

- 1 Department of Psychiatry, School of Medicine, University of Michigan, Ann Arbor, MI, USA
- 2 Child Study Center, School of Medicine, Yale University, New Haven, CT, USA
- 3 Department of Psychiatry, School of Medicine, Yale University, New Haven, CT, USA
- 4 Department of Neurobiology, School of Medicine, Yale University, New Haven, CT, USA
Striatal-enriched protein tyrosine phosphatase (STEP) has recently been implicated in several neuropsychiatric disorders with significant cognitive impairments, including Alzheimer’s disease, schizophrenia, and fragile X syndrome. A model has emerged by which STEP normally opposes the development of synaptic strengthening and that disruption in STEP activity leads to aberrant synaptic function. We review the mechanisms by which STEP contributes to the etiology of these and other neuropsychiatric disorders. These findings suggest that disruptions in STEP activity may be a common mechanism for cognitive impairments in diverse illnesses.
Introduction
Although the genetic basis of cognition is poorly understood, there is a general agreement that normal cognitive function relies on environmental, genetic, and molecular interactions (Heyes and Huber, 2000). These interactions mediate the association between specific behaviors, rewards, and punishments, enabling individuals to respond effectively to their environment. Cognitive impairments present in numerous neuropsychiatric disorders, including Alzheimer’s disease (AD; Folstein and Whitehouse, 1983), schizophrenia (SZ; Gold and Harvey, 1993), fragile X syndrome (FXS; Smith, 1993), Huntington’s disease (HD; Bourne et al., 2006), hypoxic–ischemic brain injury (Anderson and Arciniegas, 2010), and alcohol use disorders (Bates et al., 2002).
Striatal-enriched protein tyrosine phosphatase (STEP) has emerged as a critical enzyme in the signaling pathways that appear to contribute to the cognitive impairments in each of these neuropsychiatric disorders. STEP is a brain-specific phosphatase that is highly expressed within the striatum (Lombroso et al., 1991) as well as the cortex, hippocampus, and amygdala (Lombroso et al., 1993; Boulanger et al., 1995). After transcription of the Ptpn5 gene, alternative splicing produces mRNAs that encode four isoforms (STEP20, STEP38, STEP46, and STEP61); however, only STEP46 and STEP61 contain an active phosphatase domain (Sharma et al., 1995; Bult et al., 1996, 1997; Figure 1). STEP46 is expressed primarily in the cytoplasm whereas STEP61 is targeted to the postsynaptic density, extrasynaptic sites, and the endoplasmic reticulum (Lombroso et al., 1993; Boulanger et al., 1995; Goebel-Goody et al., 2009).
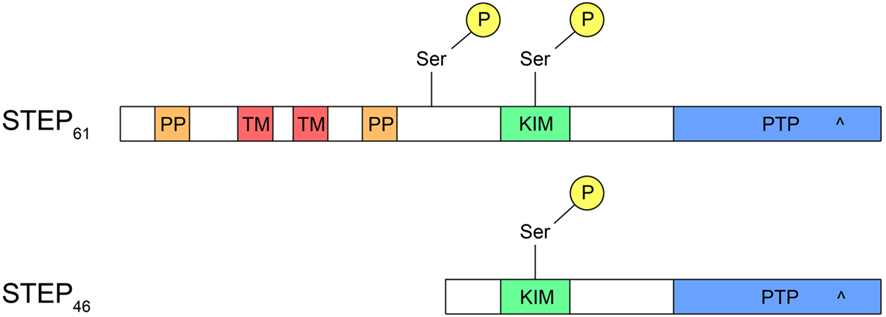
Figure 1. Structure of STEP. Both STEP46 and STEP61 contain a kinase-interacting motif (KIM) domain, necessary for binding to all substrates, and a protein tyrosine phosphatase (PTP) domain with a catalytic site (^). In addition, STEP61 has two polyproline (PP) domains, the first of which interacts with Fyn, and two transmembrane (TM) domains that target STEP61 to the endoplasmic reticulum or postsynaptic density. Whereas STEP61 has two serine (Ser) phosphorylation sites, STEP46 contains only one within the KIM domain. The additional phosphorylation site on STEP61 may facilitate proteolytic cleavage of STEP61 to produce an inactive STEP33 fragment that is unable to bind substrates due to cleavage within the KIM domain.
STEP46 and STEP61 activity are regulated by the phosphorylation of a serine (Ser) residue (Ser49 and Ser221, respectively) within a kinase-interacting motif (KIM), a binding site for all STEP substrates. Phosphorylation at this site sterically prevents STEP from associating with all of its substrates. Stimulation of glutamate N-methyl-D-aspartate receptors (NMDARs) dephosphorylates and activates STEP through a calcineurin/PP1 pathway (Paul et al., 2003; Valjent et al., 2005). When activated, STEP dephosphorylates tyrosine (Tyr) residues on its substrates, causing their inactivation, or in the case of glutamate receptors, promoting their internalization from synaptosomal surface membranes (Figure 2; only NMDAR internalization shown for clarity).
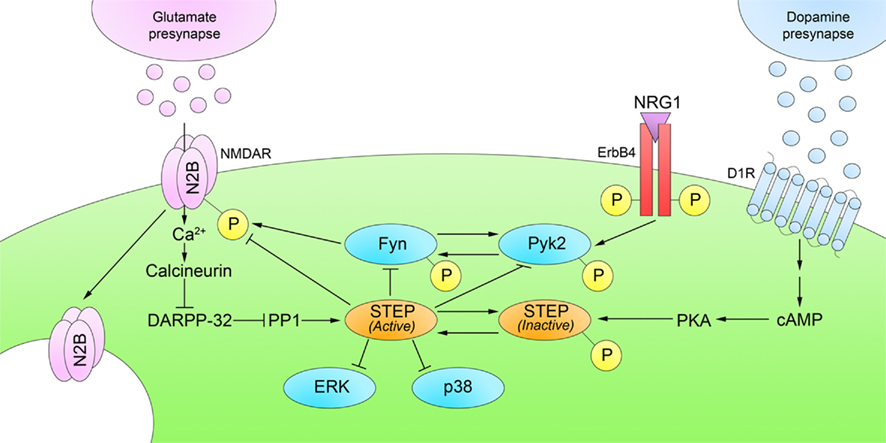
Figure 2. Regulation of STEP phosphorylation. D1R stimulation phosphorylates and inactivates STEP through a cAMP/PKA pathway. Glutamatergic stimulation of NMDARs reverses this process by dephosphorylation and activation of STEP through a calcineurin/PP1 pathway. Active STEP dephosphorylates regulatory tyrosines within STEP substrates. STEP dephosphorylation of Fyn, Pyk2, ERK1/2, and p38 leads to their inactivation; however, STEP dephosphorylation of GluN2B and GluA2 subunits results in internalization of NMDARs and AMPARs, respectively, from synaptosomal surface membranes. Only NMDARs are shown in the figure for clarity.
Substrates of STEP include the GluN2B (previously called NR2B) subunit of the NMDAR (Braithwaite et al., 2006), the GluA2 (previously called GluR2) subunit of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR; Zhang et al., 2008), as well as the kinases Fyn (Nguyen et al., 2002) and Pyk2 (Xu et al., 2010), and the mitogen-activated protein kinases (MAPKs) extracellular-regulated kinase 1/2 (ERK1/2) and p38 (Munoz et al., 2003; Paul et al., 2003; Chen et al., 2009). Stimulation of dopamine D1 receptors (D1Rs), or blockade of dopamine D2 receptors (D2Rs), activates cyclic adenosine monophosphate (cAMP)-dependent protein kinase A (PKA; Stoof and Kebabian, 1981), which phosphorylates and prevents STEP46 and STEP61 from interacting with their substrates (Paul et al., 2000). D1R stimulation can also activate a cAMP-independent cascade in which Fyn phosphorylates GluN2B, potentiates NMDAR activity, and activates ERK (Pascoli et al., 2011). These actions compliment the actions of cAMP-dependent STEP inactivation, because inactivation of STEP activates Fyn and ERK, and leads to NMDAR internalization.
Synaptic plasticity refers to a change in synaptic efficacy between neurons, which is fundamental to learning, memory, and cognition (Silva, 2003). This dynamic process requires synaptic efficacy to strengthen (long-term potentiation; LTP) or weaken (long-term depression; LTD) in response to developmental and environmental signals. Long-term potentiation is the most widely supported molecular model of memory (Bliss and Collingridge, 1993). It has recently become clear that LTP and LTD are not discrete phenomena, but rather complimentary processes with complex modulation that allow experiences to continuously reshape neural function (Malenka and Bear, 2004). As is discussed further below, STEP is currently believed to modulate both LTP and LTD through its ability to regulate internalization of NMDARs and AMPARs.
Glutamatergic transmission is essential for both LTP and LTD. While LTP is facilitated by the coordinated actions of NMDARs and AMPARs, metabotropic glutamate receptor (mGluR) activity is important to the development of LTD. Moreover, glutamatergic signaling is often modulated by dopaminergic (Centonze et al., 2001; Calabresi et al., 2007), β-adrenergic (Vanhoose and Winder, 2003), and nicotinic cholinergic stimulation (Dajas-Bailador and Wonnacott, 2004), as well as by other neuromodulators (Jin et al., 2005; Mony et al., 2009).
Striatal-enriched protein tyrosine phosphatase has recently been implicated in the pathophysiology of AD, schizophrenia, FXS, HD, hypoxic–ischemic brain injury, and other neuropsychiatric disorders. This important discovery suggests that several cognitive disorders that were previously regarded as distinct illnesses may share a common molecular pathway. Other proteins and STEP-independent mechanisms surely contribute to cognitive impairments, but this review focuses on the role of STEP in the pathogenesis of these neuropsychiatric disorders.
Alzheimer’s Disease: Amyloid Beta Leads to Increased STEP61 Activity
Alzheimer’s disease is a common form of dementia that causes progressive cognitive impairment, most notably in memory and semantic–lexical language (Huff et al., 1987). Cognitive impairments are accompanied by behavioral alterations such as agitation, dysphoria, and apathy (Mega et al., 1996). The German psychiatrist Alois Alzheimer was the first to describe the hallmark pathogenic signs of the disease in brain autopsies: neurofibrillary tangles and plaques. This observation promoted the hypothesis that AD progression is mediated by these neuroanatomical abnormalities (Hardy and Higgins, 1992). In the 1980s, tau was identified as the main constituent of neurofibrillary tangles (Goedert et al., 1989) and amyloid β (Aβ) proteins as the main constituent of plaques (Tanzi et al., 1987). These findings led to the hypothesis that hyperphosphorylated tau protein (Alonso et al., 1996) and insoluble aggregates of Aβ peptide formed from mutated amyloid precursor protein (Cai et al., 1993; Suzuki et al., 1994) were a possible mechanism underlying the pathology of AD.
The focus shifted in 1991 from extracellular deposits to a disruption in synaptic function when Terry et al. (1991) showed that cortical synaptic density correlated more strongly with cognitive impairments than plaque density. In support of the synaptic hypothesis of AD, tau has been shown to reduce anterograde transport even before it aggregates in neurofibrillary tangles (Mandelkow et al., 2003). Moreover, soluble Aβ oligomers impair synaptic function before neuronal loss occurs (Lue et al., 1999). These oligomers directly bind to and chronically activate nicotinic acetylcholine receptors (nAChRs), causing sustained increases in intracellular Ca2+ concentrations (Dougherty et al., 2003). In addition, these oligomers reduce NMDAR-dependent synaptic transmission and Ca2+ influx, resulting in the loss of synapses in rat hippocampal neurons (Cullen et al., 1996; Shankar et al., 2007). Finally, exogenous application of Aβ to neuronal cell cultures and tissue slices alters spine architecture and blocks LTP, and in vivo infusions impair cognitive function in rodents (Walsh et al., 2002; Lacor et al., 2007; Shankar et al., 2007, 2008).
The underlying mechanisms of Aβ-induced reductions in synaptic function remain a focus of intense research. Snyder et al. (2005) directly tested whether Aβ disrupts surface expression of NMDARs. They demonstrated that Aβ binds α7 nAChRs, leading to Ca2+ influx and activation of a calcineurin/PP1 pathway. PP1 dephosphorylates and activates STEP, which inhibits Fyn and promotes internalization of NMDARs and AMPARs from synaptosomal membrane surfaces (Figure 3). Chin et al. (2005) also found increased levels of STEP protein and decreased Fyn activity in the dentate gyrus of a transgenic mouse model of AD (the J20 mouse line).
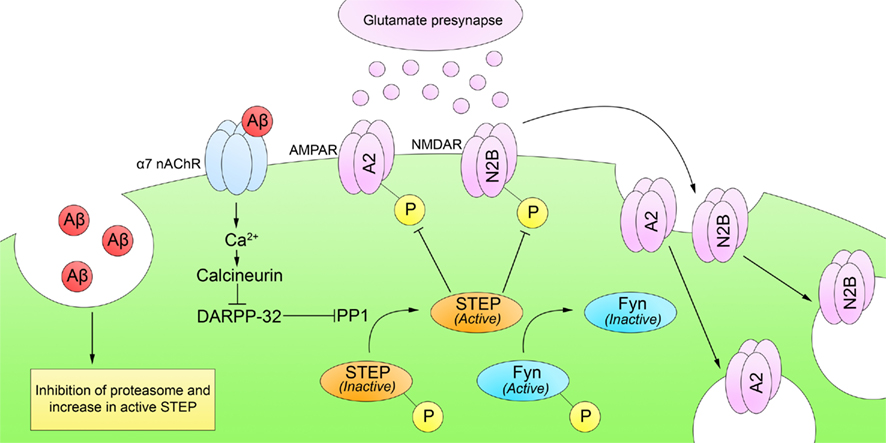
Figure 3. Striatal-enriched protein tyrosine phosphatase and Alzheimer’s disease. Cognitive impairments in AD appear before the formation of Aβ plaques, suggesting that soluble Aβ may affect synaptic function. Soluble Aβ increases both STEP levels and phosphorylation. First, soluble Aβ inhibits STEP degradation by an Aβ-mediated inhibition of the ubiquitin proteasome pathway. Second, soluble Aβ directly binds and activates α7 nAChRs, leading to Ca2+ influx and activation of calcineurin. Calcineurin/PP1 then dephosphorylates and activates STEP. Active STEP promotes the dephosphorylation of Fyn, GluA2, and GluN2B (and other substrates not depicted). Dephosphorylation of Fyn inactivates it whereas dephosphorylation of GluN2B and GluA2 leads to NMDAR and AMPAR internalization from synaptosomal surface membranes and disruption of synaptic function.
Recently, Kurup et al. (2010) demonstrated that a second pathway increases STEP61 activity in AD brains. This is due to a disruption in the normal degradation of STEP61 as a result of an Aβ-mediated inhibition of the proteasome. Because STEP61 is normally ubiquitinated and degraded by the ubiquitin proteasome pathway, inhibition of the proteasome results in substantial increases in STEP61 in brain samples from both human AD and mouse models.
If increased STEP61 levels contribute to the pathophysiology of AD, then reducing them might reverse its biochemical and cognitive deficits. Zhang et al. (2010) tested this hypothesis in a mouse model of the disease by genetically decreasing STEP levels and demonstrating restored cognitive function in 6-month-old AD mice null for STEP. Importantly, cognitive impairments improved despite a continued elevation of Aβ and phosphorylated tau. It remains to be determined whether older AD mice (i.e., 12-months old) with a similar genetic reduction in STEP also have improved cognitive functioning. It should be emphasized that STEP is one of over 400 proteins within neuronal spines, and that STEP-independent pathways certainly contribute to the pathogenesis of AD as well as the other disorders discussed in subsequent sections.
Schizophrenia: STEP Contributes to the Beneficial Effects of Neuroleptics
Schizophrenia is characterized by cognitive and emotional deterioration, with symptoms typically emerging during young adulthood. Positive symptoms include delusions, disordered thought and speech, and hallucinations. Negative symptoms, which are deficits in emotional and mental processes, include blunted affect, poverty of speech, anhedonia, and asociality. In addition to positive and negative symptoms, cognitive impairments including deficits in executive functioning predate the onset of the above symptoms and persist when these subside.
Schizophrenia was considered untreatable until the introduction of the first neuroleptic, chlorpromazine, in 1952. The mechanism of action of these “typical” antipsychotics is unknown, despite their efficacy in the treatment of SZ. The dopamine hypothesis of SZ emerged from the discovery that typical antipsychotics, such as chlorpromazine, block D2Rs, and their clinical potency is related to the affinity of antipsychotic medications for this receptor (Seeman et al., 1975; Creese et al., 1976).
Although dopamine levels are increased in the striatum (Breier et al., 1997) and decreased in the frontal cortex (Dolan et al., 1995), recent evidence suggests that the pathophysiology of SZ includes deficits in other neurotransmitter systems, in particular glutamatergic hypofunction (Jentsch and Roth, 1999). This is due in part to the fact that the dopamine hypothesis of SZ does not appear to explain the SZ-like symptoms induced by non-competitive NMDAR antagonists, such as phencyclidine and ketamine. Further support for the glutamate hypothesis comes from research in animal models and human subjects. Mice expressing 5% of the normal levels of GluN1 display SZ-like behaviors, which are attenuated by treatment with the antipsychotics haloperidol and clozapine (Mohn et al., 1999). In human patients, Pilowsky et al. (2006) used in vivo positron emission tomography to demonstrate reductions in hippocampal NMDAR binding in medication-free but not antipsychotic-treated schizophrenics. Moreover, postmortem investigation of schizophrenic brains reveals that GluN1 mRNA levels are reduced by 30% in the superior temporal cortex of cognitively impaired schizophrenics, a deficit that is correlated with antemortem severity of cognitive impairment (Humphries et al., 1996). Finally, the administration of glycine and D-serine, potentiators of NMDAR function, improves symptoms in medicated schizophrenics (Heresco-Levy et al., 1999; Kantrowitz et al., 2010).
Recent studies suggest that STEP plays a role in mediating the beneficial effects of neuroleptics. Both typical and atypical antipsychotics regulate cAMP-dependent PKA activity in the striatum and medial prefrontal cortex (Turalba et al., 2004). Recently, Carty et al. (2010) showed that both acute and subchronic administration of haloperidol, clozapine, and risperidone result in a PKA-mediated phosphorylation of STEP (Figure 4). As discussed above, this prevents STEP from interacting with its substrates. In addition, neuroleptic-mediated activation of PKA results in the phosphorylation and activation of DARPP-32. DARPP-32 is an indirect inhibitor of STEP through its inhibition of PP1, which dephosphorylates and inactivates STEP. Consequently, both PKA activation and PP1 inactivation contribute to decreased STEP activity.
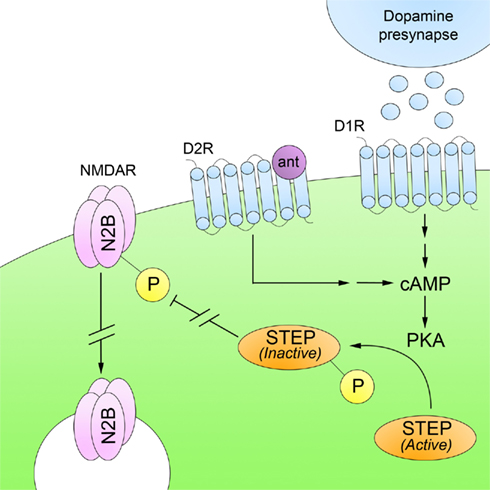
Figure 4. Striatal-enriched protein tyrosine phosphatase and schizophrenia. Deficits in glutamatergic neurotransmission are believed to underlie the pathophysiology of SZ. Administration of typical and atypical antipsychotics, which are D2R antagonists (ant), increase PKA-mediated phosphorylation of STEP. Inactivation of STEP results in increased trafficking of NMDARs to synaptosomal surface membranes. This suggests that the beneficial effects of neuroleptics are mediated, at least in part, through restoration of NMDAR levels at synaptic sites.
DARPP-32 also activates STEP substrates through STEP-independent pathways. For example, ERK is activated by DARPP-32 through the inhibition of PP1, leading to activation of upstream modulators of ERK activity, such as mitogen-activated protein kinase kinase (MEK; Valjent et al., 2005). These results suggest that neuroleptics exert their beneficial effects at least in part through D2R/PKA-mediated phosphorylation and inactivation of STEP, promoting Tyr phosphorylation of STEP substrates that include GluN2B-containing NMDARs, which now traffic to synaptosomal surface membranes.
Fragile X Syndrome: The FMRP Protein Negatively Represses STEP
Fragile X syndrome is an X-linked, monogenic neurological disorder and the leading cause of inherited mental retardation. It is characterized by hyperactivity, auditory hypersensitivity and audiogenic seizures, developmental delays, emotional dysfunction, and cognitive impairment (Visootsak et al., 2005; Koukoui and Chaudhuri, 2007). Common cognitive impairments in FXS include impaired quantitative skills, short-term memory, spatial visualization, and visual–motor coordination (Freund and Reiss, 1991).
Fragile X syndrome results from an expanded 5′-CCG repeat region in the first exon of the fragile X mental retardation 1 (Fmr1) gene (Xq27.3). The increased number of cytosines are methylated and interfere with the transcription of Fmr1 mRNA once the expansion exceeds 200 repeats (Oberle et al., 1991). The protein encoded by Fmr1, fragile X mental retardation protein (FMRP), is a cytosolic RNA-binding protein that associates with polyribosomes and the RNA-silencing complex, functioning as a negative repressor of translation (Laggerbauer et al., 2001; Li et al., 2001; Garber et al., 2006). Because FMRP and polyribosomes are present in dendritic spines, it was suggested that FMRP modulates synaptic activity through its ability to repress local translation of mRNAs (Weiler and Greenough, 1999). Synaptic activity was proposed to remove FMRP inhibitory effects, resulting in local translation of mRNAs (Bear et al., 2004). In this way, RNA-binding proteins, such as FMRP, influence synaptic plasticity by controlling activity-dependent regulation of mRNA localization and translation (Ule and Darnell, 2006).
In support of this hypothesis, FMRP is quickly synthesized and localized to dendrites after stimulation of group I mGluRs (Todd et al., 2003; Antar et al., 2004). Activation of mGluRs stimulates protein synthesis-dependent LTD in dendrites (Huber et al., 2000, 2001) and results in a rapid loss of synaptic NMDARs and AMPARs (Snyder et al., 2001). Fmr1 KO mice display increased protein synthesis (Qin et al., 2005), enhanced hippocampal LTD (Huber et al., 2002), as well as abnormal synaptic morphology, including immature development of dendritic spines (Irwin et al., 2000), delayed spine turnover and stabilization (Cruz-Martin et al., 2010), increased spine density (Comery et al., 1997), and a decreased number of functional synapses (Pfeiffer and Huber, 2007).
It is believed that FMRP regulates mGluR-dependent LTD by tightly controlling protein synthesis and that decreased FMRP expression elevates synaptic protein levels, which exaggerate mGluR-dependent LTD and disrupt synaptic morphology (Bear et al., 2004; Ronesi and Huber, 2008). In Fmr1 KO mice, mGluR-dependent LTD no longer requires new protein synthesis, suggesting that elevated levels of synaptic proteins, which would normally be inhibited by FMRP, enable LTD to persist without de novo protein synthesis (Nosyreva and Huber, 2006). For example, Fmr1 KO mice exhibit decreased surface expression of NMDARs (Eadie et al., 2010) and GluA1-containing AMPARs (Li et al., 2002; Suvrathan et al., 2010), which is most likely a consequence of uninhibited mGluR signaling (Nakamoto et al., 2007). Although it is known that Fmr1 KO exaggerates mGluR-dependent LTD by decreasing synaptic glutamate receptor expression, it is not known how this occurs.
Zhang et al. (2008) demonstrated that STEP translation is increased in the mouse hippocampus upon mGluR5 stimulation, resulting in AMPAR internalization through Tyr dephosphorylation of the GluA2 subunit, although the exact Tyr residue is not yet known (Figure 5). More recently, it was shown that STEP mRNA associates with FMRP and that STEP protein levels are elevated in Fmr1 KO mice (Goebel-Goody et al., 2010). In addition, progeny were generated by crossing Fmr1 and STEP KO mice to produce mice with lowered STEP levels despite the continued presence of the Fmr1 mutation. These mice display fewer audiogenic seizures and less spatial anxiety than Fmr1 KO mice. These results suggest that some of the deficits in animal models of FXS result from the failure of FMRP to suppress STEP translation upon mGluR stimulation, allowing increased STEP levels to inappropriately internalize NMDARs and AMPARs.
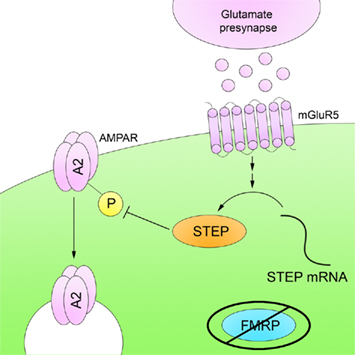
Figure 5. Striatal-enriched protein tyrosine phosphatase and fragile X syndrome. FMRP is an RNA-binding protein that represses mRNA translation in dendrites. FMRP is absent in FXS, and as a result there is a disruption in mRNA translation. STEP mRNA associates with FMRP, and STEP protein levels are increased in Fmr1 KO mice. The higher levels of active STEP protein are thought to disrupt synaptic function by dephosphorylation of STEP substrates.
The Role of STEP in Other Neuropsychiatric Disorders
Huntington’s Disease
Huntington’s disease is an autosomal dominant neurodegenerative disease caused by abnormal expansion of a CAG codon in exon 1 of the huntingtin (HTT) gene (4p16.3). The expansion results in a mutant HTT (mHTT) protein that contains excessive polyglutamine residues (The Huntington’s Disease Collaborative Research Group, 1993). Projection neurons, which are the majority of striatal neurons, are specifically affected in HD (Reiner et al., 1988). HD symptoms such as chorea, a movement disorder, and cognitive impairments have been attributed to progressive excitotoxicity of these projection neurons resulting from NMDAR activation, which destroys striatal innervation of other brain regions (DiFiglia, 1990; Fan and Raymond, 2007). Cognitive impairments common to HD patients are loss of initiative, poor perseverance, impaired judgment, and emotional blunting (Craufurd et al., 2001). In addition, attention, executive function, language comprehension, and visuospatial immediate memory are impaired as the disease progresses (Bachoud-Levi et al., 2001).
Although mHTT is present at birth in affected individuals, symptoms typically begin in middle age. Transgenic lines of mice expressing mHTT are resistant to striatal toxicity induced by quinolinic acid, an NMDAR agonist, suggesting that compensatory mechanisms in the brain protect against the toxic effect of mHTT (Hansson et al., 1999). Similar mechanisms in humans might explain why HD is a late-onset disorder.
Several lines of evidence suggest that in animal models of HD, mHTT disrupts PKA and calcineurin activity, both important regulators of STEP activity. Dopamine D1-like receptor binding in R6/2 mice is significantly reduced at 8 weeks whereas D1R mRNA is reduced by 4 weeks, well before the onset of symptoms (Cha et al., 1998). Moreover, cAMP synthesis declines in HdhQ111 mice at an early age (10 weeks) and results in decreased PKA activity and cAMP-responsive element-mediated gene transcription (Gines et al., 2003). In support of decreased D1R and PKA activity, proteins in the D1R/PKA pathway, such as DARPP-32, are also downregulated in medium spiny neurons of the striatum of transgenic HD mice (Bibb et al., 2000). In addition, calcineurin activity is decreased in R6/1 and Tet/HD94 mice (Xifro et al., 2009), which may be the result of an abnormal protein interaction between mHTT and calmodulin (Bao et al., 1996), which is required to activate calcineurin.
Saavedra et al. (2011) recently demonstrated reduced STEP levels in the striatum and cortex and increased phosphorylated STEP in the striatum, cortex, and hippocampus of several animal models of HD, including R6/1, R6/2, and Tet/HD94. Both of these events would decrease the ability of STEP to dephosphorylate its substrates. Intrastriatal injections of quinolic acid resulted in elevated phosphorylated STEP levels in R6/1 mice and sustained ERK2 signaling, suggesting that mHTT downregulates STEP, thereby inactivating it and promoting a compensatory activation of ERK2 and resistance to mHTT-induced excitotoxicity. Furthermore, intrastriatal injection of TAT–STEP, a substrate-trapping form of STEP, increases quinolinic acid-induced cell death in R6/1 mice whereas mHTT transgene shutdown reestablishes STEP expression in Tet/HD94 mice. Moreover, mHTT activates ERK1/2, promotes cell survival, and protects against mHTT-induced excitotoxicity in PC12 and striatal cultures (Apostol et al., 2006).
Resistance to NMDA toxicity, however, is correlated with the appearance of nuclear inclusions and behavioral deficits in mice transgenic for exon 1 of HTT (Hansson et al., 2001). In the YAC128 mouse model of HD, cognitive dysfunction precedes both neuropathological alterations and motor dysfunction (Van Raamsdonk et al., 2005). Moreover, impaired spatial cognition and synaptic plasticity in CA1 and the dentate gyrus occurs before the manifestation of an overt phenotype in R6/2 mice (Murphy et al., 2000). Therefore, while resistance to excitotoxicity delays neuronal loss in the early stages of HD, it also contributes to the development of synaptic dysfunction that likely contributes to the observed cognitive impairment in HD. In summary, STEP is downregulated in HD and promotes resistance to mHTT-induced excitotoxicity, most likely due in part to STEP inactivation and more sustained ERK2 activation.
Alcohol-Induced Cognitive Impairment
Alcohol-induced cognitive impairment is well-documented in alcohol use disorders, and it is believed that inhibition of NMDAR function, especially in the hippocampus, partly contributes to the ability of alcohol to disrupt memory and learning (White et al., 2000). Numerous kinases (PKA, protein kinase C, calcium-/calmodulin-dependent protein kinase II, cyclin-dependent protein kinase 5) and phosphatases (calcineurin) regulate the phosphorylation state of Ser and Tyr residues on NMDARs and contribute to the behavioral effects of alcohol (Ron, 2004). Previously, it was demonstrated that ethanol reduces Tyr phosphorylation of GluN2A/GluN2B subunits of NMDARs (Alvestad et al., 2003) and treatment with tyrosine phosphatase inhibitors reduces the ability of ethanol to inhibit Tyr phosphorylation of GluN2, suggesting that tyrosine phosphatases have an important role in mediating the effects of alcohol on NMDAR function (Ferrani-Kile et al., 2003).
Recently, it was suggested that STEP mediates aspects of the effects of alcohol on neurons. Hicklin et al. (2011) demonstrated that ethanol’s inhibition of NMDAR function (as measured by electrophysiology) was lost in STEP KO mice. The functional consequence of ethanol in inhibiting fear-conditioned responses was also attenuated in STEP KO mice. Moreover, restoring STEP in neuronal cultures and slices derived from STEP KO mice restored ethanol-induced biochemical and electrophysiological deficits. These results suggest that STEP may be required for the amnesiac effects of ethanol by dephosphorylating GluN2B subunits.
These experiments were conducted using hippocampal neurons, but acute ethanol administration in the dorsomedial striatum similarly impairs NMDAR-dependent LTP and dose-dependently promotes LTD (Yin et al., 2007). Wang et al. (2007, 2010) demonstrated that chronic administration of ethanol increases GluN2B phosphorylation and Fyn activation in the dorsomedial striatum. These results are unsurprising, however, considering the differential effects of acute and chronic ethanol treatments on NMDAR function. For example, in cortical neurons, chronic ethanol treatment has the opposite effect as acute ethanol treatment, enhancing NMDAR binding (Hu and Ticku, 1995) and potentiating NMDAR-mediated excitotoxicity (Chandler et al., 1993). Future studies should determine whether STEP mediates the effects of ethanol in the striatum as it does in the hippocampus. In addition, chronic ethanol treatments should be utilized to determine the potential role of STEP in the development of NMDAR upregulation and sensitization.
Hypoxic–Ischemic Brain Injury
Hypoxia is the reduction of blood oxygenation to the brain whereas ischemia is the reduction of blood perfusion to the brain. Hypoxic–ischemic brain injury caused by perinatal complications during childbirth or stroke is a leading cause of mental retardation and cognitive impairment. In addition, hypoxic–ischemic brain injury caused by stroke increases the risk for dementia; it is estimated that up to a third of stroke patients develop dementia within 3 months (Pohjasvaara et al., 1998). Hypoxia–ischemia results in neuronal loss from glutamate excitotoxicity and increased intracellular Ca2+ levels (Benveniste, 1991). Excessive stimulation of NMDARs leads to a massive and rapid influx of Ca2+, which generates nitric oxide and free radicals (Iadecola, 1997). In addition, intracellular changes in NMDAR subunit phosphorylation as well as MAPK activation following ischemia further exacerbate glutamate excitotoxicity and neuronal loss (Nozaki et al., 2001; Matsumoto et al., 2002). Hippocampal regions, such as CA1, are especially vulnerable to glutamate excitotoxicity (Kirino and Sano, 1984; Nitatori et al., 1995). Damage to the hippocampus is commonly observed in both vascular and senile dementia, and it is associated with cognitive impairment (Ball et al., 1985; Kril et al., 2002).
Gurd et al. (1999) were the first to report that cerebral hypoxia–ischemia results in the calpain-mediated proteolysis of STEP61 to STEP33, which is released into the cytoplasm. Soon after, Nguyen et al. (1999) demonstrated that STEP61 cleavage requires NMDAR-mediated Ca2+ influx. The cleavage site is in the middle of the substrate-binding KIM domain (Xu et al., 2009), and it was later determined that although STEP33 is catalytically active, it is unable to bind or dephosphorylate its substrates (Braithwaite et al., 2008; Xu et al., 2009). Extrasynaptic NMDAR stimulation results in the cleavage of STEP61 by calpain and the activation of p38 and cell death (Xu et al., 2009). Thus, cleavage of STEP61 to STEP33 contributes to glutamate excitotoxicity by inhibiting NMDAR internalization from synaptosomal surface membranes and promoting cell death pathways.
Future Directions
Homeostasis, a cornerstone of biological organisms, regulates physiological states to promote stability despite fluctuating conditions. Neural activity is subject to homeostatic control, optimizing activity-dependent synaptic plasticity to avoid over- or under-excitation (Turrigiano and Nelson, 2004). STEP may contribute to this homeostatic control of synaptic plasticity by providing a tonic brake on the induction of LTP (Pelkey et al., 2002). In other words, NMDAR-mediated STEP activation modulates LTP by removing glutamate receptors from synaptic membranes. Conversely, STEP may be inactivated during periods of synaptic quiescence, resulting in increased surface expression of glutamate receptors. Finally, note that in the absence of STEP, glutamate receptors would rapidly accumulate on synaptic membranes and neurons would no longer be responsive to synaptic or modulatory inputs.
Striatal-enriched protein tyrosine phosphatase is uniquely positioned to modulate LTP by internalizing NMDARs and AMPARs from the plasma membrane and curtailing the enhancement of LTP by the MAPK pathway. Because the removal of NMDARs and AMPARs is believed to be one mechanism involved in LTD, STEP can balance the degree to which synapses strengthen and weaken during learning and memory, maintaining neural homeostasis (Xiao et al., 1994). The involvement of STEP in homeoplastic synaptic regulation requires further investigation.
Elevated STEP levels and excessive internalization of NMDARs or AMPARs in AD, SZ, and FXS would result in a homeostatic imbalance, shifting synaptic plasticity toward LTD and synaptic weakening. As a result, systems critical for learning, memory, and cognition, such as those involving the hippocampus, would settle into a quiescent state, resulting in cognitive impairment. Conversely, the absence of STEP might facilitate learning. Indeed, Venkitaramani et al. (2011) demonstrated that STEP KO mice display enhanced hippocampal-dependent learning and cognitive performance. This effect parallels increased phosphorylation of STEP substrates (ERK1/2, GluN2B, Pyk2) and increased synaptosomal expression of GluN1/GluN2B-containing NMDARs and GluA1/GluA2-containing AMPARs.
Striatal-enriched protein tyrosine phosphatase levels are decreased in HD, yet HD also results in cognitive impairment. Before the onset of neuropathological abnormalities, mHTT impairs axonal transport and vesicular trafficking (Trushina et al., 2004) and represses nuclear transcription (Kegel et al., 2002). Inadequate neurotransmitter release or protein synthesis in the synapse could result in dysfunctional synaptic plasticity. For example, mHTT results in decreased glutamate neurotransmission (Usdin et al., 1999) and decreased transcription of brain-derived neurotrophic factor (Zuccato et al., 2001), a crucial modulator of synaptic plasticity (Bramham and Messaoudi, 2005). Although STEP is initially neuroprotective in preventing immediate, mHTT-induced excitotoxicity, it is unable to prevent the mHTT-induced synaptic dysfunction and eventual neurite dystrophy caused by aberrant axonal transport and nuclear transcription.
On the other hand, cognitive impairments observed after hypoxic–ischemic brain injury are caused by excitotoxicity and synaptic destruction rather than altered synaptic function. In rodents, neurotoxic lesions of the hippocampus with NMDA result in cognitive deficits, especially in spatial learning and memory (Hicks et al., 1993; Deacon et al., 2002). As previously mentioned, hypoxia–ischemia results in excitotoxicity through NMDAR-mediated cleavage of STEP61 into STEP33, which is no longer able to dephosphorylate and internalize NMDARs on the plasma membrane. Rather than decreasing STEP levels or activity, hypoxic–ischemic brain injury could be attenuated by elevating STEP levels (Braithwaite et al., 2008) or blocking the calpain cleavage site (Xu et al., 2009), thereby enhancing the ability of STEP to dephosphorylate p38 and protect neurons against excitotoxicity.
In conclusion, STEP contributes to cognitive deficits in several disorders by regulating the activity of key signaling proteins and the trafficking NMDARs and AMPARs to synaptic membranes. Although the mechanisms of impaired synaptic plasticity vary, alterations in NMDAR and AMPAR trafficking contribute to the disruption of synaptic function in each disorder. In AD, SZ, and FXS, elevated STEP activity causes excessive glutamate receptor internalization from synaptosomal surface membranes and decreased synaptic plasticity. In HD, STEP downregulation is initially neuroprotective to mHTT-induced glutamate excitotoxicity, but at the cost of disrupting synaptic plasticity and causing cognitive impairment. In hypoxic–ischemic brain injury, STEP cleavage and the loss of substrate-binding facilitates NMDAR-mediated glutamate excitotoxicity, resulting in neuronal loss, especially in the hippocampus. Although specific inhibitors are being developed, such as glutamate antagonists for schizophrenia and calpain inhibitors for ischemia–hypoxia, STEP may prove to be a novel therapeutic target for multiple disorders that impair cognitive function.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank laboratory members for helpful discussions and Dr. Marilee Ogren for critical reading of the manuscript. The work was funded by NIH grants MH091037 and MH052711 (Paul J. Lombroso).
References
Alonso, A. C., Grundke-Iqbal, I., and Iqbal, K. (1996). Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 2, 783–787.
Alvestad, R. M., Grosshans, D. R., Coultrap, S. J., Nakazawa, T., Yamamoto, T., and Browning, M. D. (2003). Tyrosine dephosphorylation and ethanol inhibition of N-methyl-D-aspartate receptor function. J. Biol. Chem. 278, 11020–11025.
Anderson, C. A., and Arciniegas, D. B. (2010). Cognitive sequelae of hypoxic-ischemic brain injury: a review. NeuroRehabilitation 26, 47–63.
Antar, L. N., Afroz, R., Dictenberg, J. B., Carroll, R. C., and Bassell, G. J. (2004). Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J. Neurosci. 24, 2648–2655.
Apostol, B. L., Illes, K., Pallos, J., Bodai, L., Wu, J., Strand, A., Schweitzer, E. S., Olson, J. M., Kazantsev, A., Marsh, J. L., and Thompson, L. M. (2006). Mutant huntingtin alters MAPK signaling pathways in PC12 and striatal cells: ERK1/2 protects against mutant huntingtin-associated toxicity. Hum. Mol. Genet. 15, 273–285.
Bachoud-Levi, A. C., Maison, P., Bartolomeo, P., Boisse, M. F., Dalla Barba, G., Ergis, A. M., Baudic, S., Degos, J. D., Cesaro, P., and Peschanski, M. (2001). Retest effects and cognitive decline in longitudinal follow-up of patients with early HD. Neurology 56, 1052–1058.
Ball, M. J., Fisman, M., Hachinski, V., Blume, W., Fox, A., Kral, V. A., Kirshen, A. J., Fox, H., and Merskey, H. (1985). A new definition of Alzheimer’s disease: a hippocampal dementia. Lancet 1, 14–16.
Bao, J., Sharp, A. H., Wagster, M. V., Becher, M., Schilling, G., Ross, C. A., Dawson, V. L., and Dawson, T. M. (1996). Expansion of polyglutamine repeat in huntingtin leads to abnormal protein interactions involving calmodulin. Proc. Natl. Acad. Sci. U.S.A. 93, 5037–5042.
Bates, M. E., Bowden, S. C., and Barry, D. (2002). Neurocognitive impairment associated with alcohol use disorders: implications for treatment. Exp. Clin. Psychopharmacol. 10, 193–212.
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377.
Benveniste, H. (1991). The excitotoxin hypothesis in relation to cerebral ischemia. Cerebrovasc. Brain Metab. Rev. 3, 213–245.
Bibb, J. A., Yan, Z., Svenningsson, P., Snyder, G. L., Pieribone, V. A., Horiuchi, A., Nairn, A. C., Messer, A., and Greengard, P. (2000). Severe deficiencies in dopamine signaling in presymptomatic Huntington’s disease mice. Proc. Natl. Acad. Sci. U.S.A. 97, 6809–6814.
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39.
Boulanger, L. M., Lombroso, P. J., Raghunathan, A., During, M. J., Wahle, P., and Naegele, J. R. (1995). Cellular and molecular characterization of a brain-enriched protein tyrosine phosphatase. J. Neurosci. 15, 1532–1544.
Bourne, C., Clayton, C., Murch, A., and Grant, J. (2006). Cognitive impairment and behavioural difficulties in patients with Huntington’s disease. Nurs. Stand. 20, 41–44.
Braithwaite, S. P., Adkisson, M., Leung, J., Nava, A., Masterson, B., Urfer, R., Oksenberg, D., and Nikolich, K. (2006). Regulation of NMDA receptor trafficking and function by striatal-enriched tyrosine phosphatase (STEP). Eur. J. Neurosci. 23, 2847–2856.
Braithwaite, S. P., Xu, J., Leung, J., Urfer, R., Nikolich, K., Oksenberg, D., Lombroso, P. J., and Shamloo, M. (2008). Expression and function of striatal enriched protein tyrosine phosphatase is profoundly altered in cerebral ischemia. Eur. J. Neurosci. 27, 2444–2452.
Bramham, C. R., and Messaoudi, E. (2005). BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog. Neurobiol. 76, 99–125.
Breier, A., Su, T. P., Saunders, R., Carson, R. E., Kolachana, B. S., de Bartolomeis, A., Weinberger, D. R., Weisenfeld, N., Malhotra, A. K., Eckelman, W. C., and Pickar, D. (1997). Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc. Natl. Acad. Sci. U.S.A. 94, 2569–2574.
Bult, A., Zhao, F., Dirkx, R. Jr., Raghunathan, A., Solimena, M., and Lombroso, P. J. (1997). STEP: a family of brain-enriched PTPs. Alternative splicing produces transmembrane, cytosolic and truncated isoforms. Eur. J. Cell Biol. 72, 337–344.
Bult, A., Zhao, F., Dirkx, R. Jr., Sharma, E., Lukacsi, E., Solimena, M., Naegele, J. R., and Lombroso, P. J. (1996). STEP61: a member of a family of brain-enriched PTPs is localized to the endoplasmic reticulum. J. Neurosci. 16, 7821–7831.
Cai, X. D., Golde, T. E., and Younkin, S. G. (1993). Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science 259, 514–516.
Calabresi, P., Picconi, B., Tozzi, A., and Di Filippo, M. (2007). Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 30, 211–219.
Carty, N., Kurup, P., Xu, J., Austin, D. R., Chen, G., and Lombroso, P. J. (2010). Striatal Enriched Protein Tyrosine Phosphatase (STEP) Mediates the Beneficial Effects of Antipsychotic Drugs. Program No. 452.21. 2010 Neuroscience Meeting Planner. Society for Neuroscience. San Diego, CA. [Online].
Centonze, D., Picconi, B., Gubellini, P., Bernardi, G., and Calabresi, P. (2001). Dopaminergic control of synaptic plasticity in the dorsal striatum. Eur. J. Neurosci. 13, 1071–1077.
Cha, J. H., Kosinski, C. M., Kerner, J. A., Alsdorf, S. A., Mangiarini, L., Davies, S. W., Penney, J. B., Bates, G. P., and Young, A. B. (1998). Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc. Natl. Acad. Sci. U.S.A. 95, 6480–6485.
Chandler, L. J., Newsom, H., Sumners, C., and Crews, F. (1993). Chronic ethanol exposure potentiates NMDA excitotoxicity in cerebral cortical neurons. J. Neurochem. 60, 1578–1581.
Chen, J., Rusnak, M., Lombroso, P. J., and Sidhu, A. (2009). Dopamine promotes striatal neuronal apoptotic death via ERK signaling cascades. Eur. J. Neurosci. 29, 287–306.
Chin, J., Palop, J. J., Puolivali, J., Massaro, C., Bien-Ly, N., Gerstein, H., Scearce-Levie, K., Masliah, E., and Mucke, L. (2005). Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 25, 9694–9703.
Comery, T. A., Harris, J. B., Willems, P. J., Oostra, B. A., Irwin, S. A., Weiler, I. J., and Greenough, W. T. (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc. Natl. Acad. Sci. U.S.A. 94, 5401–5404.
Craufurd, D., Thompson, J. C., and Snowden, J. S. (2001). Behavioral changes in Huntington disease. Neuropsychiatry Neuropsychol. Behav. Neurol. 14, 219–226.
Creese, I., Burt, D. R., and Snyder, S. H. (1976). Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 192, 481–483.
Cruz-Martin, A., Crespo, M., and Portera-Cailliau, C. (2010). Delayed stabilization of dendritic spines in fragile X mice. J. Neurosci. 30, 7793–7803.
Cullen, W. K., Wu, J., Anwyl, R., and Rowan, M. J. (1996). Beta-amyloid produces a delayed NMDA receptor-dependent reduction in synaptic transmission in rat hippocampus. Neuroreport 8, 87–92.
Dajas-Bailador, F., and Wonnacott, S. (2004). Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol. Sci. 25, 317–324.
Deacon, R. M., Bannerman, D. M., Kirby, B. P., Croucher, A., and Rawlins, J. N. (2002). Effects of cytotoxic hippocampal lesions in mice on a cognitive test battery. Behav. Brain Res. 133, 57–68.
DiFiglia, M. (1990). Excitotoxic injury of the neostriatum: a model for Huntington’s disease. Trends Neurosci. 13, 286–289.
Dolan, R. J., Fletcher, P., Frith, C. D., Friston, K. J., Frackowiak, R. S., and Grasby, P. M. (1995). Dopaminergic modulation of impaired cognitive activation in the anterior cingulate cortex in schizophrenia. Nature 378, 180–182.
Dougherty, J. J., Wu, J., and Nichols, R. A. (2003). Beta-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J. Neurosci. 23, 6740–6747.
Eadie, B. D., Cushman, J., Kannangara, T. S., Fanselow, M. S., and Christie, B. R. (2010). NMDA receptor hypofunction in the dentate gyrus and impaired context discrimination in adult Fmr1 knockout mice. Hippocampus. doi: 10.1002/hipo.20890. [Epub ahead of print].
Fan, M. M., and Raymond, L. A. (2007). N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington’s disease. Prog. Neurobiol. 81, 272–293.
Ferrani-Kile, K., Randall, P. K., and Leslie, S. W. (2003). Acute ethanol affects phosphorylation state of the NMDA receptor complex: implication of tyrosine phosphatases and protein kinase A. Brain Res. Mol. Brain Res. 115, 78–86.
Folstein, M. F., and Whitehouse, P. J. (1983). Cognitive impairment of Alzheimer disease. Neurobehav. Toxicol. Teratol. 5, 631–634.
Freund, L. S., and Reiss, A. L. (1991). Cognitive profiles associated with the fra(X) syndrome in males and females. Am. J. Med. Genet. 38, 542–547.
Garber, K., Smith, K. T., Reines, D., and Warren, S. T. (2006). Transcription, translation and fragile X syndrome. Curr. Opin. Genet. Dev. 16, 270–275.
Gines, S., Seong, I. S., Fossale, E., Ivanova, E., Trettel, F., Gusella, J. F., Wheeler, V. C., Persichetti, F., and MacDonald, M. E. (2003). Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum. Mol. Genet. 12, 497–508.
Goebel-Goody, S. M., Davies, K. D., Alvestad Linger, R. M., Freund, R. K., and Browning, M. D. (2009). Phospho-regulation of synaptic and extrasynaptic N-methyl-d-aspartate receptors in adult hippocampal slices. Neuroscience 158, 1446–1459.
Goebel-Goody, S. M., Wallis, E. D., Li, L., Hall, D. V., Royston, S., Baum, M., Naegele, J. R., and Lombroso, P. J. (2010). Loss of Striatal-Enriched Protein Tyrosine Phosphatase (STEP) Reverses Deficits in a Fragile X Syndrome Mouse Model. Program No. 452.4. 2010 Neuroscience Meeting Planner. Society for Neuroscience. San Diego, CA. [Online].
Goedert, M., Spillantini, M. G., Jakes, R., Rutherford, D., and Crowther, R. A. (1989). Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3, 519–526.
Gold, J. M., and Harvey, P. D. (1993). Cognitive deficits in schizophrenia. Psychiatr. Clin. North Am. 16, 295–312.
Gurd, J. W., Bissoon, N., Nguyen, T. H., Lombroso, P. J., Rider, C. C., Beesley, P. W., and Vannucci, S. J. (1999). Hypoxia-ischemia in perinatal rat brain induces the formation of a low molecular weight isoform of striatal enriched tyrosine phosphatase (STEP). J. Neurochem. 73, 1990–1994.
Hansson, O., Guatteo, E., Mercuri, N. B., Bernardi, G., Li, X. J., Castilho, R. F., and Brundin, P. (2001). Resistance to NMDA toxicity correlates with appearance of nuclear inclusions, behavioural deficits and changes in calcium homeostasis in mice transgenic for exon 1 of the huntington gene. Eur. J. Neurosci. 14, 1492–1504.
Hansson, O., Petersen, A., Leist, M., Nicotera, P., Castilho, R. F., and Brundin, P. (1999). Transgenic mice expressing a Huntington’s disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc. Natl. Acad. Sci. U.S.A. 96, 8727–8732.
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185.
Heresco-Levy, U., Javitt, D. C., Ermilov, M., Mordel, C., Silipo, G., and Lichtenstein, M. (1999). Efficacy of high-dose glycine in the treatment of enduring negative symptoms of schizophrenia. Arch. Gen. Psychiatry 56, 29–36.
Heyes, C., and Huber, L. (2000). The Evolution Of Cognition (Vienna series in theoretical biology). Cambridge: MIT Press.
Hicklin, T. R., Wu, P. H., Radcliffe, R. A., Freund, R. K., Goebel-Goody, S. M., Correa, P. R., Proctor, W. R., Lombroso, P. J., and Browning, M. D. (2011). Alcohol inhibition of the NMDA receptor function, long-term potentiation, and fear learning requires striatal-enriched protein tyrosine phosphatase. Proc. Natl. Acad. Sci. U.S.A. 108, 6650–6655.
Hicks, R. R., Smith, D. H., Lowenstein, D. H., Saint Marie, R., and McIntosh, T. K. (1993). Mild experimental brain injury in the rat induces cognitive deficits associated with regional neuronal loss in the hippocampus. J. Neurotrauma 10, 405–414.
Hu, X. J., and Ticku, M. K. (1995). Chronic ethanol treatment upregulates the NMDA receptor function and binding in mammalian cortical neurons. Brain Res. Mol. Brain Res. 30, 347–356.
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 7746–7750.
Huber, K. M., Kayser, M. S., and Bear, M. F. (2000). Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288, 1254–1257.
Huber, K. M., Roder, J. C., and Bear, M. F. (2001). Chemical induction of mGluR5- and protein synthesis-dependent long-term depression in hippocampal area CA1. J. Neurophysiol. 86, 321–325.
Huff, F. J., Becker, J. T., Belle, S. H., Nebes, R. D., Holland, A. L., and Boller, F. (1987). Cognitive deficits and clinical diagnosis of Alzheimer’s disease. Neurology 37, 1119–1124.
Humphries, C., Mortimer, A., Hirsch, S., and de Belleroche, J. (1996). NMDA receptor mRNA correlation with antemortem cognitive impairment in schizophrenia. Neuroreport 7, 2051–2055.
Iadecola, C. (1997). Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 20, 132–139.
Irwin, S. A., Galvez, R., and Greenough, W. T. (2000). Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb. Cortex 10, 1038–1044.
Jentsch, J. D., and Roth, R. H. (1999). The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 20, 201–225.
Jin, R., Clark, S., Weeks, A. M., Dudman, J. T., Gouaux, E., and Partin, K. M. (2005). Mechanism of positive allosteric modulators acting on AMPA receptors. J. Neurosci. 25, 9027–9036.
Kantrowitz, J. T., Malhotra, A. K., Cornblatt, B., Silipo, G., Balla, A., Suckow, R. F., D’Souza, C., Saksa, J., Woods, S. W., and Javitt, D. C. (2010). High dose D-serine in the treatment of schizophrenia. Schizophr. Res. 121, 125–130.
Kegel, K. B., Meloni, A. R., Yi, Y., Kim, Y. J., Doyle, E., Cuiffo, B. G., Sapp, E., Wang, Y., Qin, Z. H., Chen, J. D., Nevins, J. R., Aronin, N., and DiFiglia, M. (2002). Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J. Biol. Chem. 277, 7466–7476.
Kirino, T., and Sano, K. (1984). Selective vulnerability in the gerbil hippocampus following transient ischemia. Acta Neuropathol. 62, 201–208.
Koukoui, S. D., and Chaudhuri, A. (2007). Neuroanatomical, molecular genetic, and behavioral correlates of fragile X syndrome. Brain Res. Rev. 53, 27–38.
Kril, J. J., Patel, S., Harding, A. J., and Halliday, G. M. (2002). Patients with vascular dementia due to microvascular pathology have significant hippocampal neuronal loss. J. Neurol. Neurosurg. Psychiatr. 72, 747–751.
Kurup, P., Zhang, Y., Xu, J., Venkitaramani, D. V., Haroutunian, V., Greengard, P., Nairn, A. C., and Lombroso, P. J. (2010). Abeta-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. J. Neurosci. 30, 5948–5957.
Lacor, P. N., Buniel, M. C., Furlow, P. W., Clemente, A. S., Velasco, P. T., Wood, M., Viola, K. L., and Klein, W. L. (2007). Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 27, 796–807.
Laggerbauer, B., Ostareck, D., Keidel, E. M., Ostareck-Lederer, A., and Fischer, U. (2001). Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet. 10, 329–338.
Li, J., Pelletier, M. R., Perez Velazquez, J. L., and Carlen, P. L. (2002). Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol. Cell. Neurosci. 19, 138–151.
Li, Z., Zhang, Y., Ku, L., Wilkinson, K. D., Warren, S. T., and Feng, Y. (2001). The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 29, 2276–2283.
Lombroso, P. J., Murdoch, G., and Lerner, M. (1991). Molecular characterization of a protein-tyrosine-phosphatase enriched in striatum. Proc. Natl. Acad. Sci. U.S.A. 88, 7242–7246.
Lombroso, P. J., Naegele, J. R., Sharma, E., and Lerner, M. (1993). A protein tyrosine phosphatase expressed within dopaminoceptive neurons of the basal ganglia and related structures. J. Neurosci. 13, 3064–3074.
Lue, L. F., Kuo, Y. M., Roher, A. E., Brachova, L., Shen, Y., Sue, L., Beach, T., Kurth, J. H., Rydel, R. E., and Rogers, J. (1999). Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 155, 853–862.
Mandelkow, E. M., Stamer, K., Vogel, R., Thies, E., and Mandelkow, E. (2003). Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol. Aging 24, 1079–1085.
Matsumoto, S., Shamloo, M., Isshiki, A., and Wieloch, T. (2002). Persistent phosphorylation of synaptic proteins following middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 22, 1107–1113.
Mega, M. S., Cummings, J. L., Fiorello, T., and Gornbein, J. (1996). The spectrum of behavioral changes in Alzheimer’s disease. Neurology 46, 130–135.
Mohn, A. R., Gainetdinov, R. R., Caron, M. G., and Koller, B. H. (1999). Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 98, 427–436.
Mony, L., Kew, J. N., Gunthrope, M. J., and Paoletti, P. (2009). Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br. J. Pharmacol. 157, 1301–1317.
Munoz, J. J., Tarrega, C., Blanco-Aparicio, C., and Pulido, R. (2003). Differential interaction of the tyrosine phosphatases PTP-SL, STEP and HePTP with the mitogen-activated protein kinases ERK1/2 and p38alpha is determined by a kinase specificity sequence and influenced by reducing agents. Biochem. J. 372, 193–201.
Murphy, K. P., Carter, R. J., Lione, L. A., Mangiarini, L., Mahal, A., Bates, G. P., Dunnett, S. B., and Morton, A. J. (2000). Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington’s disease mutation. J. Neurosci. 20, 5115–5123.
Nakamoto, M., Nalavadi, V., Epstein, M. P., Narayanan, U., Bassell, G. J., and Warren, S. T. (2007). Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 15537–15542.
Nguyen, T. H., Liu, J., and Lombroso, P. J. (2002). Striatal enriched phosphatase 61 dephosphorylates Fyn at phosphotyrosine 420. J. Biol. Chem. 277, 24274–24279.
Nguyen, T. H., Paul, S., Xu, Y., Gurd, J. W., and Lombroso, P. J. (1999). Calcium-dependent cleavage of striatal enriched tyrosine phosphatase (STEP). J. Neurochem. 73, 1995–2001.
Nitatori, T., Sato, N., Waguri, S., Karasawa, Y., Araki, H., Shibanai, K., Kominami, E., and Uchiyama, Y. (1995). Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J. Neurosci. 15, 1001–1011.
Nosyreva, E. D., and Huber, K. M. (2006). Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J. Neurophysiol. 95, 3291–3295.
Nozaki, K., Nishimura, M., and Hashimoto, N. (2001). Mitogen-activated protein kinases and cerebral ischemia. Mol. Neurobiol. 23, 1–19.
Oberle, I., Rousseau, F., Heitz, D., Kretz, C., Devys, D., Hanauer, A., Boue, J., Bertheas, M., and Mandel, J. (1991). Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252, 1097–1102.
Pascoli, V., Besnard, A., Hervé, D., Pagès, C., Heck, N., Girault, J. A., Caboche, J., and Vanhoutte, P. (2011). Cyclic adenosine monophosphate-independent tyrosine phosphorylation of N2RB mediates cocaine-induced extracellular signal-related kinase activation. Biol. Psychiatry 69, 218–227.
Paul, S., Nairn, A. C., Wang, P., and Lombroso, P. J. (2003). NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat. Neurosci. 6, 34–42.
Paul, S., Snyder, G. L., Yokakura, H., Picciotto, M. R., Nairn, A. C., and Lombroso, P. J. (2000). The dopamine/D1 receptor mediates the phosphorylation and inactivation of the protein tyrosine phosphatase STEP via a PKA-dependent pathway. J. Neurosci. 20, 5630–5638.
Pelkey, K. A., Askalan, R., Paul, S., Kalia, L. V., Nguyen, T. H., Pitcher, G. M., Salter, M. W., and Lombroso, P. J. (2002). Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron 34, 127–138.
Pfeiffer, B. E., and Huber, K. M. (2007). Fragile X mental retardation protein induces synapse loss through acute postsynaptic translational regulation. J. Neurosci. 27, 3120–3130.
Pilowsky, L. S., Bressan, R. A., Stone, J. M., Erlandsson, K., Mulligan, R. S., Krystal, J. H., and Ell, P. J. (2006). First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol. Psychiatry 11, 118–119.
Pohjasvaara, T., Erkinjuntti, T., Ylikoski, R., Hietanen, M., Vataja, R., and Kaste, M. (1998). Clinical determinants of poststroke dementia. Stroke 29, 75–81.
Qin, M., Kang, J., Burlin, T. V., Jiang, C., and Smith, C. B. (2005). Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J. Neurosci. 25, 5087–5095.
Reiner, A., Albin, R. L., Anderson, K. D., D’Amato, C. J., Penney, J. B., and Young, A. B. (1988). Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 85, 5733–5737.
Ron, D. (2004). Signaling cascades regulating NMDA receptor sensitivity to ethanol. Neuroscientist 10, 325–336.
Ronesi, J. A., and Huber, K. M. (2008). Metabotropic glutamate receptors and fragile x mental retardation protein: partners in translational regulation at the synapse. Sci. Signal. 1, pe6.
Saavedra, A., Giralt, A., Rué, L., Xifró, X., Xu, J., Ortega, Z., Lucas, J. J., Lombroso, P. J., Alberch, J., and Pérez-Navarro, E. (2011). Striatal-enriched protein tyrosine phosphatase expression and activity in Huntington’s disease: a STEP in the resistance to excitotoxicity. J. Neurosci. 31, 8150–8162.
Seeman, P., Chau-Wong, M., Tedesco, J., and Wong, K. (1975). Brain receptors for antipsychotic drugs and dopamine: direct binding assays. Proc. Natl. Acad. Sci. U.S.A. 72, 4376–4380.
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875.
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., Brett, F. M., Farrell, M. A., Rowan, M. J., Lemere, C. A., Regan, C. M., Walsh, D. M., Sabatini, B. L., and Selkoe, D. J. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842.
Sharma, E., Zhao, F., Bult, A., and Lombroso, P. J. (1995). Identification of two alternatively spliced transcripts of STEP: a subfamily of brain-enriched protein tyrosine phosphatases. Brain Res. Mol. Brain Res. 32, 87–93.
Silva, A. J. (2003). Molecular and cellular cognitive studies of the role of synaptic plasticity in memory. J. Neurobiol. 54, 224–237.
Smith, S. E. (1993). Cognitive deficits associated with fragile X syndrome. Ment. Retard. 31, 279–283.
Snyder, E. M., Nong, Y., Almeida, C. G., Paul, S., Moran, T., Choi, E. Y., Nairn, A. C., Salter, M. W., Lombroso, P. J., Gouras, G. K., and Greengard, P. (2005). Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 8, 1051–1058.
Snyder, E. M., Philpot, B. D., Huber, K. M., Dong, X., Fallon, J. R., and Bear, M. F. (2001). Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat. Neurosci. 4, 1079–1085.
Stoof, J. C., and Kebabian, J. W. (1981). Opposing roles for D-1 and D-2 dopamine receptors in efflux of cyclic AMP from rat neostriatum. Nature 294, 366–368.
Suvrathan, A., Hoeffer, C. A., Wong, H., Klann, E., and Chattarji, S. (2010). Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 11591–11596.
Suzuki, N., Cheung, T. T., Cai, X. D., Odaka, A., Otvos, L. Jr., Eckman, C., Golde, T. E., and Younkin, S. G. (1994). An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 264, 1336–1340.
Tanzi, R. E., Gusella, J. F., Watkins, P. C., Bruns, G. A., St George-Hyslop, P., Van Keuren, M. L., Patterson, D., Pagan, S., Kurnit, D. M., and Neve, R. L. (1987). Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 235, 880–884.
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., Hansen, L. A., and Katzman, R. (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580.
The Huntington’s Disease Collaborative Research Group. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983.
Todd, P. K., Mack, K. J., and Malter, J. S. (2003). The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc. Natl. Acad. Sci. U.S.A. 100, 14374–14378.
Trushina, E., Dyer, R. B., Badger, J. D. II, Ure, D., Eide, L., Tran, D. D., Vrieze, B. T., Legendre-Guillemin, V., McPherson, P. S., Mandavilli, B. S., Van Houten, B., Zeitlin, S., McNiven, M., Aebersold, R., Hayden, M., Parisi, J. E., Seeberg, E., Dragatsis, I., Doyle, K., Bender, A., Chacko, C., and McMurray, C. T. (2004). Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 24, 8195–8209.
Turalba, A. V., Leite-Morris, K. A., and Kaplan, G. B. (2004). Antipsychotics regulate cyclic AMP-dependent protein kinase and phosphorylated cyclic AMP response element-binding protein in striatal and cortical brain regions. Neurosci. Lett. 357, 53–57.
Turrigiano, G. G., and Nelson, S. B. (2004). Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 5, 97–107.
Ule, J., and Darnell, R. B. (2006). RNA binding proteins and the regulation of neuronal synaptic plasticity. Curr. Opin. Neurobiol. 16, 102–110.
Usdin, M. T., Shelbourne, P. F., Myers, R. M., and Madison, D. V. (1999). Impaired synaptic plasticity in mice carrying the Huntington’s disease mutation. Hum. Mol. Genet. 8, 839–846.
Valjent, E., Pascoli, V., Svenningsson, P., Paul, S., Enslen, H., Corvol, J. C., Stipanovich, A., Caboche, J., Lombroso, P. J., Nairn, A. C., Greengard, P., Herve, D., and Girault, J. A. (2005). Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc. Natl. Acad. Sci. U.S.A. 102, 491–496.
Van Raamsdonk, J. M., Pearson, J., Slow, E. J., Hossain, S. M., Leavitt, B. R., and Hayden, M. R. (2005). Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington’s disease. J. Neurosci. 25, 4169–4180.
Vanhoose, A. M., and Winder, D. G. (2003). NMDA and beta1-adrenergic receptors differentially signal phosphorylation of glutamate receptor type 1 in area CA1 of hippocampus. J. Neurosci. 23, 5827–5834.
Venkitaramani, D. V., Moura, P. J., Picciotto, M. R., and Lombroso, P. J. (2011). Striatal-enriched protein tyrosine phosphatase (STEP) knockout mice have enhanced hippocampal memory. Eur. J. Neurosci. 33, 2288–2298.
Visootsak, J., Warren, S. T., Anido, A., and Graham, J. M. Jr. (2005). Fragile X syndrome: an update and review for the primary pediatrician. Clin. Pediatr. (Phila.) 44, 371–381.
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., Rowan, M. J., and Selkoe, D. J. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539.
Wang, J., Carnicella, S., Phamluong, K., Jeanblanc, J., Ronesi, J. A., Chaudhri, N., Janak, P. H., Lovinger, D. M., and Ron, D. (2007). Ethanol induces long-term facilitation of NR2B-NMDA receptor activity in the dorsal striatum: implications for alcohol drinking behavior. J. Neurosci. 27, 3593–3602.
Wang, J., Lanfranco, M. F., Gibb, S. L., Yowell, Q. V., Carnicella, S., and Ron, D. (2010). Long-lasting adaptations of the NR2B-containing NMDA receptors in the dorsomedial striatum play a crucial role in alcohol consumption and relapse. J. Neurosci. 30, 10187–10198.
Weiler, I. J., and Greenough, W. T. (1999). Synaptic synthesis of the fragile X protein: possible involvement in synapse maturation and elimination. Am. J. Med. Genet. 83, 248–252.
White, A. M., Matthews, D. B., and Best, P. J. (2000). Ethanol, memory, and hippocampal function: a review of recent findings. Hippocampus 10, 88–93.
Xiao, M. Y., Wigstrom, H., and Gustafsson, B. (1994). Long-term depression in the hippocampal CA1 region is associated with equal changes in AMPA and NMDA receptor-mediated synaptic potentials. Eur. J. Neurosci. 6, 1055–1057.
Xifro, X., Giralt, A., Saavedra, A., Garcia-Martinez, J. M., Diaz-Hernandez, M., Lucas, J. J., Alberch, J., and Perez-Navarro, E. (2009). Reduced calcineurin protein levels and activity in exon-1 mouse models of Huntington’s disease: role in excitotoxicity. Neurobiol. Dis. 36, 461–469.
Xu, J., Kurup, P., Bartos, J. A., Hell, J. W., and Lombroso, P. J. (2010). Inhibition of pyk2 Signaling by Striatal-Enriched Tyrosine Phosphatase (STEP). Program No. 452.13. 2010 Neuroscience Meeting Planner. Society for Neuroscience. San Diego, CA. [Online].
Xu, J., Kurup, P., Zhang, Y., Goebel-Goody, S. M., Wu, P. H., Hawasli, A. H., Baum, M. L., Bibb, J. A., and Lombroso, P. J. (2009). Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J. Neurosci. 29, 9330–9343.
Yin, H. H., Park, B. S., Adermark, L., and Lovinger, D. M. (2007). Ethanol reverses the direction of long-term synaptic plasticity in the dorsomedial striatum. Eur. J. Neurosci. 25, 3226–3232.
Zhang, Y., Kurup, P., Xu, J., Carty, N., Fernandez, S. M., Nygaard, H. B., Pittenger, C., Greengard, P., Strittmatter, S. M., Nairn, A. C., and Lombroso, P. J. (2010). Genetic reduction of striatal-enriched tyrosine phosphatase (STEP) reverses cognitive and cellular deficits in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. U.S.A. 107, 19014–19019.
Zhang, Y., Venkitaramani, D. V., Gladding, C. M., Kurup, P., Molnar, E., Collingridge, G. L., and Lombroso, P. J. (2008). The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. J. Neurosci. 28, 10561–10566.
Keywords: STEP, protein tyrosine phosphatase, cognition, Alzheimer’s disease, fragile X syndrome, Huntington’s disease, schizophrenia, excitotoxicity
Citation: Fitzpatrick CJ and Lombroso PJ (2011) The role of striatal-enriched protein tyrosine phosphatase (STEP) in cognition. Front. Neuroanat. 5:47. doi: 10.3389/fnana.2011.00047
Received: 29 April 2011;
Accepted: 13 July 2011;
Published online: 29 July 2011.
Edited by:
Emmanuel Valjent, Université Montpellier 1 & 2, FranceReviewed by:
Joël Bockaert, INSERM, FrancePeter Vanhoutte, Centre National de la Recherche Scientifique, France
Copyright: © 2011 Fitzpatrick and Lombroso. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Paul J. Lombroso, Laboratory of Molecular Neurobiology, 230 South Frontage Road, New Haven, CT 06520, USA. e-mail:cGF1bC5sb21icm9zb0B5YWxlLmVkdQ==