



- Department of Neurology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA
Researchers over the last decade have made substantial progress toward understanding the roles of dopamine and the basal ganglia (BG) in the control of sleep–wake behavior. In this review, we outline recent advancements regarding dopaminergic modulation of sleep through the BG and extra-BG sites. Our main hypothesis is that dopamine promotes sleep by its action on the D2 receptors in the BG and promotes wakefulness by its action on D1 and D2 receptors in the extra-BG sites. This hypothesis implicates dopamine depletion in the BG (such as in Parkinson’s disease) in causing frequent nighttime arousal and overall insomnia. Furthermore, the arousal effects of psychostimulants (methamphetamine, cocaine, and modafinil) may be linked to the ventral periaquductal gray (vPAG) dopaminergic circuitry targeting the extra-BG sleep–wake network.
Neurochemistry of the Basal Ganglia
The BG consists of four major nuclei, including the striatum (caudate and putamen), globus pallidus (GP), subthalamic nucleus (STN), and substantia nigra. Each of these nuclei differs in their neurochemistry, projection pattern, and function (see following sections). More than 95% of neurons in the striatum are projection neurons while the remaining 5% are interneurons (Wichmann and DeLong, 2006; Kreitzer, 2009). The projection neurons, which are medium spiny neurons (MSN), contain gamma amino butyric acid (GABA) as their neurotransmitter and hence are inhibitory in nature (Wichmann and DeLong, 2006). Some of the interneurons in the striatum, on the other hand, are excitatory and contain acetylcholine as their neurotransmitter (Lobo, 2009; Nambu, 2009). These cholinergic interneurons express vesicular glutamate transporter 3 (VGLUT3), and thus may also contain glutamate. The rest of the interneurons in the striatum are mostly GABAergic.
Although traditionally the GP is considered as a single nucleus, it comprises two neurochemically and functionally distinct subnuclei – the globus pallidus external (GPe) and globus pallidus internal (GPi). All the neurons in the GPe are GABAergic whereas the GPi contains both glutamatergic and GABAergic neurons (Barroso-Chinea et al., 2008). It is likely that both GABA and glutamate are present in the same neurons in the GPi, but it has never been confirmed. STN neurons are also glutamatergic, while the substantia nigra pars compacta (SNc) and pars reticulata (SNr) comprise mostly dopaminergic and GABAergic neurons, respectively (Wichmann and DeLong, 2006). In addition to these major neurotransmitters, BG neurons also contain other transmitters or neuropeptides, including enkephalin (ENK) and dynorphin in striatal MSNs; substance P, neuropeptide Y and nitric oxide in the striatal interneurons; and ENK in the GPe neurons.
Implications of the BG in Sleep–Wake Control
The BG has been implicated in a variety of motor and cognitive behaviors, including planning and other executive functions (Nambu, 2008). While these “higher level” cognitive processes are associated with the vigilance state of wakefulness, the involvement of the BG in the control of wakefulness per se has received very little attention. The BG is very likely involved in sleep regulation, considering the occurrence and high prevalence of sleep-related issues in patients with BG-related neurogenerative disorders, such as Parkinson’s disease (PD) and Huntington’s disease (HD). Disturbances of sleep are highly prevalent in PD, affecting up to 88% of patients in a community-based study (Factor et al., 1990), although a range of 55–98% has been reported across various studies (Larsen and Tandberg, 2001; Kaynak et al., 2005; Gjerstad et al., 2007). However, the interaction between PD and sleep is complex because PD patients may also have primary sleep disturbances such as rapid eye movement (REM) sleep behavior disorder, periodic limb movements during sleep and restless legs syndrome (Askenasy, 2001). Other factors such as depression, reactions to medications, nocturia, akinesia, pain, and dystonia may also underlie their sleep disturbances (Dhawan et al., 2006; Chaudhuri and Schapira, 2009). Nevertheless, insomnia and daytime sleepiness are the most frequent sleep-related complaints of PD patients. Although both sleep-onset insomnia and sleep-maintenance insomnia are reported in PD, sleep-maintenance insomnia is more common, affecting 74–88% of PD patients (Factor et al., 1990). Moreover, polysomnographic studies have demonstrated that these patients experience light and fragmented sleep. Similar to PD patients, HD patients have also been found to spend less total time in slow wave sleep (SWS) and REM sleep and have fragmentation in all states of sleep–wakefulness (S–W). In the most severe cases of HD, REM sleep has been reported to be completely absent (Arnulf et al., 2008). In addition to these clinical findings suggesting the involvement of the BG in S–W, many preclinical studies in animal models have shown that the BG may play an important role in S–W behavior and cortical activation, which will be the main focus of this review.
Neuronal Activity of the BG Across S–W Behavior
Although the activity pattern of BG neurons has been studied extensively, only a few studies have investigated BG unit activity in relation to cortical EEG or the sleep–wake states. Nevertheless, the available studies indicate that most of the BG neurons are wake-REM active, meaning they fire faster during wake and REM sleep (desynchronized EEG) than during SWS. For example, Mahon and colleagues performed intracellular recordings from striatal MSNs and showed that MSN activity critically depended upon the vigilance state. MSNs exhibited rhythmic action potential fluctuations between a DOWN state (hyperpolarized quiescent state) and an UP state (depolarized state) during SWS, coincident with cortical activity, but during wake and REM sleep they switched into high rates of discharge with “random” patterns that were not synchronized with cortical EEG UP and DOWN states (Detari et al., 1987; Mahon et al., 2003; Mahon et al., 2006). Similar to striatal MSNs, GPe neurons are more active during wake and REM sleep (Urbain et al., 2000). These findings from head-restrained animals were recently confirmed in our laboratory using freely-moving animals (S. Thankachan and J. Lu, unpublished data). This study demonstrated that ca. 50% of GPe neurons increased their firing during REM sleep and during active wakefulness. In addition, GPe neurons tend to display burst-firing regardless of S–W state. Moreover, it has been shown that GPe burst-firing is reduced following dopamine depletion in the BG in experimental animals and in PD patients (Pan and Walters, 1988; Filion and Tremblay, 1991; Filion et al., 1991; Hutchison et al., 1994; Bouali-Benazzouz et al., 2009). Finally, while SNc dopaminergic neurons do not particularly change their firing rates across S–W behavior, they switch to burst-firing mode during wake and REM sleep (Dahan et al., 2007; Monti and Monti, 2007). Burst-firing in the SNc may trigger more dopamine release in the BG during wake and REM sleep, and thus may have a stronger influence on striatal and GPe activity during wake and REM sleep than during SWS. Taken together, it appears that BG neuronal activity is lower during SWS than during wake or REM sleep, and depends upon cortical activity. In accordance with these conclusions, cortical stimulation triggered a sequence of postsynaptic responses including initially a short EPSP, then a short IPSP, and then a late EPSP with multiple spikes in all the BG nuclei including the striatum, GPe, GPi, STN, and SNr (Wilson et al., 1983; Kita, 1992; Fujimoto and Kita, 1993; Nambu et al., 2000). However, considering the numerous excitatory and inhibitory connections between the BG nuclei, it is not clear how cortical stimulations elicit the same sequence of postsynaptic events in all these nuclei. It is possible that powerful cortical drives can overpower other within-BG synaptic events. It is also possible that the latency of these events could be different in different nuclei following cortical stimulation, as none of these studies recorded neuronal activity from the different BG nuclei simultaneously following cortical stimulation. Nevertheless, it is known that neuronal activity in all the BG nuclei is strongly synchronized with cortical activity and is modulated by dopamine, serotonin and thalamic inputs during wakefulness and REM sleep.
To supplement the electrophysiological data, we used cFos as a marker for neuronal activity and studied cFos expression in the various nuclei of the BG during wake and sleep in rats. Animals were sacrificed either after spontaneous sleep (sleep group, n = 5), after 2 h of sleep deprivation (wake group, n = 5) or 2 h after intraperitoneal injection of methamphetamine (drug-induced wake group, n = 5) at a dose of 1 mg/kg body weight. The animals were sacrificed during the same circadian time to avoid any circadian influence in cFos expression. After perfusions, rat brains were sectioned at 40 μm into four series, and one series was immunohistochemically dual-labeled for tyrosine hydroxylase (TH, a marker for dopaminergic neurons) and cFos. Our data (Figure 1) showed that cFos expression in the dorsal striatum and STN was elevated during wakefulness (immediately after sleep deprivation) and during active wakefulness following administration of methamphetamine (1.0 mg/kg), compared to during sleep. The medial part of the GPe also showed a moderate increase in cFos expression during both types of wakefulness than during sleep. On the other hand, cFos expression in the ventral striatum (i.e., the nucleus of the accumbens), GPi and SNr was considerably lower during both wakefulness and sleep but slightly higher in the GPi and SNr during wakefulness (of both types) than during sleep. Finally, cFos expression was not observed in the dopaminergic neurons in the VTA, SNc and retrorubal area in any of these conditions. Collectively, these results suggest that cFos activity and neuronal firing patterns are distinct in different nuclei of the BG, and that they may play specific independent roles in S–W regulation. These differences in activity pattern can be partially explained by their differences in neurochemistry, and thus juxtacellular and dual immunolabeling studies are ultimately required for a better understanding of BG neuronal activity across S–W stages.
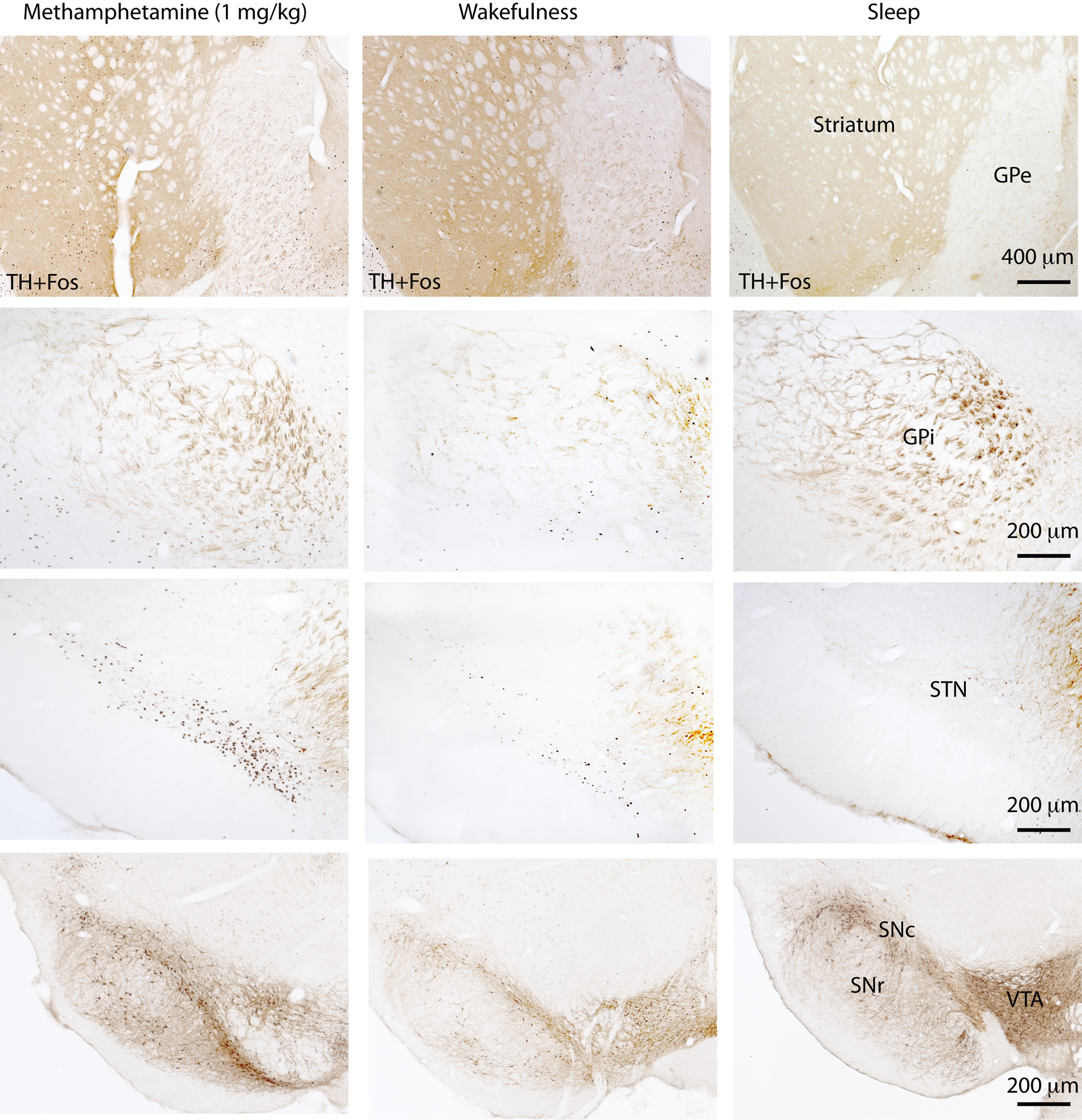
Figure 1. cFos expression in the basal ganglia 2 h after methamphetamine (1. 0 mg/kg) administration, after 2 h of wakefulness or during spontaneous sleep. Both waking states are associated with a higher number of cFos-labeled cells in the dorsal striatum, external globus pallidus (GPe), and subthalamic nucleus (STN) than the sleep state cFos expression in the internal globus pallidus (GPi) and substantia nigra pars reticulata (SNr) is low in all three conditions and indistinguishable between wakefulness and sleep. On the other hand, dopamine neurons in the ventral tegmental area (VTA), substantia nigra pars compacta (SNc) and the retrorubal area do not express cFos in any of these states.
The Effects of Neurotoxic Lesions in the BG on S–W
While the abovementioned electrophysiological and cFos studies provide evidence for the association of BG neuronal activity with the sleep–wake cycle, the lesion studies conducted in our lab and others’ established that the BG plays a significant causal role in S–W regulation. In order to study the specific roles played by different nuclei of the BG in S–W regulation, we placed small cell-specific lesions in the BG of rats using ibotenic acid (Qiu et al., 2010). As expected, we found dramatic changes in S–W amounts and architecture following discrete lesions of the various BG nuclei. Bilateral striatal lesions resulted in a significant reduction (ca. 15%) of wakefulness, and in fragmentation of both sleep and wakefulness. Sleep and wake bouts were shorter in general, but there were more frequent transitions, especially during the dark period in the animals with striatal lesions. Interestingly, this reduction in wakefulness after striatal lesions was attenuated when the lesions included the nucleus of accumbens core (NAc) (Qiu et al., 2010), suggesting that the dorsal and ventral striatum may play opposing roles in S–W regulation. Consistent with this, lesions restricted to the NAc transiently produced an increase in wakefulness (Qiu et al., 2010). Thus, these results indicate an important role for the striatum in the maintenance of wakefulness, as the dorsal striatum is one of very few structures that produces an increase in sleep and fragmentation of wakefulness after cell-specific lesions. For example, lesions of many previously-established wake-promoting cell groups, including the cholinergic basal forebrain (BF) neurons, serotonergic dorsal raphe (DR) neurons, histaminergic tuberomammillary nucleus (Parmentier et al., 2002; Gerashchenko et al., 2004; Lu et al., 2006a; Blanco-Centurion et al., 2007), noradrenergic locus coeruleus (Blanco-Centurion et al., 2007), cholinergic pedunculopontine and laterodorsal tegmental nuclei (Lu et al., 2006b) and orexin neurons in the lateral hypothalamus (Hara et al., 2001) did not produce an increase in sleep, although S–W fragmentation was observed after these lesions. Other nuclei that have been found to increase sleep following cell-specific lesions, similar to the dorsal striatum, are the medial parabrachial nucleus, the lateral hypothalamus and the dopaminergic ventral periaquaductal gray matter (vPAG) (Gerashchenko et al., 2003; Lu et al., 2006a,b).
Conversely, lesions of the GPe resulted in profound insomnia in rats with a striking 45.5% increase in total wakefulness and pronounced fragmentation of sleep and wakefulness. These S–W changes are comparable to the lesion effects produced by the only established sleep-promoting cell group, the ventrolateral preoptic area (VLPO), and thus suggest that the GPe may be another key player in the S–W control mechanism. In addition to the S–W changes, GPe lesions also produced hyperactivity and weight loss in rats. Although we were the first to report sleep changes after GPe lesions, hyperactivity and weight loss have been observed previously. We hypothesize that the weight loss following GPe lesions may be due to an oral motor disorder, based on our observations during the initial stimulatory phase of ibotenic acid injections. Since ibotenic acid is a glutamate agonist, it has a strong excitatory effect immediately following the injection (acute phase) before causing neuronal cell death. We observed capricious chewing behavior lasting over 24 h following ibotenic acid injections, and this chewing behavior did not result in food intake even in the presence of food. Following this acute phase, animals stopped eating and drinking and started losing weight abruptly. Similar effects were observed after electrolytic lesions of the lateral hypothalamus, which damages both the cell bodies and the fibers of passage, and were collectively recognized as lateral hypothalamic syndrome (Rowland and Stricker, 1982). 6-hydroxydopamine (6-OHDA) injections into the lateral hypothalamus that eliminate dopaminergic inputs to the striatum and GPe produced a similar reduction in body weight (Lenard et al., 1988). Finally, aphagia, adipsia and weight loss were observed in mice with dopamine deficiency, and these symptoms were corrected by daily administration of L-DOPA (Zhou and Palmiter, 1995). Altogether, these results strongly suggest that dopaminergic inputs to the GPe may play a key role in controlling oral motor behavior, thus affecting food/water intake. In light of these findings, it is interesting to note that PD patients, particularly in the advanced stages, also have difficulty controlling their oral motor muscles and thus have difficulty swallowing, drinking and displaying normal facial expressions. In addition to these oral motor problems, rats with GPe lesions showed other abnormal motor behaviors such as long bouts of standing and a scuffling locomotion. However, unlike S–W alterations, these motor disorders seem compensated for within 2 weeks.
In addition, we speculate that the effects of GPe lesions may mimic the symptoms of fatal familial insomnia (FFI), an autosomal dominant inherited neurodegenerative disease caused by a prions (PrC, a disease-associated prion protein) (Montagna et al., 2003). Patients with FFI exhibit total insomnia, autonomic dysregulation, rapid weight loss, abnormal motor behaviors and dementia, and die within months of disease onset. Brain autopsies showed extensive neuronal lesions, particularly in the thalamus and brainstem, and it is widely believed that the thalamic lesions underlie the insomnia in FFI (Perani et al., 1993). However, discrete neurotoxic lesions in the thalamus do not produce significant changes in the S–W cycle, motor behavior or body weight in rats (Vanderwolf and Stewart, 1988). Furthermore, infarctions in FFI patients are rarely confined to the bilateral thalamus, and thalamic strokes often include the midbrain and/or caudal hypothalamus, which may cause hypersomnia but not insomnia (Castaigne et al., 1981; Hermann et al., 2008). As mentioned earlier, rats with GPe lesions show behavioral changes that match the symptoms of FFI including insomnia, weight loss and abnormal motor behavior and these rats often die within 3–4 weeks. So, we hypothesize that pathology in the GPe may be the underlying cause of FFI. Consistent with this, hypometabolism in the BG has been reported in FFI patients (Cortelli et al., 1997).
In contrast to striatal and GPe lesions, ibotenic acid lesions in the GPi, STN or SNr did not significantly alter S–W, despite their close interactions with the striatum and GPe (Qiu et al., 2010). Furthermore, extensive lesions of the thalamus do not significantly affect S–W and motor behavior (Vanderwolf and Stewart, 1988). These results suggest that the integrity of the GPi, STN, SNr, and even thalamus may not be critical for the generation and maintenance of S–W states, although they may modulate S–W. These findings provide a basis on which a putative network for the BG control of S–W may be framed.
Proposed BG Circuitry Controlling S–W Behavior
Neuroanatomical connections between the BG and cerebral cortex are complex and characterized by many recurrent circuits (Figure 2A). On the basis of our lesion and cFos studies, we hypothesize that the recurrent interactions between the BG and cerebral cortex may influence cortical arousal and S–W behavior. Overall, there are five potential neural pathways that connect the BG and cortex: cortex-striatum-GPe-cortex; cortex-STN-GPe-cortex; cortex-STN-GPe-thalamus-cortex; cortex-striatum-GPe-GPi/SNr-thalamus-cortex; and cortex-striatum-GPe-thalamus-cortex (Figure 2A). The GPi and SNr have descending projections to the intermediate layer of the superior colliculus and midbrain extrapyramidal area (MEA), but not to the cholinergic neurons in the pedunculopontine tegmentum nucleus (PPT) (Rye et al., 1987; Steininger et al., 1992). The MEA targets the spinal-projecting neurons of the midbrain locomotor region (MLR) involved in the regulation of motor behavior (D. Sherman and J. Lu, unpublished data). Thus, this circuit may be involved in the BG control of posture and locomotion, but not of S–W regulation. The lack of sleep changes following lesions of the GPi and SNr further confirms this hypothesis. On the other hand, there are two structures within the BG-cortex pathways that contain direct cortical-projecting neurons: the GPe and the thalamus. As it has been shown that thalamic lesions do not have any significant effect on S–W, we can confine the key S–W control circuit of the BG to the striato-pallido-cortical loop (highlighted by black lines in Figure 2A). In this framework, the striatum-GPe-cortex loop may relay and enhance cortical arousal signals and thereby regulate wakefulness. This loop may also enhance and stabilize the excitatory state of the pyramidal neurons, i.e., cortical activation, which is consistent with the spectral changes in EEG recordings following lesions of the striatum and GPe. Lesions in both the striatum and GPe caused a significant shift in EEG power from the theta to the delta range during wake, NREM sleep and REM sleep, indicating that lesions in the BG resulted in a slowing down of the cortical EEG. Thus, we can conclude that the BG are involved in regulating the level of electrocortical arousal as well as behavioral arousal.
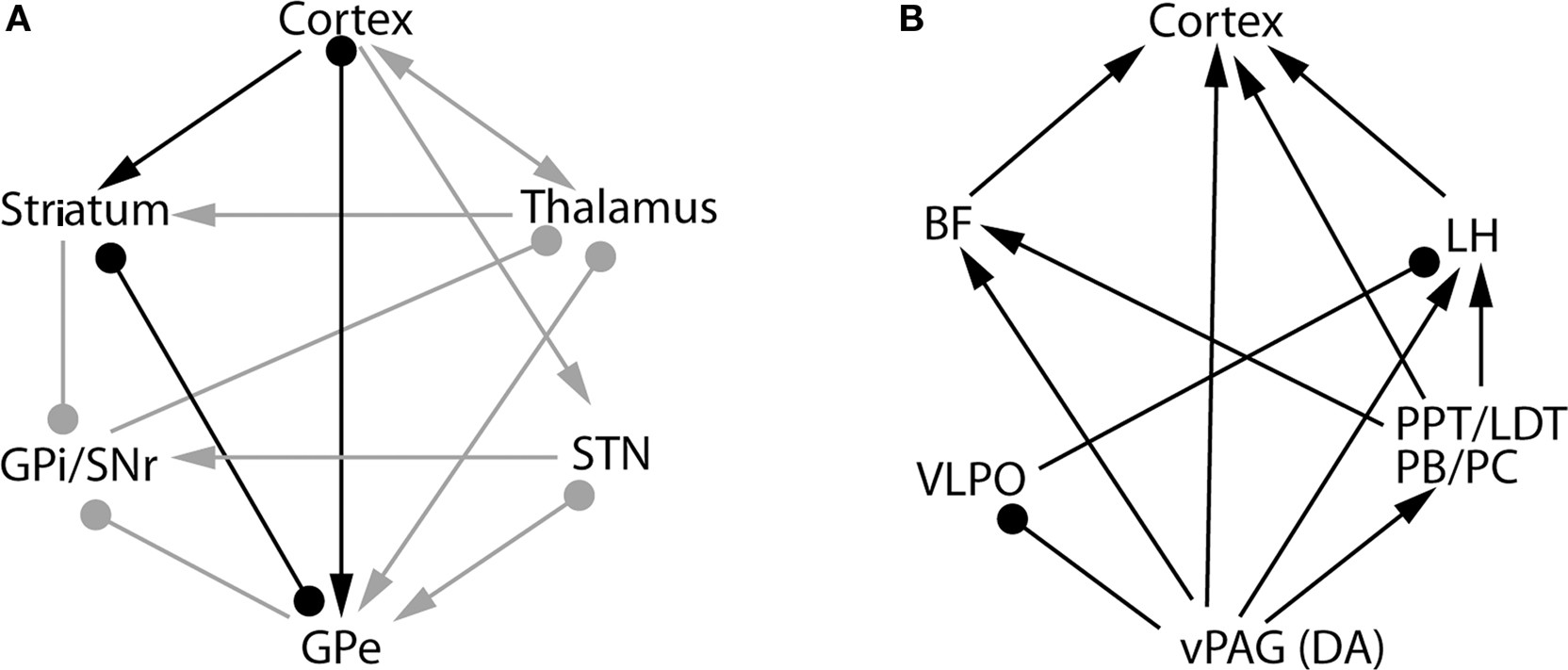
Figure 2. The basal ganglia network (A) and extra-basal ganglia network (B) hypothesized to be involved in sleep–wake control mechanisms. Several pathways connect the basal ganglia with the cerebral cortex, including the cortex-thalamus-striatum-cortex pathway, the cortex-striatum-GPe-cortex pathway, the cortex-STN-GPe-cortex and others. Of these pathways, our data indicate that the cortex-striatum-GPe-cortex network, marked by the black lines [in (A)], is involved in the control of sleep–wake behavior and cortical activation. By activating the D2 receptors in this network, dopamine from the midbrain disinhibits the GPe and promotes sleep. On the other hand, wake-active dopaminergic neurons in the vPAG promote wakefulness by inhibiting the sleep-promoting neurons in the VLPO and stimulating the wake-promoting cell groups, including the LH orexin neurons, PPT/LDT cholinergic neurons, PB/PC glutamatergic neurons and BF cholinergic neurons (B). Thus, the collective actions of dopamine in the extra-basal ganglia circuitry promote wakefulness. This network may also be responsible for the arousal effects of psychostimulants. BF, basal forebrain; vPAG, ventral periaquductal gray; PPT, pedunculopontine tegmental nucleus; LH, lateral hypothalamus; LDT, laterodorsal tegmental nucleus; PB, parabrachial nucleus; PC, precoeruleus nucleus; DA, dopamine; VLPO, ventrolateral preoptic nucleus. Arrows represents excitatory synapses; roundheads represent inhibitory synapses. The bars with both symbols represent the reciprocal connection between these nuclei with excitatory and/or inhibitory synapses on the target structure.
As mentioned previously, the GPe contains direct cortical-projecting neurons (Gritti et al., 1997; Sarter and Bruno, 2002; Furuta et al., 2004; Hur and Zaborszky, 2005). These pallidocortical projections have not been well-recognized mostly because they were previously assumed to be the part of the BF cortical projections. However, the BF cholinergic neurons (Nucleus Basalis of Mynert) located along the medial and ventral borders of the GPe seem to be distinct from the GABAergic corticopetal neurons within the GPe. This was supported by the finding that non-cholinergic, GABAergic neurons in the GPe (well within the boundaries) send ascending projections to the cortex (Gritti et al., 1997; Sarter and Bruno, 2002; Furuta et al., 2004). Interestingly, similar to the BF cortical projections, pallidocortical projections may be topographical. For example, the medial part of the GPe projects to the mPFC (Figure 3), while other parts of the GPe project to the insular, motor, and sensory cortices (Saper, 1984). In order to identify the GPe target neurons in the cortex, we injected a cocktail of tracers containing a retrograde tracer, cholera toxin subunit B (CTB), and an anterograde tracer, biotinylated dextranamide (BD), into the GPe into two rats. One week after the injections, these rats were sacrificed and the brains were harvested and processed for immunohistochemical identification of BD, CTB, and GAD 67. We found that most of the BD-labeled axon terminals (anterogradely labeled from the GPe) were in cortical layers IV-V (Figure 4C), where they formed appositions with CTB-immunoreactive (CTB-ir) neurons (Figure 4) and GAD 67-ir neurons. These CTB-ir neurons were pyramidal neurons and are presumably glutamatergic. Thus, these results suggest that GPe neurons target both GABAergic interneurons and glutamatergic pyramidal neurons in the cortex, and that these two regions are reciprocally connected. Projections to layer V are particularly important, as this layer is critical for generating EEG waves (Sanchez-Vives and McCormick, 2000).
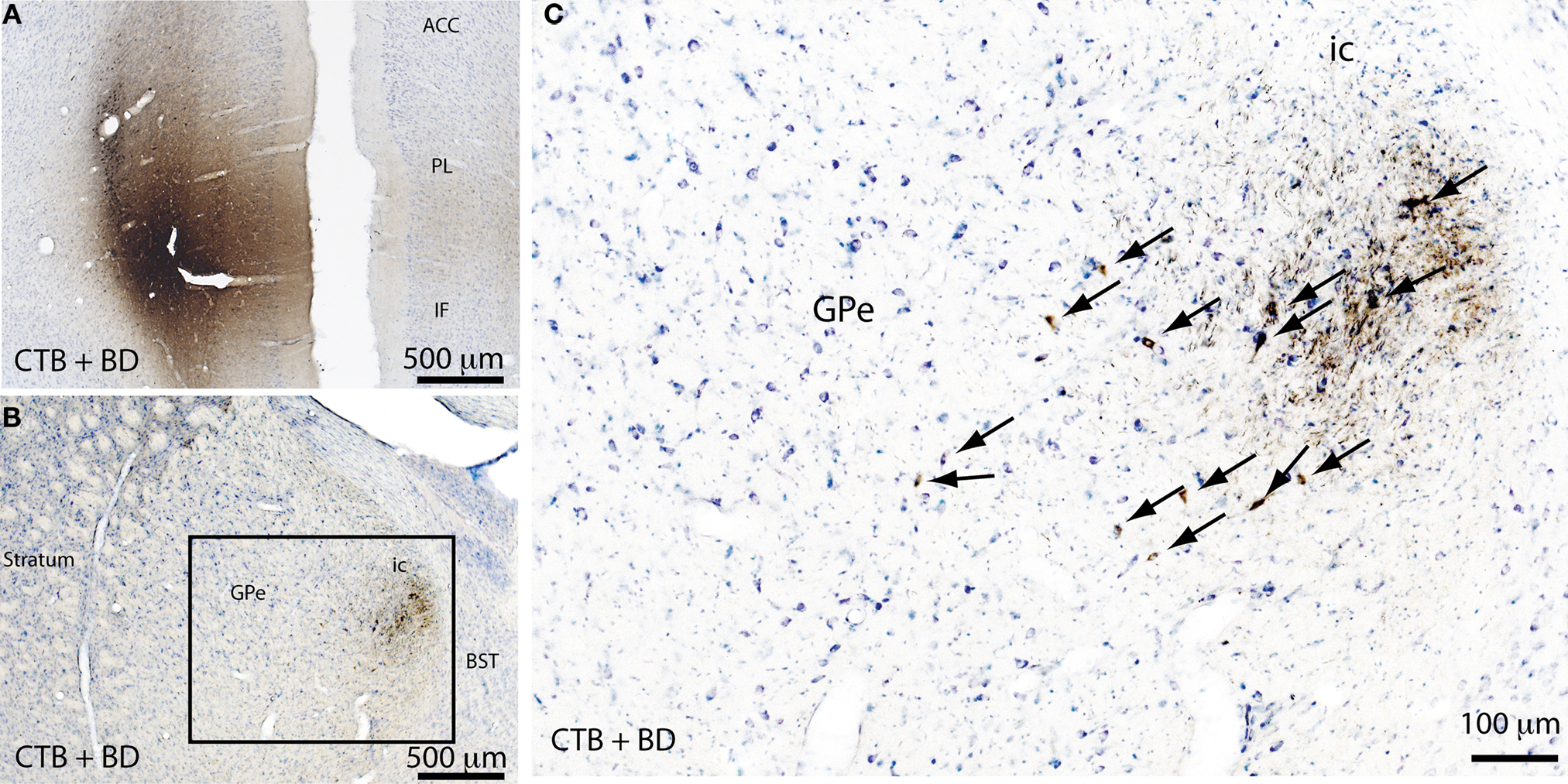
Figure 3. Pallidocortical projections. A mixture of a retrograde tracer (cholera toxin subunit B, CTB) and an anterograde tracer (biotin dextranamide, BD) were injected into the medial prefrontal cortex (mPFC) of rats to assess the pallidocortical projections. The injection site in the mPFC (A) was identified by CTB + BD immunolabeling. A large number of retrogradely-labeled (CTB-immunoreactive) neurons were found in the medial part of the GPe (B) and the same region also contained anterogradely labeled (BD-immunoreactive) neuron terminals (B,C), indicating that the GPe and mPFC are reciprocally connected. The boxed region in (B) is enlarged in (C), and arrows in (C) indicate the CTB-ir neurons in the GPe. ACC, anterior cingulate cortex; PL, prelimbic cortex; IF, infralimbic cortex; ic, internal capsule; BST, bed nucleus of stria terminalis.
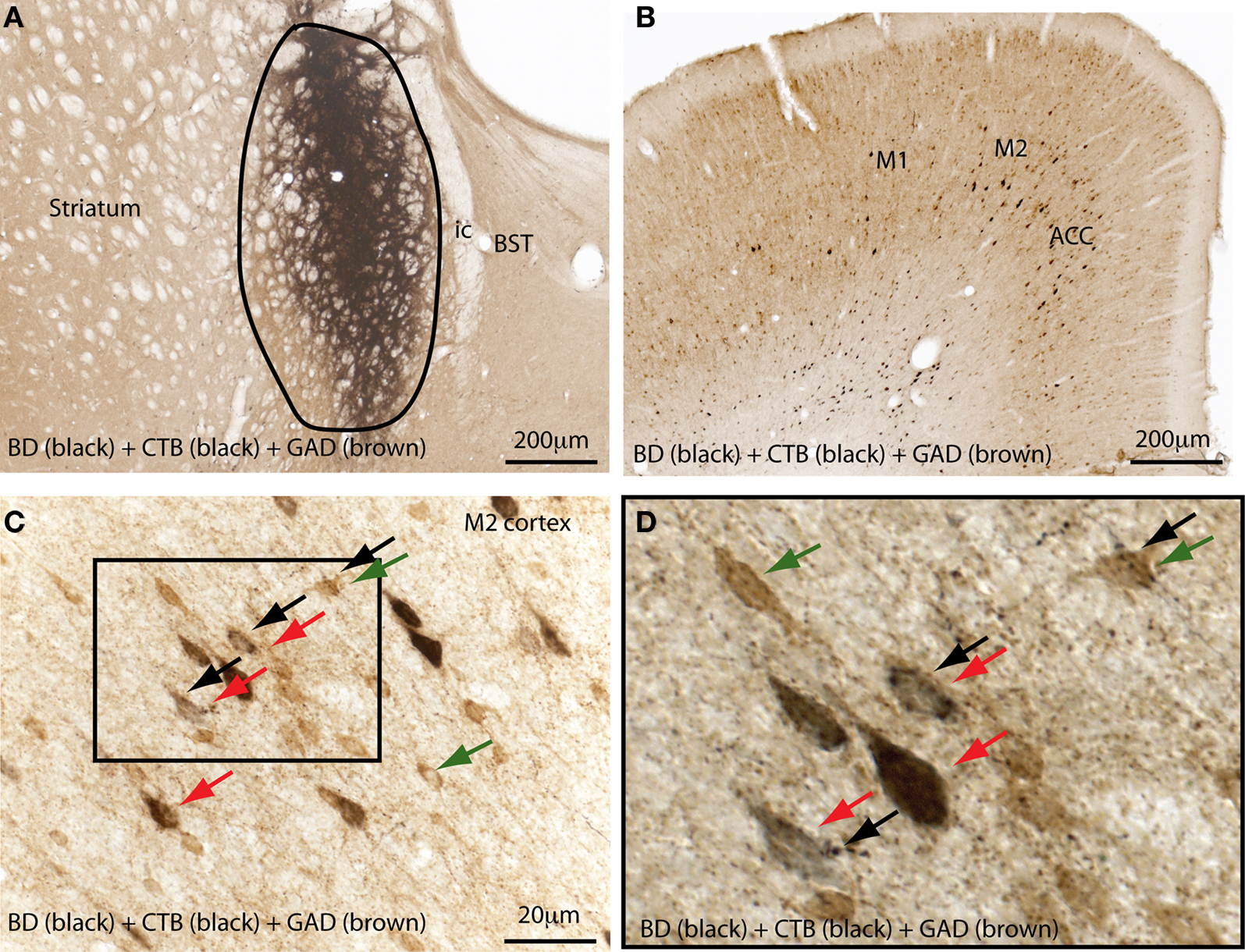
Figure 4. The external globus pallidus (GPe) targets both GABAergic and non-GABAergic neurons in the cortex. A mixture of a retrograde tracer (cholera toxin subunit B, CTB) and an anterograde tracer (biotin dextranamide, BD) was injected into the GPe (A) in a rat to characterize the target neurons of pallidal projections to the cortex. Brain sections were immunolabelled for BD, CTB, and glutamic acid decarboxylase (GAD 67). (A) Shows the injection site in the GPe. Anterogradely-labeled (BD-ir) axon terminals were mostly present in layers IV and V of the M2 and anterior cingulate cortices (B,C), where a high number of retrogradely-labeled (CTB-ir) cells were present (B). The boxed region in (C) is enlarged in (D). The BD-labeled boutons appose both the GABAergic neurons (green and black arrows) and glutamatergic pyramidal neurons (red and black arrows) in the cortex (D). Black arrows mark the axon terminal boutons; CTB-labeled neurons are stained in black color (red arrows) and GABAergic neurons are stained in brown color (green arrows). M1, primary motor cortex; M2, secondary motor cortex.
On the basis of these results, we hypothesized the possible mechanism by which the GPe contributes to S–W behavior and cortical activity. Our view is that corticopetal GABAergic GPe neurons act to suppress cortical activity regardless of S–W state and synchronize the fast cortical oscillations by targeting layer V pyramidal neurons and GABAergic interneurons (Figure 4). Once this inhibition is reduced (such as by the loss of dopamine inputs) or removed (such as in cell-body lesions), the layer V pyramidal neurons in the cerebral cortex, including the motor cortex, may switch to a more depolarized state as occurs during the wake state. This model is consistent with the observation that both lesions in the GPe and SNc (the source of dopaminergic neurons to the BG) increase wakefulness. This hypothesis is further supported by the finding that EEG delta power is increased and theta power is reduced after GPe lesions (Qiu et al., 2010), and that the EEG waves are slower in PD patients where dopamine inputs to the BG are lost (Soikkeli et al., 1991; Serizawa et al., 2008; Morita et al., 2009). Hence, it can be concluded that the GPe may be one of the many cell groups, including the basal forebrain, thalamus, lateral hypothalamus, LC, and VTA, that directly modulate cortical activity.
How does Midbrain Dopamine Modulate this Circuit?
It is well-established that the loss of dopamine inputs to the BG produces PD, and insomnia is highly prevalent among PD patients. The major source of dopamine in the BG is the midbrain dopaminergic neurons, particularly the SNc (A9 group), and these neurons primarily target the striatum, GPe and GPi. Neurotoxic lesions of the SNc in animals using 6-OHDA or NMDA agonists produced an increase in wakefulness (Lai et al., 1999; Sakata et al., 2002; Gerashchenko et al., 2006), suggesting that nigral dopaminergic inputs to the BG may promote sleep and the loss of dopaminergic inputs to the BG may underlie insomnia in PD patients. Moreover, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated animals, one of the best-known animal models of PD, displayed a significant reduction in the amount of SWS (Almirall et al., 1999; Monaca et al., 2004). Although it is believed that MPTP may damage noradrenergic and serotonergic cells in addition to dopaminergic neurons, sleep loss in the MPTP-treated animals seems to be primarily caused by the loss of nigrostriatal dopaminergic neurons. The reasons for this claim are several fold: (1) noradrenergic LC and serotonergic DR neurons have been implicated in promoting wakefulness, hence damage to these neurons would be expected to produce an increase in sleep rather than wakefulness. However, specific neurotoxic lesions of the LC or DR did not affect spontaneous sleep in rats (Lu et al., 2006b). (2) MPTP-induced insomnia cannot be attributed to extra-BG dopaminergic systems, including the VTA and vPAG, because VTA neurons have been shown to be protective against MPTP toxicity and VTA lesions do not alter S–W. On the other hand, vPAG lesions produced an increase in sleep, although vPAG neurons’ vulnerability to MPTP is not known. These findings clearly suggest that sleep loss observed after MPTP-lesions in animals may due to the loss of nigrostriatal dopaminergic neurons, and thus indicate that dopamine signaling in the BG may be involved in promoting sleep.
The view that endogenous dopamine in the BG promotes sleep appears contradictory to the popular view that dopamine may be important for wakefulness and arousal. However, it is well-known that low doses of apomorphine (100 μg/kg), a mixed D1/D2 dopamine receptor agonist, and L-DOPA induce sleep in animals and humans and trigger sleep attacks in PD patients, even during high arousal states such as when driving. However, high doses of apomorphine and L-DOPA induce arousal (Schafer and Greulich, 2000; Bonuccelli et al., 2002; Dimpfel, 2008; Hirayama et al., 2008; Garcia Ruiz, 2009). A D2 receptor antagonist has been shown to block the sleep-inducing effects of apomorphine, while a D1 receptor antagonist blocked the arousal effects (Gessa et al., 1985). In addition, systemic injections of a selective D1 agonist resulted in desynchronization of the EEG and an increase in wakefulness, whereas D1 antagonists produced a dose-dependent increase in sleep in mammals. Even in humans, a novel D1 receptor antagonist, NNC-687, increased the length of the first NREM period up to 47% over baseline and enhanced the total number, incidence, and burst-duration of sleep spindles. These evidences collectively indicate that activation of the D1 receptors may promote wakefulness and activation of the D2 receptors may promote sleep, although their specific site of action is not clear. On the basis of the current literature and our results, we hypothesize that activation of the D2 receptors in the BG promote sleep while activation of the D1 receptors in the extra-BG sites promote wakefulness, though these actions may not be exclusive.
Dopamine may act on presynaptic or postsynaptic dopaminergic receptors to bring about its effect on the target tissues. D1 and D2 dopaminergic receptors are abundantly expressed in the cerebral cortex while D1, D2, D3, and D5 receptors are expressed in the striatum and D2, D3, and D5 receptors are expressed in the GPe. Within the striatum, dopamine may exert its actions on the striatonigral neurons via D1 receptors, on striatopallidal neurons via D2 receptors, and on cholinergic interneurons via D5 receptors (Rivera et al., 2002). The D2 receptors are found to be located presynaptically on striatopallidal GABAergic terminals in the GPe, although dopamine may also exert its actions on GPe and GPi neurons via postsynaptic D2 receptors. Most GPe neurons have been shown to fire more slowly and in a bursting pattern following the loss of dopamine inputs, suggesting that the loss of dopamine inputs results in hypoactivity of the GPe neurons. Hence, it is likely that dopamine primarily acts on postsynaptic D2 receptors in the striatum and presynaptic D2 receptors in the GPe to bring about its effects on GPe neurons (Cooper and Stanford, 2001; Querejeta et al., 2001). It can therefore be inferred that the loss of D2 postsynaptic and presynaptic inhibition in the striatum and GPe would result in hyperactivity of the striatopallidal neurons, resulting in inhibition (presumably GABA-mediated) of the GPe neurons, which would then result in increased arousal. We thus hypothesized that endogenous dopamine in the BG promotes sleep by acting on the D2 receptors in the striatopallidal neurons. However, it was found that D2 receptor global knockout mice are hyperactive but show a significant reduction in wakefulness (Qu et al., 2010). Due to the wide distribution of D2 receptors in the brain, it has not been possible to identify the site where D2 receptor activation may promote wakefulness and where their absence in the D2 knockout mice may reduce wakefulness. Since our model predicts that the loss of D2 receptors in the BG may result in an increase in wakefulness, the reduced wakefulness in D2 knockout mice may be due to the absence of functional D2 receptors in the extra-BG sites, such as the cerebral cortex, BF, LH, and other wake-promoting cell groups in the brainstem and caudal hypothalamus (Figure 2B).
The VTA was thought to be such an extra-BG dopaminergic system involved in promoting wakefulness because it has direct projections to the prefrontal cortex. In line with this hypothesis, microinjection of apomorphine (D2 agonist) into the VTA produced a dose-dependent increase in behavioral and EEG sleep in rats, suggesting that VTA dopaminergic neurons may promote wakefulness, and D2-mediated inhibition of these neurons may promote sleep. However, these results are hard to reconcile with the findings that NMDA-induced VTA lesions in cats produced an increase in wakefulness rather than an increase in sleep, and orexin-saporin lesions of the VTA in rats did not produce any change in S–W. Although the loss of non-dopaminergic neurons in the VTA (which may play an opposing role in sleep regulation) cannot be overruled in these studies, we recently found that selective lesions of VTA dopaminergic neurons using 6-OHDA also did not produce an increase in sleep in rats (R. Vetrivelan and J. Lu, unpublished observations). Moreover, VTA neuronal firing rate and cFos expression (Figure 1) did not differ significantly across S–W states. Hence, the role of VTA dopaminergic neurons in promoting wakefulness is inconclusive.
However, we recently identified a novel dopaminergic cell group, the vPAG (Lu et al., 2006a), which may be critically involved in the promotion and maintenance of wakefulness. Unlike the VTA, a variety of experimental approaches confirmed the vPAG neurons as being a wake-promoting cell group. For example, vPAG neurons express cFos during wakefulness, and selective cell-specific lesions of the vPAG produced a significant (ca. 20%) reduction in wakefulness (Lu et al., 2006a). In addition, the vPAG is reciprocally connected with all the major components of the sleep–wake circuitry, including the sleep-promoting cell group, the VLPO, and with the wake-promoting cell groups, including the BF, orexinergic neurons in the lateral hypothalamus, and brainstem cholinergic (LDT) and monoaminergic (LC, DR, and median raphe) nuclei. As the above-mentioned wake-promoting cell groups express D1 and D2 receptors extensively, the vPAG may activate (via D1) or disinhibit (via D2) these groups to promote wakefulness. On the other hand, the vPAG dopaminergic neurons may inhibit the sleep-promoting VLPO neurons and, interestingly, this inhibition is mediated by alpha2 adrenergic receptors (Cornil et al., 2002; Gallopin et al., 2004). Finally, the vPAG has direct projections to the majority of the cortex (whereas the VTA projects primarily to mPFC), and thus may also directly influence cortical activity. As in the wake-promoting cell groups, this influence may be mediated by both D1 and D2 receptors. In support of this, a recent study indicated that D1 and D2 receptors act synergistically to activate cortical pyramidal neurons; that is, D1 receptors mediate direct excitation and D2 receptors mediate indirect excitation (via disinhibition of the GABAergic interneurons) of the pyramidal neurons (Xu and Yao, 2010). Thus, the vPAG may promote wakefulness by activating the above-mentioned wake-promoting cell groups and cortex via D1 and/or D2 receptors, and by simultaneously inhibiting the sleep-promoting VLPO neurons via alpha2 receptors. Furthermore, low doses of apomorphine and other dopaminergic agonists may induce its sedative effects via D2-mediated inhibition of the vPAG neurons, although evidence supporting this assertion is currently lacking.
Another important and relevant aspect of the vPAG dopaminergic system is that these neurons express dopamine transporters (DAT), which may partially explain the arousal mechanism of many psychostimulants such as methamphetamine and cocaine. These drugs may inhibit DAT-mediated reuptake of dopamine in the terminals of the vPAG neurons, increasing the postsynaptic availability of dopamine in the wake-promoting cell groups (Fig 2B) and thus increasing wakefulness. Consistent with this hypothesis, DAT knockout mice did not display hyperactivity and arousal after administration of methamphetamine, although their spontaneous wakefulness was higher than that of intact mice (Wisor et al., 2001). In contrast, animals with SNc lesions are paradoxically supersensitive to the arousal and motor effects of methamphetamine or amphetamine (Mandel and Randall, 1985; Schroeder et al., 1997, M. Qiu and J. Lu, unpublished observations). Previously, up-regulation of dopamine receptors in the BG had been proposed to be a potential mechanism of methamphetamine supersensitivity. However, this explanation is hard to reconcile with the fact that there is a very low level of dopamine in the BG after elimination of the SNc – the main dopamine source of the BG. Therefore, we hypothesize that methamphetamine supersensitivity may be due to the additive effects of (1) increased wakefulness following the loss of dopamine inputs to the BG and (2) increased wakefulness due to excess dopamine in the extra-BG sites following methamphetamine administration.
Conclusions
Research during the last decade has provided important insights into the BG circuitry controlling S–W behavior, and how dopamine may modulate this circuit. Electrophysiological, cFos and lesion studies point to a distinct role for the individual BG nuclei in the regulation of S–W, and also suggest that dopamine plays opposing roles in the BG and extra-BG networks. We hypothesize that dopamine in the BG may promote sleep via D2 receptors, while the extra-BG vPAG dopaminergic system may promote wakefulness via D1 and D2 receptors. Further studies using novel genetic methods are required to confirm this hypothesis. Developing a better understanding of the role of dopamine in S–W may shed light on the neurochemical mechanisms underlying sleep disturbances observed in patients with PD or HD, which in turn will aid in the development of novel treatment options.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by US National Institutes of Health grants – NS062727, NS061842, and NS051609 to Jun Lu; Ramalingam Vetrivelan is the recipient of a Pilot and Feasibility Award from the US National Institutes of Health-funded Boston Obesity Nutrition Research Center (P30 DK046200-17S1).
References
Almirall, H., Pigarev, I., de la Calzada, M. D., Pigareva, M., Herrero, M. T., and Sagales, T. (1999). Nocturnal sleep structure and temperature slope in MPTP treated monkeys. J. Neural Transm. 106, 1125–1134.
Arnulf, I., Nielsen, J., Lohmann, E., Schiefer, J., Wild, E., Jennum, P., Konofal, E., Walker, M., Oudiette, D., Tabrizi, S., and Durr, A. (2008). Rapid eye movement sleep disturbances in Huntington disease. Arch. Neurol. 65, 482–488.
Askenasy, J. J. (2001). Approaching disturbed sleep in late Parkinson’s disease: first step toward a proposal for a revised UPDRS. Parkinsonism Relat. Disord. 8, 123–131.
Barroso-Chinea, P., Rico, A. J., Perez-Manso, M., Roda, E., Lopez, I. P., Luis-Ravelo, D., and Lanciego, J. L. (2008). Glutamatergic pallidothalamic projections and their implications in the pathophysiology of Parkinson’s disease. Neurobiol. Dis. 31, 422–432.
Blanco-Centurion, C., Gerashchenko, D., and Shiromani, P. J. (2007). Effects of saporin-induced lesions of three arousal populations on daily levels of sleep and wake. J. Neurosci. 27, 14041–14048.
Bonuccelli, U., Colzi, A., and Del Dotto, P. (2002). Pergolide in the treatment of patients with early and advanced Parkinson’s disease. Clin. Neuropharmacol. 25, 1–10.
Bouali-Benazzouz, R., Tai, C. H., Chetrit, J., and Benazzouz, A. (2009). Intrapallidal injection of 6-hydroxydopamine induced changes in dopamine innervation and neuronal activity of globus pallidus. Neuroscience 164, 588–596.
Castaigne, P., Lhermitte, F., Buge, A., Escourolle, R., Hauw, J. J., and Lyon-Caen, O. (1981). Paramedian thalamic and midbrain infarct: clinical and neuropathological study. Ann. Neurol. 10, 127–148.
Chaudhuri, K. R., and Schapira, A. H. (2009). Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 8, 464–474.
Cooper, A. J., and Stanford, I. M. (2001). Dopamine D2 receptor mediated presynaptic inhibition of striatopallidal GABA(A) IPSCs in vitro. Neuropharmacology 41, 62–71.
Cornil, C. A., Balthazart, J., Motte, P., Massotte, L., and Seutin, V. (2002). Dopamine activates noradrenergic receptors in the preoptic area. J. Neurosci. 22, 9320–9330.
Cortelli, P., Perani, D., Parchi, P., Grassi, F., Montagna, P., De Martin, M., Castellani, R., Tinuper, P., Gambetti, P., Lugaresi, E., and Fazio, F. (1997). Cerebral metabolism in fatal familial insomnia: relation to duration, neuropathology, and distribution of protease-resistant prion protein. Neurology 49, 126–133.
Dahan, L., Astier, B., Vautrelle, N., Urbain, N., Kocsis, B., and Chouvet, G. (2007). Prominent burst firing of dopaminergic neurons in the ventral tegmental area during paradoxical sleep. Neuropsychopharmacology 32, 1232–1241.
Detari, L., Juhasz, G., and Kukorelli, T. (1987). Neuronal firing in the pallidal region: firing patterns during sleep-wakefulness cycle in cats. Electroencephalogr. Clin. Neurophysiol. 67, 159–166.
Dhawan, V., Healy, D. G., Pal, S., and Chaudhuri, K. R. (2006). Sleep-related problems of Parkinson’s disease. Age Ageing 35, 220–228.
Dimpfel, W. (2008). Pharmacological modulation of dopaminergic brain activity and its reflection in spectral frequencies of the rat electropharmacogram. Neuropsychobiology 58, 178–186.
Factor, S. A., McAlarney, T., Sanchez-Ramos, J. R., and Weiner, W. J. (1990). Sleep disorders and sleep effect in Parkinson’s disease. Mov. Disord. 5, 280–285.
Filion, M., and Tremblay, L. (1991). Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 547, 142–151.
Filion, M., Tremblay, L., and Bedard, P. J. (1991). Effects of dopamine agonists on the spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 547, 152–161.
Fujimoto, K., and Kita, H. (1993). Response characteristics of subthalamic neurons to the stimulation of the sensorimotor cortex in the rat. Brain Res. 609, 185–192.
Furuta, T., Koyano, K., Tomioka, R., Yanagawa, Y., and Kaneko, T. (2004). GABAergic basal forebrain neurons that express receptor for neurokinin B and send axons to the cerebral cortex. J. Comp. Neurol. 473, 43–58.
Gallopin, T., Luppi, P. H., Rambert, F. A., Frydman, A., and Fort, P. (2004). Effect of the wake-promoting agent modafinil on sleep-promoting neurons from the ventrolateral preoptic nucleus: an in vitro pharmacologic study. Sleep 27, 19–25.
Gerashchenko, D., Blanco-Centurion, C., Greco, M. A., and Shiromani, P. J. (2003). Effects of lateral hypothalamic lesion with the neurotoxin hypocretin-2-saporin on sleep in Long-Evans rats. Neuroscience 116, 223–235.
Gerashchenko, D., Blanco-Centurion, C. A., Miller, J. D., and Shiromani, P. J. (2006). Insomnia following hypocretin2-saporin lesions of the substantia nigra. Neuroscience 137, 29–36.
Gerashchenko, D., Chou, T. C., Blanco-Centurion, C. A., Saper, C. B., and Shiromani, P. J. (2004). Effects of lesions of the histaminergic tuberomammillary nucleus on spontaneous sleep in rats. Sleep 27, 1275–1281.
Gessa, G. L., Porceddu, M. L., Collu, M., Mereu, G., Serra, M., Ongini, E., and Biggio, G. (1985). Sedation and sleep induced by high doses of apomorphine after blockade of D-1 receptors by SCH 23390. Eur. J. Pharmacol. 109, 269–274.
Gjerstad, M. D., Wentzel-Larsen, T., Aarsland, D., and Larsen, J. P. (2007). Insomnia in Parkinson’s disease: frequency and progression over time. J. Neurol. Neurosurg. Psychiatr. 78, 476–479.
Gritti, I., Mainville, L., Mancia, M., and Jones, B. E. (1997). GABAergic and other non-cholinergic basal forebrain neurons, together with cholinergic neurons, project to the mesocortex and isocortex in the rat. J. Comp. Neurol. 383, 163–177.
Hara, J., Beuckmann, C. T., Nambu, T., Willie, J. T., Chemelli, R. M., Sinton, C. M., Sugiyama, F., Yagami, K., Goto, K., Yanagisawa, M., and Sakurai, T. (2001). Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron 30, 345–354.
Hermann, D. M., Siccoli, M., Brugger, P., Wachter, K., Mathis, J., Achermann, P., and Bassetti, C. L. (2008). Evolution of neurological, neuropsychological and sleep-wake disturbances after paramedian thalamic stroke. Stroke 39, 62–68.
Hirayama, M., Nakamura, T., Hori, N., Koike, Y., and Sobue, G. (2008). The video images of sleep attacks in Parkinson’s disease. Mov. Disord. 23, 288–290.
Hur, E. E., and Zaborszky, L. (2005). Vglut2 afferents to the medial prefrontal and primary somatosensory cortices: a combined retrograde tracing in situ hybridization study [corrected]. J. Comp. Neurol. 483, 351–373.
Hutchison, W. D., Lozano, A. M., Davis, K. D., Saint-Cyr, J. A., Lang, A. E., and Dostrovsky, J. O. (1994). Differential neuronal activity in segments of globus pallidus in Parkinson’s disease patients. Neuroreport 5, 1533–1537.
Kaynak, D., Kiziltan, G., Kaynak, H., Benbir, G., and Uysal, O. (2005). Sleep and sleepiness in patients with Parkinson’s disease before and after dopaminergic treatment. Eur. J. Neurol. 12, 199–207.
Kita, H. (1992). Responses of globus pallidus neurons to cortical stimulation: intracellular study in the rat. Brain Res. 589, 84–90.
Kreitzer, A. C. (2009). Physiology and pharmacology of striatal neurons. Annu. Rev. Neurosci. 32, 127–147.
Lai, Y. Y., Shalita, T., Hajnik, T., Wu, J. P., Kuo, J. S., Chia, L. G., and Siegel, J. M. (1999). Neurotoxic N-methyl-D-aspartate lesion of the ventral midbrain and mesopontine junction alters sleep-wake organization. Neuroscience 90, 469–483.
Larsen, J. P., and Tandberg, E. (2001). Sleep disorders in patients with Parkinson’s disease: epidemiology and management. CNS Drugs 15, 267–275.
Lenard, L., Jando, G., Karadi, Z., Hajnal, A., and Sandor, P. (1988). Lateral hypothalamic feeding mechanisms: iontophoretic effects of kainic acid, ibotenic acid and 6-hydroxydopamine. Brain Res. Bull. 20, 847–856.
Lobo, M. K. (2009). Molecular profiling of striatonigral and striatopallidal medium spiny neurons past, present, and future. Int. Rev. Neurobiol. 89, 1–35.
Lu, J., Jhou, T. C., and Saper, C. B. (2006a). Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J. Neurosci. 26, 193–202.
Lu, J., Sherman, D., Devor, M., and Saper, C. B. (2006b). A putative flip-flop switch for control of REM sleep. Nature 441, 589–594.
Mahon, S., Deniau, J. M., and Charpier, S. (2003). Various synaptic activities and firing patterns in cortico-striatal and striatal neurons in vivo. J. Physiol. Paris 97, 557–566.
Mahon, S., Vautrelle, N., Pezard, L., Slaght, S. J., Deniau, J. M., Chouvet, G., and Charpier, S. (2006). Distinct patterns of striatal medium spiny neuron activity during the natural sleep-wake cycle. J. Neurosci. 26, 12587–12595.
Mandel, R. J., and Randall, P. K. (1985). Quantification of lesion-induced dopaminergic supersensitivity using the rotational model in the mouse. Brain Res. 330, 358–363.
Monaca, C., Laloux, C., Jacquesson, J. M., Gelé, P., Maréchal, X., Bordet, R., Destée, A., and Derambure, P. (2004). Vigilance states in a parkinsonian model, the MPTP mouse. Eur. J. Neurosci. 20, 2474–2478.
Montagna, P., Gambetti, P., Cortelli, P., and Lugaresi, E. (2003). Familial and sporadic fatal insomnia. Lancet Neurol. 2, 167–176.
Monti, J. M., and Monti, D. (2007). The involvement of dopamine in the modulation of sleep and waking. Sleep Med. Rev. 11, 113–133.
Morita, A., Kamei, S., Serizawa, K., and Mizutani, T. (2009). The relationship between slowing EEGs and the progression of Parkinson’s disease. J. Clin. Neurophysiol. 26, 426–429.
Nambu, A., Tokuno, H., Hamada, I., Kita, H., Imanishi, M., Akazawa, T., Ikeuchi, Y., and Hasegawa, N. (2000). Excitatory cortical inputs to pallidal neurons via the subthalamic nucleus in the monkey. J. Neurophysiol. 84, 289–300.
Ni, Z., Bouali-Benazzouz, R., Gao, D., Benabid, A. L., and Benazzouz, A. (2000). Changes in the firing pattern of globus pallidus neurons after the degeneration of nigrostriatal pathway are mediated by the subthalamic nucleus in the rat. Eur. J. Neurosci. 12, 4338–4344.
Pan, H. S., and Walters, J. R. (1988). Unilateral lesion of the nigrostriatal pathway decreases the firing rate and alters the firing pattern of globus pallidus neurons in the rat. Synapse 2, 650–656.
Parmentier, R., Ohtsu, H., Djebbara-Hannas, Z., Valatx, J. L., Watanabe, T., and Lin, J. S. (2002). Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: evidence for the role of brain histamine in behavioral and sleep-wake control. J. Neurosci. 22, 7695–7711.
Perani, D., Cortelli, P., Lucignani, G., Montagna, P., Tinuper, P., Gallassi, R., Gambetti, P., Lenzi, G. L., Lugaresi, E., and Fazio, F. (1993). [18F]FDG PET in fatal familial insomnia: the functional effects of thalamic lesions. Neurology 43, 2565–2569.
Qiu, M. H., Vetrivelan, R., Fuller, P. M., and Lu, J. (2010). Basal ganglia control of sleep-wake behavior and cortical activation. Eur. J. Neurosci. 31, 499–507.
Qu, W. M., Xu, X. H., Yan, M. M., Wang, Y. Q., Urade, Y., and Huang, Z. L. (2010). Essential role of dopamine D2 receptor in the maintenance of wakefulness, but not in homeostatic regulation of sleep, in mice. J. Neurosci. 30, 4382–4389.
Querejeta, E., Delgado, A., Valdiosera, R., Erlij, D., and Aceves, J. (2001). Intrapallidal D2 dopamine receptors control globus pallidus neuron activity in the rat. Neurosci. Lett. 300, 79–82.
Rivera, A., Alberti, I., Martin, A. B., Narvaez, J. A., de la Calle, A., and Moratalla, R. (2002). Molecular phenotype of rat striatal neurons expressing the dopamine D5 receptor subtype. Eur. J. Neurosci. 16, 2049–2058.
Rowland, N., and Stricker, E. M. (1982). Effects of dopamine-depleting brain lesions on experimental hyperphagia in rats. Physiol. Behav. 28, 271–277.
Rye, D. B., Saper, C. B., Lee, H. J., and Wainer, B. H. (1987). Pedunculopontine tegmental nucleus of the rat: cytoarchitecture, cytochemistry, and some extrapyramidal connections of the mesopontine tegmentum. J. Comp. Neurol. 259, 483–528.
Sakata, M., Sei, H., Toida, K., Fujihara, H., Urushihara, R., and Morita, Y. (2002). Mesolimbic dopaminergic system is involved in diurnal blood pressure regulation. Brain Res. 928, 194–201.
Sanchez-Vives, M. V., and McCormick, D. A. (2000). Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat. Neurosci. 3, 1027–1034.
Saper, C. B. (1984). Organization of cerebral cortical afferent systems in the rat. II. Magnocellular basal nucleus. J. Comp. Neurol. 222, 313–342.
Sarter, M., and Bruno, J. P. (2002). The neglected constituent of the basal forebrain corticopetal projection system: GABAergic projections. Eur. J. Neurosci. 15, 1867–1873.
Schafer, D., and Greulich, W. (2000). Effects of parkinsonian medication on sleep. J. Neurol. 247, 24–27.
Schroeder, U., Kreutz, M. R., Schroeder, H., and Sabel, B. A. (1997). Amphetamine induces hypermotility in MPTP-lesioned mice. Pharmacol. Biochem. Behav. 56, 281–285.
Serizawa, K., Kamei, S., Morita, A., Hara, M., Mizutani, T., Yoshihashi, H., Yamaguchi, M., Takeshita, J., and Hirayanagi, K. (2008). Comparison of quantitative EEGs between Parkinson disease and age-adjusted normal controls. J. Clin. Neurophysiol. 25, 361–366.
Soikkeli, R., Partanen, J., Soininen, H., Pääkkönen, A., and Riekkinen P, S. r. (1991). Slowing of EEG in Parkinson’s disease. Electroencephalogr. Clin, Neurophysiol. 79, 159–165.
Steininger, T. L., Rye, D. B., and Wainer, B. H. (1992). Afferent projections to the cholinergic pedunculopontine tegmental nucleus and adjacent midbrain extrapyramidal area in the albino rat. I. Retrograde tracing studies. J. Comp. Neurol. 321, 515–543.
Urbain, N., Gervasoni, D., Souliere, F., Lobo, L., Rentero, N., Windels, F., Astier, B., Savasta, M., Fort, P., Renaud, B., Luppi, P. H., and Chouvet, G. (2000). Unrelated course of subthalamic nucleus and globus pallidus neuronal activities across vigilance states in the rat. Eur. J. Neurosci. 12, 3361–3374.
Vanderwolf, C. H., and Stewart, D. J. (1988). Thalamic control of neocortical activation: a critical re-evaluation. Brain Res. Bull. 20, 529–538.
Wichmann, T., and DeLong, M. R. (2006). “Neurotransmitters and disorders of the basal ganglia,” in Basic Neurochemistry Molecular, Cellular, and Medical Aspects, Vol. 1, eds G. J. Siegel, R. W. Albers, S. Brady, and D. Price (Burlington: Elsevier), 761–776.
Wilson, J. S., Hull, C. D., and Buchwald, N. A. (1983). Intracellular studies of the convergence of sensory input on caudate neurons of cat. Brain Res. 270, 197–208.
Wisor, J. P., Nishino, S., Sora, I., Uhl, G. H., Mignot, E., and Edgar, D. M. (2001). Dopaminergic role in stimulant-induced wakefulness. J. Neurosci. 21, 1787–1794.
Xu, T. X., and Yao, W. D. (2010). D1 and D2 dopamine receptors in separate circuits cooperate to drive associative long-term potentiation in the prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 107, 16366–16371.
Keywords: dopamine, Parkinson’s disease, insomnia, ventral periaquductal gray, globus pallidus, psychostimulants, D2 receptors
Citation: Vetrivelan R, Qiu M-H, Chang C and Lu J (2010) Role of basal ganglia in sleep–wake regulation: neural circuitry and clinical significance. Front. Neuroanat. 4:145. doi: 10.3389/fnana.2010.00145
Received: 30 June 2010;
Accepted: 20 October 2010;
Published online: 23 November 2010.
Edited by:
Jose L. Lanciego, University of Navarra, SpainReviewed by:
J. M. Monti, Clinics Hospital, UruguayAtsushi Nambu, National Institute for Physiological Sciences, Japan
Copyright: © 2010 Vetrivelan, Qiu, Chang and Lu. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Jun Lu and Ramalingam Vetrivelan, Department of Neurology, Beth Israel Deaconess Medical Center, 330 Brookline Avenue, E/CLS#707, Boston, MA 02215, USA. e-mail:amx1QGJpZG1jLmhhcnZhcmQuZWR1LA==dnJhbWFsaW5AYmlkbWMuaGFydmFyZC5lZHU=