 Javier Blesa
Javier Blesa Ines Trigo-Damas
Ines Trigo-Damas Anna Quiroga-Varela
Anna Quiroga-Varela Vernice R. Jackson-Lewis
Vernice R. Jackson-Lewis- 1Centro Integral de Neurociencias A.C., HM Puerta del Sur, Hospitales de Madrid, Móstoles and Medical School, CEU San Pablo University, Madrid, Spain
- 2Department of Medicine, Clinica Neurologica, Ospedale Santa Maria della Misericordia – Università di Perugia, Perugia, Italy
- 3Department of Pathology and Cell Biology, Columbia University, New York, NY, USA
Parkinson disease (PD) is a chronic, progressive neurological disease that is associated with a loss of dopaminergic neurons in the substantia nigra pars compacta of the brain. The molecular mechanisms underlying the loss of these neurons still remain elusive. Oxidative stress is thought to play an important role in dopaminergic neurotoxicity. Complex I deficiencies of the respiratory chain account for the majority of unfavorable neuronal degeneration in PD. Environmental factors, such as neurotoxins, pesticides, insecticides, dopamine (DA) itself, and genetic mutations in PD-associated proteins contribute to mitochondrial dysfunction which precedes reactive oxygen species formation. In this mini review, we give an update of the classical pathways involving these mechanisms of neurodegeneration, the biochemical and molecular events that mediate or regulate DA neuronal vulnerability, and the role of PD-related gene products in modulating cellular responses to oxidative stress in the course of the neurodegenerative process.
Introduction
Parkinson’s disease (PD) is associated with the selective loss of dopamine (DA) neurons in the substantia nigra pars compacta (SNpc) and DA levels in the corpus striatum of the nigrostriatal DA pathway in the brain. This loss of DA causes a deregulation in the basal ganglia circuitries that leads to the appearance of motor symptoms such as bradykinesia, resting tremor, rigidity, and postural instability as well as non-motor symptoms such as sleep disturbances, depression, and cognitive deficits (Rodriguez-Oroz et al., 2009). The exact etiology of PD still remains elusive and the precise mechanisms that cause this disease remain to be identified (Obeso et al., 2010). At the cellular level, PD is related to excess production of reactive oxygen species (ROS), to alterations in catecholamine metabolism, to modifications in mitochondrial electron transporter chain (METC) function or to enhancement of iron deposition in the SNpc. The failure of normal cellular processes that occur in relation to the aging process are also believed to contribute to the increased vulnerability of DA neurons (Schapira and Jenner, 2011; Rodriguez et al., 2014).
While the familial forms of PD, that have been described, involve mutations in a number of genes (Kieburtz and Wunderle, 2013; Trinh and Farrer, 2013), mitochondrial dysfunction, neuroinflammation and environmental factors are increasingly appreciated as key determinants of dopaminergic neuronal susceptibility in PD, and are a feature of both familial and sporadic forms of the disease (Ryan et al., 2015). In both cases, oxidative stress is thought to be the common underlying mechanism that leads to cellular dysfunction and, eventual cell death. ROS are continuously produced in vivo by all body tissues. However, oxidative stress occurs when there is an imbalance between ROS production and cellular antioxidant activity. Oxidants and superoxide radicals are produced as products of oxidative phosphorylation, making mitochondria the main site of ROS generation within the cell. ROS can affect mitochondrial DNA which can cause modulations in the synthesis of METC components like adenosine triphosphate (ATP) production as well as the leakage of ROS into the cell’s cytoplasm (Brieger et al., 2012).
Although the precise mechanism corresponding to ROS generation related to PD is still unknown, in this review, we summarize the major sources of oxidative stress generated by the DA neurons, like DA metabolism, mitochondrial dysfunction, and neuroinflammation (Figure 1).
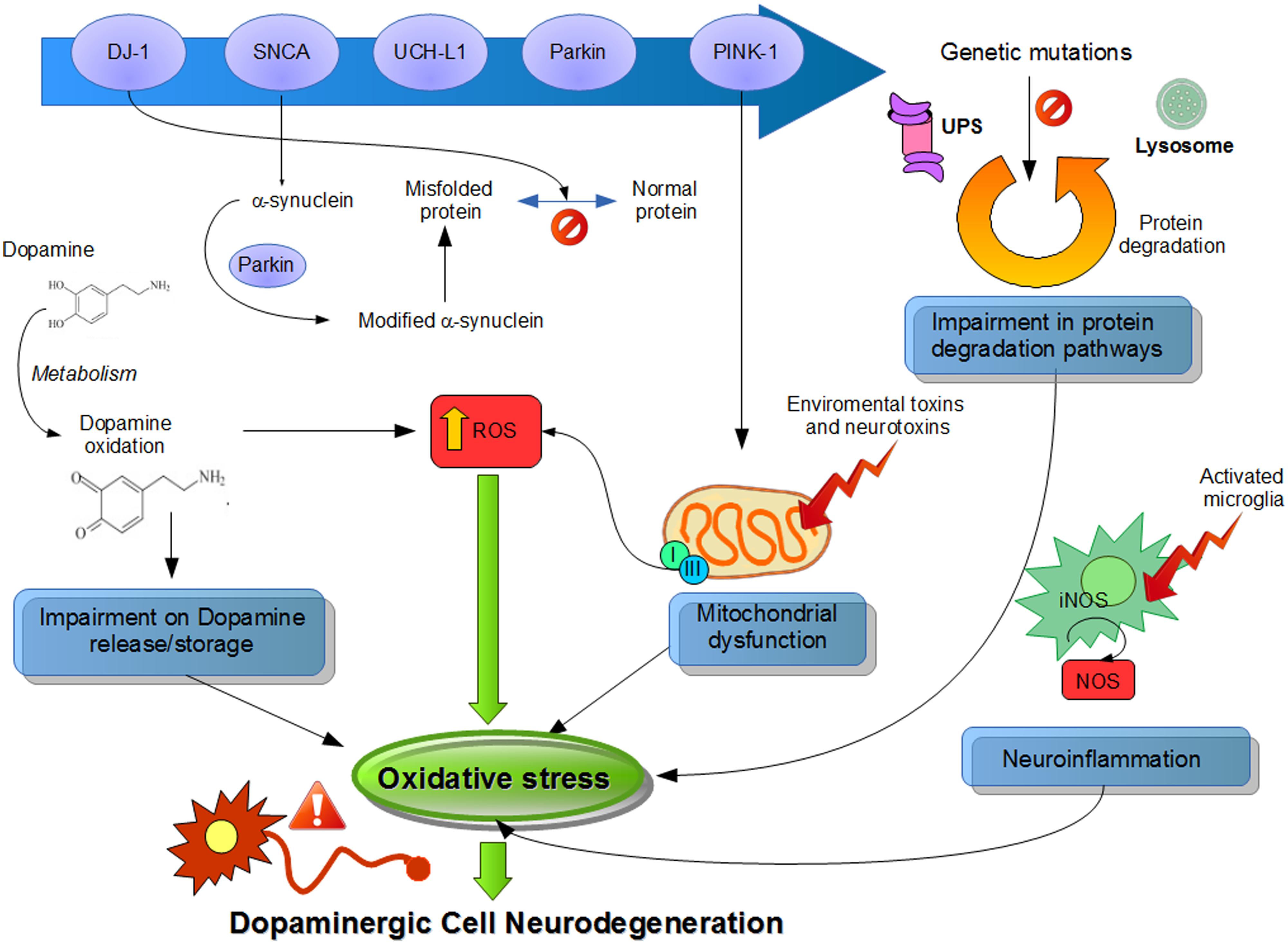
FIGURE 1. Suggested physiological processes related to pathogenesis of Parkinson’s disease (PD). Different pathways and their dysfunctions resulting from genetic modifications in PD-related genes and lead to an increased oxidative stress. Mutations or altered expression of these proteins result in mitochondrial impairment, oxidative stress, and protein misfolding. Also, dopamine metabolism may be oxidized to reactive dopamine quinones contributing to increased levels of reactive oxygen species. α-Synuclein becomes modified and accelerate its aggregation. Increased oxidative stress provokes impaired function of the UPS that degrades misfolded or damaged proteins and hereby further affecting cell survival. Environmental toxins impair mitochondrial function, increase the generation of free radicals, and lead to aggregation of proteins, including α-synuclein. Mitochondrial dysfunction by complex I inhibition affects by adding an increase in oxidative stress and a decline in ATP production, leading to damage of intracellular components and to cell death. Also, neuroinflammatory mechanisms might contribute to the cascade of consequences leading to cell death. In summary, all these several cellular mechanisms attributed to oxidative stress are implicated in the selective degeneration of dopaminergic neurons.
Dopamine Metabolism
Selective degeneration of the DA neurons of the SNpc suggests that DA itself may be a source of oxidative stress (Segura-Aguilar et al., 2014). DA is synthesized from tyrosine by tyrosine hydroxylase (TH) and aromatic amino acid decarboxylase. Following this, DA is stored in synaptic vesicles after uptake by the vesicular monoamine transporter 2 (VMAT2). However, when there is an excess amount of cytosolic DA outside of the synaptic vesicle in damaged neurons, i.e., after L-DOPA treatment, DA is easily metabolized via monoamine oxidase (MAO) or by auto-oxidation to cytotoxic ROS (Zucca et al., 2014). For example, mishandling of DA in mice with reduced VMAT2 expression was sufficient to cause DA-mediated toxicity and progressive loss of DA neurons (Caudle et al., 2007).
This oxidative process alters mitochondrial respiration and induces a change in the permeability transition pores in brain mitochondria (Berman and Hastings, 1999). Also, the auto-oxidation of DA produces electron-deficient DA quinones or DA semiquinones (Sulzer and Zecca, 2000). Some studies have demonstrated a regulatory role for quinone formation in DA neurons in the L-DOPA-treated PD model induced by neurotoxins and in methamphetamine neurotoxicity (Asanuma et al., 2003; Miyazaki et al., 2006; Ares-Santos et al., 2014). DA quinones can modify a number of PD-related proteins, such as α-synuclein (α-syn), parkin, DJ-1, Superoxide dismutase-2 (SOD2), and UCH-L1 (Belluzzi et al., 2012; Girotto et al., 2012; da Silva et al., 2013; Hauser et al., 2013; Toyama et al., 2014; Zhou et al., 2014) and have been shown to cause inactivation of the DA transporter (DAT) and the TH enzyme (Kuhn et al., 1999; Whitehead et al., 2001), as well as mitochondrial dysfunction (Lee et al., 2003), alterations of brain mitochondria (Gluck and Zeevalk, 2004) and dysfunction in Complex I activity (Jana et al., 2007, 2011; Van Laar et al., 2009). Additionally, DA quinones can be oxidized to aminochrome, whose redox-cycling leads to the generation of the superoxide radical and the depletion of cellular nicotinamide adenine dinucleotide phosphate-oxidase (NADPH), which ultimately forms the neuromelanin (Sulzer et al., 2000) known to be accumulated in the SNpc of the human brain (Ohtsuka et al., 2013, 2014; Plum et al., 2013).
Significant increases in cysteinyl adducts of L-DOPA, DA, and DOPAC have been found in substantia nigra of PD patients, suggesting the cytotoxic nature of DA oxidation (Spencer et al., 1998). Also, DA terminals actively degenerated proportionally to increased levels of DA oxidation following a single injection of DA into the striatum (Rabinovic et al., 2000). Recently, it has been shown that increased uptake of DA through the DAT in mice results in oxidative damage, neuronal loss and motor deficits (Masoud et al., 2015).
Mitochondrial Dysfunction
Mitochondrial dysfunction is closely related to increased ROS formation in PD (Schapira, 2008). Oxidative phosphorylation is the main mechanism providing energy to power neural activity in which the mitochondria use their structure, enzymes, and energy released by the oxidation of nutrients to form ATP (Hall et al., 2012). Consequently, this metabolic pathway is the main source of superoxide and hydrogen peroxide, which, at the same time, lead to propagation of free radicals contributing to the disease.
Complex I deficiencies of the respiratory chain account for the majority of unfavorable neural apoptosis generation and is considered one of the primary sources of ROS in PD. Complex I inhibition results in an enhanced production of ROS, which, in turn, will inhibit complex I. Reduction in complex I activity in the SNpc of patients with sporadic PD has been well described (Schapira et al., 1990; Hattori et al., 1991; Hattingen et al., 2009). Additionally, mitochondrial complex I deficiency in different brain regions (Mizuno et al., 1989; Parker et al., 2008), fibroblasts (Mytilineou et al., 1994), blood platelets (Krige et al., 1992; Blandini et al., 1998), skeletal muscle (Blin et al., 1994), and lymphocytes (Yoshino et al., 1992; Haas et al., 1995) of PD patients has been shown before as well.
As such, complex I inhibitors like 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or rotenone show preferential cytotoxicity to the DA neurons (Blesa and Przedborski, 2014). The mechanism by which MPTP crosses the blood–brain barrier and is oxidized to 1-methyl-4-phenylpyridinium (MPP+) is well known (Blesa and Przedborski, 2014). The MPP+ accumulates in the mitochondria where it inhibits complex I in the METC, therefore disrupting the flow of electrons along the METC, which results in decreased ATP production and increased generation of ROS (Mizuno et al., 1987). Like MPTP, rotenone is another mitochondrial complex I inhibitor. Interestingly, rotenone toxicity is involved in oxidative damage to proteins and Lewy body-like inclusions (Betarbet et al., 2000; Sherer et al., 2003a,b; Greenamyre et al., 2010). The events downstream to complex I inhibition that lead to neuronal cell death by these toxins are still unknown (Schapira, 2010).
Other evidence for mitochondrial dysfunction related to oxidative stress and DA cell damage comes from findings that mutations in genes of proteins like α-syn, parkin, DJ-1, or PINK are linked to familial forms of PD. The convergence of all of these proteins on mitochondrial dynamics uncovers a common function in the mitochondrial stress response that might provide a potential physiological basis for the pathology of PD (Norris et al., 2015; van der Merwe et al., 2015). Overall, these observations show that mutations in these genes affect mitochondrial function and integrity and, are associated with increases in oxidative stress (Zuo and Motherwell, 2013). ROS influence proteasomal, lysosomal, and mitochondrial function, which, in turn, regulate the cellular response to oxidative damage (Cook et al., 2012). The correct elimination of damaged proteins by effective proteolysis and the synthesis of new and protective proteins are vital in the preservation of brain homeostasis during periods of increased levels of ROS. Consequently, this can lead to protein misfolding (i.e., α-syn), preventing the ability of some of these proteins to be unfolded and degraded by the systems that regulate protein clearance, like the ubiquitin proteasome system or autophagy. Indeed, protein misfolding, together with the dysfunction of these protein degradation systems, may play a key role in the appearance of deleterious events implicated in the neurodegenerative process of PD (Schapira et al., 2014).
Parkin and PINK1 are localized in the mitochondria and their functions are tightly connected to the normal functioning of the mitochondria (Scarffe et al., 2014). PINK1 accumulates on the outer membrane of damaged mitochondria and recruits Parkin to the dysfunctional mitochondrion (Pickrell and Youle, 2015). In humans with parkin mutations, mitochondrial complex I activity is impaired (Müftüoglu et al., 2004). Overexpression of parkin in mice reduced DA neuronal cell loss induced by MPTP through the protection of mitochondria and the reduction of α-syn (Bian et al., 2012). On the other hand, parkin KO mice showed decreased amounts of several proteins that are involved in mitochondrial function and oxidative stress as well as increases in protein oxidation and lipid peroxidation (Palacino et al., 2004). Also, Drosophila, lacking, or deficient in parkin, exhibit mitochondrial deficits and high vulnerability to oxidative stress (Saini et al., 2010). PINK1 mutations in humans lead to mitochondrial defects and respiratory chain abnormalities (Hoepken et al., 2007; Piccoli et al., 2008). PINK1 KO in human and mouse DA neurons causes decreases in membrane potential and increases in ROS generation (Wood-Kaczmar et al., 2008). The decrease in mitochondrial membrane potential is not due to a proton leak, but to respiratory chain defects like complex I and complex III deficiency (Amo et al., 2011, 2014). Therefore, PINK1 is required for maintaining normal mitochondrial morphology of SNpc DA neurons in culture and exerts its neuroprotective effect by inhibiting ROS formation (Wang et al., 2011). In animal models, studies show that the lack of PINK1 resulted in abnormal mitochondrial morphology, loss of SNpc DA neurons, reduction in complex I activity, and enhanced vulnerability to oxidative stress (Clark et al., 2006; Kitada et al., 2007; Gautier et al., 2008). These defects can be ameliorated and rescued by the enhanced expression of parkin (Yang et al., 2006; Exner et al., 2007). This last scenario seems to involve PINK1 and Parkin in a common pathway that regulates mitochondrial physiology and cell survival in which PINK1 seems to be functioning upstream of Parkin, at least as observed in Drosophila disease models (Clark et al., 2006).
α-syn is a soluble protein that is highly enriched in the presynaptic terminals of neurons. Accumulation of α-syn as intracellular filamentous aggregates is a pathological feature of both sporadic and familial PD (Goedert et al., 2013). Accumulation of wild-type α-syn in DA neurons reduced mitochondrial complex I activity, elevated ROS production leading to cell death (Martin et al., 2006). It has been shown that α-syn inclusions elevate dendritic mitochondrial oxidative stress in DA neurons (Dryanovski et al., 2013). This mitochondrial dysfunction occurs many months before the occurrence of striatal DA loss (Subramaniam et al., 2014). The nuclear translocation of α-syn increases susceptibility of MES23.5 cells to oxidative stress (Zhou et al., 2013). Exposure to rotenone or other stimuli that promote ROS formation and mitochondrial alterations correlate well with mutant α-syn phosphorylation at Ser129 (Perfeito et al., 2014). Oxidative stress promotes uptake, accumulation, and oligomerization of extracellular α-syn in oligodendrocytes (Pukass and Richter-Landsberg, 2014) and induces posttranslational modifications of α-syn which can increase DA toxicity (Xiang et al., 2013). It has been suggested that the NADPH oxidases, which are responsible for ROS generation, could be major players in synucleinopathies (Cristóvão et al., 2012).
DJ-1 is another gene reported to cause a familial early onset PD (Puschmann, 2013). DJ-1 binds to subunits of mitochondrial complex I and regulates its activity (Hayashi et al., 2009). Although a portion of DJ-1 is present in mitochondria matrix and inter-membrane space (Zhang et al., 2005), the degree of translocation of DJ-1 into mitochondria is stimulated by oxidative stress (Canet-Avilés et al., 2004). Mitochondrial-targeted sequence-conjugated DJ-1 has been shown to be more protective against oxidative stress-induced cell death (Junn et al., 2009). DJ-1 KO mice displayed nigrostriatal DA neuron loss (Goldberg et al., 2005). Also, these DJ-1 KO mice showed altered mitochondrial respiration and morphology, reduced membrane potential, and accumulation of defective mitochondria (Irrcher et al., 2010; Krebiehl et al., 2010; Giaime et al., 2012). These defects can be reversed by DJ-1 overexpression, which points to the specific role of DJ-1 in mitochondrial function (Heo et al., 2012). Recently, following oxidative stress, DJ-1 was shown to be involved in the oxidative stress response that leads to the upregulation of the proteasome, thus inhibiting its activity and rescuing partially unfolded proteins from degradation (Moscovitz et al., 2015).
Neuroinflammation
Neuronal loss in PD is associated with chronic neuroinflammation, which is controlled primarily by microglia, the major resident immune cells in the brain (Barcia et al., 2003) and, to a lesser extent, by astrocytes and oligodendrocytes (Perry, 2012). Microglial activation has been found with a greater density in the SNpc (Lawson et al., 1990) and in the olfactory bulb of both sporadic and familial PD patients (McGeer et al., 1988; Doorn et al., 2014a,b). Additionally, activated microglia have been found in the SNpc and in the striatum of PD animal models (Pisanu et al., 2014; Stott and Barker, 2014) and have been associated with different PD-associated gene/proteins like α-syn or LRRK2 (Daher et al., 2014; Sacino et al., 2014). In response to certain environmental toxins and endogenous proteins, microglia can shift to an over-activated state and release ROS which can cause neurotoxicity (Block et al., 2007). Accumulating evidence indicates that activation of different enzymes like NADPH oxidase (NOX2) in microglia is neurotoxic not only through the production of extracellular ROS that damage neighboring neurons but also through the initiation of redox signaling in microglia that amplifies the pro-inflammatory response (Surace and Block, 2012).
Neuromelanin confers the dark pigmentation that is produced from DA oxidation and is so characteristic of the SNpc appearance. High levels of catecholamine metabolism in the midbrain are associated with increased levels of neuromelanin in the same region and, it is neuromelanin that is thought to be one of the molecules responsible for inducing chronic neuroinflammation in PD. Neuromelanin released from dying DA neurons in the SNpc activate microglia, increasing the sensitivity of DA neurons to oxidative stress-mediated cell death (Halliday et al., 2005; Li et al., 2005; Beach et al., 2007; Zhang et al., 2009). The ability of neuromelanin to interact with transition metals, especially iron, and to mediate intracellular oxidative mechanisms have received particular attention. Increased levels of iron result in increased ROS and increased oxidative stress and has been shown to be involved in aging and PD. Iron homeostasis is modulated by angiotensin in DA neurons and microglia, and glial cells play an essential role in the efficient regulation of this balance (Garrido-Gil et al., 2013).
Dopamine neurons containing neuromelanin are especially more susceptible, indicating a possible role for neuromelanin in MPTP-toxicity (Herrero et al., 1993). MPTP induces a glial response, increased levels of inflammatory cytokines and microglial activation in mice (Członkowska et al., 1996; Jackson-Lewis and Smeyne, 2005) and monkeys (Barcia et al., 2004, 2009). Angiotensin is one of the most important inflammation and oxidative stress inducers, and produces ROS by activation of the NADPH-oxidase complex. It has been suggested that the inflammatory response in the MPTP model could be mediated by brain angiotensin and microglial NADPH-derived ROS (Joglar et al., 2009). Moreover, oral treatment with NADPH oxidase antagonists mitigates the clinical and pathological features of parkinsonism in the MPTP marmoset model (Philippens et al., 2013). Also, microglia play an important role in mediating rotenone-induced neuronal degeneration through NADPH (Gao et al., 2003, 2011; Pal et al., 2014). Rotenone increased microglial activation in both the SNpc and striatum in rats (Sherer et al., 2003a), activated microglia via the NF-κB signaling pathway (Gao et al., 2013) and induced neuronal death by the microglial phagocytosis of neurons (Emmrich et al., 2013).
Parkinson’s disease-associated proteins like α-syn, parkin, LRRK2, and DJ-1 have also been reported to activate microglia (Wilhelmus et al., 2012). Extracellular α-syn released from neuronal cells is an endogenous agonist for Toll-like receptor 2 (TLR2), which activates the microglial inflammatory responses (Kim et al., 2013a). An increased number of activated microglia and increased levels of TNF-α mRNA and protein were detected in the striatum and in the SNpc of mice over-expressing WT human α-syn (Watson et al., 2012). Moreover, in α-syn KO mice, microglia secreted higher levels of proinflammatory cytokines, TNF alpha and IL-6 (interleukin-6) compared to WT mice (Austin et al., 2006). Intracerebral injection of recombinant amyloidogenic or soluble α-syn induces extensive α-syn intracellular inclusion pathology that is associated with a robust gliosis (Sacino et al., 2014). LRRK2 increases proinflammatory cytokine release from activated primary microglial cells which results in neurotoxicity (Gillardon et al., 2012). In contrast, LRRK2 inhibition attenuates microglial inflammatory responses (Moehle et al., 2012). Additionally, lipopolysaccharide induces LRRK2 up-regulation and microglial activation in mouse brains (Li et al., 2014) but they down regulated Parkin expression via NF-κB (Tran et al., 2011). Abnormal glial function is critical in parkin mutations, increasing vulnerability to inflammation-related nigral degeneration in PD (Frank-Cannon et al., 2008) and its role increases with aging (Solano et al., 2008). DJ-1 expression is up-regulated in reactive astrocytes in PD patients (Bandopadhyay et al., 2004). DJ-1 negatively regulates inflammatory responses of astrocytes and microglia by facilitating the interaction between STAT1 and its phosphatase SHP-1 (Kim et al., 2013b). Astrocyte cultures from DJ-1 KO mice treated with lipopolysaccharide have increased NO production and an up-regulation of different pro-inflammatory mediators like COX-2 and IL-6 (Waak et al., 2009).
Conclusion
The elements that potentially cause oxidative stress in PD are still unknown. DA metabolism, mitochondrial dysfunction and neuroinflammation all play critical roles in the etiology of this disease. Exposure to environmental factors or mutations in PD-associated genes of patients with either sporadic or familial PD may cause mitochondrial dysfunction that ultimately results in PD. All of these share common linkages and influence each other greatly. Limiting the early inflammatory response will reduce further both elevated oxidative stress and microglial activation that are key to slowing the death of the neurons in the SNpc. Development of potential drugs able to delay the neurodegenerative process is crucial to ameliorating the deleterious effects of oxidative stress in neurodegenerative diseases. Neuroprotective therapies will need to target multiple pathological pathways such as mitochondrial dysfunction and neuroinflammation in the next few years.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Amo, T., Saiki, S., Sawayama, T., Sato, S., and Hattori, N. (2014). Detailed analysis of mitochondrial respiratory chain defects caused by loss of PINK1. Neurosci. Lett. 580, 37–40. doi: 10.1016/j.neulet.2014.07.045
Amo, T., Sato, S., Saiki, S., Wolf, A. M., Toyomizu, M., Gautier, C. A., et al. (2011). Mitochondrial membrane potential decrease caused by loss of PINK1 is not due to proton leak, but to respiratory chain defects. Neurobiol. Dis. 41, 111–118. doi: 10.1016/j.nbd.2010.08.027
Ares-Santos, S., Granado, N., Espadas, I., Martinez-Murillo, R., and Moratalla, R. (2014). Methamphetamine causes degeneration of dopamine cell bodies and terminals of the nigrostriatal pathway evidenced by silver staining. Neuropsychopharmacology 39, 1066–1080. doi: 10.1038/npp.2013.307
Asanuma, M., Miyazaki, I., and Ogawa, N. (2003). Dopamine- or L-DOPA-induced neurotoxicity: the role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox. Res. 5, 165–176. doi: 10.1007/BF03033137
Austin, S. A., Floden, A. M., Murphy, E. J., and Combs, C. K. (2006). Alpha-synuclein expression modulates microglial activation phenotype. J. Neurosci. 26, 10558–10563. doi: 10.1523/JNEUROSCI.1799-06.2006
Bandopadhyay, R., Kingsbury, A. E., Cookson, M. R., Reid, A. R., Evans, I. M., Hope, A. D., et al. (2004). The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 127, 420–430. doi: 10.1093/brain/awh054
Barcia, C., Fernández Barreiro, A., Poza, M., Herrero, M.-T. T., and Fernandez Barreiro, A. (2003). Parkinson’s disease and inflammatory changes. Neurotox. Res. 5, 411–418. doi: 10.1007/BF03033170
Barcia, C., Ros, C. M., Carrillo, M. A., Ros, F., Gomez, A., de Pablos, V., et al. (2009). Increase of secondary processes of microglial and astroglial cells after MPTP-induced degeneration in substantia nigra pars compacta of non human primates. J. Neural Transm. Suppl. 73, 253–258. doi: 10.1007/978-3-211-92660-4_20
Barcia, C., Sánchez Bahillo, A., Fernández-Villalba, E., Bautista, V., Poza Y Poza, M., Fernández-Barreiro, A., et al. (2004). Evidence of active microglia in substantia nigra pars compacta of parkinsonian monkeys 1 year after MPTP exposure. Glia 46, 402–409. doi: 10.1002/glia.20015
Beach, T. G., Sue, L. I., Walker, D. G., Lue, L. F., Connor, D. J., Caviness, J. N., et al. (2007). Marked microglial reaction in normal aging human substantia nigra: correlation with extraneuronal neuromelanin pigment deposits. Acta Neuropathol. 114, 419–424. doi: 10.1007/s00401-007-0250–255
Belluzzi, E., Bisaglia, M., Lazzarini, E., Tabares, L. C., Beltramini, M., and Bubacco, L. (2012). Human SOD2 modification by dopamine quinones affects enzymatic activity by promoting its aggregation: possible implications for Parkinson’s disease. PLoS ONE 7:e38026. doi: 10.1371/journal.pone.0038026
Berman, S. B., and Hastings, T. G. (1999). Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson’s disease. J. Neurochem. 73, 1127–1137. doi: 10.1046/j.1471-4159.1999.0731127.x
Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V., and Greenamyre, J. T. (2000). Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 3, 1301–1306. doi: 10.1038/81834
Bian, M., Liu, J., Hong, X., Yu, M., Huang, Y., Sheng, Z., et al. (2012). Overexpression of parkin ameliorates dopaminergic neurodegeneration induced by 1- methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. PLoS ONE 7:e39953. doi: 10.1371/journal.pone.0039953
Blandini, F., Nappi, G., and Greenamyre, J. T. (1998). Quantitative study of mitochondrial complex I in platelets of parkinsonian patients. Mov. Disord. 13, 11–15. doi: 10.1002/mds.870130106
Blesa, J., and Przedborski, S. (2014). Parkinson’s disease: animal models and dopaminergic cell vulnerability. Front. Neuroanat. 8:155. doi: 10.3389/fnana.2014.00155
Blin, O., Desnuelle, C., Rascol, O., Borg, M., Peyro Saint Paul, H., Azulay, J. P., et al. (1994). Mitochondrial respiratory failure in skeletal muscle from patients with Parkinson’s disease and multiple system atrophy. J. Neurol. Sci. 125, 95–101. doi: 10.1016/0022-510X(94)90248-8
Block, M. L., Zecca, L., and Hong, J.-S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. doi: 10.1038/nrn2038
Brieger, K., Schiavone, S., Miller, F. J., and Krause, K.-H. (2012). Reactive oxygen species: from health to disease. Swiss Med. Wkly 142, w13659. doi: 10.4414/smw.2012.13659
Canet-Avilés, R. M., Wilson, M. A., Miller, D. W., Ahmad, R., McLendon, C., Bandyopadhyay, S., et al. (2004). The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. U.S.A. 101, 9103–9108. doi: 10.1073/pnas.0402959101
Caudle, W. M., Richardson, J. R., Wang, M. Z., Taylor, T. N., Guillot, T. S., McCormack, A. L., et al. (2007). Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J. Neurosci. 27, 8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007
Clark, I. E., Dodson, M. W., Jiang, C., Cao, J. H., Huh, J. R., Seol, J. H., et al. (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166. doi: 10.1038/nature04779
Cook, C., Stetler, C., and Petrucelli, L. (2012). Disruption of protein quality control in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2, a009423. doi: 10.1101/cshperspect.a009423
Cristóvão, A. C., Guhathakurta, S., Bok, E., Je, G., Yoo, S. D., Choi, D.-H., et al. (2012). NADPH oxidase 1 mediates α-synucleinopathy in Parkinson’s disease. J. Neurosci. 32, 14465–14477. doi: 10.1523/JNEUROSCI.2246-12.2012
Członkowska, A., Kohutnicka, M., Kurkowska-Jastrzebska, I., and Członkowski, A. (1996). Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson’s disease mice model. Neurodegeneration 5, 137–143. doi: 10.1006/neur.1996.0020
Daher, J. P. L., Volpicelli-Daley, L. A., Blackburn, J. P., Moehle, M. S., and West, A. B. (2014). Abrogation of α-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc. Natl. Acad. Sci. U.S.A. 111, 9289–9294. doi: 10.1073/pnas.1403215111
da Silva, F. L., Coelho Cerqueira, E., de Freitas, M. S., Gonçalves, D. L., Costa, L. T., and Follmer, C. (2013). Vitamins K interact with N-terminus α-synuclein and modulate the protein fibrillization in vitro. Exploring the interaction between quinones and α-synuclein. Neurochem. Int. 62, 103–112. doi: 10.1016/j.neuint.2012.10.001
Doorn, K. J., Goudriaan, A., Blits-Huizinga, C., Bol, J. G. J. M., Rozemuller, A. J., Hoogland, P. V. J. M., et al. (2014a). Increased amoeboid microglial density in the olfactory bulb of Parkinson’s and Alzheimer’s patients. Brain Pathol. 24, 152–165. doi: 10.1111/bpa.12088
Doorn, K. J., Moors, T., Drukarch, B., van de Berg, W., Lucassen, P. J., and van Dam, A.-M. (2014b). Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol. Commun. 2, 90. doi: 10.1186/s40478-014-0090-1
Dryanovski, D. I., Guzman, J. N., Xie, Z., Galteri, D. J., Volpicelli-Daley, L. A., Lee, V. M.-Y., et al. (2013). Calcium entry and α-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J. Neurosci. 33, 10154–10164. doi: 10.1523/JNEUROSCI.5311-12.2013
Emmrich, J. V., Hornik, T. C., Neher, J. J., and Brown, G. C. (2013). Rotenone induces neuronal death by microglial phagocytosis of neurons. FEBS J. 280, 5030–5038. doi: 10.1111/febs.12401
Exner, N., Treske, B., Paquet, D., Holmström, K., Schiesling, C., Gispert, S., et al. (2007). Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J. Neurosci. 27, 12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007
Frank-Cannon, T. C., Tran, T., Ruhn, K. A., Martinez, T. N., Hong, J., Marvin, M., et al. (2008). Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J. Neurosci. 28, 10825–10834. doi: 10.1523/JNEUROSCI.3001-08.2008
Gao, F., Chen, D., Hu, Q., and Wang, G. (2013). Rotenone directly induces BV2 cell activation via the p38 MAPK pathway. PLoS ONE 8:e72046. doi: 10.1371/journal.pone.0072046
Gao, H.-M., Liu, B., and Hong, J.-S. (2003). Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J. Neurosci. 23, 6181–6187.
Gao, H.-M., Zhou, H., Zhang, F., Wilson, B. C., Kam, W., and Hong, J.-S. (2011). HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. 31, 1081–1092. doi: 10.1523/JNEUROSCI.3732-10.2011
Garrido-Gil, P., Rodriguez-Pallares, J., Dominguez-Meijide, A., Guerra, M. J., and Labandeira-Garcia, J. L. (2013). Brain angiotensin regulates iron homeostasis in dopaminergic neurons and microglial cells. Exp. Neurol. 250, 384–396. doi: 10.1016/j.expneurol.2013.10.013
Gautier, C. A., Kitada, T., and Shen, J. (2008). Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 105, 11364–11369. doi: 10.1073/pnas.0802076105
Giaime, E., Yamaguchi, H., Gautier, C. A., Kitada, T., and Shen, J. (2012). Loss of DJ-1 does not affect mitochondrial respiration but increases ROS production and mitochondrial permeability transition pore opening. PLoS ONE 7:e40501. doi: 10.1371/journal.pone.0040501
Gillardon, F., Schmid, R., and Draheim, H. (2012). Parkinson’s disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience 208, 41–48. doi: 10.1016/j.neuroscience.2012.02.001
Girotto, S., Sturlese, M., Bellanda, M., Tessari, I., Cappellini, R., Bisaglia, M., et al. (2012). Dopamine-derived quinones affect the structure of the redox sensor DJ-1 through modifications at Cys-106 and Cys-53. J. Biol. Chem. 287, 18738–18749. doi: 10.1074/jbc.M111.311589
Gluck, M. R., and Zeevalk, G. D. (2004). Inhibition of brain mitochondrial respiration by dopamine and its metabolites: implications for Parkinson’s disease and catecholamine-associated diseases. J. Neurochem. 91, 788–795. doi: 10.1111/j.1471-4159.2004.02747.x
Goedert, M., Spillantini, M. G., Del Tredici, K., and Braak, H. (2013). 100 years of Lewy pathology. Nat. Rev. Neurol. 9, 13–24. doi: 10.1038/nrneurol.2012.242
Goldberg, M. S., Pisani, A., Haburcak, M., Vortherms, T. A., Kitada, T., Costa, C., et al. (2005). Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 45, 489–496. doi: 10.1016/j.neuron.2005.01.041
Greenamyre, J. T., Cannon, J. R., Drolet, R., and Mastroberardino, P. G. (2010). Lessons from the rotenone model of Parkinson’s disease. Trends Pharmacol. Sci. 31, 141–143. doi: 10.1016/j.tips.2009.12.006
Haas, R. H., Nasirian, F., Nakano, K., Ward, D., Pay, M., Hill, R., et al. (1995). Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann. Neurol. 37, 714–722. doi: 10.1002/ana.410370604
Hall, C. N., Klein-Flügge, M. C., Howarth, C., and Attwell, D. (2012). Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J. Neurosci. 32, 8940–8951. doi: 10.1523/JNEUROSCI.0026-12.2012
Halliday, G. M., Ophof, A., Broe, M., Jensen, P. H., Kettle, E., Fedorow, H., et al. (2005). Alpha-synuclein redistributes to neuromelanin lipid in the substantia nigra early in Parkinson’s disease. Brain 128, 2654–2664. doi: 10.1093/brain/awh584
Hattingen, E., Magerkurth, J., Pilatus, U., Mozer, A., Seifried, C., Steinmetz, H., et al. (2009). Phosphorus and proton magnetic resonance spectroscopy demonstrates mitochondrial dysfunction in early and advanced Parkinson’s disease. Brain 132, 3285–3297. doi: 10.1093/brain/awp293
Hattori, N., Tanaka, M., Ozawa, T., and Mizuno, Y. (1991). Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson’s disease. Ann. Neurol. 30, 563–571. doi: 10.1002/ana.410300409
Hauser, D. N., Dukes, A. A., Mortimer, A. D., and Hastings, T. G. (2013). Dopamine quinone modifies and decreases the abundance of the mitochondrial selenoprotein glutathione peroxidase 4. Free Radic. Biol. Med. 65, 419–427. doi: 10.1016/j.freeradbiomed.2013.06.030
Hayashi, T., Ishimori, C., Takahashi-Niki, K., Taira, T., Kim, Y., Maita, H., et al. (2009). DJ-1 binds to mitochondrial complex I and maintains its activity. Biochem. Biophys. Res. Commun. 390, 667–672. doi: 10.1016/j.bbrc.2009.10.025
Heo, J. Y., Park, J. H., Kim, S. J., Seo, K. S., Han, J. S., Lee, S. H., et al. (2012). DJ-1 null dopaminergic neuronal cells exhibit defects in mitochondrial function and structure: involvement of mitochondrial complex I assembly. PLoS ONE 7:e32629. doi: 10.1371/journal.pone.0032629
Herrero, M. T., Hirsch, E. C., Kastner, A., Ruberg, M., Luquin, M. R., Laguna, J., et al. (1993). Does neuromelanin contribute to the vulnerability of catecholaminergic neurons in monkeys intoxicated with MPTP? Neuroscience 56, 499–511. doi: 10.1016/0306-4522(93)90349-K
Hoepken, H.-H., Gispert, S., Morales, B., Wingerter, O., Del Turco, D., Mülsch, A., et al. (2007). Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol. Dis. 25, 401–411. doi: 10.1016/j.nbd.2006.10.007
Irrcher, I., Aleyasin, H., Seifert, E. L., Hewitt, S. J., Chhabra, S., Phillips, M., et al. (2010). Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 19, 3734–3746. doi: 10.1093/hmg/ddq288
Jackson-Lewis, V., and Smeyne, R. J. (2005). MPTP and SNpc DA neuronal vulnerability: role of dopamine, superoxide and nitric oxide in neurotoxicity. Minireview. Neurotox. Res. 7, 193–202. doi: 10.1007/BF03036449
Jana, S., Maiti, A. K., Bagh, M. B., Banerjee, K., Das, A., Roy, A., et al. (2007). Dopamine but not 3,4-dihydroxy phenylacetic acid (DOPAC) inhibits brain respiratory chain activity by autoxidation and mitochondria catalyzed oxidation to quinone products: implications in Parkinson’s disease. Brain Res. 1139, 195–200. doi: 10.1016/j.brainres.2006.09.100
Jana, S., Sinha, M., Chanda, D., Roy, T., Banerjee, K., Munshi, S., et al. (2011). Mitochondrial dysfunction mediated by quinone oxidation products of dopamine: implications in dopamine cytotoxicity and pathogenesis of Parkinson’s disease. Biochim. Biophys. Acta 1812, 663–673. doi: 10.1016/j.bbadis.2011.02.013
Joglar, B., Rodriguez-Pallares, J., Rodriguez-Perez, A. I., Rey, P., Guerra, M. J., and Labandeira-Garcia, J. L. (2009). The inflammatory response in the MPTP model of Parkinson’s disease is mediated by brain angiotensin: relevance to progression of the disease. J. Neurochem. 109, 656–669. doi: 10.1111/j.1471-4159.2009.05999.x
Junn, E., Jang, W. H., Zhao, X., Jeong, B. S., and Mouradian, M. M. (2009). Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J. Neurosci. Res. 87, 123–129. doi: 10.1002/jnr.21831
Kieburtz, K., and Wunderle, K. B. (2013). Parkinson’s disease: evidence for environmental risk factors. Mov. Disord. 28, 8–13. doi: 10.1002/mds.25150
Kim, C., Ho, D.-H., Suk, J.-E., You, S., Michael, S., Kang, J., et al. (2013a). Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 4, 1562. doi: 10.1038/ncomms2534
Kim, J., Choi, D., Jeong, H., Kim, J., Kim, D. W., Choi, S. Y., et al. (2013b). DJ-1 facilitates the interaction between STAT1 and its phosphatase, SHP-1, in brain microglia and astrocytes: a novel anti-inflammatory function of DJ-1. Neurobiol. Dis. 60, 1–10. doi: 10.1016/j.nbd.2013.08.007
Kitada, T., Pisani, A., Porter, D. R., Yamaguchi, H., Tscherter, A., Martella, G., et al. (2007). Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 104, 11441–11446. doi: 10.1073/pnas.0702717104
Krebiehl, G., Ruckerbauer, S., Burbulla, L. F., Kieper, N., Maurer, B., Waak, J., et al. (2010). Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS ONE 5:e9367. doi: 10.1371/journal.pone.0009367
Krige, D., Carroll, M. T., Cooper, J. M., Marsden, C. D., and Schapira, A. H. (1992). Platelet mitochondrial function in Parkinson’s disease. The Royal Kings and Queens Parkinson Disease Research Group. Ann. Neurol. 32, 782–788. doi: 10.1002/ana.410320612
Kuhn, D. M., Arthur, R. E., Thomas, D. M., and Elferink, L. A. (1999). Tyrosine hydroxylase is inactivated by catechol-quinones and converted to a redox-cycling quinoprotein: possible relevance to Parkinson’s disease. J. Neurochem. 73, 1309–1317. doi: 10.1046/j.1471-4159.1999.0731309.x
Lawson, L. J., Perry, V. H., Dri, P., and Gordon, S. (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170. doi: 10.1016/0306-4522(90)90229-W
Lee, C. S., Song, E. H., Park, S. Y., and Han, E. S. (2003). Combined effect of dopamine and MPP+ on membrane permeability in mitochondria and cell viability in PC12 cells. Neurochem. Int. 43, 147–154. doi: 10.1016/S0197-0186(02)00214-0
Li, J., Scheller, C., Koutsilieri, E., Griffiths, F., Beart, P. M., Mercer, L. D., et al. (2005). Differential effects of human neuromelanin and synthetic dopamine melanin on neuronal and glial cells. J. Neurochem. 95, 599–608. doi: 10.1111/j.1471-4159.2005.03404.x
Li, T., Yang, D., Zhong, S., Thomas, J. M., Xue, F., Liu, J., et al. (2014). Novel LRRK2 GTP-binding inhibitors reduced degeneration in Parkinson’s disease cell and mouse models. Hum. Mol. Genet. 23, 6212–6222. doi: 10.1093/hmg/ddu341
Martin, L. J., Pan, Y., Price, A. C., Sterling, W., Copeland, N. G., Jenkins, N. A., et al. (2006). Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 26, 41–50. doi: 10.1523/JNEUROSCI.4308-05.2006
Masoud, S. T., Vecchio, L. M., Bergeron, Y., Hossain, M. M., Nguyen, L. T., Bermejo, M. K., et al. (2015). Increased expression of the dopamine transporter leads to loss of dopamine neurons, oxidative stress and l-DOPA reversible motor deficits. Neurobiol. Dis. 74, 66–75. doi: 10.1016/j.nbd.2014.10.016
McGeer, P. L., Itagaki, S., Boyes, B. E., and McGeer, E. G. (1988). Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291. doi: 10.1212/WNL.38.8.1285
Miyazaki, I., Asanuma, M., Diaz-Corrales, F. J., Fukuda, M., Kitaichi, K., Miyoshi, K., et al. (2006). Methamphetamine-induced dopaminergic neurotoxicity is regulated by quinone-formation-related molecules. FASEB J. 20, 571–573. doi: 10.1096/fj.05-4996fje
Mizuno, Y., Ohta, S., Tanaka, M., Takamiya, S., Suzuki, K., Sato, T., et al. (1989). Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem. Biophys. Res. Commun. 163, 1450–1455. doi: 10.1016/0006-291X(89)91141-8
Mizuno, Y., Sone, N., and Saitoh, T. (1987). Effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 1-methyl-4-phenylpyridinium ion on activities of the enzymes in the electron transport system in mouse brain. J. Neurochem. 48, 1787–1793. doi: 10.1111/j.1471-4159.1987.tb05737.x
Moehle, M. S., Webber, P. J., Tse, T., Sukar, N., Standaert, D. G., DeSilva, T. M., et al. (2012). LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci. 32, 1602–1611. doi: 10.1523/JNEUROSCI.5601-11.2012
Moscovitz, O., Ben-Nissan, G., Fainer, I., Pollack, D., Mizrachi, L., and Sharon, M. (2015). The Parkinson’s-associated protein DJ-1 regulates the 20S proteasome. Nat. Commun. 6, 6609. doi: 10.1038/ncomms7609
Müftüoglu, M., Elibol, B., Dalmizrak, O., Ercan, A., Kulaksiz, G., Ogüs, H., et al. (2004). Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov. Disord. 19, 544–548. doi: 10.1002/mds.10695
Mytilineou, C., Werner, P., Molinari, S., Di Rocco, A., Cohen, G., and Yahr, M. D. (1994). Impaired oxidative decarboxylation of pyruvate in fibroblasts from patients with Parkinson’s disease. J. Neural Transm. Park. Dis. Dement. Sect. 8, 223–228. doi: 10.1007/BF02260943
Norris, K. L., Hao, R., Chen, L.-F., Lai, C.-H., Kapur, M., Shaughnessy, P. J., et al. (2015). Convergence of parkin, PINK1 and α-synuclein on stress-induced mitochondrial morphological remodelling. J. Biol. Chem. 290, 13862–13874. doi: 10.1074/jbc.M114.634063
Obeso, J. A., Rodriguez-Oroz, M. C., Goetz, C. G., Marin, C., Kordower, J. H., Rodriguez, M., et al. (2010). Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 16, 653–661. doi: 10.1038/nm.2165
Ohtsuka, C., Sasaki, M., Konno, K., Kato, K., Takahashi, J., Yamashita, F., et al. (2014). Differentiation of early-stage parkinsonisms using neuromelanin-sensitive magnetic resonance imaging. Parkinsonism Relat. Disord. 20, 755–760. doi: 10.1016/j.parkreldis.2014.04.005
Ohtsuka, C., Sasaki, M., Konno, K., Koide, M., Kato, K., Takahashi, J., et al. (2013). Changes in substantia nigra and locus coeruleus in patients with early-stage Parkinson’s disease using neuromelanin-sensitive MR imaging. Neurosci. Lett. 541, 93–98. doi: 10.1016/j.neulet.2013.02.012
Pal, R., Monroe, T. O., Palmieri, M., Sardiello, M., and Rodney, G. G. (2014). Rotenone induces neurotoxicity through Rac1-dependent activation of NADPH oxidase in SHSY-5Y cells. FEBS Lett. 588, 472–481. doi: 10.1016/j.febslet.2013.12.011
Palacino, J. J., Sagi, D., Goldberg, M. S., Krauss, S., Motz, C., Wacker, M., et al. (2004). Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 279, 18614–18622. doi: 10.1074/jbc.M401135200
Parker, W. D., Parks, J. K., and Swerdlow, R. H. (2008). Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 1189, 215–218. doi: 10.1016/j.brainres.2007.10.061
Perfeito, R., Lázaro, D. F., Outeiro, T. F., and Rego, A. C. (2014). Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol. Cell. Neurosci. 62C, 51–59. doi: 10.1016/j.mcn.2014.08.002
Perry, V. H. (2012). Innate inflammation in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2, a009373. doi: 10.1101/cshperspect.a009373
Philippens, I. H. C. H. M., Wubben, J. A., Finsen, B., and ‘t Hart, B. A. (2013). Oral treatment with the NADPH oxidase antagonist apocynin mitigates clinical and pathological features of parkinsonism in the MPTP marmoset model. J. Neuroimmune Pharmacol. 8, 715–726. doi: 10.1007/s11481-013-9450-z
Piccoli, C., Sardanelli, A., Scrima, R., Ripoli, M., Quarato, G., D’Aprile, A., et al. (2008). Mitochondrial respiratory dysfunction in familiar parkinsonism associated with PINK1 mutation. Neurochem. Res. 33, 2565–2574. doi: 10.1007/s11064-008-9729-2
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Pisanu, A., Lecca, D., Mulas, G., Wardas, J., Simbula, G., Spiga, S., et al. (2014). Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-γ agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol. Dis. 71C, 280–291. doi: 10.1016/j.nbd.2014.08.011
Plum, S., Helling, S., Theiss, C., Leite, R. E. P., May, C., Jacob-Filho, W., et al. (2013). Combined enrichment of neuromelanin granules and synaptosomes from human substantia nigra pars compacta tissue for proteomic analysis. J. Proteomics 94, 202–206. doi: 10.1016/j.jprot.2013.07.015
Pukass, K., and Richter-Landsberg, C. (2014). Oxidative stress promotes uptake, accumulation, and oligomerization of extracellular α-synuclein in oligodendrocytes. J. Mol. Neurosci. 52, 339–352. doi: 10.1007/s12031-013-0154-x
Puschmann, A. (2013). Monogenic Parkinson’s disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Relat. Disord. 19, 407–415. doi: 10.1016/j.parkreldis.2013.01.020
Rabinovic, A. D., Lewis, D. A., and Hastings, T. G. (2000). Role of oxidative changes in the degeneration of dopamine terminals after injection of neurotoxic levels of dopamine. Neuroscience 101, 67–76. doi: 10.1016/S0306-4522(00)00293-1
Rodriguez, M., Morales, I., Rodriguez-Sabate, C., Sanchez, A., Castro, R., Brito, J. M., et al. (2014). The degeneration and replacement of dopamine cells in Parkinson’s disease: the role of aging. Front. Neuroanat. 8:80. doi: 10.3389/fnana.2014.00080
Rodriguez-Oroz, M. C., Jahanshahi, M., Krack, P., Litvan, I., Macias, R., Bezard, E., et al. (2009). Initial clinical manifestations of Parkinson’s disease: features and pathophysiological mechanisms. Lancet Neurol. 8, 1128–1139. doi: 10.1016/S1474-4422(09)70293-5
Ryan, B. J., Hoek, S., Fon, E. A., and Wade-Martins, R. (2015). Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem. Sci. 40, 200–210. doi: 10.1016/j.tibs.2015.02.003
Sacino, A. N., Brooks, M., Thomas, M. A., McKinney, A. B., Lee, S., Regenhardt, R. W., et al. (2014). Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 111, 10732–10737. doi: 10.1073/pnas.1321785111
Saini, N., Oelhafen, S., Hua, H., Georgiev, O., Schaffner, W., and Büeler, H. (2010). Extended lifespan of Drosophila parkin mutants through sequestration of redox-active metals and enhancement of anti-oxidative pathways. Neurobiol. Dis. 40, 82–92. doi: 10.1016/j.nbd.2010.05.011
Scarffe, L. A., Stevens, D. A., Dawson, V. L., and Dawson, T. M. (2014). Parkin and PINK1: much more than mitophagy. Trends Neurosci. 37, 315–324. doi: 10.1016/j.tins.2014.03.004
Schapira, A. H. V. (2008). Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 7, 97–109. doi: 10.1016/S1474-4422(07)70327-7
Schapira, A. H. V. (2010). Complex I: inhibitors, inhibition and neurodegeneration. Exp. Neurol. 224, 331–335. doi: 10.1016/j.expneurol.2010.03.028
Schapira, A. H., Cooper, J. M., Dexter, D., Clark, J. B., Jenner, P., and Marsden, C. D. (1990). Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 54, 823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x
Schapira, A. H., and Jenner, P. (2011). Etiology and pathogenesis of Parkinson’s disease. Mov. Disord. 26, 1049–1055. doi: 10.1002/mds.23732
Schapira, A. H. V., Olanow, C. W., Greenamyre, J. T., and Bezard, E. (2014). Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: future therapeutic perspectives. Lancet 384, 545–555. doi: 10.1016/S0140-6736(14)61010-2
Segura-Aguilar, J., Paris, I., Muñoz, P., Ferrari, E., Zecca, L., and Zucca, F. A. (2014). Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 129, 898–915. doi: 10.1111/jnc.12686
Sherer, T. B., Betarbet, R., Kim, J. H., and Greenamyre, J. T. (2003a). Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci. Lett. 341, 87–90. doi: 10.1016/S0304-3940(03)00172-1
Sherer, T. B., Kim, J. H., Betarbet, R., and Greenamyre, J. T. (2003b). Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp. Neurol. 179, 9–16. doi: 10.1006/exnr.2002.8072
Solano, R. M., Casarejos, M. J., Menéndez-Cuervo, J., Rodriguez-Navarro, J. A., García de Yébenes, J., and Mena, M. A. (2008). Glial dysfunction in parkin null mice: effects of aging. J. Neurosci. 28, 598–611. doi: 10.1523/JNEUROSCI.4609-07.2008
Spencer, J. P., Jenner, P., Daniel, S. E., Lees, A. J., Marsden, D. C., and Halliwell, B. (1998). Conjugates of catecholamines with cysteine and GSH in Parkinson’s disease: possible mechanisms of formation involving reactive oxygen species. J. Neurochem. 71, 2112–2122. doi: 10.1046/j.1471-4159.1998.71052112.x
Stott, S. R. W., and Barker, R. A. (2014). Time course of dopamine neuron loss and glial response in the 6-OHDA striatal mouse model of Parkinson’s disease. Eur. J. Neurosci. 39, 1042–1056. doi: 10.1111/ejn.12459
Subramaniam, S. R., Vergnes, L., Franich, N. R., Reue, K., and Chesselet, M.-F. (2014). Region specific mitochondrial impairment in mice with widespread overexpression of alpha-synuclein. Neurobiol. Dis. 70, 204–213. doi: 10.1016/j.nbd.2014.06.017
Sulzer, D., Bogulavsky, J., Larsen, K. E., Behr, G., Karatekin, E., Kleinman, M. H., et al. (2000). Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc. Natl. Acad. Sci. U.S.A. 97, 11869–11874. doi: 10.1073/pnas.97.22.11869
Sulzer, D., and Zecca, L. (2000). Intraneuronal dopamine-quinone synthesis: a review. Neurotox. Res. 1, 181–195. doi: 10.1007/BF03033289
Surace, M. J., and Block, M. L. (2012). Targeting microglia-mediated neurotoxicity: the potential of NOX2 inhibitors. Cell. Mol. Life Sci. 69, 2409–2427. doi: 10.1007/s00018-012-1015-4
Toyama, T., Shinkai, Y., Yazawa, A., Kakehashi, H., Kaji, T., and Kumagai, Y. (2014). Glutathione-mediated reversibility of covalent modification of ubiquitin carboxyl-terminal hydrolase L1 by 1,2-naphthoquinone through Cys152, but not Lys4. Chem. Biol. Interact. 214, 41–48. doi: 10.1016/j.cbi.2014.02.008
Tran, T. A., Nguyen, A. D., Chang, J., Goldberg, M. S., Lee, J.-K., and Tansey, M. G. (2011). Lipopolysaccharide and tumor necrosis factor regulate Parkin expression via nuclear factor-kappa B. PLoS ONE 6:e23660. doi: 10.1371/journal.pone.0023660
Trinh, J., and Farrer, M. (2013). Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 9, 445–454. doi: 10.1038/nrneurol.2013.132
van der Merwe, C., Jalali Sefid Dashti, Z., Christoffels, A., Loos, B., and Bardien, S. (2015). Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: parkin, PINK1 and DJ-1. Eur. J. Neurosci. 41, 1113–1125. doi: 10.1111/ejn.12872
Van Laar, V. S., Mishizen, A. J., Cascio, M., and Hastings, T. G. (2009). Proteomic identification of dopamine-conjugated proteins from isolated rat brain mitochondria and SH-SY5Y cells. Neurobiol. Dis. 34, 487–500. doi: 10.1016/j.nbd.2009.03.004
Waak, J., Weber, S. S., Waldenmaier, A., Görner, K., Alunni-Fabbroni, M., Schell, H., et al. (2009). Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. FASEB J. 23, 2478–2489. doi: 10.1096/fj.08-125153
Wang, H.-L., Chou, A.-H., Wu, A.-S., Chen, S.-Y., Weng, Y.-H., Kao, Y.-C., et al. (2011). PARK6 PINK1 mutants are defective in maintaining mitochondrial membrane potential and inhibiting ROS formation of substantia nigra dopaminergic neurons. Biochim. Biophys. Acta 1812, 674–684. doi: 10.1016/j.bbadis.2011.03.007
Watson, M. B., Richter, F., Lee, S. K., Gabby, L., Wu, J., Masliah, E., et al. (2012). Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp. Neurol. 237, 318–334. doi: 10.1016/j.expneurol.2012.06.025
Whitehead, R. E., Ferrer, J. V., Javitch, J. A., and Justice, J. B. (2001). Reaction of oxidized dopamine with endogenous cysteine residues in the human dopamine transporter. J. Neurochem. 76, 1242–1251. doi: 10.1046/j.1471-4159.2001.00125.x
Wilhelmus, M. M. M., Nijland, P. G., Drukarch, B., de Vries, H. E., and van Horssen, J. (2012). Involvement and interplay of Parkin, PINK1, and DJ1 in neurodegenerative and neuroinflammatory disorders. Free Radic. Biol. Med. 53, 983–992. doi: 10.1016/j.freeradbiomed.2012.05.040
Wood-Kaczmar, A., Gandhi, S., Yao, Z., Abramov, A. S. Y., Miljan, E. A., Keen, G., et al. (2008). PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 3:e2455. doi: 10.1371/journal.pone.0002455
Xiang, W., Schlachetzki, J. C. M., Helling, S., Bussmann, J. C., Berlinghof, M., Schäffer, T. E., et al. (2013). Oxidative stress-induced posttranslational modifications of alpha-synuclein: specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol. Cell. Neurosci. 54, 71–83. doi: 10.1016/j.mcn.2013.01.004
Yang, Y., Gehrke, S., Imai, Y., Huang, Z., Ouyang, Y., Wang, J.-W., et al. (2006). Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. U.S.A. 103, 10793–10798. doi: 10.1073/pnas.0602493103
Yoshino, H., Nakagawa-Hattori, Y., Kondo, T., and Mizuno, Y. (1992). Mitochondrial complex I and II activities of lymphocytes and platelets in Parkinson’s disease. J. Neural Transm. Park. Dis. Dement. Sect. 4, 27–34. doi: 10.1007/BF02257619
Zhang, L., Shimoji, M., Thomas, B., Moore, D. J., Yu, S.-W., Marupudi, N. I., et al. (2005). Mitochondrial localization of the Parkinson’s disease related protein DJ-1: implications for pathogenesis. Hum. Mol. Genet. 14, 2063–2073. doi: 10.1093/hmg/ddi211
Zhang, W., Phillips, K., Wielgus, A. R., Liu, J., Albertini, A., Zucca, F. A., et al. (2009). Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: implications for progression of parkinson’s disease. Neurotox. Res. 19, 63–72. doi: 10.1007/s12640-009-9140-z
Zhou, M., Xu, S., Mi, J., Uéda, K., and Chan, P. (2013). Nuclear translocation of alpha-synuclein increases susceptibility of MES23.5 cells to oxidative stress. Brain Res. 1500, 19–27. doi: 10.1016/j.brainres.2013.01.024
Zhou, Z. D., Refai, F. S., Xie, S. P., Ng, S. H., Chan, C. H. S., Ho, P. G. H., et al. (2014). Mutant PINK1 upregulates tyrosine hydroxylase and dopamine levels, leading to vulnerability of dopaminergic neurons. Free Radic. Biol. Med. 68, 220–233. doi: 10.1016/j.freeradbiomed.2013.12.015
Zucca, F. A., Basso, E., Cupaioli, F. A., Ferrari, E., Sulzer, D., Casella, L., et al. (2014). Neuromelanin of the human substantia nigra: an update. Neurotox. Res. 25, 13–23. doi: 10.1007/s12640-013-9435-y
Keywords: mitochondrial dysfunction, dopamine, neuroinflammation, Parkinson disease, oxidative stress
Citation: Blesa J, Trigo-Damas I, Quiroga-Varela A and Jackson-Lewis VR (2015) Oxidative stress and Parkinson’s disease. Front. Neuroanat. 9:91. doi: 10.3389/fnana.2015.00091
Received: 15 April 2015; Accepted: 24 June 2015;
Published: 08 July 2015.
Edited by:
Javier DeFelipe, Cajal Institute, SpainReviewed by:
Rosario Moratalla, Cajal Institute – Spanish Research Council, SpainRubén Fernández-Santiago, Institut d’Investigacions Biomèdiques August Pi i Sunyer, Spain
Copyright © 2015 Blesa, Trigo-Damas, Quiroga-Varela and Jackson-Lewis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Javier Blesa, Centro Integral de Neurociencias A.C., HM Puerta del Sur, Hospitales de Madrid, Móstoles and Medical School, CEU San Pablo University, Avenida de Carlos V, 70, 28938 Móstoles, Madrid, Spain,amF2aWJsZXNhQGhvdG1haWwuY29t