

- 1 Departamento de Fisiología y Biofísica, Facultad de Medicina, Universidad de Buenos Aires, Buenos Aires, Argentina
- 2 Instituto Cajal, Consejo Superior Investigaciones Científicas, Madrid, Spain
- 3 Centro de Investigación Biomédica en Red, Enfermedades Neurodegenerativas, Instituto de Salud Carlos III, Madrid, Spain
Parkinson’s disease is a common neurodegenerative disorder caused by the degeneration of midbrain substantia nigra dopaminergic neurons that project to the striatum. Despite extensive investigation aimed at finding new therapeutic approaches, the dopamine precursor molecule, 3,4-dihydroxyphenyl-L-alanine (L-DOPA), remains the most effective and commonly used treatment. However, chronic treatment and disease progression lead to changes in the brain’s response to L-DOPA, resulting in decreased therapeutic effect and the appearance of dyskinesias. L-DOPA-induced dyskinesia (LID) interferes significantly with normal motor activity and persists unless L-DOPA dosages are reduced to below therapeutic levels. Thus, controlling LID is one of the major challenges in Parkinson’s disease therapy. LID is the result of intermittent stimulation of supersensitive D1 dopamine receptors located in the very severely denervated striatal neurons. Through increased coupling to Gαolf, resulting in greater stimulation of adenylyl-cyclase, D1 receptors phosphorylate DARPP-32, and other protein kinase A targets. Moreover, D1 receptor stimulation activates extracellular signal-regulated kinase and triggers a signaling pathway involving mammalian target for rapamycin and modifications of histones that results in changes in translation, chromatin modification, and gene transcription. In turn, sensitization of D1 receptor signaling causes a widespread increase in the metabolic response to D1 agonists and changes in the activity of basal ganglia neurons that correlate with the severity of LID. Importantly, different studies suggest that dyskinesias may share mechanisms with drug abuse and long term memory involving D1 receptor activation. Here we review evidence implicating D1 receptor signaling in the genesis of LID, analyze mechanisms that may translate enhanced D1 signaling into dyskinetic movements, and discuss the possibility that the mechanisms underlying LID are not unique to the Parkinson’s disease brain.
Shortly after the recognition that L-DOPA in combination with a peripheral decarboxylase inhibitor has a dramatic therapeutic effect in Parkinson’s disease, it became clear that the treatment was associated with major adverse effects like the appearance of severely incapacitating abnormal involuntary movements (Cotzias et al., 1969; Papavasiliou et al., 1972). This raised concern about the safety of the treatment and the consequences of beginning L-DOPA therapy early in the course of the disease (Fahn, 1996), but the fear that L-DOPA could be toxic was dispelled more than a decade ago (Murer et al., 1999; Olanow et al., 2004). Currently, the principal candidates for the mechanism underlying L-DOPA-induced dyskinesia (LID) are long term functional and structural changes induced in the dopamine-depleted striatum by dopamine-regulated signaling cascades (Jenner, 2008; Voon et al., 2009; Cenci and Konradi, 2010).
Clinical studies have identified several factors that increase the risk of LID: severity and/or duration of the disease, L-DOPA dose, and intermittent L-DOPA dosing (Nutt, 2000, 2008). Studies in patients indicate that initiating therapy with a selective D2-like dopamine receptor agonist is associated with a very low risk of developing dyskinesia, whereas D1-like agonists are very dyskinetogenic (Rascol et al., 2001, 2006; Holloway et al., 2004) in agreement with recent findings in animal models of Parkinson’s disease (Darmopil et al., 2009). Here we review animal studies investigating the mechanisms of LID, with a focus on rodent models and D1 receptor signaling mechanisms.
Is D1 Receptor Stimulation Necessary and Sufficient to Induce Dyskinesia?
Early studies in primates suggested that D1-like agonists are less likely than D2-like agonists to induce dyskinesia (Grondin et al., 1997; Goulet and Madras, 2000). Indeed, it has been proposed that stimulation of sensitized D2-like receptors may be necessary and sufficient to induce dyskinesia (Blanchet et al., 1995). However, based on the administration of selective D1- or D2-like receptor antagonists together with L-DOPA to MPTP-treated primates, it was concluded that neither the beneficial motor effects of L-DOPA, nor LID, can be ascribed to stimulation of one receptor family alone (Grondin et al., 1999). Currently, there is plenty of evidence that the risk of dyskinesia is very low during chronic treatment with a selective D2-like agonist in patients who have never been exposed to L-DOPA (Damier et al., 2000; Holloway et al., 2004; Rascol et al., 2006).
The discovery that rodents with a nigrostriatal lesion develop LID provided a means to perform a non-pharmacological intervention to study the mechanisms underlying it. Rats and mice with unilateral nigrostriatal lesions induced by 6-hydroxydopamine (6-OHDA) develop abnormal involuntary movements resembling “peak-dose dyskinesia” after a few challenges with L-DOPA (Andersson et al., 1999; Delfino et al., 2004; Pavon et al., 2006). Pharmacological studies show that selective D1-like agonists have more powerful dyskinetogenic effects than D2-like agonists, and D1-like antagonists are more effective at inhibiting LID than D2-like antagonists (Taylor et al., 2005; Delfino et al., 2007; Westin et al., 2007). As in patients, de novo administration of a therapeutic dose of D2-like agonist produces mild or no dyskinesia in 6-OHDA rats, but previous exposure to L-DOPA increases the dyskinetogenic effect of selective D2 receptor stimulation (Delfino et al., 2004; Larramendy et al., 2008).
Powerful evidence supporting a role for the D1R subtype in LID has been provided in studies of genetically engineered mice lacking the D1 receptor (Darmopil et al., 2009). LID is completely blocked in D1 knockouts with severe striatal dopaminergic denervation induced by 6-OHDA (Figures 1 and 2). In contrast, D2R knockouts develop LID as easily as controls. Although the latter does not rule out a possible contribution of D3R to LID expression (Bézard et al., 2003), the fact that D1R knockouts do not show abnormal involuntary movements with prolonged L-DOPA treatment suggests that D1R stimulation is essential for the development of LID (Figures 1 and 2).
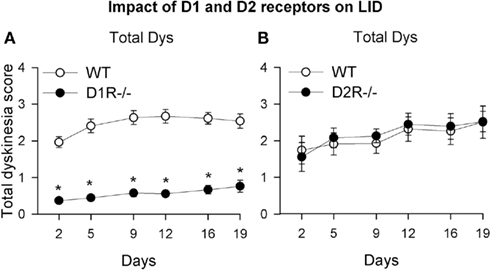
Figure 1. Genetic deletion of the D1 receptor precludes the development of LID. Effect of the genetic deletion of the D1 receptor (A) or D2 receptor (B) on scores of dyskinesia involving the forelimb, head and trunk (“total dyskinesia score”). Adult wild type and knockout mice with similar degrees of nigrostriatal denervation induced by intrastriatal administration of the toxin 6-OHDA were treated daily with L-DOPA and examined periodically for dyskinesia scores. See also Darmopil et al. (2009).

Figure 2. L-DOPA does not induce dyskinesia in D1 receptor knockout mice with severe nigrostriatal denervation. Frames extracted from videos taken from representative 6-OHDA-lesioned wild type (A), D1−/− (B), and D2−/− (C) mice, which have been treated with L-DOPA for 2 weeks. The sequence was taken 30 min after an L-DOPA challenge and the frames are 0.15 s apart from each other. Reproduced from Darmopil et al., 2009.
The possibility remains that the D3R, which is abnormally expressed in the dorsal striatum after repeated exposure to levodopa, contributes to LID expression (Bézard et al., 2003). However, there is wide divergence between studies regarding the extent of D3R upregulation and the effects of D3R preferring antagonists and partial agonists on LID (Hurley et al., 1996; Bordet et al., 1997; Quik et al., 2000; Hsu et al., 2004; Visanji et al., 2009). Moreover, D1R stimulation induces a full repertoire of dyskinetic movements resembling peak-dose dyskinesia even in the presence of D3R antagonists (Kumar et al., 2009; Mela et al., 2010), suggesting that D3R stimulation is not an obligatory requirement for LID to occur. In summary, D1R stimulation seems to be necessary for LID development and sufficient to induce dyskinetic movements. This is in agreement with data showing that, in contrast to D2-like receptor agonists (most of which have a high affinity for the D3R – Rascol, 1999), D1-like receptor agonists are markedly dyskinetogenic in monkeys rendered parkinsonian by MPTP (Doucet et al., 1996) as well as in patients (Rascol et al., 2001). Additional mechanisms involving D2 family receptors may contribute to the expression of LID, especially when dyskinetic movements are induced by D2-like agonists in animals previously exposed to L-DOPA.
LID is Associated with Changes in D1 Receptor Signaling in the Dopamine-Depleted Striatum
Early studies showing that deletion of the D1R gene blocks control of striatal prodynorphin expression and motor behavior by cocaine and amphetamine called attention to D1R signaling in behavioral sensitization to psychostimulants (Moratalla et al., 1996a). This study also suggested involvement of the direct pathway in psychostimulant sensitization, because prodynorphin is a marker of those striatal “medium spiny” neurons (MSN) that contain the D1 receptor and project to the substantia nigra. In parallel, it has been shown that repeated administration of a D1-like receptor agonist induces dyskinesia and the expression of the transcription factors FosB/ΔFosB in striatonigral MSNs (Doucet et al., 1996). Further investigation showed that LID in the 6-OHDA rat is associated with increased co-expression of FosB/ΔFosB and prodynorphin in striatal neurons (Andersson et al., 1999; see also Sgambato-Faure et al., 2005; Pavon et al., 2006). In contrast to full-length FosB proteins, which behave as immediate early genes, its truncated forms called ΔFosB show high stability and persist for long periods (McClung et al., 2004). Although ΔFosB cannot be distinguished from FosB in immunocytochemical studies, Doucet et al. (1996), Andersson et al. (1999), and Pavon et al. (2006) used Western blotting to demonstrate that it is the truncated form, ΔFosB, which is up-regulated preferentially in LID. Importantly, Andersson et al. (1999) provided causal evidence linking ΔFosB upregulation to LID, as intrastriatal infusion of fosB/ΔfosB antisense oligonucleotide inhibited the development of LID. Double in situ hybridization studies using 35S- and digoxigenin-labeled riboprobes showed that FosB/ΔFosB induction occurred in striatal neurons that contain D1R (Pavon et al., 2006), extending previous findings showing co-expression of FosB/ΔFosB with prodynorphin (Andersson et al., 1999). Interestingly, ΔFosB expression is inversely correlated with the abundance of dopamine fibers in the striatum and directly correlated with the severity of dyskinesia (Pavon et al., 2006), and it is blocked by pharmacological or genetic inactivation of the D1 receptor (Westin et al., 2007; Darmopil et al., 2009). The correlation between ΔFosB induction and the severity of LID has been corroborated in the MPTP macaque model of Parkinson’s disease, in which overexpression of a truncated form of JunD that acts as a dominant negative inhibitor of ΔFosB markedly reduces LID (Berton et al., 2009). Overall, these findings show that LID is related to specific D1R-dependent changes in gene expression in striatonigral MSNs neurons that selectively express dynorphin and D1 receptor.
The fact that L-DOPA administration to naive animals does not induce ΔFosB expression and LID suggests that denervation-induced supersensitivity of dopamine D1 receptors is at the core of the mechanism leading to LID. Although D1 receptor supersensitivity is well documented in Parkinson’s disease, D1 receptor expression does not change dramatically after nigrostriatal lesion or repeated L-DOPA administration. However, D1 receptors may be redistributed preferentially to the postsynaptic membrane and cytoplasmic compartments involved in D1R trafficking during chronic L-DOPA administration (Muriel et al., 1999; Guigoni et al., 2007; Berthet et al., 2009). Moreover, D1 receptor supersensitivity in 6-OHDA rats and Parkinson’s disease patients is related to increased levels of Gαolf (Corvol et al., 2004). Indeed, the ability of D1-like receptor agonists to recruit G proteins in striatal tissue is linearly related to dyskinesia scores in MPTP-lesioned monkeys (Aubert et al., 2005).
There is also evidence that the resulting increase in cAMP production is translated into effects on targets of the cAMP-dependent protein kinase (PKA), like the Thr34 site of DARPP-32. The striatum contains higher levels of phospho[Thr34]-DARPP-32 in dyskinetic rats than in 6-OHDA rats that have not developed dyskinesia under L-DOPA treatment (Picconi et al., 2003). Moreover, levels of phospho[Thr34]-DARPP-32 correlate with the severity of dyskinesia in a mouse model of LID (Santini et al., 2007). Similar changes in DARPP-32 phosphorylation have more recently been found in MPTP-lesioned monkeys that have developed LID (Santini et al., 2010). Importantly, selective disruption of the DARPP-32 gene in striatonigral neurons reduces LID, whereas a similar gene disruption in striatopallidal neurons has no effect (Bateup et al., 2010).
Phosphorylation at another specific target of PKA, the Ser845 in the GluR1 subunit of AMPA receptors, is also increased in highly dyskinetic mice and MPTP-lesioned monkeys, but not in control parkinsonian animals that have received L-DOPA but did not develop substantial dyskinesia (Santini et al., 2007, 2010). Moreover, there is evidence that phosphorylation at Ser845 in GluR1 is modulated by DARPP-32. Phospho[Thr34]-DARPP-32 is a potent inhibitor of protein phosphatase-1 (PP-1), so, dephosphorylation of targets of PP-1 should be reduced in LID. In agreement, DARPP-32 knockouts not only exhibit reduced LID but also show reduced phosphorylation at Ser845 in GluR1 (Santini et al., 2007).
Molecular changes related to LID have been detected in other signaling cascades associated with dopamine D1/D5 receptor function, like the extracellular signal-regulated kinase (ERK) 1/2 cascade (Figure 3). Indeed, D1R stimulation induces ERK1/2 phosphorylation in the dopamine-intact (Valjent et al., 2000; Mazzucchelli et al., 2002; Brami-Cherrier et al., 2005) and dopamine-depleted brain (Gerfen et al., 2002). More recent work established a correlation between ERK1/2 phosphorylation and LID (Pavon et al., 2006; Santini et al., 2007; Westin et al., 2007; Darmopil et al., 2009) and showed that pharmacological inhibition of the ERK1/2 signaling cascade reduces LID (Santini et al., 2007; Schuster et al., 2008; Lindgren et al., 2009). Induction of ERK1/2 phosphorylation by L-DOPA is blocked, in parallel with LID, by D1/D5 receptor antagonists (Westin et al., 2007; Santini et al., 2009a), and by genetic deletion of the specific D1 receptor subtype (Darmopil et al., 2009), further supporting the requirement for the D1R subtype and the involvement of the direct striatal outflow pathway. Finally, genetic interventions aimed at reducing the activity of Ras-GRF1, a neuronal specific activator of ERK signaling that mediates striatal ERK1/2 phosphorylation in response to D1/D5R agonists (Fasano et al., 2009), reduced LID in mice and monkeys (Fasano et al., 2010). As DARPP-32 knockouts also show reduced ERK1/2 phosphorylation, excessive inhibition of PP-1 by phospho[Thr34]-DARPP-32 may contribute to the higher levels of phospho-ERK1/2 in LID (Santini et al., 2007).
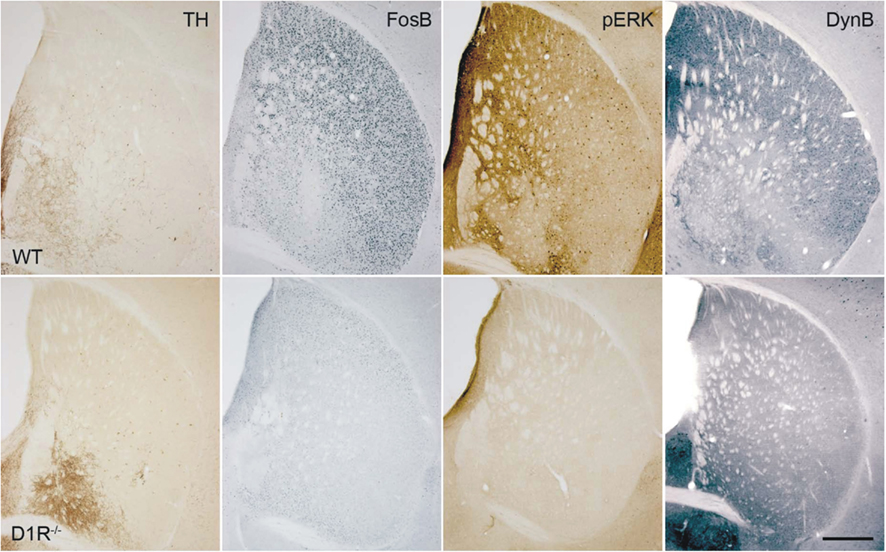
Figure 3. Genetic inactivation of dopamine D1 receptor blocks striatal molecular changes induced by chronic L-DOPA administration in hemiparkinsonian mice. Photomicrographs of adjacent coronal striatal sections from WT (top) and D1R−/− (bottom) hemiparkinsonian mice treated with L-DOPA. Sections are immunostained for TH, FosB, Dynorphin, and p-ERK. Chronic L-DOPA treatment induced marked FosB, Dynorphin, and p-ERK expression in the striatal areas that are devoid of TH-immunoreactive fibers in WT mice, but not in the striatum of D1R−/− mice. Scale bar = 500 μm.
Enhanced D1R Signaling Induces Changes in Translation and Transcription
It has been possible to follow the signaling cascade involved in LID downstream to translation and transcription mechanisms.
In the denervated striatum, but not in the normal striatum, L-DOPA administration induces phosphorylation of S6 kinase at a site specifically regulated by the mammalian target for rapamycin (mTOR). S6 kinase phosphorylates S6, a ribosomal protein, and the abundance of phosphorylated S6 kinase and S6 is linearly related to the severity of LID. L-DOPA-induced S6 phosphorylation takes place in MSNs of the direct pathway and is blocked by D1/D5R antagonists and rapamycin. Similar results were obtained with another target of mTOR, eukaryotic translation initiation factor 4E-binding protein (4E-BP), which when phosphorylated, allows formation of a multiprotein complex with a critical role in translation. Finally, inhibitors of the ERK signaling cascade prevent phosphorylation of targets of mTOR, and rapamycin, an inhibitor of mTOR signaling, reduces the development of LID (Santini et al., 2009b).
Changes downstream of the ERK1/2 signaling cascade have been detected in the MSN nucleus. Thr581 of the mitogen and stress-activated kinase-1 (MSK-1), a nuclear target of ERK1/2, and histone H3, which is modified in response to phospho[Thr581]MSK-1, are persistently activated in MSNs of the direct pathway in rodents that develop LID (Darmopil et al., 2009; Santini et al., 2009a). Induction of phospho-MSK-1 by L-DOPA was completely inhibited in animals pretreated with D1-like antagonists (Westin et al., 2007). Moreover, D1-like antagonists (Santini et al., 2009a) or genetic deletion of the D1R (Darmopil et al., 2009), reduce histone H3 activation in parallel with LID. Modified histone H3 is co-expressed with dynorphin and FosB/ΔFosB in MSNs in highly denervated striatal areas in the mouse model of LID, further supporting that histone H3 modification is related to transcriptional changes mediating LID (Darmopil et al., 2009).
Finally, some changes in transcription may be related to a switch in gene expression control from cAMP-response element binding transcription factor (CREB) dominance to ΔFosB dependence. In the intact striatum, prodynorphin transcription is mediated by CREB, but L-DOPA induction of prodynorphin transcription in parkinsonian rats is under the control of ΔFosB and JunD (Andersson et al., 2001).
Changes in D2/D3 Receptor-Mediated Signaling May Contribute to the Expression of LID
Different lines of evidence suggest that D2R modulation of striatopallidal neurons is not necessary for development of LID. In D2R knockouts with 6-OHDA striatal lesions LID develops with similar time course and intensity as in controls (Darmopil et al., 2009). Moreover, selective genetic deletion of DARPP-32 in striatopallidal neurons has no effect on the development of LID (Bateup et al., 2010). The above conclusion is reinforced by the finding that there is a very low risk of dyskinesia associated with administration of D2-like receptor agonists to patients or to drug-naïve animals with nigrostriatal lesions (Delfino et al., 2004; Holloway et al., 2004; Lundblad et al., 2005; Rascol et al., 2006). By contrast, D2R antagonists that produce tardive dyskinesia in patients, induce FosB/ΔFosB expression in striatopallidal neurons (Hiroi and Graybiel, 1996; Hiroi et al., 2002; Grande et al., 2004).
Thus, it seems likely that D2R subtype regulation of striatopallidal neurons may modulate LID expression. This is illustrated in a study by Gold et al. (2007) aimed at modulating D2 receptor signaling in LID. Modifying the expression of regulator of G-protein signaling (RSG) 9-2, a protein that inhibits D2 receptor interaction with G proteins, regulated LID: RSG 9-2 knockouts showed increased susceptibility to LID, whereas overexpression of RSG 9-2 reduced LID in MPTP-lesioned monkeys, and 6-OHDA rats. However, the effects of modifying RSG 9-2 expression on motor function were not specific to LID, suggesting that striatal D2R subtype signaling allows an increase in motor output that results in more severe dyskinesia when the main LID signaling cascade, the direct striatonigral pathway, is activated.
On the other hand, the D3R may directly modulate the main LID signaling cascade. Repeated stimulation of D1R by L-DOPA or dopamine agonists in 6-OHDA rats leads to a progressive increase in the expression of D3R in direct pathway MSNs, which parallels sensitization of the rotational response to L-DOPA (Bordet et al., 1997, 2000; see however Hurley et al., 1996; Quik et al., 2000). Similar results were obtained in MPTP-lesioned monkeys that have developed LID; further study showed that pharmacological reduction of D3R activity reduced LID (Bézard et al., 2003; see however Kumar et al., 2009; Mela et al., 2010). When co-expressed in the same cell, D1R and D3R interact synergistically in the membrane, with D3R stimulation enhancing D1-like agonist affinity and capacity to stimulate adenylyl cyclase (Fiorentini et al., 2008; Marcellino et al., 2008). Recent work suggests that direct cross-talk between D1R and D3R in the membrane reduces internalization of D1R in response to dopamine, a mechanism that may normally prevent excessive D1R activation (Berthet et al., 2009). It is worth considering whether the effects of D3R preferring drugs in LID result from their ability to modulate D1R-D3R membrane interactions rather than from their antagonist/partial agonist action at the ligand binding site.
LID is Associated with Altered Synaptic Plasticity in the Striatum
Although a causal connection has been established between enhanced D1R signaling via ERK and LID, the functional changes linking these molecular events to behavior remain poorly understood. Once established, LID is very difficult to reverse and very easy to reestablish even after a long L-DOPA-free period (Nutt, 2000). Because of this, it has been suggested that long term changes in brain circuits occur during chronic L-DOPA therapy (Calabresi et al., 2000a) and dopamine denervation (Darmopil et al., 2009). ERK’s established association with functional and structural forms of synaptic plasticity is consistent with this (Thomas and Huganir, 2004).
The critical role played by dopamine in corticostriatal long term potentiation (LTP) and LTD has been noticed 20 years ago (Calabresi et al., 1992). Early work suggested that D1R stimulation is necessary to induce LTP, whereas D2R stimulation is required for LTD (Calabresi et al., 1992, 2000b; Centonze et al., 1999, 2003; Tang et al., 2001). The mechanisms involved in LTP and LTD in direct and indirect-pathway MSNs have not been studied until very recently, with the availability of BAC transgenic mice expressing fluorescent proteins in specific neuronal populations (Wang et al., 2006; Kreitzer and Malenka, 2007; Shen et al., 2008). LTD can be readily induced in indirect pathway-MSNs via D2R-regulated endocannabinoid release. Moreover, this LTD is disrupted in animal models of Parkinson’s disease and can be restored by D2-like agonists and CB1 receptor stimulation (Kreitzer and Malenka, 2007; Shen et al., 2008). What happens in the direct pathway neurons is less clear. One group could not induce LTD in direct pathway MSNs (Kreitzer and Malenka, 2007). However, Wang et al. (2006) and Shen et al. (2008) have been able to induce LTD in D1R–MSNs with different stimulation protocols. In rodent models of Parkinson’s disease, LTD is favored over LTP in D1R–MSNs, but protocols aimed at inducing LTD result in LTP in the presence of D1-like agonists (Shen et al., 2008). The issue remains controversial and has been addressed in recent excellent reviews (Kreitzer and Malenka, 2008; Surmeier et al., 2009; Lovinger, 2010).
Concerning LID, there is evidence showing its association with altered functional plasticity in corticostriatal synapses. A pioneering slice study by Picconi et al. (2003) showed that excitatory synapses in MSNs show normal LTP in L-DOPA-treated 6-OHDA rats, and that depotentiation is selectively impaired in rats that have developed LID. As corticostriatal depotentiation is prevented by D1-like agonists, it seems likely that enhanced D1 signaling impedes depotentiation in dyskinetic rats. A deficit in depotentiation could lead to progressive enhancement of corticostriatal synaptic efficacy in the direct pathway, resulting in uncontrolled positive feedback to motor cortical areas (Figure 4). Recent in vivo studies support the idea that depotentiation of direct pathway MSNs is altered in 6-OHDA rats under chronic L-DOPA therapy (Belujon et al., 2010). Unfortunately, no study has yet taken advantage of engineered mice expressing florescent proteins in D1R- or D2R–MSNs to establish how L-DOPA impacts on plasticity in each striatal output pathway.
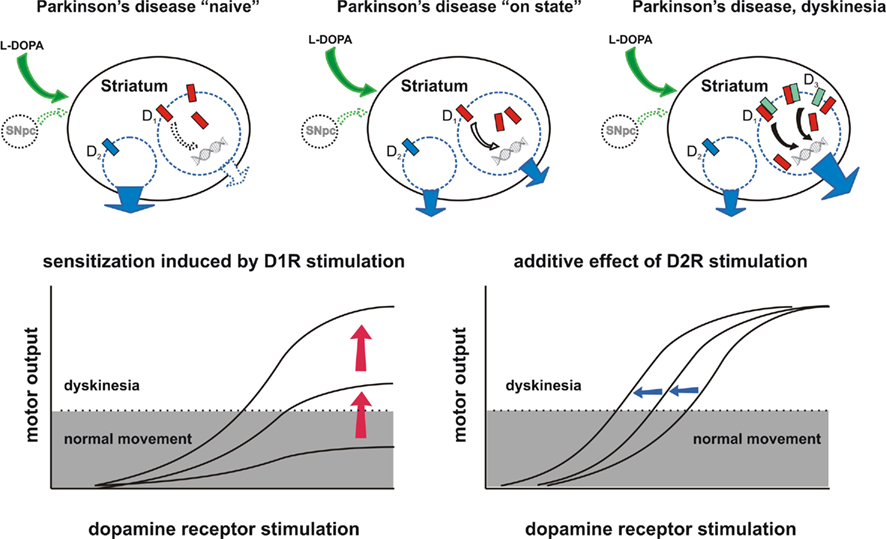
Figure 4. Mechanisms proposed to underlie LID. Top row diagrams: sensitivity of direct and indirect-pathway MSNs to dopamine receptor stimulation in a naive patient (left), in a patient that is finding benefit under L-DOPA therapy (middle), and in a dyskinetic patient (right). The naive parkinsonian brain has hyperactive and hypoactive indirect and direct pathways respectively, and may respond weakly to a first challenge with L-DOPA. Repeated D1 receptor stimulation may reinstate a balance between the direct and indirect pathways and produce clinical benefit (middle). In advanced stages of the disease (right), the complete loss of dopamine buffering capacity results in pulsatile stimulation of D1 receptors, more surface expression of D1 receptors, induction of D3 receptor expression in direct-pathway MSNs, and amplification of D1 receptor signaling, leading to dyskinesia. Bottom row diagrams: repeated pulsatile stimulation of D1 receptors in direct-pathway MSNs produces a multiplicative gain in the motor response to L-DOPA, which is reflected in a change of the slope and an increase in the maximal attainable response (left). This would progressively impel the patient to the threshold for induction of dyskinesia. D2 receptor stimulation in indirect-pathway MSNs may not drive the patient through the dyskinesia threshold, but may have an additive effect if the patient is above threshold (right). Moreover, once direct-pathway MSNs have crossed threshold, stimulation of over-expressed D3 receptors may also have multiplicative gain effects.
The ultimate mechanisms that block depotentiation have not been clarified; in fact, depotentiation itself is poorly understood. It is well known that the D1R subtype has a facilitatory effect on NMDA receptors in MSNs (Cepeda and Levine, 1998). The interaction with the NMDA receptor occurs through different mechanisms, including PKA/DARPP-32-mediated signaling and direct protein–protein interactions in the postsynaptic membrane, which result in enhanced NMDA receptor currents and NMDA receptor redistribution across dendritic spine compartments (Cepeda and Levine, 2006). In animals with LID, NMDA receptors showing altered subunit composition form complexes with D1R and these complexes are trafficked abnormally between the postsynaptic membrane and intracellular compartments or extrasynaptic membrane sites (Fiorentini et al., 2006; Gardoni et al., 2006). However, it is not clear how such receptor reorganization is related to alterations in synaptic plasticity or to other functional changes associated with LID.
An aspect of NMDA receptor involvement in LID that has not received much attention is that it also activates the ERK1/2 cascade (Thomas and Huganir, 2004). However, NMDA receptor antagonists do not modify ERK phosphorylation induced by D1-like agonists in the dopamine-depleted striatum (Gerfen et al., 2002; Rylander et al., 2009). In contrast, mGluR5 antagonists attenuate LID and reduce ERK phosphorylation (Mela et al., 2007; Rylander et al., 2009), suggesting that mGluR5 plays a more important control over ERK1/2 than the NMDA receptor in the parkinsonian condition. Thus, in LID, D1 receptor activation may result in exaggerated ERK1/2 phosphorylation through multiple mechanisms, including reduced activity of PP-1, enhanced activity of Ras-GRF1, enhanced NMDA receptor, and mGluR5 activation.
In addition to alterations in functional plasticity, indirect evidence suggests that LID is associated with structural changes in neurons. Activity-regulated cytoskeletal-associated protein (arc) is markedly up-regulated in direct pathway MSNs in rats with 6-OHDA lesion treated chronically with L-DOPA, though it is not clear that this change is selectively associated with the development of LID (Sgambato-Faure et al., 2005). Although changes in arc expression and function are frequently involved in synaptic remodeling, further work is necessary to assess such changes in LID.
LID is Associated with Widespread Functional and Structural Changes in the Basal Ganglia
There is evidence that angiogenesis and changes in the blood brain barrier take place in parallel with LID in rats with nigrostriatal lesion. This is manifest in enhanced proliferation of endothelial cells, higher total microvessel length, and loss of blood brain barrier markers across most basal ganglia structures in the denervated hemisphere (Westin et al., 2006). The effect of L-DOPA on vessels and behavior was mimicked by administration of D1-like agonists and blocked by D1-like antagonists, with drugs acting on D2 family receptors having little or opposite effects. Moreover, pharmacological inhibition of the ERK1/2 signaling cascade reduced both LID and the microvascular changes induced by L-DOPA (Lindgren et al., 2009). However, these changes in the blood brain barrier were not corroborated by functional methods in a primate model of LID (Astradsson et al., 2009), so more work is needed to understand the functional significance of these findings. Changes in the microvasculature may be secondary to the higher metabolic demands imposed by chronic D1R receptor stimulation in rats with nigrostriatal lesion (Trugman and Wooten, 1986, 1987) or result from more direct actions of dopamine agonists on blood vessels (Hirano et al., 2008).
Widespread changes in metabolism and blood flow have been detected in LID. Rat pharmacological MRI studies revealed a correlation between the severity of dyskinesia induced by D1 receptor stimulation and the magnitude of blood oxygenation level-dependent signal (BOLD) in the striatum and motor cortex (Delfino et al., 2007). Moreover, in 6-OHDA rats, the BOLD response to D1 receptor stimulation showed sensitization with repeated drug administration in both striatum and motor cortex (Delfino et al., 2007). Similarly, nuclear magnetic resonance spectroscopy (NMR) studies in reserpinized animals showed increased glutamatergic activity after L-DOPA treatment, associated with increases in striatal c-fos expression and motor activity. These responses are blocked in D1R knock out animals (Rodrigues et al., 2007), suggesting the involvement of D1 receptor. Overall, these results are consistent with clinical findings. In patients with PD, the severity of involuntary arm and hand movements correlates with increases in blood flow measured with PET in the contralateral motor cortex and basal ganglia (Brooks et al., 2000; Hirano et al., 2008). In addition, L-DOPA-induced over-activation of the motor cortex is not observed in drug-naïve hemiparkinsonian patients (Buhmann et al., 2003). An interesting observation in pharmacological MRI studies is that a D3R-preferring agonist induces regional cerebral blood volume changes similar to those induced by D1-like agonists, but only if animals have developed LID. In the absence of LID, the D3-preferring agonist has no effect (Sánchez-Pernaute et al., 2007), further suggesting that D3R stimulation acts by potentiating D1R signaling in LID.
Importantly, it seems that metabolic changes are not restricted to motor areas. A 2-deoxyglucose accumulation study conducted in MPTP-lesioned non-human primates revealed increased metabolic activity in limbic and cognitive territories of the striatum only in L-DOPA-treated animals that had developed dyskinesia (Guigoni et al., 2005).
Less is known about the electrophysiological changes associated with the widespread metabolic effects occurring in LID. A classic study by Filion et al. (1991) revealed a parallel between the development of dyskinesia after repeated administration of apomorphine to MPTP-lesioned monkeys and changes in neuronal activity in the globus pallidus. Apomorphine, a mixed D1/D2 agonist with a short plasma half-life that is known to be very dyskinetogenic in patients, did not induce dyskinesia and had small effects on the activity of pallidal neurons in monkeys before MPTP lesioning. After induction of parkinsonism, however, repeated administration of apomorphine led to growing motor side effects and growing changes in pallidal activity. More specifically, dyskinesia was associated with a marked inhibition and reduction of burst firing in the internal segment of the globus pallidus (Filion et al., 1991). This finding has been corroborated (Papa et al., 1999) and extended by showing that dyskinesia induced by D1-like agonists in the MPTP primate model has similar electrophysiological correlates (Boraud et al., 2001). Moreover, a limited number of recordings performed in patients undergoing functional neurosurgery also suggest that excessive inhibition of output neurons is involved in dyskinesia (Merello et al., 1999; Lozano et al., 2000). Overall, the data are consistent with the proposal that the gain of direct pathway striatal neurons increases in LID (Figure 4).
The studies by Filion et al. (1991) and Boraud et al. (2001) show that D2-like agonists induce dyskinesia and changes in pallidal activity similar to those produced by apomorphine. In both studies the same monkeys were repeatedly challenged with a mixed D1/D2 agonist and a selective D2-like agonist, thus the D2 agonist effects were taking place in a primed brain. Moreover, both apomorphine and D2-like receptor agonists induced increases in activity in the external pallidal segment, which could have contributed to reducing neuronal firing in the internal pallidum. Thus, D2/D3 agonist-induced dyskinesia probably shares circuit mechanisms with LID.
Although the data are consistent with the view that increased gain of direct pathway striatal neurons produces excessive inhibition of basal ganglia output, there is still no direct evidence for this. Indeed, a recent study in drug-naïve 6-OHDA rats shows that D1-like receptor stimulation produces a strong induction of immediate early genes in the striatum without restoring the response of direct pathway MSNs to cortical stimulation to control levels (Ballion et al., 2009). The possibility remains that the hypothetical increase in the gain of corticostriatal connections requires repeated exposure to L-DOPA or D1-like agonists. However, in severely denervated 6-OHDA rats, a first challenge with L-DOPA can induce dyskinesia (see Onofrio et al., 1998, for similar findings in patients). Further work correlating the activity of direct- and indirect-pathway striatal MSNs to the degree of dopamine denervation and dyskinesia scores is necessary to determine the circuit mechanisms involved in LID.
Are the Signaling Changes Occurring in LID Unique to the Parkinsonian Brain?
L-DOPA does not induce dyskinesia unless dopaminergic neurons in the substantia nigra and striatal dopaminergic markers fall by more than 90% (see for example Winkler et al., 2002). More recent work correlating the spatial pattern of striatal denervation with the molecular changes associated to LID showed that FosB/ΔFosB and prodynorphin induction is restricted to striatal areas where the density of dopaminergic fibers drops below 5% of control levels (Pavon et al., 2006; Darmopil et al., 2009). Thus, denervation supersensitivity plays a pivotal role in the development of LID by allowing the activation of non-canonical signaling cascades by D1R stimulation, suggesting this is a mechanism unique to LID. However, there is evidence that similar signaling mechanisms are activated in dopamine-intact animals during sensitization to psychostimulants and drugs of abuse. For instance, there is evidence that a D1R-activated signaling cascade involving DARPP-32 and ERK1/2 phosphorylation, induction of ΔFosB, and prodynorphin expression, modifications of histone H3, a switch in gene expression control from CREB-dependent to ΔFosB-dependent, and impaired LTD of corticostriatal synapses, is causally involved in psychostimulant sensitization and drug addiction (Moratalla et al., 1996a; Kelz et al., 1999; Valjent et al., 2000; McClung and Nestler, 2003; Brami-Cherrier et al., 2005; Stipanovich et al., 2008; Fasano et al., 2009; Kasanetz et al., 2010).
Although a point-by-point comparison between drug addiction-related and LID-related mechanisms is beyond the scope of this review, it seems clear that a large part of the signaling cascade involved in LID can be activated in an intact brain by cocaine and psychostimulants, although with much less intensity and in considerably fewer striatal neurons (Moratalla et al., 1996b; Pavon et al., 2006). This raises the question why L-DOPA cannot activate it when the brain dopamine systems are intact. The most plausible explanation is that pulsatile stimulation of D1 receptors is sufficient to activate the non-canonical components of the signaling cascade, but that the buffering capacity of even a small number of dopaminergic terminals precludes L-DOPA from producing the large variations in dopamine concentration in the extracellular space that may be necessary to drive the cascade. Indeed, L-DOPA induces slow and moderate changes in dopamine concentration in the extracellular space when presynaptic mechanisms are intact but produces faster and more marked changes in rodent models of Parkinson’s disease (Carta et al., 2006; Lindgren et al., 2010). Moreover, changes in dopamine concentration are more marked in patients with more severe parkinsonism (de la Fuente-Fernández et al., 2004). This is consistent with a recent report showing that L-DOPA does not induce dyskinesia in rats in which dopamine synthesis was knocked down but presynaptic dopaminergic terminals are preserved (Ulusoy et al., 2010). Importantly, these rats show dyskinesia in response to apomorphine, a short plasma half-life D1/D2 agonist whose effect over the dopamine receptors is not buffered by presynaptic terminals, indicating that the postsynaptic mechanisms involved in LID are functional. Moreover, the rats still do not show dyskinesia during L-DOPA administration after receiving apomorphine challenges that have been able to induce it (Ulusoy et al., 2010). In this sense, striatal TH+ neurons that appear after severe denervation and L-DOPA treatment are not able to provide enough buffering capacity to reduce dyskinesias (Darmopil et al., 2008). Overall, the data indicate that a good buffering capacity of dopamine concentration in the extracellular space may preclude sensitization to L-DOPA and attenuate the activation of already sensitized postsynaptic dyskinesia machinery.
This is not to say that supersensitivity of D1R signaling does not contribute to LID. Supersensitivity may explain why the molecular changes associated to LID are several-fold more widespread and intense than those induced by psychostimulants. However, an almost complete loss of presynaptic dopaminergic terminals (implying a loss of buffering capacity) is a prerequisite for D1R signaling supersensitivity (Pavon et al., 2006; Darmopil et al., 2009). Thus, D1R supersensitivity and the loss of buffering capacity conspire together in LID.
Does LID Share Mechanisms with Long Term Memory?
Recent findings linking D1 receptor stimulation to long term memory storage further suggest that the signaling cascade involved in LID is not unique to the parkinsonian brain, or even to the striatum. Pharmacological studies show that blockade of D1-like receptors at the time of encoding reduces the persistence of hippocampal-dependent memories (O’Carroll et al., 2006; Bethus et al., 2010). Also, intrahippocampal administration of D1-like antagonists several hours after encoding blocks retention (Bernabeu et al., 1997). An important recent study shows that a long-lasting fear memory disappears if hippocampal D1 receptors are blocked 12 h after encoding, whereas a D1-like agonist applied at the same post-training time turns a labile memory into a persistent one (Rossato et al., 2009). Thus, sequential “waves” of dopamine at different time points are critical for long term memory storage in the hippocampus.
These pharmacological observations are backed up by studies in D1R knockout mice showing that this receptor subtype is essential for spatial memory tasks (El-Ghundi et al., 1999). Moreover, D1 knockouts show spatial learning and associative learning deficits concurrent with a reduction in LTP (Granado et al., 2008; Ortiz et al., 2010). Furthermore, mice in which hippocampal D1 receptors were knocked down with a small interfering RNA show deficits in spatial learning, fear learning, classical conditioning, and LTP, ruling out effects of developmental adaptations in the D1 knockouts (Ortiz et al., 2010). Importantly, it has been shown that D1R are important for both early- and late-LTP, indicating that the D1R subtype is involved in different kinds of memory, including new protein synthesis-independent and -dependent memory (Granado et al., 2008; Ortiz et al., 2010). Further studies in vivo showed that intact D1R are required for the acquisition of associative learning and for the endogenous, non-stimulated, increase in synaptic strength produced in the hippocampus during learning (Ortiz et al., 2010), supporting the important role of D1R in learning and memory.
Elucidation of the signaling cascade mediating the role of D1R in memory formation and retention is challenging, given the likely contribution of several other neurotransmitters and modulators that can share signaling mechanisms with dopamine. However, there is evidence that EKR1/2 phosphorylation (Bekinschtein et al., 2008) and histone H3 acetylation (Levenson et al., 2004), as well as Erg1 and Arc (Granado et al., 2008; Ortiz et al., 2010) are involved in the retention of hippocampal memories. Thus, at least some components of the signaling cascade implicated in LID are also active in the hippocampus at times when D1 receptor stimulation contributes to memory persistence.
An intriguing possibility is that phasic D1R stimulation is critical for inducing components of the signaling cascade that produce physiological increases in synaptic strength. Even in the striatum, where tonic dopamine levels are relatively high, it is believed that the low affinity of D1R is not activated except during physiological surges of dopamine induced by burst firing of the dopaminergic neurons (Gonon, 1997). Thus, physiological surges of dopamine in the extracellular space could be involved in D1R promotion of memory storage in different brain areas. Longer-lasting and more marked non-physiological D1R stimulation induced by cocaine and psychostimulants in the dopamine-intact brain, or by L-DOPA in the brain that has lost the buffering capacity of striatal dopaminergic terminals, may induce abnormal plasticity and behavioral alterations.
Implications for Parkinson’s Disease Therapy
Why does pulsatile activation of the D1 receptor signaling cascade induce dyskinesia in Parkinson’s disease instead of other behavioral abnormalities like addictive behaviors or impulse control disorders? The distinctive behavior resulting from pulsatile dopaminergic stimulation in the parkinsonian brain may reflect a preferential involvement of striatal motor areas, within which the extreme but heterogeneous loss of dopamine buffering capacity may produce a patchy pattern of molecular and functional postsynaptic changes (Darmopil et al., 2009). This non-physiological patchy pattern of plasticity would distort the flow of cortical information through the direct basal ganglia pathway, giving rise to abnormal movements instead of behavioral stereotypies. In this context, over-activation of dopamine signaling cascades in areas that are not so extensively denervated in Parkinson’s disease, like the ventral striatum and frontal cortex, may lead to behavioral abnormalities that are not unique to the parkinsonian brain, like addiction to L-DOPA, pathological gambling, and other impulse control disorders (Dagher and Robbins, 2009). This hypothesis can be interrogated by studying the behavioral consequences of intermittent stimulation of dopamine receptors after manipulating dopamine buffering capacity in specific brain areas.
This view opens questions about future directions in Parkinson’s disease therapy. If the postsynaptic signaling involved in LID is not unique but part of physiological rewarding and memory storage mechanisms, targeting the non-canonical components of the signaling cascade may have undesirable consequences on behavior. In this context, increasing L-DOPA buffering capacity in the motor district of the striatum emerges as a more etiologically- and physiologically-rooted approach to the treatment of Parkinson’s disease, of which the concept of “continuous dopaminergic stimulation” (Olanow et al., 2006) can be seen as an antecedent.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. S. Darmopil and Miss I. Ruiz De Diego for their help with the figures and art work. Funded by grants from the Spanish Ministerios de Ciencia e Innovación y de Sanidad y Política Social, ISCIII, ref number, BFU2010-20664, PNSD, RedRTA (RD06/0001/1011), and CIBERNED to RM. MGM is supported by FONCYT PICT 2008 2205, CONICET PIP 77, and UBACYT M562.
References
Andersson, M., Hilbertson, A., and Cenci, M. A. (1999). Striatal fosB expression is causally linked with L-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson’s disease. Neurobiol. Dis. 6, 461–474.
Andersson, M., Konradi, C., and Cenci, M. A. (2001). cAMP response element-binding protein is required for dopamine-dependent gene expression in the intact but not the dopamine-denervated striatum. J. Neurosci. 21, 9930–9943.
Astradsson, A., Jenkins, B. G., Choi, J. K., Hallett, P. J., Levesque, M. A., McDowell, J. S., Brownell, A. L., Spealman, R. D., and Isacson, O. (2009). The blood-brain barrier is intact after levodopa-induced dyskinesias in parkinsonian primates – evidence from in vivo neuroimaging studies. Neurobiol. Dis. 35, 348–351.
Aubert, I., Guigoni, C., Håkansson, K., Li, Q., Dovero, S., Barthe, N., Bioulac, B. H., Gross, C. E., Fisone, G., Bloch, B., and Bezard, E. (2005). Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Ann. Neurol. 57, 17–26.
Ballion, B., Frenois, F., Zold, C. L., Chetrit, J., Murer, M. G., and Gonon, F. (2009). D2 receptor stimulation, but not D1, restores striatal equilibrium in a rat model of Parkinsonism. Neurobiol. Dis. 2009 35, 376–384.
Bateup, H. S., Santini, E., Shen, W., Birnbaum, S., Valjent, E., Surmeier, D. J., Fisone, G., Nestler, E. J., and Greengard, P. (2010). Distinct subclasses of medium spiny neurons differentially regulate striatal motor behaviors. Proc. Natl. Acad. Sci. U.S.A. 107, 14845–14850.
Bekinschtein, P., Cammarota, M., Katche, C., Slipczuk, L., Rossato, J. I., Goldin, A., Izquierdo, I., and Medina, J. H. (2008). BDNF is essential to promote persistence of long-term memory storage. Proc. Natl. Acad. Sci. U.S.A. 105, 2711–2716.
Belujon, P., Lodge, D. J., and Grace, A. A. (2010). Aberrant striatal plasticity is specifically associated with dyskinesia following levodopa treatment. Mov. Disord. 25, 1568–1576.
Bernabeu, R., Bevilaqua, L., Ardenghi, P., Bromberg, E., Schmitz, P., Bianchin, M., Izquierdo, I., and Medina, J. H. (1997). Involvement of hippocampal cAMP/cAMP-dependent protein kinase signaling pathways in a late memory consolidation phase of aversively motivated learning in rats. Proc. Natl. Acad. Sci. U.S.A. 94, 7041–7046.
Berthet, A., Porras, G., Doudnikoff, E., Stark, H., Cador, M., Bezard, E., and Bloch, B. (2009). Pharmacological analysis demonstrates dramatic alteration of D1 dopamine receptor neuronal distribution in the rat analog of L-DOPA-induced dyskinesia. J. Neurosci. 29, 4829–4835.
Berton, O., Guigoni, C., Li, Q., Bioulac, B. H., Aubert, I., Gross, C. E., Dileone, R. J., Nestler, E. J., and Bezard, E. (2009). Striatal overexpression of DeltaJunD resets L-DOPA-induced dyskinesia in a primate model of Parkinson disease. Biol. Psychiatry 66, 554–561.
Bethus, I., Tse, D., and Morris, R. G. (2010). Dopamine and memory: modulation of the persistence of memory for novel hippocampal NMDA receptor-dependent paired associates. J. Neurosci. 30, 1610–1618.
Bézard, E., Ferry, S., Mach, U., Stark, H., Leriche, L., Boraud, T., Gross, C., and Sokoloff, P. (2003). Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat. Med. 9, 762–767.
Blanchet, P. J., Gomez-Mancilla, B., Di Paolo, T., and Bédard, P. J. (1995). Is striatal dopaminergic receptor imbalance responsible for levodopa-induced dyskinesia? Fundam. Clin. Pharmacol. 9, 434–442.
Boraud, T., Bezard, E., Bioulac, B., and Gross, C. E. (2001). Dopamine agonist-induced dyskinesias are correlated to both firing pattern and frequency alterations of pallidal neurones in the MPTP-treated monkey. Brain 124, 546–557.
Bordet, R., Ridray, S., Carboni, S., Diaz, J., Sokoloff, P., and Schwartz, J. C. (1997). Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to levodopa. Proc. Natl. Acad. Sci. U.S.A. 94, 3363–3367.
Bordet, R., Ridray, S., Schwartz, J. C., and Sokoloff, P. (2000). Involvement of the direct striatonigral pathway in levodopa-induced sensitization in 6-hydroxydopamine-lesioned rats. Eur. J. Neurosci. 12, 2117–2123.
Brami-Cherrier, K., Valjent, E., Hervé, D., Darragh, J., Corvol, J. C., Pages, C., Arthur, S. J., Girault, J. A., and Caboche, J. (2005). Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J. Neurosci. 25, 11444–11454.
Brooks, D. J., Piccini, P., Turjanski, N., and Samuel, M. (2000). Neuroimaging of dyskinesia. Ann. Neurol. 47(Suppl. 1), S154–S158.
Buhmann, C., Glauche, V., Stürenburg, H. J., Oechsner, M., Weiller, C., and Büchel, C. (2003). Pharmacologically modulated fMRI – cortical responsiveness to levodopa in drug-naive hemiparkinsonian patients. Brain 126, 451–461.
Calabresi, P., Giacomini, P., Centonze, D., and Bernardi, G. (2000a). Levodopa-induced dyskinesia: a pathological form of striatal synaptic plasticity? Ann. Neurol. 47(Suppl. 1), S60–S68.
Calabresi, P., Gubellini, P., Centonze, D., Picconi, B., Bernardi, G., Chergui, K., Svenningsson, P., Fienberg, A. A., and Greengard, P. (2000b). Dopamine and cAMP-regulated phosphoprotein 32 kDa controls both striatal long-term depression and long-term potentiation, opposing forms of synaptic plasticity. J. Neurosci. 20, 8443–8451.
Calabresi, P., Maj, R., Pisani, A., Mercuri, N. B., and Bernardi, G. (1992). Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J. Neurosci. 12, 4224–4233.
Carta, M., Lindgren, H. S., Lundblad, M., Stancampiano, R., Fadda, F., and Cenci, M. A. (2006). Role of striatal L-DOPA in the production of dyskinesia in 6-hydroxydopamine lesioned rats. J. Neurochem. 96, 1718–1727.
Cenci, M. A., and Konradi, C. (2010). Maladaptive striatal plasticity in L-DOPA-induced dyskinesia. Prog. Brain Res. 183, 209–233.
Centonze, D., Grande, C., Saulle, E., Martin, A. B., Gubellini, P., Pavón, N., Pisani, A., Bernardi, G., Moratalla, R., and Calabresi, P. (2003). Distinct roles of D1 and D5 dopamine receptors in motor activity and striatal synaptic plasticity. J. Neurosci. 23, 8506–8512.
Centonze, D., Gubellini, P., Picconi, B., Calabresi, P., Giacomini, P., and Bernardi, G. (1999). Unilateral dopamine denervation blocks corticostriatal LTP. J. Neurophysiol. 82, 3575–3579.
Cepeda, C., and Levine, M. S. (1998). Dopamine and N-methyl-D-aspartate receptor interactions in the neostriatum. Dev. Neurosci. 20, 1–18.
Cepeda, C., and Levine, M. S. (2006). Where do you think you are going? The NMDA-D1 receptor trap. Sci. STKE 2006, pe20.
Corvol, J. C., Muriel, M. P., Valjent, E., Feger, J., Hanoun, N., Girault, J. A., Hirsch, E. C., and Hervé, D. (2004). Persistent increase in olfactory type G-protein alpha subunit levels may underlie D1 receptor functional hypersensitivity in Parkinson’s disease. J. Neurosci. 24, 7007–7014.
Cotzias, G. C., Papavasiliou, P. S., and Gellene, R. (1969). Modification of parkinsonism: chronic treatment with L-dopa. N. Engl. J. Med. 280, 337–345.
Dagher, A., and Robbins, T. W. (2009). Personality, addiction, dopamine: insights from Parkinson’s disease. Neuron 61, 502–510.
Damier, P., Tremblay, L., Féger, J., and Hirsch, E. C. (2000). Development of dyskinesias induced by treatment for Parkinson’s disease: potential role of first exposure to L-DOPA (or phenomenon of priming). Rev. Neurol. (Paris) 156, 224–235.
Darmopil, S., Martín, A. B., De Diego, I. R., Ares, S., and Moratalla, R. (2009). Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol. Psychiatry 66, 603–613.
Darmopil, S., Muñetón-Gómez, V. C., de Ceballos, M. L., Bernson, M., and Moratalla, R. (2008). Tyrosine hydroxylase cells appearing in the mouse striatum after dopamine denervation are likely to be projection neurones regulated by L-DOPA. Eur. J. Neurosci. 27, 580–592.
de la Fuente-Fernández, R., Sossi, V., Huang, Z., Furtado, S., Lu, J. Q., Calne, D. B., Ruth, T. J., and Stoessl, A. J. (2004). Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain 127, 2747–2754.
Delfino, M., Kalisch, R., Czisch, M., Larramendy, C., Ricatti, J., Taravini, I. R., Trenkwalder, C., Murer, M. G., Auer, D. P., and Gershanik, O. S. (2007). Mapping the effects of three dopamine agonists with different dyskinetogenic potential and receptor selectivity using pharmacological functional magnetic resonance imaging. Neuropsychopharmacology 32, 1911–1921.
Delfino, M. A., Stefano, A. V., Ferrario, J. E., Taravini, I. R., Murer, M. G., and Gershanik, O. S. (2004). Behavioral sensitization to different dopamine agonists in a parkinsonian rodent model of drug-induced dyskinesias. Behav. Brain Res. 152, 297–306.
Doucet, J. P., Nakabeppu, Y., Bedard, P. J., Hope, B. T., Nestler, E. J., Jasmin, B. J., Chen, J. S., Iadarola, M. J., St-Jean, M., Wigle, N., Blanchet, P., Grondin, R., and Robertson, G. S. (1996). Chronic alterations in dopaminergic neurotransmission produce a persistent elevation of deltaFosB-like protein(s) in both the rodent and primate striatum. Eur. J. Neurosci. 8, 365–381.
El-Ghundi, M., Fletcher, P. J., Drago, J., Sibley, D. R., O’Down, B. F., and George, S. R. (1999). Spatial learning deficit in dopamine D1 receptor knockout mice. Eur. J. Pharmacol. 383, 95–106.
Fasano, S., Bezard, E., D’Antoni, A., Francardo, V., Indrigo, M., Qin, L., Doveró, S., Cerovic, M., Cenci, M. A., and Brambilla, R. (2010). Inhibition of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) signaling in the striatum reverts motor symptoms associated with L-dopa-induced dyskinesia. Proc. Natl. Acad. Sci. U.S.A. 107, 21824–21829.
Fasano, S., D’Antoni, A., Orban, P. C., Valjent, E., Putignano, E., Vara, H., Pizzorusso, T., Giustetto, M., Yoon, B., Soloway, P., Maldonado, R., Caboche, J., and Brambilla, R. (2009). Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) controls activation of extracellular signal-regulated kinase (ERK) signaling in the striatum and long-term behavioral responses to cocaine. Biol. Psychiatry 66, 758–768.
Filion, M., Tremblay, L., and Bédard, P. J. (1991). Effects of dopamine agonists on the spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 547, 152–161.
Fiorentini, C., Busi, C., Gorruso, E., Gotti, C., Spano, P., and Missale, C. (2008). Reciprocal regulation of dopamine D1 and D3 receptor function and trafficking by heterodimerization. Mol. Pharmacol. 74, 59–69.
Fiorentini, C., Rizzetti, M. C., Busi, C., Bontempi, S., Collo, G., Spano, P., and Missale, C. (2006). Loss of synaptic D1 dopamine/N-methyl-D-aspartate glutamate receptor complexes in L-DOPA-induced dyskinesia in the rat. Mol. Pharmacol. 69, 805–812.
Gardoni, F., Picconi, B., Ghiglieri, V., Polli, F., Bagetta, V., Bernardi, G., Cattabeni, F., Di Luca, M., and Calabresi, P. (2006). A critical interaction between NR2B and MAGUK in L-DOPA induced dyskinesia. J. Neurosci. 26, 2914–2922.
Gerfen, C. R., Miyachi, S., Paletzki, R., and Brown, P. (2002). D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J. Neurosci. 22, 5042–5054.
Gold, S. J., Hoang, C. V., Potts, B. W., Porras, G., Pioli, E., Kim, K. W., Nadjar, A., Qin, C., LaHoste, G. J., Li, Q., Bioulac, B. H., Waugh, J. L., Gurevich, E., Neve, R. L., and Bezard, E. (2007). RGS9-2 negatively modulates L-3,4-dihydroxyphenylalanine-induced dyskinesia in experimental Parkinson’s disease. J. Neurosci. 27, 14338–14348.
Gonon, F. (1997). Prolonged and extrasynaptic excitatory action of dopamine mediated by D1 receptors in the rat striatum in vivo. J. Neurosci. 17, 5972–5978.
Goulet, M., and Madras, B. K. (2000). D(1) dopamine receptor agonists are more effective in alleviating advanced than mild parkinsonism in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated monkeys. J. Pharmacol. Exp. Ther. 292, 714–724.
Granado, N., Ortiz, O., Suárez, L. M., Martín, E. D., Ceña, V., Solís, J. M., and Moratalla, R. (2008). D1 but not D5 dopamine receptors are critical for LTP, spatial learning, and LTP-Induced arc and zif268 expression in the hippocampus. Cereb. Cortex 18, 1–12.
Grande, C., Zhu, H., Martin, A. B., Lee, M., Ortiz, O., Hiroi, N., and Moratalla, R. (2004). Chronic treatment with atypical neuroleptics induces striosomal FosB/DeltaFosB expression in rats. Biol. Psychiatry 55, 457–463.
Grondin, R., Bédard, P. J., Britton, D. R., and Shiosaki, K. (1997). Potential therapeutic use of the selective dopamine D1 receptor agonist, A-86929: an acute study in parkinsonian levodopa-primed monkeys. Neurology 49, 421–426.
Grondin, R., Doan, V. D., Grégoire, L., and Bédard, P. J. (1999). D1 receptor blockade improves L-dopa-induced dyskinesia but worsens parkinsonism in MPTP monkeys. Neurology 52, 771–776.
Guigoni, C., Doudnikoff, E., Li, Q., Bloch, B., and Bezard, E. (2007). Altered D(1) dopamine receptor trafficking in parkinsonian and dyskinetic non-human primates. Neurobiol. Dis. 26, 452–463.
Guigoni, C., Li, Q., Aubert, I., Dovero, S., Bioulac, B. H., Bloch, B., Crossman, A. R., Gross, C. E., and Bezard, E. (2005). Involvement of sensorimotor, limbic, and associative basal ganglia domains in L-3,4-dihydroxyphenylalanine-induced dyskinesia. J. Neurosci. 25, 2102–2107.
Hirano, S., Asanuma, K., Ma, Y., Tang, C., Feigin, A., Dhawan, V., Carbon, M., and Eidelberg, D. (2008). Dissociation of metabolic and neurovascular responses to levodopa in the treatment of Parkinson’s disease. J. Neurosci. 28, 4201–4209.
Hiroi, N., and Graybiel, A. M. (1996). Atypical and typical neuroleptic treatments induce distinct programs of transcription factor expression in the striatum. J. Comp. Neurol. 374, 70–83.
Hiroi, N., Martín, A. B., Grande, C., Alberti, I., Rivera, A., and Moratalla, R. (2002). Molecular dissection of dopamine receptor signaling. J Chem. Neuroanat. 23, 237–242.
Holloway, R. G., Shoulson, I., Fahn, S., Kieburtz, K., Lang, A., Marek, K., McDermott, M., Seibyl, J., Weiner, W., Musch, B., Kamp, C., Welsh, M., Shinaman, A., Pahwa, R., Barclay, L., Hubble, J., LeWitt, P., Miyasaki, J., Suchowersky, O., Stacy, M., Russell, D. S., Ford, B., Hammerstad, J., Riley, D., Standaert, D., Wooten, F., Factor, S., Jankovic, J., Atassi, F., Kurlan, R., Panisset, M., Rajput, A., Rodnitzky, R., Shults, C., Petsinger, G., Waters, C., Pfeiffer, R., Biglan, K., Borchert, L., Montgomery, A., Sutherland, L., Weeks, C., DeAngelis, M., Sime, E., Wood, S., Pantella, C., Harrigan, M., Fussell, B., Dillon, S., Alexander-Brown, B., Rainey, P., Tennis, M., Rost-Ruffner, E., Brown, D., Evans, S., Berry, D., Hall, J., Shirley, T., Dobson, J., Fontaine, D., Pfeiffer, B., Brocht, A., Bennett, S., Daigneault, S., Hodgeman, K., O’Connell, C., Ross, T., Richard, K., and Watts, A.; Parkinson Study Group. (2004). Pramipexole vs levodopa as initial treatment for Parkinson disease: a 4-year randomized 33 controlled trial. Arch. Neurol. 61, 1044–1053.
Hsu, A., Togasaki, D. M., Bezard, E., Sokoloff, P., Langston, J. W., Di Monte, D. A., and Quik, M. (2004). Effect of the D3 dopamine receptor partial agonist BP897 [N-[4-(4-(2-methoxyphenyl) piperazinyl) butyl]-2-naphthamide] on L-3,4-dihydroxyphenylalanine-induced dyskinesias and parkinsonism in squirrel monkeys. J. Pharmacol. Exp. Ther. 311, 770–777.
Hurley, M. J., Jolkkonen, J., Stubbs, C. M., Jenner, P., and Marsden, C. D. (1996). Dopamine D3 receptors in the basal ganglia of the common marmoset and following MPTP and L-DOPA treatment. Brain Res. 709, 259–264.
Jenner, P. (2008). Molecular mechanisms of L-DOPA-induced dyskinesia. Nat. Rev. Neurosci. 9, 665–677.
Kasanetz, F., Deroche-Gamonet, V., Berson, N., Balado, E., Lafourcade, M., Manzoni, O., and Piazza, P. V. (2010). Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science 328, 1709–1712.
Kelz, M. B., Chen, J., Carlezon, W.A. Jr., Whisler, K., Gilden, L., Beckmann, A. M., Steffen, C., Zhang, Y. J., Marotti, L., Self, D. W., Tkatch, T., Baranauskas, G., Surmeier, D. J., Neve, R. L., Duman, R. S., Picciotto, M. R., and Nestler, E. J. (1999). Expression of the transcription factor deltaFosB in the brain controls sensitivity to cocaine. Nature 401, 272–276.
Kreitzer, A. C., and Malenka, R. C. (2007). Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 445, 643–647.
Kreitzer, A. C., and Malenka, R. C. (2008). Striatal plasticity and basal ganglia circuit function. Neuron 60, 543–554.
Kumar, R., Riddle, L., Griffin, S. A., Grundt, P., Newman, A. H., and Luedtke, R. R. (2009). Evaluation of the D3 dopamine receptor selective antagonist PG01037 on L-DOPA-dependent abnormal involuntary movements in rats. Neuropharmacology 56, 944–955.
Larramendy, C., Taravini, I. R., Saborido, M. D., Ferrario, J. E., Murer, M. G., and Gershanik, O. S. (2008). Cabergoline and pramipexole fail to modify already established dyskinesias in an animal model of parkinsonism. Behav. Brain Res. 194, 44–51.
Levenson, J. M., O’Riordan, K. J., Brown, K. D., Trinh, M. A., Molfese, D. L., and Sweatt, J. D. (2004). Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 279, 40545–40559.
Lindgren, H. S., Andersson, D. R., Lagerkvist, S., Nissbrandt, H., and Cenci, M. A. (2010). L-DOPA-induced dopamine efflux in the striatum and the substantia nigra in a rat model of Parkinson’s disease: temporal and quantitative relationship to the expression of dyskinesia. J. Neurochem. 112, 1465–1476.
Lindgren, H. S., Ohlin, K. E., and Cenci, M. A. (2009). Differential involvement of D1 and D2 dopamine receptors in L-DOPA-induced angiogenic activity in a rat model of Parkinson’s disease. Neuropsychopharmacology 34, 2477–2488.
Lovinger, D. M. (2010). Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology 58, 951–961.
Lozano, A. M., Lang, A. E., Levy, R., Hutchison, W., and Dostrovsky, J. (2000). Neuronal recordings in Parkinson’s disease patients with dyskinesias induced by apomorphine. Ann. Neurol. 47(Suppl. 1), S141–S146.
Lundblad, M., Usiello, A., Carta, M., Håkansson, K., Fisone, G., and Cenci, M. A. (2005). Pharmacological validation of a mouse model of L-DOPA-induced dyskinesia. Exp. Neurol. 194, 66–75.
Marcellino, D., Ferré, S., Casadó, V., Cortés, A., Le Foll, B., Mazzola, C., Drago, F., Saur, O., Stark, H., Soriano, A., Barnes, C., Goldberg, S. R., Lluis, C., Fuxe, K., and Franco, R. (2008). Identification of dopamine D1-D3 receptor heteromers. Indications for a role of synergistic D1-D3 receptor interactions in the striatum. J. Biol. Chem. 283, 26016–26025.
Mazzucchelli, C., Vantaggiato, C., Ciamei, A., Fasano, S., Pakhotin, P., Krezel, W., Welzl, H., Wolfer, D. P., Pagès, G., Valverde, O., Marowsky, A., Porrazzo, A., Orban, P. C., Maldonado, R., Ehrengruber, M. U., Cestari, V., Lipp, H. P., Chapman, P. F., Pouysségur, J., and Brambilla, R. (2002). Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34, 807–820.
McClung, C. A., and Nestler, E. J. (2003). Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat. Neurosci. 6, 1208–1215.
McClung, C. A., Ulery, P. G., Perrotti, L. I., Zachariou, V., Berton, O., and Nestler, E. J. (2004). DeltaFosB: a molecular switch for long-term adaptation in the brain. Brain Res. Mol. Brain Res. 132, 146–154.
Mela, F., Marti, M., Dekundy, A., Danysz, W., Morari, M., and Cenci, M. A. (2007). Antagonism of metabotropic glutamate receptor type 5 attenuates L-DOPA-induced dyskinesia and its molecular and neurochemical correlates in a rat model of Parkinson’s disease. J. Neurochem. 101, 483–497.
Mela, F., Millan, M. J., Brocco, M., and Morari, M. (2010). The selective D(3) receptor antagonist, S33084, improves parkinsonian-like motor dysfunction but does not affect L-DOPA-induced dyskinesia in 6-hydroxydopamine hemi-lesioned rats. Neuropharmacology 58, 528–536.
Merello, M., Balej, J., Delfino, M., Cammarota, A., Betti, O., and Leiguarda, R. (1999). Apomorphine induces changes in GPi spontaneous outflow in patients with Parkinson’s disease. Mov. Disord. 14, 45–49.
Moratalla, R., Xu, M., Tonegawa, S., and Graybiel, A. M. (1996a). Cellular responses to psychomotor stimulant and neuroleptic drugs are abnormal in mice lacking the D1 dopamine receptor. Proc. Natl. Acad. Sci. U.S.A. 93, 14928–14933.
Moratalla, R., Elibol, B., Vallejo, M., and Graybiel, A. M. (1996b). Network-level changes in expression of inducible Fos-Jun proteins in the striatum during chronic cocaine treatment and withdrawal. Neuron 17, 147–156.
Murer, M. G., Raisman-Vozari, R., and Gershanik, O. (1999). Levodopa in Parkinson’s disease: neurotoxicity issue laid to rest? Drug Saf. 21, 339–352.
Muriel, M. P., Bernard, V., Levey, A. I., Laribi, O., Abrous, D. N., Agid, Y., Bloch, B., and Hirsch, E. C. (1999). Levodopa induces a cytoplasmic localization of D1 dopamine receptors in striatal neurons in Parkinson’s disease. Ann. Neurol. 46, 103–111.
Nutt, J. G. (2000). Clinical pharmacology of levodopa-induced dyskinesia. Ann. Neurol. 47(Suppl. 1), S160–S164.
Nutt, J. G. (2008). Pharmacokinetics and pharmacodynamics of levodopa. Mov. Disord. 23(Suppl. 3), S580–S584.
O’Carroll, C. M., Martin, S. J., Sandin, J., Frenguelli, B., and Morris, R. G. (2006). Dopaminergic modulation of the persistence of one-trial hippocampus dependent memory. Learn. Mem. 13, 760–769.
Olanow, C. W., Agid, Y., Mizuno, Y., Albanese, A., Bonuccelli, U., Damier, P., De Yebenes, J., Gershanik, O., Guttman, M., Grandas, F., Hallett, M., Hornykiewicz, O., Jenner, P., Katzenschlager, R., Langston, W. J., LeWitt, P., Melamed, E., Mena, M. A., Michel, P. P., Mytilineou, C., Obeso, J. A., Poewe, W., Quinn, N., Raisman-Vozari, R., Rajput, A. H., Rascol, O., Sampaio, C., and Stocchi, F. (2004). Levodopa in the treatment of Parkinson’s disease: current controversies. Mov. Disord. 19, 997–1005.
Olanow, C. W., Obeso, J. A., and Stocchi, F. (2006). Drug insight: Continuous dopaminergic stimulation in the treatment of Parkinson’s disease. Nat. Clin. Pract. Neurol. 2, 382–392.
Onofrio, C., Paci, A., and Thomas, C. (1998). Sudden appearance of invalidating dyskinesia-dystonia and off fluctuations after the introduction of levodopa in two dopaminomimetic drug naive patients with stage IV Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 65, 605–606.
Ortiz, O., Delgado-García, J. M., Espadas, I., Bahí, A., Trullas, R., Dreyer, J. L., Gruart, A., and Moratalla, R. (2010). Associative learning and CA3-CA1 synaptic plasticity are impaired in D1R null, Drd1a-/- mice and in hippocampal siRNA silenced Drd1a mice. J. Neurosci. 30, 12288–12300.
Papa, S. M., Desimone, R., Fiorani, M., and Oldfield, E. H. (1999). Internal globus pallidus discharge is nearly suppressed during levodopa-induced dyskinesias. Ann. Neurol. 46, 732–738.
Papavasiliou, P. S., Cotzias, G. C., Düby, S. E., Steck, A. J., Fehling, C., and Bell, M. A. (1972). Levodopa in Parkinsonism: potentiation of central effects with a peripheral inhibitor. N. Engl. J. Med. 286, 8–14.
Pavon, N., Martin, A. B., Mendialdua, A., and Moratalla, R. (2006). ERK phosphorylation and FosB expression are associated with L-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol. Psychiatry 59, 64–74.
Picconi, B., Centonze, D., Håkansson, K., Bernardi, G., Greengard, P., Fisone, G., Cenci, M. A., and Calabresi, P. (2003). Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat. Neurosci. 6, 501–506.
Quik, M., Police, S., He, L., Di Monte, D. A., and Langston, J. W. (2000). Expression of D(3) receptor messenger RNA and binding sites in monkey striatum and substantia nigra after nigrostriatal degeneration: effect of levodopa treatment. Neuroscience 98, 263–273.
Rascol, O. (1999). Dopamine agonists: what is the place of the newer compounds in the treatment of Parkinson’s disease? J. Neural Transm. 55, 33–45.
Rascol, O., Brooks, D. J., Korczyn, A. D., De Deyn, P. P., Clarke, C. E., Lang, A. E., and Abdalla, M. (056) Study Group. (2006). Development of dyskinesias in a 5-year trial of ropinirole and L-dopa. Mov. Disord. 21, 1844–1850.
Rascol, O., Nutt, J. G., Blin, O., Goetz, C. G., Trugman, J. M., Soubrouillard, C., Carter, J. H., Currie, L. J., Fabre, N., Thalamas, C., Giardina, W. W., and Wright, S. (2001). Induction by dopamine D1 receptor agonist ABT-431 of dyskinesia similar to levodopa in patients with Parkinson disease. Arch. Neurol. 58, 249–254.
Rodrigues, T. B., Granado, N., Ortiz, O., Cerdán, S., and Moratalla, R. (2007). Metabolic interactions between glutamatergic and dopaminergic neurotransmitter systems are mediated through D(1) dopamine receptors. J. Neurosci. Res. 85, 3284–3293.
Rossato, J. I., Bevilaqua, L. R., Izquierdo, I., Medina, J. H., and Cammarota, M. (2009). Dopamine controls persistence of long-term memory storage. Science 325, 1017–1020.
Rylander, D., Recchia, A., Mela, F., Dekundy, A., Danysz, W., and Cenci, M. A. (2009). Pharmacological modulation of glutamate transmission in a rat model of L-DOPA-induced dyskinesia: effects on motor behavior and striatal nuclear signaling. J. Pharmacol. Exp. Ther. 330, 227–235.
Sánchez-Pernaute, R., Jenkins, B. G., Choi, J. K., Iris Chen, Y. C., and Isacson, O. (2007). In vivo evidence of D3 dopamine receptor sensitization in parkinsonian primates and rodents with L-DOPA-induced dyskinesias. Neurobiol. Dis. 27, 220–227.
Santini, E., Alcacer, C., Cacciatore, S., Heiman, M., Hervé, D., Greengard, P., Girault, J. A., Valjent, E., and Fisone, G. (2009a). L-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J. Neurochem. 108, 621–633.
Santini, E., Heiman, M., Greengard, P., Valjent, E., and Fisone, G. (2009b). Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2, ra36.
Santini, E., Sgambato-Faure, V., Li, Q., Savasta, M., Dovero, S., Fisone, G., and Bezard, E. (2010). Distinct changes in cAMP and extracellular signal-regulated protein kinase signalling in L-DOPA-induced dyskinesia. PLoS ONE 5, e12322. doi: 10.1371/journal.pone.0012322
Santini, E., Valjent, E., Usiello, A., Carta, M., Borgkvist, A., and Girault, J. A. (2007). Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J. Neurosci. 27, 6995–7005.
Schuster, S., Nadjar, A., Guo, J. T., Li, Q., Ittrich, C., Hengerer, B., and Bezard, E. (2008). The 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor lovastatin reduces severity of L-DOPA-induced abnormal involuntary movements in experimental Parkinson’s disease. J. Neurosci. 28, 4311–4316.
Sgambato-Faure, V., Buggia, V., Gilbert, F., Lévesque, D., Benabid, A. L., and Berger, F. (2005). Coordinated and spatial upregulation of arc in striatonigral neurons correlates with L-dopa-induced behavioral sensitization in dyskinetic rats. J. Neuropathol. Exp. Neurol. 64, 936–947.
Shen, W., Flajolet, M., Greengard, P., and Surmeier, D. J. (2008). Dichotomous dopaminergic control of striatal synaptic plasticity. Science 321, 848–851.
Stipanovich, A., Valjent, E., Matamales, M., Nishi, A., Ahn, J. H., Maroteaux, M., Bertran-Gonzalez, J., Brami-Cherrier, K., Enslen, H., Corbillé, A. G., Filhol, O., Nairn, A. C., Greengard, P., Hervé, D., and Girault, J. A. (2008). A phosphatase cascade by which rewarding stimuli control nucleosomal response. Nature 453, 879–884.
Surmeier, D. J., Plotkin, J., and Shen, W. (2009). Dopamine and synaptic plasticity in dorsal striatal circuits controlling action selection. Curr. Opin. Neurobiol. 19, 621–628.
Tang, K., Low, M. J., Grandy, D. K., and Lovinger, D. M. (2001). Dopamine-dependent synaptic plasticity in striatum during in vivo development. Proc. Natl. Acad. Sci. U.S.A. 98, 1255–1260.
Taylor, J. L., Bishop, C., and Walker, P. D. (2005). Dopamine D1 and D2 receptor contributions to L-DOPA-induced dyskinesia in the dopamine-depleted rat. Pharmacol. Biochem. Behav. 81, 887–893.
Thomas, G. M., and Huganir, R. L. (2004). MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183.
Trugman, J. M., and Wooten, G. F. (1986). The effects of L-DOPA on regional cerebral glucose utilization in rats with unilateral lesions of the substantia nigra. Brain Res. 379, 264–274.
Trugman, J. M., and Wooten, G. F. (1987). Selective D1 and D2 dopamine agonists differentially alter basal ganglia glucose utilization in rats with unilateral 6-hydroxydopamine substantia nigra lesions. J. Neurosci. 7, 2927–2935.
Ulusoy, A., Sahin, G., and Kirik, D. (2010). Presynaptic dopaminergic compartment determines the susceptibility to L-DOPA-induced dyskinesia in rats. Proc. Natl. Acad. Sci. U.S.A. 107, 13159–13164.
Valjent, E., Corvol, J. C., Pages, C., Besson, M. J., Maldonado, R., and Caboche, J. (2000). Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J. Neurosci. 20, 8701–8709.
Visanji, N. P., Fox, S. H., Johnston, T., Reyes, G., Millan, M. J., and Brotchie, J. M. (2009). Dopamine D3 receptor stimulation underlies the development of L-DOPA-induced dyskinesia in animal models of Parkinson’s disease. Neurobiol. Dis. 35, 184–192.
Voon, V., Fernagut, P. O., Wickens, J., Baunez, C., Rodriguez, M., Pavon, N., Juncos, J. L., Obeso, J. A., and Bezard, E. (2009). Chronic dopaminergic stimulation in Parkinson’s disease: from dyskinesias to impulse control disorders. Lancet Neurol. 8, 1140–1149.
Wang, Z., Kai, L., Day, M., Ronesi, J., Yin, H. H., Ding, J., Tkatch, T., Lovinger, D. M., and Surmeier, D. J. (2006). Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron 50, 443–452.
Westin, J. E., Lindgren, H. S., Gardi, J., Nyengaard, J. R., Brundin, P., Mohapel, P., and Cenci, M. A. (2006). Endothelial proliferation and increased blood-brain barrier permeability in the basal ganglia in a rat model of 3,4-dihydroxyphenyl-L-alanine-induced dyskinesia. J. Neurosci. 26, 9448–9461.
Westin, J. E., Vercammen, L., Strome, E. M., Konradi, C., and Cenci, M. A. (2007). Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of L-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol. Psychiatry 62, 800–810.
Keywords: L-DOPA-induced dyskinesia, D1 dopamine receptor, Parkinson’s disease
Citation: Murer MG and Moratalla R (2011) Striatal signaling in L-DOPA-induced dyskinesia: common mechanisms with drug abuse and long term memory involving D1 dopamine receptor stimulation. Front. Neuroanat. 5:51. doi: 10.3389/fnana.2011.00051
Received: 11 May 2011;
Paper pending published: 06 June 2011;
Accepted: 25 July 2011;
Published online: 11 August 2011.
Edited by:
Emmanuel Valjent, Université Montpellier 1&2, FranceReviewed by:
Riccardo Brambilla, Vita-Salute San Raffaele University, ItalyM. Angela Cenci, Lund University, Sweden
Copyright: © 2011 Murer and Moratalla. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Rosario Moratalla, Instituto Cajal, Avenida Doctor Arce, 37, 20002 Madrid, Spain. e-mail:bW9yYXRhbGxhQGNhamFsLmNzaWMuZXM=