in gene therapy or pharmacological approaches can then be followed by preclinical tests in MLC mouse models, which will hopefully pave the way for much needed therapy for MLC.
5 Conclusion
This paper provides a comprehensive overview of all known MLC-causing gene variants to date. We list 151 unique variants in MLC1, 29 unique variants in GLIALCAM, 2 in GPRC5B and 1 in AQP4. 51 variants had not been discussed in literature before. This study therefore forms a valuable resource for clinicians, clinical laboratories and researchers. In addition, we have reviewed current knowledge about MLC disease mechanisms and the physiological role of MLC1, GlialCAM, AQP4 and GPRC5B. Thereby we also highlighted the knowledge gaps that should be filled with future research. Important questions are how MLC1 and GlialCAM regulate astrocyte osmoregulation, how they interplay with AQP4 in MLC and what the involvement of intracellular signalling cascades, potentially mediated by GPRC5B, is in this process. An answer to these key questions will be crucial in future efforts to develop curative therapy for MLC.
Data availability statement
All variants described in this study have been submitted to the LOVD database (www.lovd.nl): MLC1: https://databases.lovd.nl/shared/transcripts/00013671; HEPACAM: https://databases.lovd.nl/shared/transcripts/00009260; AQP4: https://databases.lovd.nl/shared/transcripts/00002726; GPRC5B: https://databases.lovd.nl/shared/transcripts/00008881. Part of this research was conducted using the UK Biobank Resource (application no. 16406).
Ethics statement
The studies involving humans were approved by the Medical Ethics Review Committee (METC) of the Amsterdam UMC. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from a by-product of routine care or industry. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
EP: Conceptualization, Data curation, Investigation, Methodology, Project administration, Visualization, Writing–original draft, Writing–review and editing. QB: Writing–review and editing, Writing–original draft. GH: Investigation, Writing–review and editing, Methodology. RvS: Writing–review and editing, Data curation, Investigation, Methodology. CS: Investigation, Writing–review and editing, Methodology. RO: Investigation, Methodology, Writing–review and editing. HvdV: Writing–review and editing, Data curation, Formal analysis, Investigation. TA: Data curation, Formal Analysis, Investigation, Methodology, Writing–review and editing. MvdK: Conceptualization, Formal Analysis, Investigation, Resources, Supervision, Writing–review and editing. RM: Conceptualization, Funding acquisition, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Supported in part by grants from ZonMW (VIDI grant 91718392) and the Dutch Rare Disease Foundation (Zeldzame Ziekten Fonds).
Acknowledgments
We thank MLC patients and their families for their participation; Nienke Postma, Timo J. ter Braak, Carola G.M. van Berkel, Gemma M. van Leeuwen and Leoni Hoogterp (Amsterdam UMC) for excellent technical assistance; Johan T. den Dunnen (Human Genetics, Leiden University Medical Center) for help with LOVD data upload. We would like to thank the many UK Biobank participants and staff.
Conflict of interest
MvdK is consultant for Calico (vanishing white matter) and co-investigator for Ionis (Alexander disease trial), without personal payment. She is the initiator and principal investigator of the Guanabenz trial (https://www.clinicaltrialsregister.eu/ctr-search/trial/2017-001438-25/NL). MvdK and TA are on patent P112686US00 “therapeutic effects of Guanabenz treatment in vanishing white matter” and patent P112686CA00 “the use of Guanabenz in the treatment of VWM”, both for the VU University Medical Center, Amsterdam, Netherlands.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1352947/full#supplementary-material
References
Abbink, T. E. M., Wisse, L. E., Jaku, E., Thiecke, M. J., Voltolini-Gonzalez, D., Fritsen, H., et al. (2019). Vanishing white matter: deregulated integrated stress response as therapy target. Ann. Clin. Transl. Neurol. 6 (8), 1407–1422. doi:10.1002/acn3.50826
Abdel-Salam, G. M., Abdel-Hamid, M. S., Ismail, S. I., Hosny, H., Omar, T., Effat, L., et al. (2016). Megalencephalic leukoencephalopathy with cysts in twelve Egyptian patients: novel mutations in MLC1 and HEPACAM and a founder effect. Metab. Brain Dis. 31 (5), 1171–1179. doi:10.1007/s11011-016-9861-7
Ain Ul Batool, S., Almatrafi, A., Fadhli, F., Alluqmani, M., Sadia, , Ali, G., et al. (2022). A homozygous missense variant in the MLC1 gene underlies megalencephalic leukoencephalopathy with subcortical cysts in large kindred: heterozygous carriers show seizure and mild motor function deterioration. Am. J. Med. Genet. A 188 (4), 1075–1082. doi:10.1002/ajmg.a.62614
Alonso-Gardon, M., Elorza-Vidal, X., Castellanos, A., La Sala, G., Armand-Ugon, M., Gilbert, A., et al. (2021). Identification of the GlialCAM interactome: the G protein-coupled receptors GPRC5B and GPR37L1 modulate megalencephalic leukoencephalopathy proteins. Hum. Mol. Genet. 30 (17), 1649–1665. doi:10.1093/hmg/ddab155
Amin, M., Vignal, C., Hamed, A. A. A., Mohammed, I. N., Elseed, M. A., Drunat, S., et al. (2022). Novel variants causing megalencephalic leukodystrophy in Sudanese families. J. Hum. Genet. 67 (3), 127–132. doi:10.1038/s10038-021-00945-7
Arnedo, T., Aiello, C., Jeworutzki, E., Dentici, M. L., Uziel, G., Simonati, A., et al. (2014b). Expanding the spectrum of megalencephalic leukoencephalopathy with subcortical cysts in two patients with GLIALCAM mutations. Neurogenetics 15 (1), 41–48. doi:10.1007/s10048-013-0381-x
Arnedo, T., Lopez-Hernandez, T., Jeworutzki, E., Capdevila-Nortes, X., Sirisi, S., Pusch, M., et al. (2014a). Functional analyses of mutations in HEPACAM causing megalencephalic leukoencephalopathy. Hum. Mutat. 35 (10), 1175–1178. doi:10.1002/humu.22622
Benfenati, V., Caprini, M., Dovizio, M., Mylonakou, M. N., Ferroni, S., Ottersen, O. P., et al. (2011). An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc. Natl. Acad. Sci. U. S. A. 108 (6), 2563–2568. doi:10.1073/pnas.1012867108
Benfenati, V., Nicchia, G. P., Svelto, M., Rapisarda, C., Frigeri, A., and Ferroni, S. (2007). Functional down-regulation of volume-regulated anion channels in AQP4 knockdown cultured rat cortical astrocytes. J. Neurochem. 100 (1), 87–104. doi:10.1111/j.1471-4159.2006.04164.x
Ben-Zeev, B., Levy-Nissenbaum, E., Lahat, H., Anikster, Y., Shinar, Y., Brand, N., et al. (2002). Megalencephalic leukoencephalopathy with subcortical cysts; a founder effect in Israeli patients and a higher than expected carrier rate among Libyan Jews. Hum. Genet. 111 (2), 214–218. doi:10.1007/s00439-002-0770-y
Binder, D. K., Oshio, K., Ma, T., Verkman, A. S., and Manley, G. T. (2004). Increased seizure threshold in mice lacking aquaporin-4 water channels. Neuroreport 15 (2), 259–262. doi:10.1097/00001756-200402090-00009
Binder, D. K., Yao, X., Zador, Z., Sick, T. J., Verkman, A. S., and Manley, G. T. (2006). Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 53 (6), 631–636. doi:10.1002/glia.20318
Boor, de G. K., Mejaski-Bosnjak, V., Brenner, C., van der Knaap, M. S., Scheper, G. C., Pronk, J. C., et al. (2006). Megalencephalic leukoencephalopathy with subcortical cysts: an update and extended mutation analysis of MLC1. Hum. Mutat. 27 (6), 505–512. doi:10.1002/humu.20332
Boor, I., Nagtegaal, M., Kamphorst, W., van d, V., Pronk, J. C., van, H. J., et al. (2007). MLC1 is associated with the dystrophin-glycoprotein complex at astrocytic endfeet. Acta Neuropathol. 114 (4), 403–410. doi:10.1007/s00401-007-0247-0
Brignone, M. S., Lanciotti, A., Camerini, S., De, N. C., Petrucci, T. C., Visentin, S., et al. (2015). MLC1 protein: a likely link between leukodystrophies and brain channelopathies. Front. Cell. Neurosci. 9, 66. doi:10.3389/fncel.2015.00106
Brignone, M. S., Lanciotti, A., Michelucci, A., Mallozzi, C., Camerini, S., Catacuzzeno, L., et al. (2022). The CaMKII/MLC1 Axis confers Ca(2+)-dependence to volume-regulated anion channels (VRAC) in astrocytes. Cells 11 (17), 2656. doi:10.3390/cells11172656
Brozova, K., Krasnicanova, H., and Rusina, R. (2019). Megalencephalic leukoencephalopathy with subcortical cysts without macrocephaly: a case study of comorbid Turner's syndrome. Clin. Neurol. Neurosurg. 184, 105400. doi:10.1016/j.clineuro.2019.105400
Bugiani, M., Dubey, M., Breur, M., Postma, N. L., Dekker, M. P., Ter Braak, T., et al. (2017). Megalencephalic leukoencephalopathy with cysts: the Glialcam-null mouse model. Ann. Clin. Transl. Neurol. 4 (7), 450–465. doi:10.1002/acn3.405
Bugiani, M., Moroni, I., Bizzi, A., Nardocci, N., Bettecken, T., Gartner, J., et al. (2003). Consciousness disturbances in megalencephalic leukoencephalopathy with subcortical cysts. Neuropediatrics 34 (4), 211–214. doi:10.1055/s-2003-42209
Bycroft, C., Freeman, C., Petkova, D., Band, G., Elliott, L. T., Sharp, K., et al. (2018). The UK Biobank resource with deep phenotyping and genomic data. Nature 562 (7726), 203–209. doi:10.1038/s41586-018-0579-z
Cao, B., Yan, H., Guo, M., Xie, H., Wu, Y., Gu, Q., et al. (2016). Ten novel mutations in Chinese patients with megalencephalic leukoencephalopathy with subcortical cysts and a long-term follow-up research. PLoS One 11 (6), e0157258. doi:10.1371/journal.pone.0157258
Capdevila-Nortes, X., Jeworutzki, E., Elorza-Vidal, X., Barrallo-Gimeno, A., Pusch, M., and Estevez, R. (2015). Structural determinants of interaction, trafficking and function in the ClC-2/MLC1 subunit GlialCAM involved in leukodystrophy. J. Physiol. 593 (18), 4165–4180. doi:10.1113/JP270467
Capdevila-Nortes, X., Lopez-Hernandez, T., Apaja, P. M., Lopez de, H. M., Sirisi, S., Callejo, G., et al. (2013b). Insights into MLC pathogenesis: GlialCAM is an MLC1 chaperone required for proper activation of volume-regulated anion currents. Hum. Mol. Genet. 22 (21), 4405–4416. doi:10.1093/hmg/ddt290
Capdevila-Nortes, X., Lopez-Hernandez, T., Apaja, P. M., Lopez de Heredia, M., Sirisi, S., Callejo, G., et al. (2013a). Insights into MLC pathogenesis: GlialCAM is an MLC1 chaperone required for proper activation of volume-regulated anion currents. Hum. Mol. Genet. 22 (21), 4405–4416. doi:10.1093/hmg/ddt290
Carvalho, J., Chennupati, R., Li, R., Gunther, S., Kaur, H., Zhao, W., et al. (2020). Orphan G protein-coupled receptor GPRC5B controls smooth muscle contractility and differentiation by inhibiting prostacyclin receptor signaling. Circulation 141 (14), 1168–1183. doi:10.1161/CIRCULATIONAHA.119.043703
Chan, H. C. S., Li, Y., Dahoun, T., Vogel, H., and Yuan, S. (2019). New binding sites, new opportunities for GPCR drug discovery. Trends Biochem. Sci. 44 (4), 312–330. doi:10.1016/j.tibs.2018.11.011
Choi, S. A., Kim, S. Y., Yoon, J., Choi, J., Park, S. S., Seong, M. W., et al. (2017). A unique mutational spectrum of MLC1 in Korean patients with megalencephalic leukoencephalopathy with subcortical cysts: p.Ala275Asp founder mutation and maternal uniparental disomy of chromosome 22. Ann. Lab. Med. 37 (6), 516–521. doi:10.3343/alm.2017.37.6.516
Cool, B. H., Chan, G. C., Lee, L., Oshima, J., Martin, G. M., and Hu, Q. (2010). A flanking gene problem leads to the discovery of a Gprc5b splice variant predominantly expressed in C57Bl/6J mouse brain and in maturing neurons. PLoS One 5 (4), e10351. doi:10.1371/journal.pone.0010351
Costei, C., Barbarosie, M., Bernard, G., Brais, B., and La Piana, R. (2021). Adult hereditary white matter diseases with psychiatric presentation: clinical pointers and MRI algorithm to guide the diagnostic process. J. Neuropsychiatry Clin. Neurosci. 33 (3), 180–193. doi:10.1176/appi.neuropsych.20110294
Dai, C. L., He, W. B., Du, J., Tan, Y. Q., Lu, G. X., and Li, W. (2017). A case of megalencephalic leukoencephalopathy with subcortical cysts type 1 was identified with a novel compound heterozygous alteration (c.135delC; c.423+2dupT) in China. Clin. Case Rep. 5 (6), 961–967. doi:10.1002/ccr3.986
Depienne, C., Bugiani, M., Dupuits, C., Galanaud, D., Touitou, V., Postma, N., et al. (2013). Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol. 12 (7), 659–668. doi:10.1016/S1474-4422(13)70053-X
De Stefano, N., Balestri, P., Dotti, M. T., Grosso, S., Mortilla, M., Morgese, G., et al. (2001). Severe metabolic abnormalities in the white matter of patients with vacuolating megalencephalic leukoencephalopathy with subcortical cysts. A proton MR spectroscopic imaging study. J. Neurol. 248 (5), 403–409. doi:10.1007/s004150170182
Devaney, J. M., Donarum, E. A., Brown, K. M., Meyer, J., Stöber, G., Lesch, K. P., et al. (2002). No missense mutation of WKL1 in a subgroup of probands with schizophrenia. Mol. Psychiatry 7, 419–423. doi:10.1038/sj.mp.4001022
Duarri, A., Lopez de Heredia, M., Capdevila-Nortes, X., Ridder, M. C., Montolio, M., Lopez-Hernandez, T., et al. (2011). Knockdown of MLC1 in primary astrocytes causes cell vacuolation: a MLC disease cell model. Neurobiol. Dis. 43 (1), 228–238. doi:10.1016/j.nbd.2011.03.015
Duarri, A., Teijido, O., Lopez-Hernandez, T., Scheper, G. C., Barriere, H., Boor, I., et al. (2008). Molecular pathogenesis of megalencephalic leukoencephalopathy with subcortical cysts: mutations in MLC1 cause folding defects. Hum. Mol. Genet. 17 (23), 3728–3739. doi:10.1093/hmg/ddn269
Dubey, M., Brouwers, E., Hamilton, E. M. C., Stiedl, O., Bugiani, M., Koch, H., et al. (2018). Seizures and disturbed brain potassium dynamics in the leukodystrophy megalencephalic leukoencephalopathy with subcortical cysts. Ann. Neurol. 83 (3), 636–649. doi:10.1002/ana.25190
Dubey, M., Bugiani, M., Ridder, M. C., Postma, N. L., Brouwers, E., Polder, E., et al. (2015). Mice with megalencephalic leukoencephalopathy with cysts: a developmental angle. Ann. Neurol. 77 (1), 114–131. doi:10.1002/ana.24307
Elorza-Vidal, X., Sirisi, S., Gaitan-Penas, H., Perez-Rius, C., Alonso-Gardon, M., Armand-Ugon, M., et al. (2018). GlialCAM/MLC1 modulates LRRC8/VRAC currents in an indirect manner: implications for megalencephalic leukoencephalopathy. Neurobiol. Dis. 119, 88–99. doi:10.1016/j.nbd.2018.07.031
Elorza-Vidal, X., Xicoy-Espaulella, E., Pla-Casillanis, A., Alonso-Gardon, M., Gaitan-Penas, H., Engel-Pizcueta, C., et al. (2020). Structural basis for the dominant or recessive character of GLIALCAM mutations found in leukodystrophies. Hum. Mol. Genet. 29 (7), 1107–1120. doi:10.1093/hmg/ddaa009
Ewald, H., and Lundorf, M. D. (2002). The missense mutation in the WKL1 gene not found in patients with bipolar affective disorder. Mol. Psychiatry 7, 340–341. doi:10.1038/sj.mp.4001002
Fields, R. D. (2008). White matter in learning, cognition and psychiatric disorders. Trends Neurosci. 31 (7), 361–370. doi:10.1016/j.tins.2008.04.001
Formaggio, F., Saracino, E., Mola, M. G., Rao, S. B., Amiry-Moghaddam, M., Muccini, M., et al. (2019). LRRC8A is essential for swelling-activated chloride current and for regulatory volume decrease in astrocytes. FASEB J. 33 (1), 101–113. doi:10.1096/fj.201701397RR
Gilbert, A., Elorza-Vidal, X., Rancillac, A., Chagnot, A., Yetim, M., Hingot, V., et al. (2021). Megalencephalic leukoencephalopathy with subcortical cysts is a developmental disorder of the gliovascular unit. Elife 10, e71379. doi:10.7554/eLife.71379
Gilbert, J., and Man, H. Y. (2017). Fundamental elements in autism: from neurogenesis and neurite growth to synaptic plasticity. Front. Cell. Neurosci. 11, 359. doi:10.3389/fncel.2017.00359
Gorospe, J. R., Singhal, B. S., Kainu, T., Wu, F., Stephan, D., Trent, J., et al. (2004). Indian Agarwal megalencephalic leukodystrophy with cysts is caused by a common MLC1 mutation. Neurology 62 (6), 878–882. doi:10.1212/01.wnl.0000115106.88813.5b
Gutmann, D. H. (1991). Chromosome 11q23.3-qter deletion and Alexander disease. Am. J. Med. Genet. 39 (2), 226–227. doi:10.1002/ajmg.1320390224
Haj-Yasein, N. N., Bugge, C. E., Jensen, V., Ostby, I., Ottersen, O. P., Hvalby, O., et al. (2015). Deletion of aquaporin-4 increases extracellular K(+) concentration during synaptic stimulation in mouse hippocampus. Brain Struct. Funct. 220 (4), 2469–2474. doi:10.1007/s00429-014-0767-z
Hamilton, E. M. C., Bertini, E., Kalaydjieva, L., Morar, B., Dojcakova, D., Liu, J., et al. (2017). UFM1 founder mutation in the Roma population causes recessive variant of H-ABC. Neurology 89 (17), 1821–1828. doi:10.1212/WNL.0000000000004578
Hamilton, E. M. C., Tekturk, P., Cialdella, F., van Rappard, D. F., Wolf, N. I., Yalcinkaya, C., et al. (2018). Megalencephalic leukoencephalopathy with subcortical cysts: characterization of disease variants. Neurology 90 (16), e1395–e1403. doi:10.1212/WNL.0000000000005334
He, Y., Wu, X., Luo, C., Wang, L., and Lin, J. (2010). Functional significance of the hepaCAM gene in bladder cancer. BMC Cancer 10, 83. doi:10.1186/1471-2407-10-83
Higashi, K., Fujita, A., Inanobe, A., Tanemoto, M., Doi, K., Kubo, T., et al. (2001). An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am. J. Physiol. Cell. Physiol. 281 (3), C922–C931. doi:10.1152/ajpcell.2001.281.3.C922
Ho, J. D., Yeh, R., Sandstrom, A., Chorny, I., Harries, W. E., Robbins, R. A., et al. (2009). Crystal structure of human aquaporin 4 at 1.8 A and its mechanism of conductance. Proc. Natl. Acad. Sci. U. S. A. 106 (18), 7437–7442. doi:10.1073/pnas.0902725106
Hoegg-Beiler, M. B., Sirisi, S., Orozco, I. J., Ferrer, I., Hohensee, S., Auberson, M., et al. (2014). Disrupting MLC1 and GlialCAM and ClC-2 interactions in leukodystrophy entails glial chloride channel dysfunction. Nat. Commun. 5, 3475. doi:10.1038/ncomms4475
Hwang, J., Park, K., Lee, G. Y., Yoon, B. Y., Kim, H., Roh, S. H., et al. (2021). Transmembrane topology and oligomeric nature of an astrocytic membrane protein, MLC1. MLC1. Open Biol. 11 (12), 210103. doi:10.1098/rsob.210103
Hyde, T. M., Ziegler, J. C., and Weinberger, D. R. (1992). Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch. Neurol. 49 (4), 401–406. doi:10.1001/archneur.1992.00530280095028
Ilyas, M., Efthymiou, S., Salpietro, V., Noureen, N., Zafar, F., Rauf, S., et al. (2020). Novel variants underlying autosomal recessive intellectual disability in Pakistani consanguineous families. BMC Med. Genet. 21 (1), 59. doi:10.1186/s12881-020-00998-z
Iossifov, I., O'Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515 (7526), 216–221. doi:10.1038/nature13908
Itoh, N., Maeda, M., Naito, Y., Narita, Y., and Kuzuhara, S. (2006). An adult case of megalencephalic leukoencephalopathy with subcortical cysts with S93L mutation in MLC1 gene: a case report and diffusion MRI. Eur. Neurol. 56 (4), 243–245. doi:10.1159/000096672
Jentsch, T. J. (2016). VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat. Rev. Mol. Cell. Biol. 17 (5), 293–307. doi:10.1038/nrm.2016.29
Jerbi Omezzine, S., Ben Ameur, H., Bousoffara, R., Ayedi, A., Hamza, H. A., and Sfar, M. T. (2008). Megalencephalic leukoencephalopathy with subcortical cysts: report of 4 new cases. J. Radiol. 89 (1), 891–894. doi:10.1016/s0221-0363(08)73877-0
Jeworutzki, E., Lopez-Hernandez, T., Capdevila-Nortes, X., Sirisi, S., Bengtsson, L., Montolio, M., et al. (2012). GlialCAM, a protein defective in a leukodystrophy, serves as a ClC-2 Cl(-) channel auxiliary subunit. Neuron 73 (5), 951–961. doi:10.1016/j.neuron.2011.12.039
Jin, S., Chen, X., Tian, Y., Jarvis, R., Promes, V., and Yang, Y. (2023). Astroglial exosome HepaCAM signaling and ApoE antagonization coordinates early postnatal cortical pyramidal neuronal axon growth and dendritic spine formation. Nat. Commun. 14 (1), 5150. doi:10.1038/s41467-023-40926-2
Jo, A. O., Ryskamp, D. A., Phuong, T. T., Verkman, A. S., Yarishkin, O., MacAulay, N., et al. (2015). TRPV4 and AQP4 channels synergistically regulate cell volume and calcium homeostasis in retinal muller glia. J. Neurosci. 35 (39), 13525–13537. doi:10.1523/JNEUROSCI.1987-15.2015
Jorgacevski, J., Zorec, R., and Potokar, M. (2020). Insights into cell surface expression, supramolecular organization, and functions of aquaporin 4 isoforms in astrocytes. Cells 9 (12), 2622. doi:10.3390/cells9122622
Jorgensen, T. H., Børglum, A. D., Mors, O., Wang, A. G., Pinaud, M., Flint, T. J., et al. (2002). Search for common haplotypes on chromosome 22q in patients with schizophrenia or bipolar disorder from the Faroe Islands. Am. J. Med. Genet. 114, 245–252. doi:10.1002/ajmg.10191
Kaganovich, M., Peretz, A., Ritsner, M., Bening Abu-Shach, U., Attali, B., and Navon, R. (2004). Is the WKL1 gene associated with schizophrenia? Am. J. Med. Genet. B Neuropsychiatr. Genet. 125B (1), 31–37. doi:10.1002/ajmg.b.20115
Kariminejad, A., Rajaee, A., Ashrafi, M. R., Alizadeh, H., Tonekaboni, S. H., Malamiri, R. A., et al. (2015). Eight novel mutations in MLC1 from 18 Iranian patients with megalencephalic leukoencephalopathy with subcortical cysts. Eur. J. Med. Genet. 58 (2), 71–74. doi:10.1016/j.ejmg.2014.12.004
Khalaf-Nazzal, R., Dweikat, I., Maree, M., Alawneh, M., Barahmeh, M., Doulani, R. T., et al. (2022). Prevalent MLC1 mutation causing autosomal recessive megalencephalic leukoencephalopathy in consanguineous Palestinian families. Brain Dev. 44 (7), 454–461. doi:10.1016/j.braindev.2022.03.009
King, L. S., Kozono, D., and Agre, P. (2004). From structure to disease: the evolving tale of aquaporin biology. Nat. Rev. Mol. Cell. Biol. 5 (9), 687–698. doi:10.1038/nrm1469
Kiriyama, T., Tanizawa, E., Hirano, M., Shinkai, T., Asai, H., Furiya, Y., et al. (2007). SPECT revealed cortical dysfunction in a patient who had genetically definite megalencephalic leukoencephalopathy with subcortical cysts. Clin. Neurol. Neurosurg. 109 (6), 526–530. doi:10.1016/j.clineuro.2007.03.012
Kitchen, P., Day, R. E., Taylor, L. H., Salman, M. M., Bill, R. M., Conner, M. T., et al. (2015). Identification and molecular mechanisms of the rapid tonicity-induced relocalization of the aquaporin 4 channel. J. Biol. Chem. 290 (27), 16873–16881. doi:10.1074/jbc.M115.646034
Koussa, S., Roukoz, H., Rizk, T., and Megarbane, A. (2005). Megalencephalic leucoencephalopathy with subcortical cysts: a study of a Lebanese family and a review of the literature. Rev. Neurol. 161 (2), 183–191. doi:10.1016/s0035-3787(05)85021-0
Koyama, S., Kawanami, T., Arawaka, S., Wada, M., and Kato, T. (2012). A Japanese adult case of megalencephalic leukoencephalopathy with subcortical cysts with a good long-term prognosis. Intern Med. 51 (5), 503–506. doi:10.2169/internalmedicine.51.6462
Kralovicova, J., and Vorechovsky, I. (2007). Global control of aberrant splice-site activation by auxiliary splicing sequences: evidence for a gradient in exon and intron definition. Nucleic Acids Res. 35 (19), 6399–6413. doi:10.1093/nar/gkm680
Kurabayashi, N., Nguyen, M. D., and Sanada, K. (2013). The G protein-coupled receptor GPRC5B contributes to neurogenesis in the developing mouse neocortex. Development 140 (21), 4335–4346. doi:10.1242/dev.099754
Lanciotti, A., Brignone, M. S., Molinari, P., Visentin, S., De, N. C., Macchia, G., et al. (2012). Megalencephalic leukoencephalopathy with subcortical cysts protein 1 functionally cooperates with the TRPV4 cation channel to activate the response of astrocytes to osmotic stress: dysregulation by pathological mutations. Hum. Mol. Genet. 21 (10), 2166–2180. doi:10.1093/hmg/dds032
Leegwater, P. A., Boor, P. K., Yuan, B. Q., van der Steen, J., Visser, A., Könst, A. A., et al. (2002). Identification of novel mutations in MLC1 responsible for megalencephalic leukoencephalopathy with subcortical cysts. Hum. Genet. 110 (3), 279–283. doi:10.1007/s00439-002-0682-x
Leegwater, P. A., Yuan, B. Q., van der Steen, J., Mulders, J., Konst, A. A., Boor, P. K., et al. (2001). Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am. J. Hum. Genet. 68 (4), 831–838. doi:10.1086/319519
Lelieveld, S. H., Reijnders, M. R. F., Pfundt, R., Yntema, H. G., Kamsteeg, E. J., De Vries, P., et al. (2016). Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 19, 1194–1196. doi:10.1038/nn.4352
Li, J., Wang, L., Guo, H., Shi, L., Zhang, K., Tang, M., et al. (2017). Targeted sequencing and functional analysis reveal brain-size-related genes and their networks in autism spectrum disorders. Mol. Psychiatry 22, 1282–1290. doi:10.1038/mp.2017.140
Liu, Y., Wang, X. R., Jiang, Y. H., Li, T., Ling, S., Wang, H. Y., et al. (2023). Interactions between the astrocytic volume-regulated anion channel and aquaporin 4 in hyposmotic regulation of vasopressin neuronal activity in the supraoptic nucleus. Cells 12 (13), 1723. doi:10.3390/cells12131723
Lopez-Hernandez, T., Ridder, M. C., Montolio, M., Capdevila-Nortes, X., Polder, E., Sirisi, S., et al. (2011a). Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am. J. Hum. Genet. 88 (4), 422–432. doi:10.1016/j.ajhg.2011.02.009
Lopez-Hernandez, T., Sirisi, S., Capdevila-Nortes, X., Montolio, M., Fernandez-Duenas, V., Scheper, G. C., et al. (2011b). Molecular mechanisms of MLC1 and GLIALCAM mutations in megalencephalic leukoencephalopathy with subcortical cysts. Hum. Mol. Genet. 20 (16), 3266–3277. doi:10.1093/hmg/ddr238
Ma, T., Yang, B., Gillespie, A., Carlson, E. J., Epstein, C. J., and Verkman, A. S. (1997). Generation and phenotype of a transgenic knockout mouse lacking the mercurial-insensitive water channel aquaporin-4. J. Clin. Invest. 100 (5), 957–962. doi:10.1172/JCI231
Mahmoud, I. G., Mahmoud, M., Refaat, M., Girgis, M., Waked, N., El Badawy, A., et al. (2014). Clinical, neuroimaging, and genetic characteristics of megalencephalic leukoencephalopathy with subcortical cysts in Egyptian patients. Pediatr. Neurol. 50 (2), 140–148. doi:10.1016/j.pediatrneurol.2013.10.008
Mancini, C., Vaula, G., Scalzitti, L., Cavalieri, S., Bertini, E., Aiello, C., et al. (2012). Megalencephalic leukoencephalopathy with subcortical cysts type 1 (MLC1) due to a homozygous deep intronic splicing mutation (c.895-226T>G) abrogated in vitro using an antisense morpholino oligonucleotide. Neurogenetics 13 (3), 205–214. doi:10.1007/s10048-012-0331-z
Masuda, T., Ueda, M., Ueyama, H., Shimada, S., Ishizaki, M., Imamura, S., et al. (2015). Megalencephalic leukoencephalopathy with subcortical cysts caused by compound heterozygous mutations in MLC1, in patients with and without subcortical cysts in the brain. J. Neurol. Sci. 351 (1-2), 211–213. doi:10.1016/j.jns.2015.03.010
Mattina, T., Perrotta, C. S., and Grossfeld, P. (2009). Jacobsen syndrome. Orphanet J. Rare Dis. 4, 9. doi:10.1186/1750-1172-4-9
Mayayo-Vallverdú, C., Ferigle, L., Vecino-Pérez, M., Lara, J., Nunes, V., and Estévez, R. (2023). Characterization of an MLC patient carrying two MLC1 variants showing radiological improvement. Brain Disord. 11, 100079. doi:10.1016/j.dscb.2023.100079
McQuillin, A., Kalsi, G., Moorey, H., Lamb, G., Mayet, S., Quested, D., et al. (2002). A novel polymorphism in exon 11 of the WKL1 gene, shows no association with schizophrenia. Eur. J. Hum. Genet. 10 (8), 491–494. doi:10.1038/sj.ejhg.5200837
Meyer, J., Huberth, A., Ortega, G., Syagailo, Y. V., Jatzke, S., Mossner, R., et al. (2001). A missense mutation in a novel gene encoding a putative cation channel is associated with catatonic schizophrenia in a large pedigree. Mol. Psychiatry 6 (3), 302–306. doi:10.1038/sj.mp.4000869
Miles, L., DeGrauw, T. J., Dinopoulos, A., Cecil, K. M., van der Knaap, M. S., and Bove, K. E. (2009). Megalencephalic leukoencephalopathy with subcortical cysts: a third confirmed case with literature review. Pediatr. Dev. Pathol. 12 (3), 180–186. doi:10.2350/08-06-0481.1
Moh, M. C., Zhang, T., Lee, L. H., and Shen, S. (2008). Expression of hepaCAM is downregulated in cancers and induces senescence-like growth arrest via a p53/p21-dependent pathway in human breast cancer cells. Carcinogenesis 29 (12), 2298–2305. doi:10.1093/carcin/bgn226
Montagna, G., Teijido, O., Eymard-Pierre, E., Muraki, K., Cohen, B., Loizzo, A., et al. (2006). Vacuolating megalencephalic leukoencephalopathy with subcortical cysts: functional studies of novel variants in MLC1. Hum. Mutat. 27 (3), 292. doi:10.1002/humu.9407
Morita, H., Imamura, A., Matsuo, N., Tatebayashi, K., Omoya, K., Takahashi, Y., et al. (2006). MR imaging and 1H-MR spectroscopy of a case of van der Knaap disease. Brain Dev. 28 (7), 466–469. doi:10.1016/j.braindev.2005.12.006
Nagelhus, E. A., Mathiisen, T. M., and Ottersen, O. P. (2004). Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience 129 (4), 905–913. doi:10.1016/j.neuroscience.2004.08.053
Nagelhus, E. A., and Ottersen, O. P. (2013). Physiological roles of aquaporin-4 in brain. Physiol. Rev. 93 (4), 1543–1562. doi:10.1152/physrev.00011.2013
Neely, J. D., Christensen, B. M., Nielsen, S., and Agre, P. (1999). Heterotetrameric composition of aquaporin-4 water channels. Biochemistry 38 (34), 11156–11163. doi:10.1021/bi990941s
Ono, J., Hasegawa, T., Sugama, S., Sagehashi, N., Hase, Y., Oku, K., et al. (1996). Partial deletion of the long arm of chromosome 11: ten Japanese children. Clin. Genet. 50 (6), 474–478. doi:10.1111/j.1399-0004.1996.tb02715.x
Passchier, E. M. J., Kerst, S., Brouwers, E., Hamilton, E. M. C., Bisseling, Q., Bugiani, M., et al. (2023). Aquaporin-4 and GPRC5B: old and new players in controlling brain oedema. Brain 146, 3444–3454. doi:10.1093/brain/awad146
Patel, H., Kumar, A., Raymond, G., and Mainali, G. (2019). Teaching NeuroImages: a rare case of Jacobsen syndrome with global diffuse hypomyelination of brain. Neurology 92 (14), e1665–e1666. doi:10.1212/WNL.0000000000007234
Patrono, C., Di Giacinto, G., Eymard-Pierre, E., Santorelli, F. M., Rodriguez, D., De Stefano, N., et al. (2003). Genetic heterogeneity of megalencephalic leukoencephalopathy and subcortical cysts. Neurology 61 (4), 534–537. doi:10.1212/01.wnl.0000076184.21183.ca
Perez-Rius, C., Folgueira, M., Elorza-Vidal, X., Alia, A., Hoegg-Beiler, M. B., Eeza, M. N. H., et al. (2019). Comparison of zebrafish and mice knockouts for Megalencephalic Leukoencephalopathy proteins indicates that GlialCAM/MLC1 forms a functional unit. Orphanet J. Rare Dis. 14 (1), 268. doi:10.1186/s13023-019-1248-5
Qiu, Z., Dubin, A. E., Mathur, J., Tu, B., Reddy, K., Miraglia, L. J., et al. (2014). SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell. 157 (2), 447–458. doi:10.1016/j.cell.2014.03.024
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Ridder, M. C., Boor, I., Lodder, J. C., Postma, N. L., Capdevila-Nortes, X., Duarri, A., et al. (2011). Megalencephalic leucoencephalopathy with cysts: defect in chloride currents and cell volume regulation. Brain 134 (Pt 11), 3342–3354. doi:10.1093/brain/awr255
Riel-Romero, R. M., Smith, C. D., and Pettigrew, A. L. (2005). Megalencephalic leukoencephalopathy with subcortical cysts in two siblings owing to two novel mutations: case reports and review of the literature. J. Child. Neurol. 20 (3), 230–234. doi:10.1177/088307380502000301
Rossi, F., Battaglini, M., Stromillo, M. L., Giorgio, A., Federico, A., and De Stefano, N. (2014). Twelve-year monitoring of a patient with megalencephalic leukoencephalopathy with subcortical cysts. Neurol. Sci. 35 (8), 1249–1253. doi:10.1007/s10072-014-1691-y
Rubie, C., Lichtner, P., Gartner, J., Siekiera, M., Uziel, G., Kohlmann, B., et al. (2003). Sequence diversity of KIAA0027/MLC1: are megalencephalic leukoencephalopathy and schizophrenia allelic disorders? Hum. Mutat. 21 (1), 45–52. doi:10.1002/humu.10145
Saijo, H., Nakayama, H., Ezoe, T., Araki, K., Sone, S., Hamaguchi, H., et al. (2003). A case of megalencephalic leukoencephalopathy with subcortical cysts (van der Knaap disease): molecular genetic study. Brain Dev. 25 (5), 362–366. doi:10.1016/s0387-7604(03)00006-8
Saini, L., Chakrabarty, B., Kumar, A., and Gulati, S. (2015). A mutation-positive child with megalencephalic leukoencephalopathy with subcortical cysts: classical imaging findings. Pediatr. Neurol. 53 (6), 547–548. doi:10.1016/j.pediatrneurol.2015.08.008
Sanchez, A., Garcia-Lareu, B., Puig, M., Prat, E., Ruberte, J., Chillon, M., et al. (2020). Cerebellar astrocyte transduction as gene therapy for megalencephalic leukoencephalopathy. Neurotherapeutics 17 (4), 2041–2053. doi:10.1007/s13311-020-00865-y
Sano, T., Kohyama-Koganeya, A., Kinoshita, M. O., Tatsukawa, T., Shimizu, C., Oshima, E., et al. (2018). Loss of GPRC5B impairs synapse formation of Purkinje cells with cerebellar nuclear neurons and disrupts cerebellar synaptic plasticity and motor learning. Neurosci. Res. 136, 33–47. doi:10.1016/j.neures.2018.02.006
Selch, S., Strobel, A., Haderlein, J., Meyer, J., Jacob, C. P., Schmitt, A., et al. (2007). MLC1 polymorphisms are specifically associated with periodic catatonia, a subgroup of chronic schizophrenia. Biol. Psychiatry 61 (10), 1211–1214. doi:10.1016/j.biopsych.2006.08.030
Shariati, G., Hamid, M., Saberi, A., Andashti, B., and Galehdari, H. (2015). Molecular prenatal diagnosis of megalencephalic leukoencephalopathy with subcortical cysts in a child from southwest of Iran. Clin. Case Rep. 3 (2), 114–117. doi:10.1002/ccr3.168
Shi, Z., Yan, H. F., Cao, B. B., Guo, M. M., Xie, H., Gao, K., et al. (2019). Identification in Chinese patients with GLIALCAM mutations of megalencephalic leukoencephalopathy with subcortical cysts and brain pathological study on Glialcam knock-in mouse models. World J. Pediatr. 15 (5), 454–464. doi:10.1007/s12519-019-00284-w
Shimada, S., Shimojima, K., Masuda, T., Nakayama, Y., Kohji, T., Tsukamoto, H., et al. (2014). MLC1 mutations in Japanese patients with megalencephalic leukoencephalopathy with subcortical cysts. Hum. Genome Var. 1, 14019. doi:10.1038/hgv.2014.19
Shukla, P., Balakrishnan, P., Agarwal, N., Ghosh, M., Kabra, M., Sharma, R., et al. (2008). Prenatal diagnosis of megalencephalic leukodystrophy. Prenat. Diagn 28 (4), 357–359. doi:10.1002/pd.1931
Shukla, P., Gupta, N., Ghosh, M., Vasisht, S., Gulati, S., Balakrishnan, P., et al. (2011). Molecular genetic studies in Indian patients with megalencephalic leukoencephalopathy. Pediatr. Neurol. 44 (6), 450–458. doi:10.1016/j.pediatrneurol.2011.01.003
Singhal, B. S., Gorospe, J. R., and Naidu, S. (2003). Megalencephalic leukoencephalopathy with subcortical cysts. J. Child. Neurol. 18 (9), 646–652. doi:10.1177/08830738030180091201
Singhal, B. S., Gursahani, R. D., Udani, V. P., and Biniwale, A. A. (1996). Megalencephalic leukodystrophy in an Asian Indian ethnic group. Pediatr. Neurol. 14 (4), 291–296. doi:10.1016/0887-8994(96)00048-3
Sirisi, S., Folgueira, M., Lopez-Hernandez, T., Minieri, L., Perez-Rius, C., Gaitan-Penas, H., et al. (2014). Megalencephalic leukoencephalopathy with subcortical cysts protein 1 regulates glial surface localization of GLIALCAM from fish to humans. Hum. Mol. Genet. 23 (19), 5069–5086. doi:10.1093/hmg/ddu231
Soni, A., Amisten, S., Rorsman, P., and Salehi, A. (2013). GPRC5B a putative glutamate-receptor candidate is negative modulator of insulin secretion. Biochem. Biophys. Res. Commun. 441 (3), 643–648. doi:10.1016/j.bbrc.2013.10.099
Soysal, Z., Okur, M., Eroz, R., Gun, E., Kocabay, K., and Besir, F. H. (2015). Megalencephalic leukoencephalopathy with subcortical cysts with homozygous mutation (C.448delc, P.Leu150 ser Fsx11) on exon 6 of Mlc1 gene. Genet. Couns. 26 (2), 233–236.
Spijker, S., Van Zanten, J. S., De Jong, S., Penninx, BWJH, Van Dyck, R., Zitman, F. G., et al. (2010). Stimulated gene expression profiles as a blood marker of major depressive disorder. Biol. Psychiatry 68, 179–186. doi:10.1016/j.biopsych.2010.03.017
Sugio, S., Tohyama, K., Oku, S., Fujiyoshi, K., Yoshimura, T., Hikishima, K., et al. (2017). Astrocyte-mediated infantile-onset leukoencephalopathy mouse model. Glia 65 (1), 150–168. doi:10.1002/glia.23084
Teijido, O., Martinez, A., Pusch, M., Zorzano, A., Soriano, E., Del Rio, J. A., et al. (2004). Localization and functional analyses of the MLC1 protein involved in megalencephalic leukoencephalopathy with subcortical cysts. Hum. Mol. Genet. 13 (21), 2581–2594. doi:10.1093/hmg/ddh291
Thalmeier, A., Dickmann, M., Giegling, I., Schneider, B., Hartmann, A. M., Maurer, K., et al. (2008). Gene expression profiling of post-mortem orbitofrontal cortex in violent suicide victims. Int. J. Neuropsychopharmacol. 11, 217–228. doi:10.1017/S1461145707007894
Tinsa, F., Farid, O., Douira, W., Rodriguez, D., Burglen, L., Boussetta, K., et al. (2009). Megalencephalic leukoencephalopathy with subcortical cysts in a Tunisian boy. J. Child. Neurol. 24 (1), 87–89. doi:10.1177/0883073808324021
Tsujino, S., Kanazawa, N., Yoneyama, H., Shimono, M., Kawakami, A., Hatanaka, Y., et al. (2003). A common mutation and a novel mutation in Japanese patients with van der Knaap disease. J. Hum. Genet. 48 (12), 605–608. doi:10.1007/s10038-003-0085-4
Turkyilmaz, A., Unver, O., Ekinci, G., and Turkdogan, D. (2019). A novel splice-site mutation on the MLC1 gene leading to exon 9 skipping and megalencephalic leukoencephalopathy with subcortical cysts in a Turkish patient. Balk. J. Med. Genet. 22 (2), 89–92. doi:10.2478/bjmg-2019-0019
Ulv Larsen, S. M., Landolt, H. P., Berger, W., Nedergaard, M., Knudsen, G. M., and Holst, S. C. (2020). Haplotype of the astrocytic water channel AQP4 is associated with slow wave energy regulation in human NREM sleep. PLoS Biol. 18 (5), e3000623. doi:10.1371/journal.pbio.3000623
van der Knaap, M. S., Abbink, T. E. M., and Min, R. (2018). “Megalencephalic leukoencephalopathy with subcortical cysts,” in GeneReviews((R)). Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephenset al. (Seattle (WA).
van der Knaap, M. S., Barth, P. G., Stroink, H., van, N. O., Arts, W. F., Hoogenraad, F., et al. (1995a). Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann. Neurol. 37 (3), 324–334. doi:10.1002/ana.410370308
van der Knaap, M. S., Barth, P. G., Vrensen, G. F., and Valk, J. (1996). Histopathology of an infantile-onset spongiform leukoencephalopathy with a discrepantly mild clinical course. Acta Neuropathol. 92 (2), 206–212. doi:10.1007/s004010050510
van der Knaap, M. S., Boor, I., and Estevez, R. (2012). Megalencephalic leukoencephalopathy with subcortical cysts: chronic white matter oedema due to a defect in brain ion and water homoeostasis. Lancet Neurol. 11 (11), 973–985. doi:10.1016/S1474-4422(12)70192-8
van der Knaap, M. S., Lai, V., Kohler, W., Salih, M. A., Fonseca, M. J., Benke, T. A., et al. (2010). Megalencephalic leukoencephalopathy with cysts without MLC1 defect. Ann. Neurol. 67 (6), 834–837. doi:10.1002/ana.21980
van der Knaap, M. S., Valk, J., Barth, P. G., Smit, L. M., van Engelen, B. G., and Tortori, D. P. (1995b). Leukoencephalopathy with swelling in children and adolescents: MRI patterns and differential diagnosis. Neuroradiology 37 (8), 679–686. doi:10.1007/BF00593394
Vellarikkal, S. K., Jayarajan, R., Verma, A., Ravi, R., Senthilvel, V., Kumar, A., et al. (2018). A founder mutation MLC1 c.736delA associated with megalencephalic leukoencephalopathy with subcortical cysts-1 in north Indian kindred. Clin. Genet. 94 (2), 271–273. doi:10.1111/cge.13251
Verkman, A. S., Ratelade, J., Rossi, A., Zhang, H., and Tradtrantip, L. (2011). Aquaporin-4: orthogonal array assembly, CNS functions, and role in neuromyelitis optica. Acta Pharmacol. Sin. 32 (6), 702–710. doi:10.1038/aps.2011.27
Vindedal, G. F., Thoren, A. E., Jensen, V., Klungland, A., Zhang, Y., Holtzman, M. J., et al. (2016). Removal of aquaporin-4 from glial and ependymal membranes causes brain water accumulation. Mol. Cell. Neurosci. 77, 47–52. doi:10.1016/j.mcn.2016.10.004
Voss, F. K., Ullrich, F., Munch, J., Lazarow, K., Lutter, D., Mah, N., et al. (2014). Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344 (6184), 634–638. doi:10.1126/science.1252826
Wang, J., Shang, J., Wu, Y., Gu, Q., Xiong, H., Ding, C., et al. (2011). Identification of novel MLC1 mutations in Chinese patients with megalencephalic leukoencephalopathy with subcortical cysts (MLC). J. Hum. Genet. 56 (2), 138–142. doi:10.1038/jhg.2010.146
Wardinsky, T. D., Weinberger, E., Pagon, R. A., Clarren, S. K., and Thuline, H. C. (1990). Partial deletion of the long arm of chromosome 11 [del(11)(q23.3----qter)] with abnormal white matter. Am. J. Med. Genet. 35 (1), 60–63. doi:10.1002/ajmg.1320350111
Wolf, N. I., and van der Knaap, M. S. (2020). Reader response: teaching NeuroImages: a rare case of Jacobsen syndrome with global diffuse hypomyelination of brain. Neurology 94 (10), 458. doi:10.1212/WNL.0000000000009070
Wu, M., Moh, M. C., and Schwarz, H. (2016). HepaCAM associates with connexin 43 and enhances its localization in cellular junctions. Sci. Rep. 6, 36218. doi:10.1038/srep36218
Xu, H., Isenmann, S., Lopez-Hernandez, T., Estevez, R., Lukacs, G. L., and Apaja, P. M. (2021). Control of membrane protein homeostasis by a chaperone-like glial cell adhesion molecule at multiple subcellular locations. Sci. Rep. 11 (1), 18435. doi:10.1038/s41598-021-97777-4
Yalcinkaya, C., Yuksel, A., Comu, S., Kilic, G., Cokar, O., and Dervent, A. (2003). Epilepsy in vacuolating megalencephalic leukoencephalopathy with subcortical cysts. Seizure 12 (6), 388–396. doi:10.1016/s1059-1311(02)00350-3
Yamamoto, T., Shimada, S., Shimojima, K., Sangu, N., Ninomiya, S., and Kubota, M. (2015). Leukoencephalopathy associated with 11q24 deletion involving the gene encoding hepatic and glial cell adhesion molecule in two patients. Eur. J. Med. Genet. 58 (9), 492–496. doi:10.1016/j.ejmg.2015.06.008
Yis, U., Scheper, G. C., Uran, N., Unalp, A., Cakmakci, H., Hiz-Kurul, S., et al. (2010). Two cases with megalencephalic leukoencephalopathy with subcortical cysts and MLC1 mutations in the Turkish population. Turk J. Pediatr. 52 (2), 179–183.
Yuzbasioglu, A., Topcu, M., Cetin Kocaefe, Y., and Ozguc, M. (2011). Novel mutations of the MLC1 gene in Turkish patients. Eur. J. Med. Genet. 54 (3), 281–283. doi:10.1016/j.ejmg.2010.11.014
Zambrano, S., Moller-Hackbarth, K., Li, X., Rodriguez, P. Q., Charrin, E., Schwarz, A., et al. (2019). GPRC5b modulates inflammatory response in glomerular diseases via NF-κB pathway. J. Am. Soc. Nephrol. 30 (9), 1573–1586. doi:10.1681/ASN.2019010089
Zhang, Q. L., Luo, C. L., Wu, X. H., Wang, C. Y., Xu, X., Zhang, Y. Y., et al. (2011). HepaCAM induces G1 phase arrest and promotes c-Myc degradation in human renal cell carcinoma. J. Cell. Biochem. 112 (10), 2910–2919. doi:10.1002/jcb.23207
Zhang, Y., Sloan, S. A., Clarke, L. E., Caneda, C., Plaza, C. A., Blumenthal, P. D., et al. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89 (1), 37–53. doi:10.1016/j.neuron.2015.11.013
Keywords: megalencephalic leukoencephalopathy with subcortical cysts, MLC1, GlialCAM, AQP4, GPRC5B, leukodystrophy, brain edema
Citation: Passchier EMJ, Bisseling Q, Helman G, van Spaendonk RML, Simons C, Olsthoorn RCL, van der Veen H, Abbink TEM, van der Knaap MS and Min R (2024) Megalencephalic leukoencephalopathy with subcortical cysts: a variant update and review of the literature. Front. Genet. 15:1352947. doi: 10.3389/fgene.2024.1352947
Received: 09 December 2023; Accepted: 29 January 2024;
Published: 29 February 2024.
Edited by:
Raúl Estévez, University of Barcelona, SpainReviewed by:
Saumel Ahmadi, Washington University in St. Louis, United StatesJanina Gburek-Augustat, University Hospital Leipzig, Germany
Tania López-Hernández, University of Barcelona, Spain
Copyright © 2024 Passchier, Bisseling, Helman, van Spaendonk, Simons, Olsthoorn, van der Veen, Abbink, van der Knaap and Min. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marjo S. van der Knaap, ms.vanderknaap@amsterdamumc.nl; Rogier Min, r.min@amsterdamumc.nl
†These authors share senior authorship