

- 1 Yerkes National Primate Research Center, Emory University, Atlanta, GA, USA
- 2 Department of Neurology, Emory University, Atlanta, GA, USA
Striatal dopamine (DA) denervation results in a significant loss of dendritic spines on medium spiny projection neurons in Parkinson’s disease. In 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated parkinsonian monkeys, spines contacted either by cortical or thalamic glutamatergic terminals are severely affected on both direct and indirect striatofugal neurons. In rodents, indirect pathway neurons appear to be more sensitive, at least in early stages of acute dopamine denervation. The remaining corticostriatal and thalamostriatal axo-spinous synapses undergo complex ultrastructural remodeling consistent with increased synaptic activity in the DA-denervated primate striatum, which may explain the pathophysiological overactivity of the corticostriatal system reported in various animal models of parkinsonism. The calcium-mediated regulation of the transcription factor myocyte enhancer factor 2 was recognized as a possible underlying mechanism for striatal spine plasticity. Future studies to determine how alterations in striatal spine plasticity contribute to the symptomatology of parkinsonism are warranted.
Introduction
Since 1891 when Cajal described dendritic spines as morphologically distinct neuronal elements (Ramon y Cajal, 1891), studies of spine development, morphogenesis and plasticity have been at the forefront of research in various laboratories using modern techniques at the electron microscopic level for three-dimensional (3D) reconstructions of individual spines (Harris and Kater, 1994; DeFelipe, 2002; Smith et al., 2009; Villalba et al., 2010), single- and two-photon microscopy used in conjunction with fluorescent molecular tools (Yuste and Bonhoeffer, 2004; Knott and Holtmaat, 2008) or neurocomputational methods (Zhang et al., 2010) to study dynamic changes in spine morphology related to pathological or physiological regulation of neuronal plasticity (Yuste and Bonhoeffer, 2001).
Despite a broad range of experimental evidence showing changes in the density or morphology of dendritic spines in a large number of neurological and psychiatric diseases, the exact contribution of spine changes to brain diseases pathophysiology remains poorly understood (Ingham et al., 1989, 1998; Harris and Kater, 1994; Fiala et al., 2002; Picconi et al., 2005; Stephens et al., 2005; Zaja-Milatovic et al., 2005; Deutch et al., 2007; Surmeier et al., 2007; Bourne and Harris, 2008; Smith and Villalba, 2008; Villalba et al., 2009, 2010).
In the striatum, the principal neuronal subtypes are the medium spiny neurons (MSNs), which represent 95% of all striatal neurons (Kemp and Powell, 1971; Chang et al., 1982). In rats, each MSN harbors approximately 5000 dendritic spines (Wickens et al., 2007). Each of these spines receives glutamatergic inputs from the cerebral cortex or specific thalamic nuclei (Kemp and Powell, 1971; Smith et al., 2004, 2009; Raju et al., 2006). Spines are also an important target of midbrain dopaminergic inputs that play a critical role in the regulation of cortical glutamatergic afferents (Smith and Bolam, 1990; Smith et al., 1994; Nicola et al., 2000). In both animal models of parkinsonism and Parkinson’s disease (PD) patients, MSNs lose as much 30–50% dendritic spines, which results in complex anatomical, neurochemical and electrophysiological changes of striatal glutamatergic transmission (Ingham et al., 1989; Stephens et al., 2005; Zaja-Milatovic et al., 2005; Smith and Villalba, 2008; Villalba et al., 2009).
The goal of this review is to provide an overall assessment of anatomical and functional evidence for spine loss in the striatum, and discuss the potential mechanisms involved in this pathophysiological phenomenon in parkinsonism. The readers are referred to other recent reviews for additional information (Deutch et al., 2007; Smith and Villalba, 2008; Smith et al., 2009).
Spine Loss in Parkinson’s Disease and PD Models
Over the past decades, it became clear that the progressive loss of striatal dopamine in PD patients and animal models of parkinsonism results in a significant loss of dendritic spines on MSNs of the dorsal striatum (Figure 1).
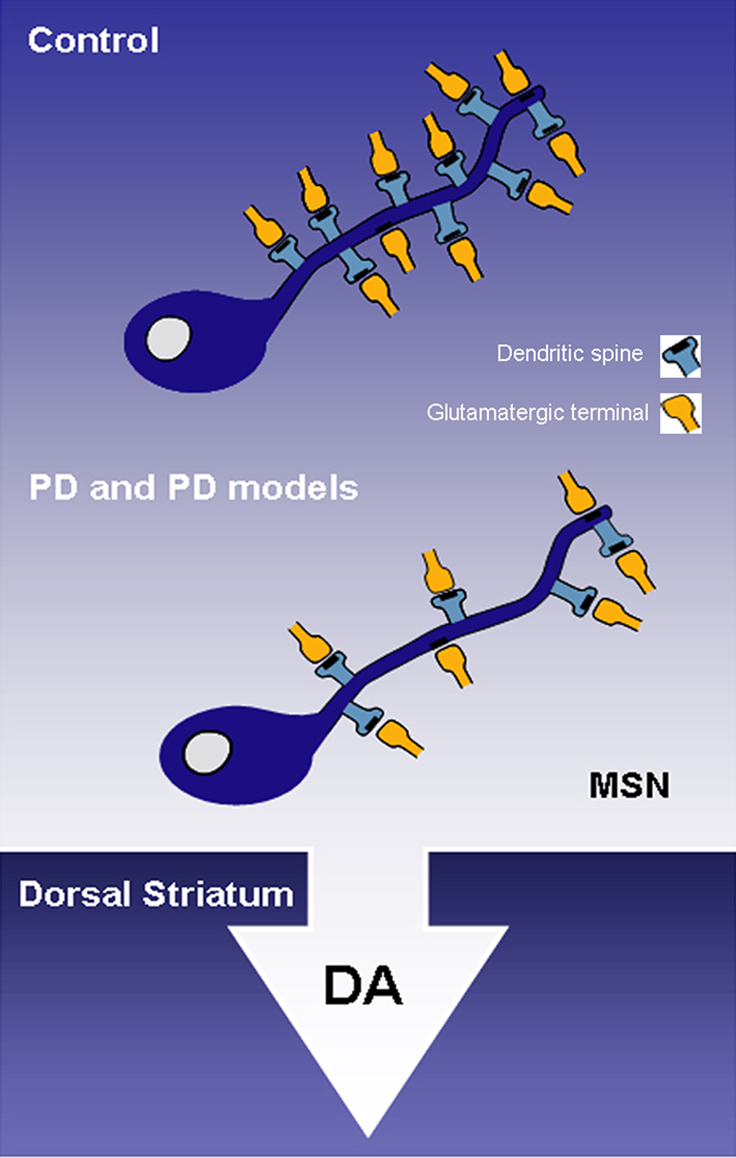
Figure 1. The progressive striatal dopaminergic depletion in Parkinson’s disease (PD) patients and animal models of PD results in a severe spine loss in medium spiny neurons (MSNs) of the dorsal striatum. DA, dopamine; MSN, medium spiny neuron; PD, Parkinson’s disease.
The first clear evidence for striatal spine loss came from studies showing that unilateral degeneration of the nigrostriatal dopaminergic system results in about 20% spine loss in the caudate-putamen complex of rats treated with 6-hydroxydopamine (6-OHDA) (Ingham et al., 1989). Since then, corresponding degrees of spine pruning have been demonstrated in postmortem striatal tissue of PD patients (Stephens et al., 2005; Zaja-Milatovic et al., 2005) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated parkinsonian monkeys (Smith and Villalba, 2008; Villalba et al., 2009). In monkeys and humans, the extent of spine loss correlates with the relative degree of striatal dopamine denervation (Zaja-Milatovic et al., 2005; Smith and Villalba, 2008; Villalba et al., 2009). For instance, in MPTP-treated parkinsonian monkeys, MSNs in striatal regions most severely dopamine-depleted, like the postcommissural putamen (i.e., the sensorimotor striatum) lose as much as 50% spines, while spine loss in less affected striatal areas like the nucleus accumbens (i.e., the limbic striatum) ranges between 20% to 25% of total striatal spines (Villalba et al., 2009) (Figure 2).
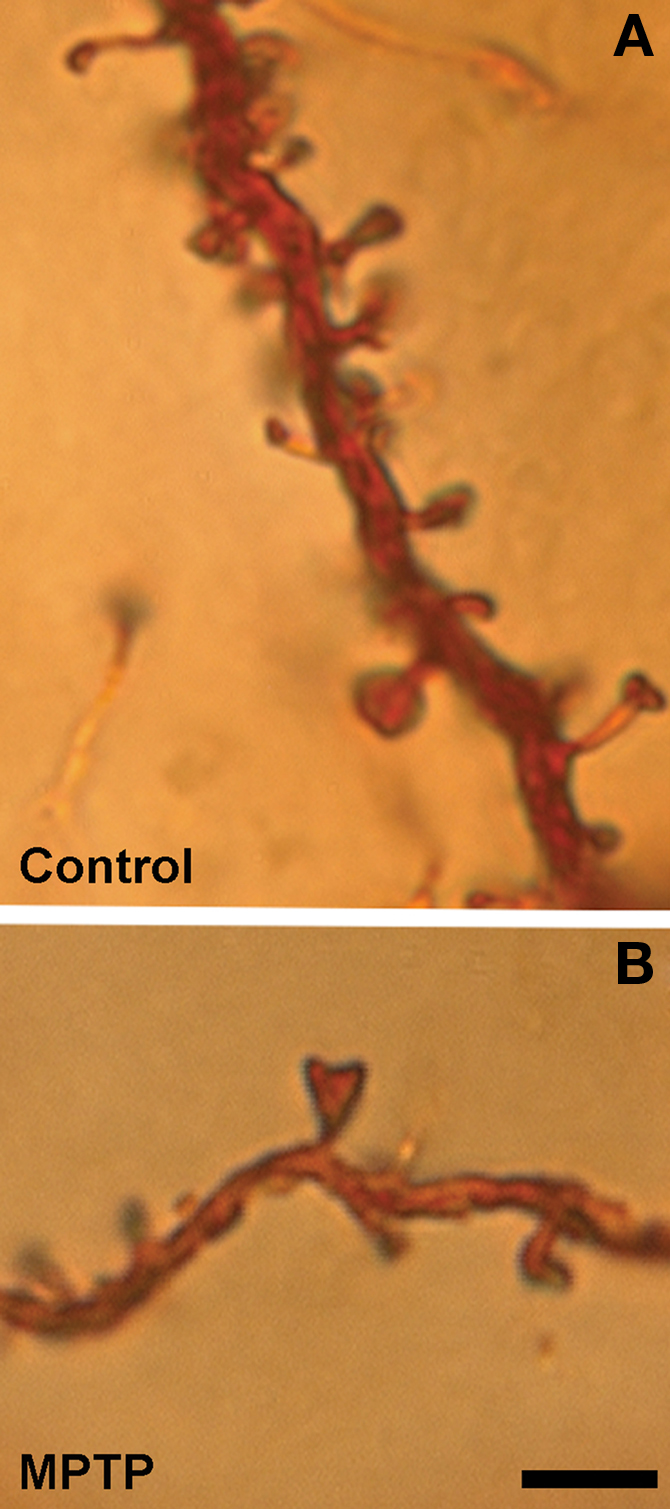
Figure 2. Dendrites from Golgi-impregnated MSNs in the caudate nucleus of a control (A) and a MPTP-treated (B) monkey. Note the dramatic spine loss on the dendrite of the MSNs from the MPTP-treated monkey compared with control. Scale bar in (B) [valid for (A)]: 5 μm.
This correlation has also been established in asymptomatic MPTP-treated monkeys with partial lesion of the nigrostriatal dopaminergic system, thereby suggesting that striatal spine loss may be an early pathology of parkinsonism that does not correlate with the severity of parkinsonian motor dysfunctions (Villalba et al., 2009).
Are Both “Direct” and “Indirect” Striatofugal Neurons Affected?
The basic model of direct and indirect pathways introduced in the late 1980s has been instrumental in our understanding of the basal ganglia circuitry, the pathophysiology of PD and the development of new therapeutic approaches for basal ganglia-related movement disorders (Albin et al., 1989; Bergman et al., 1990; Wichmann and Delong, 2007). Studies in animal models of PD suggest that striatal DA depletion results in an imbalance of activity between these two pathways in favor of an increased GABAergic striatal outflow from indirect pathway neurons combined with a decreased striatal output from direct pathway neurons, thereby leading to an overall increased tonic inhibition of thalamocortical neurons by basal ganglia outflow and resulting parkinsonian motor symptoms (Albin et al., 1989; DeLong, 1990; Obeso et al., 2000). Thus, taking into consideration that the activation of striatal MSNs rely on extrinsic glutamatergic inputs (Wilson, 1995), the possibility that spine loss affects preferentially one population of striatal MSNs over the other in parkinsonism has generated considerable interest in recent years.
Data obtained so far about this issue remain controversial. On one hand, observations gathered from reserpine-treated mice and 6-OHDA-treated rats suggested that D2-containing striatopallidal neurons, but not D1-positive striatonigral neurons, selectively lose spines following dopamine depletion (Day et al., 2006). These observations are at odds with a large number of studies in both human parkinsonians and animal models of parkinsonism showing a rather homogeneous loss of spines across large populations of Golgi-impregnated striatal MSNs (Ingham et al., 1989, 1998; Stephens et al., 2005; Zaja-Milatovic et al., 2005; Smith and Villalba, 2008; Villalba et al., 2009). Furthermore, recent data from rhesus monkeys chronically treated over many months with MPTP demonstrated a comparable loss of both D1-immunoreactive and D1-immunonegative spines in the DA-denervated putamen (Villalba et al., 2009), suggesting that spine pathogenesis affects both direct and indirect pathway striatofugal neurons in this animal model (Villalba et al., 2009). However, other monkey studies suggested a decrease in D2-positive spines accompanied with a corresponding increase in D1-immunoreactive spines in the caudate nucleus of MPTP-treated cynomolgus monkeys (Scholz et al., 2008). These apparent discrepancies may be due to the use of different animal models, different regimens of neurotoxin administration, different quantitative methods to determine striatal spine loss and different striatal regions being examined.
Does Striatal Spine Loss Affect Preferentially Cortical- Over Thalamic-Recipient Targets?
The striatum receives glutamatergic inputs from the cerebral cortex and the thalamus. Although cortical inputs target almost exclusively dendritic spines, a substantial component of the thalamostriatal system that originates from the caudal intralaminar nuclei, center median and parafascicular nuclei, target preferentially dendritic shafts, whereas inputs from rostral intralaminar, relay and associative thalamic nuclei mainly innervate dendritic spines (Smith et al., 2004, 2009; Raju et al., 2008). Although these two glutamatergic inputs cannot be differentiated from each other solely based on structural features, they can be categorized as of cortical or thalamic origin based on their specific expression in vesicular glutamate transporter 1 (vGluT1) or vGluT2, respectively (Raju et al., 2006, 2008; Smith et al., 2009). Taking advantage of these specific markers, we recently reported a significant increase in the overall density of vGluT1-positive terminals, but no significant change in the prevalence of vGluT2-positive boutons in the striatum of MPTP-treated parkinsonian monkeys (Raju et al., 2008). These findings are supported by postmortem human data showing an increased vGluT1 protein expression in the striatum of parkinsonians (Kashani et al., 2007).
At first glance, these observations are paradoxical to the fact that striatal MSNs undergo significant spine loss in parkinsonism, and that the bulk of synaptic inputs to striatal spines originate from cortical terminals (Kemp and Powell, 1971; Smith and Bolam, 1990; Raju et al., 2008). However, they are consistent with a large number of electrophysiological and neurochemical studies that suggest an increased glutamatergic transmission at corticostriatal synapses and augmented neuronal excitability in the striatum of rodent models of parkinsonism (Gubellini et al., 2002; Mallet et al., 2006, but see Day et al., 2006). Recent evidence suggests that such increased striatal activity may also occur in MPTP-treated monkeys (Liang et al., 2008). Although there is no clear explanation for this apparent discrepancy between morphological and functional data related to the corticostriatal system in parkinsonism, there are various indications that the striatum undergoes complex compensatory changes in glutamate receptors expression (Betarbet et al., 2000) and synaptic architecture that may account for increased glutamatergic transmission in parkinsonian condition (see Smith et al., 2009, for a review).
Ultrastructural Reorganization of Glutamatergic Corticostriatal and Thalamostriatal Synapses in Parkinsonism
Early rodent and human data, indeed, suggested morphological changes of asymmetric synapses consistent with increased synaptic activity in the dopamine-denervated striatum (Ingham et al., 1998; Meshul et al., 1999, 2000). These observations were recently supported an expanded by detailed 3D ultrastructural analysis (Figure 3).
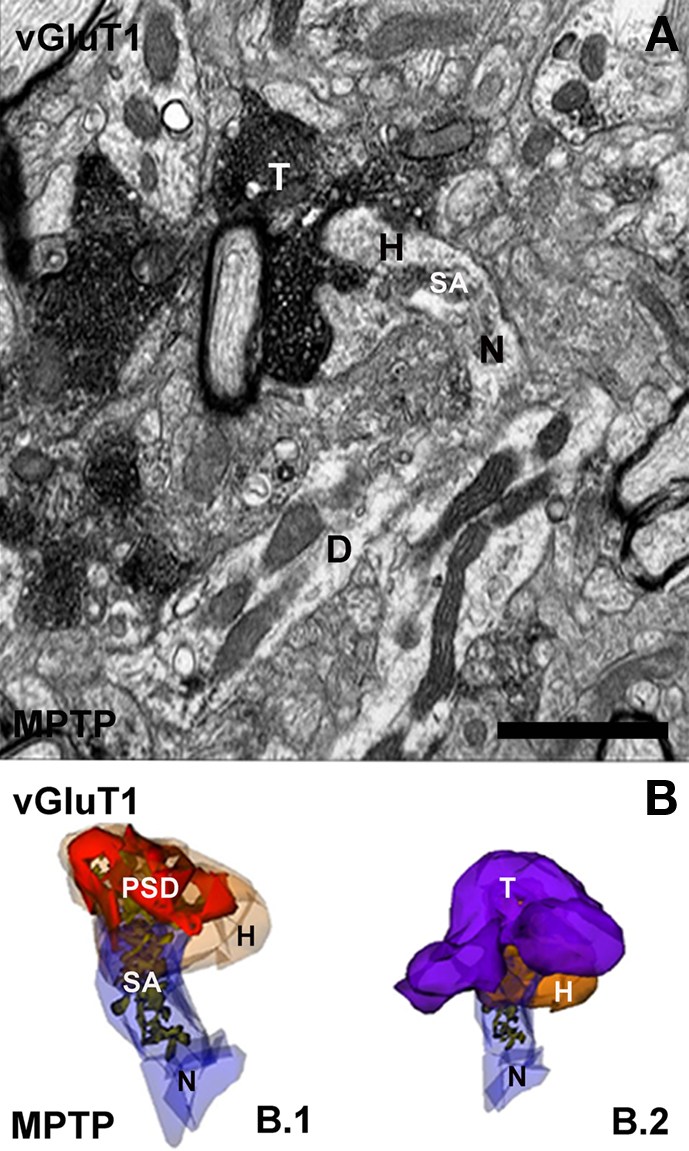
Figure 3. Electron micrograph and three-dimensional (3D) reconstruction of a corticostriatal (vGluT1-immunoreactive) axo-spinous synapse in the striatum of a MPTP-treated monkey. (A) Electron micrograph of a synapse between the spine of a MSN and a vGluT1-positive terminal (T). (B) 3D-reconstructed images from serial electron micrographs of the head (H) and neck (N) of the spine (B.1) and the axo-spine synapse (B.2) shown in (A). In (B.1), the spine is partially transparent to illustrate the complexity and distribution of the spine apparatus (SA) and the postsynaptic density (PSD). D, dendrite. Scale bar in (A): 1 μm.
Quantitative analysis of 3D-reconstructed axo-spinous cortico- and thalamostriatal analysis showed that the size of the postsynaptic density (PSD) area at both glutamatergic synapses is increased in the non-human primate model of PD, thereby suggesting that both thalamic and cortical inputs undergo plastic changes consistent with increased synaptic strength in parkinsonism (Smith et al., 2009; Villalba et al., 2010) (Figure 4).
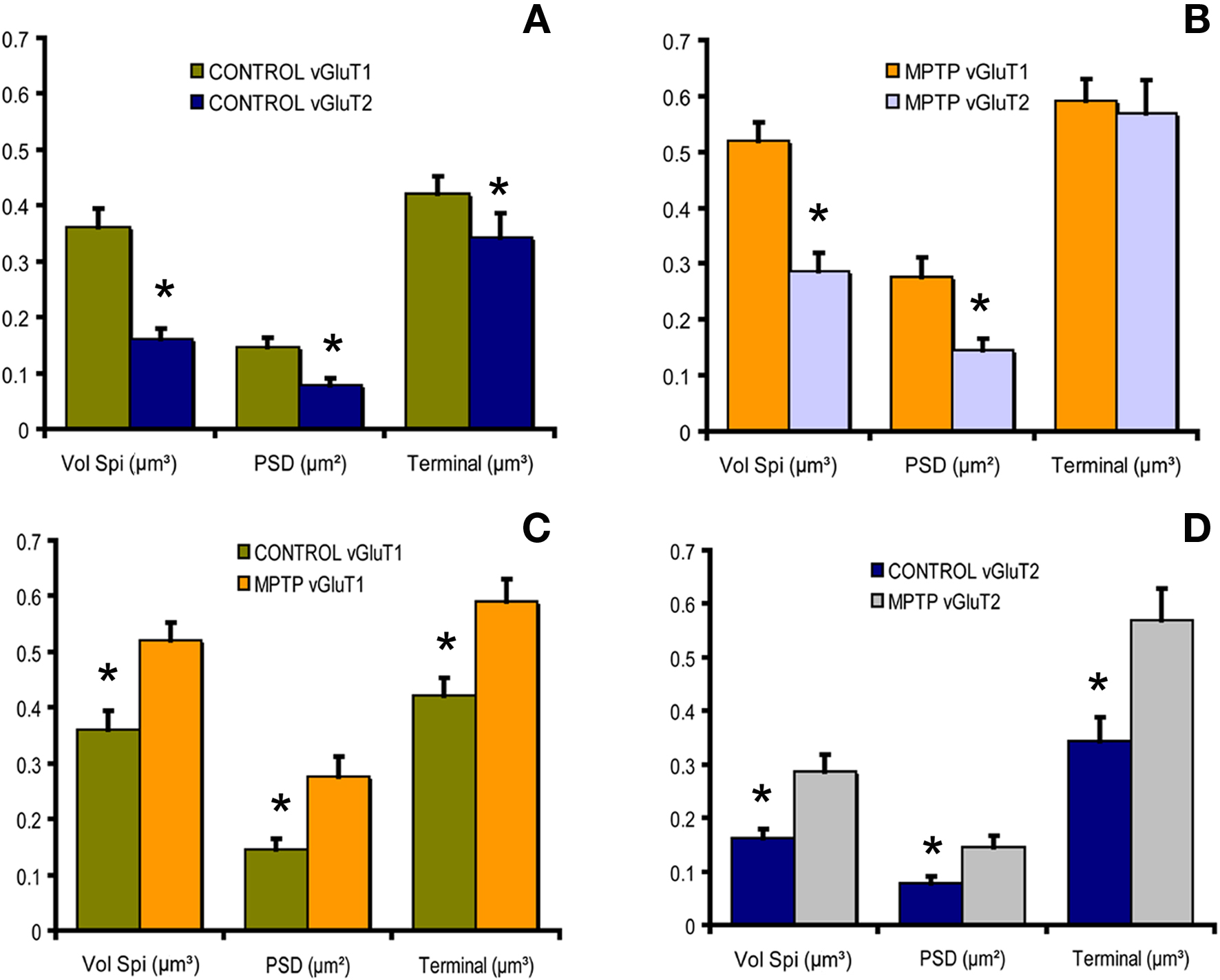
Figure 4. Histograms comparing the morphometric measurements (spine volume, PSD area, terminal volume) of structural elements at corticostriatal and thalamostriatal glutamatergic synapses using the 3D reconstruction method of serial ultrathin sections collected from 30 axo-spinous synapses in normal and MPTP-treated monkeys. (A) In control monkeys, the spine volume (Vol. Sp), the PSD areas and the size of pre-synaptic terminals at corticostriatal synapses are significantly larger than those at thalamostriatal synapses (*P < 0.001 for Vol. Sp. and PSD; *P = 0.016 for terminal; t-test). (B) The same is true for MPTP-treated monkeys, except for the pre-synaptic terminals that do not display any significant size differences in this condition. (C,D) The spine volumes, the PSD areas and the volume of vGluT1- and vGluT2-containing terminals are significantly larger in MPTP-treated parkinsonian monkeys than in controls (*P < 0.001; t-test).
Our data also revealed that the complexity and the total number of perforations at PSDs at corticostriatal and thalamostriatal glutamatergic synapses are increased in parkinsonism, a type of structural modification associated with increased synaptic efficacy and learning in the hippocampus (Nieto-Sampedro et al., 1982).
The exact mechanisms underlying these functional changes are not known, but it has been hypothesized that the compartmentalization of multiple transmission zones impedes the saturation of postsynaptic receptors and allow multiple transmitter quanta to be effective at the same postsynaptic spine, thereby enhancing the strength of individual synapses (Lisman and Harris, 1993). Thus, the extensive remodeling of PSDs at glutamatergic axo-spinous synapses in the striatum of parkinsonian monkeys provides another mechanism whereby cortical and thalamic glutamatergic afferents may increase their synaptic strength in parkinsonism (Figure 5).
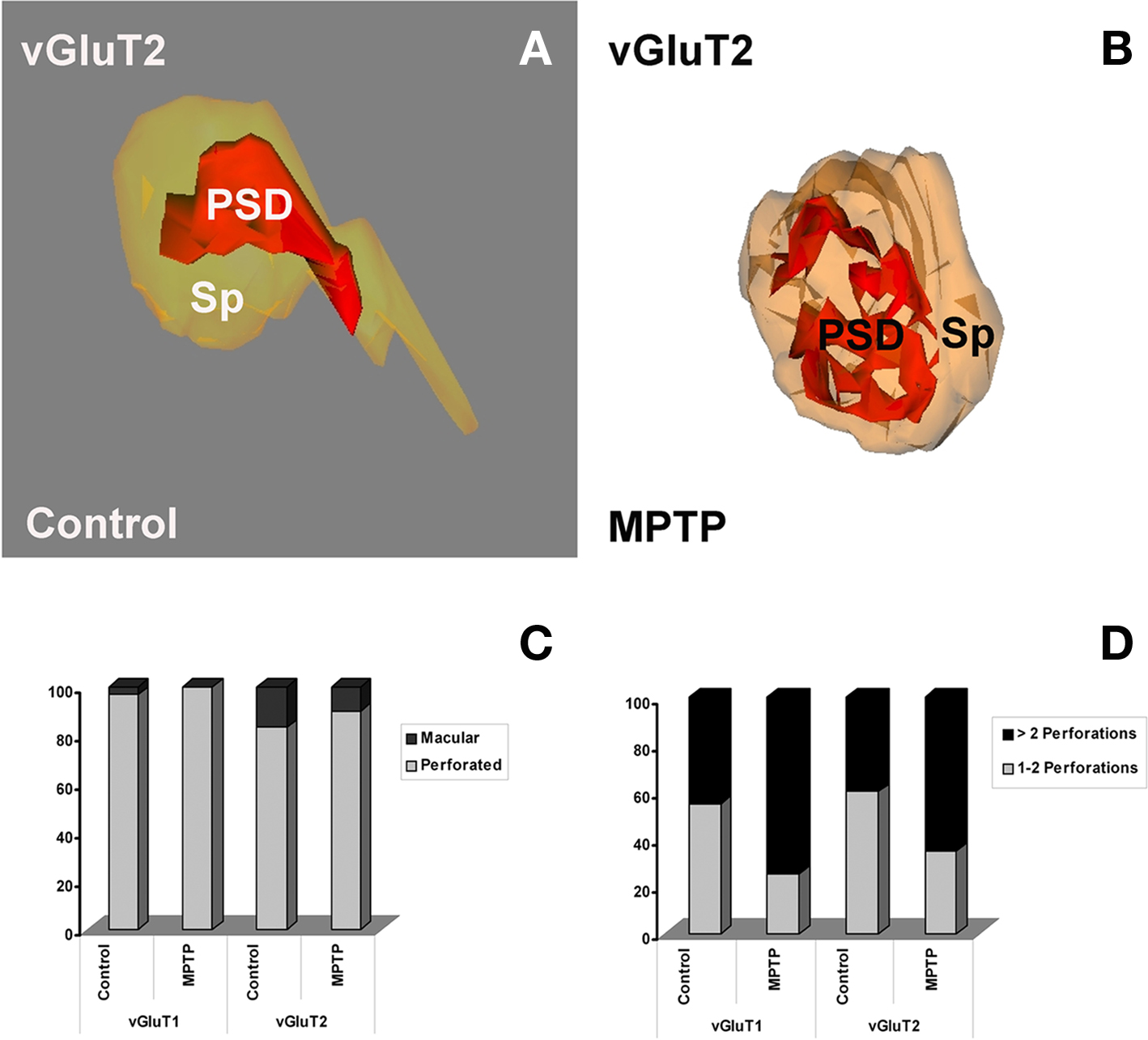
Figure 5. Morphology of three-dimensionally reconstructed dendritic spines showing different types of postsynaptic densities (PSDs). (A,B) Macular (A) and fenestrated (B) PSDs from spines contacted by vGluT2-IR terminals in control (A) and MPTP-treated (B) monkeys. (C) Quantitative analysis of the two types of PSDs (perforated vs. macular) at corticostriatal and thalamostriatal axo-spinous synapses in control and MPTP-treated monkeys. (D) Comparative analysis of the relative percentages of PSDs with different number of perforations in spines contacted by vGluT1- or vGluT2-IR terminals in control and MPTP-treated parkinsonian monkeys. The proportion of complex PSDs (more than two perforations) is significantly increased (Pearson Chi-square analysis, χ2) in MPTP-treated animals at both corticostriatal (χ2 = 4.78; P = 0.03) and thalamostriatal (χ2 = 4.34; P = 0.035) axo-spinous synapses. Sp, spine; PSD, postsynaptic density.
Another striking change observed in spines that receive vGluT1-containing cortical inputs in the striatum of MPTP-treated monkeys is the massive growth of the spine apparatus at corticostriatal synapses (Villalba et al., 2010) These changes are consistent with increased protein synthesis and increased buffering of intraspinous calcium at glutamatergic synapses in other brain regions (Fifkova et al., 1983; Bourne and Harris, 2008), thereby providing further evidence for increased corticostriatal glutamatergic transmission in parkinsonian condition (Gubellini et al., 2002).
Potential Cellular, Molecular and Genetic Mechanisms for Striatal Spine Loss in Parkinson’s Disease
Despite clear evidence for major structural and functional plastic changes in glutamatergic transmission at axo-spinous synapses in the parkinsonian striatum, the mechanisms underlying this phenomenon remain poorly understood. However, there is some indication that calcium likely contributes to this plastic event (Segal et al., 2000; Sabatini et al., 2001; Oertner and Matus, 2005; Day et al., 2006; Deutch et al., 2007; Surmeier et al., 2007). Findings in support of a calcium-regulated mechanism were recently gathered from transgenic mice with EGFP-labeled D1 or D2 DA receptors. In EGFP-D2 mice, spine loss can be prevented by genetic deletion of Cav1.3α1 subunits or the pharmacological blockade of L-type Cav1.3 channels (Day et al., 2006). Knowing that D2 dopamine receptor signaling targets only the channels that contain the Cav1.3α1 subunit (Surmeier et al., 2007), these data suggest that a dysregulation of calcium concentrations, preferentially in D2-containing striatopallidal neurons, may ultimately lead to specific spine loss on indirect pathway neurons in the rodent striatum (Day et al., 2006; Deutch et al., 2007; Surmeier et al., 2007). Further support for the involvement of calcium in this process comes from observations showing that striatal areas poor in calbindin D-28k (CaB) (Francois et al., 1994), such as the postcommissural putamen (sensorimotor striatal territory), are those that display the most severe striatal spine loss in MPTP-treated monkeys (Villalba et al., 2009). Recent data from co-culture studies suggest that the pruning of glutamatergic synapses and spines in the striatum, dependent upon calcium entry through L-type calcium channels, involves the activation of the Ca+2-dependent protein phosphatase, calcineurin and the up-regulation of the transcriptional activity of the myocyte enhancer factor 2 (MEF2). In turn, MEF2 up-regulation leads to increased expression of the Nurr77 and Arc genes linked to synaptic remodeling (Tian et al., 2010), thereby providing a signaling cascade through which striatal MSNs may undergo structural plasticity in parkinsonism (Tian et al., 2010).
It is noteworthy that MEF2 down-regulation is required for abnormal outgrowth of spines on striatal MSNs in cocaine-treated animals (Pulipparacharuvil et al., 2008). Together, these data indicate that MEF2 plays a critical role in regulating striatal spine plasticity in pathological conditions. Cholinergic signaling through M1 muscarinic receptors and Kir2 potassium channels is another essential trigger for the pruning of glutamatergic synapses in models of parkinsonism (Shen et al., 2009).
Despite the lack of direct evidence that any of the gene mutations characterized in PD underlie striatal spine loss, it is worth noting that PD-associated mutations that result in an increased kinase activity of Lrrk2 reduces neurite outgrowth in primary neuronal cultures and in the intact rat CNS, whereas LRRK2 deficiency leads to increased neurite length and branching (MacLeod et al., 2006). These findings, combined with evidence for strong Lrrk2 expression in striatal MSNs (Lee et al., 2010) and Lrrk2 enzymatic activity toward protein complexes involved in the formation of actin-enriched precursors of dendritic spines during neural development (Parisiadou et al., 2009), suggest that LRRK2 mutation in PD may contribute to striatal spine pathology in these patients.
Concluding Remarks
Striatal spine loss is a cardinal pathological feature of PD. Despite clear evidence that nigrostriatal dopaminergic lesion induces significant striatal spine loss in various animal models of PD, there is no direct evidence that dopamine denervation is the causal factor of striatal spine pathology. In fact, recent in vitro and in vivo studies have indicated that striatal spine pruning of MSNs in response to dopamine denervation is significantly attenuated by cortical lesions (Neely et al., 2007; Garcia et al., 2010), suggesting that glutamatergic cortical inputs are essential to mediate striatal spine loss in parkinsonism. Thus, whether striatal spine loss is the consequence of the lack of dopamine-mediated effects on corticostriatal glutamatergic transmission or primarily induced by dysfunction of the corticostriatal system remains to be established (Smith et al., 2009).
Another poorly understood aspect of this striatal pathology is its functional significance in PD symptomatology. Our recent observations that partially lesioned MPTP-treated monkeys with 30–40% spine loss in the sensorimotor striatum (i.e., the postcommissural putamen), do not display any significant parkinsonian symptoms suggest that the loss of spines in the motor striatum is not sufficient to elicit abnormal motor behaviors in parkinsonism (Smith et al., 2009; Villalba et al., 2009). Based on various findings from our laboratory and others showing the complex ultrastructural compensatory changes that affect remaining glutamatergic synapses in the dopamine-denervated striatum, we suggest that striatal MSNs are endowed with a high level of synaptic plasticity that allows sensorimotor-related information to be properly transmitted and integrated at the striatal level despite a major reduction in the number of corticostriatal synapses, at least early in the disease process (Smith et al., 2009; Villalba et al., 2009). Whether this level of plastic compensation also applies to non-motor striatal areas to alleviate cognitive and limbic dysfunctions remains to be established.
In conclusion, striatal spine pathology provides further evidence that early striatal dysfunctions in dopaminergic and glutamatergic transmission are key elements of the complex pathophysiology of parkinsonism.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the Yerkes Center Animal Resources Division for help with the care of MPTP-treated monkeys. Jean-Francois Pare and Susan Jenkins are also thanked for technical assistance. This work was supported by the NIH grant R01 NS 037948 and by the NCRR Yerkes Primate Center base grant RR00165.
References
Albin, R. L., Young, A. B., and Penney, J. B. (1989). The functional anatomy of basal ganglia disorders. Trends Neurosci. 12, 366–375.
Bergman, H., Wichmann, T., and DeLong, M. R. (1990). Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science 249, 1436–1438.
Betarbet, R., Porter, R. H., and Greenamyre, J. T. (2000). GluR1 glutamate receptor subunit is regulated differentially in the primate basal ganglia following nigrostriatal dopamine denervation. J. Neurochem. 74, 1166–1174.
Bourne, J. N., and Harris, K. M. (2008). Balancing structure and function at hippocampal dendritic spines. Annu. Rev. Neurosci. 31, 47–67.
Chang, H. T., Wilson, C. J., and Kitai, S. T. (1982). A Golgi study of rat neostriatal neurons: light microscopic analysis. J. Comp. Neurol. 208, 107–126.
Day, M., Wang, Z., Ding, J., An, X., Ingham, C. A., Shering, A. F., Wokosin,D., Ilijic, E., Sun, Z., Sampson, A. R., Mugnaini, E., Deutch, A. Y., Sesack, S. R., Arbuthnott, G. W., and Surmeier, D. J. (2006). Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 9, 251–259.
DeFelipe, J. (2002). Sesquicentenary of the birthday of Santiago Ramon y Cajal, the father of modern neuroscience. Trends Neurosci. 25, 481–484.
DeLong, M. R. (1990). Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 13, 281–285.
Deutch, A. Y., Colbran, R. J., and Winder, D. J. (2007). Striatal plasticity and medium spiny neuron dendritic remodeling in parkinsonism. Parkinsonism Relat. Disord. 13(Suppl. 3), S251–S258.
Fiala, J. C., Spacek, J., and Harris, K. M. (2002). Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res. Rev. 39, 29–54.
Fifkova, E., Markham, J. A., and Delay, R. J. (1983). Calcium in the spine apparatus of dendritic spines in the dentate molecular layer. Brain Res. 266, 163–168.
Francois, C., Yelnik, J., Percheron, G., and Tande, D. (1994). Calbindin D-28k as a marker for the associative cortical territory of the striatum in macaque. Brain Res. 633, 331–336.
Garcia, B. G., Neely, M. D., and Deutch, A. Y. (2010). Cortical regulation of striatal medium spiny neuron dendritic remodeling in parkinsonism: modulation of glutamate release reverses dopamine depletion-induced dendritic spine loss. Cereb. Cortex. doi: 10.1093/cercor/bhp317. [Epub ahead of print].
Gubellini, P., Picconi, B., Bari, M., Battista, N., Calabresi, P., Centonze, D., Bernardi, G., Finazzi-Agro, A., and Maccarrone, M. (2002). Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J. Neurosci. 22, 6900–6907.
Harris, K. M., and Kater, S. B. (1994). Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu. Rev. Neurosci. 17, 341–371.
Ingham, C. A., Hood, S. H., and Arbuthnott, G. W. (1989). Spine density on neostriatal neurones changes with 6-hydroxydopamine lesions and with age. Brain Res. 503, 334–338.
Ingham, C. A., Hood, S. H., Taggart, P., and Arbuthnott, G. W. (1998). Plasticity of synapses in the rat neostriatum after unilateral lesion of the nigrostriatal dopaminergic pathway. J. Neurosci. 18, 4732–4743.
Kashani, A., Betancur, C., Giros, B., Hirsch, E., and El Mestikawy, S. (2007). Altered expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in Parkinson disease. Neurobiol. Aging 28, 568–578.
Kemp, J. M., and Powell, T. P. (1971). The termination of fibres from the cerebral cortex and thalamus upon dendritic spines in the caudate nucleus: a study with the Golgi method. Philos. Trans. R. Soc. Lond. B Biol. Sci. 262, 429–439.
Knott, G., and Holtmaat, A. (2008). Dendritic spine plasticity – current understanding from in vivo studies. Brain Res. Rev. 58, 282–289.
Lee, H., Melrose, H. L., Yue, M., Pare, J. F., Farrer, M. J., and Smith, Y. (2010). Lrrk2 localization in the primate basal ganglia and thalamus: A light and electron microscopic analysis in monkeys. Exp. Neurol. 224, 438–447.
Liang, L., DeLong, M. R., and Papa, S. M. (2008). Inversion of dopamine responses in striatal medium spiny neurons and involuntary movements. J. Neurosci. 28, 7537–7547.
Lisman, J. E., and Harris, K. M. (1993). Quantal analysis and synaptic anatomy – integrating two views of hippocampal plasticity. Trends Neurosci. 16, 141–147.
MacLeod, D., Dowman, J., Hammond, R., Leete, T., Inoue, K., and Abeliovich, A. (2006). The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52, 587–593.
Mallet, N., Ballion. B., Le Moine, C., and Gonon, F. (2006). Cortical inputs and GABA interneurons imbalance projection neurons in the striatum of parkinsonian rats. J. Neurosci. 26, 3875–3884.
Meshul, C. K., Cogen, J. P., Cheng, H. W., Moore, C., Krentz, L., and McNeill, T. H. (2000). Alterations in rat striatal glutamate synapses following a lesion of the cortico- and/or nigrostriatal pathway. Exp. Neurol. 165, 191–206.
Meshul, C. K., Emre, N., Nakamura, C. M., Allen, C., Donohue, M. K., and Buckman, J. F. (1999). Time-dependent changes in striatal glutamate synapses following a 6-hydroxydopamine lesion. Neuroscience 88, 1–16.
Neely, M. D., Schmidt, D. E., and Deutch, A. Y. (2007). Cortical regulation of dopamine depletion-induced dendritic spine loss in striatal medium spiny neurons. Neuroscience 149, 457–464.
Nicola, S. M., Surmeier, J., and Malenka, R. C. (2000). Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu. Rev. Neurosci. 23, 185–215.
Nieto-Sampedro, M., Hoff, S. F., and Cotman, C. W. (1982). Perforated postsynaptic densities: probable intermediates in synapse turnover. Proc. Natl. Acad. Sci. U.S.A. 79, 5718–5722.
Obeso, J. A., Rodriguez-Oroz, M. C., Rodriguez, M., Lanciego, J. L., Artieda, J., Gonzalo, N., and Olanow, C. W. (2000). Pathophysiology of the basal ganglia in Parkinson’s disease. Trends Neurosci. 23, S8–S19.
Oertner, T. G., and Matus, A. (2005). Calcium regulation of actin dynamics in dendritic spines. Cell Calcium 37, 477–482.
Parisiadou, L., Xie, C., Cho, H. J., Lin, X., Gu, X. L., Long, C. X., Lobbestael, E., Baekelandt, V., Taymans, J. M., Sun, L., and Cai, H. (2009). Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J. Neurosci. 29, 13971–13980.
Picconi, B., Pisani, A., Barone, I., Bonsi, P., Centonze, D., Bernardi, G., and Calabresi, P. (2005). Pathological synaptic plasticity in the striatum: implications for Parkinson’s disease. Neurotoxicology 26, 779–783.
Pulipparacharuvil, S., Renthal, W., Hale, C. F., Taniguchi, M., Xiao, G., Kumar, A., Russo, S. J., Sikder, D., Dewey, C. M., Davis, M. M., Greengard, P., Nairn, A. C., Nestler, E. J., and Cowan, C. W. (2008). Cocaine regulates MEF2 to control synaptic and behavioral plasticity. Neuron 59, 621–633.
Raju, D. V., Ahern, T. H., Shah, D. J., Wright, T. M., Standaert, D. G., Hall, R. A., and Smith, Y. (2008). Differential synaptic plasticity of the corticostriatal and thalamostriatal systems in an MPTP-treated monkey model of parkinsonism. Eur. J. Neurosci. 27, 1647–1658.
Raju, D. V., Shah, D. J., Wright, T. M., Hall, R. A., and Smith, Y. (2006). Differential synaptology of vGluT2-containing thalamostriatal afferents between the patch and matrix compartments in rats. J. Comp. Neurol. 499, 231–243.
Ramon y Cajal, S. (1891). Sur la structure de l’ecorce cerebrale de quelques mammiferes. La Cellule 7, 125–176.
Sabatini, B. L., Maravall, M., and Svoboda, K. (2001). Ca2+ signaling in dendritic spines. Curr. Opin. Neurobiol. 11, 349–356.
Scholz, B., Svensson, M., Alm, H., Skold, K., Falth, M., Kultima, K., Guigoni, C., Doudnikoff E, Li, Q., Crossman, A. R., Bezard, E., and Andren, P. E. (2008). Striatal proteomic analysis suggests that first L-dopa dose equates to chronic exposure. PLoS ONE 3, e1589. doi: 10.1371/journal.pone.0001589.
Segal, I., Korkotian, I., and Murphy, D. D. (2000). Dendritic spine formation and pruning: common cellular mechanisms? Trends Neurosci. 23, 53–57.
Shen, H. W., Toda, S., Moussawi, K., Bouknight, A., Zahm, D. S., and Kalivas, P. W. (2009). Altered dendritic spine plasticity in cocaine-withdrawn rats. J. Neurosci. 29, 2876–2884.
Smith, A. D., and Bolam, J. P. (1990). The neural network of the basal ganglia as revealed by the study of synaptic connections of identified neurons. Trends Neurosci. 13, 259–265.
Smith, Y., Bennett, B. D., Bolam, J. . P, Parent, A., and Sadikot, A. F. (1994). Synaptic relationships between dopaminergic afferents and cortical or thalamic input in the sensorimotor territory of the striatum in monkey. J. Comp. Neurol. 344, 1–19.
Smith, Y., Raju, D., Nanda, B., Pare, J. F., Galvan, A., and Wichmann, T. (2009). The thalamostriatal systems: anatomical and functional organization in normal and parkinsonian states. Brain Res. Bull. 78, 60–68.
Smith, Y., Raju, D. V., Pare, J. F., and Sidibe, M. (2004). The thalamostriatal system: a highly specific network of the basal ganglia circuitry. Trends Neurosci. 27, 520–527.
Smith, Y., and Villalba, R. (2008). Striatal and extrastriatal dopamine in the basal ganglia: an overview of its anatomical organization in normal and Parkinsonian brains. Mov. Disord. 23(Suppl. 3), S534–S547.
Stephens, B., Mueller, A. J., Shering, A. F., Hood, S. H., Taggart, P., Arbuthnott, G. W., Bell, J. E., Kilford, L., Kingsbury, A. E., Daniel, S. E., and Ingham, C. A. (2005). Evidence of a breakdown of corticostriatal connections in Parkinson’s disease. Neuroscience 132, 741–754.
Surmeier, D. J., Ding, J., Day, M., Wang, Z., and Shen, W. (2007). D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 30, 228–235.
Tian, X., Kai, L., Hockberger, P. E., Wokosin, D. L., and Surmeier, D. J. (2010). MEF-2 regulates activity-dependent spine loss in striatopallidal medium spiny neurons. Mol. Cell Neurosci. 44, 94–108.
Villalba, R. M., Lee, H., and Smith, Y. (2009). Dopaminergic denervation and spine loss in the striatum of MPTP-treated monkeys. Exp. Neurol. 215, 220–227.
Villalba, R. M., Pare, J.-P., and Smith, Y. (2010). “Ultrastructural plasticity of glutamatergic corticostriatal and thalamostriatal axo-spinous synapses in parkinsonian monkeys,” in X-Triennial Meeting of the International Basal Ganglia Society (IBAGS), New Jersey, 92.
Wichmann, T., and Delong, M. R. (2007). Anatomy and physiology of the basal ganglia: relevance to Parkinson’s disease and related disorders. Handb. Clin. Neurol. 83, 1–18.
Wickens, J. R., Arbuthnott, G. W., and Shindou, T. (2007). “Simulation of GABA function in the basal ganglia: computational models of GABAergic mechanisms in basal ganglia function,” in Progress in Brain Research-GABA and the Basal Ganglia. From Molecules to Systems, eds J. M. Tepper, E. D. Abercrombie, and J. P. Bolam (Amsterdam: Elsevier), 313–330.
Wilson, C. J. (1995). “The contribution of cortical neurons to the firing pattern of striatal spiny neurons,” in Models of Information Processing in the Basal Ganglia, eds J. C. Houk, J. L. Davis, and D. G. Beiser (Cambridge, MA: The MIT Press), 20–50.
Yuste, R., and Bonhoeffer, T. (2001). Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu. Rev. Neurosci. 24, 1071–1089.
Yuste, R., and Bonhoeffer, T. (2004). Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat. Rev. Neurosci. 5, 24–34.
Zaja-Milatovic, S., Milatovic, D., Schantz, A. M., Zhang, J., Montine, K. S., Samii, A., Deutch, A. Y., and Montine, T. J. (2005). Dendritic degeneration in neostriatal medium spiny neurons in Parkinson disease. Neurology 64, 545–547.
Keywords: dendritic spine, plasticity, dopamine, glutamate, Parkinson’s disease
Citation: Villalba RM and Smith Y (2010) Striatal spine plasticity in Parkinson’s disease. Front. Neuroanat. 4:133. doi: 10.3389/fnana.2010.00133
Received: 26 July 2010;
Paper pending published: 17 August 2010;
Accepted: 29 August 2010;
Published online: 10 December 2010
Edited by:
Jose L. Lanciego, University of Navarra, SpainReviewed by:
Jose L. Lanciego, University of Navarra, SpainJosé A. Armengol, University Pablo de Olavide, Spain
Copyright: © 2010 Villalba and Smith. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Rosa M. Villalba, Yerkes National Primate Research Center, Emory University, 954 Gatewood Rd. NE, Atlanta, GA 30329, USA. e-mail:cnZpbGxhbEBlbW9yeS5lZHU=