 Haemi Choi
Haemi Choi Jeong-A. Kim
Jeong-A. Kim Kyung-Ok Cho
Kyung-Ok Cho Hyun Jung Kim
Hyun Jung Kim Min-Hyeon Park
Min-Hyeon Park- 1Department of Psychiatry, Eunpyeong St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Republic of Korea
- 2Department of Pharmacology, College of Medicine, The Catholic University of Korea, Seoul, Republic of Korea
- 3Department of Biomedicine & Health Sciences, The Catholic University of Korea, Seoul, Republic of Korea
- 4Catholic Neuroscience Institute, The Catholic University of Korea, Seoul, Republic of Korea
- 5Institute for Aging and Metabolic Diseases, The Catholic University of Korea, Seoul, Republic of Korea
- 6Department of Psychiatry, Harvard Medical School, Boston, MA, United States
- 7Division of Psychotic Disorders, McLean Hospital, Belmont, MA, United States
Multiple genome sequencing studies have identified genetic abnormalities as major causes of severe intellectual disability (ID). However, many children affected by mild ID and borderline intellectual functioning (BIF) lack a genetic diagnosis because known causative ID genetic mutations have not been identified or the role of genetic variants in mild cases is less understood. Genetic variant testing in mild cases is necessary to provide information on prognosis and risk of occurrence. In this study, we report two sibling patients who were 5 years 9 months old and 3 years 3 months old and presented to the hospital due to developmental delay. Clinical assessment and chromosomal microarray analysis were performed. The patients were diagnosed with mild intellectual disability (ID) and borderline intellectual functioning (BIF). Genetic analysis identified a loss of 12p11.22, including the OVCH1-AS1, OVCH1, and TMTC1 genes, which was the only variant that occurred in both sisters. Identical variants were found in their father with probable BIF. Neither patient presented any brain structural abnormalities or dysmorphism, and no exogenous factors or parenting problems were reported. Thus, loss of 12p11.22 may be associated with our patients’ cognitive impairment. The OVCH1, OVCH1-AS1 and TMTC1 variants identified in this study are the most likely disease-causing genes in the sisters. Our findings may expand as yet limited knowledge on mild ID and BIF causative variants, which would further support the diagnosis even if the severity is mild.
1 Introduction
Intellectual disability (ID) is characterized by significant impairments in both intellectual and adaptive functioning, which refers to the ability to learn, reason, and solve problems, and functioning in conceptual, social, and practical domains (American Psychiatric Association, 2013). According to intelligence quotient (IQ) scores, severity is classified into mild (IQ 50-69), moderate (IQ 35-49), severe (IQ 20-34) and profound (IQ 0-19). Between 1% and 3% of the global population is affected by this type of developmental disorder (Leonard and Wen, 2002; Maulik et al., 2011); of all cases, approximately 85% are classified as having mild severity (Bhasin et al., 2006). Furthermore, it commonly affects 3.2% of children and adolescents (Olusanya et al., 2020), and 30%–50% of children and adolescents have a relative risk of comorbid mental disorder associated with ID (Einfeld et al., 2011).
Individuals with an IQ of 70–85 (between ID and normal intellectual functioning) are classified as borderline intellectual functioning (BIF) (Wieland and Zitman, 2016). However, children with BIF face difficulties in school accomplishments and social participation due to limitations in learning, social skills, and emotional competencies (Baglio et al., 2016; Contena and Taddei, 2017; Predescu et al., 2020). Therefore, they run a significant risk of dropping out of school and have a higher chance of having psychiatric problems in adulthood (Douma et al., 2007; Fernell and Ek, 2010; Gigi et al., 2014; Hassiotis et al., 2019). BIF is a prevalent condition that affects approximately 12%–14% of the population (Fernell and Gillberg, 2020). Nevertheless, children with mild ID and BIF may not be recognized until they are in school and have difficulty with academics.
To date, the underlying causes of these conditions are still unclear, although the prevailing hypothesis for the etiology of ID is complex interactions between multiple genetic and environmental factors that interfere with typical brain development; for example, different kinds of variations include mutations, deletions, and copy number variants, and exogenous factors include maternal infections, delivery complications, and exposure to drugs or alcohol while pregnant (Faustman et al., 2000; van Loo and Martens, 2007; De Felice et al., 2015).
One of the strategies utilized to unravel the bases of ID is chromosomal microarrays. Chromosomal microarray analysis (CMA) is a molecular genetic tool that can identify gains and losses in the number of microscopic DNA replications that would otherwise be detected by chromosomal karyotype analysis (Batzir et al., 2015). Changes in the number of microscopic DNA replications, copy number variants (CNVs), are linked to human diseases and health (Harel and Lupski, 2018; Hu et al., 2018). CNVs are classified into common and rare CNVs according to the population frequency. A growing body of evidence suggests that rare CNVs significantly contribute to the genetic basis of ID (Di Gregorio et al., 2017; Kessi et al., 2018). Identifying disease-associated CNVs can help further elucidate the pathophysiological mechanisms of ID. Hence, CMA has been widely utilized as the initial diagnostic process for patients with ID, as it considerably improves the diagnostic yield for children affected with ID (Manning et al., 2010; Miller et al., 2010; Kearney et al., 2011; Michelson et al., 2011).
Multiple genome sequencing studies have identified that the main causes of severe ID are genetic abnormalities, with reported diagnostic results ranging from 13% to 42% in predominantly severe ID (Deciphering Developmental Disorders, 2017). For mild ID and BIF, however, the role of genetic variants is less studied but is predicted to be less important (Vissers et al., 2016). A population-based study reported that chromosomal abnormalities account for approximately 40% of severe ID and 10% of mild ID cases (Karam et al., 2015). In another population-based study, the likely causative variants in known ID genes were significantly more common in more severe ID cases than in mild ID cases (Kurki et al., 2019). The authors suggested that either the etiology of mild ID is more complex or the genetic variants that cause mild ID differ slightly from those that cause more severe ID (Kurki et al., 2019). Many children affected by mild ID or BIF lack a genetic diagnosis because known causative ID genes were not identified or the role of genetic variants in mild cases is less understood. Therefore, CNV testing in mild ID and BIF is necessary to provide information on prognosis and risk of occurrence as well as to prevent unnecessary intrusive testing.
Here, we describe two sisters with mild ID and BIF. Loss of the 12p11.22 region, including the OVCH1-AS1, OVCH1, and TMTC1 genes, was identified in both patients. Furthermore, identical CNVs were found in their father, who had probable BIF. This case highlights the importance of identifying the underlying genetic causes of mild ID and BIF.
2 Case description
2.1 Patient 1
Patient 1 is a 5-year-and 9-month-old female, who initially presented to the outpatient clinic of the Catholic University of Korea, Eunpyeong St. Mary’s Hospital at 4 years and 10 months, due to developmental delays. She was the first child born to healthy nonconsanguineous Korean parents (Figure 1). The mother’s prenatal history was unremarkable with no alcohol, smoking, or drug use during pregnancy. Patient 1 was delivered at 38 weeks of gestation via cesarean section, with a birth weight of 3.65 kg. A timeline of assessment and outcomes is outlined in Figure 2.
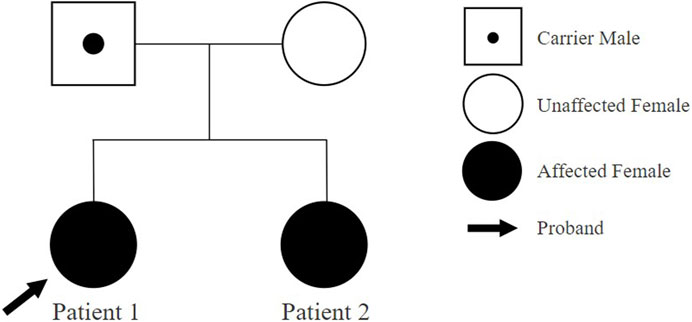
Figure 1. Family pedigree of patients.
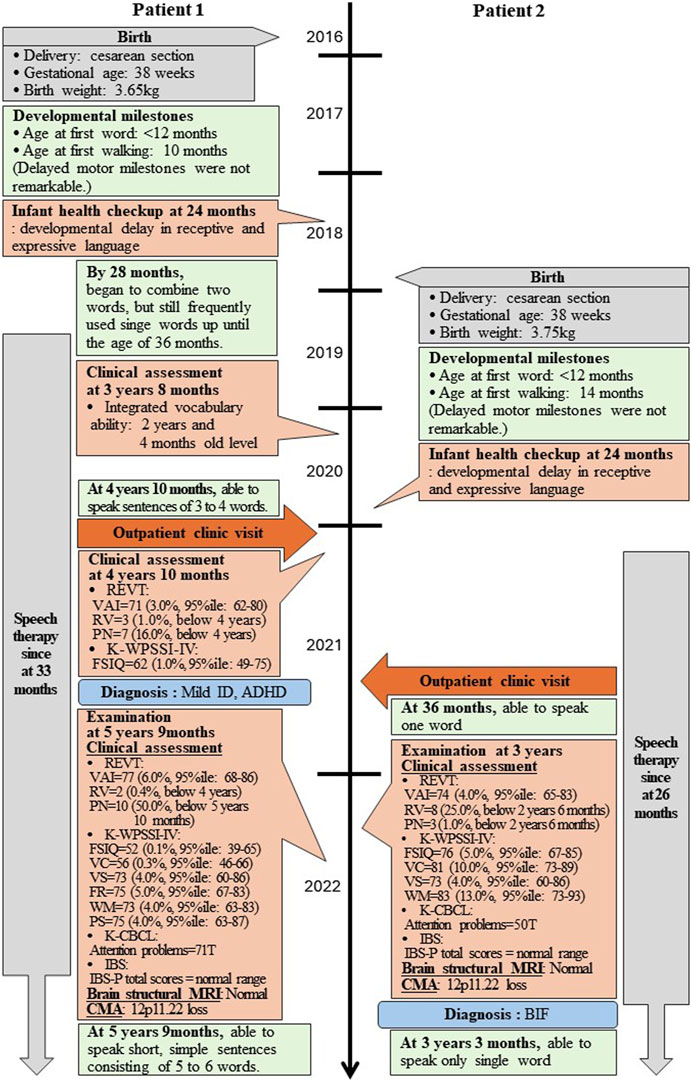
Figure 2. Timeline of two patients’ assessment and outcomes. REVT: receptive and expressive vocabulary test; VAI: vocabulary ability index; RV: receptive vocabulary; PN: picture naming; K-WPSSI-IV: Korean Wechsler preschool and primary scale of intelligence fourth edition; FSIQ: full scale intelligence quotient; VC: vocabulary comprehensive; VS: visual spatial; FR: fluid reasoning; WM: working memory; PS: processing speed; CBCL: child behavior checklist; IBS: interactive behavioral scale; MRI: magnetic resonance imaging; CMA: chromosomal microarray analysis; ID: intellectual disability; BIF: borderline intellectual functioning; ADHD: attention deficit hyperactivity disorder.
She achieved early language milestones, speaking her first word before 12 months and walking independently by 10 months, with no significant delays in motor development or seizure occurrences. However, her mother reported concerns about her language development compared to peers at 20 months. By the 24-month infant health checkup, pronounced developmental delays were noted in both receptive and expressive language; she began to form two-word phrases at 28 months but continued to use single words frequently until 36 months.
Intervention with speech therapy began at 33 months. At 3 years and 8 months, she was assessed to have an integrated vocabulary ability delayed by 1 year and 6 months. By 4 years and 10 months, symptoms of inattention and hyperactivity were observed, intensifying concerns regarding her developmental progression. A clinical psychological evaluation confirmed developmental delays in intelligence and language, with a diagnosis of mild ID characterized by a full-scale intelligence quotient (FSIQ) of 62. She also presented with attention deficit hyperactivity disorder (ADHD). At this time, her language abilities remained below the 4-year-old standard; she could form sentences of 3–4 words.
At 5 years and 9 months, she has undergone comprehensive investigation, including clinical assessment, magnetic resonance imaging (MRI), and CMA. The clinical results revealed scores of a FSIQ of 52, along with sub-scores of vocabulary comprehension (VC) at 56, visual spatial (VS) at 73, fluid reasoning (FR) at 75, working memory (WM) at 73, and processing speed (PS) at 75. Despite ongoing speech therapy initiated at 33 months, her receptive language skills still remained below 4-year-old level, while expressive skills had improved to the level of a 5-year-and 10-month-old. To date, she has been able to speak short, simple sentences consisting of 5–6 words.
The interactive behavioral scale (IBS), completed by her mother, indicated normal parental and child interactive behaviors. The family was described as having a close and supportive relationship. Clinically, she presented with no dysmorphic features, and brain MRI findings were age-appropriate.
Genetic investigations performed through CMA using an Affymetrix® Cytoscan® DX array (Affymetrix, USA) identified a 521,454 bp loss at 12p11.22 (chr12:29,554,804-30,076,258) including the OVCH1-AS1, OVCH1, and TMTC1 genes, designated as a variant of unknown significance (VUS) (Figure 3A). There were no other CNVs detected. Identical CNVs found in the patient 1 were also observed in her younger sister (Patient 2) and her father but not in her mother (Figures 3B,C).
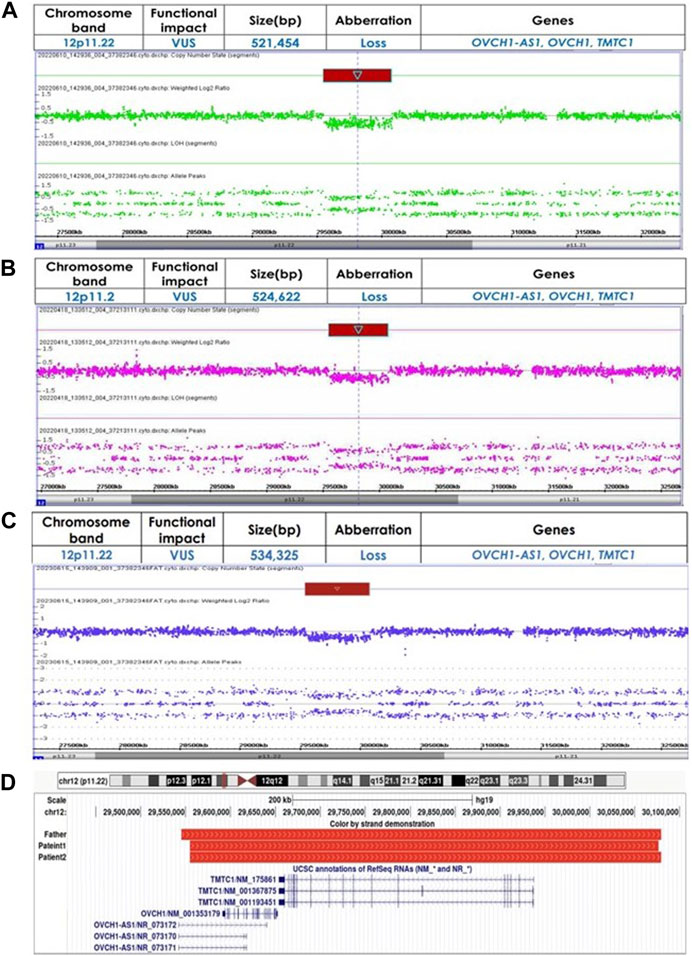
Figure 3. (A) Identified copy number variations using chromosomal microarray analysis in Patient 1 (chr12:29554804-30076258) (B) Identified copy number variations using chromosomal microarray analysis in Patient 2 (chr12:29554804-30079426) (C) Identified copy number variations using chromosomal microarray analysis in Father (chr12:29545101-30079426) (D) Chromosomal region 12p11.22 loss alignment, red bars represent CNVs identified in our patients and their father. Displayed at the bottom are the genes OVCH1-AS1, OVCH1, and TMTC1, as annotated in the UCSC Genome Browser [GRCh37/hg19 assembly].
2.2 Patient 2
Patient 2, a 3-year-and 3-month-old female, first visited the outpatient clinic of the Catholic University of Korea, Eunpyeong St. Mary’s Hospital at 3 years of age with developmental delays, alongside her older sister (Patient 1). She was born as the second child to healthy nonconsanguineous Korean parents (Figure 1). The mother denied drinking alcohol, smoking or taking drugs during pregnancy. Patient 2 was delivered at 38 weeks of gestation via cesarean section, weighing 3.75 kg. Her assessment timeline and outcomes are detailed in Figure 2.
She achieved early language milestones, speaking her first word before 12 months and walking independently by 14 months, without notable motor development delays or seizure occurrences. However, similar to her sister, she exhibited developmental delays in both receptive and expressive language skills from 20 months, which were confirmed at her 24-month infant check-up; notably, she did not begin to put two words together until 36 months.
Following an early intervention with speech therapy starting at 26 months, she was diagnosed with BIF at 3 years old, with scores of FSIQ = 76, VC = 81, VS = 73, and WM = 83. Unlike her sister, Patient 2 did not exhibit conspicuous signs of ADHD according to the child behavior checklist (CBCL). Despite ongoing speech therapy initiated at 26 months, her language skills, both receptive and expressive, appeared to be below the 30-month level. Moreover, her expressive skills were significantly lower than her receptive skills. To date, she has been still able to speak only single words.
Furthermore, the IBS results indicated that her parents’ parenting and interactive behaviors were normal. Her mother described that the family have a good relationship and are close to each other. Clinically, she presented with no dysmorphic features, and brain MRI findings were normal for her age.
Genetic investigations performed through CMA identified a 524,622bp loss at 12p11.22 (chr12:29,554,804-30,079,426) including the OVCH1-AS1, OVCH1, and TMTC1 genes, classified as a VUS (Figure 3B). There were no other CNVs in the genome. Identical CNVs found in the patient 2 were also observed in her older sister and her father (chr12:29,545,101-30,079,426, 534,325 bp loss) but not in her mother (Figures 3A,C). Figure 3D illustrates the overlapping regions of loss shared by the patients and their father.
3 Discussion
We report two individuals with 12p11.22 copy number loss involving the OVCH1-AS1, OVCH1, and TMTC1 genes. The clinical features of our patients were mild ID and BIF, but neither presented brain structural abnormalities or any dimorphism, such as facial abnormalities and short stature. Genetic analysis identified a 521 kb loss (chr12:29,554,804-30,076,258) and a 525 kb loss (chr12:29,554,804-30,079,426) at 12p11.22, which was the only variant in both sisters. Moreover, identical variants were found in their father who exhibited probable BIF. Thus, the 12p11.22 variants may be associated with our patients’ cognitive impairments.
Multiple studies have described that chromosome 12 deletions are uncommon but are linked to ID, developmental delay, growth retardation and dysmorphic features (James et al., 2005; Adam et al., 2010; Lynch et al., 2011; Labonne et al., 2016; Deng et al., 2021). Unlike previously documented CNVs, the specific abnormalities identified in our patients were not found in established databases such as the DGV, dbVar, Gene2Phenotype, or DisGeNet. However, in the DECIPHER database (Firth et al., 2009), four cases with 12p deletions, including 12p11.22 loss was reported. Notably, one case, which shared a significant deletion overlap with our patients (DECIPHER 501071, chr12:27,895,481-30,065,521), was reported to be responsible for short stature. Among these, three cases were deemed to be likely pathogenic; one patient (DECIPHER 392769, chr12:21,155,799-34,703,759) was reported to have hydrocephalus, ID, and dysmorphic features such as short stature, broad and short neck, and bulbous nose (Boilly-Dartigalongue et al., 1985); one patient (DECIPHER 395971, chr12:19,855,799-33,155,799) reported hydrocephalus, mild ID, delayed speech and language development, and dysmorphic features such as short stature, short nose, and small hand (Nagai et al., 1995); and one patient (DECIPHER 400822, chr12:14,655,799-33,155,799) reported hydrocephalus, dysarthria, and dysmorphic features such as short stature and prominent nose. All three cases presented with hydrocephalus and dysmorphic features. Among them, two cases had ID and speaking or language problems.
When comparing the phenotypes described in these earlier cases with those of our patients, there are overlapping clinical symptom, such as problems with cognitive and language functioning. However, in the present study, we did not detect any dysmorphic features or hydrocephalus in our patients. Moreover, psychiatry and clinician assessments suggested that our patients’ deficits in language development were highly likely to result from ID. In fact, a previous study confirmed that ID is associated with a higher risk of speech and language delay in children, showing that 71.3% of children with ID have speech and language delay and 93.9% of children with moderate ID have speech and language problems (Memisevic and Hadzic, 2013). Despite these similarities, the gross chromosomal structural aberrations reported in the DECIPHER cases, which only partially overlap with those of our patients, encompass additional genes. This complicates the direct correlation between these CNVs and the observed phenotypes, underscoring the critical need for further research to elucidate their functions and implications.
The 12p11.22 region likely includes the genomic loci of candidate genes for cognitive impairment. This loss covering 3 genes at 12p11.22 probably harbors the genes responsible for the ID phenotypes shared between our patients and the cases from DECIPHER. TMTC1 (transmembrane O-mannosyltransferase targeting cadherins) is one of four TMTC members, originally identified as an endoplasmic reticulum protein involved in calcium homeostasis. It has been reported to be involved in cognitive impairments related to schizophrenia (Mealer et al., 2020). Recently, TMTC1 has been described as one of the candidate genes for neurodevelopmental disorder, particularly ID (Ben-Mahmoud et al., 2023). The expression of TMTC1 mRNA is notably present in both adult and fetal brains, suggesting its relevance in these tissues (Ben-Mahmoud et al., 2023). Although TMTC1 expression in the brain is not uniformly high, it shows moderate protein expression in the cerebral cortex and is cell type enhanced in inhibitory and excitatory neurons (Human Protein Atlas, proteinatlas.org) (Sjostedt et al., 2020), highlighting its potential role in the pathogenesis of diseases affecting these brain regions. Additionally, changes TMTC1 expression, particularly in the striatum and cerebellar cortex during early life (Johnson et al., 2009), may contribute the intellectual disabilities observed (Balleine et al., 2007; Johnson et al., 2009), despite generally low levels in the cerebellar cortex and neocortex (Human Brain Transcriptome, hbatlas.org) (Johnson et al., 2009). OVCH1 (ovochymase), another gene in the 12p11.22 region, encodes oocyte extracellular polyproteins (Mino and Sawada, 2016), which were reported to be involved in early-onset myasthenia gravis (Varade et al., 2018) and familial Meniere’s disease (Skarp et al., 2022), but its function in other cells remains unknown. Given the limited expression of these genes, further research is necessary to establish a clear correlation between the genes and phenotypes, including studies on gene expression regulation and the resulting phenotypic changes.
Interestingly, no variants other than the 12p11.22 loss including OVCH1-AS1, OVCH1 and TMTC1 were found in our patients. There were no structural abnormalities in brain development according to MRI or reported exogenous factors that could affect neurodevelopment, including delivery complications, exposure to drugs or alcohol during pregnancy, and parenting problems.
In addition, their father carried identical variants in the same region encompassing the same gene loss. Although he had never been diagnosed with ID, he reported experiencing delays in his cognitive and language development. He was told as a young child that his language development lagged behind that of peers and that his capacity for comprehension was similarly slower. This suggests that he might have been unaware that he presented with BIF.
According to the previously described findings, we propose that the 12p11.22 loss may be responsible for our patients’ mild ID and BIF. Additionally, the OVCH1, OVCH1-AS1 and TMTC1 variants identified in this study are the most likely disease-causing genes in the sisters. However, no links between ID and the OVCH1 and OVCH1-AS1 genes were found in the literature; thus, TMTC1 seems to be a likely candidate gene for cognitive impairment. Hence, our findings can also be related to ID-associated variants, which would further support the diagnosis even if severity is mild.
For further study, since the father’s suspected BIF symptoms are based on self-report, an intelligence test would be helpful in clarifying his condition. Additionally, as he had struggled with BIF-related symptoms but is not currently experiencing severe difficulties, it would be helpful to follow up with the children to observe how their symptoms progress and what protective or risk factors they may have. In our study, Patient 2 was too young to complete the attention test, so there are limitations in clarifying the presence of ADHD symptoms in Patient 2. Although our study has some limitations, our findings may expand the limited knowledge on mild ID and BIF causative association in 12p11.22 loss and are highly helpful for identifying the genetic causes of ID even if symptoms are mild.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Institutional Review Board of Eunpyeong St. Mary’s Hospital, Seoul, South Korea (IRB No. PC23ZESI0148). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the patients and their parents for the publication for the publication of any potentially identifiable images or data included in this article.
Author contributions
HC: Investigation, Data curation, Formal Analysis, Visualization, Writing–original draft, Writing–review and editing, J-AK: Investigation, Data curation, Formal Analysis, Writing–original draft, K-OC: Methodology, Formal Analysis, Resources, Validation, Writing–review and editing, HK: Formal Analysis, Validation, Writing–review and editing, M-HP: Conceptualization, Formal Analysis, Resources, Funding acquisition, Project administration, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2020R1F1A1070581) and a grant from The Catholic University of Korea, Eunpyeong St. Mary’s Hospital, Research Institute of Medical Science in program year 2021 (EMBRF-2020-05).
Acknowledgments
We thank the family for their consent for publication of this study. We are grateful to Nicole de Leeuw and Jeroen Breckpot for clinical information. We also appreciate Julia Foreman at DECIPHER for assistance with case data.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adam, M. P., Mehta, A., Knight, L., Hall, D. E., and Rossi, M. R. (2010). A family with a 1.17 Mb deletion of 12q12: refining genotype-phenotype correlation. Am. J. Med. Genet. A 152A (9), 2394–2398. doi:10.1002/ajmg.a.33570
American Psychiatric Association (2013). Diagnostic and statistical manual of mental disorders, fifth edition (DSM-5). Washington, DC: American Psychiatric Association.
Baglio, G., Blasi, V., Sangiuliano Intra, F., Castelli, I., Massaro, D., Baglio, F., et al. (2016). Social competence in children with borderline intellectual functioning: delayed development of theory of mind across all complexity levels. Front. Psychol. 7, 1604. doi:10.3389/fpsyg.2016.01604
Balleine, B. W., Delgado, M. R., and Hikosaka, O. (2007). The role of the dorsal striatum in reward and decision-making. J. Neurosci. 27 (31), 8161–8165. doi:10.1523/JNEUROSCI.1554-07.2007
Batzir, N. A., Shohat, M., and Maya, I. (2015). Chromosomal microarray analysis (CMA) a clinical diagnostic tool in the prenatal and postnatal settings. Pediatr. Endocrinol. Rev. 13 (1), 448–454.
Ben-Mahmoud, A., Kishikawa, S., Gupta, V., Leach, N. T., Shen, Y., Moldovan, O., et al. (2023). A cryptic microdeletion del(12)(p11.21p11.23) within an unbalanced translocation t(7;12)(q21.13;q23.1) implicates new candidate loci for intellectual disability and Kallmann syndrome. Sci. Rep. 13 (1), 12984. doi:10.1038/s41598-023-40037-4
Bhasin, T. K., Brocksen, S., Avchen, R. N., and Van Naarden Braun, K. (2006). Prevalence of four developmental disabilities among children aged 8 years--Metropolitan Atlanta Developmental Disabilities Surveillance Program, 1996 and 2000. MMWR Surveill. Summ. 55 (1), 1–9.
Boilly-Dartigalongue, B., Riviere, D., Junien, C., Couturier, J., Toudic, L., Marie, F., et al. (1985). A new case of partial monosomy of chromosome 12,del(12)(p11.01 to p12.109) confirming the location of the gene for lactate dehydrogenase B. Ann. Genet. 28 (1), 55–57.
Contena, B., and Taddei, S. (2017). Psychological and cognitive aspects of borderline intellectual functioning. Eur. Psychol. 22, 159–166. doi:10.1027/1016-9040/a000293
Deciphering Developmental Disorders, S. (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542 (7642), 433–438. doi:10.1038/nature21062
De Felice, A., Ricceri, L., Venerosi, A., Chiarotti, F., and Calamandrei, G. (2015). Multifactorial origin of neurodevelopmental disorders: approaches to understanding complex etiologies. Toxics 3 (1), 89–129. doi:10.3390/toxics3010089
Deng, R., McCalman, M. T., Bossuyt, T. P., and Barakat, T. S. (2021). Case report: two new cases of chromosome 12q14 deletions and review of the literature. Front. Genet. 12, 716874. doi:10.3389/fgene.2021.716874
Di Gregorio, E., Riberi, E., Belligni, E. F., Biamino, E., Spielmann, M., Ala, U., et al. (2017). Copy number variants analysis in a cohort of isolated and syndromic developmental delay/intellectual disability reveals novel genomic disorders, position effects and candidate disease genes. Clin. Genet. 92 (4), 415–422. doi:10.1111/cge.13009
Douma, J. C., Dekker, M. C., de Ruiter, K. P., Tick, N. T., and Koot, H. M. (2007). Antisocial and delinquent behaviors in youths with mild or borderline disabilities. Am. J. Ment. Retard. 112 (3), 207–220. doi:10.1352/0895-8017(2007)112[207:AADBIY]2.0.CO;2
Einfeld, S. L., Ellis, L. A., and Emerson, E. (2011). Comorbidity of intellectual disability and mental disorder in children and adolescents: a systematic review. J. Intellect. Dev. Disabil. 36 (2), 137–143. doi:10.1080/13668250.2011.572548
Faustman, E. M., Silbernagel, S. M., Fenske, R. A., Burbacher, T. M., and Ponce, R. A. (2000). Mechanisms underlying Children’s susceptibility to environmental toxicants. Environ. Health Perspect. 108 (1), 13–21. doi:10.1289/ehp.00108s113
Fernell, E., and Ek, U. (2010). Borderline intellectual functioning in children and adolescents - insufficiently recognized difficulties. Acta Paediatr. 99 (5), 748–753. doi:10.1111/j.1651-2227.2010.01707.x
Fernell, E., and Gillberg, C. (2020). Borderline intellectual functioning. Handb. Clin. Neurol. 174, 77–81. doi:10.1016/B978-0-444-64148-9.00006-5
Firth, H. V., Richards, S. M., Bevan, A. P., Clayton, S., Corpas, M., Rajan, D., et al. (2009). DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 84 (4), 524–533. doi:10.1016/j.ajhg.2009.03.01
Gigi, K., Werbeloff, N., Goldberg, S., Portuguese, S., Reichenberg, A., Fruchter, E., et al. (2014). Borderline intellectual functioning is associated with poor social functioning, increased rates of psychiatric diagnosis and drug use--a cross sectional population based study. Eur. Neuropsychopharmacol. 24 (11), 1793–1797. doi:10.1016/j.euroneuro.2014.07.016
Harel, T., and Lupski, J. R. (2018). Genomic disorders 20 years on-mechanisms for clinical manifestations. Clin. Genet. 93 (3), 439–449. doi:10.1111/cge.13146
Hassiotis, A., Brown, E., Harris, J., Helm, D., Munir, K., Salvador-Carulla, L., et al. (2019). Association of borderline intellectual functioning and adverse childhood experience with adult psychiatric morbidity. Findings from a British birth cohort. BMC Psychiatry 19 (1), 387. doi:10.1186/s12888-019-2376-0
Hu, L., Yao, X., Huang, H., Guo, Z., Cheng, X., Xu, Y., et al. (2018). Clinical significance of germline copy number variation in susceptibility of human diseases. J. Genet. Genomics 45 (1), 3–12. doi:10.1016/j.jgg.2018.01.001
James, P. A., Oei, P., Ng, D., Kannu, P., and Aftimos, S. (2005). Another case of interstitial del (12) involving the proposed cardio-facio-cutaneous candidate region. Am. J. Med. Genet. Part A 136 (1), 12–16. doi:10.1002/ajmg.a.30693
Johnson, M. B., Kawasawa, Y. I., Mason, C. E., Krsnik, Z., Coppola, G., Bogdanovic, D., et al. (2009). Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62 (4), 494–509. doi:10.1016/j.neuron.2009.03.027
Karam, S. M., Riegel, M., Segal, S. L., Felix, T. M., Barros, A. J., Santos, I. S., et al. (2015). Genetic causes of intellectual disability in a birth cohort: a population-based study. Am. J. Med. Genet. A 167 (6), 1204–1214. doi:10.1002/ajmg.a.37011
Kearney, H. M., South, S. T., Wolff, D. J., Lamb, A., Hamosh, A., Rao, K. W., et al. (2011). American College of Medical Genetics recommendations for the design and performance expectations for clinical genomic copy number microarrays intended for use in the postnatal setting for detection of constitutional abnormalities. Genet. Med. 13 (7), 676–679. doi:10.1097/GIM.0b013e31822272ac
Kessi, M., Xiong, J., Wu, L., Yang, L., He, F., Chen, C., et al. (2018). Rare copy number variations and predictors in children with intellectual disability and epilepsy. Front. Neurol. 9, 947. doi:10.3389/fneur.2018.00947
Kurki, M. I., Saarentaus, E., Pietilainen, O., Gormley, P., Lal, D., Kerminen, S., et al. (2019). Contribution of rare and common variants to intellectual disability in a sub-isolate of Northern Finland. Nat. Commun. 10 (1), 410. doi:10.1038/s41467-018-08262-y
Labonne, J. D., Lee, K. H., Iwase, S., Kong, I. K., Diamond, M. P., Layman, L. C., et al. (2016). An atypical 12q24.31 microdeletion implicates six genes including a histone demethylase KDM2B and a histone methyltransferase SETD1B in syndromic intellectual disability. Hum. Genet. 135 (7), 757–771. doi:10.1007/s00439-016-1668-4
Leonard, H., and Wen, X. (2002). The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment. Retard. Dev. Disabil. Res. Rev. 8 (3), 117–134. doi:10.1002/mrdd.10031
Lynch, S. A., Foulds, N., Thuresson, A. C., Collins, A. L., Anneren, G., Hedberg, B. O., et al. (2011). The 12q14 microdeletion syndrome: six new cases confirming the role of HMGA2 in growth. Eur. J. Hum. Genet. 19 (5), 534–539. doi:10.1038/ejhg.2010.215
Manning, M., Hudgins, L., Professional, P., and Guidelines, C. (2010). Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet. Med. 12 (11), 742–745. doi:10.1097/GIM.0b013e3181f8baad
Maulik, P. K., Mascarenhas, M. N., Mathers, C. D., Dua, T., and Saxena, S. (2011). Prevalence of intellectual disability: a meta-analysis of population-based studies. Res. Dev. Disabil. 32 (2), 419–436. doi:10.1016/j.ridd.2010.12.018
Mealer, R. G., Williams, S. E., Daly, M. J., Scolnick, E. M., Cummings, R. D., and Smoller, J. W. (2020). Glycobiology and schizophrenia: a biological hypothesis emerging from genomic research. Mol. Psychiatry. 25 (12), 3129–3139. doi:10.1038/s41380-020-0753-1
Memisevic, H., and Hadzic, S. (2013). Speech and language disorders in children with intellectual disability in Bosnia and Herzegovina. Disability, CBR Inclus. Develop. 24 (2), 92–99.
Michelson, D. J., Shevell, M. I., Sherr, E. H., Moeschler, J. B., Gropman, A. L., and Ashwal, S. (2011). Evidence report: genetic and metabolic testing on children with global developmental delay: report of the quality standards subcommittee of the American academy of neurology and the practice committee of the child neurology society. Neurology 77 (17), 1629–1635. doi:10.1212/WNL.0b013e3182345896
Miller, D. T., Adam, M. P., Aradhya, S., Biesecker, L. G., Brothman, A. R., Carter, N. P., et al. (2010). Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 86 (5), 749–764. doi:10.1016/j.ajhg.2010.04.006
Mino, M., and Sawada, H. (2016). Follicle cell trypsin-like protease HrOvochymase: Its cDNA cloning, localization, and involvement in the late stage of oogenesis in the ascidian Halocynthia roretzi. Mol. Reprod. Dev. 83 (4), 347–358. doi:10.1002/mrd.22627
Nagai, T., Nishimura, G., Kato, R., Hasegawa, T., Ohashi, H., and Fukushima, Y. (1995). Del(12)(p11.21p12.2) associated with an asphyxiating thoracic dystrophy or chondroectodermal dysplasia-like syndrome. Am. J. Med. Genet. 55 (1), 16–18. doi:10.1002/ajmg.1320550106
Olusanya, B. O., Wright, S. M., Nair, M. K. C., Boo, N. Y., Halpern, R., Kuper, H., et al. (2020). Global burden of childhood epilepsy, intellectual disability, and sensory impairments. Pediatrics 146 (1), e20192623. doi:10.1542/peds.2019-2623
Predescu, E., Sipos, R., Costescu, C. A., Ciocan, A., and Rus, D. I. (2020). Executive functions and emotion regulation in attention-deficit/hyperactivity disorder and borderline intellectual disability. J. Clin. Med. 9 (4), 986. doi:10.3390/jcm9040986
Sjostedt, E., Zhong, W., Fagerberg, L., Karlsson, M., Mitsios, N., Adori, C., et al. (2020). An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367 (6482), eaay5947. doi:10.1126/science.aay5947
Skarp, S., Korvala, J., Kotimaki, J., Sorri, M., Mannikko, M., and Hietikko, E. (2022). New genetic variants in CYP2B6 and SLC6A support the role of oxidative stress in Familial Meniere’s disease. Genes (Basel). 13 (6). doi:10.3390/genes13060998
van Loo, K. M., and Martens, G. J. (2007). Genetic and environmental factors in complex neurodevelopmental disorders. Curr. Genomics 8 (7), 429–444. doi:10.2174/138920207783591717
Varade, J., Wang, N., Lim, C. K., Zhang, T., Zhang, Y., Liu, X., et al. (2018). Novel genetic loci associated HLA-B*08:01 positive myasthenia gravis. J. Autoimmun. 88, 43–49. doi:10.1016/j.jaut.2017.10.002
Vissers, L. E., Gilissen, C., and Veltman, J. A. (2016). Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 17 (1), 9–18. doi:10.1038/nrg3999
Keywords: cognitive impairment, neurodevelopmental disorder, 12p deletion, CNV, chromosomal microarray analysis
Citation: Choi H, Kim J-A, Cho K-O, Kim HJ and Park M-H (2024) Case Report: Intellectual disability and borderline intellectual functioning in two sisters with a 12p11.22 loss. Front. Genet. 15:1355823. doi: 10.3389/fgene.2024.1355823
Received: 14 December 2023; Accepted: 19 March 2024;
Published: 02 April 2024.
Edited by:
Lawrence Todd Reiter, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Rincic Martina, University of Zagreb, CroatiaMuzammil Ahmad Khan, Gomal University, Pakistan
Copyright © 2024 Choi, Kim, Cho, Kim and Park. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min-Hyeon Park, neominnie00@daum.net