 Emanuela Paldino1Antonella Cardinale1Vincenza D’Angelo2Ilaria Sauve1
Emanuela Paldino1Antonella Cardinale1Vincenza D’Angelo2Ilaria Sauve1 Carmela Giampà1,3
Carmela Giampà1,3 Francesca R. Fusco1*
Francesca R. Fusco1*- 1Laboratory of Neuroanatomy, Santa Lucia Foundation IRCCS Hospital, Rome, Italy
- 2Department of Neuroscience, University of Rome Tor Vergata, Rome, Italy
- 3Department of Anatomy and Cell Biology, Catholic University, Rome, Italy
Poly (ADP-ribose) polymerases (PARPs) are enzymes that catalyze ADP-ribose units transfer from NAD to their substrate proteins. It has been observed that PARP-1 is able to increase both post-ischemic and excitotoxic neuronal death. In fact, we have previously shown that, INO-1001, a PARP-1 inhibitor, displays a neuroprotective effect in the R6/2 model of Huntington’s disease (HD). In this study, we investigated the effects of PARP-1-inhibition on modulation of phosphorylated c-AMP response element binding protein (pCREB) and CREB-binding protein (CBP) localization in the different striatal neuronal subsets. Moreover, we studied the neurodegeneration of those interneurons that are particularly vulnerable to HD such as parvalbuminergic and calretininergic, and of other subclasses of interneurons that are known to be resistant, such as cholinergic and somatostatinergic interneurons. Transgenic mice were treated with INO-1001 (10 mg/Kg daily) starting from 4 weeks of age. Double-label immunofluorescence was performed to value the distribution of CBP in ubiquitinated Neuronal intranuclear inclusions (NIIs) in the striatum. INO-1001-treated and saline-treated brain sections were incubated with: goat anti-choline acetyl transferase; goat anti-nitric oxide synthase; mouse anti-parvalbumin and mouse anti-calretinin. Morphometric evaluation and cell counts were performed. Our study showed that the PARP inhibitor has a positive effect in sparing parvalbumin and calretinin-containing interneurons of the striatum, where CREB was upregulated. Moreover, INO-1001 promoted CBP localization into the nuclei of the R6/2 mouse. The sum of our data corroborates the previous observations indicating PARP inhibition as a possible therapeutic tool to fight HD.
Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder, which is typically characterized by psychiatric disturbances, motor dysfunction and cognitive decline (Wilson et al., 1987). In HD, a gene located on the short arm of chromosome 4 named IT15 is mutated. IT15 encodes for the protein huntingtin (The Huntington’s Disease Research Collaborative Group, 1993), which mutation leads to a CAG expansion beyond the normal 10–35 triplet repeat range (Albin and Tagle, 1995). From a neuropathological point of view, a dramatic degeneration of neurons in the striatal part of the basal ganglia occurs in HD, accounting for the progressively severe motor dysfunction (Vonsattel et al., 1985; Hedreen et al., 1991; Storey et al., 1992).
Aside from the projection neurons, HD also affects interneurons such as parvalbumin and calretinin-containing neurons. These interneurons appear to degenerate at almost the same rate as the projection neurons (Ferrer et al., 1994). Conversely, the degeneration process relatively spares other subclasses of striatal interneurons, such as somatostatin-NPY and cholinergic interneurons.
Mutated huntingtin impairs the function of cAMP response element-binding protein (CREB; Steffan et al., 2000; Sugars and Rubinsztein, 2003; Sugars et al., 2004), suggesting that inhibition of CREB-mediated gene transcription could play an important role in HD (Kazantsev et al., 1999; Steffan et al., 2000; Nucifora et al., 2001; Mantamadiotis et al., 2002; Jiang et al., 2003). Accordingly, it was observed that cAMP cerebrospinal fluid levels are reduced in HD patients (Cramer et al., 1984) and that CREB-regulated gene transcription is down regulated in the R6/2 HD transgenic mouse (Luthi-Carter et al., 2000; Wyttenbach et al., 2001).
Of note, we previously observed that in parvalbumin-containing neurons, levels of activated CREB are markedly reduced after excitotoxic lesions, which could, at least in part, account for their selective vulnerability (Giampà et al., 2006).
In HD, the N-terminal fragment of mutated huntingtin forms ubiquitinated aggregates named neuronal intranuclear inclusions (NIIs; DiFiglia et al., 1997). These aggregates were shown to be able to interact with several transcription factors, thereby impairing their functions (Martindale et al., 1998; Gutekunst et al., 1999).
CREB binding protein (CBP) is a transcriptional co-activator that was shown to mediate neuronal survival signals (Bonni et al., 1999; Walton and Dragunow, 2000). Moreover, it was observed that NIIs sequestrate CBP in the R6/2 HD mice. Therefore, striatal function is impaired by the interaction between mutated huntingtin and CBP, causing a disruption of transcription, and leading to toxicity for the neuron.
Recently, we showed that a poly (ADP-ribose) polymerase 1 (PARP-1) inhibitor was neuroprotective in the R6/2 mouse model of HD, where sparing of striatal neurons was associated with an increased level of pCREB (Cardinale et al., 2015).
PARP-1, a 116-kD protein, member of PARP family, is a nuclear enzyme, consists of three main domains: the N-terminal DNA-binding domain (DBD), the automodification domain (AMD) and the C-terminal catalytic domain (Kameshita et al., 1984; Kurosaki et al., 1987) involved in the poly(ADP-ribosylation) reaction. The relationship between the inappropriate activation of PARP-1 and neurodegeneration has been demonstrated (Strosznajder et al., 2005; Kauppinen et al., 2011; Martire et al., 2013). In fact, the abnormal activation of PARP induces the release and the translocation of apoptosis-inducing factor (AIF) from the mitochondria to the nucleus causing a programmed cell death, caspase-independent, named parthanatos (Wang et al., 2009).
Many authors have focused the attention on PARP-1 inhibition by common inhibitors used in clinical for cancer treatment, able to restore the physiological cell functions such as mitochondrial activity, or the regulation of transcription factors including p53 required for neuronal survival (Martire et al., 2015).
In an earlier study, our group had shown that, in the R6/2 mouse of HD, the beneficial effects exerted by phosphodiesterase inhibitors on phenotype and on projection neurons sparing was associated to a rescue of parvalbumin positive interneurons and to an inhibition of CBP sequestration into NIIs (Giampà et al., 2009).
The aim of the present study was to deepen our knowledge of the effects of PARP-1-inhibition. In particular, we aimed at investigating the effects of PARP-1 inhibition on CBP localization, by reducing CBP sequestration into the NIIs, and thus decreasing cellular toxicity resulting from it. Moreover, we studied the effects of the compound on calretinin and parvalbumin containing striatal cells, two subsets of interneurons that are particularly vulnerable to HD. In summary, our aim was to verify if the modulation of CBP activity (Oliveira et al., 2006), on one hand, and the sparing of selected striatal interneurons (Torres et al., 1994), on the other, were associated with the previously described neuroprotective effects and the rescue motor deficits in HD mice.
Materials and Methods
Animals and PARP-1 Inhibitor Administration
All studies were performed in accordance with European Communities Council Directive of 24 November 1986(86/609/EEC) as adopted by Santa Lucia Foundation Animal care and Use committee. The Santa Lucia Foundation Animal Care and Use committee approved this study.
The animal model of HD, R6/2, was employed for this study. Transgenic R6/2 mice were obtained by ovarian transplantation of hemizygote females X B6CBAF1/J males, provided by Jackson laboratories (Bar Harbor, ME, USA) and the F1 mice were used to perform all the experiments. Animals were genotyped by PCR assay of DNA obtained from tail tissue. After genotyping at 4 weeks of age, mice were weaned and the treatment started. The treatment was continued until sacrifice. The criterion for euthanasia was that the mouse was not capable to right itself when placed on a side (after 13th week of age). Twenty-four mice per group were used. The study groups were composed by: R6/2 and wild type mice treated with 0.9% saline administered by intraperitoneal injection and R6/2 and wild type mice with INO-1001 dissolved in saline (10 mg/Kg/day). INO-1001 was diluted immediately before use and administered twice in the same day. Mice were housed five in each cage under standard conditions with ad libitum access to food and water.
Tissue Processing
Experimental animals were transcardially perfused under deep anesthesia with Zoletil and Rampun (500–800 mg/Kg), followed by 60 ml of 4% paraformaldehyde in saline solution. Brains were removed and post fixed overnight at 4°C, cryoprotected in 10% sucrose and 20% glycerol 0.1 M phosphate buffer (PB) with 0.02% sodium azide for 48 h at 4°C. Subsequently, all brains were sectioned on sliding microtome at 40 μm thickness to obtain serial sections.
Immunohistochemical Studies
Analysis of NIIs in Neurons Expressing CBP
Double-label immunofluorescences were carried out to evaluate the distribution of ubiquitinated NIIs according to CREB binding protein (CBP) expression levels in the striatum. Coronal brain sections of mice treated with PARP-1 inhibitor and vehicle were incubated with a cocktail of anti-CBP antibody (rabbit anti-CBP, Immunological Sciences, RM, Italy) and a mouse antibody against mutant huntingtin protein (Clone mEM48, Merk Millipore Corporation, Darmstadt, Germany) at 1:200 dilution in a 0.1 M PB solution containing 0.3% Triton X-100 for 72 h at 4°C, except for CBP protein which needs the antigen retrieval method. Antigen retrieval was performed in Citrate Buffer (pH 6) for 20 min at 80°C. After that, sections were retained in this buffer solution while allowing it to cool at room temperature. Sections were rinsed three times for 5 min at room temperature and subsequently incubated with the primary antibody against CBP. The immunohistochemical staining was completed with the streptavidin-biotin amplification and the goat anti-mouse secondary antibody for EM48 (Jackson Immunoresearch, West Grove, PA, USA) for 2 h at room temperature at 1:100 dilution in a 0.1 M PB solution containing 0.3% Triton X-100. Sections were mounted on gelatin-coated slides, cover slipped with GEL-MOUNT (Sigma-Aldrich, Italy) and a confocal laser scanning microscope (Zeiss LSM 510) was used to acquire all of the images. Three separate fields (dorsolateral, central and medial each 1 mm in diameter) on each hemisphere in each of three rostro caudally spaced sections in each of four mice per group were examined. NIIs have been quantified and measured by using the Java image processing and analysis program; ImageJ. Cells of interest were selected using the freehand tool. From the Analyze menu, measurements were set “Mean Grey Value, Area and Min and Max Grey Value” were selected. Finally, the mean values of all measures were obtained. Analysis of NIIs size and number was performed by the operator in blind to treatment.
Striatal Interneurons Characterization
A single immunological staining for striatal interneurons markers, counterstained with Neurotrace fluorescent Nissl (green fluorescence) and DAPI was performed. INO-1001 and saline-treated brain sections were incubated with goat anti-choline acetyl transferase (ChAT; Nova biological, CA, USA); goat anti-nitric oxide synthase (NOS; Sigma, St. Louis, MO, USA); mouse anti-parvalbuminergic (PARV, Chemicon International, Inc., Temecula, CA, USA) and mouse anti-calretinin (CALR; Chemicon International, Inc., Temecula, CA, USA).
All primary antibodies were used at a 1:200 dilution, in 0.1 M PB containing 0.3% Triton X-100 for 72 h at 4°C. Section were rinsed three times for 5 min at room temperature and subsequently incubated with secondary antibodies: donkey anti-mouse and donkey anti-goat (Jackson Immunoresearch, West Grove, PA, USA) for 2 h at room temperature at 1:100 dilution in a 0.1 M PB solution containing 0.3% Triton X-100. Subsequently sections were mounted on slides, cover slipped with GEL-MOUNT and examined under an epi-illumination fluorescence microscope (Zeiss Axioskop 2). The confocal laser scanner microscopy (Zeiss LSM700) was used to acquire images.
Study of Parvalbuminergic Neurons
Peroxidase-antiperoxidase diaminobenzidine tetrahydrochloride single-label immunohistochemistry for parvalbumin (PARV) was performed to identify and count parvalbuminergic interneurons in the striatum. Serial sections from rostral neostriatum through the level of anterior commissure (interaural 4.66 mm/Bregma 0.86 mm to interaural 3.34 mm/Bregma −0, 0.46 mm) for three animals per groups, were incubated with mouse anti-PARV at 1:200 dilution in 0.1 M PB solution containing 0.3% Triton X-100 for 72 h at 4°C. Subsequently sections were incubated with mouse peroxidase-antiperoxidase complex diluted 1:100 in 0.1 M PB solution with 0.3% Triton X-100 at room temperature for 1 h. After peroxidase-antiperoxidase incubation, sections were incubated in Tris-Hcl buffer containing 10 mg diaminobenzidine tetrahydrochloride for 5 min, adding 15 μl of 3% hydrogen peroxidase. The peroxidase-antiperoxidase diaminobenzidine tetrahydrochloride-labeled sections were then washed in distillated water, placed in 0.1 M PB, mounted on gelatin-coated slides, dried, dehydrated and coverslipped. PARV positive cells count was performed using NeurolucidaTM Stereo Investigator software (Zeiss, Rochester, NY, USA).
pCREB Expression and Quantification
The dual-label immunofluorescence was employed to assess the expression pattern of phosphorylated CREB in the striatal interneurons population.
Sections were incubated with a cocktail of rabbit anti-pCREB (Millipore Corporation, RM, Italy) and one of the striatal interneuron markers: ChAT, NOS, CALR and PARV. Tissue was mounted on slides, coverslipped with GEL-MOUNT and the confocal laser scanner microscopy (Zeiss LSM700). For these experiments, parameters set on sections of control mice (Wt), such as laser %, pinhole and gain master, were equal during images acquisition.
Statistical Analysis
Single and Double Immunolabeling
All the collected images have been quantified by using the Java image processing and analysis program ImageJ. Cells of interest were selected using the freehand tool. From the Analyze menu, Set measurements Mean “Grey Value”, “Area” and “Min and Max Grey Value” were selected. The region characterized by absence of fluorescence was considered in the background and it was subtracted. Finally, the mean values with SEM were obtained for all measures. ANOVA analysis available in the software Stat version 12 and GraphPad Prism version 7.0 was performed. P values of less than 0.05 were considered statistically significant.
Results
INO-1001 Prevents the Sequestration of CBP
CBP localization and expression in R6/2 transgenic mice treated with saline or INO-1001 were investigated. A specific antibody against mutated huntingtin revealed NIIs in R6/2 mice and the CBP immunoreactivity in each group was analyzed. Figures 1A–F show the physiological (mostly nuclear) distribution of CBP in wild type mice, which exhibit normal morphological features and vitality. A strong depletion of CBP was observed in the nuclei of saline-treated R6/2 mice (Figures 1G–I).
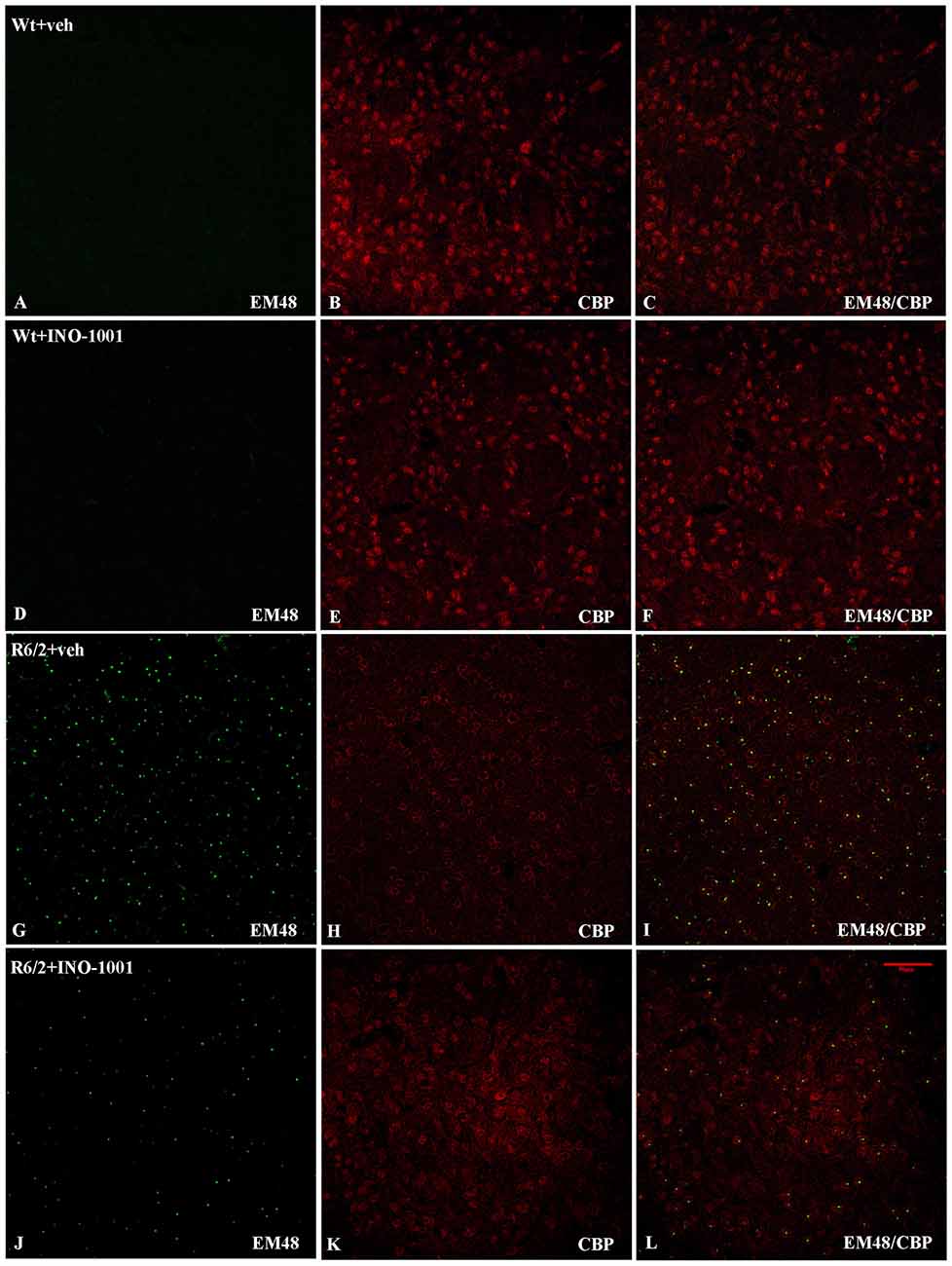
Figure 1. Double-label immunofluorescence for CREB-binding protein (CBP) and Neuronal intranuclear inclusions (NIIs). The double label immunostaining was performed in Wt mice (A–C), INO-1001 treated Wt (D–F), vehicle treated R6/2 mice (G–I) and INO-1001 treated mice (J–L). CBP is labeled in red and NIIs in green. NIIs are not found in the WT groups (A,D). In (J) please observe the low density, reduced size and immunolabeling intensity of NIIs in R6/2 treated with INO- 1001, and the CBP expression pattern similar to Wt groups.
NIIs Are Reduced after PARP Inhibition
As expected, wild type mice striata did not show any immunoreactivity for NIIs. On the contrary, striatal brains of 11-week-old R6/2 mice showed, as previously demonstrated, an elevated number of NIIs [31] (Figures 1G–I). As mentioned above, the NIIs positive nuclei did not show CBP expression in saline-treated R6/2 mice compared to INO-1001-treated mice (Figures 1J–L). Moreover, the analysis of all saline-treated R6/2 revealed that the NIIs area and immunoreaction intensity were decreased compared to cells of INO-1001 treated mice (Figures 2A,B). Daily administration of PARP-1 inhibitor showed a reduced NIIs density, with a significant decrease in NIIs size. Treatment promoted the CBP expression restoration, showing a CBP staining pattern in R6/2 mice brains sections that was comparable to that of wild type animals.
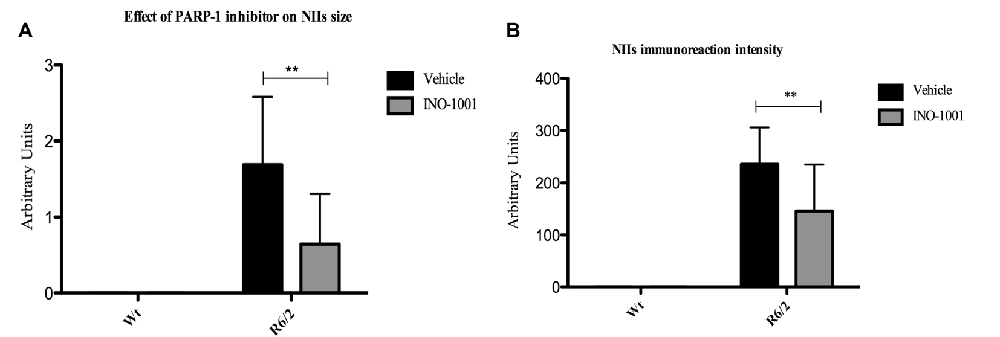
Figure 2. The effect of Poly (ADP-ribose) polymerase-1 (PARP-1) inhibitor on NIIs size and intensity. Two-way ANOVA performed on data obtained by vehicle and INO-1001 treated R6/2 mice revealed a statistically significant effect of treatment on NIIs size and intensity. (A) Bonferroni analysis showed a significant decrease of NIIs size p < 0.001 (F(1,28) = 7.019; P = 0.0031) and intensity in mice treated with PARP-1 inhibitor respect to vehicle treated R6/2 p < 0.001 (F(1,28) = 4.96; P = 0.0060; Panel B).
INO-1001 Prevents the Loss of Parvalbumin- and Calretinin Containing Interneurons
Striatal PARV interneurons of wild-type animals administered either saline or INO-1001 were medium sized with axons with very dense arborizations (Figure 3). The density of PARV interneurons was markedly reduced in the vehicle-treated R6/2 mice, with a smaller cell body and fewer or no arborizations. INO-1001 increased the density of PARV interneurons in the R6/2 mice compared to saline treatment (Figure 3). The Anova test revealed a significant effect of the treatment. Bonferroni analysis showed no effect on the number of PARV interneurons in the wild-type mice treated with INO-1001 and a protective effect on the striatal parvalbumin-containing neurons of R6/2 mice.
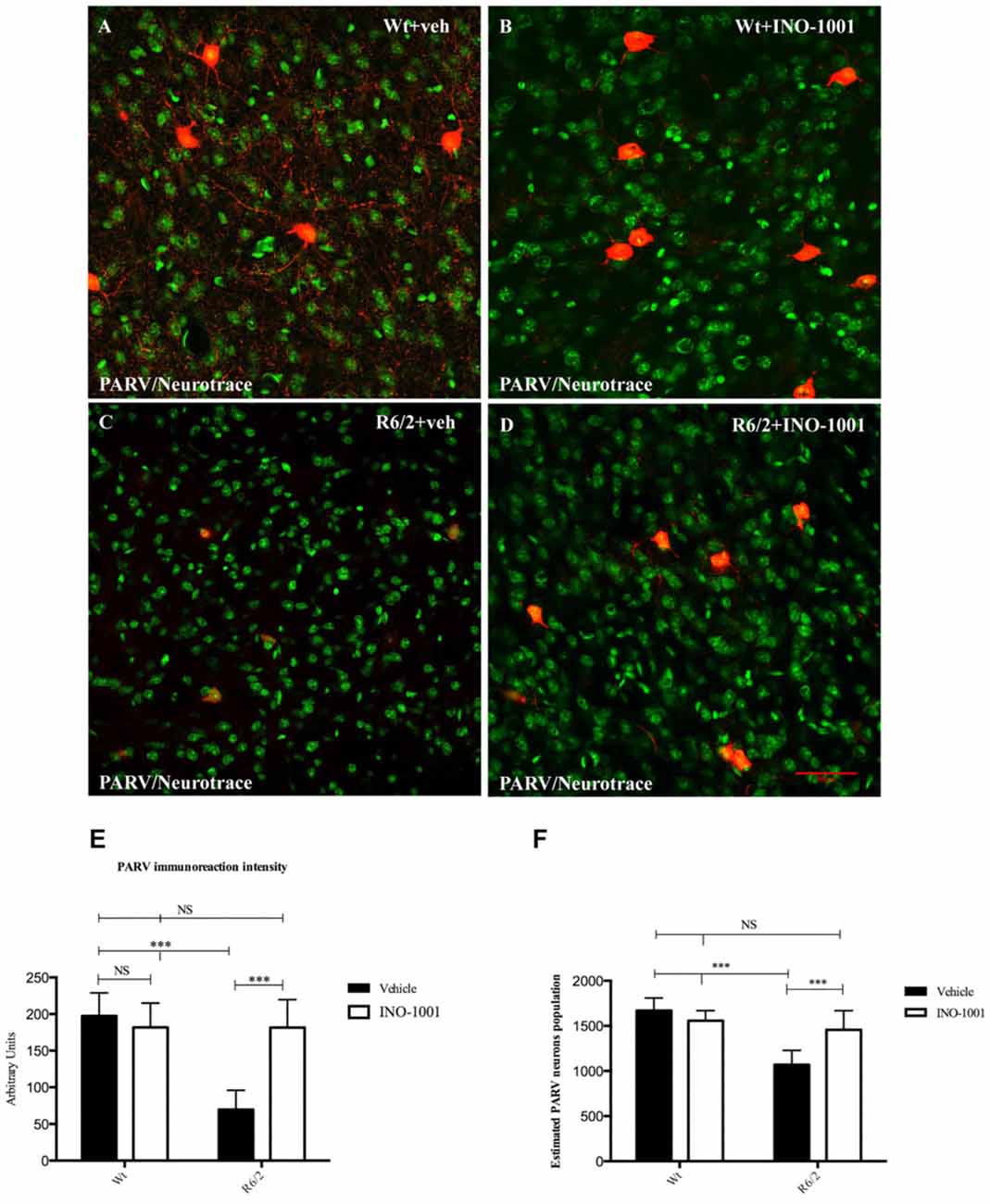
Figure 3. Confocal laser scanning microscopy images of double-label immunofluorescence for PV (visualized in red-Cy3 fluorescence) and the Nissl-like fluorescent marker Neurotrace (visualized in green fluorescence) (A–D). Graphs show (E) the marked effect of INO-1001 administration in R6/2 mice striatal PARV neurons. Analyzed data revealed the significant effect of PARP-1 inhibitor (p < 0.0001) in promoting the parvalbumin-containing neurons protection. INO-1001 treatment had no effect in Wt mice. Densities of all interneuron subtypes markers were compared by means of two-way ANOVA with Bonferroni analysis (F(1,28) = 6.041; p = 0.0004). (E–F) Single-label diaminobenzidine tetrahydrochloride immunohistochemistry for PV in the dorsolateral striata of vehicle-treated (E) and INO-1001-treated (F) R6/2 mice. Graph (F) shows the statistically significant effect of INO-1001 daily administration on the most vulnerable striatal interneurons subtype in R6/2 mice.
Striatal interneurons were identified by the specific immunoreactivity markers ChAT, NOS, CALR and PARV. Collected data confirmed the previous observations about the survival of specific striatal interneurons subtypes in R6/2 mice and a specific vulnerability of CALR and PARV positive interneurons in the late states of HD. The single immunolabeling study showed an equal distribution of ChAT and NOS interneurons in all experimental groups (data not shown), compared to PARV and CALR positive neurons, which exhibited a dramatically reduced number, and immunoreactivity intensity, in saline- treated R6/2 mice. However, INO-1001 treatment promoted a significant increase in number of both PARV and CALR interneurons and arborizations (Figure 4). The density of PARV and CALR interneurons was comparable to that of wild type mice.
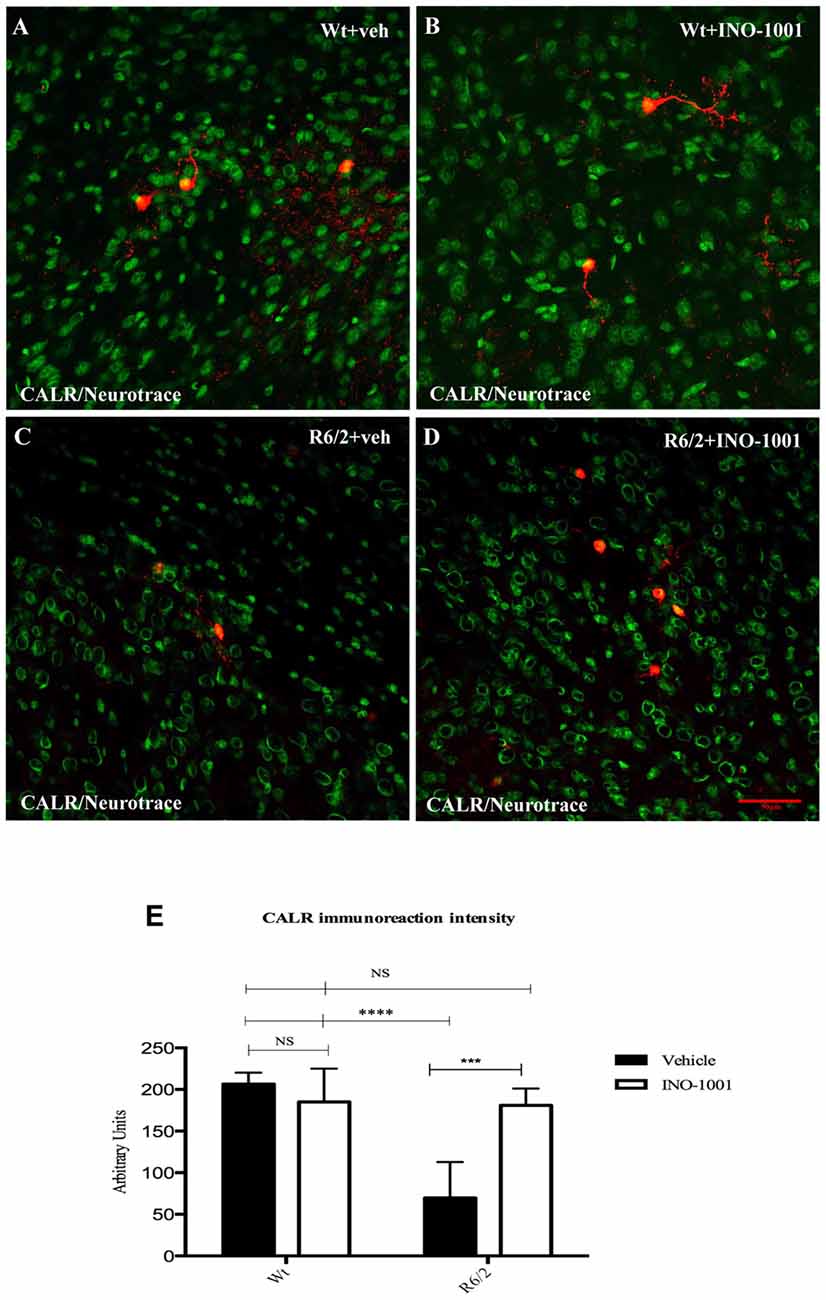
Figure 4. Confocal laser scanning microscopy images of double-label immunofluorescence for CALR (visualized in red-Cy3 fluorescence) and the Nissl-like fluorescent marker Neurotrace (visualized in green fluorescence) (A–D). The graph (E) shows the marked effect of INO-1001 administration in R6/2 mice striatal CALR neurons. Analyzed data revealed the significant effect of PARP-1 inhibitor in promoting the calretinin-containing neurons protection. INO-1001 treatment had no effect in Wt mice. Densities of all interneuron subtypes markers were compared by means of two-way ANOVA with Bonferroni analysis (F(1,28) = 12.56; P = 0.0001).
Analysis of pCREB Expression Levels in the Interneurons Subtypes
PARP-1 inhibitor, INO-1001, promoted a rescue in pCREB expression in R6/2 mice compared to saline-treated mice [25]. The intensity of phosphorylated CREB immunoreaction product in the interneurons subtypes was investigated in all experimental groups. The present study shows that pCREB expression was decreased in CALR interneurons of saline treated R6/2 mice, where it was up regulated by INO-1001 treatment (Figure 5). A comparable result was obtained in PARV neurons, where INO-1001 significantly increased pCREB content in the R6/2 mice (Figure 6).
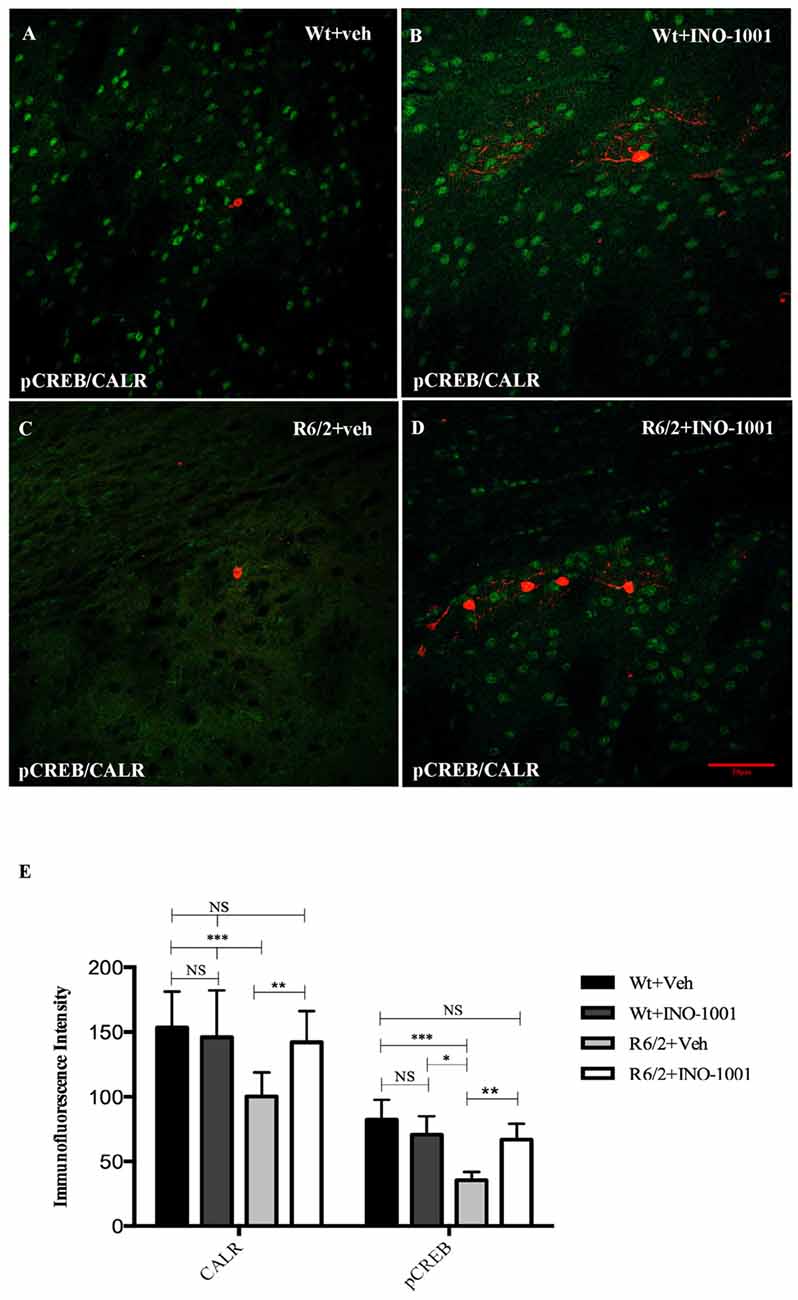
Figure 5. Confocal images of double-label immunofluorescence for phosphorylated c-AMP response element binding protein (pCREB) and Calretinin. pCREB is showed in green CY-2 fluorescence, Calretinin is labeled in red Cy-3 (A–D). Graph (E) shows the immunoreaction intensity relative to pCREB and Calretinin in each animal groups. A two-way ANOVA with genotype and treatment as main factors revealed that R6/2 mice had a significant impairment in pCREB expression levels in the Calretinin positive interneurons R6/2 compared to vehicle and INO-1001-treated wild-type (F(1,28) = 29.625; P = 0.0001). INO-1001 treatment restored pCREB expression in Calretinin-containing neurons in a genotype dependent fashion F(1,28) = 21.523; P = 0.0001.
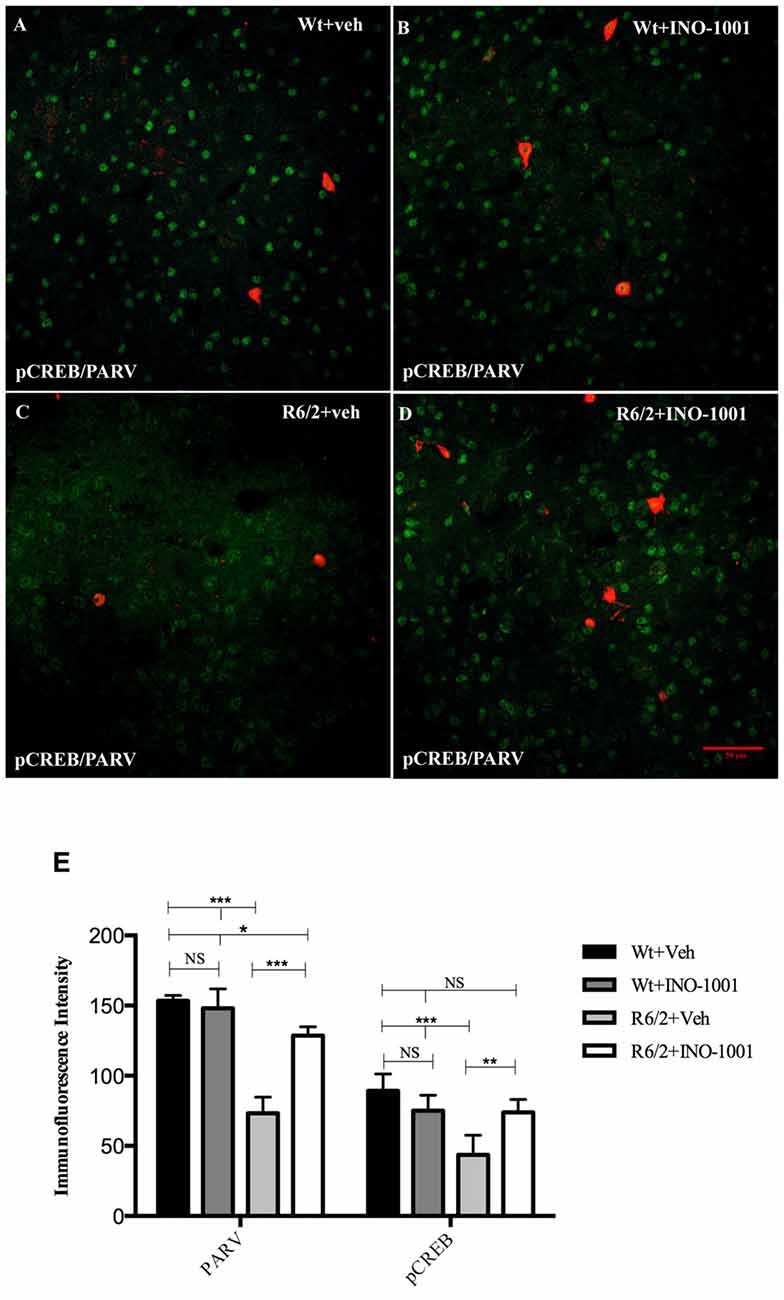
Figure 6. Confocal images of double-label immunofluorescence for pCREB and Parvalbumin. pCREB is labeled in green CY-2 fluorescence, Parvalbumin in red Cy-3 (A–D). Graph shows the immunoreaction intensity relative to pCREB and Parvalbumin in the different experimental animal groups (E). pCREB content is lower in the vulnerable parvalbuminergic interneurons of saline treated R6/2 mice respect to R6/2 treated with INO-1001 (F(1,28) = 67.837; P = 0.0000) that increases pCREB and parvalbumin expression levels in a genotype dependent fashion (significant genotype × treatment interaction pCREB F(1,28) = 32.889; P = 0.0010; PARV F(1,28) = 57.123; P = 0.0000).
Discussion
Our findings demonstrate that chronic treatment with the PARP inhibitor INO-1001 has a protective effect for striatal parvalbumin and calretinin-containing neurons in terms of neurodegeneration, and prevents CBP sequestration into NIIs.
Neurodegeneration affects neuronal subpopulations of the striatum in different ways. In fact, PARV-positive GABAergic interneurons display a cell vulnerability that is comparable to that of projection neurons (Ferrer et al., 1994; Liang et al., 2005). Notably, projection neurons and interneurons are different in their sensitivity to both ischemic and excitotoxic insults (Roberts and DiFiglia, 1989; Chesselet et al., 1990). However, the molecular basis of such different behavior remains to be fully elucidated. (Beal et al., 1989; Figueredo-Cardenas et al., 1994). One possibility that our group had investigated was that some interneurons tendency to survive longer (such as cholinergic interneurons) was related to their ability to maintain sufficient levels of BDNF (Fusco et al., 2003).
Striatal parvalbumin interneurons are GABAergic neurons that have a spiny varicose dendrites and indented nuclei (Celio, 1990; Bennett and Bolam, 1994) and axons with typically dense collateral arborizations (Cicchetti and Parent, 1996) and because they fire phasically at high frequency in response to cortical stimulation, they are considered fast-firing neurons (Kita, 1993; Kawaguchi et al., 1995). Striatal PV neurons are considered interneurons because of their morphological characteristics, which include a medium-sized cell body, aspiny varicose dendrites and a strongly indented nucleus (Celio, 1990; Bennett and Bolam, 1994).
GABAergic PV-positive interneurons only constitute 3%–5% of the cells in the rodent neostriatum (Kawaguchi et al., 1995), and yet they are able to exert a very powerful inhibitory control on the activity of cortical projecting neurons (Koos and Tepper, 1999). Cepeda et al. (2013) described how such a small population of neurons plays a pivotal role in HD. Indeed, by means of optogenetics and electrophysiology, they described that parvalbumin neurons are an important source for increased frequency of spontaneous GABA synaptic activity in the striatum.
Interestingly, these neurons are not only as vulnerable as projection neurons to HD but they also tend to form NIIs to the same extent as projection neurons, whereas other types of more resistant interneurons, such as cholinergic and somatostatin interneurons, form NIIs less frequently (Kosinski et al., 1999). Thus, the observation that PARP inhibition significantly spared the striatal PARV interneurons confirms the beneficial effect of the treatment on HD pathology.
However, a sparing of parvalbumin neurons could be desirable not only for HD, but also in other hyperkinetic neurological disorders. Indeed, striatal parvalbumin expression is decreased in a dystonic hamster model (Bode et al., 2017). Thus, disorders where parvalbumin interneurons fast spiking activity is involved such as dystonia could benefit from such neuroprotective effect (Reiner et al., 2003).
Calretinin is a calcium-binding protein containing a structural domain that binds calcium (Henzi et al., 2009). A protective role for calretinin has been postulated, as it was observed that CALR neurons are spared in a 6-OHDA-lesion model (Tsuboi et al., 2000) and also in HD patients (Massouh et al., 2008). Moreover, CALR was shown to be correlated to an impaired neurogenesis in adult HD (Fedele et al., 2011). In a recent report, Dong et al. (2012) demonstrated that CALR can interact with mutant huntingtin and that it can decrease cytotoxicity caused by mutant huntingtin in cellular models of HD.
We observed that CALR neurons were rescued by INO-1001 treatment, supporting the idea that functionally active CALR interneurons may participate in the survival of striatum in HD.
The observation that PARP-inhibition was effective in decreasing the size of NIIs was in agreement with our previous report about the reduced frequency of NIIs in the INO-1001 treated mice. The role of NIIs in HD has been discussed for many years now, and the question whether aggregates could be the basis of clinical manifestations and neurodegeneration in HD still remains unsolved (Davies and Scherzinger, 1997; Kim and Tanzi, 1998; Saudou et al., 1998; Sisodia, 1998). It is conceivable that HD aggregates could trigger cellular dysfunction leading to the death of those neurons in which they are contained. Indeed, NIIs formation can impair cellular functions by their ability to sequester proteins and transcription factors, thereby causing a dysfunction of a number of cellular transcriptional mechanisms (Maat-Schieman et al., 1999; Perutz, 1999; Preisinger et al., 1999; Meade et al., 2000). However, there is no clear demonstration that aggregates are the direct cause of HD neuronal death. Moreover, Saudou et al. demonstrated that neuronal death caused by the mutation is not directly related to NIIs formation in cultured cells transfected with mutant huntingtin. Additionally, Reiner and co-workers (Reiner et al., 1988) observed that the presence of NIIs was related to a prolonged survival in chimeric R6/2 mice.
However, our studies have shown that every treatment that proved beneficial for HD phenotype and neurodegeneration (DeMarch et al., 2008) was also effective in decreasing the frequency and the size of NIIs. Moreover, a toxic effect for aggregates was demonstrated. Therefore, polyQ aggregation could be considered as a therapeutic target for HD (Bates, 2003).
Transcriptional responses to cAMP are mediated by the interaction of CREB with CBP, and cAMP activated transcription is regulated by CREB phosphorylation (Azuma et al., 1996).
However, CBP is not only a transcriptional coactivator for CREB (Arany et al., 1994). Indeed, CBP is able to acetylate promoter proximal nucleosome histones, which results in an augmented availability of DNA for other important factors (Ogryzko et al., 1996). Of note, several studies clearly demonstrated that histone acetyltransferase activity could be altered by mutated huntingtin. Moreover, the enzyme dysfunction could be at the basis of the transcription deregulation occurring in HD. Interestingly, histone deacetylase inhibitors have proven to display a beneficial effect in several HD models (Sadri-Vakili and Cha, 2006).
CBP is abundant in the nuclei of 100% of wild type cells, but only in 18% of HD mutant cells (Gines et al., 2003). The nuclear localization of CBP is therefore a feature of normal cells, and its displacement away from the nucleus and sequestration into the aggregates accounts for the cellular dysfunction in HD. The phenomenon described is indeed typical of HD pathology, as it has been observed in several HD models, from cell cultures to human patients (Nucifora et al., 2001). An association between the depletion of CBP and mutant huntingtin-mediated cellular toxicity was previously demonstrated (Jiang et al., 2006).
Therefore, the physiological role of CBP is markedly impaired in HD and it participates in the neuronal degeneration not only via the altered CREB function but also through the detrimental effect on the mechanism of histone acetylation. Correspondingly, the improved functioning of CBP might be related not only to the restored CREB functions, but also to rescued physiological functioning of CBP per se.
Accordingly, we are reporting that INO-1001 is able to prevent CBP segregation into the aggregates. This confirms our recent report on neuroprotection exerted by INO-1001 in the R6/2 mice, and supporting the idea that reinstating the nuclear localization of CBP is able to participate to the correct cellular functioning. In line with our earlier report, we can establish that there is a strong correlation between CBP recruitment in NIIs and neuronal death.
Thus, we can speculate that the reinstated CREB function, along with the physiological localization of CBP in the nucleus, can concur in keeping adequate BNDF expression levels that are necessary to protect striatal neurons from HD neurodegeneration (Bemelmans et al., 1999; Pérez-Navarro et al., 2000).
We also provide the first evidence that parvalbumin and calretinin-containing interneuron are rescued by PARP inhibition in the R6/2 mouse model of HD. Therefore, these results demonstrate the possibility of ameliorating HD neuropathology by chronic peripheral administration of INO-1001 and, more generally, underline the potential therapeutic value of PARP inhibitors in HD.
Author Contributions
FRF designed the experimental plan and wrote the article. CG performed experiments and statistical analysis. EP performed experiments and wrote the the results. IS performed experiments. AC performed experiments. VDA performed experiments.
Funding
GR-2010-2316475—Italian Department of Health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer NM declared a shared affiliation, though no other collaboration, with one of the authors VD to the handling Editor, who ensured that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
We thank Prof. James H. Lynch for style editing.
References
Albin, R. L., and Tagle, D. A. (1995). Genetics and molecular biology of Huntington’s disease. Trends Neurosci. 18, 11–14. doi: 10.1016/0166-2236(95)93943-R
Arany, Z., Sellers, W. R., Livingston, D. M., and Eckner, R. (1994). E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell 77, 799–800. doi: 10.1016/0092-8674(94)90127-9
Azuma, Y., Ogita, K., and Yoneda, Y. (1996). Particular nuclear transcription factors responsive to systemic administration of kainic acid in murine brain. Neurochem. Int. 29, 289–299. doi: 10.1016/0197-0186(95)00157-3
Bates, G. (2003). Huntingtin aggregation and toxicity in Huntington’s disease. Lancet 361, 1642–1644. doi: 10.1016/s0140-6736(03)13304-1
Beal, M. F., Kowall, N. W., Swartz, K. J., Ferrante, R. J., and Martin, J. B. (1989). Differential sparing of somatostatin-neuropeptide Y and cholinergic neurons following striatal excitotoxin lesions. Synapse 3, 38–47. doi: 10.1002/syn.890030106
Bemelmans, A. P., Horellou, P., Pradier, L., Brunet, I., Colin, P., and Mallet, J. (1999). Brain-derived neurotrophic factor-mediated protection of striatal neurons in an excitotoxic rat model of Huntington’s disease, as demonstrated by adenoviral gene transfer. Hum. Gene Ther. 10, 2987–2997. doi: 10.1089/10430349950016393
Bennett, B. D., and Bolam, J. P. (1994). Synaptic input and output of parvalbuminimmunoreactive neurons in the neostriatum of the rat. Neuroscience 62, 707–719. doi: 10.1016/0306-4522(94)90471-5
Bode, C., Richter, F., Spröte, C., Brigadski, T., Bauer, A., Fietz, S., et al. (2017). Altered postnatal maturation of striatal GABAergic interneurons in a phenotypic animal model of dystonia. Exp. Neurol. 287, 44–53. doi: 10.1016/j.expneurol.2016.10.013
Bonni, A., Brunet, A., West, A. E., Datta, S. R., Takasu, M. A., and Greenberg, M. E. (1999). Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286, 1358–1362. doi: 10.1126/science.286.5443.1358
Cardinale, A., Paldino, E., Giampà, C., Bernardi, G., and Fusco, F. R. (2015). PARP-1 inhibition is neuroprotective in the R6/2 mouse model of Huntington’s disease. PLoS One 7:e0134482. doi: 10.1371/journal.pone.0134482
Celio, M. R. (1990). Calbindin D-28k and parvalbumin in the rat nervous system. Neuroscience 35, 375–475. doi: 10.1016/0306-4522(90)90091-h
Cepeda, C., Galvan, L., Holley, S. M., Rao, S. P., André, V. M., Botelho, E. P., et al. (2013). Multiple sources of striatal inhibition are differentially affected in Huntington’s disease mouse models. J. Neurosci. 33, 7393–7406. doi: 10.1523/JNEUROSCI.2137-12.2013
Chesselet, M. F., Gonzales, C., Lin, C. S., Polsky, K., and Jin, B. K. (1990). Ischemic damage in the striatum of adult gerbils: relative sparing of somatostatinergic and cholinergic interneurons contrasts with loss of efferent neurons. Exp. Neurol. 110, 209–218. doi: 10.1016/0014-4886(90)90032-n
Cicchetti, F., and Parent, A. (1996). Striatal interneurons in Huntington’s disease: selective increase in the density of calretinin-immunoreactive medium-sized neurons. Mov. Disord. 11, 619–626. doi: 10.1002/mds.870110605
Cramer, H., Warter, J. M., and Renaud, B. (1984). Analysis of neurotransmitter metabolites and adenosine 3′,5′-monophosphate in the CSF of patients with extrapyramidal motor disorders. Adv. Neurol. 40, 431–435.
Davies, S. W., and Scherzinger, E. (1997). Nuclear inclusions in Huntington’s disease. Trends Cell Biol. 7:422. doi: 10.1016/s0962-8924(97)88136-6
DeMarch, Z., Giampà, C., Patassini, S., Bernardi, G., and Fusco, F. R. (2008). Beneficial effects of rolipram in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 30, 375–387. doi: 10.1016/j.nbd.2008.02.010
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
Dong, G., Gross, K., Qiao, F., Ferguson, J., Callegari, E. A., Rezvani, K., et al. (2012). Calretinin interacts with huntingtin and reduces mutant huntingtin-caused cytotoxicity. J. Neurochem. 123, 437–446. doi: 10.1111/j.1471-4159.2012.07919.x
Fedele, V., Roybon, L., Nordström, U., Li, J. Y., and Brundin, P. (2011). Neurogenesis in the R6/2 mouse model of Huntington’s disease is impaired at the level of NeuroD1. Neuroscience 173, 76–81. doi: 10.1016/j.neuroscience.2010.08.022
Ferrer, I., Oliver, B., Russi, A., Casas, R., and Rivera, R. (1994). Parvalbumin and calbindin-D28k immunocytochemistry in human neocortical epileptic foci. J. Neurol. Sci. 123, 18–25. doi: 10.1016/0022-510x(94)90198-8
Figueredo-Cardenas, G., Anderson, K. D., Chen, Q., Veenman, C. L., and Reiner, A. (1994). Relative survival of striatal projection neurons and interneurons after intrastriatal injection of quinolinic acid in rats. Exp. Neurol. 129, 37–56. doi: 10.1006/exnr.1994.1145
Fusco, F. R., Zuccato, C., Tartari, M., Martorana, A., De March, Z., Giampa, C., et al. (2003). Co-localization of brain-derived neurotrophic factor (BDNF) and wild-type huntingtin in normal and quinolinic acid-lesioned rat brain. Eur. J. Neurosci. 18, 1093–1102. doi: 10.1046/j.1460-9568.2003.02844.x
Giampà, C., DeMarch, Z., D’Angelo, V., Morello, M., Martorana, A., Sancesario, G., et al. (2006). Striatal modulation of cAMP-response-element-binding protein (CREB) after excitotoxic lesions: implications with neuronal vulnerability in Huntington’s disease. Eur. J. Neurosci. 23, 11–20. doi: 10.1111/j.1460-9568.2005.04545.x
Giampà, C., Middei, S., Patassini, S., Borreca, A., Marullo, F., Laurenti, D., et al. (2009). Phosphodiesterase type IV inhibition prevents sequestration of CREB binding protein, protects striatal parvalbumin interneurons and rescues motor deficits in the R6/2 mouse model of Huntington’s disease. Eur. J. Neurosci. 29, 902–910. doi: 10.1111/j.1460-9568.2009.06649.x
Gines, S., Seong, I. S., Fossale, E., Ivanova, E., Trettel, F., Gusella, J. F., et al. (2003). Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum. Mol. Genet. 12, 497–508. doi: 10.1093/hmg/12.5.497
Gutekunst, C. A., Li, S. H., Yi, H., Mulroy, J. S., Kuemmerle, S., Jones, R., et al. (1999). Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J. Neurosci. 19, 2522–2534.
Hedreen, J. C., Peyser, C. E., Folstein, S. E., and Ross, C. A. (1991). Neuronal loss in layers V and VI of cerebral cortex in Huntington’s disease. Neurosci. Lett. 133, 257–261. doi: 10.1016/0304-3940(91)90583-f
Henzi, T., Blum, W. V., Pfefferli, M., Kawecki, T. J., Salicio, V., and Schwaller, B. (2009). SV40-induced expression of calretinin protects mesothelial cells from asbestos cytotoxicity and may be a key factor contributing to mesothelioma pathogenesis. Am. J. Pathol. 174, 2324–2336. doi: 10.2353/ajpath.2009.080352
Jiang, H., Nucifora, F. C. Jr., Ross, C. A., and DeFranco, D. B. (2003). Cell death triggered by polyglutamine-expanded huntingtin in a neuronal cell line is associated with degradation of CREB-binding protein. Hum. Mol. Genet. 12, 1–12. doi: 10.1093/hmg/12.1.1
Jiang, H., Poirier, M. A., Liang, Y., Pei, Z., Weiskittel, C. E., Smith, W. W., et al. (2006). Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis. 23, 543–551. doi: 10.1016/j.nbd.2006.04.011
Kameshita, I., Matsuda, Z., Taniguchi, T., and Shizuta, Y. (1984). Poly (ADP-Ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain the DNA-binding domain and the automodification domain. J. Biol. Chem. 259, 4770–4776.
Kauppinen, T. M., Suh, S. W., Higashi, Y., Berman, A. E., Escartin, C., Won, S. J., et al. (2011). Poly(ADP-ribose) polymerase-1 modulates microglial responses to amyloid β. J. Neuroinflammation 8:152. doi: 10.1186/1742-2094-8-152
Kawaguchi, Y., Wilson, C. J., Augood, S. J., and Emson, P. C. (1995). Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 18, 527–535. doi: 10.1016/0166-2236(95)98374-8
Kazantsev, A., Preisinger, E., Dranovsky, A., Goldgaber, D., and Housman, D. (1999). Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl. Acad. Sci. U S A 96, 11404–11409. doi: 10.1073/pnas.96.20.11404
Kim, T. W., and Tanzi, R. E. (1998). Neuronal intranuclear inclusions in polyglutamine diseases: nuclear weapons or nuclear fallout? Neuron 21, 657–659. doi: 10.1016/s0896-6273(00)80581-4
Kita, H. (1993). GABAergic circuits of the striatum. Prog. Brain Res. 99, 51–72. doi: 10.1016/s0079-6123(08)61338-2
Koos, T., and Tepper, J. M. (1999). Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat. Neurosci. 2, 467–472. doi: 10.1038/8138
Kosinski, C. M., Cha, J. H., Young, A. B., Mangiarini, L., Bates, G., Schiefer, J., et al. (1999). Intranuclear inclusions in subtypes of striatal neurons in Huntington’s disease transgenic mice. Neuroreport 10, 3891–3896. doi: 10.1097/00001756-199912160-00031
Kurosaki, T., Ushiro, H., Mitsuuchi, Y., Suzuki, S., Matsuda, M., Matsuda, Y., et al. (1987). Primary structure of human poly(ADP-ribose) synthetase as deduced from cDNA sequence. J. Biol. Chem. 262, 15990–15997.
Liang, Z. Q., Wang, X. X., Wang, Y., Chuang, D. M., DiFiglia, M., Chase, T. N., et al. (2005). Susceptibility of striatal neurons to excitotoxic injury correlates with basal levels of Bcl-2 and the induction of P53 and c-Myc immunoreactivity. Neurobiol. Dis. 20, 562–573. doi: 10.1016/j.nbd.2005.04.011
Luthi-Carter, R., Strand, A., Peters, N. L., Solano, S. M., Hollingsworth, Z. R., Menon, A. S., et al. (2000). Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum. Mol. Genet. 9, 1259–1271. doi: 10.1093/hmg/9.9.1259
Maat-Schieman, M. L., Dorsman, J. C., Smoor, M. A., Siesling, S., Van Duinen, S. G., Verschuuren, J. J., et al. (1999). Distribution of inclusions in neuronal nuclei and dystrophic neurites in Huntington disease brain. J. Neuropathol. Exp. Neurol. 58, 129–137. doi: 10.1097/00005072-199902000-00003
Mantamadiotis, T., Lemberger, T., Bleckmann, S. C., Kern, H., Kretz, O., Martin, V. A., et al. (2002). Disruption of CREB function in brain leads to neurodegeneration. Nat. Genet. 31, 47–54. doi: 10.1038/ng882
Martindale, D., Hackam, A., Wieczorek, A., Ellerby, L., Wellington, C., McCutcheon, K., et al. (1998). Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat. Genet. 18, 150–154. doi: 10.1038/ng0298-150
Martire, S., Fuso, A., Rotili, D., Tempera, I., Giordano, C., De Zottis, I., et al. (2013). PARP-1 modulates amyloid β peptide-induced neuronal damage. PLoS One 2011:e72169. doi: 10.1371/journal.pone.0072169
Martire, S., Mosca, L., and d’Erme, M. (2015). PARP-1 involvement in neurodegeneration: a focus on Alzheimer’s and Parkinson’s diseases. Mech Ageing Dev. 146–148, 53–64. doi: 10.1016/j.mad.2015.04.001
Massouh, M., Wallman, M. J., Pourcher, E., and Parent, A. (2008). The fate of the large striatal interneurons expressing calretinin in Huntington’s disease. Neurosci. Res. 62, 216–224. doi: 10.1016/j.neures.2008.08.007
Meade, C. A., Figueredo-Cardenas, G., Fusco, F., Nowak, T. S. Jr., Pulsinelli, W. A., and Reiner, A. (2000). Transient global ischemia in rats yields striatal projection neuron and interneuron loss resembling that in Huntington’s disease. Exp. Neurol. 166, 307–323. doi: 10.1006/exnr.2000.7530
Nucifora, F. C. Jr., Sasaki, M., Peters, M. F., Huang, H., Cooper, J. K., Yamada, M., et al. (2001). Interference by huntingtin and atrophin-1 with cbpmediated transcription leading to cellular toxicity. Science 291, 2423–2428. doi: 10.1126/science.1056784
Ogryzko, V. V., Schiltz, R. L., Russanova, V., Howard, B. H., and Nakatani, Y. (1996). The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87, 953–959. doi: 10.1016/s0092-8674(00)82001-2
Oliveira, A. M. M., Abel, T., Brindle, P. K., and Wood, M. A. (2006). Differential role for CBP and p300 CREB binding domain in motor skill learning. Behav. Neurosci. 120, 724–729. doi: 10.1037/0735-7044.120.3.724
Pérez-Navarro, E., Canudas, A. M., Akerund, P., Alberch, J., and Arenas, E. (2000). Brain-derived neurotrophic factor, neurotrophin-3 and neurotrophin- 4/5 prevent the death of striatal projection neurons in a rodent model of Huntington’s disease. J. Neurochem. 75, 2190–2199. doi: 10.1046/j.1471-4159.2000.0752190.x
Perutz, M. F. (1999). Glutamine repeats and neurodegenerative diseases. Brain Res. Bull. 50:467. doi: 10.1016/s0361-9230(99)00137-9
Preisinger, E., Jordan, B. M., Kazantsev, A., and Housman, D. (1999). Evidence for a recruitment and sequestration mechanism in Huntington’s disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 1029–1034. doi: 10.1098/rstb.1999.0455
Reiner, A., Albin, R. L., Anderson, K. D., D’Amato, C. J., Penney, J. B., and Young, A. B. (1988). Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. U S A 85, 5733–5737. doi: 10.1073/pnas.85.15.5733
Reiner, A., Dragatsis, I., Zeitlin, S., and Goldowitz, D. (2003). Wild-type huntingtin plays a role in brain development and neuronal survival. Mol. Neurobiol. 28, 259–275. doi: 10.1385/MN:28:3:259
Roberts, R. C., and DiFiglia, M. (1989). Short- and long-term survival of large neurons in the excitotoxic lesioned rat caudate nucleus: a light and electron microscopic study. Synapse 3, 363–371. doi: 10.1002/syn.890030410
Sadri-Vakili, G., and Cha, J. H. (2006). Histone deacetylase inhibitors: a novel therapeutic approach to Huntington’s disease (complex mechanism of neuronal death). Curr. Alzheimer Res. 3, 403–408. doi: 10.2174/156720506778249407
Saudou, F., Finkbeiner, S., Devys, D., and Greenberg, M. E. (1998). Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95, 55–66. doi: 10.1016/s0092-8674(00)81782-1
Sisodia, S. S. (1998). Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell. Mol. Neurobiol. 95, 1–4.
Steffan, J. S., Kazantsev, A., Spasic-Boskovic, O., Greenwald, M., Zhu, Y. Z., Gohler, H., et al. (2000). The Huntington’s disease protein interacts with p53 and CREB- binding protein and represses transcription. Proc. Natl. Acad. Sci. U S A 97, 6763–6768. doi: 10.1073/pnas.100110097
Storey, E., Kowall, N. W., Finn, S. F., Mazurek, M. F., and Beal, M. F. (1992). The cortical lesion of Huntington’s disease: further neurochemical characterization and reproduction of some of the histological and neurochemical features by N-methyl-D-aspartate lesions of rat cortex. Ann. Neurol. 32, 526–534. doi: 10.1002/ana.410320408
Strosznajder, R. P., Jesko, H., and Zambrzycka, A. (2005). Poly(ADP-ribose) polymerase: the nuclear target in signal transduction and its role in brain ischemia-reperfusion injury. Mol. Neurobiol. 31, 149–167. doi: 10.1385/mn:31:1-3:149
Sugars, K. L., Brown, R., Cook, L. J., Swartz, J., and Rubinsztein, D. C. (2004). Decreased cAMP response element-mediated transcription: an early event in exon 1 and full-length cell models of Huntington’s disease that contributes to polyglutamine pathogenesis. J. Biol. Chem. 279, 4988–4999. doi: 10.1074/jbc.M310226200
Sugars, K. L., and Rubinsztein, D. C. (2003). Transcriptional abnormalities in Huntington disease. Trends Genet. 19, 233–238. doi: 10.1016/s0168-9525(03)00074-x
The Huntington’s Disease Research Collaborative Group. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s disease collaborative research group. Cell 72, 971–983.
Torres, E. M., Perry, T. A., Blockland, A., Wilkinson, L. S., Wiley, R. G., Lappi, D. A., et al. (1994). Behavioural, histochemical and biochemical consequences of selective immunolesions in discrete regions of the basal forebrain cholinergic system. Neuroscience 63, 95–122. doi: 10.1016/0306-4522(94)90010-8
Tsuboi, K., Kimber, T. A., and Shults, C. W. (2000). Calretinin-containing axons and neurons are resistant to an intrastriatal 6-hydroxydopamine lesion. Brain Res. 866, 55–64. doi: 10.1016/s0006-8993(00)02219-8
Vonsattel, J. P., Myers, R. H., Stevens, T. J., Ferrante, R. J., Bird, E. D., and Richardson, E. P. Jr. (1985). Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 44, 559–577. doi: 10.1097/00005072-198511000-00003
Walton, M. R., and Dragunow, I. (2000). Is CREB a key to neuronal survival? Trends Neurosci. 23, 48–53. doi: 10.1016/s0166-2236(99)01500-3
Wang, Y., Dawson, V. L., and Dawson, T. M. (2009). Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp. Neurol. 218, 193–202. doi: 10.1016/j.expneurol.2009.03.020
Wilson, R. S., Como, P. G., Garron, D. C., Klawans, H. L., Barr, A., and Klawans, D. (1987). Memory failure in Huntington’s disease. J. Clin. Exp. Neuropsychol. 9, 147–154. doi: 10.1080/01688638708405354
Wyttenbach, A., Swartz, J., Kita, H., Thykjaer, T., Carmichael, J., Bradley, J., et al. (2001). Polyglutamine expansions cause decreased CREmediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington’s disease. Hum. Mol. Genet. 10, 1829–1845. doi: 10.1093/hmg/10.17.1829
Keywords: Huntington’s disease, neurodegeneration, PARP-1 inhibition, CBP, pCREB, interneurons, parvalbumin, calretinin
Citation: Paldino E, Cardinale A, D’Angelo V, Sauve I, Giampà C and Fusco FR (2017) Selective Sparing of Striatal Interneurons after Poly (ADP-Ribose) Polymerase 1 Inhibition in the R6/2 Mouse Model of Huntington’s Disease. Front. Neuroanat. 11:61. doi: 10.3389/fnana.2017.00061
Received: 13 March 2017; Accepted: 07 July 2017;
Published: 02 August 2017.
Edited by:
Francesco Fornai, University of Pisa, ItalyReviewed by:
Vittorio Maglione, IRCCS Neuromed, ItalyNicola B. Mercuri, University of Rome Tor Vergata, Italy
Copyright © 2017 Paldino, Cardinale, D’Angelo, Sauve, Giampà and Fusco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca R. Fusco, Zi5mdXNjb0Boc2FudGFsdWNpYS5pdA==