 Aaron D. Levy
Aaron D. Levy Mitchell H. Omar
Mitchell H. Omar Anthony J. Koleske
Anthony J. Koleske- 1Interdepartmental Neuroscience Program, Yale University, New Haven, CT, USA
- 2Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT, USA
- 3Department of Neurobiology, Yale University, New Haven, CT, USA
Dendritic spines are the receptive contacts at most excitatory synapses in the central nervous system. Spines are dynamic in the developing brain, changing shape as they mature as well as appearing and disappearing as they make and break connections. Spines become much more stable in adulthood, and spine structure must be actively maintained to support established circuit function. At the same time, adult spines must retain some plasticity so their structure can be modified by activity and experience. As such, the regulation of spine stability and remodeling in the adult animal is critical for normal function, and disruption of these processes is associated with a variety of late onset diseases including schizophrenia and Alzheimer’s disease. The extracellular matrix (ECM), composed of a meshwork of proteins and proteoglycans, is a critical regulator of spine and synapse stability and plasticity. While the role of ECM receptors in spine regulation has been extensively studied, considerably less research has focused directly on the role of specific ECM ligands. Here, we review the evidence for a role of several brain ECM ligands and remodeling proteases in the regulation of dendritic spine and synapse formation, plasticity, and stability in adults.
Dendritic Spines are Highly Structured Postsynaptic Signaling Compartments
Dendritic spines are protrusions from the dendrite shaft of neurons that comprise the receptive contact at most excitatory synapses in the brain (Gray, 1959a,b; Harris and Kater, 1994; Hering and Sheng, 2001). Ultrastructurally, dendritic spines are composed of a thin neck supported by unbranched filamentous actin (F-actin) and a bulbous head containing a network of branched F-actin (Korobova and Svitkina, 2010; Tønnesen et al., 2014). The spine head also contains the membrane-associated postsynaptic density (PSD), a highly organized network of neurotransmitter receptors, adhesion receptors, scaffolding proteins, and downstream signaling molecules (Harris and Stevens, 1989; Kennedy, 1994, 1997; Hunt et al., 1996; Walikonis et al., 2000; Sheng and Kim, 2011; Harris and Weinberg, 2012). Scaffolding proteins and cell adhesion molecules (CAMs) connect the PSD to the spine actin cytoskeleton. Adhesion molecules also connect to both the presynaptic partner and the extracellular matrix (ECM) in and around the synaptic cleft (Figure 1A). These and other dendritic spine proteins regulate actin filament formation, turnover, and stability, thereby controlling dendritic spine structure.
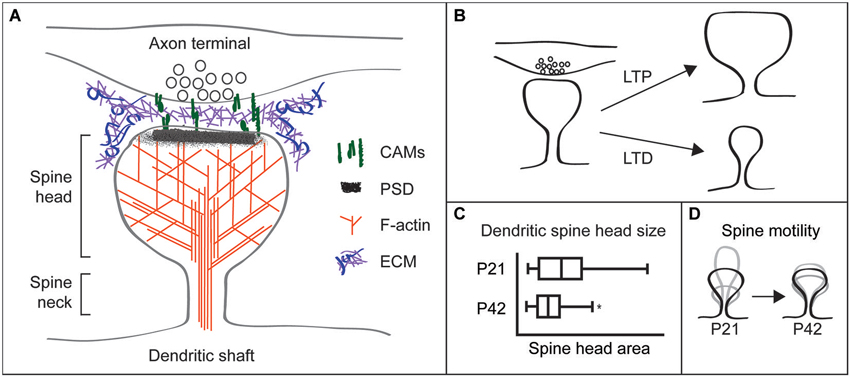
Figure 1. Dendritic spines are highly structured and plastic synaptic specializations. (A) Schematic of a dendritic spine apposed to a presynaptic terminal. The spine head and neck are supported by a network of filamentous (F)-actin. Postsynaptic cell adhesion molecules (CAMs) connect to the postsynaptic density (PSD) and F-actin in the spine, and extend from the spine to associate with CAMs on the presynaptic terminal. Perisynapic and putative synaptic cleft extracellular matrix (ECM) may interact with multiple CAMs and other cell surface receptors. (B) Spine structural changes accompany synaptic plasticity, with long-term potentiation (LTP) increasing spine head size and long-term depression (LTD) decreasing head size. (C) Mouse hippocampal CA1 neuron spine head sizes were obtained from electron microscopy of the stratum radiatum. Spine head size and its variance decrease as animals mature from P21 (juvenile) to P42 (adult). Figure modified with permission from Kerrisk et al. (2013). (D) Spine motility, defined as changes in spine length over time, is high in juvenile animals. By contrast, spines from adult animals are relatively immotile.
Spines have a unique structure that is intrinsic to their function. The thin spine neck, ~100–300 nm in diameter (Harris and Stevens, 1989; Tønnesen et al., 2014), restricts diffusion to compartmentalize biochemical and electrical postsynaptic signaling (Majewska et al., 2000; Yuste et al., 2000; Sabatini et al., 2002; Noguchi et al., 2005; Carter and Sabatini, 2008; Harvey et al., 2008; Higley and Sabatini, 2012; Takasaki and Sabatini, 2014; Tønnesen et al., 2014). This compartmentalization enables molecular modifications specific to individual spines and synapses, including changes in synaptic efficacy and spine shape and size. Overall spine head size varies considerably among spines, from ~200–1400 nm in diameter (Harris and Stevens, 1989; Tønnesen et al., 2014). Spine size correlates with synaptic strength and larger spines commonly contain larger PSDs with more AMPA-type glutamate receptors and appose axon terminals with larger readily-releasable pools of neurotransmitter (Harris and Stevens, 1988, 1989; Matsuzaki et al., 2001). Therefore, large spines are more likely to produce strong excitatory postsynaptic currents and have greater influence on neuronal firing and network signaling.
Dendritic Spine Structure is Dynamic and Regulated by Activity and Development
Recent advances in imaging and single synapse stimulation techniques have revealed that the size and transmission properties of individual dendritic spines can be altered rapidly in response to synaptic activity. Use of glutamate uncaging at individual spines has shown that long-term activity-dependent synaptic strengthening or weakening, also known as long-term potentiation (LTP) and long-term depression (LTD), respectively, occur at discrete synapses and are associated with changes in spine size. High frequency synaptic stimulation that causes LTP promotes spine head enlargement, while low frequency stimulation that causes LTD results in spine head shrinkage (Matsuzaki et al., 2004; Nägerl et al., 2004; Zhou et al., 2004; Oh et al., 2013; Figure 1B). Furthermore, smaller spines are more likely to be lost following LTD-inducing stimulation paradigms (Bastrikova et al., 2008).
Experiments using longitudinal transcranial imaging of individual cortical spines support these ex vivo studies. Manipulating sensory input alters the likelihood that dendritic spines will or will not be lost (spine stability) over days, weeks, and months (Oray et al., 2004; Zuo et al., 2005a,b; Lai et al., 2012). Additionally, in vivo imaging experiments in mouse models show that stress and genetic abnormalities disrupt normal spine structural dynamics and stability (Pan et al., 2010; Liston et al., 2013). Excitingly, studies using imaging probes that report the activity of specific signaling pathways are beginning to elucidate the signaling events that underlie long-term changes in spine size and signaling properties (Murakoshi et al., 2011; Murakoshi and Yasuda, 2012; Lai and Ip, 2013; Oh et al., 2013; Zhai et al., 2013).
Spine structural plasticity is also heavily influenced by developmental stage. Juvenile animals have increased variance in spine head size (Sfakianos et al., 2007; Kerrisk et al., 2013; Figure 1C) and much more dynamic spine motility relative to spines in adult animals (Dunaevsky et al., 1999; Trachtenberg et al., 2002; Majewska and Sur, 2003; Holtmaat et al., 2005; Figure 1D). Furthermore, higher levels of spine formation and loss occur in adolescent mice vs. adults (Grutzendler et al., 2002).
While the age-dependent loss of spine plasticity has been reproducibly observed, the mechanisms that underlie this phenomenon are not well understood. Multiple synaptic proteins and signaling events differ between juvenile and adult animals as well as between wild type and disease-model animals, which might help to explain differences in spine stability (Scheetz and Constantine-Paton, 1994; Wu et al., 2009; Gundelfinger et al., 2010; Charrier et al., 2012; Akbik et al., 2013; Koleske, 2013). These observations do not, however, directly address whether or how specific pairing of pre- and post-synaptic compartments induces the machinery and mechanisms that confer increased synapse and dendritic spine stability. While it is a difficult experimental question to address, insights into this question are crucial to understanding neurological disorders and how we can gain control of synaptic flexibility.
Brain Disorders Involve Loss of Dendritic Spine Stability
Loss of dendritic spine stability in adulthood underlies several major neurological and psychiatric disorders, which are accompanied by perceptual, cognitive, memory, and behavioral deficits. For instance, cortical neurons in patients with Alzheimer’s disease, Parkinson’s disease, and other neurodegenerative disorders or dementia have decreased synapse and spine densities (Catalá et al., 1988; Katzman, 1989; Terry et al., 1991; Scheff and Price, 2003). Schizophrenia patients also have reduced cortical spine densities (Garey et al., 1998; Glantz and Lewis, 2000), and medium spiny neurons in Huntington’s disease patients show spine densities that are increased earlier and reduced later in disease progression (Ferrante et al., 1991). Whether spine loss causes disease or results from other problems is unknown, but disrupted network connectivity via spine loss may underlie the cognitive deficits that occur in these patients. These observations demonstrate the importance of dendritic spine stability for normal brain function and suggest that a deeper comprehension of spine stabilization mechanisms could lead to a better understanding of these diseases and possibly new therapeutic approaches.
Extracellular Matrix Receptors Control Dendritic Spine Stability and Remodeling
Several studies demonstrate that specific ECM receptors can regulate dendritic spine stability and remodeling. Brain ECM is composed of secreted proteins and proteoglycans that assemble into cross-linked meshworks to provide structural support to the surrounding cells (Barros et al., 2011; Dansie and Ethell, 2011; Wlodarczyk et al., 2011; Soleman et al., 2013). The brain ECM forms a gel that surrounds neurons and glia, including the space adjacent to and between synapses (Nicholson and Syková, 1998). There, pre- and postsynaptic CAMs associate with one another and with the ECM to initiate and maintain synaptic contact (Bukalo and Dityatev, 2012; Missler et al., 2012). These transmembrane cell adhesion proteins connect to the intracellular dendritic spine actin network and influence the activities of actin regulatory molecules, thereby controlling spine shape (Huntley et al., 2002; Washbourne et al., 2004; Lin and Koleske, 2010; Benson and Huntley, 2012; Cheadle and Biederer, 2012; Sloniowski and Ethell, 2012; Koleske, 2013). Many adhesion molecules also influence synaptic transmission, a key regulator of spine structure (Chan et al., 2006, 2007; Huang et al., 2006; Shi and Ethell, 2006; Bukalo and Dityatev, 2012).
Integrin adhesion receptors are a major family of ECM receptors. Engagement of ECM by integrins triggers changes in cell morphology and motility powered by actin cytoskeletal rearrangements in diverse cell types (Horwitz et al., 1986; Tamkun et al., 1986; Otey and Burridge, 1990; Tawil et al., 1993; Wang et al., 1993; Chong et al., 1994; Gumbiner, 1996; Schwartz and Horwitz, 2006; Schwartz, 2010). Integrins are crucial in the brain as well, where they mediate processes such as migration, axonal outgrowth and pathfinding, and synaptic plasticity (DeFreitas et al., 1995; Chan et al., 2006, 2007; Shi and Ethell, 2006; Belvindrah et al., 2007a,b). Integrin signaling also modulates spine head size and stability during adolescence in mice (Warren et al., 2012; Kerrisk et al., 2013).
Other ECM receptors also function in the brain during adulthood, where they may stabilize spines. For example, dystroglycan, part of the dystrophin glycoprotein complex, plays important roles in axonal pathfinding (Wright et al., 2012) and synapse formation (Sato et al., 2008), but also associates with mature inhibitory synapses and modulates synaptic plasticity (Lévi et al., 2002; Satz et al., 2010; Pribiag et al., 2014). ApoER2, a receptor for the ECM protein reelin, is expressed from late embryonic periods through adulthood, where it is essential for proper migration of cortical neurons in development (Hack et al., 2007) but also plays roles in synapse maintenance and plasticity (Beffert et al., 2005, 2006; Trotter et al., 2011). These and other ECM receptor studies provide strong evidence that ECM regulates dendritic spine stability and remodeling.
Studying ECM-Mediated Control of Spine Structure Poses Unique Difficulties
Extracellular matrix molecules at synapses are likely candidates for regulators of synapse and dendritic spine stability. While studies have identified ECM receptors important for neuronal function and dendritic spine morphology, they often fail to identify the critical ECM ligands that drive these important processes. This failure may be partially due to the inherent difficulty of studying ECM components. Extracellular matrix molecules are secreted, so in the brain where many different cell types are intermingled, cell origin and site of function can be difficult to identify. The heterogenous cell population in the brain also complicates purification of ECM molecules from specific cell types. Additionally, many ECM molecules are large, such as laminins (800 kDa), and can have multiple interaction domains from the same molecule driving distinct pathways (Colognato and Yurchenco, 2000). Extracellular matrix biochemical activities are also altered by covalent modification and/or proteolytic processing, which can be triggered by synaptic activity (Nedivi et al., 1993; Qian et al., 1993; Sung et al., 1993; Szklarczyk et al., 2002; Chen et al., 2008; Horejs et al., 2014). Furthermore, it can be difficult to disentangle possible functions of ECM components in spine stabilization, e.g., providing extracellular rigidity, mediating spine-ECM adhesions, and/or inducing intracellular signaling cascades (Figure 2).
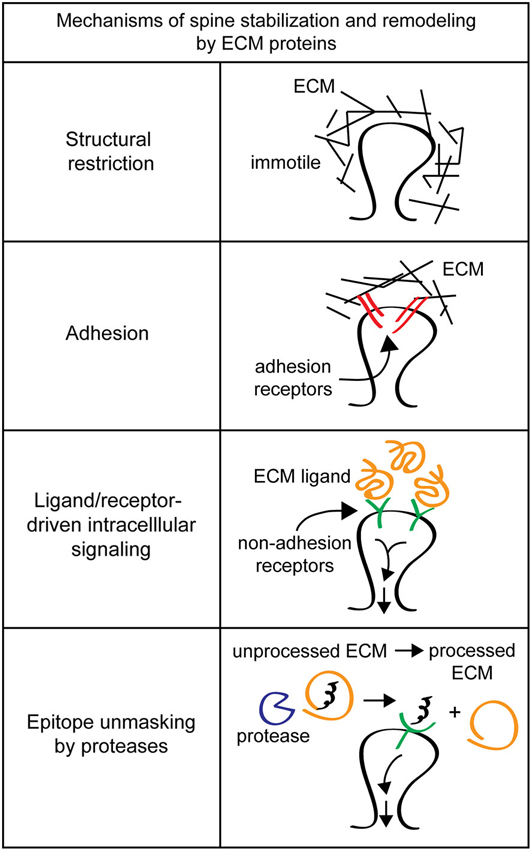
Figure 2. Mechanisms of spine stabilization and remodeling by extracellular matrix (ECM) proteins. Extracellular matrix components can stabilize and remodel dendritic spines by a variety of different mechanisms. Structural restriction: ECM components such as chondroitin sulfate proteoglycans (CSPGs) can form a matrix around dendritic spines to provide extracellular rigidity and physically restrict spine motion. Adhesion: classical ECM proteins such as fibronectin and RGD-containing proteins can act as adhesion substrates and bind to integrin adhesion receptors to remodel spines. Ligand/receptor-driven intracellular signaling: ECM proteins like reelin function as ligands for non-adhesion receptors to drive intracellular signaling cascades that regulate spine remodeling and formation. Epitope unmasking by proteases: extracellular proteases such as tissue plasminogen activator (tPA) and the matrix metalloproteinase (MMPs) can cleave ECM proteins to reveal cryptic ligands that drive intracellular signaling to change spine morphology.
Regardless of the challenges posed by studying the roles of ECM in dendritic spine and synapse stability, emerging evidence indicates that specific ECM components are key regulators of dendritic spine and synapse structure, plasticity, and stability. Here, we review the evidence that specific ECM components and their interaction partners control dendritic spine and synapse structure and how remodeling of the ECM may contribute to dendritic spine plasticity and stability in adults.
ECM Proteins are Key Regulators of Dendritic Spine and Synapse Stability and Remodeling
Chondroitin Sulfate Proteoglycans Restrict Functional Plasticity and Stabilize Spines
Chondroitin sulfate proteoglycans (CSPGs), including the lecticans (aggrecan, neurocan, versican and brevican), phosphacan, and leucine-rich CSPGs, are major components of the mature brain ECM. Each CSPG consists of a multi-domain protein core, important for interactions with other ECM molecules, as well as multiple glycosaminoglycan (GAG) side chains that can be degraded by the bacterial enzyme chondroitinaseABC (chABC). The GAG chains are critical for many CSPG functions (Galtrey and Fawcett, 2007), and the pattern of sulfation can define the specific response of the CSPG to signaling partners (Gama et al., 2006). Some CSPGs, notably brevican (Yamada et al., 1994), also exist in non-proteoglycan forms, and loss of CSPG protein core genes is associated with neurological disease (Cichon et al., 2011; Mühleisen et al., 2012). Many CSPGs assemble to form dense peri-neuronal nets (PNNs) around inhibitory neurons (Kwok et al., 2011), which can be identified by staining for GAG epitopes. In addition, a subset of excitatory neurons are also surrounded by more diffuse CSPGs (Wegner et al., 2003).
While the role of specific CSPG core proteins in dendritic spine structure and plasticity has not been extensively studied, a wealth of evidence indicates that the GAG chains of CSPGs restrict circuit plasticity in vivo, particularly in the visual system. In the primary visual cortex of rodents and other mammals, cells that receive geniculocortical inputs representing both eyes typically respond more strongly to stimulation of one eye, a phenomenon called ocular dominance (OD). Monocular deprivation (MD) enforced by closing one eye increases the proportion of cells that respond to stimulation of the open eye while reducing the proportion that respond to the closed eye, but only during a critical period for OD plasticity from P19–P35 (Wiesel and Hubel, 1963; Gordon and Stryker, 1996). Chondroitin sulfate proteoglycan expression in primary visual cortex increases through this critical period, and rearing mice in the dark, which delays critical period closure, also delays the developmental increase in CSPGs. This suggests that CSPG expression may be causally linked to the age-dependent loss of plasticity (Pizzorusso et al., 2002). Indeed, while MD normally cannot induce OD plasticity in adult rats after the critical period, MD can shift OD in adult rats that have had chABC injected directly into primary visual cortex (Pizzorusso et al., 2002). In a similar critical period plasticity paradigm, fear memories can be robustly erased by extinction training only during an early critical period, but chABC degradation of PNNs in the amygdala reinstates the ability to erase fear memories in adult rats (Gogolla et al., 2009). In addition, mice lacking the CSPGs neurocan or brevican have deficits in LTP maintenance without other apparent developmental defects, suggesting a role for CSPGs in adults (Zhou et al., 2001; Brakebusch et al., 2002). These results demonstrate that CSPGs are critical for the functional stability of neuronal circuits in vivo.
Chondroitin sulfate proteoglycans normally stabilize dendritic spines. The physiological changes induced by MD are associated with a reduction in spine density of layer II/III visual cortical neurons responsive to the deprived eye (Mataga et al., 2004; Pizzorusso et al., 2006). This loss of spines can be rescued by opening the deprived eye and closing the previously open eye, but only in juvenile animals. However, chABC treatment reinstates this plasticity in adult animals, demonstrating that CSPGs normally stabilize existing spines (Pizzorusso et al., 2006). Loss of CSPGs also enhances spine motility, measured as the magnitude of fluctuations in spine length over time (Figure 1C). Spine motility decreases with age (Majewska and Sur, 2003), but chABC treatment of adult visual cortex in vivo and of hippocampal organotypic slices in vitro enhances spine motility (Orlando et al., 2012; de Vivo et al., 2013), reverting spines to a more immature phenotype. This is similar to the effect of MD, which also increases spine motility (Oray et al., 2004). These results demonstrate that CSPGs stabilize dendritic spine structure and movement (Figures 3A,B).
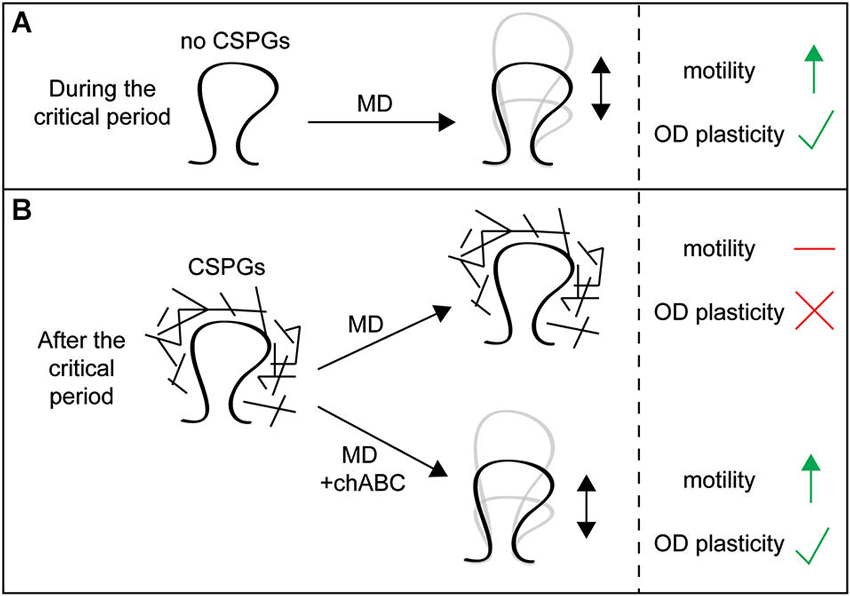
Figure 3. Chondroitin sulfate proteoglycans around spines restrict spine dynamics and functional plasticity. (A) In juvenile animals during the critical period, CSPG expression is low and visual monocular deprivation (MD) can increase spine motility in primary visual cortex and drive changes in ocular dominance (OD) plasticity. (B) In adult animals after the critical period, CSPG expression is high and MD can no longer increase spine motility or drive OD plasticity. However, treatment with chondroitinaseABC (chABC) to degrade CSPG glycosaminoglycan (GAG) chains allows MD to once again increase spine motility and drive OD plasticity in adults, demonstrating that CSPGs restrict spine remodeling and functional plasticity in adult animals.
Chondroitin sulfate proteoglycans interact with interneurons in PNNs and the development of inhibitory circuits is associated with closure of the critical period, suggesting the effects of chABC treatment on OD plasticity and spine stability may reflect alterations of inhibitory circuits (Pizzorusso et al., 2002). However, emerging evidence indicates that CSPGs can also act directly on spines, independently of PNNs and GABAergic neurons. Orlando et al. (2012) have shown that microinjection of chABC into the stratum radiatum of hippocampal slices, which lacks PNNs but has diffuse CSPG staining, increases CA1 pyramidal neuron spine motility and the number of spines with outgrowths from their heads, mimicking the effects of chABC bath application. This demonstrates that CSPGs normally stabilize spine structure and reduce spine head outgrowths independently of PNNs and inhibitory function (Orlando et al., 2012). These increases in both motility and spine head outgrowth with chABC application require β1 integrin function, which has been shown to be involved in dendrite and spine stability (Warren et al., 2012). While CSPGs interact with and inhibit integrin function (Wu et al., 2002; Tan et al., 2011), whether these specific interactions regulate spine stability is unclear, and should be the target of future studies.
RGD Peptides, and Possibly Fibronectin, Regulate Dendritic Spine Remodeling
In the brain, there is little expression of most of the fibrous ECM proteins such as fibronectin, vitronectin, and the collagens that are major ECM components in other tissues (Ruoslahti, 1996a). Fibronectin mRNA and protein can be detected at low levels in discrete populations of neurons and astroglia, and its expression is increased in the hippocampus of adult animals by kainic acid treatment (Hoffman et al., 1998), but very little is known about the function of fibronectin in the brain. Instead, researchers have more commonly used synthetic peptides common to fibronectin and other matrix proteins that carry an arginine-glycine-aspartate (RGD) motif critical for binding to integrins and for adhesion (Ruoslahti and Pierschbacher, 1987; Ruoslahti, 1996b). RGD peptides have also been shown to evoke changes in synaptic plasticity and structural stability. For example, RGD peptides block the maintenance phase of LTP (Staubli et al., 1990; Bahr et al., 1997; Chun et al., 2001) and potentiate NMDA receptor (NMDAR) currents (Lin et al., 2003; Bernard-Trifilo et al., 2005), demonstrating that RGD-containing proteins may be involved in adult plasticity. RGD peptides also regulate structural stability in mature neurons, as treatment of 14 DIV cultured hippocampal neurons with RGD peptides induces an elongation of existing spines and causes filopodia formation. These changes can be blocked by NMDAR and CaMKII antagonists, suggesting that integrins regulate the stability of dendritic spines via NMDARs and CaMKII in vitro (Shi and Ethell, 2006). To be clear, studies using RGD peptides do not demonstrate that any specific RGD-containing ECM protein functions in the brain, as many extracellular proteins have RGD motifs, but they strongly suggest the involvement of integrin receptors in these diverse processes. In addition to traditional “outside-in” integrin activation described above, integrin adhesion can be activated by intracellular signaling partners in an “inside-out” mechanism (Calderwood, 2004; Moser et al., 2009). Inside-out signaling is active but has not been well studied in neurons, and may help explain changes in integrin-mediated ECM contact with changes in neuronal activity. For example, it has recently been shown that reelin signals through its receptors ApoER2 and VLDLR to promote integrin α5β1 adhesion to fibronectin by an inside-out mechanism to control neuronal positioning during cortical development (Sekine et al., 2012). More work needs to be done in the future to establish which RGD-containing brain proteins have effects on synaptic signaling, plasticity and spine structure, and how these signaling mechanisms interact with inside-out signaling pathways.
Reelin Enhances Glutamatergic Transmission and Plasticity and may Stabilize Spines
Reelin is a 385 kDa secreted ECM protein that is a key regulator of neuronal migration in development (Tissir and Goffinet, 2003; D’Arcangelo, 2014). However, even after neurons have reached their proper destination, reelin continues to modulate synaptic signaling pathways to control dendritic spine structure and synaptic plasticity. Reelin is expressed by layer I and II GABAergic interneurons, primarily Cajal-Retzius cells (Rodriguez et al., 2000), which project to other cortical layers where they secrete reelin into the ECM. Upon release, reelin surrounds and adheres to dendritic shafts and spines of cortical pyramidal cells (Rodriguez et al., 2000; Pappas et al., 2001), suggesting that it might regulate spine structure (Rodriguez et al., 2000). Indeed, younger (P21-P31) heterozygous reelin+/− mice, which have grossly normal neuron positioning but only half the level of reelin (Liu et al., 2001; Pappas et al., 2001), have significantly reduced dendritic spine density and altered spine morphology (Liu et al., 2001; Niu et al., 2008; Iafrati et al., 2014). Interestingly, by adulthood reelin+/− mice exhibit only minimal spine density loss compared to wild type, suggesting that compensatory mechanisms promote additional spine development when reelin levels are reduced (Ventruti et al., 2011).
In adult animals, reelin continues to promote synaptic function and regulate spine morphology. Adult reelin+/− mice have reduced levels of synaptic signaling molecules (Ventruti et al., 2011) as well as deficits in excitatory postsynaptic responses, LTP, and LTD (Qiu et al., 2006a), while addition of recombinant reelin to hippocampal slices or direct injection into the ventricles significantly enhances hippocampal LTP (Beffert et al., 2005; Pujadas et al., 2010; Rogers et al., 2011). Recombinant reelin also increases NMDA and AMPA currents in cultured hippocampal slices and primary hippocampal cultures (Chen et al., 2005; Qiu et al., 2006b; Groc et al., 2007; Qiu and Weeber, 2007). These results together demonstrate that reelin is both necessary and sufficient for adult plasticity and glutamatergic signaling. Reelin is also sufficient to promote spine remodeling, as postnatal overexpression of reelin in the mouse forebrain increases spine head size as well as the number of spines with multiple synaptic contacts, while leaving spine density unchanged. Turning off this reelin overexpression decreases spine size and density, indicating that in some cases reelin may also interact with spine stability mechanisms (Pujadas et al., 2010). In addition, injection of recombinant reelin into the ventricles of adult mice increases hippocampal CA1 spine density (Rogers et al., 2011), suggesting that reelin may also promote spine formation in adults. Interestingly, injection of reelin leads to an hours-long transient increase in reelin levels (Rogers et al., 2011), while genetic overexpression would cause a constant increase, suggesting the timing and duration of reelin expression may be important for its effect on spines. Together, these results indicate that in different contexts, reelin promotes synaptic transmission and plasticity and modulates spine dynamics and stability. Further work on stability would benefit from a conditional reelin knockout mouse that could be used to test the necessity of reelin for spine stability in adult animals.
Reelin levels also appear to affect disease pathology in humans. For example, reelin expression is reduced approximately 50% in patients with schizophrenia (Impagnatiello et al., 1998; Berretta, 2012) and reelin haploinsufficiency in mice causes increased neuron packing density, decreased GAD67 levels, reduced pre-pulse inhibition and loss of dendritic spines, all features associated with schizophrenia pathology (Tueting et al., 1999; Liu et al., 2001). Reelin may also be neuroprotective against Alzheimer’s disease, as it has been shown to interact with soluble amyloid-β42, protect against amyloid-β42-induced spine loss and neuron death in cultured neurons, and reduce amyloid plaque development and memory loss in J20 Alzheimer’s model mice (Pujadas et al., 2014). Together, these data show that reelin, which has important roles in neuron development and positioning, also plays critical roles in late onset diseases after development is complete.
Agrin Promotes Filopodia and Dendritic Spine Formation
Agrin is best known for its prominent role in development of the vertebrate neuromuscular junction (NMJ) synapse, where it is deposited by motor neurons to induce acetylcholine receptor clustering in the muscle (Gautam et al., 1996; Glass et al., 1996; Sanes and Lichtman, 2001). Agrin is also widely expressed in the brain, with the highest levels of agrin expression coinciding with the peak period of synaptic development (O’Connor et al., 1994; Cohen et al., 1997). Indeed, antisense-mediated knockdown of agrin inhibits synapse development in cultured neurons (Ferreira, 1999; Bose et al., 2000). In contrast to knockdown systems, however, cultured agrin−/− neurons do not exhibit synaptic deficits (Li et al., 1999; Serpinskaya et al., 1999), suggesting that compensatory mechanisms may arise in the absence of endogenous agrin (Bose et al., 2000). Interestingly, knockdown of agrin in both mature (McCroskery et al., 2009) and immature (McCroskery et al., 2006) neuronal cultures reduces dendritic filopodia number, and agrin overexpression or clustering in immature cultured rat and mouse hippocampal neurons is sufficient to induce filopodia in vitro (Annies et al., 2006; McCroskery et al., 2006). As filopodia are precursors for dendritic spines (Ziv and Smith, 1996), these results support a role for agrin in promoting synapse and spine formation even in mature neurons. Indeed, agrin−/− mice in which perinatal lethality is rescued by muscle-specific agrin re-expression exhibit decreased cortical dendritic spine density (Ksiazek et al., 2007).
A role for agrin in spine and synapse stability is also supported by studies of neurotrypsin (also called motopsin or Prss12), an extracellular protease whose only known substrate is agrin (Gschwend et al., 1997; Molinari et al., 2002; Reif et al., 2007; Stephan et al., 2008). neurotrypsin−/− mice have reduced CA1 apical dendritic spine density (Mitsui et al., 2009). Neurotrypsin is released from presynaptic neurons in response to NMDAR-mediated activity and cleaves agrin at the synapse, suggesting that neurotrypsin might be involved in activity-dependent plasticity. Indeed, while neurons in hippocampal slices from adult neurotrypsin−/− mice have normal electrophysiological LTP, LTP-inducing stimuli fail to induce the formation of new filopodia in the knockouts, suggesting that neurotrypsin is required for some aspects of structural plasticity that accompany LTP in adult animals. Interestingly, a soluble cleavage fragment of agrin produced by neurotrypsin can rescue the loss of LTP-induced filopodia formation in neurotrypsin−/− mice (Matsumoto-Miyai et al., 2009; Figure 4). These results suggest a role for neurotrypsin and agrin in supporting new spine formation following LTP induction protocols in mature animals. Further work should address the molecular mechanisms downstream of agrin cleavage that promote filopodia formation and whether and how these new filopodia form functional dendritic spines and synapses.
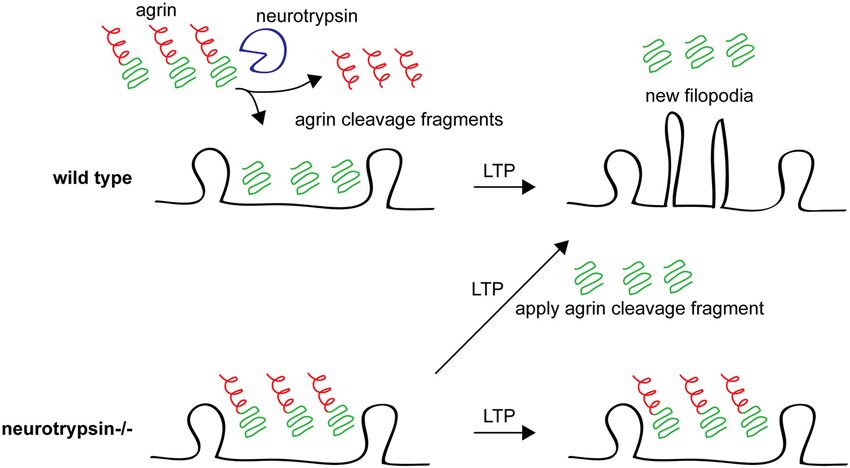
Figure 4. Agrin cleavage by neurotrypsin plays an important role in filopodia formation following LTP. In wild type animals after an LTP stimulus, agrin is cleaved by neurotrypsin (top left) and the agrin fragment promotes growth of new dendritic filopodia (top right). In neurotrypsin knockout mice, agrin cannot be cleaved (bottom left) and new filopodia are not formed in response to an LTP-inducing stimulus (bottom right). However, application of a soluble recombinant neurotrypsin-dependent agrin cleavage fragment rescues this phenotype, promoting new filopodia growth after LTP even in neurotrypsin knockout hippocampal slices. See Matsumoto-Miyai et al. (2009).
Tenascins are Required for Synaptic Plasticity and may Interact with Spines
The tenascins are a family of ECM proteins that oligomerize through a tenascin association domain and interact with other ECM proteins and receptors through tenascin’s EGF-like and fibronectin type III-repeats (Jones and Jones, 2000). Tenascin R (TNR) and tenascin C (TNC) are both expressed in the brain, TNR exclusively so, where they play roles in synaptic plasticity.
Tenascin R is required for normal plasticity, synaptic transmission, and behavior. Tenascin R knockout mice have impaired hippocampal LTP but normal LTD, increased basal synaptic transmission, and anxiety and motor deficits (Bukalo et al., 2001; Saghatelyan et al., 2001; Freitag et al., 2003; Gurevicius et al., 2004). Tenascin R is mainly associated with CSPGs in PNNs around inhibitory interneurons (Brückner et al., 2000), where it crosslinks some CSPG family members (Aspberg et al., 1997). Tenascin R’s affect on LTP is due to it carrying the human natural killer-1 (HNK1) carbohydrate epitope (Kruse et al., 1985) which normally interferes with γ-aminobutyric acid type B GABAB receptor function. GABAB receptors block GABAA receptor-mediated inhibition by reducing presynaptic GABA release through a retrograde mechanism (Saghatelyan et al., 2001, 2003). Therefore the loss of HNK1 with tenascin-R−/− disinhibits GABAB receptors, allowing them to block GABAA-mediated inhibition and increase excitatory transmission (Saghatelyan et al., 2000, 2001; Nikonenko et al., 2003), raising the threshold for LTP induction (Bukalo et al., 2007).
A key role for TNR in spine stability has been described in the GABAergic granule cells of the olfactory bulb, which have non-conventional dendritic spines that serve both pre- and post-synaptic functions. Tenascin R is expressed and deposited in the olfactory bulb only in adults. Granule cells born in adult tenascin-R−/− mice have reduced spine density and reduced migration to the olfactory bulb, while granule cells born in juvenile animals are normal. The reduction in spine density is not secondary to migration defects, as interfering with TNR function after wild type adult-born granule cells have migrated to the olfactory bulb produces a similar reduction in spine density (David et al., 2013). These results demonstrate that TNR regulates the strength of inhibitory contacts onto excitatory neurons to alter adult synaptic plasticity and also regulates spine stability on a subset of inhibitory interneurons.
Tenascin C plays a role in modulating hippocampal plasticity. Tenascin C expression is high early in development but decreases through adolescence and is very low in adults (Ferhat et al., 1996). However, TNC expression can be transiently induced in adult animals by stimuli that cause LTP (Nakic et al., 1998), suggesting a role for TNC in plasticity. Indeed, tenascin-C−/− mice have reduced hippocampal CA1 LTP and lack CA1 LTD, though LTP in other regions of the hippocampus is normal (Evers et al., 2002; Strekalova et al., 2002). The specific role for TNC in neuron structure has been understudied, although one study suggests that TNC knockout causes redistribution of stubby dendritic spines in cortex away from primary dendrites and toward higher order dendrites (Irintchev et al., 2005). Further studies will undoubtedly reveal more detailed functions for TNC in spine formation, plasticity, and stability.
Laminins Organize and Maintain Synapses
Laminins are large, secreted, heterotrimeric glycoproteins made up of alpha (α), beta (β), and gamma (γ) subunits that interact with numerous transmembrane proteins, including integrin receptors, α-dystroglycan, and basal CAM/Lutheran (Horwitz et al., 1985; Buck and Horwitz, 1987; Smalheiser and Schwartz, 1987; Gehlsen et al., 1988; Ignatius and Reichardt, 1988; Gee et al., 1993; Henry and Campbell, 1996; El Nemer et al., 1998; Kikkawa et al., 2007; Aumailley, 2013; Yousif et al., 2013). Multiple α, β, and γ genes have been identified and they can combine to form over a dozen distinct heterotrimers (Aumailley et al., 2005; Aumailley, 2013). Each of the three subunits of laminin have an N-terminal short arm region, which mediates interactions with transmembrane receptors and other ECM molecules, and a coiled-coil domain, which mediates heterotrimerization. The α subunits also have a C-terminal globular domain that engages with cell surface receptors, including several integrins and α-dystroglycan (Colognato and Yurchenco, 2000; Aumailley, 2013).
Early experiments revealed that laminins can promote neurite growth from various cultured neuronal cells (Manthorpe et al., 1983; Liesi et al., 1984; Lander et al., 1985). Subsequently, Sanes and colleagues identified key roles for laminins at the NMJ where specific laminin subunits control development, maturation, and stability of the synapse (Sanes, 1982; Hunter et al., 1989a,b; Martin et al., 1995; Patton et al., 1997, 2001; Nishimune et al., 2008; Samuel et al., 2012). For example, β2 subunit-containing laminins are produced by the muscle and localize to the center of the synapse to direct acetylcholine receptor clustering (Martin et al., 1995). Additionally, an interaction between the β2 subunit and a presynaptic voltage-gated calcium channel maintains active zone organization (Nishimune et al., 2004). The α4 and α5 laminin subunits also play roles at the NMJ where they signal through the ECM receptor dystroglycan to promote postsynaptic maturation. The combined loss of laminins α4 and α5 results in smaller, much less elaborate synapses, and loss of α5 alone from the muscle causes a delay in synapse maturation (Nishimune et al., 2008). Interestingly, loss of laminin α4 also causes premature aging at the NMJ, accelerating age-related phenotypes by several months (Samuel et al., 2012). This work and additional evidence in the peripheral nervous system established laminins as major players in synapse formation and maintenance.
Recent evidence also supports roles for laminins in maintaining synapse structure and stability in the central nervous system. Mice lacking the laminin β2 subunit have disrupted hippocampal synapse structure, including misaligned pre- and postsynaptic partners and increased PSD length (Egles et al., 2007). Co-culturing experiments indicate that β2 laminin is produced by postsynaptic neurons in this system. In the hippocampus, kainic acid injection to induce excitotoxic injury degrades laminin γ1 and causes neuron death. These effects are absent in mice lacking the protease tissue plasminogen activator (tPA) and can be blocked with inhibitors of plasmin, an extracellular protease that is the substrate of tPA and degrades laminins. Importantly, adding a laminin antibody to disrupt laminin-neuron interactions can restore neuronal sensitivity to excitotoxic insult in tPA-deficient mice (Chen and Strickland, 1997). Furthermore, plasmin-mediated laminin degradation is associated with reduced LTP (Nakagami et al., 2000), although specific effects on dendritic spine size or stability have not been investigated.
The roles of laminins in the brain are not as well characterized as their roles in the peripheral nervous system. Nonetheless, these observations suggest that laminins function at synapses to maintain neuronal stability and synapse structure and function. More work is necessary to describe functions of specific laminin subunits in the brain as well as the receptors that mediate CNS laminin:neuronal interactions.
Netrin:DCC Signaling Regulates Spine Morphology and LTP
Netrins are laminin-related proteins that play diverse conserved roles in neuronal morphogenesis and stability (Ishii et al., 1992; Serafini et al., 1994, 1996; Barallobre et al., 2000; Adler et al., 2006; Colón-Ramos et al., 2007; DeNardo et al., 2012; Smith et al., 2012). In mice and humans, the netrin family consists of three secreted molecules, netrins 1, 3, and 4, and two membrane-bound, GPI-anchored proteins, netrin G1 and G2. Receptors for secreted netrins include deleted in colorectal cancer (DCC), the UNC5 family of proteins, and specific integrin receptors (Chan et al., 1996; Keino-Masu et al., 1996; Leonardo et al., 1997; Yebra et al., 2003; Stanco et al., 2009). Interestingly, netrins share homology with the short arm regions of the β or γ subunits of laminin (Lai Wing Sun et al., 2011) and netrin 4 binds the short arm of laminin γ1 and γ3 subunits to form netrin:laminin complexes and disrupt laminin:laminin interactions (Schneiders et al., 2007).
Recent work suggests netrin:DCC interactions might regulate synapse structure and function in the brain. Loss of DCC after initial development causes smaller dendritic spine head size and impairs learning and LTP. Also, Netrin-1 and DCC co-fractionate from synapses of mature rats, and DCC is present at spines of CA1 pyramidal neurons in mature (60 DIV) cultured hippocampal slices (Horn et al., 2013). While this suggests that netrin can regulate both spine morphology and synaptic transmission in adulthood, further study is needed to understand the roles of netrins at synapses in the adult CNS.
ECM Proteases Regulate Spine and Synapse Stability and Remodeling
Tissue Plasminogen Activator Creates a Permissive Environment for Spine Destabilization
Tissue plasminogen activator (tPA) is a secreted extracellular serine protease best known for its role in cleaving and activating plasminogen into the active protease plasmin to prevent blood clots in the circulatory system (Collen, 1999). In the CNS, tPA is expressed and secreted widely (Sappino et al., 1993; Strickland, 2001), though its activity is located primarily in neurons of the hippocampus, amygdala, cerebellum and hypothalamus (Sappino et al., 1993; Baranes et al., 1998; Lochner et al., 2006). Tissue plasminogen activator was first identified as an activity-dependent immediate early gene strongly induced in rat hippocampus after seizures or LTP (Qian et al., 1993), suggesting a role for the protease in adult plasticity. Indeed, tPA−/− mice have deficits specifically in hippocampal LTP maintenance (Frey et al., 1996; Huang et al., 1996) with no problems in short term potentiation paradigms like pre-pulse facilitation or early-phase LTP. Tissue plasminogen activator is also sufficient to support late-phase LTP, as genetic overexpression of tPA enhances LTP proportional to the amount of gene overexpression (Madani et al., 1999). Tissue plasminogen activator can also regulate plasticity in other systems, including cerebellar motor learning (Seeds et al., 2003) and striatal LTD (Calabresi et al., 2000), and tPA−/− mice are resistant to chemically-induced synaptic potentiation (Huang et al., 1996; Baranes et al., 1998).
Tissue plasminogen activator is a key regulator of dendritic spine stability during plasticity, both in the visual system and in response to stress. Tissue plasminogen activator becomes activated in binocular primary visual cortex during MD, and tPA knockout blocks MD-induced OD plasticity shifts. Importantly, this loss of plasticity can be rescued by recombinant tPA (Mataga et al., 2002), demonstrating a critical role for tPA in OD plasticity. In addition, MD upregulates spine motility, and this effect can be mimicked by direct application of tPA or plasmin to visual cortex. Importantly, the increased motility induced by MD occludes that caused by plasmin application, suggesting that plasmin and MD function in the same pathway to permit MD-induced structural plasticity (Oray et al., 2004). In addition, while 4 days of MD causes spine pruning in visual cortex during the OD critical period, this spine loss is blocked in tPA−/− mice, indicating that tPA is also required for spine pruning in response to MD (Mataga et al., 2004). Chronic stress can also cause spine loss in the hippocampus and amygdala. Plasminogen is activated around dendritic spines by chronic stress, and knockout of tPA or plasminogen blocks stress-induced spine loss (Pawlak et al., 2005; Bennur et al., 2007). These results suggest that tPA negatively regulates spine stability and that its activation creates a permissive environment that destabilizes spines and promotes their loss.
One open question in the field is what substrates of tPA promote plasticity in each of these paradigms. Classically, tPA cleaves plasminogen to make the active protease plasmin, which can then degrade ECM targets. Indeed, plasmin can degrade laminin and the CSPG phosphacan to regulate LTP and hippocampal mossy fiber outgrowth (Nakagami et al., 2000; Wu et al., 2000). Plasmin also likely degrades ECM proteins in the MD paradigm, but its exact targets are unknown (Oray et al., 2004). However, it is important to note that tPA certainly has other non-ECM targets in the brain. For example, the loss of late LTP in tPA and plasminogen knockouts is mostly due to reduced proBDNF cleavage to create the mature form of BDNF (Pang et al., 2004). In addition, tPA can cleave the NR1 subunit of the NMDAR to potentiate NMDAR currents (Nicole et al., 2001). It is clear from these varied results that tPA and plasmin target a variety of ECM and non-ECM proteins to regulate synaptic and structural plasticity. It will be important to clearly identify the critical tPA targets in each experimental paradigm to better understand the role of tPA in dendritic spine regulation.
Matrix Metalloproteinases Play Diverse Roles in Dendritic Spine Remodeling in Disease, Development, and Plasticity
Matrix metalloproteinases (MMPs) are a large class of secreted and transmembrane proteases that can degrade many ECM proteins, transmembrane receptors, and other signaling proteins (Visse and Nagase, 2003). Matrix metalloproteinases were initially identified as critical for brain function because the mRNA for TIMP1, an endogenous inhibitor of MMPs, is upregulated in a kainic acid-induced epilepsy model, suggesting an activity-dependent role for MMPs in epilepsy (Nedivi et al., 1993; Rivera et al., 1997; Jaworski et al., 1999). In the kindling model of epilepsy (Morimoto et al., 2004), MMP9 knockout delays seizure onset while MMP9 overexpression speeds onset (Wilczynski et al., 2008). In addition, MMP2 and MMP9 are expressed in neurons and glia and are upregulated by kainate treatment (Szklarczyk et al., 2002), and other MMPs may also be expressed after injury or certain stimulations (Bilousova et al., 2006; Meighan et al., 2006). Importantly, kainate-induced seizures cause hippocampal spine loss that is blocked in MMP9−/− mice (Wilczynski et al., 2008).
MMP9 activity is also important in another pathophysiological condition, Fragile X syndrome (FXS). Mice with FXS have more long and thin spines than wild type mice, especially early in development. Treatment of FXS mice or hippocampal cultures derived from these mice with minocycline to inhibit MMP9 can normalize spine morphology (Bilousova et al., 2009), suggesting that hyperactive MMP9 in development prevents spine maturation. Indeed, MMP9 has recently been shown directly to be hyperactive in FXS mice, and disruption of the MMP9 gene in FXS mice normalizes the spine, behavioral, and signal transduction defects associated with FXS (Sidhu et al., 2014). These results demonstrate that pathophysiological activation of MMP9 can promote changes in dendritic spine morphology associated with disease (Figure 5A).
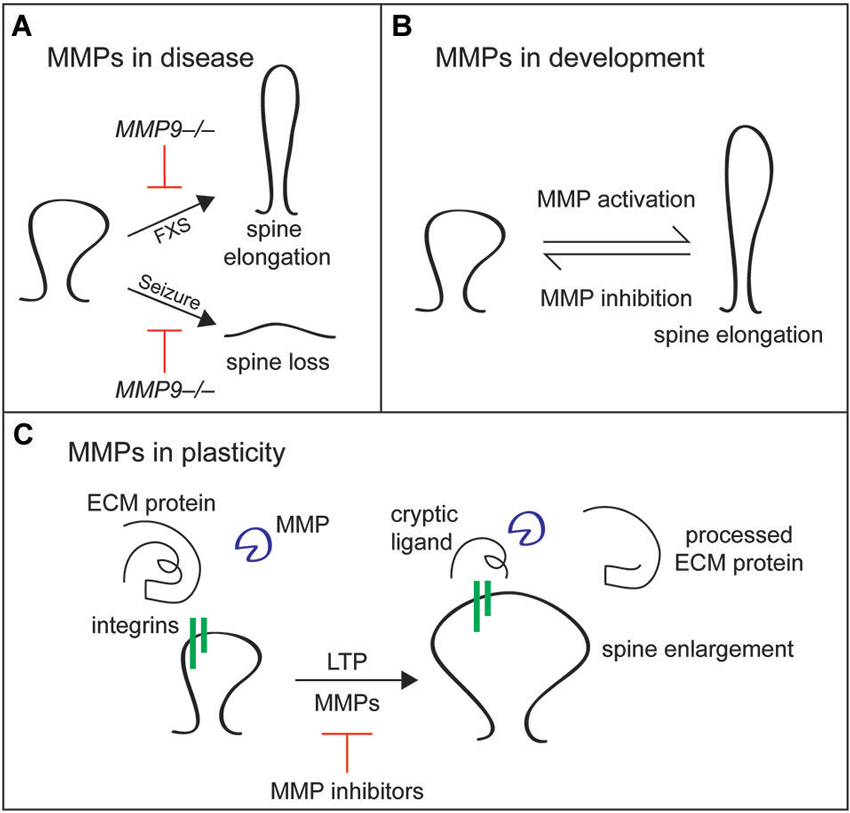
Figure 5. Matrix metalloproteinases play diverse roles in dendritic spine remodeling in disease, development, and plasticity. (A) In Fragile X syndrome (FXS), MMP9 is hyperactive and dendritic spines are elongated early in development. MMP9 loss of function or inhibition with minocycline normalizes these spine defects. In epilepsy models, seizure-induced spine loss is blocked by inhibition of MMP activity. (B) In development, activation of MMPs, particularly MMP9, causes spine thinning and elongation, while MMP inhibition promotes the maturation of filopodia into mature, mushroom shaped spines. (C) Matrix metalloproteinases play a different role in adult plasticity, cleaving unknown ECM proteins to reveal cryptic integrin ligands that drive spine enlargement during LTP. In addition to changes in spine size, MMP activity is required for the maintenance phase of LTP.
Matrix metalloproteinases also play roles in developmental processes such as spine protrusion and early maturation. For example, bath application of activated MMP9 to young hippocampal cultures or organotypic slices and overexpression of activated MMP9 cause dendritic spines to become longer and thinner (Michaluk et al., 2011). Similarly, treatment of maturing hippocampal cultures with MMP7 causes spine elongation (Bilousova et al., 2006). By contrast, MMP inhibition of cultured neurons promotes maturation of thin filopodial spines into mature mushroom-shaped spines (Tian et al., 2007; Bilousova et al., 2009: Figure 5B). Matrix metalloproteinase activation promotes spine elongation at least in part through cleavage of intercellular cell adhesion molecule 5 (ICAM5). Full length ICAM5 is found in immature neurons and is cleaved by MMPs to release a soluble extracellular domain that promotes filopodial elongation (Tian et al., 2007). Soluble ICAM5 also increases AMPA receptor expression and cofilin phosphorylation, phenotypes that are associated with spine maturation and depend on β1 integrin (Conant et al., 2011; Lonskaya et al., 2013). Interestingly, ICAM5 localization in cortical neurons shifts during synapse development from predominately dendritic filopodia and spines to predominately the dendritic shaft, and this developmental shift is blocked in MMP9−/− mice (Kelly et al., 2014). These data indicate that ICAM5 is an important substrate of MMP9 during synaptogenesis. Together, these results show that MMPs have central roles in dendritic spine development and maturation.
It is important to consider that the effects of MMPs on dendritic spines can differ greatly depending on the method of MMP manipulation and the maturity of the system. The MMP-dependent elongation of spines discussed above is dependent on manipulation of MMP activity in young or maturing systems or under pathophysiological conditions and requires general application of MMP-affecting drugs for long periods of time. In more mature systems and with local application of MMPs during plasticity events, MMP activity has the opposite effect. For example, local application of active MMP9 to dendritic spines in acute hippocampal slices is by itself sufficient to potentiate synapses and increase spine volume, the same changes that are caused by theta-burst pairing, which induces LTP. Notably, MMP9-induced potentiation and spine enlargement are occluded by prior theta-burst pairing, suggesting that MMP9 activation and LTP induction function in the same pathway to consolidate spine enlargement and LTP (Wang et al., 2008). Similarly, chemical LTP induction in mature cultured neurons increases spine head size of smaller spines in an MMP-dependent manner (Szepesi et al., 2014). In agreement with these findings, MMP9 is required for maintenance of LTP and LTP-induced spine volume increase in acute hippocampal slices from adult animals (Nagy et al., 2006; Wang et al., 2008), and inhibition of MMPs 3 and 9 blocks acquisition of spatial learning in adult animals (Meighan et al., 2006). Importantly, many of these acute phenotypes in mature systems depend on integrin β1 function (Nagy et al., 2006; Wang et al., 2008), suggesting that MMPs may reveal cryptic integrin ligands in the ECM that maintain spine structural plasticity in mature neurons (Figure 5C). Given the diverse effects of MMP targeting treatments on spine development, plasticity, and maintenance, further studies should address the molecular basis for the differential effects of MMP manipulation in both young and mature systems (Dziembowska and Wlodarczyk, 2012; Stawarski et al., 2014). In addition, the specific ECM molecules that signal through integrins are unknown, and future studies will hopefully link proteolysis of specific proteins by MMPs with specific changes in dendritic spines to understand the signaling mechanisms involved in MMP-mediated dendritic spine remodeling.
Conclusion
Precise regulation of dendritic spine and synapse formation, plasticity, and stability is essential for proper circuit and brain function. Emerging evidence indicates that ECM proteins, their receptors, and ECM proteases are major physiological regulators of spines and synapses (Table 1). Extracellular matrix molecules are potent regulators of the actin cytoskeleton, which dictates dendritic spine morphology and powers dynamic changes in dendritic spine shape. Moreover, the ECM surrounds neurons and its composition is influenced greatly by synaptic activity, making it an ideal substrate to influence spine and synapse structure and physiology.

Table 1. Role(s) of ECM proteins in synaptic plasticity and/or regulation of spine structure.
Future studies in this field will be critical to identify the molecules that signal through ECM receptors such as integrins to control spine stability and plasticity. Elucidating these molecules and the mechanisms by which they function is essential to understand how differential stability and plasticity are achieved in adulthood vs. development, and in healthy individuals vs. those with neurodegenerative or late-onset psychiatric disease. Only then can we target these mechanisms therapeutically to gain control of synaptic flexibility and stability.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank A. Scherer for his careful critiques and comments on this manuscript during its preparation.
References
Adler, C. E., Fetter, R. D., and Bargmann, C. I. (2006). UNC-6/Netrin induces neuronal asymmetry and defines the site of axon formation. Nat. Neurosci. 9, 511–518. doi: 10.1038/nn1666
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Akbik, F. V., Bhagat, S. M., Patel, P. R., Cafferty, W. B. J., and Strittmatter, S. M. (2013). Anatomical plasticity of adult brain is titrated by Nogo Receptor 1. Neuron 77, 859–866. doi: 10.1016/j.neuron.2012.12.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Annies, M., Bittcher, G., Ramseger, R., Löschinger, J., Wöll, S., Porten, E., et al. (2006). Clustering transmembrane-agrin induces filopodia-like processes on axons and dendrites. Mol. Cell. Neurosci. 31, 515–524. doi: 10.1016/j.mcn.2005.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aspberg, A., Miura, R., Bourdoulous, S., Shimonaka, M., Heinegârd, D., Schachner, M., et al. (1997). The C-type lectin domains of lecticans, a family of aggregating chondroitin sulfate proteoglycans, bind tenascin-R by protein-protein interactions independent of carbohydrate moiety. Proc. Natl. Acad. Sci. U S A 94, 10116–10121. doi: 10.1073/pnas.94.19.10116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aumailley, M. (2013). The laminin family. Cell Adh. Migr. 7, 48–55. doi: 10.4161/cam.22826
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aumailley, M., Bruckner-Tuderman, L., Carter, W. G., Deutzmann, R., Edgar, D., Ekblom, P., et al. (2005). A simplified laminin nomenclature. Matrix Biol. 24, 326–332. doi: 10.1016/j.matbio.2005.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bahr, B. A., Staubli, U., Xiao, P., Chun, D., Ji, Z. X., Esteban, E. T., et al. (1997). Arg-Gly-Asp-Ser-selective adhesion and the stabilization of long-term potentiation: pharmacological studies and the characterization of a candidate matrix receptor. J. Neurosci. 17, 1320–1329.
Barallobre, M. J., Del Rio, J. A., Alcantara, S., Borrell, V., Aguado, F., Ruiz, M., et al. (2000). Aberrant development of hippocampal circuits and altered neural activity in netrin 1-deficient mice. Development 127, 4797–4810.
Baranes, D., Lederfein, D., Huang, Y. Y., Chen, M., Bailey, C. H., and Kandel, E. R. (1998). Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron 21, 813–825. doi: 10.1016/s0896-6273(00)80597-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barros, C. S., Franco, S. J., and Müller, U. (2011). Extracellular matrix: functions in the nervous system. Cold Spring Harb. Perspect. Biol. 3:a005108. doi: 10.1101/cshperspect.a005108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bastrikova, N., Gardner, G. A., Reece, J. M., Jeromin, A., and Dudek, S. M. (2008). Synapse elimination accompanies functional plasticity in hippocampal neurons. Proc. Natl. Acad. Sci. U S A 105, 3123–3127. doi: 10.1073/pnas.0800027105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beffert, U., Nematollah Farsian, F., Masiulis, I., Hammer, R. E., Yoon, S. O., Giehl, K. M., et al. (2006). ApoE receptor 2 controls neuronal survival in the adult brain. Curr. Biol. 16, 2446–2452. doi: 10.1016/j.cub.2006.10.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beffert, U., Weeber, E. J., Durudas, A., Qiu, S., Masiulis, I., Sweatt, J. D., et al. (2005). Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron 47, 567–579. doi: 10.1016/j.neuron.2005.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belvindrah, R., Graus-Porta, D., Goebbels, S., Nave, K. A., and Müller, U. (2007a). Beta1 integrins in radial glia but not in migrating neurons are essential for the formation of cell layers in the cerebral cortex. J. Neurosci. 27, 13854–13865. doi: 10.1523/JNEUROSCI.4494-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belvindrah, R., Hankel, S., Walker, J., Patton, B. L., and Müller, U. (2007b). Beta1 integrins control the formation of cell chains in the adult rostral migratory stream. J. Neurosci. 27, 2704–2717. doi: 10.1523/jneurosci.2991-06.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bennur, S., Shankaranarayana Rao, B. S., Pawlak, R., Strickland, S., McEwen, B. S., and Chattarji, S. (2007). Stress-induced spine loss in the medial amygdala is mediated by tissue-plasminogen activator. Neuroscience 144, 8–16. doi: 10.1016/j.neuroscience.2006.08.075
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benson, D. L., and Huntley, G. W. (2012). Synapse adhesion: a dynamic equilibrium conferring stability and flexibility. Curr. Opin. Neurobiol. 22, 397–404. doi: 10.1016/j.conb.2011.09.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bernard-Trifilo, J. A., Kramár, E. A., Torp, R., Lin, C.-Y., Pineda, E. A., Lynch, G., et al. (2005). Integrin signaling cascades are operational in adult hippocampal synapses and modulate NMDA receptor physiology. J. Neurochem. 93, 834–849. doi: 10.1111/j.1471-4159.2005.03062.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berretta, S. (2012). Extracellular matrix abnormalities in schizophrenia. Neuropharmacology 62, 1584–1597. doi: 10.1016/j.neuropharm.2011.08.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bilousova, T. V., Dansie, L., Ngo, M., Aye, J., Charles, J. R., Ethell, D. W., et al. (2009). Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J. Med. Genet. 46, 94–102. doi: 10.1136/jmg.2008.061796
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bilousova, T. V., Rusakov, D. A., Ethell, D. W., and Ethell, I. M. (2006). Matrix metalloproteinase-7 disrupts dendritic spines in hippocampal neurons through NMDA receptor activation. J. Neurochem. 97, 44–56. doi: 10.1111/j.1471-4159.2006.03701.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bose, C. M., Qiu, D., Bergamaschi, A., Gravante, B., Bossi, M., Villa, A., et al. (2000). Agrin controls synaptic differentiation in hippocampal neurons. J. Neurosci. 20, 9086–9095.
Brakebusch, C., Seidenbecher, C. I., Asztely, F., Rauch, U., Matthies, H., Meyer, H., et al. (2002). Brevican-deficient mice display impaired hippocampal CA1 long-term potentiation but show no obvious deficits in learning and memory. Mol. Cell. Biol. 22, 7417–7427. doi: 10.1128/mcb.22.21.7417-7427.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brückner, G., Grosche, J., Schmidt, S., Härtig, W., Margolis, R. U., Delpech, B., et al. (2000). Postnatal development of perineuronal nets in wild-type mice and in a mutant deficient in tenascin-R. J. Comp. Neurol. 428, 616–629. doi: 10.1002/1096-9861(20001225)428:4<616::aid-cne3>3.0.co;2-k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buck, C. A., and Horwitz, A. F. (1987). Cell surface receptors for extracellular matrix molecules. Annu. Rev. Cell Biol. 3, 179–205. doi: 10.1146/annurev.cb.03.110187.001143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bukalo, O., and Dityatev, A. (2012). Synaptic cell adhesion molecules. Adv. Exp. Med. Biol. 970, 97–128. doi: 10.1007/978-3-7091-0932-8_5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bukalo, O., Schachner, M., and Dityatev, A. (2001). Modification of extracellular matrix by enzymatic removal of chondroitin sulfate and by lack of tenascin-R differentially affects several forms of synaptic plasticity in the hippocampus. Neuroscience 104, 359–369. doi: 10.1016/s0306-4522(01)00082-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bukalo, O., Schachner, M., and Dityatev, A. (2007). Hippocampal metaplasticity induced by deficiency in the extracellular matrix glycoprotein tenascin-R. J. Neurosci. 27, 6019–6028. doi: 10.1523/jneurosci.1022-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Calabresi, P., Napolitano, M., Centonze, D., Marfia, G. A., Gubellini, P., Teule, M. A., et al. (2000). Tissue plasminogen activator controls multiple forms of synaptic plasticity and memory. Eur. J. Neurosci. 12, 1002–1012. doi: 10.1046/j.1460-9568.2000.00991.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Calderwood, D. A. (2004). Integrin activation. J. Cell Sci. 117, 657–666. doi: 10.1242/jcs.01014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carter, A., and Sabatini, B. (2008). “Spine calcium signaling,” in Dendrites, eds G. Stuart, N. Spruston and M. Hausser 2nd Edn. (New York: Oxford University Press), 287–308.
Catalá, I., Ferrer, I., Galofré, E., and Fábregues, I. (1988). Decreased numbers of dendritic spines on cortical pyramidal neurons in dementia. A quantitative Golgi study on biopsy samples. Hum. Neurobiol. 6, 255–259.
Chan, C.-S., Levenson, J. M., Mukhopadhyay, P. S., Zong, L., Bradley, A., Sweatt, J. D., et al. (2007). Alpha3-integrins are required for hippocampal long-term potentiation and working memory. Learn. Mem. 14, 606–615. doi: 10.1101/lm.648607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chan, C.-S., Weeber, E. J., Zong, L., Fuchs, E., Sweatt, J. D., and Davis, R. L. (2006). Beta 1-integrins are required for hippocampal AMPA receptor-dependent synaptic transmission, synaptic plasticity and working memory. J. Neurosci. 26, 223–232. doi: 10.1523/jneurosci.4110-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chan, S. S., Zheng, H., Su, M. W., Wilk, R., Killeen, M. T., Hedgecock, E. M., et al. (1996). UNC-40, a C. elegans homolog of DCC (Deleted in Colorectal Cancer), is required in motile cells responding to UNC-6 netrin cues. Cell 87, 187–195. doi: 10.1016/s0092-8674(00)81337-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Charrier, C., Joshi, K., Coutinho-Budd, J., Kim, J.-E., Lambert, N., de Marchena, J., et al. (2012). Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation. Cell 149, 923–935. doi: 10.1016/j.cell.2012.03.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cheadle, L., and Biederer, T. (2012). The novel synaptogenic protein Farp1 links postsynaptic cytoskeletal dynamics and transsynaptic organization. J. Cell Biol. 199, 985–1001. doi: 10.1083/jcb.201205041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Y., Beffert, U., Ertunc, M., Tang, T.-S., Kavalali, E. T., Bezprozvanny, I., et al. (2005). Reelin modulates NMDA receptor activity in cortical neurons. J. Neurosci. 25, 8209–8216. doi: 10.1523/jneurosci.1951-05.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Z. L., and Strickland, S. (1997). Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell 91, 917–925. doi: 10.1016/s0092-8674(00)80483-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Z.-L., Yu, H., Yu, W.-M., Pawlak, R., and Strickland, S. (2008). Proteolytic fragments of laminin promote excitotoxic neurodegeneration by up-regulation of the KA1 subunit of the kainate receptor. J. Cell Biol. 183, 1299–1313. doi: 10.1083/jcb.200803107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chong, L. D., Traynor-Kaplan, A., Bokoch, G. M., and Schwartz, M. A. (1994). The small GTP-binding protein Rho regulates a phosphatidylinositol 4-phosphate 5-kinase in mammalian cells. Cell 79, 507–513. doi: 10.1016/0092-8674(94)90259-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chun, D., Gall, C. M., Bi, X., and Lynch, G. (2001). Evidence that integrins contribute to multiple stages in the consolidation of long term potentiation in rat hippocampus. Neuroscience 105, 815–829. doi: 10.1016/s0306-4522(01)00173-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cichon, S., Mühleisen, T. W., Degenhardt, F. A., Mattheisen, M., Miró, X., Strohmaier, J., et al. (2011). Genome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorder. Am. J. Hum. Genet. 88, 372–381. doi: 10.1016/j.ajhg.2011.01.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cohen, N. A., Kaufmann, W. E., Worley, P. F., and Rupp, F. (1997). Expression of agrin in the developing and adult rat brain. Neuroscience 76, 581–596. doi: 10.1016/s0306-4522(96)00345-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Colognato, H., and Yurchenco, P. D. (2000). Form and function: the laminin family of heterotrimers. Dev. Dyn. 218, 213–234. doi: 10.1002/(sici)1097-0177(200006)218:2<213::aid-dvdy1>3.0.co;2-r
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Colón-Ramos, D. A., Margeta, M. A., and Shen, K. (2007). Glia promote local synaptogenesis through UNC-6 (netrin) signaling in C. elegans. Science 318, 103–106. doi: 10.1126/science.1143762
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conant, K., Lonskaya, I., Szklarczyk, A., Krall, C., Steiner, J., Maguire-Zeiss, K., et al. (2011). Methamphetamine-associated cleavage of the synaptic adhesion molecule intercellular adhesion molecule-5. J. Neurochem. 118, 521–532. doi: 10.1111/j.1471-4159.2010.07153.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dansie, L. E., and Ethell, I. M. (2011). Casting a net on dendritic spines: the extracellular matrix and its receptors. Dev. Neurobiol. 71, 956–981. doi: 10.1002/dneu.20963
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D’Arcangelo, G. (2014). Reelin in the years: controlling neuronal migration and maturation in the Mammalian brain. Adv. Neurosci. 2014:19. doi: 10.1155/2014/597395
David, L. S., Schachner, M., and Saghatelyan, A. (2013). The extracellular matrix glycoprotein tenascin-R affects adult but not developmental neurogenesis in the olfactory bulb. J. Neurosci. 33, 10324–10339. doi: 10.1523/jneurosci.5728-12.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
DeFreitas, M. F., Yoshida, C. K., Frazier, W. A., Mendrick, D. L., Kypta, R. M., and Reichardt, L. F. (1995). Identification of integrin alpha 3 beta 1 as a neuronal thrombospondin receptor mediating neurite outgrowth. Neuron 15, 333–343. doi: 10.1016/0896-6273(95)90038-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
DeNardo, L. A., de Wit, J., Otto-Hitt, S., and Ghosh, A. (2012). NGL-2 regulates input-specific synapse development in CA1 pyramidal neurons. Neuron 76, 762–775. doi: 10.1016/j.neuron.2012.10.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Vivo, L., Landi, S., Panniello, M., Baroncelli, L., Chierzi, S., Mariotti, L., et al. (2013). Extracellular matrix inhibits structural and functional plasticity of dendritic spines in the adult visual cortex. Nat. Commun. 4:1484. doi: 10.1038/ncomms2491
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dunaevsky, A., Tashiro, A., Majewska, A. K., Mason, C. A., and Yuste, R. (1999). Developmental regulation of spine motility in the mammalian central nervous system. Proc. Natl. Acad. Sci. U S A 96, 13438–13443. doi: 10.1073/pnas.96.23.13438
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dziembowska, M., and Wlodarczyk, J. (2012). MMP9: a novel function in synaptic plasticity. Int. J. Biochem. Cell Biol. 44, 709–713. doi: 10.1016/j.biocel.2012.01.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Egles, C., Claudepierre, T., Manglapus, M. K., Champliaud, M.-F., Brunken, W. J., and Hunter, D. D. (2007). Laminins containing the beta2 chain modulate the precise organization of CNS synapses. Mol. Cell. Neurosci. 34, 288–298. doi: 10.1016/j.mcn.2006.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
El Nemer, W., Gane, P., Colin, Y., Bony, V., Rahuel, C., Galactéros, F., et al. (1998). The Lutheran blood group glycoproteins, the erythroid receptors for laminin, are adhesion molecules. J. Biol. Chem. 273, 16686–16693. doi: 10.1074/jbc.273.27.16686
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Evers, M. R., Salmen, B., Bukalo, O., Rollenhagen, A., Bösl, M. R., Morellini, F., et al. (2002). Impairment of L-type Ca2+ channel-dependent forms of hippocampal synaptic plasticity in mice deficient in the extracellular matrix glycoprotein tenascin-C. J. Neurosci. 22, 7177–7194.
Ferhat, L., Chevassus au Louis, N., Jorquera, I., Niquet, J., Khrestchatisky, M., Ben-Ari, Y., et al. (1996). Transient increase of tenascin-C in immature hippocampus: astroglial and neuronal expression. J. Neurocytol. 25, 53–66. doi: 10.1007/bf02284785
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferrante, R. J., Kowall, N. W., and Richardson, E. P. (1991). Proliferative and degenerative changes in striatal spiny neurons in Huntington’s disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J. Neurosci. 11, 3877–3887.
Ferreira, A. (1999). Abnormal synapse formation in agrin-depleted hippocampal neurons. J. Cell Sci. 112(Pt. 24), 4729–4738.
Freitag, S., Schachner, M., and Morellini, F. (2003). Behavioral alterations in mice deficient for the extracellular matrix glycoprotein tenascin-R. Behav. Brain Res. 145, 189–207. doi: 10.1016/s0166-4328(03)00109-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frey, U., Muller, M., and Kuhl, D. (1996). A different form of long-lasting potentiation revealed in tissue plasminogen activator mutant mice. J. Neurosci. 16, 2057–2063.
Galtrey, C. M., and Fawcett, J. W. (2007). The role of chondroitin sulfate proteoglycans in regeneration and plasticity in the central nervous system. Brain Res. Rev. 54, 1–18. doi: 10.1016/j.brainresrev.2006.09.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gama, C. I., Tully, S. E., Sotogaku, N., Clark, P. M., Rawat, M., Vaidehi, N., et al. (2006). Sulfation patterns of glycosaminoglycans encode molecular recognition and activity. Nat. Chem. Biol. 2, 467–473. doi: 10.1038/nchembio810
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garey, L. J., Ong, W. Y., Patel, T. S., Kanani, M., Davis, A., Mortimer, A. M., et al. (1998). Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J. Neurol. Neurosurg. Psychiatr. 65, 446–453. doi: 10.1136/jnnp.65.4.446
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gautam, M., Noakes, P. G., Moscoso, L., Rupp, F., Scheller, R. H., Merlie, J. P., et al. (1996). Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 85, 525–535. doi: 10.1016/s0092-8674(00)81253-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gee, S. H., Blacher, R. W., Douville, P. J., Provost, P. R., Yurchenco, P. D., and Carbonetto, S. (1993). Laminin-binding protein 120 from brain is closely related to the dystrophin-associated glycoprotein, dystroglycan and binds with high affinity to the major heparin binding domain of laminin. J. Biol. Chem. 268, 14972–14980.
Gehlsen, K. R., Dillner, L., Engvall, E., and Ruoslahti, E. (1988). The human laminin receptor is a member of the integrin family of cell adhesion receptors. Science 241, 1228–1229. doi: 10.1126/science.2970671
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glantz, L. A., and Lewis, D. A. (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 57, 65–73. doi: 10.1001/archpsyc.57.1.65
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glass, D. J., Bowen, D. C., Stitt, T. N., Radziejewski, C., Bruno, J., Ryan, T. E., et al. (1996). Agrin acts via a MuSK receptor complex. Cell 85, 513–523. doi: 10.1016/s0092-8674(00)81252-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gogolla, N., Caroni, P., Lüthi, A., and Herry, C. (2009). Perineuronal nets protect fear memories from erasure. Science 325, 1258–1261. doi: 10.1126/science.1174146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gordon, J. A., and Stryker, M. P. (1996). Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J. Neurosci. 16, 3274–3286.
Gray, E. G. (1959a). Axo-somatic and axo-dendritic synapses of the cerebral cortex: an electron microscope study. J. Anat. 93, 420–433.
Gray, E. G. (1959b). Electron microscopy of synaptic contacts on dendrite spines of the cerebral cortex. Nature 183, 1592–1593. doi: 10.1038/1831592a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Groc, L., Choquet, D., Stephenson, F. A., Verrier, D., Manzoni, O. J., and Chavis, P. (2007). NMDA receptor surface trafficking and synaptic subunit composition are developmentally regulated by the extracellular matrix protein Reelin. J. Neurosci. 27, 10165–10175. doi: 10.1523/jneurosci.1772-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grutzendler, J., Kasthuri, N., and Gan, W.-B. (2002). Long-term dendritic spine stability in the adult cortex. Nature 420, 812–816. doi: 10.1038/nature01276
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gschwend, T. P., Krueger, S. R., Kozlov, S. V., Wolfer, D. P., and Sonderegger, P. (1997). Neurotrypsin, a novel multidomain serine protease expressed in the nervous system. Mol. Cell. Neurosci. 9, 207–219. doi: 10.1006/mcne.1997.0616
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gumbiner, B. M. (1996). Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell 84, 345–357. doi: 10.1016/s0092-8674(00)81279-9
Gundelfinger, E. D., Frischknecht, R., Choquet, D., and Heine, M. (2010). Converting juvenile into adult plasticity: a role for the brain’s extracellular matrix. Eur. J. Neurosci. 31, 2156–2165. doi: 10.1111/j.1460-9568.2010.07253.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gurevicius, K., Gureviciene, I., Valjakka, A., Schachner, M., and Tanila, H. (2004). Enhanced cortical and hippocampal neuronal excitability in mice deficient in the extracellular matrix glycoprotein tenascin-R. Mol. Cell. Neurosci. 25, 515–523. doi: 10.1016/j.mcn.2003.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hack, I., Hellwig, S., Junghans, D., Brunne, B., Bock, H. H., Zhao, S., et al. (2007). Divergent roles of ApoER2 and Vldlr in the migration of cortical neurons. Development 134, 3883–3891. doi: 10.1242/dev.005447
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harris, K. M., and Kater, S. B. (1994). Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu. Rev. Neurosci. 17, 341–371. doi: 10.1146/annurev.neuro.17.1.341
Harris, K. M., and Stevens, J. K. (1988). Dendritic spines of rat cerebellar Purkinje cells: serial electron microscopy with reference to their biophysical characteristics. J. Neurosci. 8, 4455–4469.
Harris, K. M., and Stevens, J. K. (1989). Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J. Neurosci. 9, 2982–2997.
Harris, K. M., and Weinberg, R. J. (2012). Ultrastructure of synapses in the mammalian brain. Cold Spring Harb. Perspect. Biol. 4:a005587. doi: 10.1101/cshperspect.a005587
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harvey, C. D., Yasuda, R., Zhong, H., and Svoboda, K. (2008). The spread of Ras activity triggered by activation of a single dendritic spine. Science 321, 136–140. doi: 10.1126/science.1159675
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Henry, M. D., and Campbell, K. P. (1996). Dystroglycan: an extracellular matrix receptor linked to the cytoskeleton. Curr. Opin. Cell Biol. 8, 625–631. doi: 10.1016/s0955-0674(96)80103-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hering, H., and Sheng, M. (2001). Dendritic spines: structure, dynamics and regulation. Nat. Rev. Neurosci. 2, 880–888. doi: 10.1038/35104061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Higley, M. J., and Sabatini, B. L. (2012). Calcium signaling in dendritic spines. Cold Spring Harb. Perspect. Biol. 4:a005686. doi: 10.1101/cshperspect.a005686
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoffman, K. B., Pinkstaff, J. K., Gall, C. M., and Lynch, G. (1998). Seizure induced synthesis of fibronectin is rapid and age dependent: implications for long-term potentiation and sprouting. Brain Res. 812, 209–215. doi: 10.1016/s0006-8993(98)00727-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Holtmaat, A. J. G. D., Trachtenberg, J. T., Wilbrecht, L., Shepherd, G. M., Zhang, X., Knott, G. W., et al. (2005). Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45, 279–291. doi: 10.1016/j.neuron.2005.01.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Horejs, C.-M., Serio, A., Purvis, A., Gormley, A. J., Bertazzo, S., Poliniewicz, A., et al. (2014). Biologically-active laminin-111 fragment that modulates the epithelial-to-mesenchymal transition in embryonic stem cells. Proc. Natl. Acad. Sci. U S A 111, 5908–5913. doi: 10.1073/pnas.1403139111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Horn, K. E., Glasgow, S. D., Gobert, D., Bull, S.-J., Luk, T., Girgis, J., et al. (2013). DCC expression by neurons regulates synaptic plasticity in the adult brain. Cell Rep. 3, 173–185. doi: 10.1016/j.celrep.2012.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Horwitz, A., Duggan, K., Buck, C., Beckerle, M. C., and Burridge, K. (1986). Interaction of plasma membrane fibronectin receptor with talin—a transmembrane linkage. Nature 320, 531–533. doi: 10.1038/320531a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Horwitz, A., Duggan, K., Greggs, R., Decker, C., and Buck, C. (1985). The cell substrate attachment (CSAT) antigen has properties of a receptor for laminin and fibronectin. J. Cell Biol. 101, 2134–2144. doi: 10.1083/jcb.101.6.2134
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, Y. Y., Bach, M. E., Lipp, H. P., Zhuo, M., Wolfer, D. P., Hawkins, R. D., et al. (1996). Mice lacking the gene encoding tissue-type plasminogen activator show a selective interference with late-phase long-term potentiation in both Schaffer collateral and mossy fiber pathways. Proc. Natl. Acad. Sci. U S A 93, 8699–8704. doi: 10.1073/pnas.93.16.8699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, Z., Shimazu, K., Woo, N. H., Zang, K., Müller, U., Lu, B., et al. (2006). Distinct roles of the beta 1-class integrins at the developing and the mature hippocampal excitatory synapse. J. Neurosci. 26, 11208–11219. doi: 10.1523/jneurosci.3526-06.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hunt, C. A., Schenker, L. J., and Kennedy, M. B. (1996). PSD-95 is associated with the postsynaptic density and not with the presynaptic membrane at forebrain synapses. J. Neurosci. 16, 1380–1388.
Hunter, D. D., Porter, B. E., Bulock, J. W., Adams, S. P., Merlie, J. P., and Sanes, J. R. (1989a). Primary sequence of a motor neuron-selective adhesive site in the synaptic basal lamina protein S-laminin. Cell 59, 905–913. doi: 10.1016/0092-8674(89)90613-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hunter, D. D., Shah, V., Merlie, J. P., and Sanes, J. R. (1989b). A laminin-like adhesive protein concentrated in the synaptic cleft of the neuromuscular junction. Nature 338, 229–234. doi: 10.1038/338229a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huntley, G. W., Gil, O., and Bozdagi, O. (2002). The cadherin family of cell adhesion molecules: multiple roles in synaptic plasticity. Neuroscientist 8, 221–233. doi: 10.1177/1073858402008003008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iafrati, J., Orejarena, M. J., Lassalle, O., Bouamrane, L., Gonzalez-Campo, C., and Chavis, P. (2014). Reelin, an extracellular matrix protein linked to early onset psychiatric diseases, drives postnatal development of the prefrontal cortex via GluN2B-NMDARs and the mTOR pathway. Mol. Psychiatry 19, 417–426. doi: 10.1038/mp.2013.66
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ignatius, M. J., and Reichardt, L. F. (1988). Identification of a neuronal laminin receptor: an Mr 200K/120K integrin heterodimer that binds laminin in a divalent cation-dependent manner. Neuron 1, 713–725. doi: 10.1016/0896-6273(88)90170-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Impagnatiello, F., Guidotti, A. R., Pesold, C., Dwivedi, Y., Caruncho, H., Pisu, M. G., et al. (1998). A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc. Natl. Acad. Sci. U S A 95, 15718–15723. doi: 10.1073/pnas.95.26.15718
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Irintchev, A., Rollenhagen, A., Troncoso, E., Kiss, J. Z., and Schachner, M. (2005). Structural and functional aberrations in the cerebral cortex of tenascin-C deficient mice. Cereb. Cortex 15, 950–962. doi: 10.1093/cercor/bhh195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ishii, N., Wadsworth, W. G., Stern, B. D., Culotti, J. G., and Hedgecock, E. M. (1992). UNC-6, a laminin-related protein, guides cell and pioneer axon migrations in C. elegans. Neuron 9, 873–881. doi: 10.1016/0896-6273(92)90240-e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jaworski, J., Biedermann, I. W., Lapinska, J., Szklarczyk, A., Figiel, I., Konopka, D., et al. (1999). Neuronal excitation-driven and AP-1-dependent activation of tissue inhibitor of metalloproteinases-1 gene expression in rodent hippocampus. J. Biol. Chem. 274, 28106–28112. doi: 10.1074/jbc.274.40.28106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jones, F. S., and Jones, P. L. (2000). The tenascin family of ECM glycoproteins: structure, function and regulation during embryonic development and tissue remodeling. Dev. Dyn. 218, 235–259. doi: 10.1002/(sici)1097-0177(200006)218:2<235::aid-dvdy2>3.3.co;2-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Katzman, R. (1989). Alzheimer’s disease is a degenerative disorder. Neurobiol. Aging 10, 581–582; discussion 588–590. doi: 10.1016/0197-4580(89)90137-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keino-Masu, K., Masu, M., Hinck, L., Leonardo, E. D., Chan, S. S., Culotti, J. G., et al. (1996). Deleted in Colorectal Cancer (DCC) encodes a netrin receptor. Cell 87, 175–185. doi: 10.1016/s0092-8674(00)81336-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kelly, E. A., Tremblay, M.-È., Gahmberg, C. G., Tian, L., and Majewska, A. K. (2014). Subcellular localization of intercellular adhesion molecule-5 (telencephalin) in the visual cortex is not developmentally regulated in the absence of matrix metalloproteinase-9. J. Comp. Neurol. 522, 676–688. doi: 10.1002/cne.23440
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kennedy, M. B. (1994). The biochemistry of synaptic regulation in the central nervous system. Annu. Rev. Biochem. 63, 571–600. doi: 10.1146/annurev.biochem.63.1.571
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kennedy, M. B. (1997). The postsynaptic density at glutamatergic synapses. Trends Neurosci. 20, 264–268. doi: 10.1016/s0166-2236(96)01033-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kerrisk, M. E., Greer, C. A., and Koleske, A. J. (2013). Integrin α3 is required for late postnatal stability of dendrite arbors, dendritic spines and synapses and mouse behavior. J. Neurosci. 33, 6742–6752. doi: 10.1523/JNEUROSCI.0528-13.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kikkawa, Y., Sasaki, T., Nguyen, M. T., Nomizu, M., Mitaka, T., and Miner, J. H. (2007). The LG1–3 tandem of laminin alpha5 harbors the binding sites of Lutheran/basal cell adhesion molecule and alpha3beta1/alpha6beta1 integrins. J. Biol. Chem. 282, 14853–14860. doi: 10.1074/jbc.m611706200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koleske, A. J. (2013). Molecular mechanisms of dendrite stability. Nat. Rev. Neurosci. 14, 536–550. doi: 10.1038/nrn3486
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Korobova, F., and Svitkina, T. (2010). Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol. Biol. Cell 21, 165–176. doi: 10.1091/mbc.e09-07-0596
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kruse, J., Keilhauer, G., Faissner, A., Timpl, R., and Schachner, M. (1985). The J1 glycoprotein–a novel nervous system cell adhesion molecule of the L2/HNK-1 family. Nature 316, 146–148. doi: 10.1038/316146a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ksiazek, I., Burkhardt, C., Lin, S., Seddik, R., Maj, M., Bezakova, G., et al. (2007). Synapse loss in cortex of agrin-deficient mice after genetic rescue of perinatal death. J. Neurosci. 27, 7183–7195. doi: 10.1523/jneurosci.1609-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kwok, J. C. F., Dick, G., Wang, D., and Fawcett, J. W. (2011). Extracellular matrix and perineuronal nets in CNS repair. Dev. Neurobiol. 71, 1073–1089. doi: 10.1002/dneu.20974
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lai, C. S. W., Franke, T. F., and Gan, W.-B. (2012). Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature 483, 87–91. doi: 10.1038/nature10792
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lai, K.-O., and Ip, N. Y. (2013). Structural plasticity of dendritic spines: the underlying mechanisms and its dysregulation in brain disorders. Biochim. Biophys. Acta 1832, 2257–2263. doi: 10.1016/j.bbadis.2013.08.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lai Wing Sun, K., Correia, J. P., and Kennedy, T. E. (2011). Netrins: versatile extracellular cues with diverse functions. Development 138, 2153–2169. doi: 10.1242/dev.044529
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lander, A. D., Fujii, D. K., and Reichardt, L. F. (1985). Purification of a factor that promotes neurite outgrowth: isolation of laminin and associated molecules. J. Cell Biol. 101, 898–913. doi: 10.1083/jcb.101.3.898
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leonardo, E. D., Hinck, L., Masu, M., Keino-Masu, K., Ackerman, S. L., and Tessier-Lavigne, M. (1997). Vertebrate homologues of C. elegans UNC-5 are candidate netrin receptors. Nature 386, 833–838. doi: 10.1038/386833a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lévi, S., Grady, R. M., Henry, M. D., Campbell, K. P., Sanes, J. R., and Craig, A. M. (2002). Dystroglycan is selectively associated with inhibitory GABAergic synapses but is dispensable for their differentiation. J. Neurosci. 22, 4274–4285.
Li, Z., Hilgenberg, L. G., O’Dowd, D. K., and Smith, M. A. (1999). Formation of functional synaptic connections between cultured cortical neurons from agrin-deficient mice. J. Neurobiol. 39, 547–557. doi: 10.1002/(sici)1097-4695(19990615)39:4<547::aid-neu8>3.0.co;2-c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liesi, P., Dahl, D., and Vaheri, A. (1984). Neurons cultured from developing rat brain attach and spread preferentially to laminin. J. Neurosci. Res. 11, 241–251. doi: 10.1002/jnr.490110304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, B., Arai, A. C., Lynch, G., and Gall, C. M. (2003). Integrins regulate NMDA receptor-mediated synaptic currents. J. Neurophysiol. 89, 2874–2878. doi: 10.1152/jn.00783.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, Y.-C., and Koleske, A. J. (2010). Mechanisms of synapse and dendrite maintenance and their disruption in psychiatric and neurodegenerative disorders. Annu. Rev. Neurosci. 33, 349–378. doi: 10.1146/annurev-neuro-060909-153204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liston, C., Cichon, J. M., Jeanneteau, F., Jia, Z., Chao, M. V., and Gan, W.-B. (2013). Circadian glucocorticoid oscillations promote learning-dependent synapse formation and maintenance. Nat. Neurosci. 16, 698–705. doi: 10.1038/nn.3387
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, W. S., Pesold, C., Rodriguez, M. A., Carboni, G., Auta, J., Lacor, P., et al. (2001). Down-regulation of dendritic spine and glutamic acid decarboxylase 67 expressions in the reelin haploinsufficient heterozygous reeler mouse. Proc. Natl. Acad. Sci. U S A 98, 3477–3482. doi: 10.1073/pnas.051614698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lochner, J. E., Honigman, L. S., Grant, W. F., Gessford, S. K., Hansen, A. B., Silverman, M. A., et al. (2006). Activity-dependent release of tissue plasminogen activator from the dendritic spines of hippocampal neurons revealed by live-cell imaging. J. Neurobiol. 66, 564–577. doi: 10.1002/neu.20250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lonskaya, I., Partridge, J., Lalchandani, R. R., Chung, A., Lee, T., Vicini, S., et al. (2013). Soluble ICAM-5, a product of activity dependent proteolysis, increases mEPSC frequency and dendritic expression of GluA1. PLoS One 8:e69136. doi: 10.1371/journal.pone.0069136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Madani, R., Hulo, S., Toni, N., Madani, H., Steimer, T., Muller, D., et al. (1999). Enhanced hippocampal long-term potentiation and learning by increased neuronal expression of tissue-type plasminogen activator in transgenic mice. EMBO J. 18, 3007–3012. doi: 10.1093/emboj/18.11.3007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Majewska, A. K., Brown, E., Ross, J., and Yuste, R. (2000). Mechanisms of calcium decay kinetics in hippocampal spines: role of spine calcium pumps and calcium diffusion through the spine neck in biochemical compartmentalization. J. Neurosci. 20, 1722–1734.
Majewska, A. K., and Sur, M. (2003). Motility of dendritic spines in visual cortex in vivo: changes during the critical period and effects of visual deprivation. Proc. Natl. Acad. Sci. U S A 100, 16024–16029. doi: 10.1073/pnas.2636949100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Manthorpe, M., Engvall, E., Ruoslahti, E., Longo, F. M., Davis, G. E., and Varon, S. (1983). Laminin promotes neuritic regeneration from cultured peripheral and central neurons. J. Cell Biol. 97, 1882–1890. doi: 10.1083/jcb.97.6.1882
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martin, P. T., Ettinger, A. J., and Sanes, J. R. (1995). A synaptic localization domain in the synaptic cleft protein laminin beta 2 (s-laminin). Science 269, 413–416. doi: 10.1126/science.7618109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mataga, N., Mizuguchi, Y., and Hensch, T. K. (2004). Experience-dependent pruning of dendritic spines in visual cortex by tissue plasminogen activator. Neuron 44, 1031–1041. doi: 10.1016/j.neuron.2004.11.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mataga, N., Nagai, N., and Hensch, T. K. (2002). Permissive proteolytic activity for visual cortical plasticity. Proc. Natl. Acad. Sci. U S A 99, 7717–7721. doi: 10.1073/pnas.102088899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsumoto-Miyai, K., Sokolowska, E., Zurlinden, A., Gee, C. E., Lüscher, D., Hettwer, S., et al. (2009). Coincident pre- and postsynaptic activation induces dendritic filopodia via neurotrypsin-dependent agrin cleavage. Cell 136, 1161–1171. doi: 10.1016/j.cell.2009.02.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsuzaki, M., Ellis-Davies, G. C., Nemoto, T., Miyashita, Y., Iino, M., and Kasai, H. (2001). Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 4, 1086–1092. doi: 10.1038/nn736
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsuzaki, M., Honkura, N., Ellis-Davies, G. C. R., and Kasai, H. (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766. doi: 10.1038/nature02617
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McCroskery, S., Bailey, A., Lin, L., and Daniels, M. P. (2009). Transmembrane agrin regulates dendritic filopodia and synapse formation in mature hippocampal neuron cultures. Neuroscience 163, 168–179. doi: 10.1016/j.neuroscience.2009.06.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McCroskery, S., Chaudhry, A., Lin, L., and Daniels, M. P. (2006). Transmembrane agrin regulates filopodia in rat hippocampal neurons in culture. Mol. Cell. Neurosci. 33, 15–28. doi: 10.1016/j.mcn.2006.06.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meighan, S. E., Meighan, P. C., Choudhury, P., Davis, C. J., Olson, M. L., Zornes, P. A., et al. (2006). Effects of extracellular matrix-degrading proteases matrix metalloproteinases 3 and 9 on spatial learning and synaptic plasticity. J. Neurochem. 96, 1227–1241. doi: 10.1111/j.1471-4159.2005.03565.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Michaluk, P., Wawrzyniak, M., Alot, P., Szczot, M., Wyrembek, P., Mercik, K., et al. (2011). Influence of matrix metalloproteinase MMP-9 on dendritic spine morphology. J. Cell Sci. 124, 3369–3380. doi: 10.1242/jcs.090852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Missler, M., Südhof, T. C., and Biederer, T. (2012). Synaptic cell adhesion. Cold Spring Harb. Perspect. Biol. 4:a005694. doi: 10.1101/cshperspect.a005694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mitsui, S., Osako, Y., Yokoi, F., Dang, M. T., Yuri, K., Li, Y., et al. (2009). A mental retardation gene, motopsin/neurotrypsin/prss12, modulates hippocampal function and social interaction. Eur. J. Neurosci. 30, 2368–2378. doi: 10.1111/j.1460-9568.2009.07029.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Molinari, F., Rio, M., Meskenaite, V., Encha-Razavi, F., Augé, J., Bacq, D., et al. (2002). Truncating neurotrypsin mutation in autosomal recessive nonsyndromic mental retardation. Science 298, 1779–1781. doi: 10.1126/science.1076521
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morimoto, K., Fahnestock, M., and Racine, R. J. (2004). Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog. Neurobiol. 73, 1–60. doi: 10.1016/j.pneurobio.2004.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moser, M., Legate, K. R., Zent, R., and Fässler, R. (2009). The tail of integrins, talin and kindlins. Science 324, 895–899. doi: 10.1126/science.1163865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mühleisen, T. W., Mattheisen, M., Strohmaier, J., Degenhardt, F., Priebe, L., Schultz, C. C., et al. (2012). Association between schizophrenia and common variation in neurocan (NCAN), a genetic risk factor for bipolar disorder. Schizophr. Res. 138, 69–73. doi: 10.1016/j.schres.2012.03.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murakoshi, H., Wang, H., and Yasuda, R. (2011). Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 472, 100–104. doi: 10.1038/nature09823
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murakoshi, H., and Yasuda, R. (2012). Postsynaptic signaling during plasticity of dendritic spines. Trends Neurosci. 35, 135–143. doi: 10.1016/j.tins.2011.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nägerl, U. V., Eberhorn, N., Cambridge, S. B., and Bonhoeffer, T. (2004). Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron 44, 759–767. doi: 10.1016/j.neuron.2004.11.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagy, V., Bozdagi, O., Matynia, A., Balcerzyk, M., Okulski, P., Dzwonek, J., et al. (2006). Matrix metalloproteinase-9 is required for hippocampal late-phase long-term potentiation and memory. J. Neurosci. 26, 1923–1934. doi: 10.1523/jneurosci.4359-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nakagami, Y., Abe, K., Nishiyama, N., and Matsuki, N. (2000). Laminin degradation by plasmin regulates long-term potentiation. J. Neurosci. 20, 2003–2010.
Nakic, M., Manahan-Vaughan, D., Reymann, K. G., and Schachner, M. (1998). Long-term potentiation in vivo increases rat hippocampal tenascin-C expression. J. Neurobiol. 37, 393–404. doi: 10.1002/(sici)1097-4695(19981115)37:3<393::aid-neu5>3.3.co;2-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nedivi, E., Hevroni, D., Naot, D., Israeli, D., and Citri, Y. (1993). Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature 363, 718–722. doi: 10.1038/363718a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicholson, C., and Syková, E. (1998). Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 21, 207–215. doi: 10.1016/s0166-2236(98)01261-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicole, O., Docagne, F., Ali, C., Margaill, I., Carmeliet, P., MacKenzie, E. T., et al. (2001). The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat. Med. 7, 59–64. doi: 10.1038/83358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nikonenko, A., Schmidt, S., Skibo, G., Brückner, G., and Schachner, M. (2003). Tenascin-R-deficient mice show structural alterations of symmetric perisomatic synapses in the CA1 region of the hippocampus. J. Comp. Neurol. 456, 338–349. doi: 10.1002/cne.10537
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nishimune, H., Sanes, J. R., and Carlson, S. S. (2004). A synaptic laminin-calcium channel interaction organizes active zones in motor nerve terminals. Nature 432, 580–587. doi: 10.1038/nature03112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nishimune, H., Valdez, G., Jarad, G., Moulson, C. L., Müller, U., Miner, J. H., et al. (2008). Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. J. Cell Biol. 182, 1201–1215. doi: 10.1083/jcb.200805095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niu, S., Yabut, O., and D’Arcangelo, G. (2008). The Reelin signaling pathway promotes dendritic spine development in hippocampal neurons. J. Neurosci. 28, 10339–10348. doi: 10.1523/JNEUROSCI.1917-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Noguchi, J., Matsuzaki, M., Ellis-Davies, G. C. R., and Kasai, H. (2005). Spine-neck geometry determines NMDA receptor-dependent Ca2+ signaling in dendrites. Neuron 46, 609–622. doi: 10.1016/j.neuron.2005.03.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O’Connor, L. T., Lauterborn, J. C., Gall, C. M., and Smith, M. A. (1994). Localization and alternative splicing of agrin mRNA in adult rat brain: transcripts encoding lsoforms that aggregate acetylcholine receptors are not restricted to cholinergic regions. J. Neurosci. 14, 1141–1152.
Oh, W. C., Hill, T. C., and Zito, K. (2013). Synapse-specific and size-dependent mechanisms of spine structural plasticity accompanying synaptic weakening. Proc. Natl. Acad. Sci. U S A 110, E305–E312. doi: 10.1073/pnas.1214705110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oray, S., Majewska, A. K., and Sur, M. (2004). Dendritic spine dynamics are regulated by monocular deprivation and extracellular matrix degradation. Neuron 44, 1021–1030. doi: 10.1016/j.neuron.2004.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Orlando, C., Ster, J., Gerber, U., Fawcett, J. W., and Raineteau, O. (2012). Perisynaptic chondroitin sulfate proteoglycans restrict structural plasticity in an integrin-dependent manner. J. Neurosci. 32, 18009–18017, 18017a. doi: 10.1523/JNEUROSCI.2406-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Otey, C. A., and Burridge, K. (1990). Patterning of the membrane cytoskeleton by the extracellular matrix. Semin. Cell Biol. 1, 391–399.
Pan, F., Aldridge, G. M., Greenough, W. T., and Gan, W.-B. (2010). Dendritic spine instability and insensitivity to modulation by sensory experience in a mouse model of fragile X syndrome. Proc. Natl. Acad. Sci. U S A 107, 17768–17773. doi: 10.1073/pnas.1012496107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pang, P. T., Teng, H. K., Zaitsev, E., Woo, N. T., Sakata, K., Zhen, S., et al. (2004). Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 306, 487–491. doi: 10.1126/science.1100135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pappas, G. D., Kriho, V., and Pesold, C. (2001). Reelin in the extracellular matrix and dendritic spines of the cortex and hippocampus: a comparison between wild type and heterozygous reeler mice by immunoelectron microscopy. J. Neurocytol. 30, 413–425. doi: 10.1023/A:1015017710332
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patton, B. L., Cunningham, J. M., Thyboll, J., Kortesmaa, J., Westerblad, H., Edström, L., et al. (2001). Properly formed but improperly localized synaptic specializations in the absence of laminin alpha4. Nat. Neurosci. 4, 597–604. doi: 10.1038/88414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patton, B. L., Miner, J. H., Chiu, A. Y., and Sanes, J. R. (1997). Distribution and function of laminins in the neuromuscular system of developing, adult and mutant mice. J. Cell Biol. 139, 1507–1521. doi: 10.1083/jcb.139.6.1507
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pawlak, R., Rao, B. S. S., Melchor, J. P., Chattarji, S., McEwen, B., and Strickland, S. (2005). Tissue plasminogen activator and plasminogen mediate stress-induced decline of neuronal and cognitive functions in the mouse hippocampus. Proc. Natl. Acad. Sci. U S A 102, 18201–18206. doi: 10.1073/pnas.0509232102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pizzorusso, T., Medini, P., Berardi, N., Chierzi, S., Fawcett, J. W., and Maffei, L. (2002). Reactivation of ocular dominance plasticity in the adult visual cortex. Science 298, 1248–1251. doi: 10.1126/science.1072699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pizzorusso, T., Medini, P., Landi, S., Baldini, S., Berardi, N., and Maffei, L. (2006). Structural and functional recovery from early monocular deprivation in adult rats. Proc. Natl. Acad. Sci. U S A 103, 8517–8522. doi: 10.1073/pnas.0602657103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pribiag, H., Peng, H., Shah, W. A., Stellwagen, D., and Carbonetto, S. (2014). Dystroglycan mediates homeostatic synaptic plasticity at GABAergic synapses. Proc. Natl. Acad. Sci. U S A 111, 6810–6815. doi: 10.1073/pnas.1321774111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pujadas, L., Gruart, A., Bosch, C., Delgado, L., Teixeira, C. M., Rossi, D., et al. (2010). Reelin regulates postnatal neurogenesis and enhances spine hypertrophy and long-term potentiation. J. Neurosci. 30, 4636–4649. doi: 10.1523/JNEUROSCI.5284-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pujadas, L., Rossi, D., Andrés, R., Teixeira, C. M., Serra-Vidal, B., Parcerisas, A., et al. (2014). Reelin delays amyloid-beta fibril formation and rescues cognitive deficits in a model of Alzheimer’s disease. Nat. Commun. 5:3443. doi: 10.1038/ncomms4443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qian, Z., Gilbert, M. E., Colicos, M. A., Kandel, E. R., and Kuhl, D. (1993). Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature 361, 453–457. doi: 10.1038/361453a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qiu, S., Korwek, K. M., Pratt-Davis, A. R., Peters, M., Bergman, M. Y., and Weeber, E. J. (2006a). Cognitive disruption and altered hippocampus synaptic function in Reelin haploinsufficient mice. Neurobiol. Learn. Mem. 85, 228–242. doi: 10.1016/j.nlm.2005.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qiu, S., and Weeber, E. J. (2007). Reelin signaling facilitates maturation of CA1 glutamatergic synapses. J. Neurophysiol. 97, 2312–2321. doi: 10.1152/jn.00869.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qiu, S., Zhao, L. F., Korwek, K. M., and Weeber, E. J. (2006b). Differential reelin-induced enhancement of NMDA and AMPA receptor activity in the adult hippocampus. J. Neurosci. 26, 12943–12955. doi: 10.1523/jneurosci.2561-06.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reif, R., Sales, S., Hettwer, S., Dreier, B., Gisler, C., Wölfel, J., et al. (2007). Specific cleavage of agrin by neurotrypsin, a synaptic protease linked to mental retardation. FASEB J. 21, 3468–3478. doi: 10.1096/fj.07-8800com
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rivera, S., Tremblay, E., Timsit, S., Canals, O., Ben-Ari, Y., and Khrestchatisky, M. (1997). Tissue inhibitor of metalloproteinases-1 (TIMP-1) is differentially induced in neurons and astrocytes after seizures: evidence for developmental, immediate early gene and lesion response. J. Neurosci. 17, 4223–4235.
Rodriguez, M. A., Pesold, C., Liu, W. S., Kriho, V., Guidotti, A., Pappas, G. D., et al. (2000). Colocalization of integrin receptors and reelin in dendritic spine postsynaptic densities of adult nonhuman primate cortex. Proc. Natl. Acad. Sci. U S A 97, 3550–3555. doi: 10.1073/pnas.97.7.3550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rogers, J. T., Rusiana, I., Trotter, J., Zhao, L., Donaldson, E., Pak, D. T. S., et al. (2011). Reelin supplementation enhances cognitive ability, synaptic plasticity and dendritic spine density. Learn. Mem. 18, 558–564. doi: 10.1101/lm.2153511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rogers, J. T., Zhao, L., Trotter, J. H., Rusiana, I., Peters, M. M., Li, Q., et al. (2013). Reelin supplementation recovers sensorimotor gating, synaptic plasticity and associative learning deficits in the heterozygous reeler mouse. J. Psychopharmacol. 27, 386–395. doi: 10.1177/0269881112463468
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ruoslahti, E. (1996a). Brain extracellular matrix. Glycobiology 6, 489–492. doi: 10.1093/glycob/6.5.489
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ruoslahti, E. (1996b). RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 12, 697–715. doi: 10.1146/annurev.cellbio.12.1.697
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ruoslahti, E., and Pierschbacher, M. D. (1987). New perspectives in cell adhesion: RGD and integrins. Science 238, 491–497. doi: 10.1126/science.2821619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sabatini, B. L., Oertner, T. G., and Svoboda, K. (2002). The life cycle of Ca(2+) ions in dendritic spines. Neuron 33, 439–452. doi: 10.1016/s0896-6273(02)00573-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saghatelyan, A. K., Dityatev, A., Schmidt, S., Schuster, T., Bartsch, U., and Schachner, M. (2001). Reduced perisomatic inhibition, increased excitatory transmission and impaired long-term potentiation in mice deficient for the extracellular matrix glycoprotein tenascin-R. Mol. Cell. Neurosci. 17, 226–240. doi: 10.1006/mcne.2000.0922
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saghatelyan, A. K., Gorissen, S., Albert, M., Hertlein, B., Schachner, M., and Dityatev, A. (2000). The extracellular matrix molecule tenascin-R and its HNK-1 carbohydrate modulate perisomatic inhibition and long-term potentiation in the CA1 region of the hippocampus. Eur. J. Neurosci. 12, 3331–3342. doi: 10.1046/j.1460-9568.2000.00216.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saghatelyan, A. K., Snapyan, M., Gorissen, S., Meigel, I., Mosbacher, J., Kaupmann, K., et al. (2003). Recognition molecule associated carbohydrate inhibits postsynaptic GABA(B) receptors: a mechanism for homeostatic regulation of GABA release in perisomatic synapses. Mol. Cell. Neurosci. 24, 271–282. doi: 10.1016/s1044-7431(03)00163-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Samuel, M. A., Valdez, G., Tapia, J. C., Lichtman, J. W., and Sanes, J. R. (2012). Agrin and synaptic laminin are required to maintain adult neuromuscular junctions. PLoS One 7:e46663. doi: 10.1371/journal.pone.0046663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sanes, J. R. (1982). Laminin, fibronectin and collagen in synaptic and extrasynaptic portions of muscle fiber basement membrane. J. Cell Biol. 93, 442–451. doi: 10.1083/jcb.93.2.442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sanes, J. R., and Lichtman, J. W. (2001). Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat. Rev. Neurosci. 2, 791–805. doi: 10.1038/35097557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sappino, A. P., Madani, R., Huarte, J., Belin, D., Kiss, J. Z., Wohlwend, A., et al. (1993). Extracellular proteolysis in the adult murine brain. J. Clin. Invest. 92, 679–685. doi: 10.1172/jci116637
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sato, S., Omori, Y., Katoh, K., Kondo, M., Kanagawa, M., Miyata, K., et al. (2008). Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation. Nat. Neurosci. 11, 923–931. doi: 10.1038/nn.2160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Satz, J. S., Ostendorf, A. P., Hou, S., Turner, A., Kusano, H., Lee, J. C., et al. (2010). Distinct functions of glial and neuronal dystroglycan in the developing and adult mouse brain. J. Neurosci. 30, 14560–14572. doi: 10.1523/jneurosci.3247-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scheetz, A. J., and Constantine-Paton, M. (1994). Modulation of NMDA receptor function: implications for vertebrate neural development. FASEB J. 8, 745–752.
Scheff, S. W., and Price, D. A. (2003). Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiol. Aging 24, 1029–1046. doi: 10.1016/j.neurobiolaging.2003.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schneiders, F. I., Maertens, B., Böse, K., Li, Y., Brunken, W. J., Paulsson, M., et al. (2007). Binding of netrin-4 to laminin short arms regulates basement membrane assembly. J. Biol. Chem. 282, 23750–23758. doi: 10.1074/jbc.m703137200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schwartz, M. A. (2010). Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb. Perspect. Biol. 2:a005066. doi: 10.1101/cshperspect.a005066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schwartz, M. A., and Horwitz, A. R. (2006). Integrating adhesion, protrusion, and contraction during cell migration. Cell 125, 1223–1225. doi: 10.1016/j.cell.2006.06.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seeds, N. W., Basham, M. E., and Ferguson, J. E. (2003). Absence of tissue plasminogen activator gene or activity impairs mouse cerebellar motor learning. J. Neurosci. 23, 7368–7375.
Sekine, K., Kawauchi, T., Kubo, K.-I., Honda, T., Herz, J., Hattori, M., et al. (2012). Reelin controls neuronal positioning by promoting cell-matrix adhesion via inside-out activation of integrin α5β1. Neuron 76, 353–369. doi: 10.1016/j.neuron.2012.07.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Serafini, T., Colamarino, S. A., Leonardo, E. D., Wang, H., Beddington, R., Skarnes, W. C., et al. (1996). Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell 87, 1001–1014. doi: 10.1016/s0092-8674(00)81795-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Serafini, T., Kennedy, T. E., Galko, M. J., Mirzayan, C., Jessell, T. M., and Tessier-Lavigne, M. (1994). The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell 78, 409–424. doi: 10.1016/0092-8674(94)90420-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Serpinskaya, A. S., Feng, G., Sanes, J. R., and Craig, A. M. (1999). Synapse formation by hippocampal neurons from agrin-deficient mice. Dev. Biol. 205, 65–78. doi: 10.1006/dbio.1998.9112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sfakianos, M. K., Eisman, A., Gourley, S. L., Bradley, W. D., Scheetz, A. J., Settleman, J., et al. (2007). Inhibition of Rho via Arg and p190RhoGAP in the postnatal mouse hippocampus regulates dendritic spine maturation, synapse and dendrite stability and behavior. J. Neurosci. 27, 10982–10992. doi: 10.1523/jneurosci.0793-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sheng, M., and Kim, E. (2011). The postsynaptic organization of synapses. Cold Spring Harb. Perspect. Biol. 3:a005678. doi: 10.1101/cshperspect.a005678
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shi, Y., and Ethell, I. M. (2006). Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2+/calmodulin-dependent protein kinase II-mediated actin reorganization. J. Neurosci. 26, 1813–1822. doi: 10.1523/jneurosci.4091-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sidhu, H., Dansie, L. E., Hickmott, P. W., Ethell, D. W., and Ethell, I. M. (2014). Genetic removal of matrix metalloproteinase 9 rescues the symptoms of fragile x syndrome in a mouse model. J. Neurosci. 34, 9867–9879. doi: 10.1523/jneurosci.1162-14.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sloniowski, S., and Ethell, I. M. (2012). Looking forward to EphB signaling in synapses. Semin. Cell Dev. Biol. 23, 75–82. doi: 10.1016/j.semcdb.2011.10.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smalheiser, N. R., and Schwartz, N. B. (1987). Cranin: a laminin-binding protein of cell membranes. Proc. Natl. Acad. Sci. U S A 84, 6457–6461. doi: 10.1073/pnas.84.18.6457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smith, C. J., Watson, J. D., VanHoven, M. K., Colón-Ramos, D. A., and Miller, D. M. (2012). Netrin (UNC-6) mediates dendritic self-avoidance. Nat. Neurosci. 15, 731–737. doi: 10.1038/nn.3065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Soleman, S., Filippov, M. A., Dityatev, A., and Fawcett, J. W. (2013). Targeting the neural extracellular matrix in neurological disorders. Neuroscience 253, 194–213. doi: 10.1016/j.neuroscience.2013.08.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stanco, A., Szekeres, C., Patel, N., Rao, S., Campbell, K., Kreidberg, J. A., et al. (2009). Netrin-1-alpha3beta1 integrin interactions regulate the migration of interneurons through the cortical marginal zone. Proc. Natl. Acad. Sci. U S A 106, 7595–7600. doi: 10.1073/pnas.0811343106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Staubli, U., Vanderklish, P., and Lynch, G. (1990). An inhibitor of integrin receptors blocks long-term potentiation. Behav. Neural Biol. 53, 1–5. doi: 10.1016/0163-1047(90)90712-f
Stawarski, M., Stefaniuk, M., and Wlodarczyk, J. (2014). Matrix metalloproteinase-9 involvement in the structural plasticity of dendritic spines. Front. Neuroanat. 8:68. doi: 10.3389/fnana.2014.00068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stephan, A., Mateos, J. M., Kozlov, S. V., Cinelli, P., Kistler, A. D., Hettwer, S., et al. (2008). Neurotrypsin cleaves agrin locally at the synapse. FASEB J. 22, 1861–1873. doi: 10.1096/fj.07-100008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strekalova, T., Sun, M., Sibbe, M., Evers, M., Dityatev, A., Gass, P., et al. (2002). Fibronectin domains of extracellular matrix molecule tenascin-C modulate hippocampal learning and synaptic plasticity. Mol. Cell. Neurosci. 21, 173–187. doi: 10.1006/mcne.2002.1172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strickland, S. (2001). Tissue plasminogen activator in nervous system function and dysfunction. Thromb. Haemost. 86, 138–143.
Sung, U., O’Rear, J. J., and Yurchenco, P. D. (1993). Cell and heparin binding in the distal long arm of laminin: identification of active and cryptic sites with recombinant and hybrid glycoprotein. J. Cell Biol. 123, 1255–1268. doi: 10.1083/jcb.123.5.1255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szepesi, Z., Hosy, E., Ruszczycki, B., Bijata, M., Pyskaty, M., Bikbaev, A., et al. (2014). Synaptically released matrix metalloproteinase activity in control of structural plasticity and the cell surface distribution of GluA1-AMPA receptors. PLoS One 9:e98274. doi: 10.1371/journal.pone.0098274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szklarczyk, A., Lapinska, J., Rylski, M., McKay, R. D. G., and Kaczmarek, L. (2002). Matrix metalloproteinase-9 undergoes expression and activation during dendritic remodeling in adult hippocampus. J. Neurosci. 22, 920–930.
Takasaki, K., and Sabatini, B. L. (2014). Super-resolution 2-photon microscopy reveals that the morphology of each dendritic spine correlates with diffusive but not synaptic properties. Front. Neuroanat. 8:29. doi: 10.3389/fnana.2014.00029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tamkun, J. W., DeSimone, D. W., Fonda, D., Patel, R. S., Buck, C., Horwitz, A. F., et al. (1986). Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell 46, 271–282. doi: 10.1016/0092-8674(86)90744-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tan, C. L., Kwok, J. C. F., Patani, R., Ffrench-Constant, C., Chandran, S., and Fawcett, J. W. (2011). Integrin activation promotes axon growth on inhibitory chondroitin sulfate proteoglycans by enhancing integrin signaling. J. Neurosci. 31, 6289–6295. doi: 10.1523/jneurosci.0008-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tawil, N., Wilson, P., and Carbonetto, S. (1993). Integrins in point contacts mediate cell spreading: factors that regulate integrin accumulation in point contacts vs. focal contacts. J. Cell Biol. 120, 261–271. doi: 10.1083/jcb.120.1.261
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. doi: 10.1002/ana.410300410
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tian, L., Stefanidakis, M., Ning, L., Van Lint, P., Nyman-Huttunen, H., Libert, C., et al. (2007). Activation of NMDA receptors promotes dendritic spine development through MMP-mediated ICAM-5 cleavage. J. Cell Biol. 178, 687–700. doi: 10.1083/jcb.200612097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tissir, F., and Goffinet, A. M. (2003). Reelin and brain development. Nat. Rev. Neurosci. 4, 496–505. doi: 10.1038/nrn1113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tønnesen, J., Katona, G., Rózsa, B., and Nägerl, U. V. (2014). Spine neck plasticity regulates compartmentalization of synapses. Nat. Neurosci. 17, 678–685. doi: 10.1038/nn.3682
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trachtenberg, J. T., Chen, B. E., Knott, G. W., Feng, G., Sanes, J. R., Welker, E., et al. (2002). Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420, 788–794. doi: 10.1038/nature01273
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trotter, J. H., Klein, M., Jinwal, U. K., Abisambra, J. F., Dickey, C. A., Tharkur, J., et al. (2011). ApoER2 function in the establishment and maintenance of retinal synaptic connectivity. J. Neurosci. 31, 14413–14423. doi: 10.1523/jneurosci.3135-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tueting, P., Costa, E., Dwivedi, Y., Guidotti, A., Impagnatiello, F., Manev, R., et al. (1999). The phenotypic characteristics of heterozygous reeler mouse. Neuroreport 10, 1329–1334. doi: 10.1097/00001756-199904260-00032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ventruti, A., Kazdoba, T. M., Niu, S., and D’Arcangelo, G. (2011). Reelin deficiency causes specific defects in the molecular composition of the synapses in the adult brain. Neuroscience 189, 32–42. doi: 10.1016/j.neuroscience.2011.05.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Visse, R., and Nagase, H. (2003). Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function and biochemistry. Circ. Res. 92, 827–839. doi: 10.1161/01.res.0000070112.80711.3d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walikonis, R. S., Jensen, O. N., Mann, M., Provance, D. W., Mercer, J. A., and Kennedy, M. B. (2000). Identification of proteins in the postsynaptic density fraction by mass spectrometry. J. Neurosci. 20, 4069–4080.
Wang, X.-B., Bozdagi, O., Nikitczuk, J. S., Zhai, Z. W., Zhou, Q., and Huntley, G. W. (2008). Extracellular proteolysis by matrix metalloproteinase-9 drives dendritic spine enlargement and long-term potentiation coordinately. Proc. Natl. Acad. Sci. U S A 105, 19520–19525. doi: 10.1073/pnas.0807248105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, N., Butler, J. P., and Ingber, D. E. (1993). Mechanotransduction across the cell surface and through the cytoskeleton. Science 260, 1124–1127. doi: 10.1126/science.7684161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Warren, M. S., Bradley, W. D., Gourley, S. L., Lin, Y.-C., Simpson, M. A., Reichardt, L. F., et al. (2012). Integrin β1 signals through Arg to regulate postnatal dendritic arborization, synapse density and behavior. J. Neurosci. 32, 2824–2834. doi: 10.1523/JNEUROSCI.3942-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Washbourne, P., Dityatev, A., Scheiffele, P., Biederer, T., Weiner, J. A., Christopherson, K. S., et al. (2004). Cell adhesion molecules in synapse formation. J. Neurosci. 24, 9244–9249. doi: 10.1523/jneurosci.3339-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wegner, F., Härtig, W., Bringmann, A., Grosche, J., Wohlfarth, K., Zuschratter, W., et al. (2003). Diffuse perineuronal nets and modified pyramidal cells immunoreactive for glutamate and the GABA(A) receptor alpha1 subunit form a unique entity in rat cerebral cortex. Exp. Neurol. 184, 705–714. doi: 10.1016/s0014-4886(03)00313-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wiesel, T. N., and Hubel, D. H. (1963). Single cell responses in striate cortex of kittens deprived of vision in one eye. J. Neurophysiol. 26, 1003–1017.
Wilczynski, G. M., Konopacki, F. A., Wilczek, E., Lasiecka, Z., Gorlewicz, A., Michaluk, P., et al. (2008). Important role of matrix metalloproteinase 9 in epileptogenesis. J. Cell Biol. 180, 1021–1035. doi: 10.1083/jcb.200708213
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wlodarczyk, J., Mukhina, I., Kaczmarek, L., and Dityatev, A. (2011). Extracellular matrix molecules, their receptors, and secreted proteases in synaptic plasticity. Dev. Neurobiol. 71, 1040–1053. doi: 10.1002/dneu.20958
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wright, K. M., Lyon, K. A., Leung, H., Leahy, D. J., Ma, L., and Ginty, D. D. (2012). Dystroglycan organizes axon guidance cue localization and axonal pathfinding. Neuron 76, 931–944. doi: 10.1016/j.neuron.2012.10.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, S. H., Arévalo, J. C., Sarti, F., Tessarollo, L., Gan, W.-B., and Chao, M. V. (2009). Ankyrin repeat-rich membrane spanning/kidins220 protein regulates dendritic branching and spine stability in vivo. Dev. Neurobiol. 69, 547–557. doi: 10.1002/dneu.20723
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, Y., Chen, L., Zheng, P.-S., and Yang, B. B. (2002). beta 1-Integrin-mediated glioma cell adhesion and free radical-induced apoptosis are regulated by binding to a C-terminal domain of PG-M/versican. J. Biol. Chem. 277, 12294–12301. doi: 10.1074/jbc.m110748200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, Y. P., Siao, C. J., Lu, W., Sung, T. C., Frohman, M. A., Milev, P., et al. (2000). The tissue plasminogen activator (tPA)/plasmin extracellular proteolytic system regulates seizure-induced hippocampal mossy fiber outgrowth through a proteoglycan substrate. J. Cell Biol. 148, 1295–1304. doi: 10.1083/jcb.148.6.1295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamada, H., Watanabe, K., Shimonaka, M., and Yamaguchi, Y. (1994). Molecular cloning of brevican, a novel brain proteoglycan of the aggrecan/versican family. J. Biol. Chem. 269, 10119–10126.
Yebra, M., Montgomery, A. M. P., Diaferia, G. R., Kaido, T., Silletti, S., Perez, B., et al. (2003). Recognition of the neural chemoattractant Netrin-1 by integrins alpha6beta4 and alpha3beta1 regulates epithelial cell adhesion and migration. Dev. Cell 5, 695–707. doi: 10.1016/s1534-5807(03)00330-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yousif, L. F., Di Russo, J., and Sorokin, L. (2013). Laminin isoforms in endothelial and perivascular basement membranes. Cell Adh. Migr. 7, 101–110. doi: 10.4161/cam.22680
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yuste, R., Majewska, A. K., and Holthoff, K. (2000). From form to function: calcium compartmentalization in dendritic spines. Nat. Neurosci. 3, 653–659. doi: 10.1038/76609
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhai, S., Ark, E. D., Parra-Bueno, P., and Yasuda, R. (2013). Long-distance integration of nuclear ERK signaling triggered by activation of a few dendritic spines. Science 342, 1107–1111. doi: 10.1126/science.1245622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, X. H., Brakebusch, C., Matthies, H., Oohashi, T., Hirsch, E., Moser, M., et al. (2001). Neurocan is dispensable for brain development. Mol. Cell. Biol. 21, 5970–5978. doi: 10.1128/mcb.21.17.5970-5978.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, Q., Homma, K. J., and Poo, M.-M. (2004). Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44, 749–757. doi: 10.1016/j.neuron.2004.11.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ziv, N. E., and Smith, S. J. (1996). Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron 17, 91–102. doi: 10.1016/s0896-6273(00)80283-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zuo, Y., Lin, A., Chang, P., and Gan, W. B. (2005a). Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron 46, 181–189. doi: 10.1016/j.neuron.2005.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zuo, Y., Yang, G., Kwon, E., and Gan, W.-B. (2005b). Long-term sensory deprivation prevents dendritic spine loss in primary somatosensory cortex. Nature 436, 261–265. doi: 10.1038/nature03715
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: extracellular matrix, dendritic spine, chondroitin sulfate proteoglycans, agrin, reelin, extracellular proteases, RGD peptide, integrins
Citation: Levy AD, Omar MH and Koleske AJ (2014) Extracellular matrix control of dendritic spine and synapse structure and plasticity in adulthood. Front. Neuroanat. 8:116. doi: 10.3389/fnana.2014.00116
Received: 05 August 2014; Accepted: 29 September 2014;
Published online: 20 October 2014.
Edited by:
Nicolas Heck, University Pierre and Marie Curie, FranceReviewed by:
George W. Huntley, Icahn School of Medicine at Mount Sinai, USALeszek Kaczmarek, Nencki Institute, Poland
Constanze I. Seidenbecher, Leibniz Institute for Neurobiology, Germany
Copyright © 2014 Levy, Omar and Koleske. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony J. Koleske, Department of Molecular Biophysics and Biochemistry, Yale University, 333 Cedar Street, Sterling Hall of Medicine CE-33, New Haven, CT 06420-8024, USA e-mail:YW50aG9ueS5rb2xlc2tlQHlhbGUuZWR1
†These authors have contributed equally to this work.