 Sergi Ferré1*
Sergi Ferré1* César Quiroz1
César Quiroz1 Marco Orru1 Xavier Guitart1
Marco Orru1 Xavier Guitart1 Gemma Navarro2,3
Gemma Navarro2,3 Antonio Cortés2,3
Antonio Cortés2,3 Vicent Casadó2,3
Vicent Casadó2,3 Enric I. Canela2,3
Enric I. Canela2,3 Carme Lluis2,3 and Rafael Franco2,3,4
Carme Lluis2,3 and Rafael Franco2,3,4
- 1 National Institute on Drug Abuse, Intramural Research Program, National Institutes of Health, U.S. Department of Health and Human Services, Baltimore, MD, USA
- 2 Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas, University of Barcelona, Barcelona, Spain
- 3 Faculty of Biology, Department of Biochemistry and Molecular Biology, University of Barcelona, Barcelona, Spain
- 4 Centro de Investigación Médica Aplicada, Universidad de Navarra, Pamplona, Spain
A very significant density of adenosine A2A receptors (A2ARs) is present in the striatum, where they are preferentially localized postsynaptically in striatopallidal medium spiny neurons (MSNs). In this localization A2ARs establish reciprocal antagonistic interactions with dopamine D2 receptors (D2Rs). In one type of interaction, A2AR and D2R are forming heteromers and, by means of an allosteric interaction, A2AR counteracts D2R-mediated inhibitory modulation of the effects of NMDA receptor stimulation in the striatopallidal neuron. This interaction is probably mostly responsible for the locomotor depressant and activating effects of A2AR agonist and antagonists, respectively. The second type of interaction involves A2AR and D2R that do not form heteromers and takes place at the level of adenylyl cyclase (AC). Due to a strong tonic effect of endogenous dopamine on striatal D2R, this interaction keeps A2AR from signaling through AC. However, under conditions of dopamine depletion or with blockade of D2R, A2AR-mediated AC activation is unleashed with an increased gene expression and activity of the striatopallidal neuron and with a consequent motor depression. This interaction is probably the main mechanism responsible for the locomotor depression induced by D2R antagonists. Finally, striatal A2ARs are also localized presynaptically, in cortico-striatal glutamatergic terminals that contact the striato-nigral MSN. These presynaptic A2ARs heteromerize with A1 receptors (A1Rs) and their activation facilitates glutamate release. These three different types of A2ARs can be pharmacologically dissected by their ability to bind ligands with different affinity and can therefore provide selective targets for drug development in different basal ganglia disorders.
Postsynaptic Striatal Adenosine A2A Receptors
A very significant density of adenosine A2A receptors (A2ARs) is present in the striatum (Rosin et al., 1998; Hettinger et al., 1998; Schiffmann et al., 2007; Quiroz et al., 2009), where they are preferentially localized postsynaptically in the soma and dendrites of GABAergic striatopallidal. These neurons also show a high density of dopamine D2 receptors (D2Rs) and there is clear evidence for the existence of postsynaptic mechanisms in the control of glutamatergic neurotransmission to the enkephalinergic medium spiny neuron (MSN) by at least two reciprocal antagonistic interactions between A2ARs and D2Rs (Ferré et al., 2008). In one type of interaction, stimulation of A2AR counteracts the D2R-mediated inhibitory modulation of NMDA receptor (NMDAR)-mediated effects, which include modulation of Ca2+ influx, transition to the up-state and neuronal firing (Azdad et al., 2009; Higley and Sabatini, 2010; Figure 1). This interaction seems to be mostly responsible for the locomotor depressant and activating effects of A2AR agonists and antagonists, respectively (Ferré et al., 2008; Orru et al., 2011), which correlates with the results of behavioral experiments showing that A2AR activation or blockade decreases or increases, respectively, the motor effects elicited by D2R activation (Ferré et al., 2008).
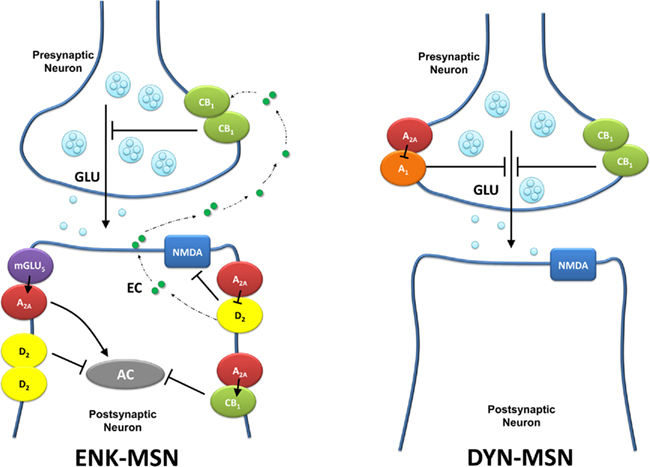
Figure 1. Schematic representation of the different subpopulation of striatal A2ARs. Presynaptic A2ARs are localized in glutamatergic terminals that contact the dynorphynergic medium spiny neuron (DYN-MSN), where they form heteromers with A1Rs. Postsynaptic A2ARs are localized in the enkephalinergic medium spiny neuron (ENK-MSN), where they from heteromers with D2Rs, CB1Rs, and mGlu5Rs. AC, adenylyl cyclase; EC, endocannabinoid; GLU, glutamate.
Initially, the main mechanism responsible for this A2AR–D2R interaction was attributed to what it was described as an “intramembrane interaction,” by which activation of A2AR could decrease the affinity of an adjacent D2R for agonists in striatal membrane preparations (Ferré et al., 1991). It was afterward hypothesized that this kind of intramembrane interaction was a biochemical property of receptor heteromers with important functional implications (Zoli et al., 1993). A receptor heteromer is now defined as a macromolecular complex composed of at least two (functional) receptor units with biochemical properties that are demonstrably different from those of its individual components (Ferré et al., 2009). The term “intramembrane interaction” is now known as “allosteric interaction in the receptor heteromer,” which is defined as an intermolecular interaction by which binding of a ligand to one of the receptor units in the receptor heteromer changes the binding properties of another receptor unit (Ferré et al., 2009). Another definition recently introduced in the field of receptor heteromers is “biochemical fingerprint,” which is a biochemical characteristic of a receptor heteromer that can be used for its identification, even in a native tissue (Ferré et al., 2009). The introduction of this concept is important in view of the difficulty in demonstrating receptor heteromers in native tissues. Biophysical techniques, such as bioluminescence and fluorescence resonance energy transfer (BRET and FRET) techniques can be easily applied in artificial cell systems to demonstrate receptor heteromerization (Bouvier, 2001), but not in native tissues. Recent technological advances, using receptor labeling with selective fluorescent ligands, have allowed the demonstration of receptor homomers with time-resolved FRET in a native tissue (oxytocin receptor homomers in mammary glands; Albizu et al., 2010). However, this required the use of high quantities of a tissue with high expression of the receptor under study (Albizu et al., 2010).
The A2AR–D2R allosteric interaction, in fact, constitutes a biochemical fingerprint of the A2AR–D2R heteromer, since it depends on the proper quaternary structure of the heteromer. Thus, it has been recently shown that disruption of an electrostatic interaction between identified intracellular domains of the A2AR and D2R leads to a significant modification of the quaternary structure of the heteromer and to the disappearance of the A2AR–D2R allosteric interaction (Borroto-Escuela et al., 2010a; Navarro et al., 2010). The electrostatic interaction in the A2AR–D2R heteromer involves an arginine-rich epitope of the third intracellular loop (3IL) of the D2R and a phosphorylated residue localized in the C terminus of the A2AR (Woods and Ferré, 2005; Navarro et al., 2010). It is important to point out that this electrostatic interaction is not directly involved in the A2AR–D2R heteromer interface, which seems to be mostly determined by direct interactions between transmembrane domains (Borroto-Escuela et al., 2010b; Navarro et al., 2010).
A closer look to recent electrophysiological experiments (Azdad et al., 2009; Higley and Sabatini, 2010) indicates that, although useful as a biochemical fingerprint, the allosteric interaction in the receptor heteromer does not play a main role in the antagonistic A2AR–D2R-mediated functional interaction. In the study by Azdad et al. (2009), the D2R-mediated response consisted on the counteraction of NMDAR-mediated increase in firing rate by enkephalinergic MSNs (analyzed by patch-clamp experiments in identified striatal D2R-expressing MSNs). In this experimental setting, application of an A2AR agonist did not produce any significant effect on its own, but completely blocked the D2R-mediated response. Remarkably, this interaction was dependent on the integrity of the quaternary structure of the A2AR–D2R heteromer. Thus, the counteracting effect of the A2AR agonist disappeared after the application of peptides that selectively disrupted the intracellular electrostatic interaction (Azdad et al., 2009). Importantly, the counteracting effect of the A2AR agonist was detected in the presence of a high concentration of the D2R agonist, which should be able to surmount a decrease in the affinity of the D2R caused by A2AR occupation (Ferré et al., 1991). Therefore, although it might still be involved, the allosteric interaction, which leads to a lower affinity of D2R for dopamine when adenosine is activating A2AR, does not seem to be the main mechanism underlying the A2AR–D2R functional interaction in the A2AR–D2R heteromer.
The same intracellular arginine-rich epitope of the D2R that is involved in the electrostatic interaction with A2AR in the A2AR–D2R heteromer has been demonstrated to bind to calmodulin and also to be fundamental for the activation of Gi/o proteins (Bofill-Cardona et al., 2000; Navarro et al., 2009). Since calmodulin binding to the same epitope of the D2R impairs its ability to signal through Gi/o proteins (Bofill-Cardona et al., 2000), it is likely that binding of the C terminus of the A2AR to the same epitope reduces the capacity of the D2R to bind calmodulin and to signal through Gi/o proteins. In fact, it has recently been shown that the binding of calmodulin to the A2AR–D2R heteromer is occurring within the proximal portion of the A2AR but not with the D2R (Navarro et al., 2009). It is possible that agonist binding to the A2AR induces a conformational change in the A2AR–D2R heteromer that causes an even further impairment in the coupling of the D2R to the Gi/o protein. Thus, it seems that, in the A2AR–D2R heteromer, D2R does not signal through Gi/o proteins or that its main signaling is by a G-protein-independent mechanism. However, the recent study by Higley and Sabatini (2010) suggests that the D2R-mediated inhibitory modulation of NMDAR-mediated Ca2+ signaling in the enkephalinergic MSN is mediated by PKA and, therefore, most probably related to the ability of D2R to couple to Gi/o and to inhibit adenylyl cyclase (AC). Interestingly, in these experiments (and in agreement with the experiments by Azdad et al., 2009), an A2AR agonist did not produce any significant effect on its own, but counteracted the effect of a D2R agonist. Thus, although Higley and Sabatini (2010) suggested that this interaction between A2AR and D2R takes place at the AC level, it shows similar characteristics to the A2AR–D2R heteromer-dependent interaction. In summary, A2AR–D2R heteromers seem to play a key role in the modulation of NMDAR-mediated signaling in the enkephalinergic MSN, but the molecular mechanisms involved in these A2AR–D2R–NMDAR interactions are yet to be determined.
In addition to the antagonistic A2AR–D2R receptor interaction in the A2AR–D2R heteromer, D2R stimulation impedes A2AR to signal through AC (Kull et al., 1999; Chen et al., 2001; Hillion et al., 2002; Håkansson et al., 2006; Figure 1). This D2R–A2AR interaction takes place at the second messenger level, and stimulation of Gi/o-coupled D2R counteracts the effects of Gs/olf-coupled A2AR (Ferré et al., 2007, 2008). Due to a strong tonic effect of endogenous dopamine on striatal D2R, this interaction keeps A2AR from signaling through AC. However, under conditions of dopamine depletion or with pharmacological D2R blockade, A2AR-mediated signaling through the cAMP–PKA cascade may be unleashed. Antagonism of D2R is biochemically associated with a significant increase in the phosphorylation of PKA-dependent substrates, which increases gene expression and the activity of the enkephalinergic MSN, producing locomotor depression (reviewed in Ferré et al., 2008). This appears to be the main mechanism responsible for the locomotor depression induced by D2R antagonists. Thus the motor depressant and most biochemical effects induced by pharmacologic blockade of D2R may be counteracted by pharmacological blockade of A2AR (Chen et al., 2001; Håkansson et al., 2006).
The two reciprocal antagonistic interactions, A2AR toward D2R (A2R–D2R) and D2R toward A2AR (D2R–A2AR), take place simultaneously in the same cell, which suggest that are most likely mediated by the existence of at least two different populations of postsynaptic striatal A2AR in the enkephalinergic MSN (Ferré et al., 2008). One population would be forming heteromers with D2R and would determine that A2AR stimulation inhibits D2R-mediated signaling (A2AR–D2R interaction), while another population would not be forming heteromers with D2R and would determine that D2R stimulation inhibits A2AR-mediated signaling (D2R–A2AR interaction). This second population of postsynaptic A2AR would either not form heteromers or would form heteromers with other receptors, such as glutamate mGlu5 receptors (mGlu5Rs; Ferré et al., 2002; Figure 1). Importantly, heteromerization of A2AR with mGlu5R is associated with a synergistic effect upon A2AR and mGlu5R co-activation at the level of AC and MAPK, providing a physiological mechanism by which A2AR can overcome the D2R–A2AR interaction (Ferré et al., 2002; Nishi et al., 2003). Co-stimulation of A2AR and mGlu5R in vivo, with the central administration of selective agonists, allowed A2AR to get rid of the inhibitory effect of the D2R and signal through the cAMP–PKA cascade (Ferré et al., 2002). Since this A2AR–D2R–mGlu5R interaction could be demonstrated in animal models of Parkinson’s disease (Popoli et al., 2001; Kachroo et al., 2005), it was postulated that co-administration of A2AR and mGlu5R antagonists could be useful as a therapeutic strategy in this disease (Popoli et al., 2001).
Still a third population of postsynaptic A2AR would form heteromers with cannabinoid CB1 receptors (CB1Rs; Carriba et al., 2007; Figure 1). In this heteromer, activation of A2AR is necessary to allow CB1R-mediated signaling. Thus, in a human neuroblastoma cell line, CB1R-mediated inhibition of AC activity was found to be completely dependent on A2AR co-activation (Carriba et al., 2007). Similarly, several biochemical effects of CB1R agonists in primary striatal cell cultures and striatal slices have been shown to depend on A2AR co-activation (Yao et al., 2003; Andersson et al., 2005). Accordingly, Tebano et al. (2009) reported that the depression of synaptic transmission induced by a CB1R agonist in cortico-striatal slices was prevented by A2AR antagonists and also by the conditional genetic blockade of striatal postsynaptic A2AR. The permissive effect of A2AR toward CB1R function did not seem to occur presynaptically, as the ability of the CB1R agonist to increase the R2/R1 ratio under a protocol of paired-pulse stimulation was not modified by an A2AR antagonist (Tebano et al., 2009). These results would predict that A2AR antagonists should produce similar behavioral effects than CB1R antagonists and, in fact, pharmacological or genetic inactivation of A2ARs reduce the motor depressant, cataleptic, and rewarding effects of CB1R agonists (Soria et al., 2004; Andersson et al., 2005; Carriba et al., 2007; Justinova et al., 2011). Significantly, it has been recently reported that low doses of an A2AR antagonist (MSX-3) reduce in squirrel monkeys self-administration of THC and anandamide, but not cocaine (Justinova et al., 2011).
Although the studies just mentioned indicate that the motor (depressant) effects of CB1R agonists might depend on adenosine A2A receptor signaling, a recent study by Lerner et al. (2010) suggested quite the opposite, that CB1R signaling mediates the locomotor-activating effects of A2AR antagonists. Thus, pharmacological or genetic inactivation of CB1R reduced the locomotor activation induced by an A2AR antagonist in mice habituated to the testing environment (Lerner et al., 2010). The mechanistic explanation of this interaction is related to the previously reported D2R agonist-mediated endocannabinoid release by the enkephalinergic MSN, which by retrograde signaling would inhibit glutamate release by stimulating CB1R localized in glutamatergic terminals. This would lead to a decreased stimulation of the striatopallidal MSN, which would produce locomotor activation (Kreitzer and Malenka, 2007). In fact, Kreitzer and Malenka (2007) advocated that, instead of direct postsynaptic effects, such as the previously mentioned D2R-mediated modulation of NMDAR-mediated signaling (Azdad et al., 2009; Higley and Sabatini, 2010), this indirect and endocannabinoid-mediated presynaptic effect is the main mechanism by which D2R stimulation produces inhibition of the enkephalinergic MSN function. According to Lerner et al. (2010), an A2AR antagonist would then produce locomotor activation by disinhibiting a tonic A2AR-mediated inhibition of D2R-mediated endocannabinoid release. However, this hypothesis would predict that CB1R agonists and antagonists should produce locomotor activation and depression, respectively, and that CB1R blockade should counteract the motor effects of D2 receptor agonists. This is the opposite of what has been reported in previous studies (for a recent review, see Ferré et al., 2010). To reevaluate the findings by Lerner et al. (2010) we studied in detail the effects of pharmacological interactions between A2AR antagonists and CB1R antagonists on the locomotor activity in rats not habituated to the testing environment (Orru et al., submitted). Whereas we could indeed reproduce the results by Lerner et al. (2010) showing that a CB1R antagonist significantly decreases the locomotor effects induced by an A2AR antagonist, we found that the CB1R antagonist also produces a comparable decrease in locomotion in vehicle-treated animals (statistical analysis indicated that the locomotor effects of A2AR and CB1R antagonists were not interrelated). It was therefore the use of habituated animals (which display very low locomotor activity in the testing environment) what masked the depressant effect of CB1R antagonist in the vehicle-treated animals in the study by Lerner et al. (2010).
In addition to the three populations of postsynaptic striatal A2AR so far reported, there is also experimental evidence for a potentially more complex picture, which includes the possibility of receptor heteromultimers. Thus, using a new biophysical/based technology, sequential resonance energy transfer (SRET), and bimolecular fluorescence complementation plus BRET, evidence for A2AR–CB1R–D2R and A2AR–D2R–mGlu5R heteromers in transfected cells has been recently obtained (Carriba et al., 2008; Cabello et al., 2009; Navarro et al., 2010). Mutation experiments indicated that the interactions of the intracellular domains of the CB1R receptor with A2AR and D2R are fundamental for the correct formation of the quaternary structure needed for the function (MAPK signaling) of the A2AR–CB1R–D2R heteromers. It should be noted that the analysis of MAPK signaling in striatal slices of CB1R KO mice and wild-type littermates supports the existence of A2AR–CB1R–D2R receptor heteromers in the brain (Navarro et al., 2010). Despite the stoichiometry of the different populations of postsynaptic striatal A2AR heteromers (and homomers) is not known, taking into account the very high density of A2ARs and D2Rs in the enkephalinergic MSM, we postulate that A2AR and D2R homomers and A2AR–D2R heteromers are the most common receptor populations, followed by combinations of those populations with CB1R and mGlu5R.
It is also of importance to mention that there is also evidence for the existence of A2AR receptors, also co-localized with D2Rs, in the somatodendritic and nerve terminal regions of the cholinergic striatal interneurons and that their interactions modulate acetylcholine release (James and Richardson, 1993; Jin et al., 1993; Preston et al., 2000; Tozzi et al., 2011). The study by Jin et al. (1993) showed evidence for an antagonistic A2AR–D2AR interaction in the modulation of striatal acetylcholine release. Thus, A2AR stimulation counteracted the ability of D2R activation to inhibit acetylcholine release. Similarly, a recent study showed that A2AR blockade potentiates D2R-mediated modulation of acetylcholine release (Tozzi et al., 2011), again indicating the existence of an antagonistic A2AR–D2R interaction and, probably, A2AR–A2AR heteromers in striatal cholinergic interneurons.
Presynaptic Striatal Adenosine A2A Receptors
Striatal A2ARs are not only localized postsynaptically but also presynaptically, in glutamatergic terminals, where they heteromerize with A1 receptors (A1Rs) and where they perform a fine-tuned modulation of glutamate release (Ciruela et al., 2006; Quiroz et al., 2009; Figure 1). Thus, A1R–A2AR heteromers seem to work as a concentration-dependent switch (Ferré et al., 2007), with adenosine acting primarily at A1Rs at low concentrations, and at both A1Rs and A2ARs at higher concentrations. Activation of the A1R in the A1R–A2AR heteromer produces inhibition of glutamate release, while the additional activation of the A2AR produces the opposite effect, on a mechanism that seems to involve an allosteric modulation in the receptor heteromer and interactions at the G protein level (Ciruela et al., 2006; Ferré et al., 2007). Interestingly, presynaptic A2ARs are preferentially localized in glutamatergic terminals of cortico-striatal afferents to the dynorphinergic MSN (Quiroz et al., 2009). Apart from morphological evidence provided by immunohistochemical and electron microscopy experiments, patch-clamp experiments in identified enkephalinergic and dynorphinergic MSNs provided a functional demonstration of the segregation of striatal presynaptic A2ARs. Thus, an A2AR agonist and an A2AR receptor antagonist significantly increased and decreased, respectively, the amplitude of excitatory postsynaptic currents induced by the intrastriatal stimulation of glutamatergic afferents measured in identified enkephalinergic, but not dynorphinergic MSNs. Mean-variance analysis indicated a presynaptic locus for the A2AR-mediated modulation (Quiroz et al., 2009). Thus, there seems to be a selective A2AR-mediated modulation of glutamate release to the dynorphinergic MSN, which is in disagreement with the recently proposed role of postsynaptic A2ARs in the modulation of glutamate release to the enkephalinergic MSN (Lerner et al., 2010).
The powerful modulatory role of presynaptic A2ARs on striatal glutamate release was first demonstrated with in vivo microdialysis experiments by Popoli et al. (1995), who showed that the striatal perfusion of an A2AR agonist produced a very pronounced increase in the basal striatal extracellular concentrations of glutamate. Also intrastriatal perfusion of an A2AR antagonist through a microdialysis probe could significantly counteract striatal glutamate release induced by cortical electrical stimulation in the orofacial premotor cortex (Quiroz et al., 2009). A striking unexpected finding was that the counteraction of glutamate release was also accompanied by a complete counteraction of the jaw movements induced by the cortical electrical stimulation, demonstrating the very important role of presynaptic A2ARs in the control of cortico-striatal glutamatergic neurotransmission. By combining cortical electrical stimulation and recording of EMG activity of the mastication muscles, a power correlation coefficient (PCC) was established as a quantitative in vivo measure of cortico-striatal neurotransmission (Quiroz et al., 2009). PCC was shown to be significantly and dose dependently decreased by the systemic administration of an A2AR receptor antagonist. PCC could therefore be used as a method to screen the presynaptic effect of A2AR antagonists (see below).
According to the widely accepted functional basal circuitry model (Obeso et al., 2002; DeLong and Wichmann, 2007), blockade of postsynaptic striatal A2AR in the A2AR–D2R heteromer, localized in the enkephalinergic MSN should potentiate spontaneous or psychostimulant-induced motor activation. On the other hand, according to the same model, blockade of presynaptic striatal A2AR localized in the cortico-striatal glutamatergic terminals that make synaptic contact with the dynorphinergic MSN should decrease motor activity. The clear locomotor-activating effects of systemically administered A2AR antagonists could be explained by the significantly higher density of postsynaptic versus presynaptic striatal A2AR and to a stronger influence of a tonic adenosine and A2AR-mediated modulation of the enkephalinergic versus dynorphinergic MSNs under basal conditions. The results by Shen et al. (2008) about the differential effects of A2AR antagonists on psychostimulant-induced locomotor activation in WT versus conditional striatal postsynaptic A2AR KO mice (potentiation versus counteraction, respectively) support this hypothesis. As previously suggested (Ferré et al., 2007), activation of presynaptic A2ARs seems to be highly dependent on the level of adenosine generated upon cortico-striatal glutamatergic input.
Striatal D2Rs are also localized presynaptically, in dopaminergic and glutamatergic terminals (Higley and Sabatini, 2010), giving the frame for the existence of interactions with A2ARs at least in those terminals establishing contact with the dynorphinergic MSN. The experimental evidence suggest that there is also a presynaptic D2R–A2AR interaction by which D2R activation tonically inhibits the ability of endogenous adenosine to produce an A2AR-mediated increase in the basal extracellular levels of glutamate. Thus, dopamine denervation significantly potentiates A2AR agonist-mediated stimulation of glutamate release (Tanganelli et al., 2004). This has the biochemical characteristics of an interaction between A2ARs and D2Rs at the AC level and not forming A2AR–D2R heteromers. Furthermore, results Rodrigues 2005 have also demonstrated the existence of mGlu5Rs in striatal glutamatergic terminals co-localized with A2ARs and which facilitate glutamate release in a synergistic manner. The interplay between adenosine- and dopamine-mediated actions at the presynaptic level is therefore affected by the occurrence of mGlu5Rs.
The presynaptic localization of CB1Rs in striatal glutamatergic terminals is well established, and therefore they can be co-localized with A2AR in terminals establishing contact with the dynorphinergic MSN (Ferré et al., 2010). The existence of A2AR–CB1R heteromers in striatal glutamatergic terminals which could mediate the reinforcing effects of cannabinoids has been recently postulated (Ferré et al., 2010; Justinova et al., 2011). However, a recent study by Martire et al. (2011) indicates that cannabinoid/adenosine functional interactions result from an interaction at the second messenger level. In the frame of heteromerization A2AR activation should facilitate the Gi/o-mediated effect of CB1R activation measured, as inhibition of glutamate release. Nevertheless, Martire et al. (2011), by studying extracellular field potentials recordings in cortico-striatal slices and superfused striatal nerve terminals, very convincingly showed that, instead, A2AR activation prevents CB1R-mediated inhibition of glutamate release. These results indicate that regulation of glutamate release by cannabinoids is not dependent on presynaptic A2AR–CB1R heteromers.
In summary, a great amount of available data indicates that, presynaptically, A2ARs form heteromers mostly with A1Rs. In addition, there seems to be a population of A2ARs not forming heteromers but establishing antagonistic interactions with D2Rs and CB1Rs and synergistic interactions with mGlu5Rs. Apart from co-expression, at this moment we do not know the variables that determine the ability of A2ARs to bind to different receptors to form different pre and postsynaptic heteromers. Thus, D2Rs are also localized presynaptically, but yet they do not seem to form heteromers with A2ARs. A2ARs could bind with more affinity to A1Rs than to D2Rs or particular scaffolding proteins could favor a particular A2AR heteromer. All these are questions still need to be answered.
Targeting Striatal Pre and Postsynaptic A2A Receptors
A surprising yet fundamental finding of a recent study is that several A2AR antagonists previously thought as being pharmacologically similar present different striatal pre and postsynaptic profiles (Orru et al., 2011). Six compounds already known as selective A2AR antagonists were first screened for their ability to block striatal pre and postsynaptic A2ARs with in vivo models. Locomotor activation was used to evaluate postsynaptic activity while PCC counteraction was used to determine presynaptic activity (see above). SCH-442416 and KW-6002, showed preferential pre and postsynaptic profiles, respectively, and four compounds, MSX-3, SCH-420814, SCH-58261, and ZM-241385, showed mixed pre–postsynaptic profiles. Combining in vivo microdialysis with cortical electrical stimulation was used as an additional in vivo evaluation of presynaptic activity of A2AR antagonists. In agreement with its preferential presynaptic profile, SCH-442416 significantly counteracted striatal glutamate release induced by cortical stimulation at a dose that strongly counteracted PCC but did not induce locomotor activation. On the other hand, according to its preferential postsynaptic profile, KW-6002 did not modify striatal glutamate release induced by cortical stimulation at a dose that produced a pronounced locomotor activation but did not counteract PCC.
Another important finding of the study by Orru et al. (2011) was that at least part of these pharmacological differences between A2AR antagonists could be explained by the ability of pre and postsynaptic A2AR to form different receptor heteromers, with A1R and D2R, respectively (see above). Radioligand-binding experiments were performed in cells stably expressing A2AR, A2AR–D2R heteromers, or A1R–A2AR heteromers to determine possible differences in the affinity of these different A2ARs for A2AR antagonists. Co-expression with A1R did not significantly modify the affinity of A2ARs for the different ligands, but co-expression with D2Rs decreased the affinity of all compounds, with the exception of KW-6002 (Orru et al., 2011). The structural changes in the A2AR induced by heteromerization with the D2R could be detected not only by antagonists but also by agonist binding. Indeed, the affinity of the selective A2AR agonist CGS-21680 was reduced in cells co-transfected with D2Rs. When trying to explain the differential action of SCH-442416 observed in vivo, it is interesting to note that this compound in particular showed a much higher affinity for the A2AR in a presynaptic-like than in a postsynaptic-like context. In fact, the affinity of A2AR for SCH-442416 in cells expressing A2AR–D2R heteromers was markedly reduced (40 times higher B50 values in competitive-inhibition experiments with [3H]ZM-241385 in cells expressing A2AR–D2R than A1R–A2AR heteromers).
The decrease in affinity upon co-expression with D2Rs was much less pronounced for ZM-241385, SCH-58261, MSX2, or SCH-420814, for which the affinity was reduced from two to about ninefold (Orru et al., 2011). Taking into account that these A2AR antagonists behaved qualitatively similar than the A2AR agonist CGS-21680 in terms of binding to A1R–A2AR and A2AR–D2R heteromers, it was expected that these four compounds compete equally for the binding of the endogenous agonist at pre and at postsynaptic sites. This would fit with the in vivo data, which showed that these compounds have a non-preferred pre–postsynaptic profile. Yet, KW-6002 was the only antagonist whose affinity was not significantly different in cells expressing A2AR, A1R–A2AR heteromers, or A2AR–D2R heteromers. Thus, KW-6002 showed the best relative affinity for A2AR–D2R heteromers of all compounds, which can at least partially explain its preferential postsynaptic profile. Experiments performed with the non-selective adenosine receptor antagonist caffeine also showed a correlation between the in vivo data and the in vitro preference for postsynaptic A2AR-containing heteromers. It was previously reported that in transfected mammalian cells the affinity of A2AR for the non-selective adenosine receptor antagonist caffeine did not change when co-transfected with D2R, but it was significantly decreased (about 10 times) when co-transfected with A1R (Ciruela et al., 2006). As predicted, caffeine did not significantly reduce PCC at doses that produce pronounced motor activation (Zanoveli et al., in preparation).
It must be pointed out that to say that SCH-442416 is a selective presynaptic A2AR antagonist is an oversimplification. In fact, the in vitro data indicated that SCH-442416 binds equally well to the A2AR not forming heteromers than to the A2AR in the A1R–A2AR heteromer. Therefore, according to the previous description of the different populations of striatal A2ARs, SCH-442416 should also be effective at counteracting D2R antagonist-induced motor depression. In fact, at doses that are not producing locomotor activation (but that reduce PCC), SCH-442416 significantly counteracts the locomotor depression induced by the D2R antagonist raclopride (Orru et al., submitted). On the other hand, KW-6002 produced the same locomotor activation with or without co-administration with raclopride, in agreement with its ability to block the three populations of A2AR studied so far in vitro, A2AR, A2A–D2R, and A1R–A2AR. Importantly, KW-6002 also produced the same locomotor activation when co-administered with the A2AR agonist CGS-21680, while SCH-442416, at the same dose that counteracted the depressant effect of raclopride, did not significantly counteract the depressant effect of CGS-21680. These results, therefore agree with the hypothesis that the subpopulation of postsynaptic A2AR forming heteromers with D2R are mainly responsible for both the locomotor activation and depression induced by A2AR antagonists and agonists, respectively. In summary, SCH-442416 can be considered as a compound that at relatively low doses not only binds preferentially to presynaptic A2ARs localized in cortico-striatal glutamatergic terminals (Orru et al., 2011), but also to a subpopulation of postsynaptic A2ARs most probably not forming heteromers with D2Rs, but which function is tonically inhibited by D2Rs activated by endogenous dopamine. Interestingly, [11C]SCH-442416 has been used in rats, monkeys, and humans as a PET radioligand and shown to nicely label striatal A2ARs (Moresco et al., 2005; Schiffmann et al., 2007; Brooks et al., 2010). The low doses used in PET experiments indicate that [11C]SCH-442416 is mostly labeling presynaptic A2ARs and postsynaptic A2ARs that do not form heteromers with D2Rs. The use of [11C]SCH-442416 and other less selective radioligand in combination with cold SCH-442416 could allow the identification of the different populations of A2ARs in the human brain. The picture is still incomplete, and a further evaluation of the affinity of A2AR antagonists for A2AR–mGlu5R and A2AR–CB1 heteromers (and of heterotrimers) is needed. Nevertheless, the information so far available is very valuable to attempt the design of more efficient A2AR antagonists to be used in basal ganglia disorders.
A2A Receptor Heteromers as Targets for Drug Development
The results of the above mentioned studies support the notion that receptor heteromers may be used as selective targets for drug development. Main reasons are the very specific neuronal localization of receptor heteromers (even more specific than receptor subtypes themselves), and a differential ligand affinity of a receptor depending on its partner (or partners) in the receptor heteromer. Striatal A2AR-containing heteromers become particularly interesting targets, eventually useful for a variety of neuropsychiatric disorders. Blocking postsynaptic A2ARs in the enkephalinergic MSN should be beneficial for Parkinson’s disease because it would decrease the activity of the indirect striatal efferent pathway. On the one hand, one benefit would come from potentiating the effect of l-dopa or other dopamine receptor agonists on the D2R-mediated signaling in the A2AR–D2R heteromer. On the other hand, blockade of A2ARs not forming heteromers with D2Rs (but antagonistically interacting with D2R at the AC level) should counteract the effects of the disinhibited A2AR signaling. However, blocking presynaptic A2AR in glutamatergic terminals contacting dynorphinergic MSN (either forming or not heteromers with A1R) should decrease glutamatergic transmission through the direct striatal efferent pathway, thus decreasing motor activity and, therefore, decreasing the antiparkinsonian efficacy of A2AR antagonists. The most convenient A2AR antagonist to treat Parkinson’s disease patients would have more affinity for postsynaptic than for presynaptic receptors. Additionally, a selective blockade of presynaptic A2ARs should be useful in dyskinetic disorders such as Huntington’s disease and could also be useful in obsessive–compulsive disorders and drug addiction. Effective treatment of l-dopa-induced dyskinesia using “presynaptic” A2AR antagonists would be a possibility to explore.
The results by Orru et al. (2011) give a mechanistic explanation to the already reported antiparkinsonian activity of KW-6002 and suggest that SCH-442416 could be useful for the treatment of dyskinetic disorders, obsessive–compulsive disorders and in drug addiction. Medicinal chemistry and in silico modeling should help in elucidating the molecular properties that determine the particular pharmacological profile of SCH-442416 and KW-6002, which may be used as lead compounds to obtain, respectively, more effective antidyskinetic and antiparkinsonian compounds.
Acknowledgments
Work supported with the intramural funds of the National Institute on Drug Abuse.
References
Albizu, L., Cottet, M., Kralikova, M., Stoev, S., Seyer, R., Brabet, I., Roux, T., Bazin, H., Bourrier, E., Lamarque, L., Breton, C., Rives, M. L., Newman, A., Javitch, J., Trinquet, E., Manning, M., Pin, J. P., Mouillac, B., and Durroux, T. (2010). Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat. Chem. Biol. 6, 587–594.
Andersson, M., Usiello, A., Borgkvist, A., Pozzi, L., Dominguez, C., Fienberg, A. A., Svenningsson, P., Fredholm, B. B., Borrelli, E., Greengard, P., and Fisone, G. (2005). Cannabinoid action depends on phosphorylation of dopamine- and cAMP-regulated phosphoprotein of 32 kDa at the protein kinase A site in striatal projection neurons. J. Neurosci. 25, 8432–8438.
Azdad, K., Gall, D., Woods, A. S., Ledent, C., Ferré, S., and Schiffmann, S. N. (2009). Dopamine D2 and adenosine A2A receptors regulate NMDA-excitation in accumbens neurons through A2A-D2 receptor heteromerization. Neuropsychopharmacology 34, 972–986.
Bofill-Cardona, E., Kudlacek, O., Yang, Q., Ahorn, H., Freissmuth, M., and Nanoff, C. (2000). Binding of calmodulin to the D2-dopamine receptor reduces receptor signaling by arresting the G protein activation switch. J. Biol. Chem. 275, 32672–32680.
Borroto-Escuela, D. O., Marcellino, D., Narvaez, M., Flajolet, M., Heintz, N., Agnati, L., Ciruela, F., and Fuxe, K. (2010a). A serine point mutation in the adenosine A2AR C-terminal tail reduces receptor heteromerization and allosteric modulation of the dopamine D2R. Biochem. Biophys. Res. Commun. 394, 222–227.
Borroto-Escuela, D. O., Romero-Fernandez, W., Tarakanov, A. O., Gómez-Soler, M., Corrales, F., Marcellino, D., Narvaez, M., Frankowska, M., Flajolet, M., Heintz, N., Agnati, L. F., Ciruela, F., and Fuxe, K. (2010b). Characterization of the A2AR-D2R interface: focus on the role of the C-terminal tail and the transmembrane helices. Biochem. Biophys. Res. Commun. 402, 801–807.
Bouvier, M. (2001). Oligomerization of G-protein-coupled transmitter receptors. Nat. Rev. Neurosci. 2, 274–286.
Brooks, D. J., Papapetropoulos, S., Vandenhende, F., Tomic, D., He, P., Coppell, A., and O’Neill, G. (2010). An open-label, positron emission tomography study to assess adenosine A2A brain receptor occupancy of vipadenant (BIIB014) at steady-state levels in healthy male volunteers. Clin. Neuropharmacol. 33, 55–60.
Cabello, N., Gandia, J., Bertarelli, D. C., Watanabe, M., Lluis, C., Franco, R., Ferré, S., Lujan, R., and Ciruela, F. (2009). Metabotropic glutamate type 5, dopamine D2 and adenosine A2A receptors form higher-order oligomers in living cells. J. Neurochem. 109, 1497–1507.
Carriba, P., Navarro, G., Ciruela, F., Ferré, S., Casado, V., Agnati, L. F., Cortes, A., Mallol, J., Fuxe, K., Canela, E. I., Lluis, C., and Franco, R. (2008). Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat. Methods 5, 727–733.
Carriba, P., Ortiz, O., Patkar, K., Justinova, Z., Stroik, J., Themann, A., Müller, C., Woods, A. S., Hope, B. T., Ciruela, F., Casadó, V., Canela, E. I., Lluis, C., Goldberg, S. R., Moratalla, R., Franco, R., and Ferré, S. (2007). Striatal adenosine A2A and cannabinoid CB1 receptors form functional heteromeric complexes that mediate the motor effects of cannabinoids. Neuropsychopharmacology 32, 2249–2259.
Chen, J. F., Moratalla, R., Impagnatiello, F., Grandy, D. K., Cuellar, B., Rubinstein, M., Beilstein, M. A., Hackett, E., Fink, J. S., Low, M. J., Ongini, E., and Schwarzschild, M. A. (2001). The role of the D(2) dopamine receptor (D(2)R) in A(2A) adenosine receptor (A(2A)R)-mediated behavioral and cellular responses as revealed by A(2A) and D(2) receptor knockout mice. Proc. Natl. Acad. Sci. U.S.A. 98, 1970–1975.
Ciruela, F., Casado, V., Rodrigues, R. J., Luján, R., Burgueno, J., Canals, M., Borycz, J., Rebola, N., Goldberg, S. R., Mallol, J., Cortés, A., Canela, E. I., López-Giménez, J. F., Milligan, G., Lluis, C., Cunha, R. A., Ferré, S., and Franco, R. (2006). Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J. Neurosci. 26, 2080–2087.
DeLong, M. R., and Wichmann, T. (2007). Circuits and circuit disorders of the basal ganglia. Arch. Neurol. 64, 20–24.
Ferré, S., Baler, R., Bouvier, M., Caron, M. G., Devi, L. A., Durroux, T., Fuxe, K., George, S. R., Javitch, J. A., Lohse, M. J., Mackie, K., Milligan, G., Pfleger, K. D. G., Pin, J.-P., Volkow, N., Waldhoer, M., Woods, A. S., and Franco, R. (2009). Building a new conceptual framework for receptor heteromers. Nat. Chem. Biol. 5, 131–134.
Ferré, S., Ciruela, F., Woods, A. S., Lluis, C., and Franco, R. (2007). Functional relevance of neurotransmitter receptor heteromers in the central nervous system. Trends Neurosci. 30, 440–446.
Ferré, S., Karcz-Kubicha, M., Hope, B. T., Popoli, P., Burgueno, J., Casado, V., Fuxe, K., Lluis, C., Goldberg, S. R., Franco, R., and Ciruela, F. (2002). Synergistic interaction between adenosine A2A and glutamate mGlu5 receptors: implications for striatal neuronal function. Proc. Natl. Acad. Sci. U.S.A. 99, 11940–11945.
Ferré, S., Lluis, C., Justinova, Z., Quiroz, C., Orru, M., Navarro, G., Canela, E. I., Franco, R., and Goldberg, S. R. (2010). Adenosine-cannabinoid receptor interactions. Implications for striatal function. Br. J. Pharmacol. 160, 443–453.
Ferré, S., Quiroz, C., Woods, A. S., Cunha, R., Popoli, P., Ciruela, F., Lluis, C., Franco, R., Azdad, K., and Schiffmann, S. N. (2008). An update on adenosine A2A-dopamine D2 receptor interactions. Implications for the function of G protein-coupled receptors. Curr. Pharm. Des. 14, 1468–1474.
Ferré, S., von Euler, G., Johansson, B., Fredholm, B. B., and Fuxe, K. (1991). Stimulation of high affinity adenosine A-2 receptors decreases the affinity of dopamine D-2 receptors in rat striatal membranes. Proc. Natl. Acad. Sci. U.S.A. 88, 7238–7241.
Håkansson, K., Galdi, S., Hendrick, J., Snyder, G., Greengard, P., and Fisone, G. (2006). Regulation of phosphorylation of the GluR1 AMPA receptor by dopamine D2 receptors. J. Neurochem. 96, 482–488.
Hettinger, B. D., Lee, A., Linden, J., and Rosin, D. L. (1998). Ultrastructural localization of adenosine A2A receptors suggests multiple cellular sites for modulation of GABAergic neurons in rat striatum. J. Comp. Neurol. 431, 331–346.
Higley, M. J., and Sabatini, B. L. (2010). Competitive regulation of synaptic Ca2( influx by D2 dopamine and A2A adenosine receptors. Nat. Neurosci. 13, 958–966.
Hillion, J., Canals, M., Torvinen, M., Casado, V., Scott, R., Terasmaa, A., Hansson, A., Watson, S., Olah, M. E., Mallol, J., Canela, E. I., Zoli, M., Agnati, L. F., Ibanez, C. F., Lluis, C., Franco, R., Ferré, S., and Fuxe, K. (2002). Coaggregation, cointernalization and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J. Biol. Chem. 277, 18091–18097.
James, S., and Richardson, P. J. (1993). The subcellular distribution of [3H]-CGS 21680 binding sites in the rat striatum: copurification with cholinergic nerve terminals. Neurochem. Int. 23, 115–122.
Jin, S., Johansson, B., and Fredholm, B. B. (1993). Effects of adenosine A1 and A2 receptor activation on electrically evoked dopamine and acetylcholine release from rat striatal slices. J. Pharmacol. Exp. Ther. 267, 801–808.
Justinova, Z., Ferré, S., Redhi, G. H., Mascia, P., Stroik, J., Quarta, D., Yasar, S., Muller, C. E., Franco, R., and Goldberg, S. R. (2011). Reinforcing and neurochemical effects of cannabinoid CB1 receptor agonists, but not cocaine, are altered by an adenosine A2A receptor antagonist. Addict. Biol. 16, 405–415.
Kachroo, A., Orlando, L. R., Grandy, D. K., Chen, J. F., Young, A. B., and Schwarzschild, M. A. (2005). Interactions between metabotropic glutamate 5 and adenosine A2A receptors in normal and parkinsonian mice. J. Neurosci. 25, 10414–10419.
Kreitzer, A. C., and Malenka, R. C. (2007). Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 445, 643–647.
Kull, B., Ferré, S., Arslan, G., Svenningsson, P., Fuxe, K., Owman, C., and Fredholm, B. B. (1999). Reciprocal interactions between adenosine A2A and dopamine D2 receptors in CHO cells co-transfected with the two receptors. Biochem. Pharmacol. 58, 1035–1045.
Lerner, T. N., Horne, E. A., Stella, N., and Kreitzer, A. C. (2010). Endocannabinoid signaling mediates psychomotor activation by adenosine A2A antagonists. J. Neurosci. 30, 2160–2164.
Martire, A., Tebano, M. T., Chiodi, V., Ferreira, S. G., Cunha, R. A., Köfalvi, A., and Popoli, P. (2011). Pre-synaptic adenosine A2A receptors control cannabinoid CB1 receptor-mediated inhibition of striatal glutamatergic neurotransmission. J. Neurochem. 116, 273–280.
Moresco, R. M., Todde, S., Belloli, S., Simonelli, P., Panzacchi, A., Rigamonti, M., Galli-Kienle, M., and Fazio, F. (2005). In vivo imaging of adenosine A2A receptors in rat and primate brain using [11C]SCH442416. Eur. J. Nucl. Med. Mol. Imaging 32, 405–413.
Navarro, G., Aymerich, M. S., Marcellino, D., Cortes, A., Casado, V., Mallol, J., Canela, E. I., Agnati, L. F., Woods, A. S., Fuxe, K., Lluis, C., Lanciego, J. L., Ferré, S., and Franco, R. (2009). Interactions between calmodulin, adenosine A2A and dopamine D2 receptors. J. Biol. Chem. 284, 28058–28068.
Navarro, G., Ferré, S., Cordomi, A., Moreno, E., Mallol, J., Casadó, V., Cortés, A., Hoffmann, H., Ortiz, J., Canela, E. I., Lluís, C., Pardo, L., Franco, R., and Woods, A. S. (2010). Interactions between intracellular domains as key determinants of the quaternary structure and function of receptor heteromers. J. Biol. Chem. 285, 27346–27359.
Nishi, A., Liu, F., Matsuyama, S., Hamada, M., Higashi, H., Nairn, A. C., and Greengard, P. (2003). Metabotropic mGlu5 receptors regulate adenosine A2A receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 100, 1322–1327.
Obeso, J. A., Rodríguez-Oroz, M. C., Rodríguez, M., Arbizu, J., and Giménez-Amaya, J. M. (2002). The basal ganglia and disorders of movement: pathophysiological mechanisms. News Physiol. Sci. 17, 51–55.
Orru, M., Bakešová, J., Brugarolas, M., Quiroz, C., Beaumont, V., Goldberg, S. R, Lluís, C., Cortés, A., Franco, R., Casadó, V., Canela, E. I., and Ferré, S. (2011). Striatal pre- and postsynaptic profile of adenosine A(2A) receptor antagonists. PLoS ONE 6, e16088. doi:10.1371/journal.pone.0016088
Popoli, P., Betto, P., Reggio, R., and Ricciarello, G. (1995). Adenosine A2A receptor stimulation enhances striatal extracellular glutamate levels in rats. Eur. J. Pharmacol. 287, 215–217.
Popoli, P., Pezzola, A., Torvinen, M., Reggio, R., Pintor, A., Scarchili, L., Fuxe, K., and Ferré, S. (2001). The selective mGlu5 receptor agonist CHPG inhibits quinpirole-induced turning in 6-hydroxydopamine-lesioned rats and modulates the binding characteristics of dopamine D2 receptors in the rat striatum: interactions with adenosine A2A receptors. Neuropsychopharmacology 25, 505–513.
Preston, Z., Lee, K., Widdowson, L., Freeman, T. C., Dixon, A. K., and Richardson, P. J. (2000). Adenosine receptor expression and function in rat striatal cholinergic interneurons. Br. J. Pharmacol. 130, 886–890.
Quiroz, C., Lujan, R., Uchigashima, M., Simoes, A. P., Lerner, T. N., Borycz, J., Kachroo, A., Canas, P. M., Orru, M., Schwarzschild, M. A., Rosin, D. L., Kreitzer, A. C., Cunha, R. A., Watanabe, M., and Ferré, S. (2009). Key modulatory role of presynaptic adenosine A2A receptors in cortical neurotransmission to the striatal direct pathway. ScientificWorldJournal 9, 1321–1344.
Rodrigues, R. J., Alfaro, T. M., Rebola, N., Oliveira, C. R., and Cunha, R. A.. (2005). Co-localization and functional interaction between adenosine A(2A) and metabotropic group 5 receptors in glutamatergic nerve terminals of the rat striatum. J. Neurochem. 92, 433–441.
Rosin, D. L., Robeva, A., Woodard, R. L., Guyenet, P. G., and Linden, J. (1998). Immunohistochemical localization of adenosine A2A receptors in the rat central nervous system. J. Comp. Neurol. 401, 163–186.
Schiffmann, S. N., Fisone, G., Moresco, R., Cunha, R., and Ferré, S. (2007). Adenosine A2A receptors and basal ganglia physiology. Prog. Neurobiol. 83, 277–292.
Shen, H. Y., Coelho, J. E., Ohtsuka, N., Canas, P. M., Day, Y. J., Huang, Q. Y., Rebola, N., Yu, L., Boison, D., Cunha, R. A., Linden, J., Tsien, J. Z., and Chen, J. F. (2008). A critical role of the adenosine A2A receptor in extrastriatal neurons in modulating psychomotor activity as revealed by opposite phenotypes of striatum and forebrain A2A receptor knock-outs. J. Neurosci. 28, 2970–2975.
Soria, G., Castañé, A., Ledent, C., Parmentier, M., Maldonado, R., and Valverde, O. (2004). Adenosine A2A receptors are involved in physical dependence and place conditioning induced by THC. Eur. J. Neurosci. 20, 2203–2213.
Tanganelli, S., Sandager-Nielsen, K., Ferraro, L., Antonelli, T., Kehr, J., Franco, R., Ferré, S., Agnati, L. F., Fuxe, K., and Scheel-Krüger, J. (2004). Striatal plasticity at the network level. Focus on adenosine A2A and D2 interactions in models of Parkinson’s disease. Parkinsonism Relat. Disord. 10, 273–280.
Tebano, M. T., Martire, A., Chiodi, V., Pepponi, R., Ferrante, A., Domenici, M. R., Frank, C., Chen, J. F., Ledent, C., and Popoli, P. (2009). Adenosine A2A receptors enable the synaptic effects of cannabinoid CB1 receptors in the rodent striatum. J. Neurochem. 110, 1921–1930.
Tozzi, A., de Iure, A., Di Filippo, M., Tantucci, M., Costa, C., Borsini, F., Ghiglieri, V., Giampà, C., Fusco, F. R., Picconi, B., and Calabresi, P. (2011). The distinct role of medium spiny neurons and cholinergic interneurons in the D2/A2A receptor interaction in the striatum: implications for Parkinson’s disease. J. Neurosci. 31, 1850–1862.
Woods, A. S., and Ferré, S. (2005). The amazing stability of the arginine-phosphate electrostatic interaction. J. Proteome Res. 4, 1397–1402.
Yao, L., Fan, P., Jiang, Z., Mailliard, W. S., Gordon, A. S., and Diamond, I. (2003). Addicting drugs utilize a synergistic molecular mechanism in common requiring adenosine and Gi-beta gamma dimers. Proc. Natl. Acad. Sci. U.S.A. 100, 14379–14384.
Keywords: adenosine A2A receptor, striatum, receptor heteromers, dopamine receptors, cannabinoid receptors
Citation: Ferré S, Quiroz C, Orru M, Guitart X, Navarro G, Cortés A, Casadó V, Canela EI, Lluis C and Franco R (2011) Adenosine A2A receptors and A2A receptor heteromers as key players in striatal function. Front. Neuroanat. 5:36. doi: 10.3389/fnana.2011.00036
Received: 02 May 2011; Paper pending published: 17 May 2011;
Accepted: 08 June 2011; Published online: 17 June 2011.
Edited by:
Emmanuel Valjent, Université Montpellier 1 and 2, FranceReviewed by:
Jose L. Lanciego, University of Navarra, SpainJulie Perroy, Centre National de la Recherche Scientifique, France
Copyright: © 2011 Ferré, Quiroz, Orru, Guitart, Navarro, Cortés, Casadó, Canela, Lluis and Franco. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Sergi Ferré, National Institute on Drug Abuse, Intramural Research Program, 251 Bayview Boulevard, Baltimore, MD 21224, USA. e-mail:c2ZlcnJlQGludHJhLm5pZGEubmloLmdvdg==