 Cristina Guardia-Laguarta
Cristina Guardia-Laguarta Estela Area-Gomez2
Estela Area-Gomez2 Serge Przedborski
Serge Przedborski- 1Departments of Pathology, Columbia University Medical Center, New York, NY, USA
- 2Departments of Neurology, Columbia University Medical Center, New York, NY, USA
- 3Departments of Genetics and Development, Columbia University Medical Center, New York, NY, USA
α-synuclein (α-syn) is one of the genes that when mutated or overexpressed causes Parkinson’s Disease (PD). Initially, it was described as a synaptic terminal protein and later was found to be localized at mitochondria. Mitochondria-associated membranes (MAM) have emerged as a central endoplasmic reticulum (ER) subcellular compartments where key functions of the cell occur. These domains, enriched in cholesterol and anionic phospholipids, are where calcium homeostasis, lipid transfer, and cholesterol metabolism are regulated. Some proteins, related to mitochondrial dynamics and function, are also localized to this area. Several neurodegenerative diseases have shown alterations in MAM functions and resident proteins, including Charcot Marie-Tooth and Alzheimer’s disease (AD). We have recently reported that MAM function is downregulated in cell and mouse models of PD expressing pathogenic mutations of α-syn. This review focuses on the possible role of α-syn in these cellular domains and the early pathogenic features of PD that could be explained by α-syn-MAM disturbances.
Parkinson Disease Background
Parkinson disease (PD) is the second most prevalent neurodegenerative disease after Alzheimer disease (AD). Its main symptoms are resting tremors, rigidity, slowness of voluntary movements, freezing, and postural instability. Histopathologically, this disease is characterized by (a) a significant loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc; Braak et al., 2003) and (b) the accumulation of intracytoplasmic aggregates called Lewy bodies, composed mainly of alpha-synuclein protein (α-syn; Spillantini et al., 1997, 1998). This aggregation occurs at the SNpc and other cerebral areas such as locus ceruleus, nucleus basalis, hypothalamus, cerebral cortex, and autonomic nervous system (Maroteaux et al., 1988; Parkinson, 2002).
The majority of the PD cases are sporadic with only less than 10% of the cases related to mutations in genes such as PARK2, PARK7, PINK1, LRRK2 or SNCA (Polymeropoulos et al., 1997; Krüger et al., 1998; Zarranz et al., 2004). Among these, mutations or duplication in SNCA, which codifies for α-syn, have been shown to cause autosomal dominant forms of familial PD (Krüger et al., 1998; Singleton et al., 2003; Zarranz et al., 2004; Schon and Przedborski, 2011).
Subcellular Localization α-Synuclein
α-syn is a 140 aa protein, highly expressed in nervous tissues, that was identified as the precursor protein for the non–beta amyloid component of AD plaques (Uéda et al., 1993). Despite numerous research efforts, its main function remains unknown.
The majority of α-syn is soluble and resides in the cytoplasm. However, many researchers have demonstrated that α-syn, upon a yet unknown stimulus, is capable of binding to membranes and changes its N-terminal domain conformation upon this interaction (Eliezer et al., 2001; Jao et al., 2004, 2008). In vitro, α-syn binds preferentially to anionic phospholipids and liposomes of high curvature (Davidson et al., 1998; Fortin et al., 2004; Auluck et al., 2010). In the cell, these membrane regions are called lipid raft domains, which are detergent resistant membranes (DRM) with unique molecular characteristics (Simons and Toomre, 2000). Initially, lipid rafts were believed to form only at the plasma membrane; however, many authors have shown that these domains can also be localized intracellularly (Hayashi and Fujimoto, 2010).
In an effort to understand the function of this protein, many groups have reported several subcellular localizations for α-syn. In the last decades, multiple research data have shown α-syn located at pre-synaptic terminals (Kahle et al., 2000), participating in the regulation of the synaptic pool size and neurotransmitter release (Iwai et al., 1995; Masliah et al., 1996; Abeliovich et al., 2000; Murphy et al., 2000; Cabin et al., 2002; Gitler et al., 2008).
More recently, α-syn has been reported to bind to mitochondria (Li et al., 2007; Cole et al., 2008; Devi et al., 2008; Parihar et al., 2008; Zhang et al., 2008). This binding is especially significant in the striatum, substantia nigra (SNpc), and cortex of PD brains (Devi et al., 2008). Supporting these results, a recent study describes the existence of an N-terminal sequence in α-syn that could work as a mitochondrial targeting sequence (Devi et al., 2008). Moreover, α-syn binding to membranes is favored by the presence of cardiolipin (Zigoneanu et al., 2012), a lipid specific of the mitochondria membrane.
Mitochondria and α-Synuclein in the Pathogenesis of PD
Supporting α-syn localization to the mitochondria, PD patients, and cellular models containing pathogenic mutations of this protein show a deficit in mitochondrial functionality (Hsu et al., 2000; Schon and Przedborski, 2011), and, in particular, a significant decrease in complex I activity (Devi et al., 2008). In fact, the decrease in complex I activity is also present in PD brains and cellular models containing mutations in other genes related to the disease.
Moreover, exposure to a contaminant called 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine (MPTP), which is an inhibitor of the mitochondrial complex I, provokes parkinsonism symptoms and loss of dopaminergic neurons (Langston et al., 1983; Dauer and Przedborski, 2003). Additionally, injection of another complex I inhibitor, rotenone, caused a similar phenotype (Betarbet et al., 2000). All these data supports a role for mitochondrial dysfunction in the pathogenesis of PD.
In addition to complex I dysfunction, pathogenic mutations of α-syn have also been shown to interact with and reduce the activity of complex IV (Elkon et al., 2002). Also, there is data that relates the age-related accumulation of non-aggregated α-syn to mitochondria with a reduced dopamine phenotype in the SNpc (Chu and Kordower, 2007). Studies in one of the transgenic models of PD bearing the α-syn A53T mutation, show not only complex I inhibition, but also damaged mitochondrial DNA and aberrant mitochondrial dynamics (Martin et al., 2006; Chinta et al., 2010; Choubey et al., 2011).
It is widely known that mitochondria are dynamic organelles that undergo fusion and fission continuously (Chan, 2006). Mitochondrial movement is especially dramatic in neurons, where mitochondria travel along the axons to provide the terminals with ATP and other metabolites. Perturbations of this flux of mitochondria throughout the cell body cause defects in cell viability. Curiously, alterations in mitochondrial dynamics have been extensively reported in numerous neurodegenerative diseases, i.e., AD (Wang et al., 2008, 2009), PD (Yu et al., 2011; Cooper et al., 2012) and Charcot Marie-Tooth (Baloh et al., 2007; Chen and Chan, 2009). For instance, in the case of PD, the mitochondrial protein PINK1, whose mutation causes PD, is known to interact with the proteins Miro and Milton, both microtubule-associated proteins (Weihofen et al., 2009). Moreover, research data showed that aggregates of wild-type α-syn disrupt the mitochondrial trafficking of cargoes (Galvin et al., 1999; Lee et al., 2006).
An alternative consequence of the deregulation of mitochondrial trafficking is the alteration of the mitochondrial quality control. Spare or damaged mitochondria are degraded by mitophagy, a process in which the cell has to be able to differentiate healthy from damaged mitochondria. An indication of healthy mitochondria is a high membrane potential and low reactive oxygen species (Twig and Shirihai, 2011). The primary mechanism for degrading or minimizing damaged mitochondria is fusion and fission (Schon and Przedborski, 2011). During these processes, the cell “neutralize” unhealthy mitochondrial content, mixing it with other healthy organelles. In the case of neurodegenerative diseases, such as PD, where the fusion/fission machinery is altered, the accumulation of “bad” mitochondria can lead to the disease. More specifically, some authors link those dysfunctional mitochondria that cannot reach axonal extremes in PD with an increased expression of α-syn and aggregation (Lee et al., 2002). This could be the cause of the accumulation of mitochondrial mutations observed in the SNpc of PD patients that leads to a loss of dopaminergic neurons (Bender et al., 2006; Kraytsberg et al., 2006).
α-Synuclein is Localized at Mitochondria-Associated Membranes
Trying to answer this question, we revisited the exact cellular localization of α-syn. We have recently described a more accurate localization of α-syn (Guardia-Laguarta et al., 2014). Our data shows the existence of a subpopulation of α-syn that resides at the mitochondria-associated membranes or MAM. This region interconnects the endoplasmic reticulum (ER) and the mitochondria and is responsible for specific cellular functions. These membranes are composed of intracellular lipid rafts. This result is compelling, as previous works show that α-syn has an affinity for lipid rafts (Fortin et al., 2004) and negatively-charged membranes (Davidson et al., 1998) and could possibly explain the studies describing α-syn as a mitochondrial protein (Li et al., 2007; Cole et al., 2008; Devi et al., 2008; Parihar et al., 2008; Shavali et al., 2008). The lack of appropriate markers for MAM and the technical difficulty in fractionating this kind of membrane because of its association with the ER could explain previous results (Area-Gomez et al., 2012).
MAM is a subcompartment of the ER that is connected to the mitochondria (Figure 1; Rusiñol et al., 1994; Csordás et al., 2006; Hayashi et al., 2009). It is involved in a number of core cellular functions; i.e., calcium homeostasis (Csordás et al., 2010), cholesterol metabolism (Rusiñol et al., 1994), and phospholipid transfer from the ER to the mitochondria (Vance, 1990). Specifically, MAM has been described as the residence of several proteins related to phospholipid regulation (phosphatidylserine synthase 2: PTDSS2), cholesterol metabolism (acyl-CoA:cholesterol acyltransferase) (Rusiñol et al., 1994), and calcium transport from the ER to the mitochondria (the type 3 inositol 1,4,5-triphosphate receptor, IP3R3) (Hayashi and Fujimoto, 2010). Notably, mitochondrial distribution and dynamics are influenced by the physical connections formed by MAM (Rizzuto et al., 1998; Levine and Rabouille, 2005; Csordás et al., 2006; Hayashi et al., 2009; Friedman et al., 2011; Rowland and Voeltz, 2012). During mitochondrial fission, ER tubules appear to “embrace” mitochondria and mark sites of mitochondrial division (Friedman et al., 2011). In addition, isolated MAM from different tissues have been shown to be enriched in proteins related to the control of mitochondrial dynamics (e.g., FIS1, MFN2, and DRP1). Finally, MAM also contain some proteins involved in apoptosis (e.g., VDAC1 [voltage-dependent anion channel 1], BAX and BID (Garofalo et al., 2005; Ciarlo et al., 2010)). Indeed, calcium release at ER-mitochondrial contacts, which is important for ATP production, could be responsible for sensitizing mitochondria to apoptosis (Iwasawa et al., 2011; Tabas and Ron, 2011). The alteration of mitochondrial-ER contacts can cause deregulation of the calcium signal which results in inappropriate protein folding, metabolic alterations, and apoptosis (Csordás and Hajnóczky, 2009; Bui et al., 2010).
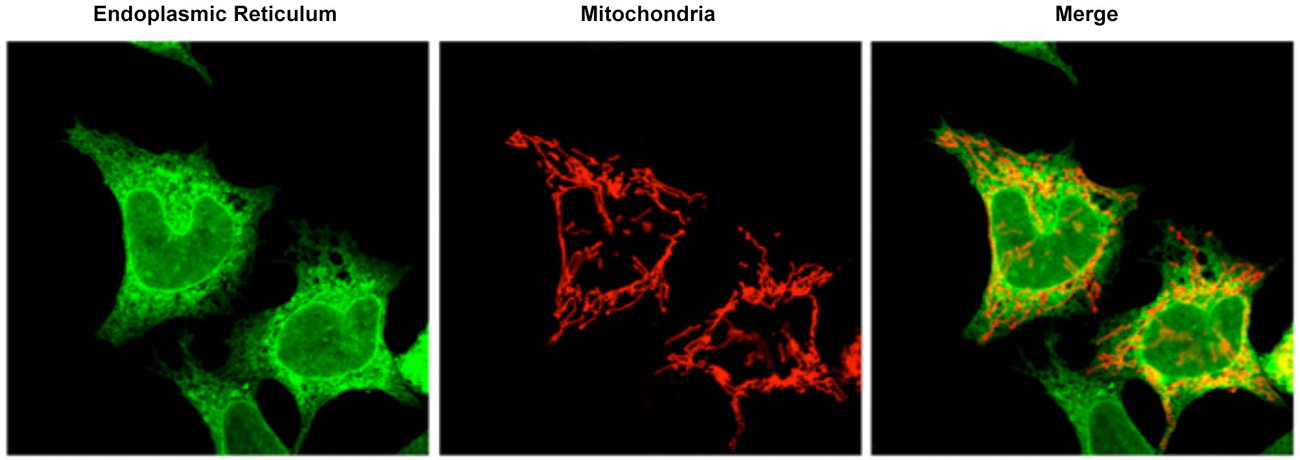
Figure 1. Confocal microscopy image representative of the ER mitochondrial connections in M17 dopaminergic cell line. ER is labeled with GFP-Sec61-β (green) and mitochondria labeled with pDsRed2-mito (red). Merge image indicates the colocalization of both organelles.
Could the Pathogenesis of PD be Explained by Early Alteration in MAM Function?
Scorrano’s group made the first correlation between MAM disturbance and disease when they described Mfn2 as a MAM resident protein that participates as a scaffold between ER and mitochondria. Mutations of Mfn2 cause Charcot Marie Tooth type 2a (de Brito and Scorrano, 2008). More recently it has been shown that presenilin-1, presenilin-2, and γ-secretase activity—all key factors associated with the pathogenicity of AD—are highly enriched in the MAM (Area-Gomez et al., 2009). Moreover, mutation or ablation of these γ-secretase components provokes a significant upregulation of several activities located at the MAM (Area-Gomez et al., 2012). Similarly, we have also shown that mutations in α-syn cause an alteration in the regulation of MAM function (Guardia-Laguarta et al., 2014). Supporting this observation, several groups have reported alterations in the lipidic composition of membranes from PD brains (Fabelo et al., 2011). These data suggest that these molecular alterations would change thermodynamic properties, organization, and signal transduction in the PD brain.
MAM regulates the homeostasis of cholesterol through the acyl-coA cholesterol acyltransferase (ACAT) activity. ACAT is the enzyme responsible of the conversion of free cholesterol to cholesteryl esters that eventually will be stored as lipid droplets. Therefore, ACAT activity regulates the amount of free cholesterol in cellular membranes. Cholesterol regulation alterations in PD have been extensively reported in the literature (de Lau et al., 2006; Huang et al., 2007; Hu et al., 2008). Interestingly, α-syn contains two cholesterol binding domains that play a role in the regulation of its binding to membranes and perhaps aggregation (Fantini and Yahi, 2013). In fact, α-syn transgenic mice treated with statins (cholesterol-lowering drug) showed a significant reduction in α-syn aggregation (Bar-On et al., 2008). Finally, numerous reports have described the interaction of α-syn and lipid droplets (Cole et al., 2002; De Franceschi et al., 2009). Taking all of this into account, it may well be that the cholesterol alterations in PD are a consequence of a MAM dysfunction caused by mutations in α-syn, a MAM protein.
The transfer of calcium between ER and mitochondria via MAM is a highly regulated process that controls the whole calcium homeostasis in the cell (Rizzuto et al., 2009; Csordás et al., 2010). As in many other neurodegenerative diseases, calcium homeostasis is altered in PD patients and animal models. In neurons, these alterations result in excitotoxic events that may eventually cause cell death (Rizzuto et al., 2009). Brini’s group was the first to show that α-syn is involved in the regulation of calcium homeostasis by altering the ER-mitochondria communication (Calì et al., 2012). Hodge and Colombini (1997) show that VDAC, a voltage-dependent calcium channel that controls mitochondrial calcium levels and mitochondrial function (and is localized in MAM), is decreased in nigral neurons positive for α-syn. Finally, recent evidence shows that increased Parkin expression improves calcium transfer through MAM; implying that Parkin mutations that cause PD could be detrimental for maintaining healthy levels of calcium (Calì et al., 2013).
Oxidative stress has been considered one of the main factors in the pathogenesis of PD (Kidd, 2000; Jenner, 2003). Increased levels of lipid hydroperoxydes have been found in SNpc and midbrain from PD patients (Yoritaka et al., 1996). Indeed, oxidative damage, lipoxidation of proteins like α-syn and oxidative DNA damage have been found in early-stages of PD (Dalfó et al., 2005), indicating a role for oxidative stress in the disease. As a response to this insult, it has been suggested that the SNpc suffers an increase in the turnover of membrane phospholipid synthesis that may be behind the specificity of neuronal death in the SNpc in PD (Ross et al., 2001). Again, the results point to an early imbalance in a very basic function of the cell, as it is the phospholipid transfer, controlled by MAM membranes that, over time, cause a disabling neurodegenerative process.
It is also well known that ER-mitochondria connections regulate mitochondrial dynamic processes (Csordás et al., 2006; Hayashi et al., 2009; Friedman et al., 2011; Rowland and Voeltz, 2012). It has been previously described that mutations in α-syn increase mitochondrial fragmentation (Kamp et al., 2010; Nakamura et al., 2011). Correlating this to MAM dysfunction, we have confirmed this fragmented phenotype in our mutant cells (Guardia-Laguarta et al., 2014). Nevertheless, it is possible that the fragmentation observed when α-syn is mutated is not due to defects on the fusion/fission mitochondrial machinery but rather to MAM alteration (Guardia-Laguarta et al., 2014).
Next, autophagy, a strictly regulated mechanism, is altered in PD (Chinta et al., 2010). Actually, PINK1 and Parkin, are known to be part of the mitochondrial autophagy cascade, or mitophagy. There is also evidence of the relation of α-syn with mitophagy: first it was described that transgenic animals expressing A53T α-syn present alterations in mitophagy (Chinta et al., 2010). The same result was found in yeast expressing wild-type α-syn (Sampaio-Marques et al., 2012). Interestingly, it has been reported that the autophagosomes, a key step during autophagy pathway, are formed at the MAM boundaries (Hamasaki et al., 2013).
Finally, consistent with other authors (Calì et al., 2012), our results suggest that pathogenic mutations result in a lower binding of α-syn to MAM. Therefore, it is possible that a certain amount of wild-type α-syn is necessary to maintain normal function, and so, mutation in α-syn does not cause a toxic gain-of-function, but rather a loss of “relevant” function in mitochondrial morphology maintenance and in some of the main MAM functions.
Future Questions
While we believe that our data help create a new way of thinking about PD pathogenesis, many questions need further research to properly address this new “MAM hypothesis”.
First, α-syn was initially described as a protein with a perinuclear and pre-synaptic dual localization, hence the name. The perinuclear localization is in agreement with our data because MAM, as part of the ER, is known to be enriched around the nucleus (de Brito and Scorrano, 2010). However, in order to satisfy both the pre-synaptic and this new MAM localization, the ER-mitochondria domains should also be present at these synaptic terminals. Notably, some authors have shown that ER-mitochondrial connections (McNulty, 1980) and known MAM markers (Mavlyutov et al., 2012) can be found closely juxtaposed to synaptic membranes. Moreover, similar connections between ER and mitochondria were also observed in ganglion cell membranes close to nerve endings (Watanabe and Burnstock, 1976; Taxi and Eugène, 1995).
Second, our data does not address the question of whether the toxic effect of α-syn is due to its aggregation tendency or to its soluble state, or caused by the overexpression of the protein (Narhi et al., 1999; Goldberg and Lansbury, 2000; Ostrerova-Golts et al., 2000). Furthermore, none of the experiments carried out reveal any aggregation of α-syn in dopaminergic cells lines or tissues. It may well be that only monomeric α-syn in MAM initiate the cascade of events that ultimately leads to mitochondrial dysfunction and dopaminergic cell loss. Equally possible is that α-syn binding to MAM triggers the aggregation of this protein into oligomers.
Finally, our work focuses only on the relationship between α-syn and MAM. Whether MAM dysfunction is an event also related to mutations in other PD genes requires additional investigation. Nonetheless, cellular symptoms caused by pathogenic mutations in these other genes are practically identical to those provoked by mutations in α-syn.
Conclusions
Our data suggest that MAM alteration may play an important role during the progression of PD pathology as an early event that may cause an imbalance in basic functions of the cell. Our “MAM hypothesis” helps reconcile many of the cellular symptoms seen in PD over time, such the accumulation of unhealthy mitochondria, altered autophagy, dysfunctional calcium levels, increased lipid droplets, and altered phospholipid species that lead to neurodegeneration. In addition, the localization of α-syn in MAM may help reconcile questions regarding the role of both the ER and mitochondria in the pathogenesis of PD, and may explain some of the features of DA neuron degeneration, i.e.: the deregulation of calcium homeostasis and mitochondrial dysfunction.
We hypothesize that a more detailed study of other functions that are located in MAM will reveal other alterations related to PD progression and that MAM alteration could be a good pre-symptomatic predictor of future PD pathology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Marc Tambini for valuable comments and corrections on the manuscript. This work was supported by the U.S. Department of Defense (W911NF-12-1-9159 to EAS), the J. Willard and Alice S. Marriott Foundation (to EAS), the Lucien Coté Early Investigator Award in Clinical Genetics from the Parkinson’s disease Foundation (PDF-CEI-1364 and PDF-CEI-1240, to CG-L) and a research grant from the American Parkinson Disease Association (to CG-L).
References
Abeliovich, A., Schmitz, Y., Fariñas, I., Choi-Lundberg, D., Ho, W. H., Castillo, P. E., et al. (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252. doi: 10.1016/s0896-6273(00)80886-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Area-Gomez, E., de Groof, A. J. C., Boldogh, I., Bird, T. D., Gibson, G. E., Koehler, C. M., et al. (2009). Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 175, 1810–1816. doi: 10.2353/ajpath.2009.090219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Area-Gomez, E., Del Carmen Lara Castillo, M., Tambini, M. D., Guardia-Laguarta, C., de Groof, A. J. C., Madra, M., et al. (2012). Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 31, 4106–4123. doi: 10.1038/emboj.2012.202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Auluck, P. K., Caraveo, G., and Lindquist, S. (2010). α-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell Dev. Biol. 26, 211–233. doi: 10.1146/annurev.cellbio.042308.113313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baloh, R. H., Schmidt, R. E., Pestronk, A., and Milbrandt, J. (2007). Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J. Neurosci. 27, 422–430. doi: 10.1523/jneurosci.4798-06.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bar-On, P., Crews, L., Koob, A. O., Mizuno, H., Adame, A., Spencer, B., et al. (2008). Statins reduce neuronal alpha-synuclein aggregation in in vitro models of Parkinson’s disease. J. Neurochem. 105, 1656–1667. doi: 10.1111/j.1471-4159.2008.05254.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bender, A., Krishnan, K. J., Morris, C. M., Taylor, G. A., Reeve, A. K., Perry, R. H., et al. (2006). High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517. doi: 10.1038/ng1769
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V., and Greenamyre, J. T. (2000). Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 3, 1301–1306. doi: 10.1038/81834
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Braak, H., Del Tredici, K., Rüb, U., de Vos, R. A. I., Jansen Steur, E. N. H., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/s0197-4580(02)00065-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bui, M., Gilady, S. Y., Fitzsimmons, R. E. B., Benson, M. D., Lynes, E. M., Gesson, K., et al. (2010). Rab32 modulates apoptosis onset and Mitochondria-Associated Membrane (MAM) properties. J. Biol. Chem. 285, 31590–31602. doi: 10.1074/jbc.M110.101584
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cabin, D. E., Shimazu, K., Murphy, D., Cole, N. B., Gottschalk, W., McIlwain, K. L., et al. (2002). Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 22, 8797–8807.
Calì, T., Ottolini, D., Negro, A., and Brini, M. (2012). α-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 287, 17914–17929. doi: 10.1074/jbc.M111.302794
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Calì, T., Ottolini, D., Negro, A., and Brini, M. (2013). Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim. Biophys. Acta 1832, 495–508. doi: 10.1016/j.bbadis.2013.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chan, D. C. (2006). Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 22, 79–99. doi: 10.1146/annurev.cellbio.22.010305.104638
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, H., and Chan, D. C. (2009). Mitochondrial dynamics–fusion, fission, movement and mitophagy–in neurodegenerative diseases. Hum. Mol. Genet. 18, R169–R176. doi: 10.1093/hmg/ddp326
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chinta, S. J., Mallajosyula, J. K., Rane, A., and Andersen, J. K. (2010). Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 486, 235–239. doi: 10.1016/j.neulet.2010.09.061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choubey, V., Safiulina, D., Vaarmann, A., Cagalinec, M., Wareski, P., Kuum, M., et al. (2011). Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 286, 10814–10824. doi: 10.1074/jbc.M110.132514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chu, Y., and Kordower, J. H. (2007). Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson’s disease? Neurobiol. Dis. 25, 134–149. doi: 10.1016/j.nbd.2006.08.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ciarlo, L., Manganelli, V., Garofalo, T., Matarrese, P., Tinari, A., Misasi, R., et al. (2010). Association of fission proteins with mitochondrial raft-like domains. Cell Death Differ. 17, 1047–1058. doi: 10.1038/cdd.2009.208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cole, N. B., Dieuliis, D., Leo, P., Mitchell, D. C., and Nussbaum, R. L. (2008). Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp. Cell Res. 314, 2076–2089. doi: 10.1016/j.yexcr.2008.03.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cole, N. B., Murphy, D. D., Grider, T., Rueter, S., Brasaemle, D., and Nussbaum, R. L. (2002). Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J. Biol. Chem. 277, 6344–6352. doi: 10.1074/jbc.m108414200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cooper, O., Seo, H., Andrabi, S., Guardia-Laguarta, C., Graziotto, J., Sundberg, M., et al. (2012). Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 4:141ra90. doi: 10.1126/scitranslmed.3003985
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Csordás, G., and Hajnóczky, G. (2009). SR/ER-mitochondrial local communication: calcium and ROS. Biochim. Biophys. Acta 1787, 1352–1362. doi: 10.1016/j.bbabio.2009.06.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Csordás, G., Renken, C., Várnai, P., Walter, L., Weaver, D., Buttle, K. F., et al. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921. doi: 10.1083/jcb.200604016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Csordás, G., Várnai, P., Golenár, T., Roy, S., Purkins, G., Schneider, T. G., et al. (2010). Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 39, 121–132. doi: 10.1016/j.molcel.2010.06.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dalfó, E., Portero-Otín, M., Ayala, V., Martínez, A., Pamplona, R., and Ferrer, I. (2005). Evidence of oxidative stress in the neocortex in incidental Lewy body disease. J. Neuropathol. Exp. Neurol. 64, 816–830. doi: 10.1097/01.jnen.0000179050.54522.5a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dauer, W., and Przedborski, S. (2003). Parkinson’s disease: mechanisms and models. Neuron 39, 889–909. doi: 10.1016/S0896-6273(03)00568-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davidson, W. S., Jonas, A., Clayton, D. F., and George, J. M. (1998). Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449. doi: 10.1074/jbc.273.16.9443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Brito, O. M., and Scorrano, L. (2010). An intimate liaison: spatial organization of the endoplasmic reticulum-mitochondria relationship. EMBO J. 29, 2715–2723. doi: 10.1038/emboj.2010.177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Franceschi, G., Frare, E., Bubacco, L., Mammi, S., Fontana, A., and de Laureto, P. P. (2009). Molecular insights into the interaction between alpha-synuclein and docosahexaenoic acid. J. Mol. Biol. 394, 94–107. doi: 10.1016/j.jmb.2009.09.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Lau, L. M. L., Koudstaal, P. J., Hofman, A., and Breteler, M. M. B. (2006). Serum cholesterol levels and the risk of Parkinson’s disease. Am. J. Epidemiol. 164, 998–1002. doi: 10.1093/aje/kwj283
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100. doi: 10.1074/jbc.M710012200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eliezer, D., Kutluay, E., Bussell, R., and Browne, G. (2001). Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 307, 1061–1073. doi: 10.1006/jmbi.2001.4538
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Elkon, H., Don, J., Melamed, E., Ziv, I., Shirvan, A., and Offen, D. (2002). Mutant and wild-type alpha-synuclein interact with mitochondrial cytochrome C oxidase. J. Mol. Neurosci. 18, 229–238. doi: 10.1385/jmn:18:3:229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fabelo, N., Martín, V., Santpere, G., Marín, R., Torrent, L., Ferrer, I., et al. (2011). Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol. Med. 17, 1107–1118. doi: 10.2119/molmed.2011.00119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fantini, J., and Yahi, N. (2013). The driving force of alpha-synuclein insertion and amyloid channel formation in the plasma membrane of neural cells: key role of ganglioside-and cholesterol-binding domains. Adv. Exp. Med. Biol. 991, 15–26. doi: 10.1007/978-94-007-6331-9_2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fortin, D. L., Troyer, M. D., Nakamura, K., Kubo, S., Anthony, M. D., and Edwards, R. H. (2004). Lipid rafts mediate the synaptic localization of alpha-synuclein. J. Neurosci. 24, 6715–6723. doi: 10.1523/jneurosci.1594-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Friedman, J. R., Lackner, L. L., West, M., DiBenedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. doi: 10.1126/science.1207385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Galvin, J. E., Uryu, K., Lee, V. M., and Trojanowski, J. Q. (1999). Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains α-, β- and γ-synuclein. Proc. Natl. Acad. Sci. U S A 96, 13450–13455. doi: 10.1073/pnas.96.23.13450
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garofalo, T., Giammarioli, A. M., Misasi, R., Tinari, A., Manganelli, V., Gambardella, L., et al. (2005). Lipid microdomains contribute to apoptosis-associated modifications of mitochondria in T cells. Cell Death Differ. 12, 1378–1389. doi: 10.1038/sj.cdd.4401672
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gitler, A. D., Bevis, B. J., Shorter, J., Strathearn, K. E., Hamamichi, S., Su, L. J., et al. (2008). The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. U S A 105, 145–150. doi: 10.1073/pnas.0710685105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goldberg, M. S., and Lansbury, P. T. (2000). Is there a cause-and-effect relationship between alpha-synuclein fibrillization and Parkinson’s disease? Nat. Cell Biol. 2, E115–E119. doi: 10.1038/35017124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guardia-Laguarta, C., Area-Gomez, E., Rüb, C., Liu, Y., Magrané, J., Becker, D., et al. (2014). α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259. doi: 10.1523/JNEUROSCI.2507-13.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hamasaki, M., Furuta, N., Matsuda, A., Nezu, A., Yamamoto, A., Fujita, N., et al. (2013). Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393. doi: 10.1038/nature11910
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hayashi, T., and Fujimoto, M. (2010). Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol. Pharmacol. 77, 517–528. doi: 10.1124/mol.109.062539
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hayashi, T., Rizzuto, R., Hajnoczky, G., and Su, T. P. (2009). MAM: more than just a housekeeper. Trends Cell Biol. 19, 81–88. doi: 10.1016/j.tcb.2008.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hodge, T., and Colombini, M. (1997). Regulation of metabolite flux through voltage-gating of VDAC channels. J. Membr. Biol. 157, 271–279. doi: 10.1007/s002329900235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hsu, L. J., Sagara, Y., Arroyo, A., Rockenstein, E., Sisk, A., Mallory, M., et al. (2000). α-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 157, 401–410. doi: 10.1016/s0002-9440(10)64553-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hu, G., Antikainen, R., Jousilahti, P., Kivipelto, M., and Tuomilehto, J. (2008). Total cholesterol and the risk of Parkinson disease. Neurology 70, 1972–1979. doi: 10.1212/01.wnl.0000312511.62699.a8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, X., Chen, H., Miller, W. C., Mailman, R. B., Woodard, J. L., Chen, P. C., et al. (2007). Lower low-density lipoprotein cholesterol levels are associated with Parkinson’s disease. Mov. Disord. 22, 377–381. doi: 10.1002/mds.21290
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iwai, A., Masliah, E., Yoshimoto, M., Ge, N., Flanagan, L., de Silva, H. A., et al. (1995). The precursor protein of non-A β component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 14, 467–475. doi: 10.1016/0896-6273(95)90302-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iwasawa, R., Mahul-Mellier, A.-L., Datler, C., Pazarentzos, E., and Grimm, S. (2011). Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 30, 556–568. doi: 10.1038/emboj.2010.346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jao, C. C., Der-Sarkissian, A., Chen, J., and Langen, R. (2004). Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc. Natl. Acad. Sci. U S A 101, 8331–8336. doi: 10.1073/pnas.0400553101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jao, C. C., Hegde, B. G., Chen, J., Haworth, I. S., and Langen, R. (2008). Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. U S A 105, 19666–19671. doi: 10.1073/pnas.0807826105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jenner, P. (2003). Oxidative stress in Parkinson’s disease. Ann. Neurol. 53(Suppl. 3), S26–S36; discussion S36–S38. doi: 10.1002/ana.10483
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kahle, P. J., Neumann, M., Ozmen, L., Muller, V., Jacobsen, H., Schindzielorz, A., et al. (2000). Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J. Neurosci. 20, 6365–6373.
Kamp, F., Exner, N., Lutz, A. K., Wender, N., Hegermann, J., Brunner, B., et al. (2010). Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 29, 3571–3589. doi: 10.1038/emboj.2010.223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kidd, P. M. (2000). Parkinson’s disease as multifactorial oxidative neurodegeneration: implications for integrative management. Altern. Med. Rev. 5, 502–529.
Kraytsberg, Y., Kudryavtseva, E., McKee, A. C., Geula, C., Kowall, N. W., and Khrapko, K. (2006). Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 38, 518–520. doi: 10.1038/ng1778
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krüger, R., Kuhn, W., Müller, T., Woitalla, D., Graeber, M., Kösel, S., et al. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Langston, J. W., Ballard, P., Tetrud, J. W., and Irwin, I. (1983). Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980. doi: 10.1126/science.6823561
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, H.-J., Choi, C., and Lee, S.-J. (2002). Membrane-bound α-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem. 277, 671–678. doi: 10.1074/jbc.m107045200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, H.-J., Khoshaghideh, F., Lee, S., and Lee, S.-J. (2006). Impairment of microtubule-dependent trafficking by overexpression of α-synuclein. Eur. J. Neurosci. 24, 3153–3162. doi: 10.1111/j.1460-9568.2006.05210.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levine, T., and Rabouille, C. (2005). Endoplasmic reticulum: one continuous network compartmentalized by extrinsic cues. Curr. Opin. Cell Biol. 17, 362–368. doi: 10.1016/j.ceb.2005.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, W.-W., Yang, R., Guo, J.-C., Ren, H.-M., Zha, X.-L., Cheng, J.-S., et al. (2007). Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 18, 1543–1546. doi: 10.1097/wnr.0b013e3282f03db4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maroteaux, L., Campanelli, J. T., and Scheller, R. H. (1988). Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815.
Martin, L. J., Pan, Y., Price, A. C., Sterling, W., Copeland, N. G., Jenkins, N. A., et al. (2006). Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 26, 41–50. doi: 10.1523/jneurosci.4308-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Masliah, E., Iwai, A., Mallory, M., Uéda, K., and Saitoh, T. (1996). Altered presynaptic protein NACP is associated with plaque formation and neurodegeneration in Alzheimer’s disease. Am. J. Pathol. 148, 201–210.
Mavlyutov, T. A., Epstein, M. L., Liu, P., Verbny, Y. I., Ziskind-Conhaim, L., and Ruoho, A. E. (2012). Development of the sigma-1 receptor in C-terminals of motoneurons and colocalization with the N,N′-dimethyltryptamine forming enzyme, indole-N-methyl transferase. Neuroscience 206, 60–68. doi: 10.1016/j.neuroscience.2011.12.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McNulty, J. A. (1980). Ultrastructural obsevations on synaptic ribbons in the pineal organ of the goldfish. Cell Tissue Res. 210, 249–256. doi: 10.1007/bf00237613
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murphy, D. D., Rueter, S. M., Trojanowski, J. Q., and Lee, V. M. (2000). Synucleins are developmentally expressed and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 20, 3214–3220.
Nakamura, K., Nemani, V. M., Azarbal, F., Skibinski, G., Levy, J. M., Egami, K., et al. (2011). Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein α-synuclein. J. Biol. Chem. 286, 20710–20726. doi: 10.1074/jbc.M110.213538
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Narhi, L., Wood, S. J., Steavenson, S., Jiang, Y., Wu, G. M., Anafi, D., et al. (1999). Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J. Biol. Chem. 274, 9843–9846. doi: 10.1074/jbc.274.14.9843
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ostrerova-Golts, N., Petrucelli, L., Hardy, J., Lee, J. M., Farer, M., and Wolozin, B. (2000). The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J. Neurosci. 20, 6048–6054.
Parihar, M. S., Parihar, A., Fujita, M., Hashimoto, M., and Ghafourifar, P. (2008). Mitochondrial association of alpha-synuclein causes oxidative stress. Cell. Mol. Life Sci. 65, 1272–1284. doi: 10.1007/s00018-008-7589-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Parkinson, J. (2002). An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 14, 223–236; discussion 222. doi: 10.1176/appi.neuropsych.14.2.223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rizzuto, R., Marchi, S., Bonora, M., Aguiari, P., Bononi, A., De Stefani, D., et al. (2009). Ca(2+) transfer from the ER to mitochondria: when, how and why. Biochim. Biophys. Acta 1787, 1342–1351. doi: 10.1016/j.bbabio.2009.03.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rizzuto, R., Pinton, P., Carrington, W., Fay, F. S., Fogarty, K. E., Lifshitz, L. M., et al. (1998). Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766. doi: 10.1126/science.280.5370.1763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ross, B. M., Mamalias, N., Moszczynska, A., Rajput, A. H., and Kish, S. J. (2001). Elevated activity of phospholipid biosynthetic enzymes in substantia nigra of patients with Parkinson’s disease. Neuroscience 102, 899–904. doi: 10.1016/s0306-4522(00)00501-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rowland, A. A., and Voeltz, G. K. (2012). Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat. Rev. Mol. Cell Biol. 13, 607–625. doi: 10.1038/nrm3440
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rusiñol, A. E., Cui, Z., Chen, M. H., and Vance, J. E. (1994). A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 269, 27494–27502.
Sampaio-Marques, B., Felgueiras, C., Silva, A., Rodrigues, M., Tenreiro, S., Franssens, V., et al. (2012). SNCA (α-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy. Autophagy 8, 1494–1509. doi: 10.4161/auto.21275
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schon, E. A., and Przedborski, S. (2011). Mitochondria: the next (neurode)generation. Neuron 70, 1033–1053. doi: 10.1016/j.neuron.2011.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shavali, S., Brown-Borg, H. M., Ebadi, M., and Porter, J. (2008). Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci. Lett. 439, 125–128. doi: 10.1016/j.neulet.2008.05.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Simons, K., and Toomre, D. (2000). Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31–39. doi: 10.1038/35036052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). α-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi: 10.1126/science.1090278
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U S A 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). α-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tabas, I., and Ron, D. (2011). Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190. doi: 10.1038/ncb0311-184
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taxi, J., and Eugène, D. (1995). Effects of axotomy, deafferentation and reinnervation on sympathetic ganglionic synapses: a comparative study. Int. Rev. Cytol. 159, 195–263. doi: 10.1016/s0074-7696(08)62108-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Twig, G., and Shirihai, O. S. (2011). The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 14, 1939–1951. doi: 10.1089/ars.2010.3779
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uéda, K., Fukushima, H., Masliah, E., Xia, Y., Iwai, A., Yoshimoto, M., et al. (1993). Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U S A 90, 11282–11286. doi: 10.1073/pnas.90.23.11282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vance, J. E. (1990). Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256.
Wang, X., Su, B., Fujioka, H., and Zhu, X. (2008). Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am. J. Pathol. 173, 470–482. doi: 10.2353/ajpath.2008.071208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, X., Su, B., Zheng, L., Perry, G., Smith, M. A., and Zhu, X. (2009). The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 109(Suppl. 1), 153–159. doi: 10.1111/j.1471-4159.2009.05867.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watanabe, H., and Burnstock, G. (1976). Junctional subsurface organs in frog sympathetic ganglion cells. J. Neurocytol. 5, 125–136. doi: 10.1007/bf01176186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weihofen, A., Thomas, K. J., Ostaszewski, B. L., Cookson, M. R., and Selkoe, D. J. (2009). Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 48, 2045–2052. doi: 10.1021/bi8019178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yoritaka, A., Hattori, N., Uchida, K., Tanaka, M., Stadtman, E. R., and Mizuno, Y. (1996). Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc. Natl. Acad. Sci. U S A 93, 2696–2701. doi: 10.1073/pnas.93.7.2696
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, W., Sun, Y., Guo, S., and Lu, B. (2011). The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum. Mol. Genet. 20, 3227–3240. doi: 10.1093/hmg/ddr235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zarranz, J. J., Alegre, J., Gómez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. doi: 10.1002/ana.10795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, L., Zhang, C., Zhu, Y., Cai, Q., Chan, P., Uéda, K., et al. (2008). Semi-quantitative analysis of alpha-synuclein in subcellular pools of rat brain neurons: an immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Res. 1244, 40–52. doi: 10.1016/j.brainres.2008.08.067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zigoneanu, I. G., Yang, Y. J., Krois, A. S., Haque, M. E., and Pielak, G. J. (2012). Interaction of α-synuclein with vesicles that mimic mitochondrial membranes. Biochim. Biophys. Acta 1818, 512–519. doi: 10.1016/j.bbamem.2011.11.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: alpha-synuclein, Parkinson’s disease, mitochondria-associated membranes, endoplasmic reticulum, phospholipid
Citation: Guardia-Laguarta C, Area-Gomez E, Schon EA and Przedborski S (2015) Novel subcellular localization for α-synuclein: possible functional consequences. Front. Neuroanat. 9:17. doi: 10.3389/fnana.2015.00017
Received: 30 October 2014; Accepted: 04 February 2015;
Published online: 23 February 2015.
Edited by:
Javier Blesa, Columbia University, USAReviewed by:
Benjamin Dehay, Institute of Neurodegenerative Diseases - CNRS UMR5293, FranceJacqueline Burré, Weill Cornell Medical College, USA
Copyright © 2015 Guardia-Laguarta, Area-Gomez, Schon and Przedborski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Guardia-Laguarta, Departments of Pathology, Columbia University Medical Center, Room P&S 4-401, Motor Neuron Center, 630W 168th Street, New York, NY 10032, USA e-mail:Y2cyNjQ1QGN1bWMuY29sdW1iaWEuZWR1