


- 1 Laboratorio de Circuitos Corticales (Centro de Tecnología Biomédica), Universidad Politécnica de Madrid, Madrid, Spain
- 2 Departamento de Neurobiología Funcional y de Sistemas, Instituto Cajal (CSIC), Madrid, Spain
Neurofibrillary tangles (NFT) represent one of the main neuropathological features in the cerebral cortex associated with Alzheimer’s disease (AD). This neurofibrillary lesion involves the accumulation of abnormally hyperphosphorylated or abnormally phosphorylated microtubule-associated protein tau into paired helical filaments (PHF-tau) within neurons. We have used immunocytochemical techniques and confocal microscopy reconstructions to examine the distribution of PHF-tau-immunoreactive (ir) cells, and their perisomatic GABAergic and glutamatergic innervations in the hippocampal formation and adjacent cortex of AD patients. Furthermore, correlative light and electron microscopy was employed to examine these neurons and the perisomatic synapses. We observed two patterns of staining in PHF-tau-ir neurons, pattern I (without NFT) and pattern II (with NFT), the distribution of which varies according to the cortical layer and area. Furthermore, the distribution of both GABAergic and glutamatergic terminals around the soma and proximal processes of PHF-tau-ir neurons does not seem to be altered as it is indistinguishable from both control cases and from adjacent neurons that did not contain PHF-tau. At the electron microscope level, a normal looking neuropil with typical symmetric and asymmetric synapses was observed around PHF-tau-ir neurons. These observations suggest that the synaptic connectivity around the perisomatic region of these PHF-tau-ir neurons was apparently unaltered.
Introduction
In the brain of patients with Alzheimer’s disease (AD) the microtubule-associated protein tau is abnormally hyperphosphorylated or abnormally phosphorylated on sites that are not found in normal adult human tau isolated from postmortem brain samples (Matsuo et al., 1994 ). In this state, tau is the major protein subunit of the paired helical filaments (PHF) that form neurofibrillary tangles (NFT), a hallmark of this disease (Kidd, 1963 ; Brion et al., 1985 ; Grundke-Iqbal et al., 1986 ; Kosik et al., 1986 ; Goedert and Spillantine, 2006 ). In AD brains, PHF-tau not only accumulates in NFT (Grundke-Iqbal et al., 1986 ) but also in the cytosol, mixed with other diffusely scattered abnormal fibers (Iqbal et al., 1986 ; Bancher et al., 1989 ). This diffuse cytoplasmic PHF-tau reactivity has been postulated to reflect an altered neuronal state that precedes the formation of compacted NFT (Tolnay and Probst, 1999 ).
NFT were first described ultrastructurally in the 1960s, showing them to be bundles of abnormal cytoplasmic filaments that accumulate in nerve cell bodies and their processes (reviewed in Goedert and Spillantine, 2006 ; Kidd, 1963 , 1964 ; Terry, 1963 ; Terry et al., 1964 ; ). It was subsequently shown that the manner in which neurofibrillary lesions appear follows a stereotypical pattern that has been used to define six neuropathological stages (Braak and Braak, 1991 , 1995 ; Braak et al., 2006 ; Alafuzoff et al., 2008 ). In general, the most strongly affected cortical areas are the CA1 field, the subiculum and the entorhinal cortex (Hyman et al., 1984 ; Price et al., 1991 ; Morrison and Hof, 2002 ). NFT are first observed in the transentorhinal and entorhinal cortex, and incipiently in CA1 (stages I and II; transentorhinal stages), before these regions become more extensively affected and NFT appear in the subiculum at stages III and IV (the limbic stages). Finally, the main feature of stages V and VI is the development of abundant NFT in neocortical association areas (isocortical stages).
NFT accumulation is thought to be highly specific since there are areas where tangles are relatively infrequent, such as the pre- and parasubiculum or the dentate gyrus (Hyman et al., 1984 , 1986 ; Arendt et al., 1998 ; Braak et al., 2006 ). According to their affinity for silver staining methods (Alzheimer, 1911 ) or by applying specific immunocytochemical techniques, various morphologically subtypes of NFT have been described(reviewed in Tolnay and Probst, 1999 ; Bancher et al., 1989 ; Braak et al., 1994 ; Braak and Braak, 1995 ; Shimazaki et al., 2005 ). However, three main subtypes have general been reported that seem to correspond to various evolutionary stages of the lesions. The earliest stage is termed the pre-tangle stage and it is characterized by the accumulation of PHF-tau protein in the somatodendritic domain of affected neurons. These neurons are non-argyrophilic and therefore, they can only be detected with anti-PHF-tau antibodies. The next stage involves the appearance of classic intracellular NFT, while in the final stage ghost NFT accumulate, defined as NFT located “freely” in the neuropil with no relationship to a neuronal soma (Bancher et al., 1989 ; Braak et al., 1994 ). Both, classical and ghost NFT are easily identified with silver staining techniques.
NFT seem to specifically affect certain neuronal types (Braak and Braak, 1987 ; Braak et al., 1989 ; Iwamoto and Emson, 1991 ; Hyman and Gomez-Isla, 1994 ; Morrison et al., 1998 ; Braak et al., 2000 ). For example, most PHF-tau-immunoreactive (-ir) neurons in the neocortex and hippocampal formation are relatively large pyramidal neurons, whereas spiny stellate cells, small pyramidal cells, as well as non-spiny interneurons are reported to be remarkably resistant to neurofibrillary lesions (Hirano and Zimmerman, 1962 ; Mountjoy et al., 1983 ; Pearson et al., 1985 ; Braak and Braak, 1986 ; Braak et al., 1989 ). However, the appearance of NFT in particular neurochemical types of non-pyramidal cells has also been highlighted (Roberts et al., 1985 ; Iwamoto and Emson, 1991 ). It is thought that neurons that contain NFT may survive for decades but probably with functional deficits (Braak et al., 2006 ). However, as far as we know there are no studies addressing the synaptic innervation of different types of PHF-tau-ir cells. Thus, one of the main goals of the present study was to examine the changes to the pericellular innervation of PHF-tau-ir neurons in the hippocampal formation and adjacent cortex of AD patients.
Since NFT seem to affect mainly pyramidal neurons, it is important to remember that different portions of the pyramidal cell are innervated by different types of GABAergic interneurons (Houser et al., 1984 ; DeFelipe and Farinas, 1992 ; Jones, 1993 ; Freund and Buzsaki, 1996 ; Somogyi et al., 1998 ). For example, double-bouquet cells innervate the dendritic spines and shafts of apical collateral branches and the basal dendrites of pyramidal neurons. By contrast, basket cells innervate the soma and proximal dendrites, and chandelier cells innervate the axon initial segments, two cell types that are thought to exert a strong influence on the output of pyramidal cells. In particular, chandelier cells are thought to control the generation and back propagation of action potentials, as well as participating in complex activities such as the synchronization of firing patterns in large populations of pyramidal cells in different states of consciousness (see Miles et al., 1996 ; Klausberger et al., 2004 ; Cobb and Davies, 2005 ; Howard et al., 2005 ). In addition, dendritic spines and shafts represent the only postsynaptic sites on pyramidal cells that establish synapses with axon terminals forming asymmetric glutamatergic (excitatory) synapses (Spruston 2008 ; Bourne and Harris, 2007 ; ). However, both glutamatergic and GABAergic terminals are typically found in the perisomatic region, although only GABAergic terminals establish axo-somatic synapses (Alonso-Nanclares et al., 2004 ; Merchan-Perez et al., 2009 ). Nevertheless, the tight spatial association of excitatory axo-dendritic synapses with axo-somatic inhibitory terminals around the perisomatic regions of pyramidal cells has recently been proposed to be responsible for the non-synaptic glutamatergic interactions observed in electrophysiological and pharmacological studies (Ren et al., 2007 ), probably due to neurotransmitter spillover (Merchan-Perez et al., 2009 ).
Since in patients at different stages of the disease, the number of neurons that express PHF-tau in different cortical areas is very variable, we simply set out to study the perisomatic innervation of these neurons independent of the individual pathological characteristics. Therefore, we used dual immunocytochemistry and confocal microscopy reconstructions to examine the distribution of GABAergic and glutamatergic axon terminals in the perisomatic region and proximal processes of PHF-tau-ir pyramidal cells. Accordingly, in these cells we studied the distribution of the GABA transporter 1 (GAT-1: Minelli et al., 1995 ; DeFelipe and Gonzalez-Albo, 1998 ), the vesicular GABA transporter (VGAT: Chaudhry et al., 1998 ; Dumoulin et al., 1999 ; Minelli et al., 2003 ) and the vesicular glutamate transporter 1 (VGLUT-1: Ni et al., 1996 ; Bellocchio et al., 1998 ; Fremeau et al., 2001 ; Kaneko and Fujiyama, 2002 ; Varoqui et al., 2002 ; Alonso-Nanclares et al., 2004 ). In addition, antibodies against the calcium binding protein, parvalbumin (PV), were used to examine whether PHF-tau-ir pyramidal neurons are innervated by specific types of axon terminals from GABAergic interneurons, especially since PV immunocytochemistry has been shown to label a subpopulation of GABAergic neurons that include basket cells and chandelier cells (DeFelipe et al., 1989 ). Furthermore, we examined to what extent PV-ir neurons were affected by neurofibrillary lesions. Finally, there is very little information regarding the perisomatic innervation of PHF-tau-ir neurons at the ultrastuctural level. Indeed, it is possible that innervation of PHF-tau-ir pyramidal neurons is affected and that this innervation varies according to the type of neurofibrillary alteration. Thus, we used a correlative light and electron microscopy method to examine the perisomatic synapses around PHF-tau-ir neurons.
Materials and Methods
Human brain tissue was obtained at autopsy from two sources: from eight patients with AD [aged 80–94: 84 ± 7, mean ± standard deviation (SD)]; and from control human brain tissue from seven individuals (aged 23–69: 53 ± 17, mean ± SD) who died in an accident and were free of any neurological or known psychiatric illness (Table 1 ). The AD brain tissues were obtained from the Instituto de Neuropatología (Dr. I. Ferrer, Servicio de Anatomía Patológica, IDIBELL-Hospital Universitario de Bellvitge, Barcelona, Spain) and from the Banco de Tejidos Fundación CIEN (Dr. A. Rábano, área de Neuropatología, Centro Alzheimer, Fundación Reina Sofia, Madrid, Spain). The control human brains were obtained from the Servicio de Patología Forense (Dr. R. Alcaraz, Instituto Vasco de Medicina Legal, Bilbao, Spain). Following a neuropathological examination, the AD stages were defined according to Braak and Braak (1991 : Table 2 ). Control cases (C1–C7) were associated with different scores for the amyloid and neurofibrillary pathology. While C1 and C5–C7 had a Braak score of zero for both pathologies, C2–C3 had a small number (+) of PHF-tau-ir neurons, whereas C4 had many (+++). Finally, while C2 developed numerous (+++) plaques immunostained for Aβ, there were no such plaques in cases C3–C4 (see Figures 4 and 8 ). In all cases, the time between death and tissue processing was between 1.5 and 3 h, and the brain samples were obtained following the guidelines and approval by the Institutional Ethical Committee. The tissue from these human brains has been used in previous studies (e.g., Arellano et al., 2004 ).
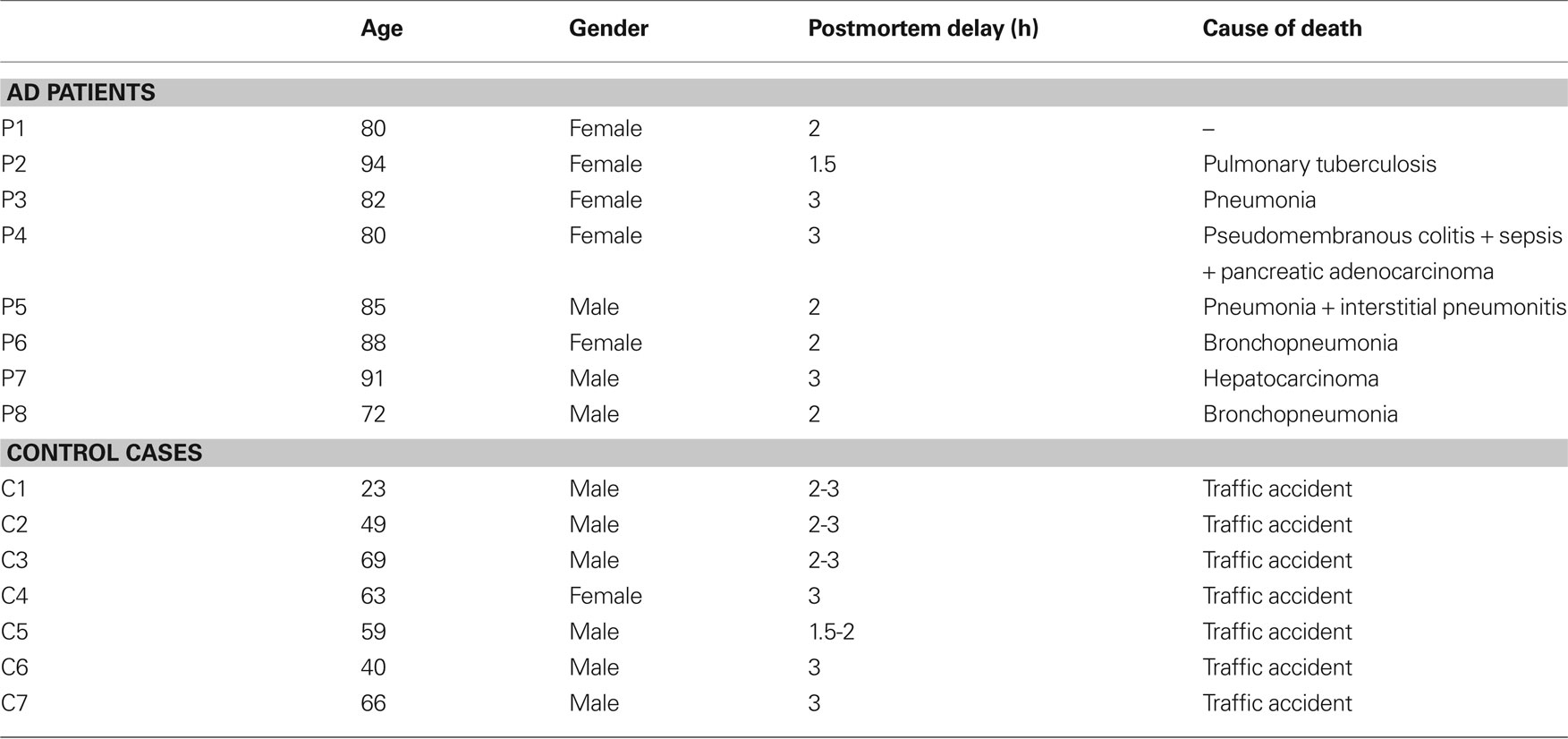
Table 1. Summary of case data.
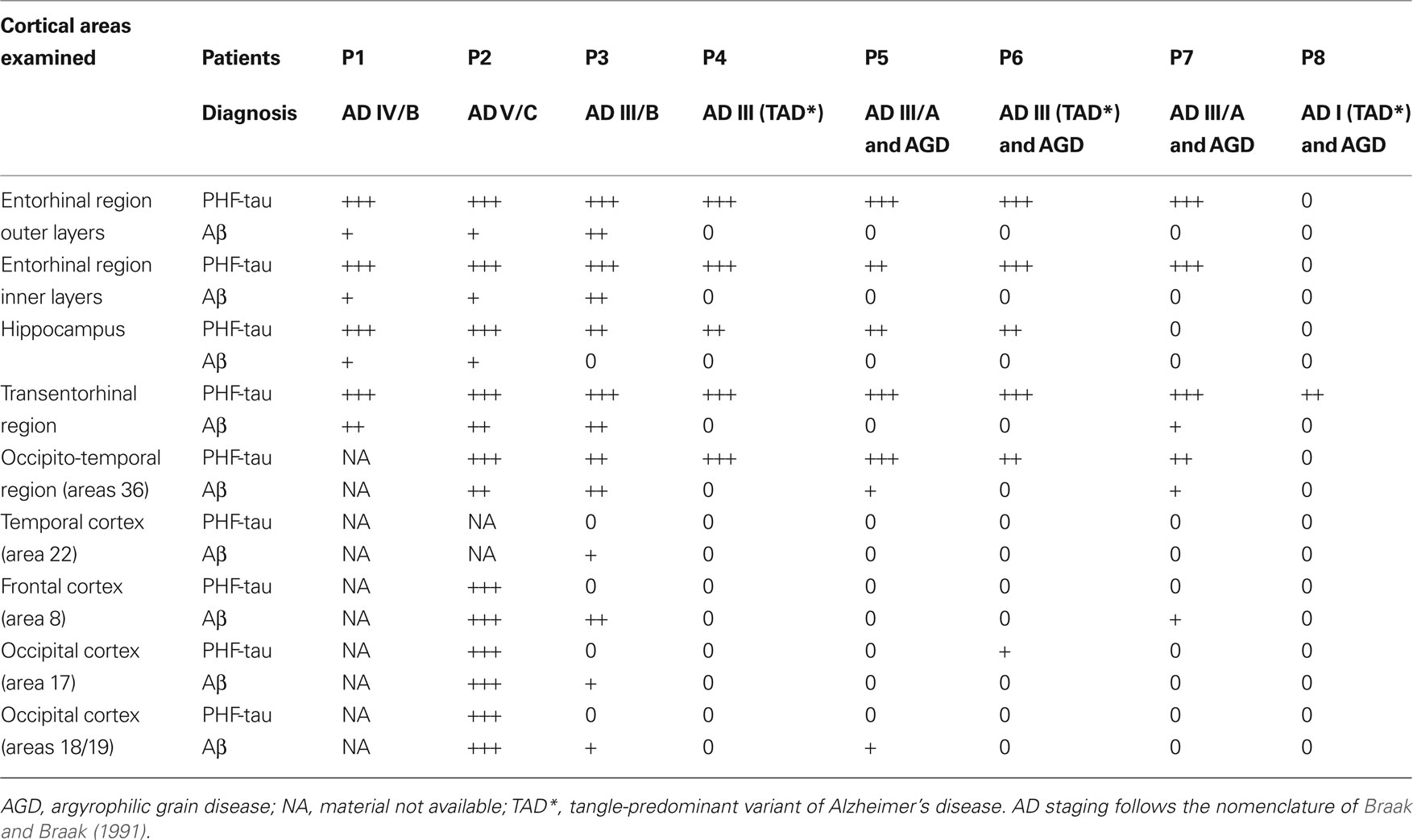
Table 2. Neuropathological assessment of the accumulation of NFT (immunostained for PHF-tau) and plaques (immunostained for Aβ). The overall amount of NFT and Aβ is graded and labeled zero (0) where no discernible change is detected, (+) with a small, (++) moderate and (+++) large amount of NFT or Aβ.
Upon removal, the brain tissue was immediately fixed in cold 4% paraformaldehyde in phosphate buffer (PB: 0.1 M, pH 7.4), and after 2 h, the tissue was cut into small blocks and post-fixed in the same fixative for 24–48 h at 4°C. After fixation, all the specimens were immersed in graded sucrose solutions and they were stored in a cryoprotectant solution at −20°C. Serial sections (50 μm) of the cortical tissue were obtained using a vibratome, and the sections from each region and case were batch-processed for immunocytochemical staining. The sections immediately adjacent to those stained immunocytochemically were Nissl-stained in order to identify the cortical areas and the laminar boundaries.
Immunohistochemistry
Free-floating sections were pre-treated in 1% H2O2 for 30 min to remove endogenous peroxidase activity, and subsequently they were blocked for 1 h in PB with 0.25% Triton-X and 3% normal goat or horse serum (for polyclonal antisera and monoclonal antibodies, respectively: Vector Laboratories Inc., Burlingame, CA, USA). Single immunohistochemistry was performed with the following polyclonal antisera and monoclonal antibodies: GABA transporter 1 (GAT-1, 1:500, AB1570: Chemicon International, Temecula, CA, USA); vesicular GABA transporter (VGAT, 1:2,000, 131003: Synaptic Systems, Goettingen, Germany); vesicular glutamate transporter 1 (VGLUT-1, 1:5,000, AB5905: Chemicon); parvalbumin (PV, 1:1,000, AB11427: Abcam, Cambridge, United Kingdom); and an anti-human PHF-tau monoclonal antibody (clone AT8, 1:100, MN1020: Thermo Scientific; Waltham, MA, USA). This anti-PHF-tau antibody has been shown to detect PHF-tau doubly phosphorylated (we shall refer to as PHF-tau) at both Ser202 and Thr205 (Porzig et al., 2007 ). The sections were incubated overnight at 4°C with the antibodies described above and the following day, the sections were rinsed and incubated for 2 h with biotinylated goat anti-rabbit IgG (1:200, BA-1000: Vector Laboratories) (GAT-1, VGAT and PV immunostaining), biotinylated goat anti-guinea-pig IgG (1:200, BA-7000: Vector Laboratories) (VGLUT-1 immunostaining) or biotinylated horse anti-mouse IgG (1:200, BA-2000: Vector Laboratories) (PHF-tau immunostaining) antibodies. The sections were then incubated for 1 h in an avidin–biotin peroxidase complex (Vectastain ABC Elite PK6100, Vector), and finally with the 3,3′-diaminobenzidine tetrahydrochloride chromogen (DAB: Sigma-Aldrich, St. Louis, MO, USA). After staining, the sections were dehydrated, cleared with xylene and coverslipped. In addition, adjacent sections to those stained for PHF-tau were processed immunocytochemically as above to visualize amyloid beta (Aβ) plaques using a mouse antibody against Aβ (clone 6F/3D 1:50, M0872: Dako, Glostrup, Denmark). Control sections were processed as above but without the primary antibodies and no significant staining was seen under these conditions. Furthermore, to rule out possible false positive staining as a consequence of the AT8 antibody recognizing non-phosphorylated tau rather than the hyperphosphorylated tau at high concentrations, we carried out an experiment with serial dilutions of this PHF-tau antibody up to 1:20,000. We observed a progressive decrease in the staining intensity of all the elements. No separate elements ceased staining at different dilutions and no staining was observed at dilutions above 1:10,000.
Dual Fluorescence Immunohistochemistry and Histochemistry
To ascertain the relationship between neurons immunocytochemically stained for PHF-tau and neurons histochemically labeled for Thioflavine-S to visualize NFT, some sections were stained using the same anti-PHF-tau antibody at the dilutions and incubation times indicated above. Thereafter, the sections were incubated for 2 h at room temperature with the Alexa fluor conjugated goat-anti mouse 594 antibody (1:2,000, A-11005, Molecular Probes, Eugene, OR, USA). After rinsing the sections in PB, they were incubated for 10 min in a 1% solution of Thioflavine-S and they were then rinsed for 5 min in 100%, 70% and then 50% ethanol. After rinsing in PB, the sections were treated with Autofluorescence Eliminator Reagent (2160, Chemicon) to reduce lipofucsin like-autofluorescence without adversely affecting other fluorescent labeling in the sections.
To examine the pericellular innervation of PHF-tau-ir neurons, some sections were double stained with a mixture of antibodies GAT-1/PHF-tau, VGAT/PHF-tau, PV/PHF-tau, or VGLUT-1/PHF-tau. The same primary antibodies, dilutions, and incubation times as indicated above were used. Free-floating sections were incubated overnight at 4°C in a solution containing the primary antibodies and then for 2 h at room temperature with a biotinylated goat anti-rabbit antibody for GAT-1/PHF-tau, VGAT/PHF-tau, and PV/PHF-tau, or in a biotinylated goat anti-guinea pig antibody for VGLUT-1/PHF-tau (1:200: Vector). After rinsing in PB, the sections were incubated for 2 h at room temperature with streptavidin coupled to Alexa fluor 488 (1:2,000, S-32354, Molecular Probes) and with Alexa fluor goat-anti mouse 594. After rinsing in PB, the sections were treated with Autofluorescence Eliminator Reagent.
Finally, the sections were washed and mounted with ProLong Gold Antifade Reagent (Invitrogen Corporation, Carlsbad, CA, USA), and they were examined on a LeicaDMI 6000B confocal laser scanning system equipped with an argon/krypton mixed gas laser with excitation peaks at 488 and 594 nm. The fluorescence of Alexa 488 and 594 was recorded through separate channels. We obtained both single optical sections and image stacks (z-step 0.14 μm) that consisted of 19–43 image planes. Both 40× oil-immersion (NA, 1.25, refraction index, 1.52) and 63X oil-immersion (NA, 1.40, refraction index, 1.52) lens were used, in some cases with a calculated optimal zoom factor 3.2.
Quantitative and Statistical Analyses
A BX51 Olympus microscope equipped with a motorized stage and the Neurolucida package (MicroBrightField, Williston, VT, USA) was used to estimate the density of PHF-tau-ir neurons with different patterns of staining. Using the Neurolucida package, the hippocampal formation and adjacent cortex were traced in contiguous Nissl-stained sections with the 4× objective, and the complete surface of the previously defined counting fields was scanned with a 40× objective in successive and non-overlapping frames of 17,250 μm2. All the PHF-tau-ir neurons were recorded and classified as displaying a type I or II immunostaining pattern (see Results). In Table 3 , the areas of the cortical regions examined in each control and AD patient cases are shown.
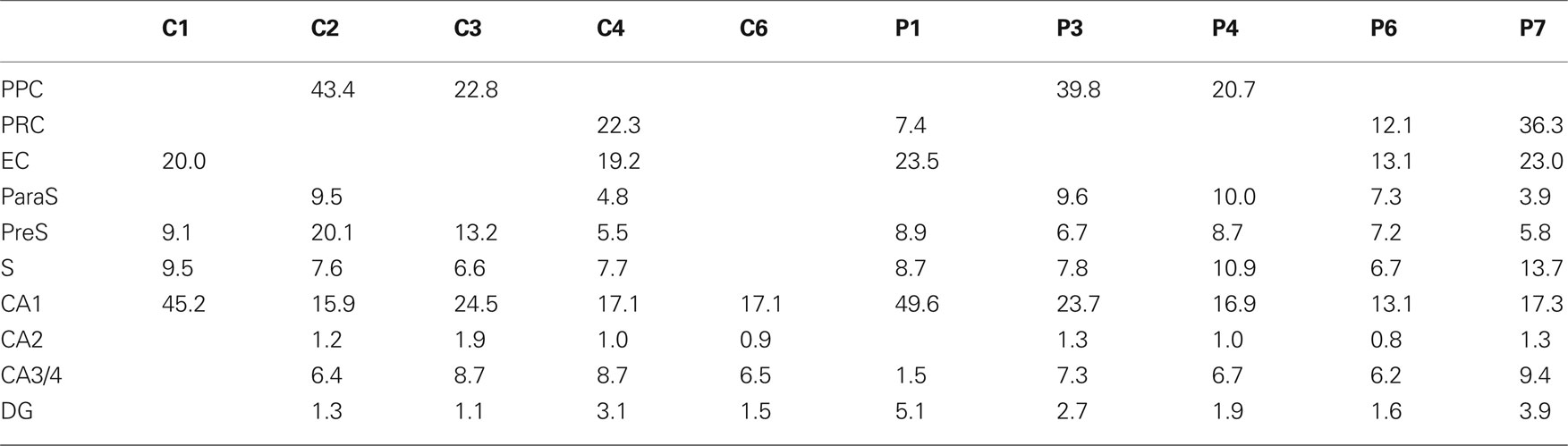
Table 3. Total areas (mm2) analyzed in control and AD patients.
To estimate the number of perisomatic GABAergic terminals around PHF-tau-ir neurons and around neurons that do not accumulate PHF-tau, we quantified the number of VGAT-ir boutons associated with these neurons in the CA1 field and the supragranular layers of the parahippocampal gyrus (PHG; which includes entorhinal and perirhinal cortex) in sections from AD patient P7 stained for VGAT/PHF-tau. In this way, such comparisons could be used as internal controls to eliminate the effects of age and pathological progression. We chose brain sections from patient P7 because the immunostaining was of the best quality. Moreover, to minimize possible differences due to cellular variability, we selected neurons (n = 20 per immunocytochemical pattern of staining and cortical region) with similar somatic parameters. Sections were examined on a Zeiss LSM 710 confocal laser scanning microscope and single optical sections (physical size 67.41 μm × 67.41 μm, logical size 1024 × 1024 pixels) were obtained with a 63× oil-immersion lens (NA, 1.40, refraction index, 1.45) and an optical zoom factor 2.0. Because of the limited penetration of the antibodies in the sections, the images were taken from the superficial part of the section (2–3 μm in depth from the surface). Using the Neurolucida 7.1 Confocal module (MicroBrightfield, Inc., Williston, VT, USA), it was possible to draw and calculate the perimeter of the cells and mark the labeled boutons around them. After drawing the perimeter, the red channel of the confocal microscope system (which corresponded to PHF-tau staining) was removed to carry out a blind study of the perisomatic innervation. The results are presented as the number of VGAT-ir boutons per 100 μm of the somatic perimeter and the data are presented as the mean ± SD. Statistical comparisons were carried out with the aid of GraphPad Prism statistical package (Prism, San Diego, CA, USA). A two-way analysis of variance (ANOVA), followed by Bonferroni post-test were used to analyze the differences in the number of VGAT-ir boutons around the three types of neurons (PHF-tau-negative, type I and II PHF-tau-ir) in the different regions (CA1 field and parahippocampal gyrus).
Finally, the proportion of PV-ir neurons that were also labeled for PHF-tau was estimated in at least two sections from the hippocampal formation of four patients (P1, P3, P4, and P7), and in that of a single control (C7), all of which were processed for dual fluorescence immunohistochemistry. These sections were examined on a BX51 Olympus fluorescence microscope.
Electron Microscopy
Sections adjacent to those used for histopathological assessment and some sections immunostained for PHF-tau were processed for electron microscopy. Immunohistochemistry for PHF-tau followed the same protocol as that laid out above, except that the pre-treatment in 1% H2O2 only lasted for 10 min and Triton-X was excluded from the buffers. Once the immunohistochemical staining had been completed, the sections were post-fixed in 2% glutaraldehyde in PB for 1 h, treated with 1% osmium tetroxide for 40 min, dehydrated and flat embedded in Araldite resin. Plastic-embedded sections were studied using correlative light and electron microscopy as described in detail elsewhere (DeFelipe and Fairen, 1993 ). Briefly, sections were photographed under the light microscope and then serially cut into semithin (2 μm thick) sections with a Reichert ultramicrotome. The semithin sections were stained with 1% toluidine blue in 1% borax, they were examined under the light microscope and then photographed to locate the region of interest. Selected semithin sections were further sectioned into serial ultrathin sections (50–70 nm thick) with a diamond knife using a Reichert ultramicrotome. The ultrathin sections were collected on formvar-coated single-slot nickel grids, and stained with uranyl acetate and lead citrate. Digital pictures were captured at different magnifications in a Jeol 150 transmission electron microscope (JEOL USA, Inc., MA, USA) equipped with a SIS Megaview III CCD digital camera.
Results
A Note on the Nomenclature of the Human Hippocampal Regions
The hippocampus proper (Cornu Ammonis fields) is commonly subdivided into the CA1, CA2, CA3, and CA4 fields, according to the nomenclature of Lorente de Nó (1934) . Ammon’s horn neurons within the granule cell layer constitute the CA4 field. However, as pointed out by Insausti and Amaral (2004) , the absence of clear cytoarchitectonic features or connections that distinguish CA3 neurons from those of the CA4 make it more convenient to consider these fields simply as the CA3, thereby including the neurons of Ammon’s horn in this region. Thus, for the sake of clarity we denominate this region CA3/CA4. In addition, we will use the nomenclature of Insausti and Amaral, (2004) when considering the hippocampal formation and adjacent cortex: dentate gyrus (DG); hippocampus proper (CA1, CA2, and CA3/4); subicular complex (subdivided in subiculum [Sub], presubiculum [preS], parasubiculum [paraS]); entorhinal cortex (EC); perirhinal cortex (PRC); and posterior parahippocampal cortex (PPC). Finally, CA1 was subdivided into three equal portions: proximal, medial, and distal with respect to the DG.
Distribution and Staining Pattern of PHF-tau-ir Neurons
We studied the distribution and pattern of PHF-tau-ir neurons, and their relationship with Aβ plaques, in order to obtain more detailed information on the pathological characteristics of the different patients examined.
Using a variety of silver and immunohistochemical staining techniques, the pathological changes of the immunoreactive neurons were classified and a morphological series was established to reflect the sequence of changes believed to be experienced by the individual nerve cell as the disease progresses (Braak et al., 1994 ). In general, early stages are characterized by abnormal delicate fibrillary inclusions within affected neurons. These fibers aggregate into large bundles that finally fill the entire neuronal cytoplasm as classic NFT. In the present work, we classified PHF-tau-ir neurons as type I or II, based on previous studies (Bancher et al., 1989 and Braak et al., 1994 ). In sections immunocytochemically stained with the anti-PHF-tau antibody, numerous neurons were labeled in all cortical areas of the hippocampal formation and adjacent cortex examined (Figure 1 ). Type I corresponds to Stage 0–1 in Bancher et al. (1989) and Group 1–2 of Braak et al. (1994) , while type II corresponds to Stage 2–3 of Bancher et al. (1989) and Group 3–5 of Braak et al. (1994) . The type I pattern was characterized by the diffuse cytoplasmic staining (with no NFT: Figures 2 and 3 ) of neurons with a normal morphology, but with dendrites and proximal axons that were often strongly stained, displaying Golgi-like labeling. In neurons with a type II pattern, the amount of somatic cytoplasm occupied by the NFT varied (Figures 2 and 3 ). The dendritic arbor of the neurons had relatively little NFT and it usually displayed numerous dendritic processes. Neurons whose cytoplasm was full of NFT have very few dendritic processes, suggesting they were undergoing neuronal atrophy.
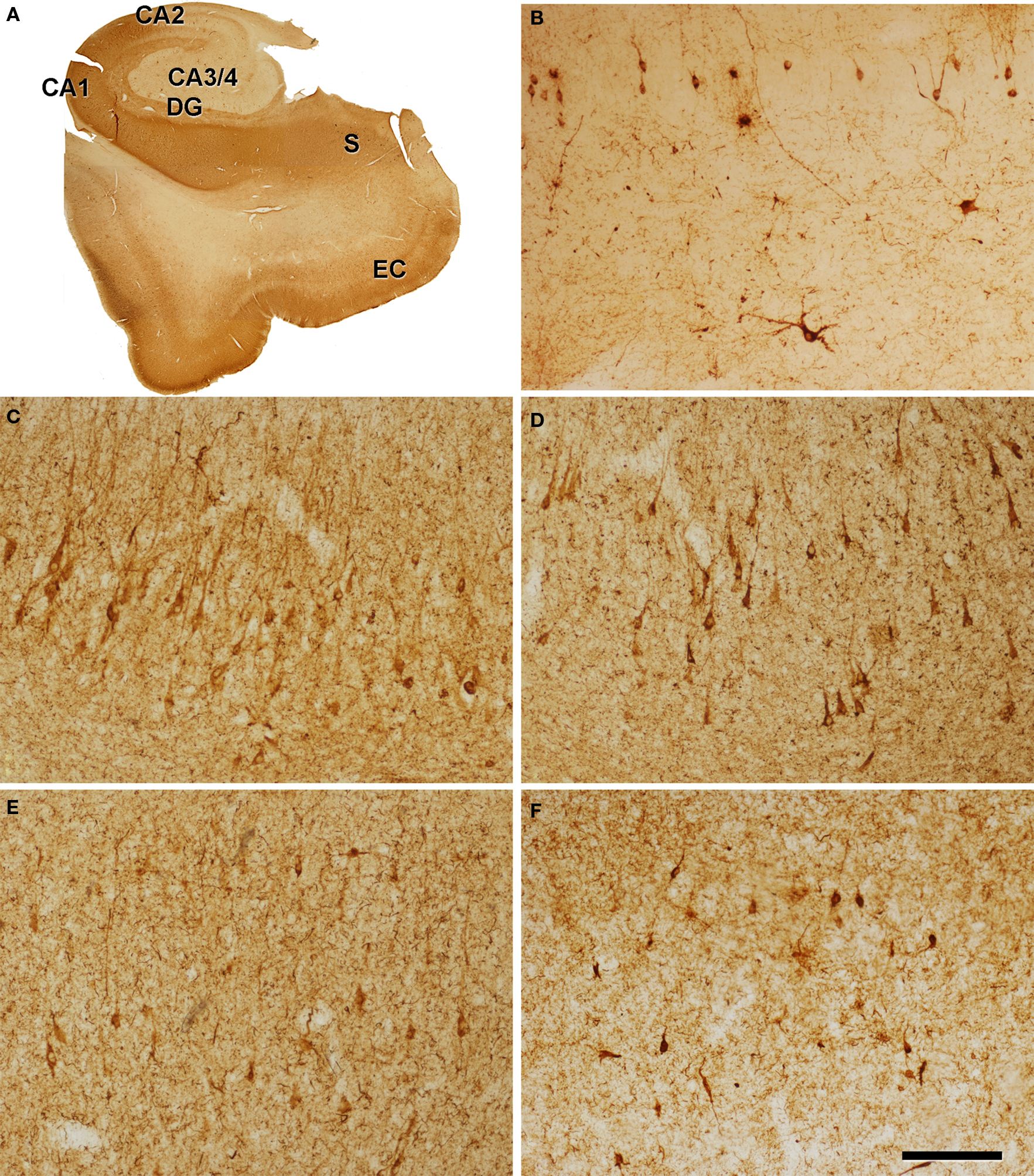
Figure 1. Hippocampal formation from patient P7 immunostained for PHF-tau. (A) Low-power photomicrograph. (B–F) Photomicrographs illustrating different areas of the hippocampal formation: hilus of the dentate gyrus [DG] (B), CA2 (C), CA1 (D), subiculum [S] (E), and entorhinal cortex [EC] (F). Scale bar: 1.8 mm in (A); 170 μm in (B–F).
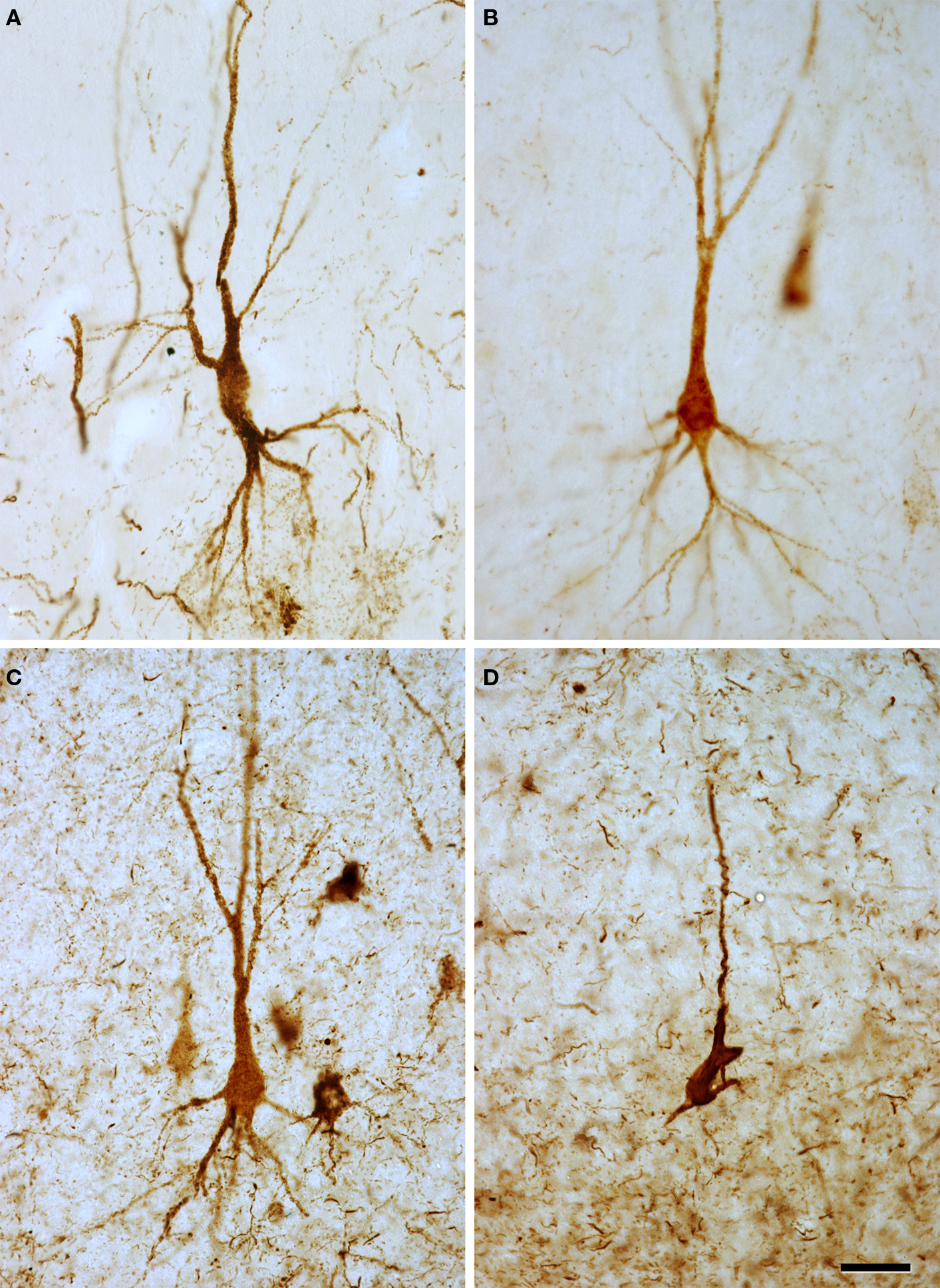
Figure 2. High-power microphotographs to illustrate the morphological appearance of PHF-tau-ir neurons. (A) Pyramidal neuron from the CA1 field of control case C2. (B,C) Pyramidal neurons from the CA2 field of patients P4 and P3, respectively. (D) Pyramidal neuron from the entorhinal cortex of patient P1. (A–C) Correspond to pattern I of PHF-tau staining, which is characterized by a diffuse cytoplasmic staining and the neurons display a normal morphology. (D) Correspond to pattern II of PHF-tau staining, which is characterized by the presence of NFT. These neurons appear to have fewer processes probably due to neuronal atrophy. Scale bar: 37.5 μm.
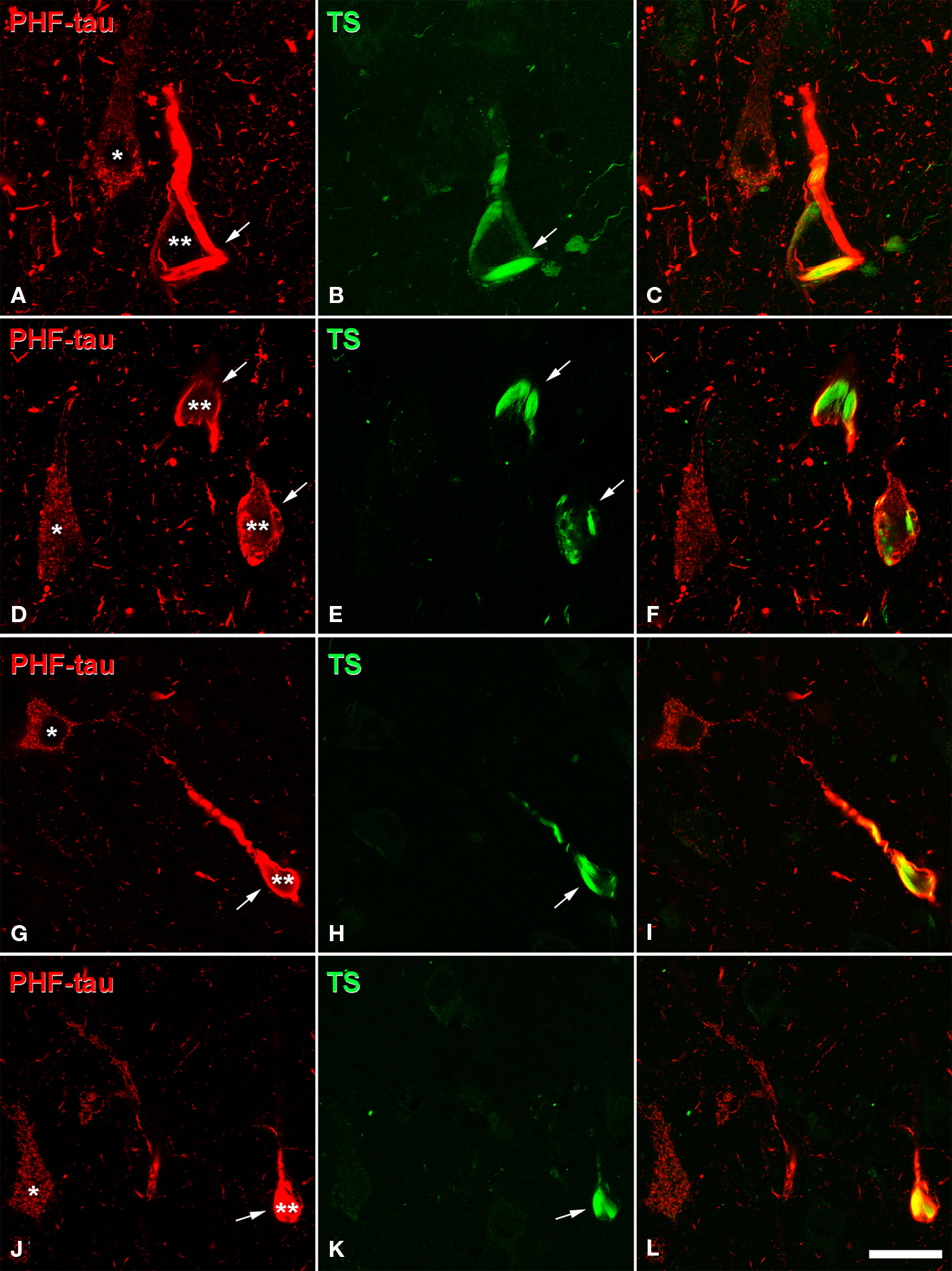
Figure 3. Confocal microscopy of sections double stained with an anti-PHF-tau antibody (red) and Thioflavine-S (TS, green), which labels PHF-tau forming NFT. (A,B), (D,E), (G,H), and (J,K) pairs of confocal images from the same section and field in the CA1 hippocampal region of AD patient P1, P7, P4, and P3, respectively. Panels (C,F,I,L) were obtained by combining these images (A) and (B), (D) and (E), (G) and (H), (J) and (K), respectively. Note that type I PHF-tau-ir neurons (one asterisk) were free of NFT, whereas type II neurons (two asterisks) contained NFT (arrows). Scale bar: (A–C), 21 μm; (D–F), 24 μm; (G–L), 31.5 μm.
Since PHF-tau-ir neurons were also present in control tissue (see Discussion), we plotted all PHF-tau-ir neurons in the hippocampal formation and adjacent cortex from both control (Figure 4 ) and AD patients (Figure 5 ). Moreover, we quantified the density of PHF-tau-ir neurons with type I and II staining in the different regions (Figure 6 ). In general, a lower density of PHF-tau-ir cells was found in control tissue than in that from AD patients in all the fields studied, with the exception of control case C4 in which a similar or higher density than in AD cases was evident in the Sub, CA1, and CA2. The density of PHF-tau-ir neurons varied considerably between different AD patients. For example, while patient P1 was diagnosed at AD stage IV (the most severe stage of all the cases studied), this patient only had the highest density of PHF-tau-ir neurons in the preS when compared with the rest of the control and AD tissue (Figure 6 A). Patient P4 had much lower densities in all the hippocampal fields than P3, P6, and P7 patients, even though each was considered as stage AD III (Table 2 ). In addition, patients P6 and P7 showed a relatively high density of PHF-tau-ir neurons in the DG even though granular cells do not typically develop neurofibrillary lesions (Braak and Braak, 1991 ). Interestingly, these two patients also suffered from argyrophilic grain disease (AGD), which is a neurodegenerative disease often associated to other neurodegenerative disorders and that is characterized by the presence of argyrophilic grains in several cortical regions. It has been suggested that AGD may enhance the pathological alterations in several neurodegenerative disorders, including AD, particularly in the DG (Thal et al., 2005 ; Ferrer et al., 2008 ; Josephs et al., 2008 ). Thus, the high density of PHF-tau-ir neurons in the DG in cases P6 and P7 might be explained by the co-existence of AGD.
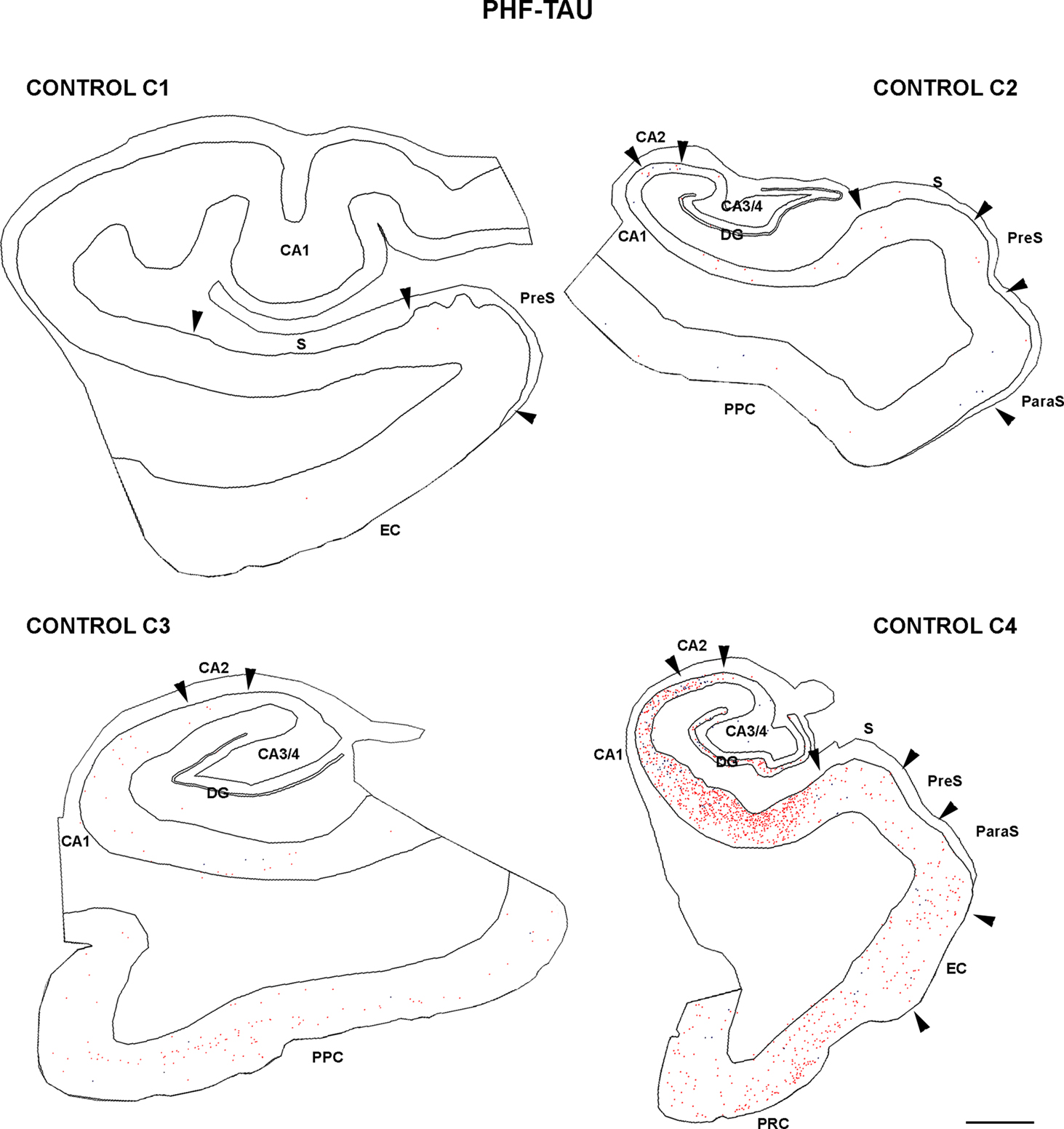
Figure 4. Drawings based on Neurolucida plots made with a 40× objective to illustrate the distribution of PHF-tau-ir neurons displaying type I (blue dots) and type II (red dots) patterns of staining in the hippocampal formation and adjacent cortex from control cases. Borders between the different cytoarchitectonic regions are indicated by arrowheads. Scale bar: 1,000 μm. Abbreviations as in Figure 5 .
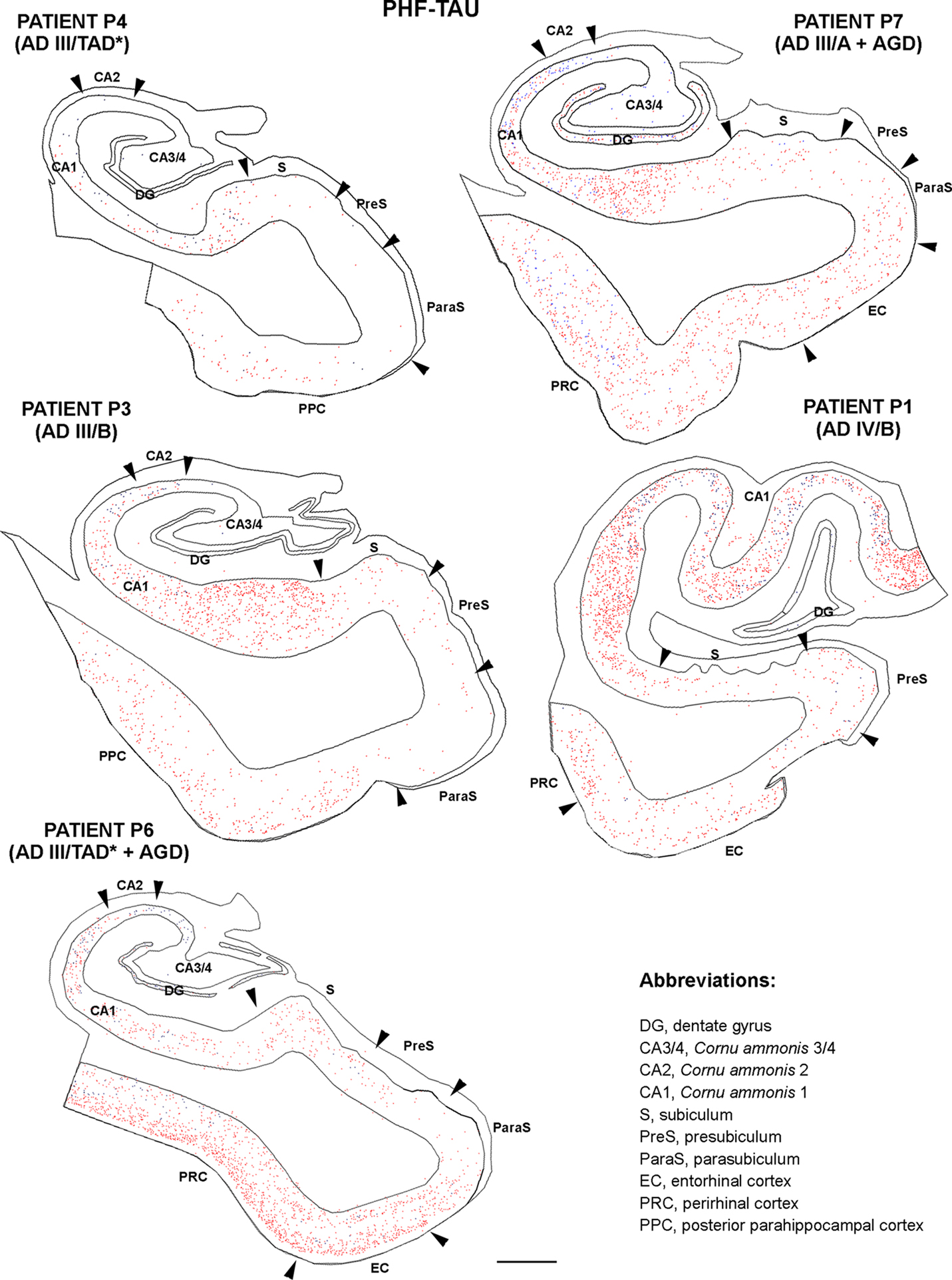
Figure 5. Drawings based on Neurolucida plots made with a 40× objective showing the distribution of PHF-tau-ir neurons showing type I (blue dots) and type II (red dots) staining in the hippocampal formation and adjacent cortex from AD cases. Borders between the different cytoarchitectonic regions are indicated by arrowheads. Scale bar: 1,000 μm.
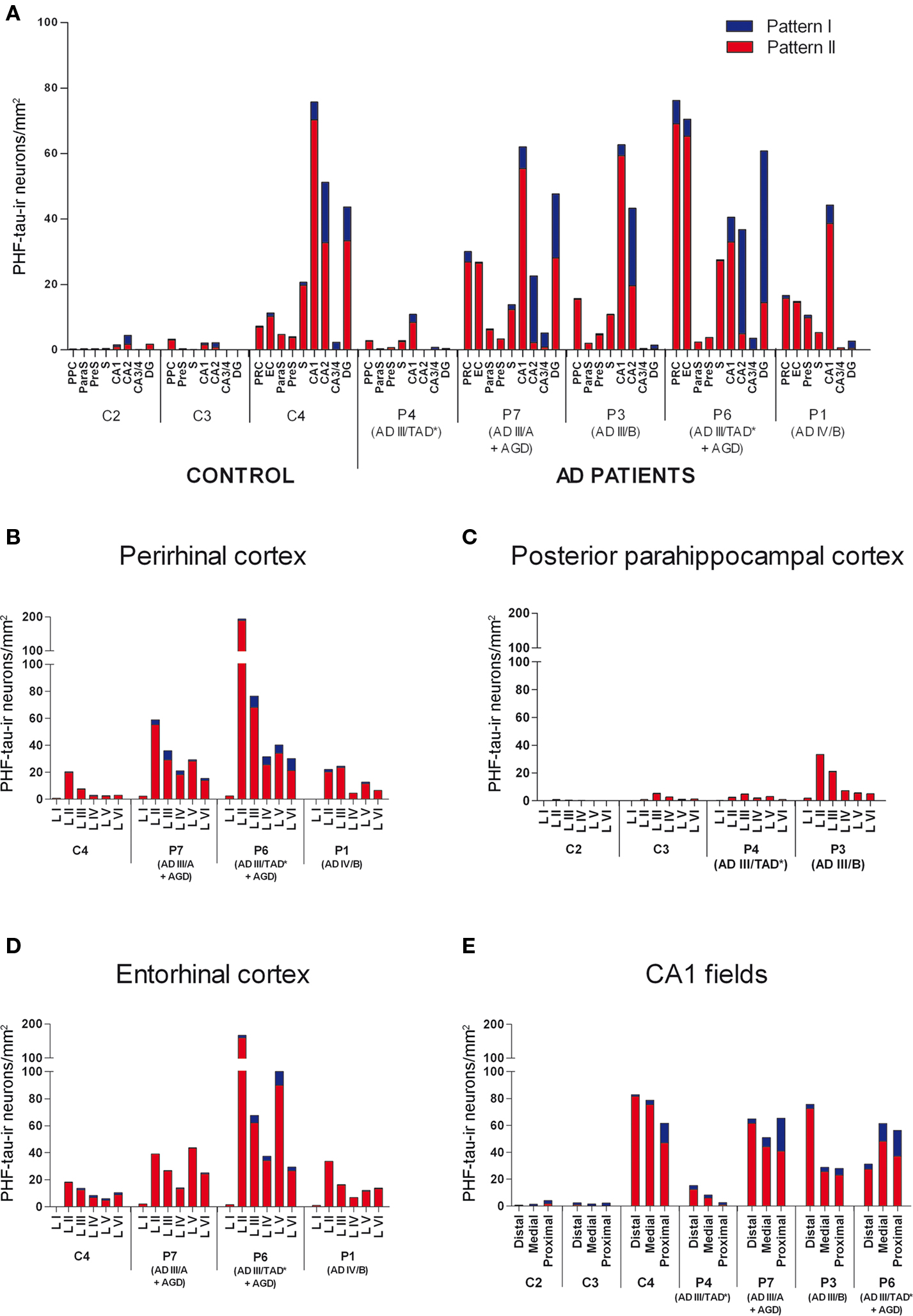
Figure 6. (A) Graphs showing the density of PHF-tau-ir neurons with type I and II staining in control and AD patients in different regions of the hippocampal formation and adjacent cortex: granular layer of dentate gyrus (DG); pyramidal layer of hippocampus proper (CA1, CA2, and CA3/4); pyramidal layer of subiculum (S), presubiculum (preS), parasubiculum (paraS), entorhinal cortex (EC), perirhinal cortex (PRC), and posterior parahippocampal cortex (PPC). (B–E) Graphs showing the density of PHF-tau-ir neurons with type I and II staining in control and AD patients in the different layers of the PRC, PPC, and EC, and in the pyramidal cell layer of the distal, medial and proximal portions of CA1, respectively.
The distribution by layers revealed that both in control and AD tissue, the highest density of PHF-tau-ir neurons was found in layers II-III and occasionally in layer V of the PPC, PRC, and EC. In CA1, CA2, CA3/4, and DG, virtually all PHF-tau-ir neurons were located in the pyramidal layer of the CA fields and in the granular layer of the DG (Figure 6 ).
The percentage of neurons displaying type I staining increased from the adjacent cortex of the hippocampal formation (PPC or PRC and EC) towards the DG, with the highest proportion of these neurons found in the field CA3/4 (Figure 7 ). The widest variability of these neurons was observed in the DG: 76% of neurons in case P3 and 78% of neurons in case P6 displayed type I staining, whereas in P7 and C4 (control case) this figure was 41 and 24%, respectively (Figure 7 ).
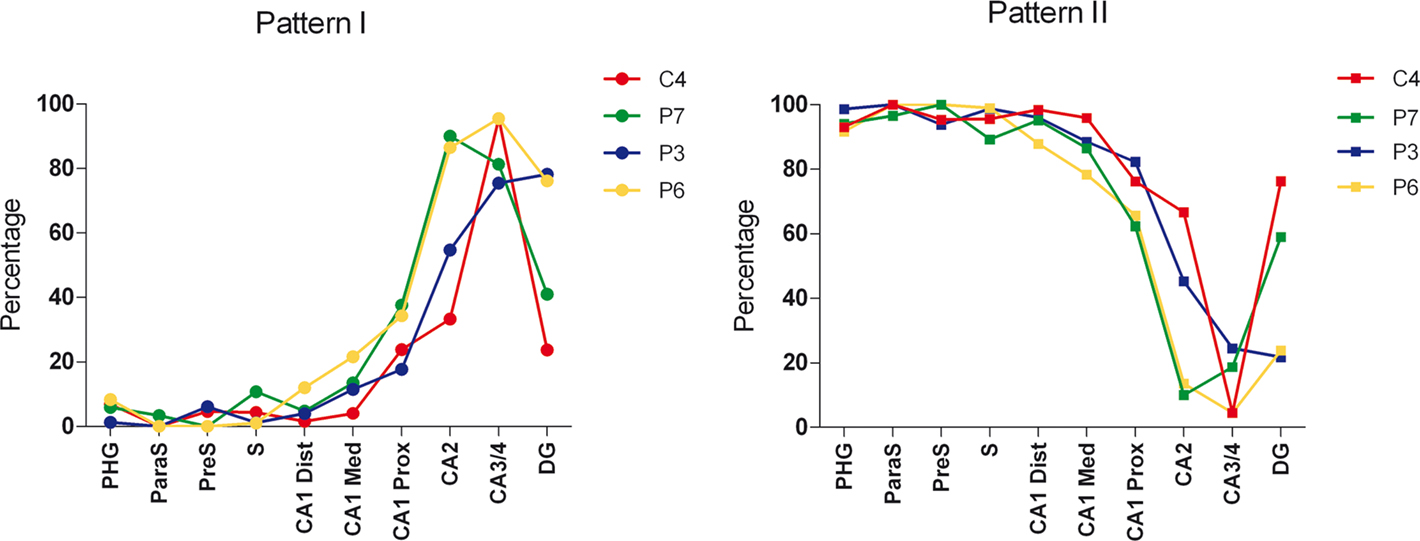
Figure 7. Graphs showing the percentage of PHF-tau-ir neurons displaying type I and II staining in the hippocampal formation and adjacent cortex of control and AD patients. Dentate gyrus (DG); CA3/4, CA2, CA1 proximal (CA1 Prox), CA1 medial (CA1 Med), CA1 Distal (CA1 Dist), subiculum (S), presubiculum (preS), parasubiculum (paraS) and parahippocampal gyrus (PHG) that includes the entorhinal cortex, perirhinal cortex, or posterior parahippocampal cortex.
Finally, adjacent sections stained for PHF-tau were processed to visualize Aβ plaques to find out whether the density and distribution of PHF-tau-ir neurons was related to the density of Aβ plaques in tissue from both control (Figure 8 ) and AD patients (Figure 9 ). Comparing the distribution of PHF-tau-ir neurons and Aβ plaques revealed no such correlation since there were regions where the density of both PHF-tau-ir neurons and Aβ plaques was very high whereas in other regions a high density of PHF-tau-ir neurons coincided with a region with few Aβ plaques and vice versa.
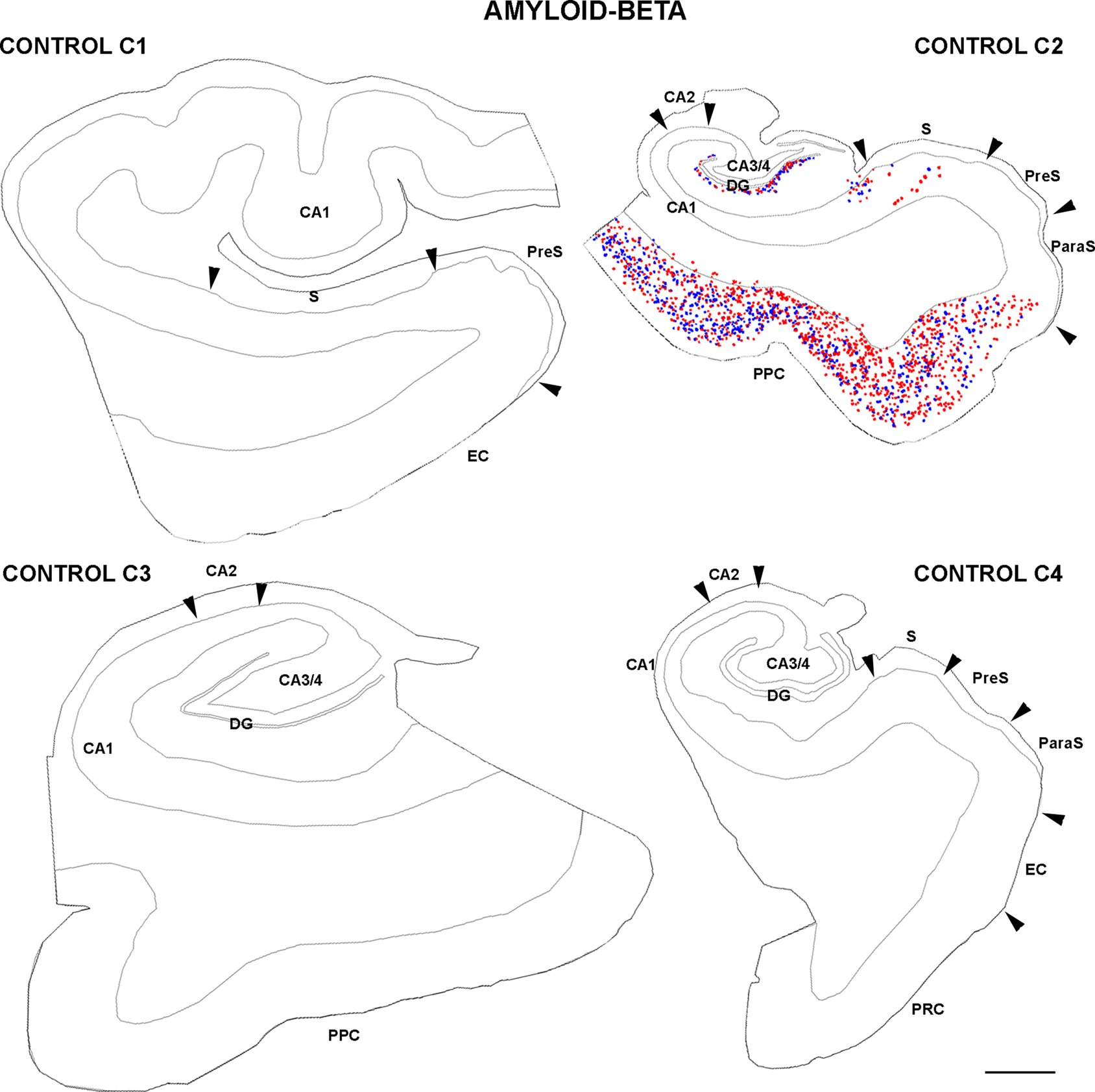
Figure 8. Drawings based on Neurolucida plots made with a 40× objective showing the distribution of diffuse, cored neuritic, and non-cored neuritic plaques (red, green, and blue circles, respectively; see Garcia-Marin et al., 2009 ) in the hippocampal formation and adjacent cortex from control cases. Borders between the different cortical cytoarchitectonic regions are indicated by arrowheads. Abbreviations as in Figure 5 . Scale bar: 1,000 μm.
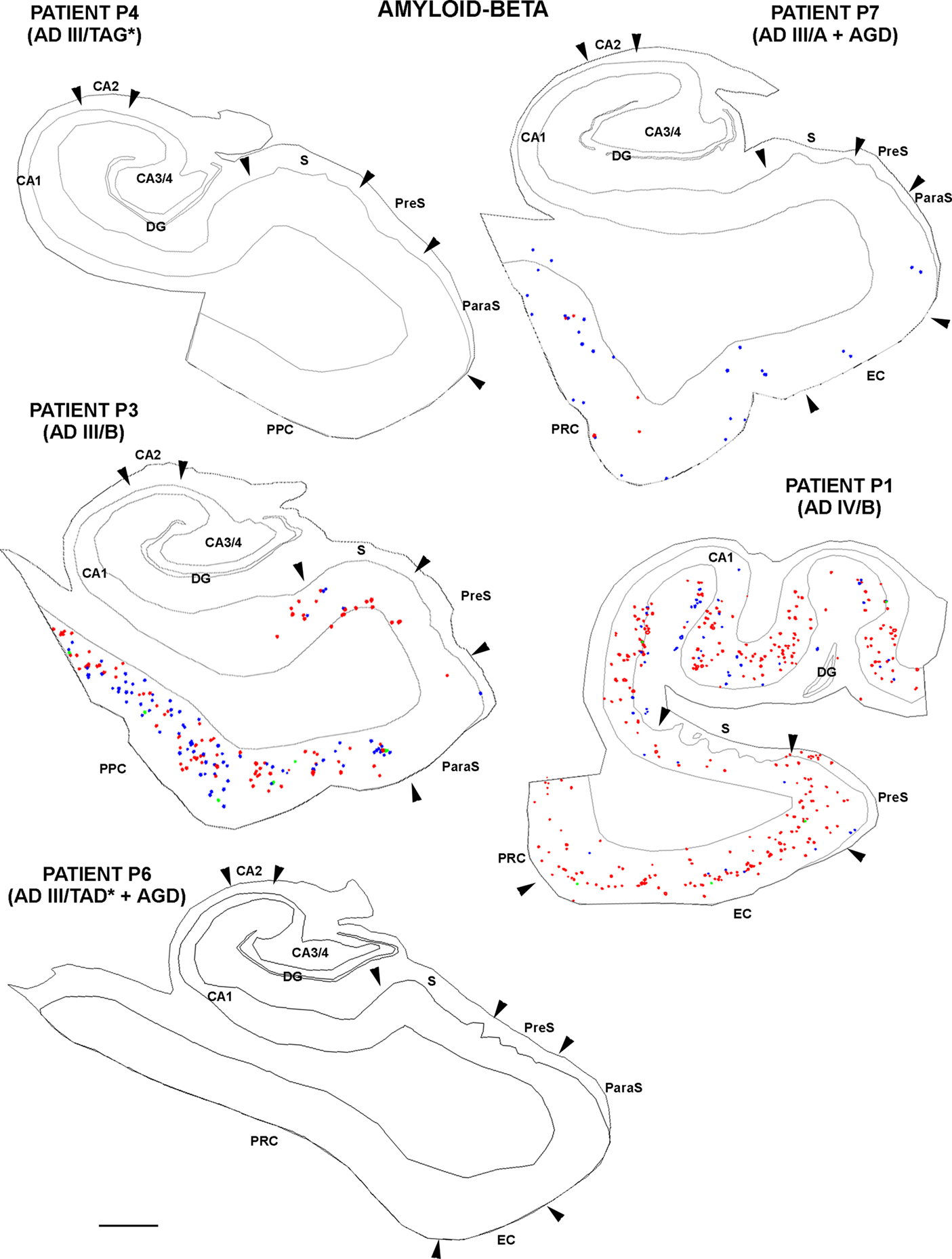
Figure 9. Drawings based on Neurolucida plots made with a 40× objective showing the distribution of diffuse, cored neuritic and non-cored neuritic plaques (red, green, and blue circles, respectively; see Garcia-Marin et al., 2009 ) in the hippocampal formation and adjacent cortex from AD cases. Borders between the different cortical cytoarchitectonic regions are indicated by arrowheads. Abbreviations as in Figure 5 . Scale bar: 1,000 μm.
Dual Immunofluorescence Studies
Since the main objective of this study was to examine the perisomatic innervation of neurons showing different patterns of PHF-tau staining, irrespective of the neuropathological stage of the patient, we used dual immunofluorescence to determine whether the expression of PHF-tau induced or was related to any changes in the affected neurons. While it is well-established that only GABAergic terminals form axo-somatic synapses with pyramidal cell bodies, both glutamatergic and GABAergic terminals are typically found in the perisomatic region (Merchan-Perez et al., 2009 ). For this reason, we used dual immunofluorescence to label GABAergic (GAT-1, VGAT, and PV) and glutamatergic (VGLUT-1) axon terminals and to examine the innervation in the perisomatic region, proximal dendrites, and axon initial segment of PHF-tau stained neurons in different cortical areas (PPC, PRC, EC, paraS, preS, Sub, CA1, CA2, CA3/4, and DG) in tissue from control and AD patients. Control cases were simply used to verify that postmortem factors did not alter the staining of the perisomatic terminals typically described in experimental animals and human biopsy tissue (e.g., Alonso-Nanclares and DeFelipe, 2005 ; Merchan-Perez et al., 2009 ). Indeed, the staining pattern of perisomatic terminals was identical to that published previously in both experimental animals and human biopsy tissue. Thus, we considered that the human tissue used here was suitable for the purpose of the study. Because of the limited penetration of the antibodies, we only studied those PHF-tau-ir cells that were situated in the superficial part of the section where both GABAergic and glutamatergic terminals could be clearly labeled.
GABAergic innervation of PHF-tau-ir cells
The pericellular staining of GAT-1, VGAT, and PV around PHF-tau-ir neurons was similar to that found around PHF-tau-negative cells from both control and AD tissue. For example, we examined the innervation of the soma and proximal dendrites by GAT-1 and VGAT-ir terminals of PHF-tau-ir cells with type I staining in detail (n = 364). Normal innervation was apparent in all the regions studied and the perisomatic innervation was indistinguishable between PHF-tau-ir cells and PHF-tau-negative cells (Figures 10 A–F). Remarkably, innervation of the soma and proximal dendrites by GAT-1-ir and VGAT-ir axon terminals was apparently intact even in PHF-tau-ir neurons displaying the type II pattern (n = 382 cells, Figure 11 ). To quantify these observations, we estimated the number of VGAT-ir boutons around PHF-tau-ir and PHF-tau negative neurons (Figures 10 J,K) in two regions (CA1 field and PHG) of patient P7. As shown in Table 4 , the number of perisomatic terminals did not differ significantly around PHF-tau-ir and PHF-tau negative neurons.
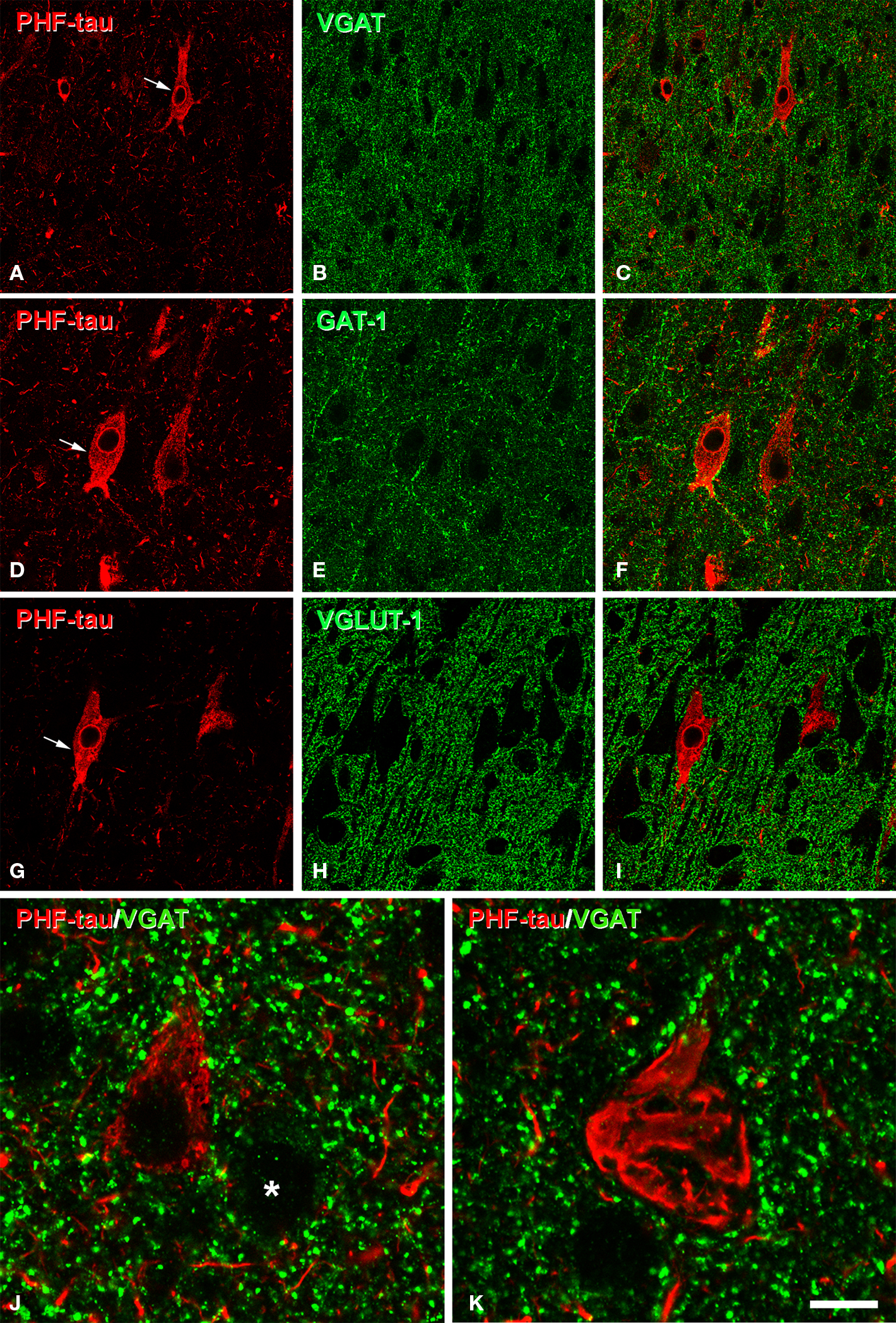
Figure 10. (A,B), (D,E) (G,H), pairs of confocal images from the same section and field showing the distribution of GABAergic (VGAT-ir and GAT-1-ir) and glutamatergic (VGLUT-1-ir) perisomatic axon terminals (green) around the cell body of PHF-tau-negative neurons and PHF-tau-ir neurons with type I staining (red) in the CA2 of patient P7 (A,B), CA1 of patient P7 (D,E), and CA1 of patient P3 (G,H). Arrows indicate some type I PHF-tau-ir neurons. Panels (C,F) and (I) were obtained by combining images (A) and (B), (D) and (E), and (G) and (H), respectively. (J),(K), high magnification confocal images from the entorhinal cortex of patient P7 to illustrate perisomatic VGAT-ir boutons around PHF-tau-negative neurons (asterisk in (J)) and pattern I (J) and pattern II PHF-tau-ir neurons (K). Scale bar: (A–C), 67 μm; (D–I), 43 μm; (J,K), 9 μm.
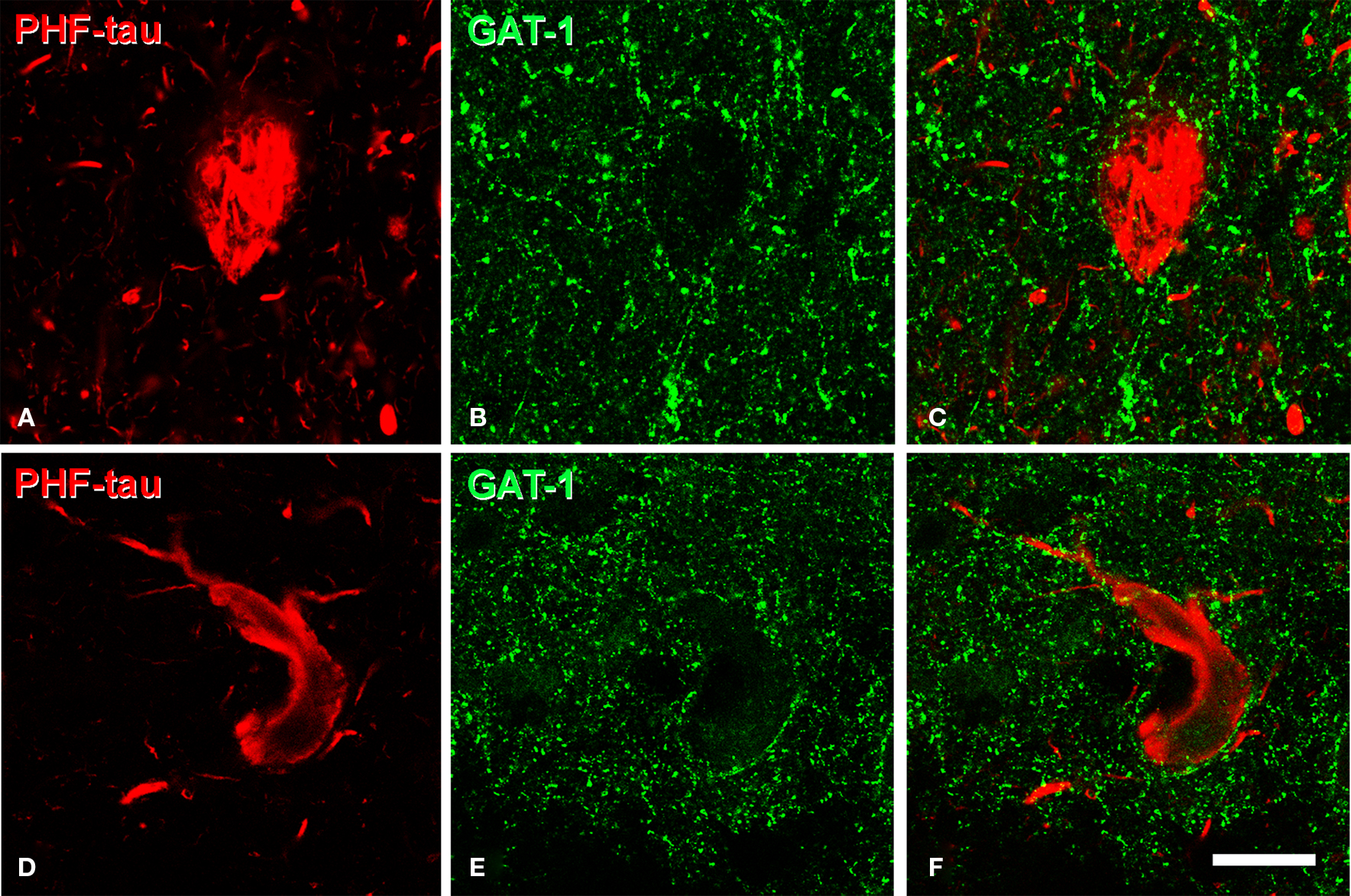
Figure 11. (A,B), (D,E), pairs of confocal images from the same section and field to illustrate the presence of GAT-1-ir axon terminals (green) around the cell bodies of PHF-tau-ir neurons with type II staining (red) in the entorhinal cortex (A–C) and subiculum (D–F) of patients P7 and P1, respectively. Panels (C) and (F) were obtained by combining images (A) and (B), and (D) and (E), respectively. Scale bar: 20 μm.

Table 4. Comparison of the number of VGAT-immunoreactive perisomatic boutons per 100 μm of the perimeter (mean ± SD) of PHF-tau-negative neurons, and of type I and II PHF-tau-ir neurons, in CA1 field and parahippocampal gyrus (PHG; that includes the entorhinal and the perirhinal cortex) of patient P7.
Similarly, the axon initial segment innervated by GAT-1 and VGAT stained chandelier terminals also appeared unaltered in PHF-tau-ir cells displaying type I (n = 10 cells) or II staining (n = 14 cells; Figures 12 A–F).
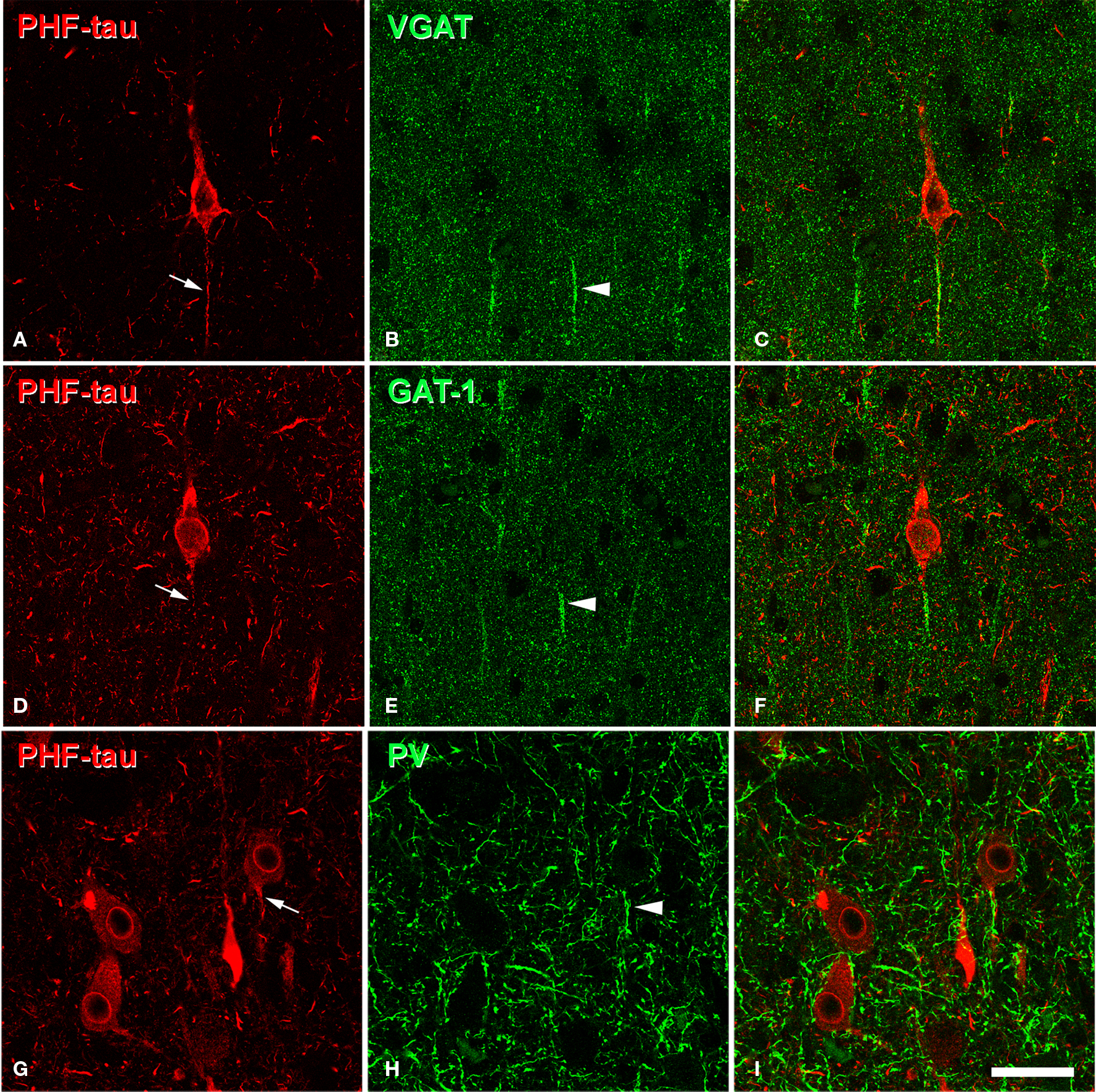
Figure 12. (A,B), (D,E), (G,H), pairs of confocal images from the same section and field to illustrate chandelier cell axon terminals (arrowhead) around the axon initial segment (arrow) of PHF-tau-ir neurons (red) with type II staining in the CA1 field (A,B) and subiculum (D,E) of patient P3, and in CA1 of patient P7 (G,H). Panels (C), (F), and (I) were obtained by combining images (A) and (B), (D) and (E), and (G) and (H), respectively. Chandelier cell axon terminals were labeled using antibodies against VGAT, GAT-1 or PV (green). Scale bar: 43 μm.
To further study the pericellular innervation of PHF-tau-ir cells, we also used dual immunofluorescence for PV and PHF-tau. It is well-established that PV-ir neurons represent a large subpopulation of cortical GABAergic interneurons that includes basket and chandelier cells (DeFelipe, 1993 ). These cells provide the main GABAergic innervation to the soma, proximal dendrites and axon initial segments of pyramidal cells. We again found that the innervation of the soma and the axon initial segment of type I and II stained PHF-tau-ir cells by PV-ir axon terminals (basket and chandelier terminals) was apparently unaltered (Figures 12 G–I and 13 ). In addition, the vast majority of PV-ir cells did not contain PHF-tau (Figures 13 A–C and G–I). To quantify this observation, we counted the number of PV-ir neurons that were also PHF-tau-ir in the hippocampal formation. Of the 3943 PV-ir neurons analyzed (36 in DG; 49 in CA3/4; 396 in CA1; 1120 in Sub; and 2342 in EC), only two cells were PHF-tau-ir (one in the stratum oriens and one in CA3/4). Thus, GABAergic innervation of the soma, proximal dendrites and axon initial segments apparently remains unaltered in AD patients.
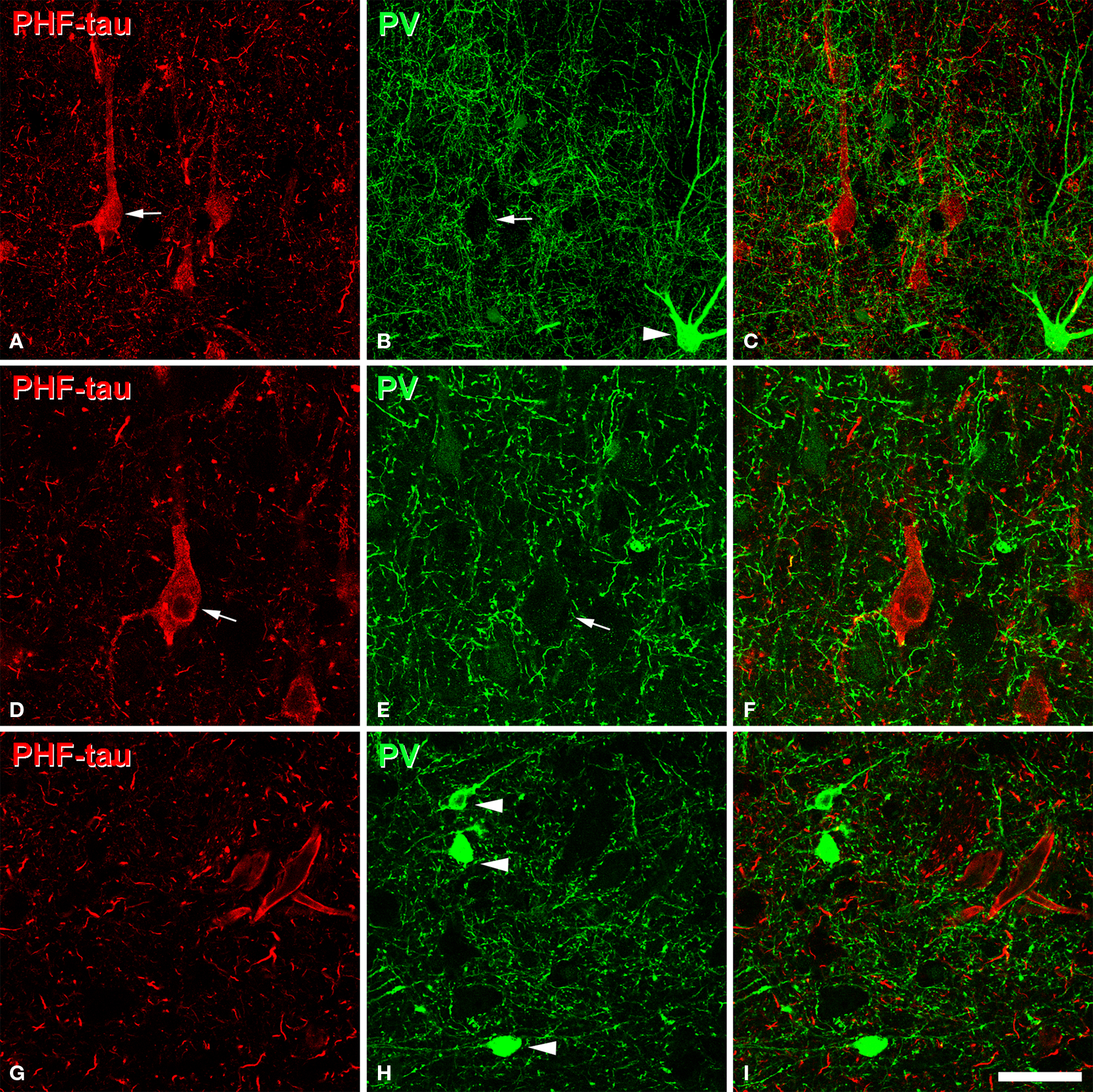
Figure 13. (A,B), pairs of confocal images (stacks of 10 optical sections; step size: 1. 04 μm) to illustrate that PV-ir axon terminals (green) are preserved around the somata of PHF-tau-ir neurons (red) in CA1 (patient P7). (D,E), pairs of single confocal sections showing an PHF-tau-ir pyramidal cell (also indicated in (A) with an arrow) to illustrate its perisomatic innervation by PV-ir axon terminals at higher magnification. (G,H), pairs of single confocal sections showing three PV-ir neurons (arrowhead), none of them are labeled for PHF-tau. Panels (C), (F), and (I) were obtained after combining images (A) and (B), (D) and (E), and (G) and (H), respectively. Scale bar: (A–C), 67 μm; (D–I), 43 μm.
Glutamatergic terminals around PHF-tau-ir cells
We studied the distribution of VGLUT-1-ir glutamatergic axon terminals around PHF-tau-ir cells and like the innervation of GABAergic terminals, the distribution of the VGLUT-1-ir axon terminals was also apparently normal around PHF-tau-ir cells. We examined PHF-tau-ir cells showing a type I (n = 95 cells) and II (n = 103 cells) pattern in detail, comparing them to the perisomatic innervation observed on neurons with no PHF-tau in control tissue or that from AD patients. Again the perisomatic innervation appeared to be similar around PHF-tau-ir and negative neurons (Figures 10 G–I).
Electron Microscopy
The ultrastructure of PHF-tau-ir neurons and the surrounding neuropil was examined using correlative light and electron microscopy (DeFelipe and Fairen, 1993 ). Briefly, 100 μm thick plastic sections containing type I or II stained PHF-tau-ir neurons were first cut into 2 μm semithin sections. Thereafter, selected 2 μm semithin sections were cut into ultrathin sections for electron microscopy. In this way, it was possible the systematically and accurately localize the stained cells and surrounding neuropil in the successive steps of the procedure (Figure 14 ).
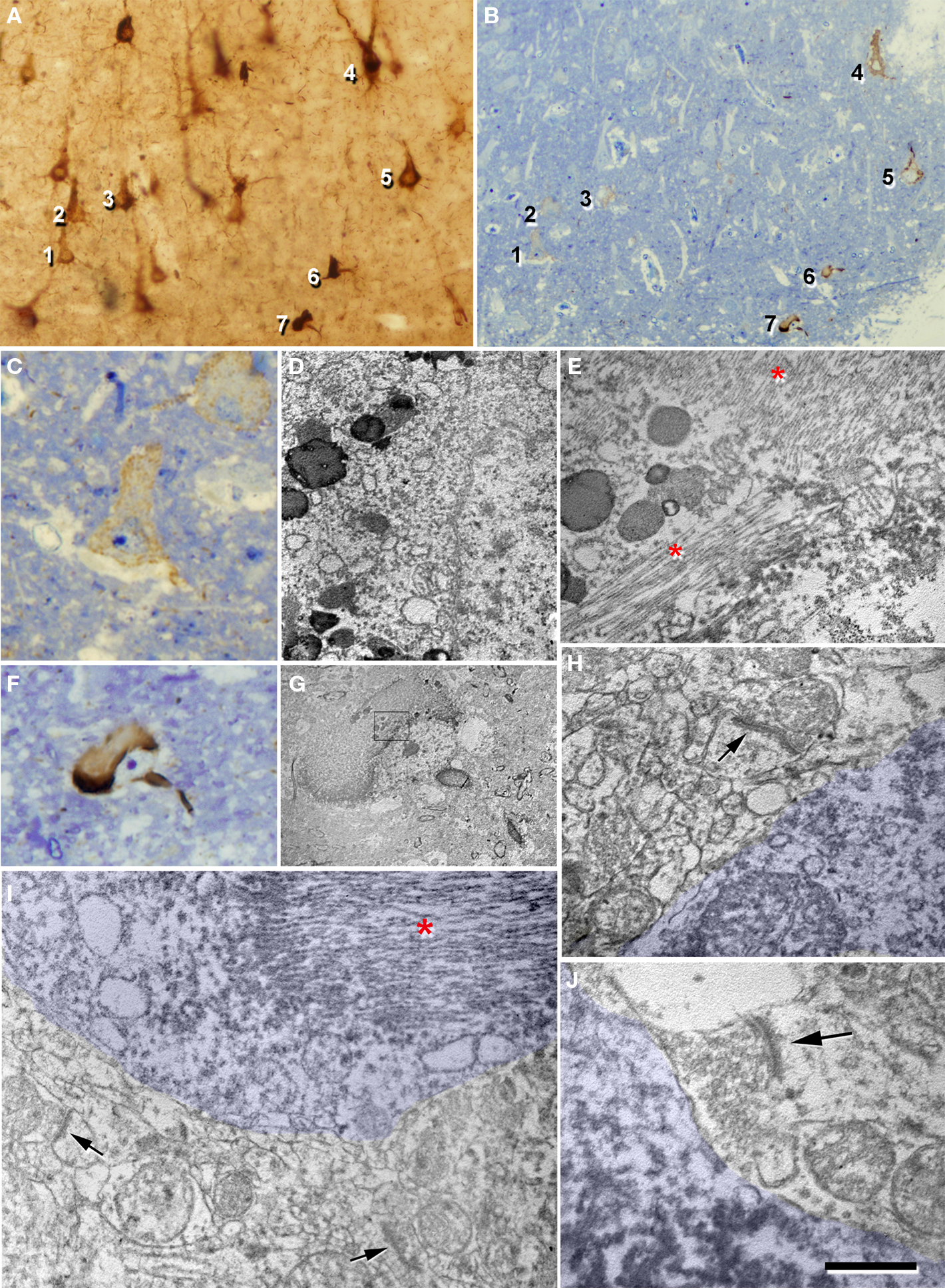
Figure 14. Correlative light and electron microscopy of PHF-tau-ir neurons in the pyramidal layer of CA1 from patient P1. (A) Photomicrograph of a plastic-embedded 100 μm-thick section immunostained for PHF-tau. (B) Photomicrograph of a 2-μm-thick semithin section stained with 1% toluidine blue obtained from the section shown in (A). (C), (F), higher magnification of cells 1 (pattern I of PHF-tau-immunostaining) and 7 (pattern II of PHF-tau-immunostaining), respectively, also shown in (A) and (B). (D), (E,G), electron micrographs of neurons 1 and 7, respectively, obtained after the resectioning of the semithin section shown in (B). The cytoplasm of neuron 1 (D) has a normal appearance, whereas the cytoplasm of neuron 7 (G) and (E) has NFT of paired helical filaments stained for PHF-tau (asterisks in E and I). (H,I,J) Electron micrographs illustrating the neuropil around PHF-tau-ir cells. Arrows indicate some normal looking synaptic contacts situated in close proximity of the PHF-tau-ir cell bodies which have been pseudo-colored in blue. Scale bar: (A), 104 μm; (B), 95 μm; (C,F), 23 μm; (D), 1.5 μm; (E), 1.1 μm; (G), 10 μm; (H), 0.6 μm; (I), 0.8 μm; (J), 0.5 μm.
A total of 16 neurons displaying type I PHF-tau staining were studied by correlative light and electron microscopy and bundles of PHF-tau filaments did not appear to be present in these neurons at the light microscopy or ultrastructural level. Rather, the PHF-tau staining in their cytoplasm consisted of an electron dense granular deposition formed by small particles that were responsible for the homogeneous brownish staining observed under the light microscope. Other ultrastructural characteristics of the cytoplasm in the labeled cells were indistinguishable from those of neurons that did not appear to contain PHF-tau. As a consequence, we considered these neurons to be normal (Figures 14 C,D). Nevertheless, the preservation of the tissue was not optimal and as such we were unable to analyze the ultrastructure of the cytoplasm in greater detail. Hence, it remains possible that these neurons suffer some alterations in their cytoplasm that passed unnoticed.
We also examined 21 neurons displaying type II PHF-tau staining. In these neurons abnormally hyperphosphorylated PHF-tau formed NFT of PHF arranged in relatively large bundles that were packed into the cytoplasm of the soma and proximal dendrites. These bundles were often so large and densely packed that cytoplasmic organelles were completely replaced with these PHF-tau-ir bundles. That is, the regions of the soma or dendrites that contained these bundles were basically lacking other elements or cytoplasmic organelles. However, the rest of the cytoplasm that was not occupied by these abnormal bundles was apparently unaltered (Figures 14 E–G).
Finally, when we analyzed the neuropil surrounding PHF-tau-ir neurons it had a normal aspect, and no reactive glial processes nor abnormal looking dendrite or axonal profiles were observed. Furthermore, the typical symmetric synapses innervating the somata of pyramidal cells were preserved on the PHF-tau-ir neurons with either type I or II staining. In addition, in the neuropil adjacent to these cells normal looking symmetric and asymmetric synapses were also observed (Figures 14 H–J and 15 ). These observations suggest that the synaptic connectivity around these neurons was apparently unaltered.
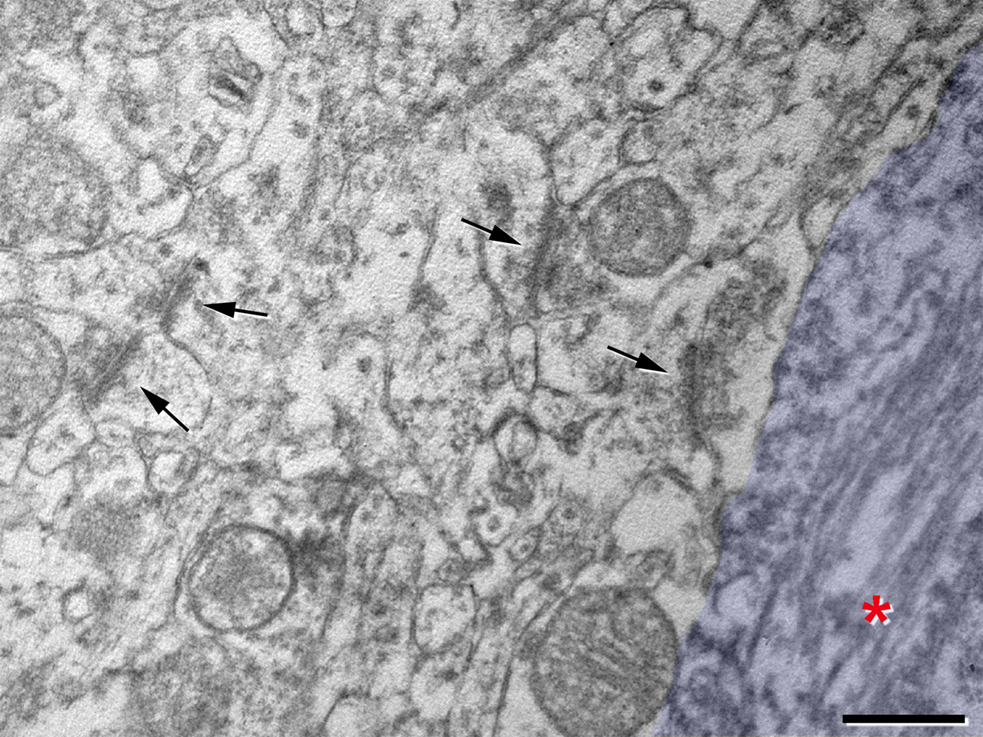
Figure 15. High magnification electron micrograph illustrating the neuropil around the PHF-tau-ir cell 7 also shown in Figure 14 , where the soma has been pseudo-colored in blue. Asterisk, paired helical filaments stained for PHF-tau. Arrows indicate some synaptic contacts in the neuropil. Scale bar: 0.5 μm.
Discussion
There were three main findings from the present study. First, we observed two main patterns of PHF-tau staining in neurons, type I and II patterns, the distribution of which varies according to the cortical layer and area. Second, the distribution of both GABAergic and glutamatergic terminals around the soma and proximal processes of PHF-tau-ir neurons does not seem to be altered, and it is indistinguishable from that in control tissue or in adjacent neurons that do not contain PHF-tau. Third, at the electron microscope level, neurons with type I PHF-tau staining did not develop bundles of PHF-tau filaments and the cytoplasm was apparently normal. However, PHF-tau in type stained II neurons formed NFT of PHF arranged in large bundles that were packed into the cytoplasm of the soma and proximal dendrites. Normal looking symmetric and asymmetric synapses were also observed in the adjacent neuropil.
Distribution of PHF-tau-ir Neurons in the Hippocampal Formation and Adjacent Cortex
In general, the percentage of type I and II PHF-tau-ir neurons in the hippocampal formation and adjacent cortex was similar between the different patients (except in CA2 and the DG), and there were numerous PHF-tau-ir neurons in both AD patients and in control tissue from some individuals. As far as we know, this is the first attempt to quantify the distribution of different patterns of PHF-tau staining in the human hippocampal formation and adjacent cortex in control and AD cases. Thus, we cannot compare these results with those from previous studies. It seems likely that type I staining is due to the accumulation of unpolymerized PHF-tau whereas the type II pattern is due to the accumulation of polymerized PHF-tau into NFT. Whether pattern I represents a pre-tangle stage and therefore precedes pattern II, or if this just occurs in a subpopulation of these neurons representing an independent stage is unclear. The amount of somatic cytoplasm occupied by NFT in type II stained neurons is rather variable. Since there are clearly fewer labeled dendritic processes in neurons with more NFT, it seems that atrophy progresses as type II stained neurons accumulate NFT. However, further studies using intracellular injection of Lucifer Yellow, for example, or other markers to visualize the complete dendritic arbor of the neurons will be necessary to ascertain whether neurons with type II staining have fewer dendritic branches than those showing a type I pattern. Whatever the relationship between the type I and II pattern, these observations further emphasize that neurons selectively accumulate PHF-tau in a region dependent manner, either as NFT or in the unpolymerized form.
Most affected neurons in regions other than the DG can be recognized as pyramidal neurons as would be expected given the large number of neurons affected and the fact that these neurons represent the vast majority of cortical neurons. However, PV-ir neurons were rarely PHF-tau-ir and they represent a large subpopulation of GABAergic interneurons (Celio, 1986 ; DeFelipe, 1993 ; Burkhalter, 2008 ). Although we cannot rule out that PV/PHF-tau-ir neurons are lost by cell death, it seems unlikely given the preservation of the basket and chandelier axon terminals that originate from neurons expressing PV (see below), even in cortical regions with abundant NFT. Thus, basket and chandelier cells are unaffected by tau pathologies. In addition, pyramidal cells located in different cortical regions and layers of the hippocampal formation and adjacent cortex project to different targets (Insausti and Amaral, 2004 ; Duvernoy, 2005 ). Since the density of PHF-tau-ir pyramidal neurons varies in function of both the cortical region and cortical layer, this further emphasizes that the malfunction of synaptic circuits in which PHF-tau-ir neurons participate is probably very specific (see also reviewed in Morrison and Hof, 2007 ; Lewis et al., 1987 ; Hof and Morrison, 1990 ; Hof et al., 1990 , 1995 ; Arnold et al., 1991 ; Gomez-Isla et al., 1996 ; ).
Pericellular Innervation of PHF-tau-ir Neurons
At both the confocal and ultrastructural levels, the perisomatic distribution of glutamatergic and GABAergic terminals apparently remains unaltered in all PHF-tau-ir neurons studied in the different hippocampal regions examined. Although only GABAergic terminals establish axo-somatic synapses, both glutamatergic and GABAergic terminals are typically found in the perisomatic region (Merchan-Perez et al., 2009 ). This is in line with other studies suggesting that GABAergic circuits in the cerebral cortex of AD patients are relatively well preserved when compared to circuits that use other neurotransmitters (Young, 1987 ; Lowe et al., 1988 ; Reinikainen et al., 1988 ; Ferrer et al., 1991 ; Hof et al., 1991 ; Nägga et al., 1999 ; Bell et al., 2003 ; Rissman et al., 2007 ). However, other studies indicated possible alterations to the PV-ir basket and chandelier cells in the cerebral cortex of AD patients that are responsible for the GABAergic pericellular innervation of pyramidal cells (reviewed in Jones, 1993DeFelipe et al., 1989 ; Freund and Buzsaki, 1996 ; DeFelipe, 1999 ). In general, it seems that alterations to PV-ir cells are not homogeneous throughout the cerebral cortex of AD patients. For example, the subpopulation of PV-ir cells apparently remains unaltered in the prefrontal and inferior temporal cortex (Hof et al., 1991 ) and in the temporal cortex (Ferrer et al., 1991 ), whereas PV-ir cells appear to be more vulnerable in the hippocampal formation of AD patients (Brady and Mufson, 1997 ). Indeed, a 60% decrease in the number of PV-ir interneurons was observed in the DG/CA4, CA1 and CA2 but not in the CA3, Sub or preS (Brady and Mufson, 1997 ). A loss of PV-ir neurons in different layers of the EC has also been reported (Solodkin et al., 1996 ; Mikkonen et al., 1999 ).
A decrease in PV-ir chandelier terminals has been described in layer II of the temporal cortex (Fonseca et al., 1993 ), as well as a decrease in the intensity of PV staining in the neuropil (Solodkin et al., 1996 ). However, this decrease in PV-ir terminals was thought to be related to the loss of the pyramidal target neurons rather than to a loss of PV-ir neurons. Nevertheless, the relationship between the PV-ir presynaptic terminals and their postsynaptic targets, either PHF-tau-ir or negative, has yet not been studied specifically. Thus, one of the goals of this study was to examine the pericellular innervation and the neuropil surrounding PHF-tau-ir neurons. We did not observe any loss of GABAergic or glutamatergic axon terminals around the soma of PHF-tau-ir neurons. The fact that the presynaptic terminals of basket and chandelier cells were still present around type I PHF-tau-ir neurons was expected as these neurons look normal. However, the innervation of type II PHF-tau-ir neurons by these terminals was more surprising since many of these neurons are atrophied. Therefore, it seems that the connections between GABAergic basket/chandelier cells and PHF-tau-ir neurons remain unaltered. Furthermore, our study shows that PV-ir interneurons rarely express PHF-tau in the hippocampal formation of different types of AD patients, supporting the idea that this subpopulation of GABAergic cells is in general well preserved. However, alterations in the synaptic connections may exist at the molecular or physiological level, and further studies on animal models will be necessary to resolve this question.
Finally, the discrepancy observed between different laboratories regarding the loss or preservation of GABAergic neurons may be explained by differences in tissue processing, in the methods of analysis (quantitative versus qualitative studies), and/or in the postmortem delay, together with the clinical variability between patients and control tissue.
Significance of PHF-tau
One of the striking findings in relation to the neuropathological aspects of AD is the fact that abundant Aβ plaques and/or NFT may also be found in non-demented elderly and young control individuals. Thus, there is a poor correlation between the degree of cognitive impairment and the presence of Aβ plaques and NFT, in accordance with other pathological findings such as neuronal loss (Crystal et al., 1988 ; Katzman et al., 1988 ; Price et al., 1991 ; Arriagada et al., 1992 ; Dickson et al., 1992 ; see Price et al., 2009 for a recent study). Indeed, in the present study, two of the seven control cases (C2 and C4) displayed dense accumulations of Aβ plaques or PHF-tau-ir neurons, respectively. Thus, the development of Aβ plaques and NFT seems to be a natural aging process in certain individuals, although it is greatly exacerbated or accelerated in AD.
There is some controversy about the significance of the intracellular accumulation of NFT (for a recent review see Hernandez and Avila, 2008 ). Initially, it was thought that the presence of NFT was intimately linked to cell death and areas that were more severely damaged reflected the areas with more NFT in the human hippocampal formation (Hyman et al., 1984 ). These areas were the Sub, layer II and layer IV of the EC. It was proposed that the presence of NFT is associated with impaired axonal transport which would culminate in neuronal death (Stamer et al., 2002 ; Terwel et al., 2002 ; Cente et al., 2006 ; Smith et al., 2007 ). However, NFT accumulation might represent a compensatory strategy (at least initially) to protect neurons against oxidative stress or toxic cytosolic PHF-tau (Alonso Adel et al., 2006 ; Nunomura et al., 2006 ; Hernandez and Avila, 2007 ; Iqbal et al., 2008 ). Studies on transgenic mouse models have demonstrated that the presence of NFT does not necessarily lead to cell death, and it was shown that neuronal loss in the DG preceded the formation of NFT (Spires et al., 2006 ). In addition, NFT were found in the striatum without any evidence of neuronal loss. In the same mouse model, caspase activation occurs in neurons containing NFT indicative of apoptotic death (Spires-Jones et al., 2008 ). However, this caspase activation seems to be insufficient to cause acute neuronal death as these neurons do not express either morphological or biochemical changes typically found in classical apoptosis. Thus, it was proposed that forming NFT could be an anti-apoptotic strategy that would delay neuronal death even after caspase activation (Spires-Jones et al., 2008 ).
Relationship Between Tauopathies and Synapses
Irrespective of whether PHF-tau-ir neurons finally die or not, their perisomatic innervation seems to be unaltered. In other words, it seems that these perisomatic axons do not degenerate, nor are they eliminated by glial phagocytosis (“synaptic stripping”: see Blinzinger and Kreutzberg, 1968 ; Moran and Graeber, 2004 ) due to the expression of PHF-tau, at least at the stages of the disease examined in the present work. In addition, a normal looking neuropil with typical symmetric and asymmetric synapses were also observed in the neuropil adjacent to these cells. However, we cannot conclude that the normal activity of the cell is not compromised. That is, we do not know if the synapses established with PHF-tau-ir somata and dendrites are functionally normal at the presynaptic level and/or at the postsynaptic level. Indeed, the dendritic arbor of type II stained neurons seems to be atrophied and thus their function must be altered even though synaptic connections are still present in the perisomatic region. Thus, PHF-tau accumulation could cause synaptic dysfunction in the absence of clear morphological changes.
Tauopathies certainly lead to cognitive decline or dementia in the absence of Aβ and therefore, neurofibrillary lesions may be sufficient to explain this cognitive decline. Thus, what is the contribution of Aβ plaques to this process? Aβ plaques induce local morphological alterations to the dendrites in contact with Aβ (Tsai et al., 2004 ; Spires et al., 2005 ; Knafo et al., 2009a ,b ). These alterations include the sprouting of spines on dendrites contacting Aβ plaques, the loss of dendritic spines and the thinning of dendritic shafts passing through Aβ plaques. Therefore, the loss or morphological alteration of spines provoked by Aβ plaques in the neuropil may be related to local alterations, mostly to axo-spinous glutamatergic synapses. Furthermore, we recently found that neurons in contact with Aβ plaques appear to be normal at the ultrastructural level in the APP/PS1 mouse model of AD, although the membrane of the neuronal somata in contact with the Aβ plaque lacked GABAergic axo-somatic synapses in both the APP/PS1 mouse and in AD patients (Garcia-Marin et al., 2009 ). In addition, the portion of the soma of these neurons that was not in contact with the Aβ plaques established typical synapses, whereas synapses are absent from the region in direct contact with the Aβ plaques. Thus, direct contact of the Aβ plaque with the cell body does not induce perisomatic disconnection further away from the contact domain. Based on these findings, we proposed that dendrites are more susceptible to the toxic effect of Aβ than the cell body, which should clearly have important consequences on cortical circuits. Furthermore, we found a significant decrease in the frequency of large spines in Aβ plaque-free regions of the DG in the APP/PS1 mice model of AD (Knafo et al., 2009a ,b ). Because large spines are considered to be the physical traces of long-term memory (Kasai et al., 2003 ; Bourne and Harris, 2007 ), a widespread decrease in the frequency of large spines probably contributes to the cognitive impairments observed in this AD model. Whether this also occurs in AD patients is not yet known, although partial GABAergic disconnection at the level of the somata of neurons in contact with the Aβ plaques has been shown to occur both in the mice model of AD and in AD patients. Since the distribution of Aβ plaques and PHF-tau-ir neurons coincided in some regions but not in others, their local densities are rather variable and they have different impact on microcircuits. Thus, to better understand the correlation between the presence of Aβ plaques and NFT with cognitive impairment, this relationship should be studied in greater detail, at the level of the microanatomical changes that occur in the different cortical regions of AD patients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that should be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Jesús ávila and Ricardo Insausti for their helpful comments on the manuscript. We also wish to thank A.I. García for technical assistance, C. Hernández and B. García for assistance with the confocal microscopy and Dr. R. Alcaraz (Servicio de Patología Forense, Instituto Vasco de Medicina Legal, Bilbao, Spain), Dr. I. Ferrer (Instituto de Neuropatología, Servicio de Anatomía Patológica, IDIBELL-Hospital Universitario de Bellvitge, Universidad de Barcelona, Hospitalet de Llobregat, Spain) and Dr. A. Rábano (área de Neuropatología, Banco de Tejidos Fundación CIEN, Centro Alzheimer, Fundación Reina Sofia, Madrid, Spain) for supplying the brain tissue. This work was supported by grants from the following entities: CIBERNED (CB06/05/0066), Fundación CIEN (Financiación de Proyectos de Investigación de Enfermedad de Alzheimer y enfermedades relacionadas 2008), Fundación Caixa (BM05-47-0), the EU 6th Framework Program (PROMEMORIA LSHM-CT-2005-512012), the Spanish Ministerio de Educación Ciencia e Innovación (BFU2006-13395 and SAF2009-09394), and the Spanish Ministerio de Ciencia y Tecnología (FPU AP20050690 to Lidia Blazquez-Llorca).
References
Alafuzoff, I., Arzberger, T., Al-Sarraj, S., Bodi, I., Bogdanovic, N., Braak, H., Bugiani, O., Del Tredici, K., Ferrer, I., Gelpi, E., Giaccone, G., Graeber, M. B., Ince, P., Kamphorst, W., King, A., Korkolopoulou, P., Kovacs, G. G., Larionov, S., Meyronet, D., Monoranu, C., Parchi, P., Patsouris, E., Roggendorf, W., Seilhean, D., Tagliavini, F., Stadelmann, C., Streichenberger, N., Thal, D. R., Wharton, S. B., and Kretzschmar, H. (2008). Staging of neurofibrillary pathology in Alzheimer’s disease: a study of the BrainNet Europe Consortium. Brain Pathol. 18, 484–496.
Alonso Adel, C., Li, B., Grundke-Iqbal, I., and Iqbal, K. (2006). Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc. Natl. Acad. Sci. U.S.A. 103, 8864–8869.
Alonso-Nanclares, L., and DeFelipe, J. (2005). Vesicular glutamate transporter 1 immunostaining in the normal and epileptic human cerebral cortex. Neuroscience 134, 59–68.
Alonso-Nanclares, L., Minelli, A., Melone, M., Edwards, R. H., DeFelipe, J., and Conti, F. (2004). Perisomatic glutamatergic axon terminals: a novel feature of cortical synaptology revealed by vesicular glutamate transporter 1 immunostaining. Neuroscience 123, 547–556.
Alzheimer, A. (1911). Über eine eigenartige Krankheitsfälle des späten Alters. Z. Gesamte Neurol Psychiatr. 4, 356–385.
Arellano, J. I., Munoz, A., Ballesteros-Yanez, I., Sola, R. G., and DeFelipe, J. (2004). Histopathology and reorganization of chandelier cells in the human epileptic sclerotic hippocampus. Brain 127, 45–64.
Arendt, T., Bruckner, M. K., Gertz, H. J., and Marcova, L. (1998). Cortical distribution of neurofibrillary tangles in Alzheimer’s disease matches the pattern of neurons that retain their capacity of plastic remodelling in the adult brain. Neuroscience 83, 991–1002.
Arnold, S. E., Hyman, B. T., Flory, J., Damasio, A. R., and Van Hoesen, G. W. (1991). The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb. Cortex 1, 103–116.
Arriagada, P. V., Growdon, J. H., Hedley-Whyte, E. T., and Hyman, B. T. (1992). Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639.
Bancher, C., Brunner, C., Lassmann, H., Budka, H., Jellinger, K., Wiche, G., Seitelberger, F., Grundke-Iqbal, I., Iqbal, K., and Wisniewski, H. M. (1989). Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res. 477, 90–99.
Bell, K. F., de Kort, G. J., Steggerda, S., Shigemoto, R., Ribeiro-da-Silva, A., and Cuello, A. C. (2003). Structural involvement of the glutamatergic presynaptic boutons in a transgenic mouse model expressing early onset amyloid pathology. Neurosci. Lett. 353, 143–147.
Bellocchio, E. E., Hu, H., Pohorille, A., Chan, J., Pickel, V. M., and Edwards, R. H. (1998). The localization of the brain-specific inorganic phosphate transporter suggests a specific presynaptic role in glutamatergic transmission. J. Neurosci. 18, 8648–8659.
Blinzinger, K., and Kreutzberg, G. (1968). Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z. Zellforsch. Mikrosk. Anat. 85, 145–157.
Bourne, J., and Harris, K. M. (2007). Do thin spines learn to be mushroom spines that remember? Curr. Opin. Neurobiol. 17, 381–386.
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H., and Del Tredici, K. (2006). Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404.
Braak, E., Braak, H., and Mandelkow, E. M. (1994). A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 87, 554–567.
Braak, H., and Braak, E. (1986). Ratio of pyramidal cells versus non-pyramidal cells in the human frontal isocortex and changes in ratio with ageing and Alzheimer’s disease. Prog. Brain Res. 70, 185–212.
Braak, H., and Braak, E. (1987). Projection neurons of basolateral amygdaloid nuclei develop meganeurites in juvenile and adult human neuronal ceroid lipofuscinosis. Clin. Neuropathol. 6, 116–119.
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259.
Braak, H., and Braak, E. (1995). Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 16, 271–278.
Braak, H., Braak, E., and Kalus, P. (1989). Alzheimer’s disease: areal and laminar pathology in the occipital isocortex. Acta Neuropathol. 77, 494–506.
Braak, H., Del Tredici, K., Schultz, C., and Braak, E. (2000). Vulnerability of select neuronal types to Alzheimer’s disease. Ann. N. Y. Acad. Sci. 924, 53–61.
Brady, D. R., and Mufson, E. J. (1997). Parvalbumin-immunoreactive neurons in the hippocampal formation of Alzheimer’s diseased brain. Neuroscience 80, 1113–1125.
Brion, J. P., Couck, A. M., Passareiro, E., and Flament-Durand, J. (1985). Neurofibrillary tangles of Alzheimer’s disease: an immunohistochemical study. J. Submicrosc. Cytol. 17, 89–96.
Burkhalter, A. (2008). Many specialists for suppressing cortical excitation. Front. Neurosci. 2, 155–167.
Celio, M. R. (1986). Parvalbumin in most gamma-aminobutyric acid-containing neurons of the rat cerebral cortex. Science 231, 995–997.
Cente, M., Filipcik, P., Pevalova, M., and Novak, M. (2006). Expression of a truncated tau protein induces oxidative stress in a rodent model of tauopathy. Eur. J. Neurosci. 24, 1085–1090.
Chaudhry, F. A., Reimer, R. J., Bellocchio, E. E., Danbolt, N. C., Osen, K. K., Edwards, R. H., and Storm-Mathisen, J. (1998). The vesicular GABA transporter, VGAT, localizes to synaptic vesicles in sets of glycinergic as well as GABAergic neurons. J. Neurosci. 18, 9733–9750.
Cobb, S. R., and Davies, C. H. (2005). Cholinergic modulation of hippocampal cells and circuits. J. Physiol. 562, 81–88.
Crystal, H., Dickson, D., Fuld, P., Masur, D., Scott, R., Mehler, M., Masdeu, J., Kawas, C., Aronson, M., and Wolfson, L. (1988). Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology 38, 1682–1687.
DeFelipe, J. (1993). Neocortical neuronal diversity: chemical heterogeneity revealed by colocalization studies of classic neurotransmitters, neuropeptides, calcium-binding proteins, and cell surface molecules. Cereb. Cortex 3, 273–289.
DeFelipe, J., and Fairen, A. (1993). A simple and reliable method for correlative light and electron microscopic studies. J. Histochem. Cytochem. 41, 769–772.
DeFelipe, J., and Farinas, I. (1992). The pyramidal neuron of the cerebral cortex: morphological and chemical characteristics of the synaptic inputs. Prog. Neurobiol. 39, 563–607.
DeFelipe, J., and Gonzalez-Albo, M. C. (1998). Chandelier cell axons are immunoreactive for GAT-1 in the human neocortex. Neuroreport 9, 467–470.
DeFelipe, J., Hendry, S. H., and Jones, E. G. (1989). Visualization of chandelier cell axons by parvalbumin immunoreactivity in monkey cerebral cortex. Proc. Natl. Acad. Sci. U.S.A. 86, 2093–2097.
Dickson, D. W., Crystal, H. A., Mattiace, L. A., Masur, D. M., Blau, A. D., Davies, P., Yen, S. H., and Aronson, M. K. (1992). Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol. Aging 13, 179–189.
Dumoulin, A., Rostaing, P., Bedet, C., Levi, S., Isambert, M. F., Henry, J. P., Triller, A., and Gasnier, B. (1999). Presence of the vesicular inhibitory amino acid transporter in GABAergic and glycinergic synaptic terminal boutons. J. Cell. Sci. 112(Pt 6), 811–823.
Duvernoy, H. M. (2005). The Human Hippocampus: Functional Anatomy, Vascularization and Serial Sections with MRI. Berlin, Heidelberg: Springer-Verlag.
Ferrer, I., Santpere, G., and van Leeuwen, F. W. (2008). Argyrophilic grain disease. Brain 131, 1416–1432.
Ferrer, I., Soriano, E., Tunon, T., Fonseca, M., and Guionnet, N. (1991). Parvalbumin immunoreactive neurons in normal human temporal neocortex and in patients with Alzheimer’s disease. J. Neurol. Sci. 106, 135–141.
Fonseca, M., Soriano, E., Ferrer, I., Martinez, A., and Tunon, T. (1993). Chandelier cell axons identified by parvalbumin-immunoreactivity in the normal human temporal cortex and in Alzheimer’s disease. Neuroscience 55, 1107–1116.
Fremeau, R. T. Jr., Troyer, M. D., Pahner, I., Nygaard, G. O., Tran, C. H., Reimer, R. J., Bellocchio, E. E., Fortin, D., Storm-Mathisen, J., and Edwards, R. H. (2001). The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron 31, 247–260.
Garcia-Marin, V., Blazquez-Llorca, L., Rodriguez, J. R., Boluda, S., Muntane, G., Ferrer, I., and DeFelipe, J. (2009). Diminished perisomatic GABAergic terminals on cortical neurons adjacent to amyloid plaques. Front. Neuroanat. 3, 28.
Goedert, M., and Spillantine, M. G. (2006). “Neurodegenerative alpha-synucleinopathies and tauopathies.” in Basic Neurochemistry: Molecular, Cellular, and Medical Aspects, eds G. J. Siegel, R. W. Albers, S. T. Brady, and D. L. Price (Burlington, MA: Elsevier Academic Press), 745–760.
Gomez-Isla, T., Price, J. L., McKeel, D. W. Jr., Morris, J. C., Growdon, J. H., and Hyman, B. T. (1996). Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci. 16, 4491–4500.
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917.
Hernandez, F., and Avila, J. (2008). Tau aggregates and tau pathology. J. Alzheimers Dis. 14, 449–452.
Hirano, A., and Zimmerman, H. M. (1962). Alzheimer’s neurofibrillary changes. A topographic study. Arch. Neurol. 7, 227–242.
Hof, P. R., Cox, K., and Morrison, J. H. (1990). Quantitative analysis of a vulnerable subset of pyramidal neurons in Alzheimer’s disease: I. Superior frontal and inferior temporal cortex. J. Comp. Neurol. 301, 44–54.
Hof, P. R., Cox, K., Young, W. G., Celio, M. R., Rogers, J., and Morrison, J. H. (1991). Parvalbumin-immunoreactive neurons in the neocortex are resistant to degeneration in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 50, 451–462.
Hof, P. R., Giannakopoulos, P., Vickers, J. C., Bouras, C., and Morrison, J. H. (1995). The morphologic and neurochemical basis of dementia: aging, hierarchical patterns of lesion distribution and vulnerable neuronal phenotype. Rev. Neurosci. 6, 97–124.
Hof, P. R., and Morrison, J. H. (1990). Quantitative analysis of a vulnerable subset of pyramidal neurons in Alzheimer’s disease: II. Primary and secondary visual cortex. J. Comp. Neurol. 301, 55–64.
Houser, C. R., Barber, R. P., and Vaughn, J. E. (1984). Immunocytochemical localization of glutamic acid decarboxylase in the dorsal lateral vestibular nucleus: evidence for an intrinsic and extrinsic GABAergic innervation. Neurosci. Lett. 47, 213–220.
Howard, A., Tamas, G., and Soltesz, I. (2005). Lighting the chandelier: new vistas for axo-axonic cells. Trends Neurosci. 28, 310–316.
Hyman, B. T., and Gomez-Isla, T. (1994). Alzheimer’s disease is a laminar, regional, and neural system specific disease, not a global brain disease. Neurobiol. Aging 15, 353–354.
Hyman, B. T., Van Hoesen, G. W., Damasio, A. R., and Barnes, C. L. (1984). Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science 225, 1168–1170.
Hyman, B. T., Van Hoesen, G. W., Kromer, L. J., and Damasio, A. R. (1986). Perforant pathway changes and the memory impairment of Alzheimer’s disease. Ann. Neurol. 20, 472–481.
Insausti, R., and Amaral, D. G. (2004). “Hippocampal formation,” in The Human Nervous System, eds G. Paxinos, and J. K. Mai (San Diego, CA: Elsevier Academic Press), 872–915.
Iqbal, K., Alonso Adel, C., and Grundke-Iqbal, I. (2008). Cytosolic abnormally hyperphosphorylated tau but not paired helical filaments sequester normal MAPs and inhibit microtubule assembly. J. Alzheimers Dis. 14, 365–370.
Iqbal, K., Grundke-Iqbal, I., Zaidi, T., Merz, P. A., Wen, G. Y., Shaikh, S. S., Wisniewski, H. M., Alafuzoff, I., Winblad, B. (1986). Defective brain microtubule assembly in Alzheimer’s disease. Lancet 2, 421–426.
Iwamoto, N., and Emson, P. C. (1991). Demonstration of neurofibrillary tangles in parvalbumin-immunoreactive interneurones in the cerebral cortex of Alzheimer-type dementia brain. Neurosci. Lett. 128, 81–84.
Jones, E. G. (1993). GABAergic neurons and their role in cortical plasticity in primates. Cereb. Cortex 3, 361–372.
Josephs, K. A., Whitwell, J. L., Parisi, J. E., Knopman, D. S., Boeve, B. F., Geda, Y. E., Jack, C. R. Jr., Petersen, R. C., and Dickson, D. W. (2008). Argyrophilic grains: a distinct disease or an additive pathology? Neurobiol. Aging 29, 566–573.
Kaneko, T., and Fujiyama, F. (2002). Complementary distribution of vesicular glutamate transporters in the central nervous system. Neurosci. Res. 42, 243–250.
Kasai, H., Matsuzaki, M., Noguchi, J., Yasumatsu, N., and Nakahara, H. (2003). Structure-stability-function relationships of dendritic spines. Trends Neurosci. 26, 360–368.
Katzman, R., Terry, R., DeTeresa, R., Brown, T., Davies, P., Fuld, P., Renbing, X., and Peck, A. (1988). Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann. Neurol. 23, 138–144.
Kidd, M. (1963). Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature 197, 192–193.
Klausberger, T., Marton, L. F., Baude, A., Roberts, J. D., Magill, P. J., and Somogyi, P. (2004). Spike timing of dendrite-targeting bistratified cells during hippocampal network oscillations in vivo. Nat. Neurosci. 7, 41–47.
Knafo, S., Alonso-Nanclares, L., Gonzalez-Soriano, J., Merino-Serrais, P., Fernaud-Espinosa, I., Ferrer, I., and DeFelipe, J. (2009a). Widespread changes in dendritic spines in a model of Alzheimer’s disease. Cereb. Cortex 19, 586–592.
Knafo, S., Venero, C., Merino-Serrais, P., Fernaud-Espinosa, I., Gonzalez-Soriano, J., Ferrer, I., Santpere, G., and DeFelipe, J. (2009b). Morphological alterations to neurons of the amygdala and impaired fear conditioning in a transgenic mouse model of Alzheimer’s disease. J. Pathol. 219, 41–51.
Kosik, K. S., Joachim, C. L., and Selkoe, D. J. (1986). Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 83, 4044–4048.
Lewis, D. A., Campbell, M. J., Terry, R. D., and Morrison, J. H. (1987). Laminar and regional distributions of neurofibrillary tangles and neuritic plaques in Alzheimer’s disease: a quantitative study of visual and auditory cortices. J. Neurosci. 7, 1799–1808.
Lorente de Nó, R. (1934). Studies on the structure of the cerebral cortex. II. Continuation of the study of the ammonic system. J. Psychol. Neurol. 46, 113–177.
Lowe, S. L., Francis, P. T., Procter, A. W., Palmer, A. M., Davison, A. N., and Bowen, D. M. (1988). Gamma-aminobutyric acid concentration in brain tissue at two stages of Alzheimer’s disease. Brain 111(Pt 4), 785–799.
Matsuo, E. S., Shin, R. W., Billingsley, M. L., Van deVoorde, A., O’Connor, M., Trojanowski, J. Q., Lee, V. M. (1994). Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron 13, 989–1002.
Merchan-Perez, A., Rodriguez, J. R., Ribak, C. E., and DeFelipe, J. (2009). Proximity of excitatory and inhibitory axon terminals adjacent to pyramidal cell bodies provides a putative basis for nonsynaptic interactions. Proc. Natl. Acad. Sci. U.S.A. 106, 9878–9883.
Mikkonen, M., Alafuzoff, I., Tapiola, T., Soininen, H., and Miettinen, R. (1999). Subfield- and layer-specific changes in parvalbumin, calretinin and calbindin-D28K immunoreactivity in the entorhinal cortex in Alzheimer’s disease. Neuroscience 92, 515–532.
Miles, R., Toth, K., Gulyas, A. I., Hajos, N., and Freund, T. F. (1996). Differences between somatic and dendritic inhibition in the hippocampus. Neuron 16, 815–823.
Minelli, A., Alonso-Nanclares, L., Edwards, R. H., DeFelipe, J., and Conti, F. (2003). Postnatal development of the vesicular GABA transporter in rat cerebral cortex. Neuroscience 117, 337–346.
Minelli, A., Brecha, N. C., Karschin, C., DeBiasi, S., and Conti, F. (1995). GAT-1, a high-affinity GABA plasma membrane transporter, is localized to neurons and astroglia in the cerebral cortex. J. Neurosci. 15, 7734–7746.
Moran, L. B., and Graeber, M. B. (2004). The facial nerve axotomy model. Brain Res. Brain Res. Rev. 44, 154–178.
Morrison, B. M., Hof, P. R., and Morrison, J. H. (1998). Determinants of neuronal vulnerability in neurodegenerative diseases. Ann. Neurol. 44, S32–S44.
Morrison, J. H., and Hof, P. R. (2002). Selective vulnerability of corticocortical and hippocampal circuits in aging and Alzheimer’s disease. Prog. Brain Res. 136, 467–486.
Morrison, J. H., and Hof, P. R. (2007). Life and death of neurons in the aging cerebral cortex. Int. Rev. Neurobiol. 81, 41–57.
Mountjoy, C. Q., Roth, M., Evans, N. J., and Evans, H. M. (1983). Cortical neuronal counts in normal elderly controls and demented patients. Neurobiol. Aging 4, 1–11.
Nägga, K., Bogdanovic, N., and Marcusson, J. (1999). GABA transporters (GAT-1) in Alzheimer’s disease. J. Neural Transm. 106, 1141–1149.
Ni, B., Du, Y., Wu, X., DeHoff, B. S., Rosteck, P. R. Jr., and Paul, S. M. (1996). Molecular cloning, expression, and chromosomal localization of a human brain-specific Na(+)-dependent inorganic phosphate cotransporter. J. Neurochem. 66, 2227–2238.
Nunomura, A., Castellani, R. J., Zhu, X., Moreira, P. I., Perry, G., and Smith, M. A. (2006). Involvement of oxidative stress in Alzheimer disease. J. Neuropathol. Exp. Neurol. 65, 631–641.
Pearson, R. C., Esiri, M. M., Hiorns, R. W., Wilcock, G. K., and Powell, T. P. (1985). Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 82, 4531–4534.
Porzig, R., Singer, D., and Hoffmann, R. (2007). Epitope mapping of mAbs AT8 and Tau5 directed against hyperphosphorylated regions of the human tau protein. Biochem. Biophys. Res. Commun. 358, 644–649.
Price, J. L., Davis, P. B., Morris, J. C., and White, D. L. (1991). The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer’s disease. Neurobiol. Aging 12, 295–312.
Price, J. L., McKeel, D. W. Jr., Buckles, V. D., Roe, C. M., Xiong, C., Grundman, M., Hansen, L. A., Petersen, R. C., Parisi, J. E., Dickson, D. W., Smith, C. D., Davis, D. G., Schmitt, F. A., Markesbery, W. R., Kaye, J., Kurlan, R., Hulette, C., Kurland, B. F., Higdon, R., Kukull, W., and Morris, J. C. (2009). Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol. Aging 30, 1026–1036.
Reinikainen, K. J., Paljarvi, L., Huuskonen, M., Soininen, H., Laakso, M., and Riekkinen, P. J. (1988). A post-mortem study of noradrenergic, serotonergic and GABAergic neurons in Alzheimer’s disease. J. Neurol. Sci 84, 101–116.
Ren, K., King, M. A., Liu, J., Siemann, J., Altman, M., Meyers, C., Hughes, J. A., and Meyer, E. M. (2007). The alpha7 nicotinic receptor agonist 4OH-GTS-21 protects axotomized septohippocampal cholinergic neurons in wild type but not amyloid-overexpressing transgenic mice. Neuroscience 148, 230–237.
Rissman, R. A., De Blas, A. L., and Armstrong, D. M. (2007). GABA(A) receptors in aging and Alzheimer’s disease. J. Neurochem. 103, 1285–1292.
Roberts, G. W., Crow, T. J., and Polak, J. M. (1985). Location of neuronal tangles in somatostatin neurones in Alzheimer’s disease. Nature 314, 92–94.
Shimazaki, M., Nakano, H., and Kobayashi, K. (2005). Correlation between tau phosphorylation sites and tangle morphology in Alzheimer’s disease. Psychogeriatrics 5, 22–35.
Smith, K. D., Kallhoff, V., Zheng, H., and Pautler, R. G. (2007). In vivo axonal transport rates decrease in a mouse model of Alzheimer’s disease. Neuroimage 35, 1401–1408.
Solodkin, A., Veldhuizen, S. D., and Van Hoesen, G. W. (1996). Contingent vulnerability of entorhinal parvalbumin-containing neurons in Alzheimer’s disease. J. Neurosci. 16, 3311–3321.
Somogyi, P., Tamas, G., Lujan, R., and Buhl, E. H. (1998). Salient features of synaptic organisation in the cerebral cortex. Brain Res. Brain Res. Rev. 26, 113–135.
Spires, T. L., Meyer-Luehmann, M., Stern, E. A., McLean, P. J., Skoch, J., Nguyen, P. T., Bacskai, B. J., and Hyman, B. T. (2005). Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J. Neurosci. 25, 7278–7287.
Spires, T. L., Orne, J. D., SantaCruz, K., Pitstick, R., Carlson, G. A., Ashe, K. H., and Hyman, B. T. (2006). Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am. J. Pathol. 168, 1598–1607.
Spires-Jones, T. L., de Calignon, A., Matsui, T., Zehr, C., Pitstick, R., Wu, H. Y., Osetek, J. D., Jones, P. B., Bacskai, B. J., Feany, M. B., Carlson, G. A., Ashe, K. H., Lewis, J., and Hyman, B. T. (2008). In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J. Neurosci. 28, 862–867.
Spruston, N. (2008). Pyramidal neurons: dendritic structure and synaptic integration. Nat. Rev. Neurosci. 9, 206–221.
Stamer, K., Vogel, R., Thies, E., Mandelkow, E., and Mandelkow, E. M. (2002). Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 156, 1051–1063.
Terry, R. D. (1963). The fine structure of neurofibrillary tangles in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 22, 629–642.
Terry, R. D., Gonatas, N. K., and Weiss, M. (1964). Ultrastructural studies in Alzheimer’s presenile dementia. Am. J. Pathol. 44, 269–297.
Terwel, D., Dewachter, I., and Van, Leuven F. (2002). Axonal transport, tau protein, and neurodegeneration in Alzheimer’s disease. Neuromolecular. Med. 2, 151–165.
Thal, D. R., Schultz, C., Botez, G., Del Tredici, K., Mrak, R. E., Griffin, W. S., Wiestler, O. D., Braak, H., and Ghebremedhin, E. (2005). The impact of argyrophilic grain disease on the development of dementia and its relationship to concurrent Alzheimer’s disease-related pathology. Neuropathol. Appl. Neurobiol. 31, 270–279.
Tolnay, M., and Probst, A. (1999). REVIEW: tau protein pathology in Alzheimer’s disease and related disorders. Neuropathol. Appl. Neurobiol. 25, 171–187.
Tsai, J., Grutzendler, J., Duff, K., and Gan, W. B. (2004). Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat. Neurosci. 7, 1181–1183.
Varoqui, H., Schafer, M. K., Zhu, H., Weihe, E., and Erickson, J. D. (2002). Identification of the differentiation-associated Na + /PI transporter as a novel vesicular glutamate transporter expressed in a distinct set of glutamatergic synapses. J. Neurosci. 22, 142–155.
Keywords: basket terminals, chandelier terminals, GABAergic axons, glutamatergic axons, neurofibrillary tangles
Citation: Blazquez-Llorca L, Garcia-Marin V and DeFelipe J (2010) Pericellular innervation of neurons expressing abnormally hyperphosphorylated tau in the hippocampal formation of Alzheimer’s disease patients. Front. Neuroanat. 4:20. doi: 10.3389/fnana.2010.00020
Received: 25 March 2010;
Paper pending published: 18 April 2010;
Accepted: 28 April 2010;
Published online: 24 June 2010
Edited by:
Patrick R. Hof, Mount Sinai School of Medicine, USAReviewed by:
Gregory Elder, The Mount Sinai Medical Center, USAStephen D. Ginsberg, Nathan Kline Institute, USA
Copyright: © 2010 Blazquez-Llorca, Garcia-Marin and DeFelipe. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence:
Javier DeFelipe, Laboratorio de Circuitos Corticales (Centro de Tecnología Biomédica), Universidad Politécnica de Madrid, Campus Montegancedo S/N, Pozuelo de Alarcón, 28223 Madrid, Spain; Instituto Cajal (CSIC), Avenida Doctor Arce 37, 28002 Madrid, Spain. e-mail:ZGVmZWxpcGVAY2FqYWwuY3NpYy5lcw==;
Lidia Blazquez-Llorca and Virginia Garcia-Marin have contributed equally to this work.