 Xin Xu
Xin Xu Eric C. Miller
Eric C. Miller Lucas Pozzo-Miller
Lucas Pozzo-Miller- Department of Neurobiology, Civitan International Research Center, The University of Alabama at Birmingham, Birmingham, AL, USA
Spines are small cytoplasmic extensions of dendrites that form the postsynaptic compartment of the majority of excitatory synapses in the mammalian brain. Alterations in the numerical density, size, and shape of dendritic spines have been correlated with neuronal dysfunction in several neurological and neurodevelopmental disorders associated with intellectual disability, including Rett syndrome (RTT). RTT is a progressive neurodevelopmental disorder associated with intellectual disability that is caused by loss of function mutations in the transcriptional regulator methyl CpG-binding protein 2 (MECP2). Here, we review the evidence demonstrating that principal neurons in RTT individuals and Mecp2-based experimental models exhibit alterations in the number and morphology of dendritic spines. We also discuss the exciting possibility that signaling pathways downstream of brain-derived neurotrophic factor (BDNF), which is transcriptionally regulated by MeCP2, offer promising therapeutic options for modulating dendritic spine development and plasticity in RTT and other MECP2-associated neurodevelopmental disorders.
Introduction
Espinas dendríticas are small cytoplasmic extensions emerging from the dendrites of neurons that were first described in the cerebellum and cerebrum of birds and mammals by Santiago Ramón y Cajal at the end of the 19th century (Ramón y Cajal, 1888, 1891b, 1896; as cited in Yuste, 2010). Cajal had already envisioned that dendritic spines are contacted by axons at synapses (Ramón y Cajal, 1891a, 1894), and used this arrangement as the main example in support of his Neuronal Doctrine (Ramón y Cajal, 1933). With the aid of electron microscopy and confocal fluorescence microscopy, it is now well established that spines are the postsynaptic sites of most excitatory synapses in the brain, receiving inputs from glutamatergic axons (Bhatt et al., 2009; Yuste, 2010; Shirao and Gonzalez-Billault, 2013). Despite their small size (∼1 μm in diameter), proper dendritic spine formation is critical for brain function. Numerous proteins, including neurotransmitter and neuropeptide receptors, signaling kinases, and phosphatases, as well as ion channels are expressed in dendritic spines, where they participate in excitatory synaptic transmission and activity-dependent synaptic plasticity, and ultimately in learning and memory (Sala and Segal, 2014). During development and throughout adulthood, the numerical density and morphology of individual spines are critical for the fine-tuning of neuronal and synaptic excitability, allowing the initial establishment and activity-dependent remodeling of connectivity of neuronal circuits (Luebke et al., 2010).
The morphology of dendritic spines is highly variable, and by defining the biochemical and electrical properties of the postsynaptic compartment, it contributes to the strength and plasticity of excitatory synapses (Luebke et al., 2010). Spines have been broadly classified into three morphological types: stubby, mushroom and thin (Peters and Kaiserman-Abramof, 1969). Mushroom spines have a large head that is connected to the parent dendrite through a narrow neck. Stubby spines do not have a noticeable neck and are most common during postnatal development (Chapleau et al., 2009b; Rochefort and Konnerth, 2012). These two types of large spines are referred to as “memory spines,” because they are stable, persist for longer periods of time, and are the postsynaptic side of strong excitatory synapses (Trachtenberg et al., 2002; Kasai et al., 2003). Conversely, thin spines have a thin, long neck, and a small bulbous head, are highly motile, unstable, and often short-lived, usually representing weak or silent synapses (Rochefort and Konnerth, 2012). Because thin spines are more plastic than large spines and have the potential to become stable spines, they have been dubbed “learning spines” (Grutzendler et al., 2002; Trachtenberg et al., 2002; Kasai et al., 2003; Holtmaat et al., 2005). It should be noted that thin protrusions longer than thin spines and without a noticeable head are called dendritic filopodia, and are more numerous than spines in developing neurons. Dendritic filopodia are transient and highly motile protrusions that can receive synaptic input and develop into mature spines, thus initiating synaptogenesis (Fiala et al., 1998; Luebke et al., 2010).
Following the well-known relationship between form and function in biological systems, recent in vitro and in vivo studies have demonstrated that the morphology of spines relates closely to the function and plasticity of the synapses they belong to (Yuste et al., 2000; Trachtenberg et al., 2002; Mizrahi et al., 2004; Segal, 2005; Kasai et al., 2010). For example, the volume of the spine head is directly proportional to the area of the postsynaptic density and the number of synaptic vesicles docked at the presynaptic active zone (Harris and Stevens, 1989; Schikorski and Stevens, 1999), the number of postsynaptic receptors (Nusser et al., 1998), and hence to the size of synaptic currents and synaptic strength (Yuste and Bonhoeffer, 2001; Luebke et al., 2010). Two-photon uncaging of glutamate on large spines evoked larger postsynaptic currents mediated by AMPA receptors than uncaging on small spines (Matsuzaki et al., 2001; Kasai et al., 2010). Such a structure-function relationship is also evident in the intracellular Ca2+ signals within spines triggered by afferent synaptic activity (Yuste et al., 2000; Nimchinsky et al., 2002; Bloodgood and Sabatini, 2007). Together with structural changes in response to afferent synaptic stimulation (Murphy and Segal, 1996; Srivastava and Penzes, 2011), all these findings support the long held view that dendritic spines are the morphological substrate of neuronal plasticity and learning and memory (Sala and Segal, 2014). In support of this notion, induction of long-term potentiation (LTP) leads to spine enlargement (Matsuzaki et al., 2004; Park et al., 2006), whereas long-term depression (LTD) causes spine shrinkage (Nagerl et al., 2004; Zhou et al., 2004; Hoogenraad and Akhmanova, 2010; Penzes et al., 2011b).
The relationship between dendritic spines and cognitive abilities was noted in early studies, when the term “spine dysgenesis” was coined by Dominick Purpura (Huttenlocher, 1970; Marin-Padilla, 1972; Purpura, 1974). Such anomalies in the morphology – and likely function – of dendritic spines have been described in several neurological disorders associated with cognitive decline, including typical aging, Alzheimer’s and Huntington diseases, schizophrenia, neurodevelopmental intellectual disabilities, and autism spectrum disorders (Fiala et al., 2002; Fukuda et al., 2005; Zhao et al., 2007; Bourgeron, 2009; Chapleau et al., 2009b; Garey, 2010; Penzes et al., 2011a; Levenga and Willemsen, 2012).
Dendritic Spine Dysgenesis in Rett Syndrome
Rett syndrome (RTT) is an X-linked progressive autism spectrum disorder associated with intellectual disability that affects girls during early childhood (∼1:15,000 birth worldwide; Neul and Zoghbi, 2004; Chapleau et al., 2009b). The disorder is characterized by a seemingly typical development for 6 to 18 months followed by regression and onset of a variety of neurological features, including motor impairments, loss of acquired language, intellectual disability, seizures, and anxiety (Chahrour and Zoghbi, 2007). The majority of RTT individuals carry loss-of-function mutations in MECP2, the gene encoding methyl CpG-binding protein 2 (MeCP2), a global transcriptional regulator that binds to methylated CpG sites in promoter regions of DNA (Amir et al., 1999; Chahrour et al., 2008). Emerging evidence indicates that RTT results from a deficit in synaptic maturation in the brain, and that MeCP2 plays a critical role in neuronal and synaptic maturation and pruning during development (Cohen et al., 2003; Calfa et al., 2011b), as well as in the function of established neuronal networks in adulthood (McGraw et al., 2011).
Pyramidal neurons in the cortex and hippocampus of RTT individuals have dendrites with atypical morphology (Belichenko et al., 1994; Armstrong et al., 1995; Chapleau et al., 2009a; Figure 1A). Two different mouse models lacking Mecp2 (Chen et al., 2001; Guy et al., 2001) have reduced dendritic complexity (Fukuda et al., 2005; Nguyen et al., 2012; Stuss et al., 2012), and decreased dendritic spine density and motility in cortical and hippocampal neurons (Belichenko et al., 2009; Tropea et al., 2009; Landi et al., 2011; Chapleau et al., 2012; Castro et al., 2014). On the other hand, Mecp2308 mice expressing truncated MeCP2 (Shahbazian et al., 2002), which have impaired synaptic plasticity and hippocampal-dependent learning and memory and other RTT-related neurological deficits, do not show any dendritic or synaptic anomalies neither in cortical nor hippocampal neurons (Moretti et al., 2006). The reduced dendritic spine density, along with a decrease in the proportion of mushroom spines, is also present in primary hippocampal neurons (Chao et al., 2007; Baj et al., 2014) and hippocampal slice cultures (Figure 1B) prepared from newborn Mecp2 knockout (KO) pups, as well as in neurons derived from induced pluripotent stem cells obtained from RTT individuals (Marchetto et al., 2010).
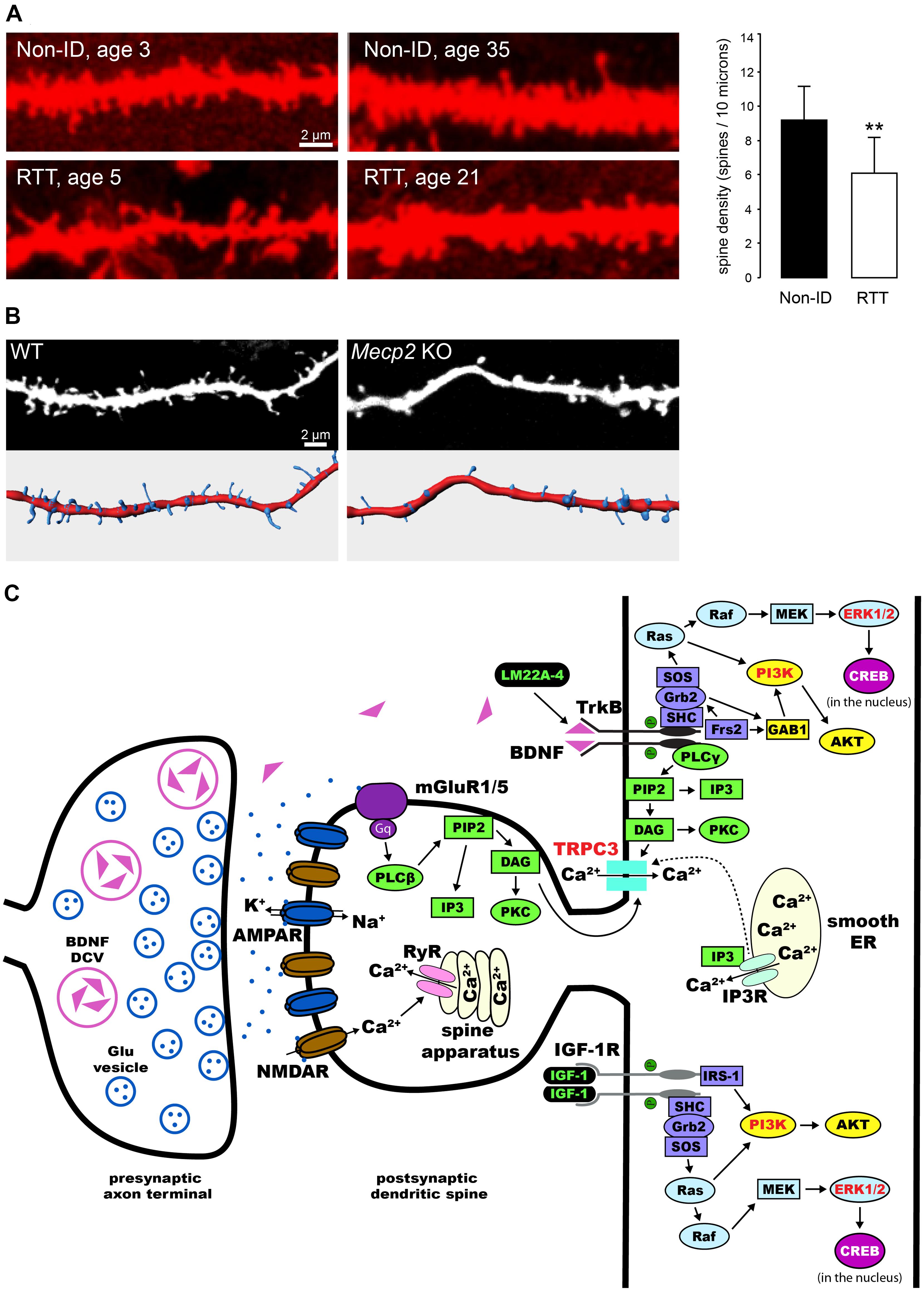
FIGURE 1. Dendritic spine dysgenesis in Rett syndrome, and intracellular signaling cascades involved in spine plasticity mediated by BDNF and IGF-1. (A) Confocal images of human CA1 pyramidal neurons in hippocampal sections from autopsy material labeled with DiI. Neurons from RTT individuals have lower dendritic spine density than those from typically developing individuals (Non-ID, non-intellectually disabled). **P < 0.01 (adapted from Chapleau et al., 2009a). (B) Confocal images of apical dendritic segments (top) of eYFP-expressing CA1 pyramidal neurons in 11 days in vitro hippocampal slice cultures prepared from postnatal day-5 wildtype (WT) and Mecp2 knockout (KO) mice, and their corresponding surface-rendered reconstructions (bottom). (C) Schematic diagram of an exemplary excitatory synapse on a dendritic spine of a pyramidal neuron in the hippocampus. We highlight the intracellular signaling cascades that mediate the effects of BDNF and IGF-1 on structural plasticity of spines. TrkB receptors are activated upon binding of BDNF, leading to dimerization and auto-phosphorylation. This process allows for the binding of adaptor proteins to their intracellular domain, and the subsequent activation of Ras/ERK, PI3K, and PLCγ (reviewed by Huang and Reichardt, 2003). All these pathways have been implicated in the effects of BDNF on dendritic spines (highlighted in red, see text for references). Potential therapies for the treatment of RTT act on these pathways (highlighted in green, see text for details and references): LM22A-4 binds and activates TrkB receptors directly (Massa et al., 2010); activation of IGF-1 receptors triggers the PI3K and Ras/ERK signaling pathways (Zheng and Quirion, 2004). DCV, dense core vesicle; Glu, glutamate; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; NMDAR, N-methyl-D-aspartate receptor; mGluR, metabotropic glutamate receptor; ER, endoplasmic reticulum; RyR, ryanodine receptor; PIP2, phosphatidylinositol 4,5 bisphosphate; DAG, diacylglycerol; IP3, inositol triphosphate; IP3R, IP3 receptor; PKC, protein kinase C; SH-2, src homology domain 2; SHC, SH-2-containing protein; Grb2, growth factor receptor-binding protein 2; GAB1, Grb2-associated-binding protein 1; SOS, nucleotide exchange factor son-of-sevenless; Frs2, fibroblast growth factor receptor substrate 2; AKT, protein kinase B; Ras, rat sarcoma proto-oncogenic G-protein; Raf, proto-oncogenic serine/threonine protein kinase; MAPK, mitogen-activated protein kinase; MEK, MAPK kinase; cAMP, cyclic adenosine monophosphate; CREB, cAMP response element-binding protein; IGF-1R, IGF-1 receptor; IRS-1, insulin receptor substrate 1.
The spine dysgenesis phenotype in pyramidal neurons of the hippocampus in Mecp2 KO mice has revealed unexpected complexities. CA1 and CA3 pyramidal neurons have lower spine density only in neonatal (postnatal day-7) Mecp2 KOs, well before excitatory synapse expansion. Spine density reaches wildtype (WT) levels a week later (postnatal day-15), and is maintained at WT levels throughout the symptomatic stage (postnatal day-40 to 60). Quantitative electron microscopy confirmed that the density of asymmetric spine synapses in CA1 stratum radiatum of Mecp2 KOs is comparable to that of WT mice (Calfa et al., 2011a; Chapleau et al., 2012). This developmental progression of the spine density phenotype is also reflected in the density of excitatory synapses imaged as VGLUT1-PSD95 immunofluorescent puncta, which is lower in area CA1 of 2 week-old Mecp2 null mice, but comparable to WT levels at 5 weeks of age (Chao et al., 2007). Altogether, these data demonstrate that proper MeCP2 functioning is required for dendritic spine formation during early postnatal development, and that a secondary compensatory mechanism seems to take place during atypical development in Mecp2 KOs. A couple of possibilities exist as to the extent of the compensatory mechanisms necessary to bring spine density to WT levels in hippocampal neurons. One possibility is that enhanced hippocampal network activity in Mecp2 KOs promotes dendritic spine formation (Calfa et al., 2011a). A second possibility is that deranged homeostatic plasticity promotes spinogenesis, while still affecting pyramidal neuron function (Blackman et al., 2012; Qiu et al., 2012).
Consistent with a model that tightly regulated MeCP2 levels are necessary during brain development and adulthood, overexpression of Mecp2 in vitro or in the MECP2 duplication mouse model (Mecp2Tg1) either increased (Jugloff et al., 2005; Chao et al., 2007; Jiang et al., 2013) or decreased (Zhou et al., 2006; Chapleau et al., 2009a; Cheng et al., 2014) dendritic complexity, spine density, and the density of excitatory synapses. Table 1 sumarizes all the published work on dendritic spines in RTT and experimenal models based on MeCP2 loss-of-function.
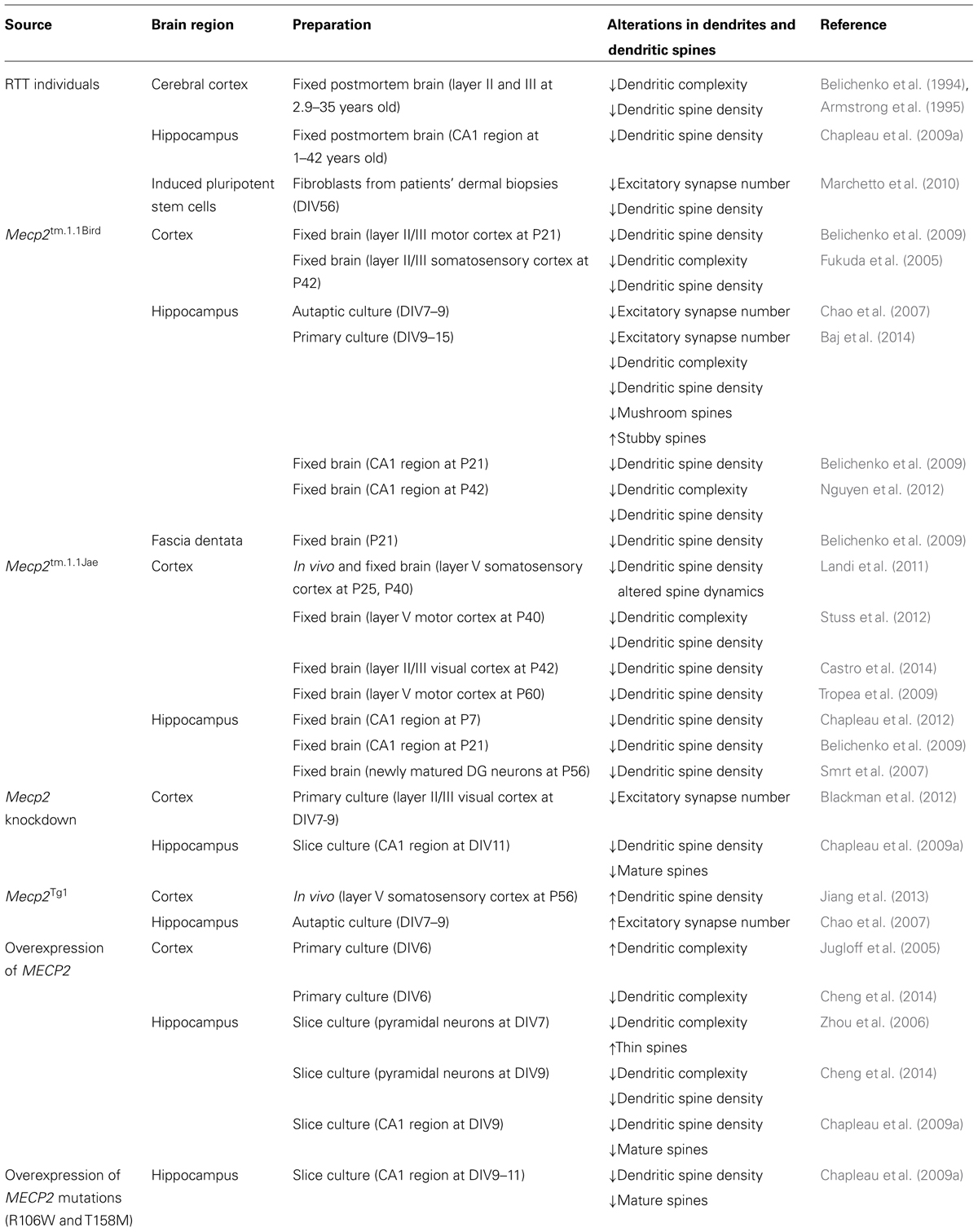
TABLE 1. Dendritic spine dysgenesis in RTT individuals and MeCP2-deficient cells and mice.
Role of BDNF in Dendritic Spine Formation and Plasticity: A Potential Therapy for RTT
MeCP2 regulates the expression of thousands of genes, including brain-derived neurotrophic factor (Bdnf; Chen et al., 2003; Martinowich et al., 2003; Zhou et al., 2006). BDNF is well known to promote neuronal and synaptic maturation (Carvalho et al., 2008), increase dendritic spine density, and enhance synaptic plasticity and learning and memory (Figurov et al., 1996; Luine and Frankfurt, 2013). MeCP2 binds to the Bdnf promoter and modulates Bdnf expression in an activity-dependent manner (Chen et al., 2003; Martinowich et al., 2003; Zhou et al., 2006; Chahrour and Zoghbi, 2007). Lower Bdnf mRNA and BDNF protein levels, as well as impaired BDNF trafficking and activity-dependent release, have been highlighted as pathophysiological mechanisms of RTT disease progression (Chang et al., 2006; Wang et al., 2006; Ogier et al., 2007; Li et al., 2012; Xu et al., 2014). Indeed, overexpression of BDNF rescues several cellular and behavioral deficits in Mecp2 KO mice (Chang et al., 2006; Chahrour and Zoghbi, 2007). These studies indicate that BDNF plays a critical role in neurological impairments in MeCP2-deficient mice.
Several studies have demonstrated that BDNF participates in synaptic plasticity, and is critical for dendritic spine formation and maturation during development (Poo, 2001; Tyler et al., 2002a; Tanaka et al., 2008; Cohen-Cory et al., 2010; Vigers et al., 2012). For example, exogenously applied BDNF increases spine density in cultured hippocampal neurons and CA1 pyramidal neurons in slice cultures (Tyler and Pozzo-Miller, 2001; Ji et al., 2005). In addition, BDNF shifts the proportions of morphological types of spines in hippocampal slice cultures (Tyler and Pozzo-Miller, 2003; Chapleau et al., 2008). Moreover, overexpression of the Bdnf gene in cultured hippocampal neurons rescued the dendritic atrophy caused by shRNA-mediated Mecp2 knockdown (Larimore et al., 2009). These effects of BDNF on dendritic spines are mediated by the tropomyosin related kinase B (TrkB) receptor (Tyler and Pozzo-Miller, 2001), and subsequent activation of extracellular signal-regulated kinase (ERK; Alonso et al., 2004), phosphatidylinositol 3-kinase (PI3K; Kumar et al., 2005), and phospholipase C-γ (PLC-γ), which leads to the opening of canonical transient receptor potential (TRPC) channels containing the TRPC3 subunit (Amaral and Pozzo-Miller, 2007; Chapleau et al., 2009b; Li et al., 2012; Luine and Frankfurt, 2013; Figure 1C).
Activity-dependent release of endogenously expressed (native) BDNF also modulates spine morphology in conjunction with spontaneous neurotransmitter release (Tyler et al., 2002b; Tyler and Pozzo-Miller, 2003; Tanaka et al., 2008). In addition, proper secretory trafficking of BDNF is essential for its actions on dendritic spine development and plasticity. The human BDNF gene has a single nucleotide polymorphism – a methionine (met) substitution for valine (val) at codon 66 – that impairs BDNF trafficking and its activity-dependent release, resulting in cognitive dysfunction in the general population (Egan et al., 2003; Chen et al., 2004), as well as more severe neurological symptoms in RTT individuals (Zeev et al., 2009). Consistently, dendritic complexity is reduced in dentate granule cells of Val66Met knock-in mice (Chen et al., 2006). Therefore, expression of this BDNF polymorphism might lead to deleterious effects on dendritic spine density and morphology.
The main limitation of BDNF-based therapies for neurological disorders, including RTT, is its poor blood-brain barrier permeability. Synthetic BDNF-loop mimetics with selective TrkB agonist activity are exciting alternatives (Massa et al., 2010; Kajiya et al., 2014). Indeed, systemic treatment with LM22A-4 rescues respiratory deficits in female Mecp2 heterozygous mice (Schmid et al., 2012), and prevents spine loss in striatal medium-spiny neurons in a mouse model of Huntington’s, rescuing their motor deficits (Simmons et al., 2013).
Other intriguing substitutes for BDNF are insulin-like growth factor-1 (IGF-1) and its active tripeptide ([1–3]IGF-1, also known as glypromate, GPE), a hormone widely expressed in the CNS during brain development that promotes neuronal survival as well as synaptic maturation (D’Ercole, 1996; O’Kusky et al., 2000; Tropea et al., 2009). Indeed, systemic treatment of male Mecp2 KO mice with [1–3]IGF-1 significantly increased activity of signaling pathways downstream of TrkB and improved several RTT-like symptoms and increased dendritic spine density in cortical neurons (Tropea et al., 2009), effects that are all recapitulated by full-length IGF-1 (Castro et al., 2014). These effects are due to the activation of IGF-1 receptors directly by IGF-1, and indirectly by [1–3]IGF-1, which does not bind to the IGF-1 receptor but rather increases the expression of IGF-1 (Carlsson-Skwirut et al., 1989; Corvin et al., 2012). It should be noted that full-length IGF-1 worsened a metabolic syndrome in Mecp2 KO mice, and did not affect dendritic spine density in hippocampal neurons (Pitcher et al., 2013). The safety and efficacy of recombinant human full-length IGF-1 (mecasermin) in a Phase-1 clinical trial in RTT individuals have been recently reported (Khwaja et al., 2014). The [1–3]IGF-1 analog glycyl-L-methylprolyl-L-glutamic acid (NNZ-2566; Neuren Pharmaceuticals) is in a Phase-2 clinical trial in RTT individuals.
Conclusion
Activity-dependent plasticity of dendritic spines includes both the formation of new spines and their maturation from thin, filipodia-like protrusions to “memory spines” that accompany excitatory synapse formation during brain development, as well as the structural remodeling of already existing spines. Alterations in neuronal circuitry are due to, or at least reflected by, deficits in dendritic spine structure and function. Dendritic spine anomalies have been identified in multiple brain regions in RTT and Mecp2-based mouse models. Since BDNF promotes the formation, maintenance, and activity-dependent sculpting of dendritic spines, and plays a critical role in neurological dysfunction in RTT, it emerges as one of the most exciting therapeutic agents for RTT. Thus, treatments that target the BDNF receptor TrkB and/or its downstream signaling pathways stand out as strong candidates to improve not only the spine dysgenesis phenotype, but also other synaptic plasticity deficits in RTT and other neurodevelopmental disorders caused by impaired BDNF availability.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH grants NS-065027 and HD-074418 (to Lucas Pozzo-Miller). We are indebted to Chris Chapleau, Wei Li, and Mary Phillips for thoughtful discussions.
References
Alonso, M., Medina, J. H., and Pozzo-Miller, L. (2004). ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn. Mem. 11, 172–178. doi: 10.1101/lm.67804
Amaral, M. D., and Pozzo-Miller, L. (2007). TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J. Neurosci. 27, 5179–5189. doi: 10.1523/JNEUROSCI.5499-06.2007
Amir, R. E., Van Den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Armstrong, D., Dunn, J. K., Antalffy, B., and Trivedi, R. (1995). Selective dendritic alterations in the cortex of Rett syndrome. J. Neuropathol. Exp. Neurol. 54, 195–201. doi: 10.1097/00005072-199503000-00006
Baj, G., Patrizio, A., Montalbano, A., Sciancalepore, M., and Tongiorgi, E. (2014). Developmental and maintenance defects in Rett syndrome neurons identified by a new mouse staging system in vitro. Front. Cell. Neurosci. 8:18. doi: 10.3389/fncel.2014.00018
Belichenko, P. V., Oldfors, A., Hagberg, B., and Dahlstrom, A. (1994). Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport 5, 1509–1513. doi: 10.1097/00001756-199407000-00025
Belichenko, P. V., Wright, E. E., Belichenko, N. P., Masliah, E., Li, H. H., Mobley, W. C.,et al. (2009). Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J. Comp. Neurol. 514, 240–258. doi: 10.1002/cne.22009
Bhatt, D. H., Zhang, S., and Gan, W. B. (2009). Dendritic spine dynamics. Annu. Rev. Physiol. 71, 261–282. doi: 10.1146/annurev.physiol.010908.163140
Blackman, M. P., Djukic, B., Nelson, S. B., and Turrigiano, G. G. (2012). A critical and cell-autonomous role for MeCP2 in synaptic scaling up. J. Neurosci. 32, 13529–13536. doi: 10.1523/JNEUROSCI.3077-12.2012
Bloodgood, B. L., and Sabatini, B. L. (2007). Ca2+ signaling in dendritic spines. Curr. Opin. Neurobiol. 17, 345–351. doi: 10.1016/j.conb.2007.04.003
Bourgeron, T. (2009). A synaptic trek to autism. Curr. Opin. Neurobiol. 19, 231–234. doi: 10.1016/j.conb.2009.06.003
Calfa, G., Hablitz, J. J., and Pozzo-Miller, L. (2011a). Network hyperexcitability in hippocampal slices from Mecp2 mutant mice revealed by voltage-sensitive dye imaging. J. Neurophysiol. 105, 1768–1784. doi: 10.1152/jn.00800.2010
Calfa, G., Percy, A. K., and Pozzo-Miller, L. (2011b). Experimental models of Rett syndrome based on Mecp2 dysfunction. Exp. Biol. Med. (Maywood) 236, 3–19. doi: 10.1258/ebm.2010.010261
Carlsson-Skwirut, C., Lake, M., Hartmanis, M., Hall, K., and Sara, V. R. (1989). A comparison of the biological activity of the recombinant intact and truncated insulin-like growth factor 1 (IGF-1). Biochim. Biophys. Acta 1011, 192–197. doi: 10.1016/0167-4889(89)90209-7
Carvalho, A. L., Caldeira, M. V., Santos, S. D., and Duarte, C. B. (2008). Role of the brain-derived neurotrophic factor at glutamatergic synapses. Br. J. Pharmacol. 153(Suppl. 1), S310–S324. doi: 10.1038/sj.bjp.0707509
Castro, J., Garcia, R. I., Kwok, S., Banerjee, A., Petravicz, J., Woodson, J.,et al. (2014). Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc. Natl. Acad. Sci. U.S.A. 111, 9941–9946. doi: 10.1073/pnas.1311685111
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J.,et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437. doi: 10.1016/j.neuron.2007.10.001
Chang, Q., Khare, G., Dani, V., Nelson, S., and Jaenisch, R. (2006). The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49, 341–348. doi: 10.1016/j.neuron.2005.12.027
Chao, H. T., Zoghbi, H. Y., and Rosenmund, C. (2007). MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 56, 58–65. doi: 10.1016/j.neuron.2007.08.018
Chapleau, C. A., Boggio, E. M., Calfa, G., Percy, A. K., Giustetto, M., and Pozzo-Miller, L. (2012). Hippocampal CA1 pyramidal neurons of Mecp2 mutant mice show a dendritic spine phenotype only in the presymptomatic stage. Neural Plast. 2012, 976164. doi: 10.1155/2012/976164
Chapleau, C. A., Calfa, G. D., Lane, M. C., Albertson, A. J., Larimore, J. L., Kudo, S.,et al. (2009a). Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol. Dis. 35, 219–233. doi: 10.1016/j.nbd.2009.05.001
Chapleau, C. A., Larimore, J. L., Theibert, A., and Pozzo-Miller, L. (2009b). Modulation of dendritic spine development and plasticity by BDNF and vesicular trafficking: fundamental roles in neurodevelopmental disorders associated with mental retardation and autism. J. Neurodev. Disord 1, 185–196. doi: 10.1007/s11689-009-9027-6
Chapleau, C. A., Carlo, M. E., Larimore, J. L., and Pozzo-Miller, L. (2008). The actions of BDNF on dendritic spine density and morphology in organotypic slice cultures depend on the presence of serum in culture media. J. Neurosci. Methods 169, 182–190. doi: 10.1016/j.jneumeth.2007.12.006
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331. doi: 10.1038/85906
Chen, W. G., Chang, Q., Lin, Y., Meissner, A., West, A. E., Griffith, E. C.,et al. (2003). Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889. doi: 10.1126/science.1086446
Chen, Z. Y., Jing, D., Bath, K. G., Ieraci, A., Khan, T., Siao, C. J.,et al. (2006). Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 314, 140–143. doi: 10.1126/science.1129663
Chen, Z. Y., Patel, P. D., Sant, G., Meng, C. X., Teng, K. K., Hempstead, B. L.,et al. (2004). Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J. Neurosci. 24, 4401–4411. doi: 10.1523/JNEUROSCI.0348-04.2004
Cheng, T. L., Wang, Z., Liao, Q., Zhu, Y., Zhou, W. H., Xu, W.,et al. (2014). MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev. Cell 28, 547–560. doi: 10.1016/j.devcel.2014.01.032
Cohen, D. R., Matarazzo, V., Palmer, A. M., Tu, Y., Jeon, O. H., Pevsner, J.,et al. (2003). Expression of MeCP2 in olfactory receptor neurons is developmentally regulated and occurs before synaptogenesis. Mol. Cell. Neurosci. 22, 417–429. doi: 10.1016/S1044-7431(03)00026-5
Cohen-Cory, S., Kidane, A. H., Shirkey, N. J., and Marshak, S. (2010). Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev. Neurobiol. 70, 271–288. doi: 10.1002/dneu.20774
Corvin, A. P., Molinos, I., Little, G., Donohoe, G., Gill, M., Morris, D. W.,et al. (2012). Insulin-like growth factor 1 (IGF1) and its active peptide (1-3)IGF1 enhance the expression of synaptic markers in neuronal circuits through different cellular mechanisms. Neurosci. Lett. 520, 51–56. doi: 10.1016/j.neulet.2012.05.029
D’Ercole, A. J. (1996). Insulin-like growth factors and their receptors in growth. Endocrinol. Metab. Clin. North. Am. 25, 573–590. doi: 10.1016/S0889-8529(05)70341-8
Egan, M. F., Kojima, M., Callicott, J. H., Goldberg, T. E., Kolachana, B. S., Bertolino, A.,et al. (2003). The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112, 257–269. doi: 10.1016/S0092-8674(03)00035-7
Fiala, J. C., Feinberg, M., Popov, V., and Harris, K. M. (1998). Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J. Neurosci. 18, 8900–8911.
Fiala, J. C., Spacek, J., and Harris, K. M. (2002). Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res. Brain Res. Rev. 39, 29–54. doi: 10.1016/S0165-0173(02)00158-3
Figurov, A., Pozzo-Miller, L. D., Olafsson, P., Wang, T., and Lu, B. (1996). Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381, 706–709. doi: 10.1038/381706a0
Fukuda, T., Itoh, M., Ichikawa, T., Washiyama, K., and Goto, Y. (2005). Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. J. Neuropathol. Exp. Neurol. 64, 537–544.
Garey, L. (2010). When cortical development goes wrong: schizophrenia as a neurodevelopmental disease of microcircuits. J. Anat. 217, 324–333. doi: 10.1111/j.1469-7580.2010.01231.x
Grutzendler, J., Kasthuri, N., and Gan, W. B. (2002). Long-term dendritic spine stability in the adult cortex. Nature 420, 812–816. doi: 10.1038/nature01276
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326. doi: 10.1038/85899
Harris, K. M., and Stevens, J. K. (1989). Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J. Neurosci. 9, 2982–2997.
Holtmaat, A. J., Trachtenberg, J. T., Wilbrecht, L., Shepherd, G. M., Zhang, X., Knott, G. W.,et al. (2005). Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45, 279–291. doi: 10.1016/j.neuron.2005.01.003
Hoogenraad, C. C., and Akhmanova, A. (2010). Dendritic spine plasticity: new regulatory roles of dynamic microtubules. Neuroscientist 16, 650–661. doi: 10.1177/1073858410386357
Huang, E. J., and Reichardt, L. F. (2003). Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642. doi: 10.1146/annurev.biochem.72.121801.161629
Ji, Y., Pang, P. T., Feng, L., and Lu, B. (2005). Cyclic AMP controls BDNF-induced TrkB phosphorylation and dendritic spine formation in mature hippocampal neurons. Nat. Neurosci. 8, 164–172. doi: 10.1038/nn1381
Jiang, M., Ash, R. T., Baker, S. A., Suter, B., Ferguson, A., Park, J.,et al. (2013). Dendritic arborization and spine dynamics are abnormal in the mouse model of MECP2 duplication syndrome. J. Neurosci. 33, 19518–19533. doi: 10.1523/JNEUROSCI.1745-13.2013
Jugloff, D. G., Jung, B. P., Purushotham, D., Logan, R., and Eubanks, J. H. (2005). Increased dendritic complexity and axonal length in cultured mouse cortical neurons overexpressing methyl-CpG-binding protein MeCP2. Neurobiol. Dis. 19, 18–27. doi: 10.1016/j.nbd.2004.11.002
Kajiya, M., Takeshita, K., Kittaka, M., Matsuda, S., Ouhara, K., Takeda, K.,et al. (2014). BDNF mimetic compound LM22A-4 regulates cementoblast differentiation via the TrkB-ERK/Akt signaling cascade. Int. Immunopharmacol. 19, 245–252. doi: 10.1016/j.intimp.2014.01.028
Kasai, H., Fukuda, M., Watanabe, S., Hayashi-Takagi, A., and Noguchi, J. (2010). Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 33, 121–129. doi: 10.1016/j.tins.2010.01.001
Kasai, H., Matsuzaki, M., Noguchi, J., Yasumatsu, N., and Nakahara, H. (2003). Structure-stability-function relationships of dendritic spines. Trends Neurosci. 26, 360–368. doi: 10.1016/S0166-2236(03)00162-0
Khwaja, O. S., Ho, E., Barnes, K. V., O’Leary, H. M., Pereira, L. M., Finkelstein, Y.,et al. (2014). Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 111, 4596–4601. doi: 10.1073/pnas.1311141111
Kumar, V., Zhang, M. X., Swank, M. W., Kunz, J., and Wu, G. Y. (2005). Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J. Neurosci. 25, 11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005
Landi, S., Putignano, E., Boggio, E. M., Giustetto, M., Pizzorusso, T., and Ratto, G. M. (2011). The short-time structural plasticity of dendritic spines is altered in a model of Rett syndrome. Sci. Rep. 1, 45. doi: 10.1038/srep00045
Larimore, J. L., Chapleau, C. A., Kudo, S., Theibert, A., Percy, A. K., and Pozzo-Miller, L. (2009). Bdnf overexpression in hippocampal neurons prevents dendritic atrophy caused by Rett-associated MECP2 mutations. Neurobiol. Dis. 34, 199–211. doi: 10.1016/J.Nbd.2008.12.011
Levenga, J., and Willemsen, R. (2012). Perturbation of dendritic protrusions in intellectual disability. Prog. Brain Res. 197, 153–168. doi: 10.1016/B978-0-444-54299-1.00008-X
Li, W., Calfa, G., Larimore, J., and Pozzo-Miller, L. (2012). Activity-dependent BDNF release and TRPC signaling is impaired in hippocampal neurons of Mecp2 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 109, 17087–17092. doi: 10.1073/pnas.1205271109
Luebke, J. I., Weaver, C. M., Rocher, A. B., Rodriguez, A., Crimins, J. L., Dickstein, D. L.,et al. (2010). Dendritic vulnerability in neurodegenerative disease: insights from analyses of cortical pyramidal neurons in transgenic mouse models. Brain Struct. Funct. 214, 181–199. doi: 10.1007/s00429-010-0244–242
Luine, V., and Frankfurt, M. (2013). Interactions between estradiol, BDNF and dendritic spines in promoting memory. Neuroscience 239, 34–45. doi: 10.1016/j.neuroscience.2012.10.019
Marchetto, M. C., Carromeu, C., Acab, A., Yu, D., Yeo, G. W., Mu, Y.,et al. (2010). A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539. doi: 10.1016/j.cell.2010.10.016
Marin-Padilla, M. (1972). Structural abnormalities of the cerebral cortex in human chromosomal aberrations: a Golgi study. Brain Res. 44, 625–629. doi: 10.1016/0006-8993(72)90324-1
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y.,et al. (2003). DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302, 890–893. doi: 10.1126/science.1090842
Massa, S. M., Yang, T., Xie, Y., Shi, J., Bilgen, M., Joyce, J. N.,et al. (2010). Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J. Clin. Invest. 120, 1774–1785. doi: 10.1172/JCI41356
Matsuzaki, M., Ellis-Davies, G. C., Nemoto, T., Miyashita, Y., Iino, M., and Kasai, H. (2001). Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 4, 1086–1092. doi: 10.1038/nn736
Matsuzaki, M., Honkura, N., Ellis-Davies, G. C., and Kasai, H. (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766. doi: 10.1038/nature02617
McGraw, C. M., Samaco, R. C., and Zoghbi, H. Y. (2011). Adult neural function requires MeCP2. Science 333, 186. doi: 10.1126/science.1206593
Mizrahi, A., Crowley, J. C., Shtoyerman, E., and Katz, L. C. (2004). High-resolution in vivo imaging of hippocampal dendrites and spines. J. Neurosci. 24, 3147–3151. doi: 10.1523/JNEUROSCI.5218-03.2004
Moretti, P., Levenson, J. M., Battaglia, F., Atkinson, R., Teague, R., Antalffy, B.,et al. (2006). Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 26, 319–327. doi: 10.1523/JNEUROSCI.2623-05.2006
Murphy, D. D., and Segal, M. (1996). Regulation of dendritic spine density in cultured rat hippocampal neurons by steroid hormones. J. Neurosci. 16, 4059–4068.
Nagerl, U. V., Eberhorn, N., Cambridge, S. B., and Bonhoeffer, T. (2004). Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron 44, 759–767. doi: 10.1016/j.neuron.2004.11.016
Neul, J. L., and Zoghbi, H. Y. (2004). Rett syndrome: a prototypical neurodevelopmental disorder. Neuroscientist 10, 118–128. doi: 10.1177/1073858403260995
Nguyen, M. V., Du, F., Felice, C. A., Shan, X., Nigam, A., Mandel, G.,et al. (2012). MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J. Neurosci. 32, 10021–10034. doi: 10.1523/JNEUROSCI.1316-12.2012
Nimchinsky, E. A., Sabatini, B. L., and Svoboda, K. (2002). Structure and function of dendritic spines. Annu. Rev. Physiol. 64, 313–353. doi: 10.1146/annurev.physiol.64.081501.160008
Nusser, Z., Lujan, R., Laube, G., Roberts, J. D., Molnar, E., and Somogyi, P. (1998). Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron 21, 545–559. doi: 10.1016/S0896-6273(00)80565-6
Ogier, M., Wang, H., Hong, E., Wang, Q., Greenberg, M. E., and Katz, D. M. (2007). Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J. Neurosci. 27, 10912–10917. doi: 10.1523/JNEUROSCI.1869-07.2007
O’Kusky, J. R., Ye, P., and D’Ercole, A. J. (2000). Insulin-like growth factor-I promotes neurogenesis and synaptogenesis in the hippocampal dentate gyrus during postnatal development. J. Neurosci. 20, 8435–8442.
Park, M., Salgado, J. M., Ostroff, L., Helton, T. D., Robinson, C. G., Harris, K. M.,et al. (2006). Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron 52, 817–830. doi: 10.1016/j.neuron.2006.09.040
Penzes, P., Cahill, M. E., Jones, K. A., Vanleeuwen, J. E., and Woolfrey, K. M. (2011a). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293. doi: 10.1038/nn.2741
Penzes, P., Woolfrey, K. M., and Srivastava, D. P. (2011b). Epac2-mediated dendritic spine remodeling: implications for disease. Mol. Cell. Neurosci. 46, 368–380. doi: 10.1016/j.mcn.2010.11.008
Peters, A., and Kaiserman-Abramof, I. R. (1969). The small pyramidal neuron of the rat cerebral cortex. The synapses upon dendritic spines. Z. Zellforsch. Mikrosk. Anat. 100, 487–506. doi: 10.1007/BF00344370
Pitcher, M. R., Ward, C. S., Arvide, E. M., Chapleau, C. A., Pozzo-Miller, L., Hoeflich, A.,et al. (2013). Insulinotropic treatments exacerbate metabolic syndrome in mice lacking MeCP2 function. Hum. Mol. Genet. 22, 2626–2633. doi: 10.1093/hmg/ddt111
Poo, M. M. (2001). Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2, 24–32. doi: 10.1038/35049004
Purpura, D. P. (1974). Dendritic spine “dysgenesis” and mental retardation. Science 186, 1126–1128. doi: 10.1126/science.186.4169.1126
Qiu, Z., Sylwestrak, E. L., Lieberman, D. N., Zhang, Y., Liu, X. Y., and Ghosh, A. (2012). The Rett syndrome protein MeCP2 regulates synaptic scaling. J. Neurosci. 32, 989–994. doi: 10.1523/JNEUROSCI.0175-11.2012
Ramón y Cajal, S. (1888). Estructura de los centros nerviosos de las aves. Rev. Trim. Histol. Norm. Pat. 1, 1–10.
Ramón y Cajal, S. (1891a). Significación fisiológica de las expansiones protoplásmicas y nerviosas de la sustancia gris. Rev. De Cienc. Med. De Barc. 22, 23.
Ramón y Cajal, S. (1891b). Sur la structure de l’ecorce cerebrale de quelques mamiferes. La Cellule 7, 124–176.
Ramón y Cajal, S. (1894). La fine structure des centres nerveaux. the Croonian Lecture. Proc. R. Soc. Lond. 55, 443–468. doi: 10.1098/rspl.1894.0063
Ramón y Cajal, S. (1896). Les espines collaterales des cellules du cerveau colorees au bleu de methylene. Rev. Trim. Microgr. 1, 5–19.
Ramón y Cajal, S. (1933). Neuronismo o Reticularismo? Las Pruebas Objetivas De La Unidad Anatómica De Las Celulas Nerviosas. Madrid: Instituto Cajal.
Rochefort, N. L., and Konnerth, A. (2012). Dendritic spines: from structure to in vivo function. EMBO Rep. 13, 699–708. doi: 10.1038/embor.2012.102
Sala, C., and Segal, M. (2014). Dendritic spines: the locus of structural and functional plasticity. Physiol. Rev. 94, 141–188. doi: 10.1152/physrev.00012.2013
Schikorski, T., and Stevens, C. F. (1999). Quantitative fine-structural analysis of olfactory cortical synapses. Proc. Natl. Acad. Sci. U.S.A. 96, 4107–4112. doi: 10.1073/pnas.96.7.4107
Schmid, D. A., Yang, T., Ogier, M., Adams, I., Mirakhur, Y., Wang, Q.,et al. (2012). A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J. Neurosci. 32, 1803–1810. doi: 10.1523/JNEUROSCI.0865-11.2012
Segal, M. (2005). Dendritic spines and long-term plasticity. Nat. Rev. Neurosci. 6, 277–284. doi: 10.1038/nrn1649
Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J.,et al. (2002). Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35, 243–254. doi: 10.1016/S0896-6273(02)00768-7
Shirao, T., and Gonzalez-Billault, C. (2013). Actin filaments and microtubules in dendritic spines. J. Neurochem. 126, 155–164. doi: 10.1111/jnc.12313
Simmons, D. A., Belichenko, N. P., Yang, T., Condon, C., Monbureau, M., Shamloo, M.,et al. (2013). A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington’s disease. J. Neurosci. 33, 18712–18727. doi: 10.1523/JNEUROSCI.1310-13.2013
Smrt, R. D., Eaves-Egenes, J., Barkho, B. Z., Santistevan, N. J., Zhao, C., Aimone, J. B.,et al. (2007). Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol. Dis. 27, 77–89. doi: 10.1016/j.nbd.2007.04.005
Srivastava, D. P., and Penzes, P. (2011). Rapid estradiol modulation of neuronal connectivity and its implications for disease. Front. Endocrinol. (Lausanne) 2:77. doi: 10.3389/fendo.2011.00077
Stuss, D. P., Boyd, J. D., Levin, D. B., and Delaney, K. R. (2012). MeCP2 mutation results in compartment-specific reductions in dendritic branching and spine density in layer 5 motor cortical neurons of YFP-H mice. PLoS ONE 7:e31896. doi: 10.1371/journal.pone.0031896
Tanaka, J., Horiike, Y., Matsuzaki, M., Miyazaki, T., Ellis-Davies, G. C., and Kasai, H. (2008). Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science 319, 1683–1687. doi: 10.1126/science.1152864
Trachtenberg, J. T., Chen, B. E., Knott, G. W., Feng, G., Sanes, J. R., Welker, E.,et al. (2002). Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420, 788–794. doi: 10.1038/nature01273
Tropea, D., Giacometti, E., Wilson, N. R., Beard, C., Mccurry, C., Fu, D. D.,et al. (2009). Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 106, 2029–2034. doi: 10.1073/pnas.0812394106
Tyler, W. J., Alonso, M., Bramham, C. R., and Pozzo-Miller, L. D. (2002a). From acquisition to consolidation: on the role of brain-derived neurotrophic factor signalining in hippocampal-dependent learning. Learn. Mem. 9, 224–237. doi: 10.1101/lm.51202
Tyler, W. J., Perrett, S. P., and Pozzo-Miller, L. D. (2002b). The role of neurotrophins in neurotransmitter release. Neuroscientist 8, 524–531. doi: 10.1177/1073858402238511
Tyler, W. J., and Pozzo-Miller, L. D. (2001). BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J. Neurosci. 21, 4249–4258.
Tyler, W. J., and Pozzo-Miller, L. (2003). Miniature synaptic transmission and BDNF modulate dendritic spine growth and form in rat CA1 neurones. J. Physiol. 553, 497–509. doi: 10.1113/jphysiol.2003.052639
Vigers, A. J., Amin, D. S., Talley-Farnham, T., Gorski, J. A., Xu, B., and Jones, K. R. (2012). Sustained expression of brain-derived neurotrophic factor is required for maintenance of dendritic spines and normal behavior. Neuroscience 212, 1–18. doi: 10.1016/j.neuroscience.2012.03.031
Wang, H., Chan, S. A., Ogier, M., Hellard, D., Wang, Q., Smith, C.,et al. (2006). Dysregulation of brain-derived neurotrophic factor expression and neurosecretory function in Mecp2 null mice. J. Neurosci. 26, 10911–10915. doi: 10.1523/JNEUROSCI.1810-06.2006
Xu, X., Kozikowski, A. P., and Pozzo-Miller, L. (2014). A selective histone deacetylase-6 inhibitor improves BDNF trafficking in hippocampal neurons from Mecp2 knockout mice: implications for Rett syndrome. Front. Cell. Neurosci. 8:68. doi: 10.3389/fncel.2014.00068
Yuste, R. (2010). Dendritic Spines. Cambridge, MA: The MIT Press. doi: 10.7551/mitpress/9780262013505.001.0001
Yuste, R., and Bonhoeffer, T. (2001). Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu. Rev. Neurosci. 24, 1071–1089. doi: 10.1146/annurev.neuro.24.1.1071
Yuste, R., Majewska, A., and Holthoff, K. (2000). From form to function: calcium compartmentalization in dendritic spines. Nat. Neurosci. 3, 653–659. doi: 10.1038/76609
Zeev, B. B., Bebbington, A., Ho, G., Leonard, H., De Klerk, N., Gak, E.,et al. (2009). The common BDNF polymorphism may be a modifier of disease severity in Rett syndrome. Neurology 72, 1242–1247. doi: 10.1212/01.wnl.0000345664.72220.6a
Zhao, X., Pak, C., Smrt, R. D., and Jin, P. (2007). Epigenetics and Neural developmental disorders: Washington DC, September 18 and 19, 2006. Epigenetics 2, 126–134. doi: 10.4161/epi.2.2.4236
Zheng, W. H., and Quirion, R. (2004). Comparative signaling pathways of insulin-like growth factor-1 and brain-derived neurotrophic factor in hippocampal neurons and the role of the PI3 kinase pathway in cell survival. J. Neurochem. 89, 844–852. doi: 10.1111/j.1471-4159.2004.02350.x
Zhou, Q., Homma, K. J., and Poo, M. M. (2004). Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44, 749–757. doi: 10.1016/j.neuron.2004.11.011
Keywords: MeCP2, BDNF, excitatory synapse, spine density, hippocampus, organotypic slice cultures, TrkB, autism spectrum disorder
Citation: Xu X, Miller EC and Pozzo-Miller L (2014) Dendritic spine dysgenesis in Rett syndrome. Front. Neuroanat. 8:97. doi: 10.3389/fnana.2014.00097
Received: 19 July 2014; Accepted: 25 August 2014;
Published online: 10 September 2014.
Edited by:
Nicolas Heck, University Pierre and Marie Curie, FranceReviewed by:
Craig M. Powell, University of Texas Southwestern Medical Centre, USASerena Dudek, National Institute of Environmental Health Sciences, USA
Copyright © 2014 Xu, Miller and Pozzo-Miller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucas Pozzo-Miller, Department of Neurobiology, Civitan International Research Center, The University of Alabama at Birmingham, SHEL-1002, 1825 University Boulevard, Birmingham, AL, 35294-2182, USA e-mail:bHVjYXNwbUB1YWIuZWR1