 Anita K. Roopun
1
Anita K. Roopun
1 Fiona E.N. LeBeau
1
Fiona E.N. LeBeau
1 James Rammell
1
James Rammell
1 Mark O. Cunningham
1
Mark O. Cunningham
1 Roger D. Traub
2
Roger D. Traub
2 Miles A. Whittington
1*
Miles A. Whittington
1*
- 1 Institute of Neuroscience, The Medical School, Newcastle University, Newcastle, UK
- 2 Department of Physical Sciences, International Business Machines Thomas J. Watson Research Center, Yorktown Heights, NY, USA
Acetylcholine is the primary neuromodulator involved in cortical arousal in mammals. Cholinergic modulation is involved in conscious awareness, memory formation and attention – processes that involve intercommunication between different cortical regions. Such communication is achieved in part through temporal structuring of neuronal activity by population rhythms, particularly in the beta and gamma frequency ranges (12–80 Hz). Here we demonstrate, using in vitro and in silico models, that spectrally identical patterns of beta2 and gamma rhythms are generated in primary sensory areas and polymodal association areas by fundamentally different local circuit mechanisms: Glutamatergic excitation induced beta2 frequency population rhythms only in layer 5 association cortex whereas cholinergic neuromodulation induced this rhythm only in layer 5 primary sensory cortex. This region-specific sensitivity of local circuits to cholinergic modulation allowed for control of the extent of cortical temporal interactions. Furthermore, the contrasting mechanisms underlying these beta2 rhythms produced a high degree of directionality, favouring an influence of association cortex over primary auditory cortex.
Introduction
The neocortex generates population rhythms via activity within – and interactions between – local circuits. Defined frequency bands correlate to distinct behavioural states, for example neocortical gamma oscillations (30–80 Hz) are involved in sensory representation (Singer and Gray, 1995 ), selective attention (Tiitinen et al., 1993 ) and short term memory (Tallon-Baudry et al., 1998 ). They facilitate synchrony of neuronal activity over distance (Traub et al., 1996 ) and thus the formation of functional connections following sensory input (Womelsdorf et al., 2007 ). In contrast beta rhythms play a role in higher order sensory processing and sensorimotor control. They are generated by motor and sensorimotor cortex (MacKay and Mendonça, 1995 ; Fairhall et al., 2007 ) and may provide a common temporal framework for comparing actual and expected sensory and motor events (Baker, 2007 ; Donner et al., 2007 ). Their accompanying network dynamics are also favourable for establishing longer range temporal cortical interactions than gamma rhythms alone (Kopell et al., 2000 ).
While gamma rhythms facilitate interareal interactions predominantly within specific sensory modalities (e.g. the visual system), beta rhythms are involved in interactions across different modalities. Visual and motor areas synchronise at beta frequencies during startle responses (Roelfsema et al., 1997 ) and beta rhythms are involved in polymodal sensory response coordination (von Stein et al., 1999 ). It is these longer range, polymodal cortical interactions that appear absent during nonREM sleep (Massimini et al., 2005 ) – a state associated with a reduction in cortical cholinergic excitation (Steriade, 2004 ). In general, acetylcholine has been shown to be important for cognitive function, in particular, the capacity for focused attention and the storage of sensory stimuli during short and long-term memory formation (Muir et al., 1992 ; Sarter and Bruno, 1997 ). There is evidence for cholinergic modulation of top-down processes involved in cognition, where higher (associational) areas influence the pattern of activation of primary sensory cortices (Robbins, 2005 ). However, acetylcholine receptor expression is higher in primary sensory and limbic cortical areas relative to association areas of cortex (Mesulam, 1995 ).
To investigate the mechanisms underlying the influence of acetylcholine on cortico-critical interactions we turn here to in vitro and in silico models. Such models have demonstrated that, for glutamatergic excitation, neocortical gamma rhythms are generated predominantly in layers II/III by fast spiking, GABAergic interneurons in heterogeneous neuronal networks formed by both electrical and chemical synapses (Cunningham et al., 2004 ). This type of population rhythm is seen alone in this model in primary auditory cortex, but also manifests concurrently with beta2 rhythms (20–30 Hz) in association (parietal) cortex. In this model, with glutamatergic excitation, the beta2 rhythm seen is generated by different cortical neuronal interactions to those seen for gamma rhythms. Beta2 oscillations occur in deep rather than superficial laminae (Roopun et al., 2006 , 2008 ), and occur as a population phenomenon in a manner not dependent on chemical synaptic transmission. Instead, gap junctional connectivity between electrically active layer 5 intrinsically bursting (IB) cell compartments (e.g. their axons) provides both the drive for axonal bursting and a mean to synchronise this bursting within the population.
The rodent horizontal cortical slice is an ideal reduced model with which to study the effects of cholinergic neuromodulation on the basic properties of above dynamic processes. The juxtaposition of parietal (associational) and primary auditory cortices allows for the modelling of communication between primary and higher-order sensory areas to address how top-down (rather than reciprocal) interactions may occur. The previous in vitro studies described above are used here as a starting point to compare effects of cholinergic neuromodulation on local cortical dynamics at gamma and beta2 frequencies. We show that near-identical spectral patterns of rhythms are generated in the two areas by cholinergic (primary auditory) or glutamatergic (parietal) excitation. However, the beta2 rhythm, while generated in each case by layer V IB neurons, arose from starkly contrasting underlying mechanisms. Coexpression of glutamatergic and cholinergic beta2 rhythms generated highly directed coherence between parietal and auditory areas in a manner dependent on this mechanistic mismatch.
Materials and Methods
Slice Preparation
Adult male Wistar rats (∼150 g) were anaesthetised with isoflurane, followed by an intramuscular injection of xylazine (∼10 mg/kg) and ketamine (∼100 mg/kg), before perfusion with oxygenated, sucrose-containing artificial cerebrospinal fluid (sACSF). All procedures were carried out in accordance with the UK Home Office Animals (Scientific procedures) Act. Horizontal slices of cortex (450 μM thick), containing 1° auditory cortex (Au1) and adjacent polymodal association cortex (par2) were maintained at the interface between humidified carbogen gas (95% O2/5% CO2) and normal ACSF that contained (in mM): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 1.2 CaCl2, 24 NaHCO3 and 10 glucose at 34°C. All surgical procedures were in accordance with regulations of the UK Animals (Scientific Procedures) Act, 1986. All drugs were bath-applied: Carbachol (15–20 μM), Atropine (100 nM), carbenoxolone (100 μM) and d-tubocurarine (20 μM) were obtained from Sigma-Aldrich (Poole, UK). Gabazine (500 nM), kainate (400 nM), 4-(4-aminophenyl)-1,2-dihydro-1-methyl-2-propylcarbamoyl-6,7-methylenedioxyphthalazine (SYM2206, 25 μM) were obtained from Tocris Cookson (Bristol, UK).
Data Acquisition and Analysis
Extracellular field potential recordings were taken with micropipettes (2–5 MΩ) filled with ACSF. Intracellular recordings use pipettes with 2 M potassium acetate (50–100 MΩ). Extracellular data were band pass filtered at 0.002–0.4 kHz, with intracellular DC recordings low-passed filtered at 2.5 kHz. Stable oscillations were recorded 2 h after either carbachol or kainate application. Power spectra were derived from Fourier analysis of 60-s epochs of data and results presented as mean ± standard error of mean (SEM). Spike and synaptic event detection was performed on the basis of peak transient deflections from mean membrane voltage. Cross correlograms, phase, synchrony and coherence measures were derived using scripts written in Matlab (The Mathworks, Natick, USA). Interaction between primary and association cortices was performed using a Granger causality test. Sixty seconds epochs of unfiltered data were downsampled to 0.5 kHz and split into 2 s epochs. Each epoch was used to construct a bivariate autoregressive model (order 20) and the best model for all epochs used estimate causality using the BSMART toolbox (Cui et al., 2008 ). Statistical significance was measured by paired/unpaired t-tests and one-way analysis of variance (ANOVA) tests. Results were deemed statistically significantly if P < 0.05.
Simulation Methods
We used the cortical column simulation program described in Traub et al. (2005) , with certain modifications: (a) there were now 2,000 tufted IB pyramidal cells, and 90 superficial and 100 deep neurogliaform interneurons were added. As before, there were 90 superficial and 100 deep low-threshold spiking (LTS) interneurons, along with 200 tufted regular spiking (RS) layer 5 pyramidal cells and 500 nontufted RS layer 6 pyramidal cells. Superficial and deep axoaxonic and basket cells, all fast-spiking, were present as before; (b) we did not include gap junctions between principal neurons (pyramidal + stellate) and ectopic spikes in these cells were suppressed, in accord with the experimental absence of observed spikelets in auditory cortical principal cells during cholinergically induced beta2; (c) the density of dendritic gap junctions between LTS interneurons was high, with mean index 5.67 for superficial LTS cells and 8 for deep LTS cells. Gap junction conductances were 3 and 4 nS, respectively; (d) the density of synaptic connections between deep LTS interneurons and tufted IB cells was increased, so that each of the latter received input from 60 deep LTS cells (and 20 superficial LTS cells, as before). Unitary LTS IPSCs in pyramidal cells decayed with time constant 30 ms, as before, but now had peak conductances of 0.2 nS (superficial) and 0.5 nS (deep); (e) the density of transient (A-type) K+ conductance on the axons of IB pyramids was made high (420 mS/cm2), in order to limit axonal bursting; (f) M-type and AHP-type K+ conductances were reduced in tufted IB cells, in accordance with presumed effects of carbachol, and high-threshold gCa was also reduced; (g) tufted IB and RS pyramidal cells were depolarized with injected currents, again according to presumed effects of carbachol.
Results
Cholinergic Neuromodulation Generates Oscillations in Primary Auditory Cortex, but not Association Cortex
Glutamatergic excitation of primary auditory cortex (Au1) generates a population oscillation with a single modal spectral peak at gamma frequencies in layers II/III and coexistent layer II/III gamma and layer V beta2 rhythms in adjacent association cortex (par2) (Figure 1 A, see also Cunningham et al., 2004 ; Roopun et al., 2006 ). In contrast, cholinergic neuromodulation (bath applied carbachol) generated coexistent gamma and beta2 rhythms in Au1 but failed to generate any population rhythms in par2 at concentrations up to 0.1 mM. Figure 1 A shows example power spectra from layer IV – allowing visualisation of the field potentials originating in adjacent superficial (layer III) and deep (layer V) layers concurrently for comparison. Robust beta2 oscillations were generated with peak power in layer V of Au1 (358 ± 82 μV2, n = 8) and mean frequency of 25 ± 2 Hz, a value not significantly different from the layer V beta2 rhythm generated by kainate in par2 (P > 0.1, n = 8). Cholinergic gamma oscillations had peak power in layer II/III of Au1 (796 ± 38 μV2, n = 8), with mean frequency of 39 ± 4 Hz (n = 8). While power values were the same for layer II/III gamma rhythms in par2 generated by kainate and Au1 generated by carbachol (P > 0.05, n = 8), there was a small but significant difference in frequency: par2 gamma (kainate) frequency was 43 ± 4 Hz (P < 0.05, n = 8).
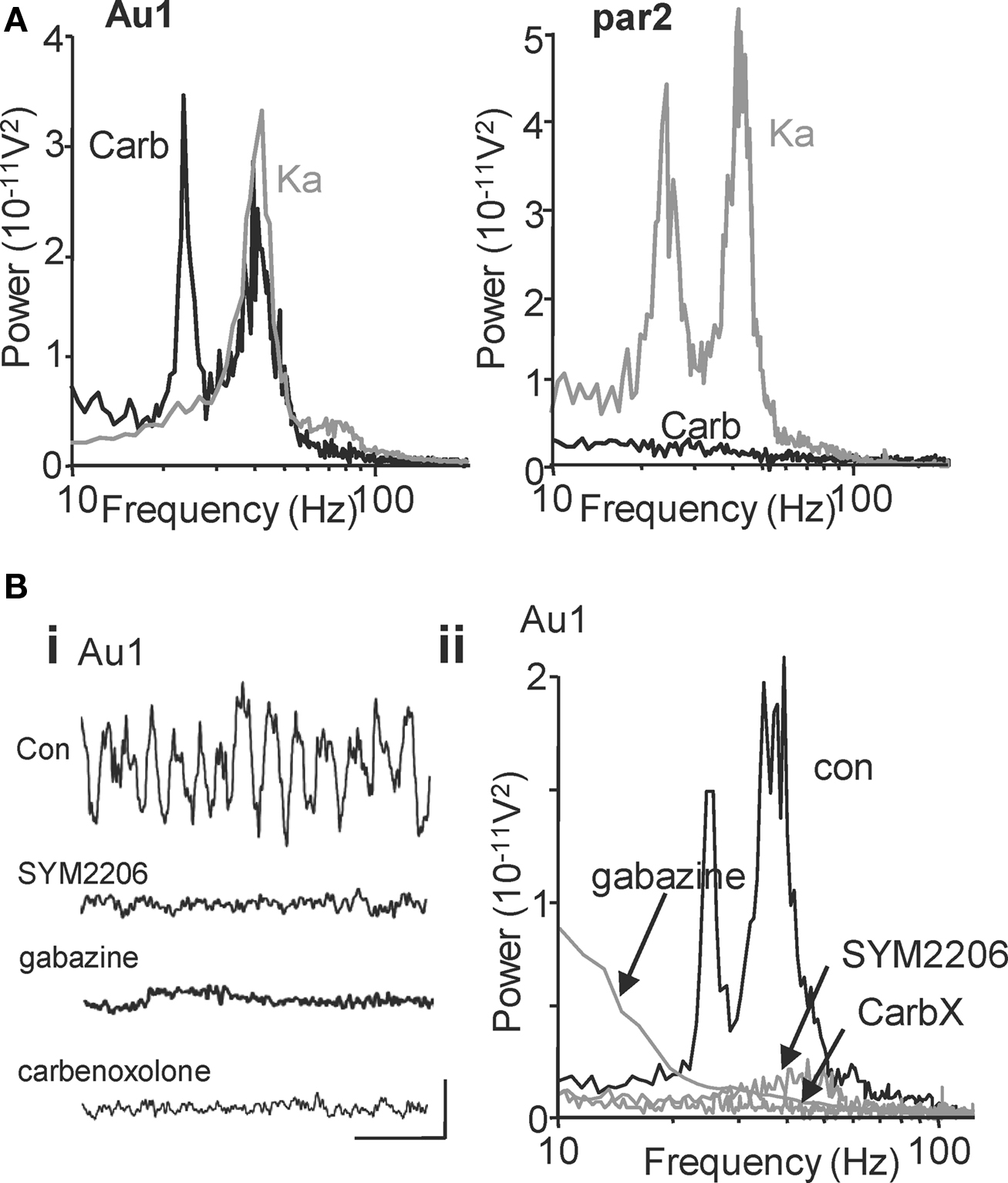
Figure 1. Cholinergic neuromodulation generates beta2 and gamma oscillations in primary auditory cortex: Spectral but not mechanistic similarity to glutamatergic rhythms in association cortex. (A) Power spectra of local field potential activity in layer IV of primary auditory cortex (Au1) induced by cholinergic neuromodulation (carb, black line) or glutamatergic excitation (Ka, grey line). Layer IV recordings reveal field potential activity originating in both adjacent superficial (layer III) and deep (layer V) layers and thus permit direct visual comparison (e.g. Roopun et al., 2006 ). Note both manipulations generate gamma rhythms but only cholinergic modulation induces a beta rhythm. In contrast, power spectra of local field potential activity in layer IV of association cortex (par2, right graph) show no rhythm generation by cholinergic modulation (carb, black line), but a spectrally similar pattern of gamma and beta2 rhythms induced by glutamatergic excitation (Ka, grey line). (B) Auditory cortical beta rhythms depend on excitatory and inhibitory synaptic activity in contrast to those seen in par2 (Roopun et al., 2006 ). (Bi) Five-hundred milliseconds traces showing extracellular activity in the presence of carbachol. Scale bars: 100 μV, 100 ms. (Bii) Power spectra of activity from layer IV in the 1° auditory cortex after carbachol (15 μM) application (black trace) and after 60 min of SYM2206 (25 μM) application (grey line, top graph), gabazine application (500 nM, grey line, middle graph) and carbenoxolone (100 μM, grey line bottom graph).
Also, in contrast to kainate-induced parietal cortex gamma and beta2 rhythms, separation of layers I–III from LV and LVI abolished the auditory cortex gamma and beta2 rhythms (gamma power before and after lesion: 4.2 × 10−11 V2 vs. 0.3 × 10−11 V2, P < 0.001; beta2 power before and after lesion: 3.6 × 10−11 V2 vs. 0.2 × 10−11 V2, P < 0.001, n = 5) indicating that neurons in LIV, or connections between neurons passing through LIV, were required for both frequencies in auditory cortex. Lesion at the level of layer 2 abolished gamma rhythms but preserved the layer V beta2 rhythm suggesting that the circuitry underlying gamma rhythm generation was not required for the beta2 rhythm generation (see also Figure 3 ).
Differential Pharmacology of Primary Auditory and Association Cortex Beta Oscillations Points to Fundamentally Different Underlying Mechanisms
In order to further profile possible differences in underlying mechanism for concurrent gamma/beta2 rhythms in primary sensory and association areas, we used pharmacological tools to examine the roles of AMPA- and GABAA receptor-mediated synaptic communication and the possible dependence of gap junction-mediated intercellular communication. Blockade of AMPA receptors with SYM2206 (25 μM) reduced mean carbachol-induced Au1 gamma power from 255 ± 45 to 60 ± 7 μV2 (n = 8, P < 0.05, Figure 1 Bi) but also removed the modal beta2 peak in spectra (202 ± 18 μV2 was reduced to 29 ± 8 μV2, n = 6, P < 0.05). Similarly, while kainate-induced beta rhythms in par2 were actually increased by GABAA receptor blockade, this frequency was abolished alongside the gamma rhythm in Au1 when induced by carbachol. Mean control power in the cholinergic, Au1 gamma band of 413 ± 63 μV2 was reduced to 95 ± 14 μV2 on application of gabazine (500 nM) (n = 6, P < 0.05, Figure 1 Bii). Mean control beta2 power of 156 ± 7 μV2 was reduced to 47 ± 12 μV2 (n = 6, P < 0.05). Both cholinergic Au1 gamma and beta2 rhythms were sensitive to the gap junction blocker carbenoxolone (Figure 1 Bii), as seen for kainate-induced par2 rhythms.
Primary Auditory Beta Rhythms are Generated by Local Circuits Containing Low-Threshold Spiking Interneurons
During cholinergic Au1 field beta2 rhythms LTS cells in cortical layers V and IV, fired at beta2 frequency, 24 ± 2 Hz, during carbachol-induced beta2 rhythms in Au1 (n = 4, Figure 2 A), often with brief spike bursts on each beta period (Figures 2 Aii,Aiii). In contrast, fast spiking interneurons in LIV/V (FS cells) fired much faster than the local field potential beta2 rhythm (at 59 ± 5Hz, n = 4, Figure 2 A).
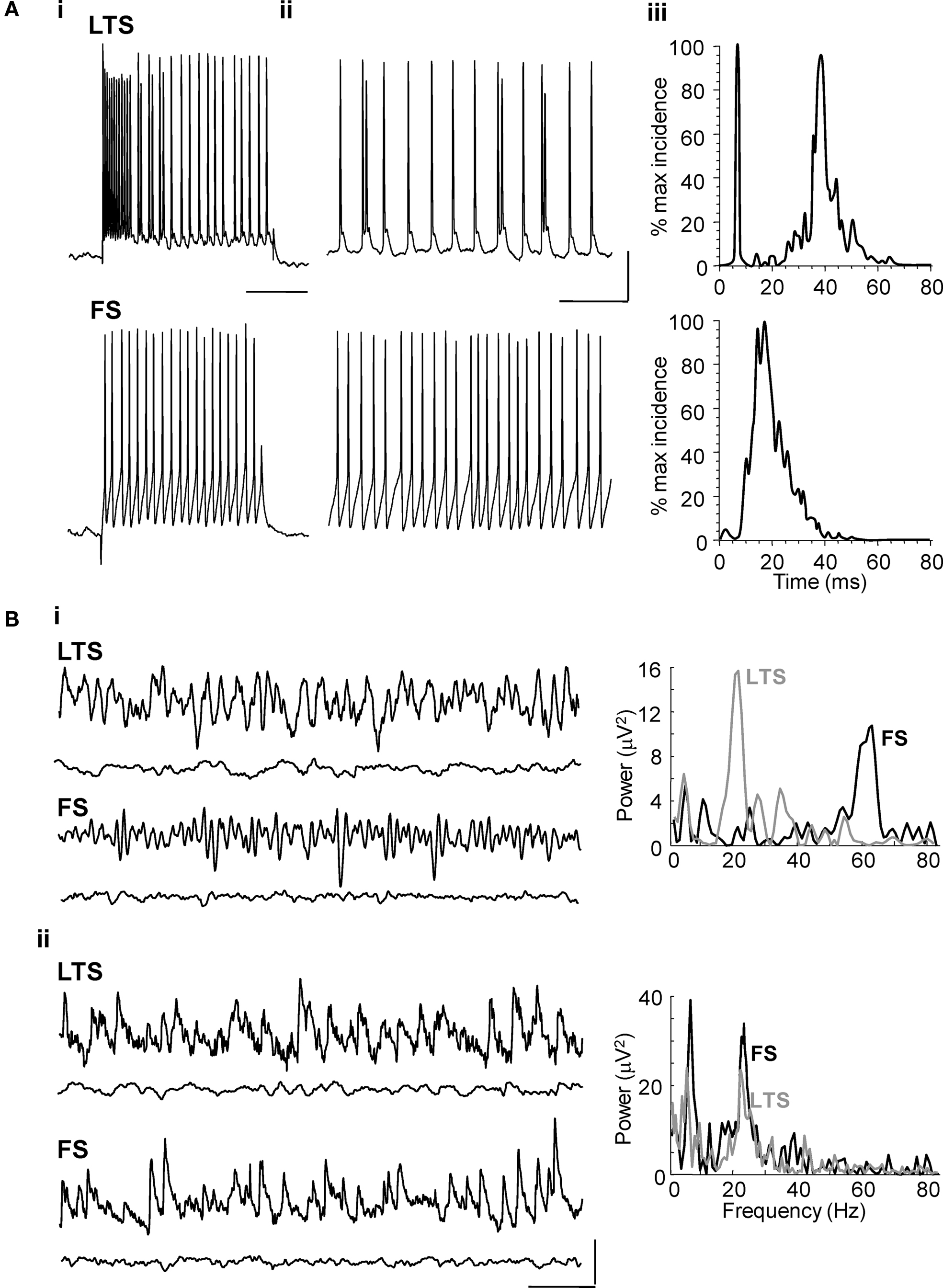
Figure 2. Layer V LTS interneuron – but not FS interneuron – outputs are at beta frequency during cholinergically driven Au1 rhythms. (A) Intracellular recordings from layer IV/V interneurons in the primary auditory cortex. (Ai) Current steps of 0.2 nA characterised the layer V cells as LTS or FS interneurons in experiment. (Aii) The firing rate of an LTS cell and an FS cell at resting membrane potential (RMP = −62 and −56 mV, respectively) during carbachol-induced oscillations. (Aiii) Corresponding spike-frequency histograms from 60 s epochs of activity in each cell type. Note only LTS cells generate outputs at beta frequency. Scale bar for current step: 10 mV, for on-going spike generation 15 mV, 100 ms. (B) Excitatory and inhibitory synaptic inputs to LTS and FS interneurons in layer V. (Bi) Example traces showing inhibitory postsynaptic potentials at −30 mV membrane potential in LTS and FS cells (upper traces) during cholinergic beta rhythms in Au1. Inhibitory inputs match, in frequency, the different outputs from these two cell types as shown by the inter-event time histograms on the right. Lower traces show behaviour of each cell type in the presence of gabazine (500 nM). (Bii) Example traces showing excitatory synaptic inputs to an LTS and FS interneuron during population beta rhythm (upper traces). Both neuron subtypes receive compound EPSPs at the population beta frequency. Lower traces show behaviour of each cell type in the presence of SYM2206 (25 μM). Scale bars 2 mV, 100 ms.
Inhibitory synaptic inputs to LTS and FS cells also showed a stark contrast. While LTS cells received IPSPs at a frequency (25 ± 1 Hz) not significantly different to the beta2 field potential oscillation (n = 3, P > 0.05, Figure 2 Bi), FS cells received smaller, IPSPs with faster kinetics at much higher frequencies (Figure 2 Bi). Both interneuron subtypes received trains of synaptic excitatory potentials at beta2 frequency (Figure 2 Bii) suggesting common drive from local excitatory principal cells (see below). Furthermore, targeting FS cell excitation selectively with the muscarinic antagonist atropine (100 nM) reduced gamma oscillation power but not beta2 oscillation power (Figure 3 A, P > 0.5 control vs. atropine for beta2 rhythm, P < 0.001 control vs. atropine for gamma rhythms, n = 5). In contrast, targeting LTS cells selectively with the nicotinic receptor antagonist d-Tubocurarine (10 μM) had the opposite effect – sparing gamma rhythms and reducing beta2 rhythms (Figure 3 B, P < 0.01 control vs. d-Tubocurarine for beta2 rhythms, P > 0.1 for gamma rhythms, n = 5). These data, along with the observations of beta2 frequency inputs and outputs in LTS cells, suggest this subclass of interneuron was involved in Au1 beta generation at the local circuit level.
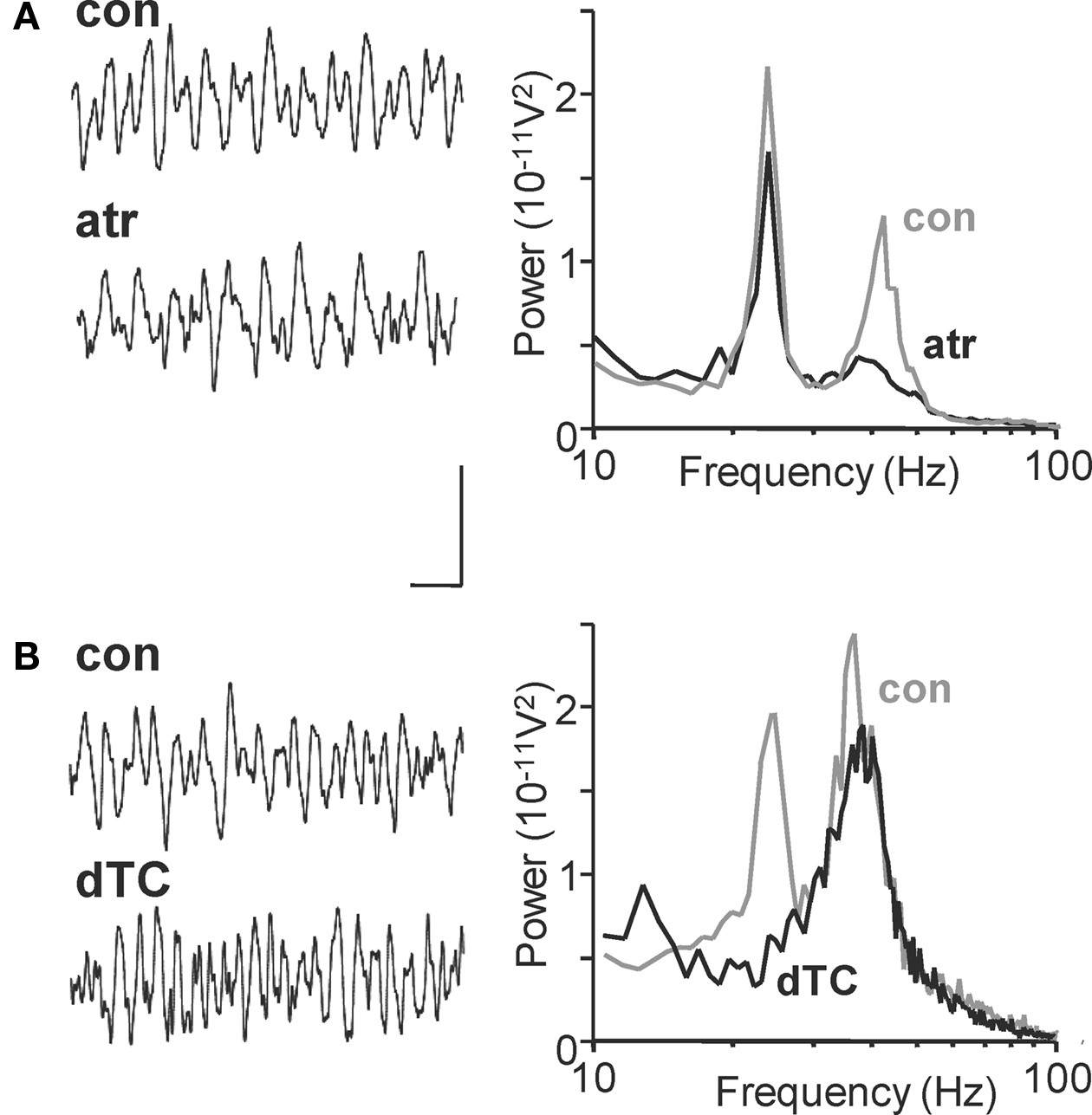
Figure 3. Nicotinic and muscarinic cholinoceptors differentiate between layer II/III gamma and layer 5 beta2 cholinergic rhythms in Au1. (A) Example traces of field potential activity from layer IV in the 1° auditory cortex after carbachol (15 μM) application (con) and subsequent atropine (100 nM) application (atr). Spectra on the right show pooled frequency content from n = 5 experiments showing selective blockade of gamma rhythms with muscarinic receptor blockade (black trace). (B) Corresponding 500 ms recordings of layer IV extracellular activity in the presence of carbachol (con) and after application of d-Tubocurarine (10 μM, dTC). Spectra show pooled activity from n = 5 60 s epochs of field recording illustrating the selective blockade of beta rhythms with nicotinic receptor antagonism. Scale bar for traces = 100 μV, 100 ms.
Cholinergic Beta Oscillations Accompany Strong Layer 5 Pyramidal Cell Outputs
In par2, kainate-induced beta2 oscillations are generated by intense beta2 frequency axonal spike bursts in a gap junctionally coupled plexus of axons of IB neurons in layer V. Very little output was seen in layer V RS cells in comparison (6.1 ± 3.9 Hz, P < 0.05 IB vs. RS spike rate). During carbachol-induced oscillations in Au1 IB cells fired at 20.1 ± 4.6 Hz (n = 4, Figure 4 A), a rate not significantly different from IB spiking in par2 with kainate drive (P > 0.1). However, the profile of spike generation, as recorded in cell somata, was different. No evidence for the rhythmic barrages of spikelets seen in par2 beta rhythms was seen for Au1 beta rhythms (Roopun et al., 2006 ). Layer V RS cells in Au1 also fired close to beta2 field potential frequency on average (19.2 ± 4.3 Hz), and both cell types revealed strong beta2 frequency IPSPs on depolarisation to −30 mV membrane potential (Figure 4 A). Kainate failed to generate beta frequency outputs in either cell type in layer V of Au1 (Figure 4 B), indicating that cholinergic neuromodulation was critical for the generation of outputs, patterned at beta frequencies, from primary auditory cortex.
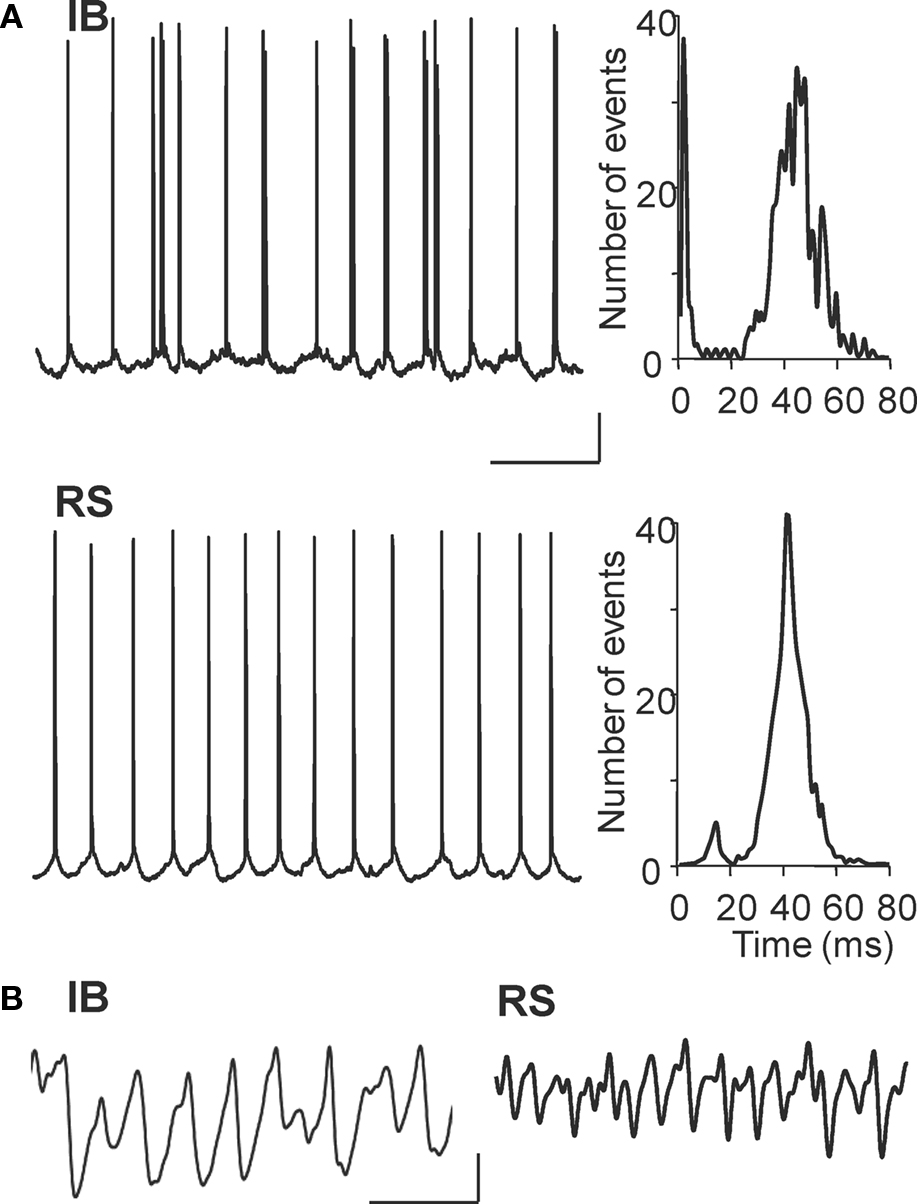
Figure 4. Cholinergic neuromodulation generates beta frequency outputs from layer V principal cells in Au1. (A) Example intracellular recordings from a layer V IB cell and RS cell at rmp during carbachol-induced beta2 oscillations in the 1° auditory cortex. Corresponding event histograms on the right show interspike intervals for both cells correspond to the population beta2 rhythm in experiment. (B) Example recordings from IB and RS cells at −30 mV membrane potential. Both cell types demonstrate prominent beta2 frequency inhibitory postsynaptic potentials, though the higher gamma frequency activity is also evident in RS cells. Scale bars 100 ms, 20 mV (A), 100 ms, 5 mV/5 nS (B).
A Local Circuit of IB and LTS Neurons is Necessary and Sufficient for Cholinergic Beta2 Rhythms
Experimental data above suggested a prominent role for predominantly IB principal cells exciting LTS interneurons, and receiving feedback inhibition from these neurons. However, the lack of specific tools allowing study of each proposed network component in isolation, without disturbing the rhythm itself, required testing this core circuit with biologically realistic computational models. Our network model was able to reproduce, and account for, each of the cellular firing patterns, synaptic inputs, and receptor and gap junction pharmacology observed during cholinergic beta2 rhythms in Au1 (Figure 5 , cf. Figures 1 , 2 , 4 ). Although the network model contained many cell types, its basic principles were straightforward. Superficial and layer 4 neurons (other than LTS interneurons) did not participate in simulated beta2 generation. FS interneurons fired significantly faster than the beta2 rhythm (as in experiments) and produced an essentially tonic inhibition in other cells. Deep RS pyramidal cells participated in simulated beta2 but were not essential to its generation (Figure 5 C).
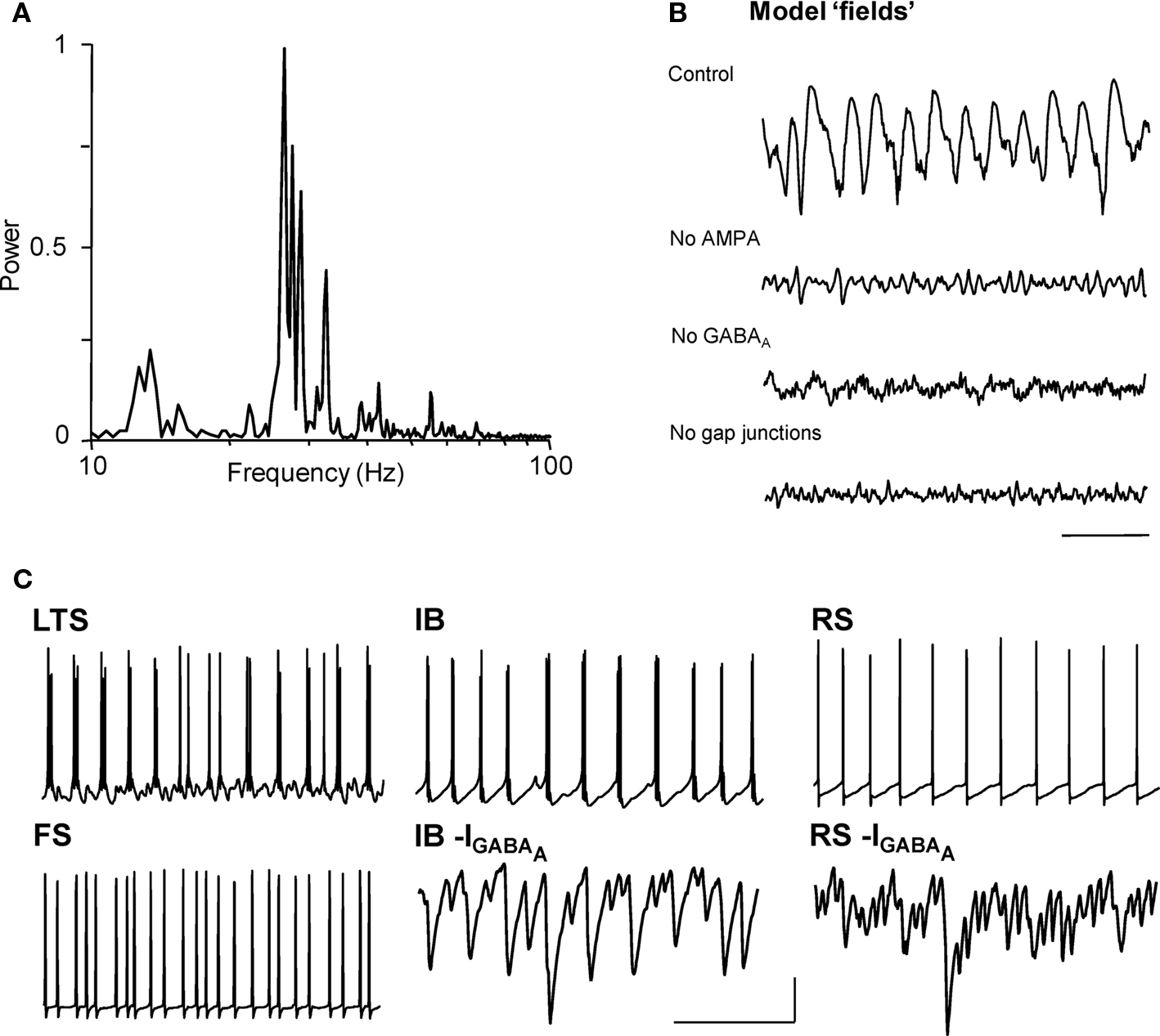
Figure 5. Model predicts synaptic connectivity between layer V pyramidal cells and gap junctionally coupled LTS cells is necessary and sufficient for cholinergic beta2 rhythm generation. (A) Model ‘field’ potential (mean synaptic activity) spectrum from excited, interconnected IB, RS, FS and LTS cell network shows a prominent beta2 frequency peak. (B) The pharmacology of model beta2 rhythms is the same as that in experiment (cf. Figure 1 ). Scale bar 100 ms. Note gap junctions are only present in the model between LTS interneurons. (C) The model reproduces the pattern of pyramidal cell inhibitory inputs (GABAA receptor-mediated postsynaptic currents, shown inverted) and spike outputs faithfully and demonstrates that only LTS interneurons, and not FS interneurons, generate a beta2 frequency output. Scale bars 20 mV (somatic membrane potential traces), 5 nS (synaptic currents), 100 ms.
The model predicted that the key circuit components underlying the synaptic nature of the rhythm were the layer 5 IB pyramids, and deep and superficial LTS interneurons. Connectivity between these cell types is strong (Silberberg and Markram, 2007 ), with IB cells synaptically exciting LTS interneurons (hence the need for AMPA receptors, Figure 5 B), and LTS reciprocally inhibiting IB cells (and each other) with IPSCs having slow kinetics that were tuned to the beta2 frequency – thus the need for GABAA receptors. While the precise nature of each cell’s intrinsic properties were not essential for beta2 generation, these synaptic components certainly were.
The model did not use gap junctions between pyramidal cells (unlike our model of nonsynaptic beta2 in association cortex; Roopun et al., 2006 ), and indeed we found that activity in the pyramidal cell axonal plexus was disruptive of simulated cholinergic beta2 (not shown). However, electrical coupling between LTS interneurons – precedented in cortical circuits with relevance to rhythm generation (e.g. Gibson et al., 1999 ; Beierlein et al., 2000 ) – was essential for stability of the rhythm, and removing them led to the loss of field oscillations (Figure 5 B), even though individual neurons continued to fire.
The Cholinergic, Auditory Beta Rhythm is Essential for Intercommunication with Association Cortex
In order to address the consequences of selective generation of beta2 rhythms in Au1 with cholinergic neuromodulation we recorded concurrently from layer IV of both par2 and Au1 in conditions where both regions generated gamma rhythms but only par2 generated beta2 rhythms (glutamate excitation via kainate), and conditions where both regions generated gamma and beta2 rhythms (kainate and carbachol together). In the absence of cholinergic neuromodulation auditory field potentials only weakly correlated with field potentials in par2 (Figure 6 A). Peak correlation was seen with a lag between association and primary sensory cortex of approximately one gamma period (21 ± 3 ms, n = 4). Granger causality estimates – in this case the extent to which previous behaviour in the population activity of one cortical area influences the present population activity in the other – showed only very weak influence of auditory cortex on association cortex at all frequencies from 1 to 100 Hz (Figure 6 B). However, in the reverse direction, a causal relationship was suggested between par2 population activity and that in Au1 at gamma frequencies (G-score 0.15 ± 0.04, n = 4).
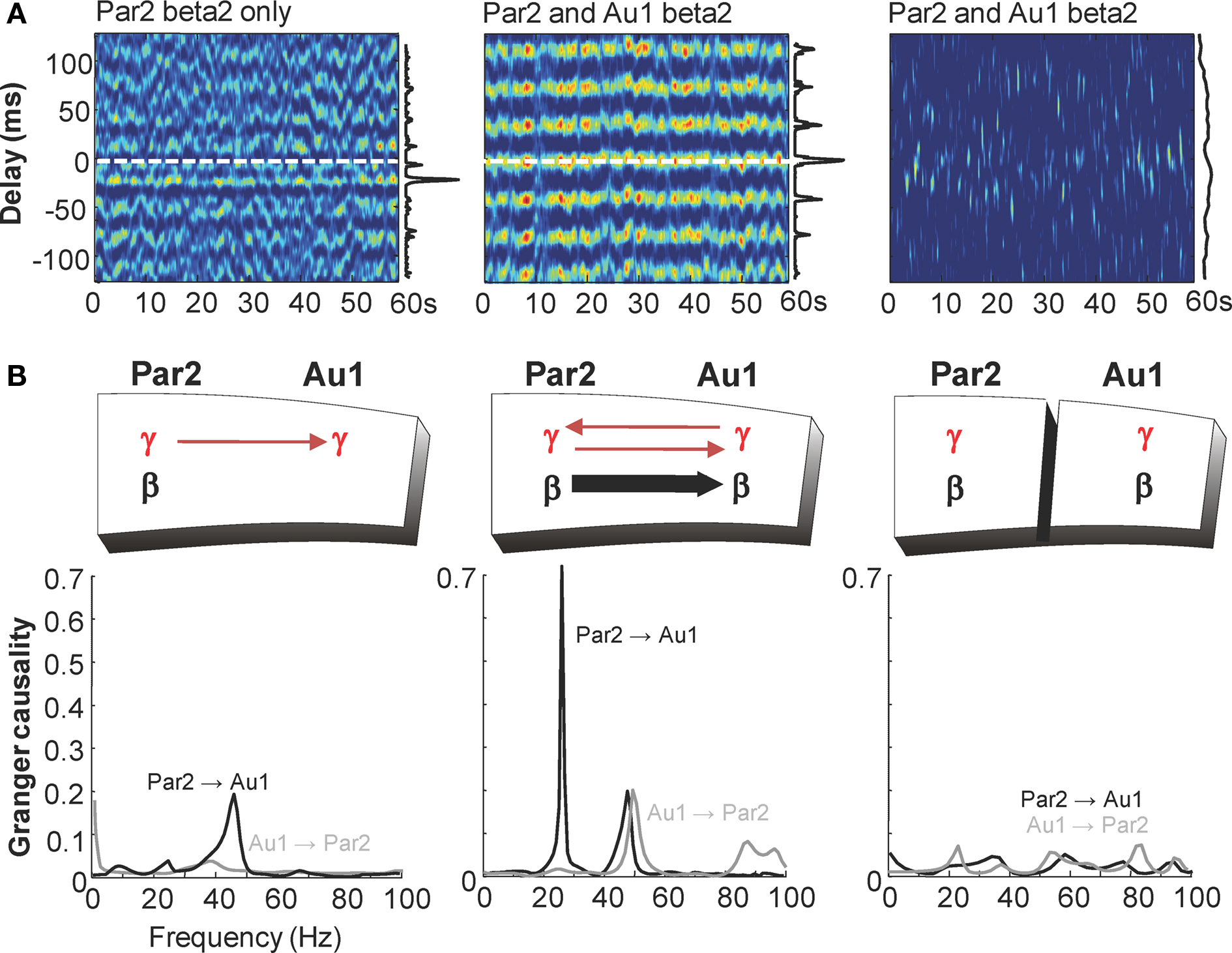
Figure 6. Cholinergic neuromodulation is essential for Au1-par2 coherence at beta2 frequencies. (A) Windowed cross correlograms of 60 s epochs of field potential recordings from layer 5 of association cortex (par2) and primary auditory cortex (Au1) in conditions where beta2 rhythms are generated only in par2 (kainate bath application), and present in both par2 and Au1 (kainate and carbachol bath application) in intact slice and with full-thickness cut between par2 and Au1. Vertical graphs along side show pooled histograms of phase relationship measured as the position of the peak in each cross correlation window. Note the peak phase relationship corresponds to gamma frequencies with par2 beta alone and beta frequency when both areas generate this rhythm. Separation of areas abolishes phase relationships (B). Granger causality estimates for concurrent field potentials in LIV par2 and Au1 in conditions where both areas generate gamma rhythms but only par2 generates beta rhythms (kainate bath application). The lack of influence of Au1 on par2 (grey line) contrasts with influence of par2 on Au1 at gamma frequencies (black line). Middle graph shows Granger causality estimates for concurrent field potentials in LIV par2 and Au1 in conditions where Au1 and par2 generate both beta2 and gamma rhythms (kainate and carbachol bath application). Interactions at gamma frequencies become bidirectional and par2 now Granger-causes beta2 rhythms in Au1 (black line). These directed interactions are absent when par2 and Au1 are physically separated (right graph).
In the additional presence of cholinergic neuromodulation, the predominant correlation between field potentials in the two areas was at beta2 frequency, with near-zero millisecond synchrony (par2 lead, 0.5 ± 1 ms, n = 4, Figure 6 A). The presence of carbachol had a striking influence on Granger causality estimates (Figure 6 B). Interactions in the gamma frequency range became reciprocal (G-score par2-Au1 0.16 ± 0.04, Au1-par2 0.21 ± 0.04) and a large increase in causality at beta2 frequency was seen when considering the influence of par2 on Au1 (beta2 G-score kainate only 0.03 ± 0.01, kainate and carbachol 0.65 ± 0.10, n = 4, P < 0.01). This interaction was highly directional, with negligible causality seen at beta2 frequencies from Au1 to par2. Physical separation of par2 from Au1 preserved each area’s rhythms but abolished directed coherence estimates between regions, suggesting functional connectivity between par2 and Au1 was essential for generating this pattern of temporal interaction.
Discussion
These data demonstrate three basic phenomena associated with intercortical communication at beta frequencies using in vitro and in silico models:
(1) Despite spectral similarity, beta rhythms are induced in primary sensory and association areas by distinct modes of excitation: Primary sensory beta rhythms required cholinergic neuromodulation, whereas associational cortical beta rhythms required tonic glutamatergic excitation.
(2) The local circuit mechanisms underlying primary sensory and association cortical beta rhythms were markedly different: Primary sensory beta rhythms were generated predominantly by chemical synaptic interactions between layer V principal cells and LTS interneurons. Computational models predicted that fast synaptic excitation and inhibition between these elements of the local circuit were vital to generate cholinergic, primary sensory beta2 rhythms. In contrast, association cortical beta rhythms were almost completely inert to chemical synaptic activity and involved gap junctional communication specifically between IB neurons (Roopun et al., 2006 ). While cholinergic beta was also sensitive to gap junctions the computational model predicted a role for LTS–LTS gap junctions and not those between intrinsically bursting neurons.
(3) In the presence of cholinergic neuromodulation, these different mechanisms of beta generation suggest directed temporal communication in which ‘top-down’ (i.e. from higher to lower order sensory cortices) interactions may predominate.
Cholinergic activity is closely associated with cortical arousal (Jasper and Tessier, 1971 ). It is associated with enhanced sensory processing in the cortex (Gil et al., 1997 ), enhanced top-down interactions (Robbins, 2005 ), and is proposed to influence discrete functional networks underlying conscious awareness and attention (Perry et al., 1999 ; Kimura, 2000 ). While the two main subtypes of cholinoceptor – nicotinic and muscarinic – may work in tandem to enhance sensory responses (McCormick and Prince, 1985 ; Gil et al., 1997 ), there is evidence for selectivity of effects in some instances. For example, the activation of nicotinic receptors facilitates afferent inputs to the cortex, whereas the activation of muscarinic receptors is important for enhancing intra-laminar connections (Kimura, 2000 ). Muscarinic receptor activation is associated with gamma frequency neocortical rhythms (Rodriguez et al., 2004 ; Langmead et al., 2008 ). In contrast, enhanced cortical activation by nicotine is associated with increased visual attention and working memory (Ernst et al., 2001 ; Levin, 2002 ), and an increase in beta rhythm generation (Ferger and Kuschinsky, 1994 ).
This apparently differential involvement of nicotinic and muscarinic receptors in beta and gamma rhythms, respectively, was also seen in the models used in the present study (Figure 3 ) and was used to probe the mechanism underlying cholinergically induced beta rhythms in auditory cortex. Blockade of GABAA receptors abolished the auditory beta rhythm, demonstrating an absolute dependence on synaptic inhibition. However, unlike gamma rhythms, fast spiking interneurons in beta-generating deep layers did not spike at the population frequency, and the kinetics of IPSPs onto principal cells were slower than those associated with fast spiking interneurons during the beta rhythm. Instead, the major inhibitory neuron involved in local circuits generating beta appeared to be the LTS cell. These cells generated strong outputs at the population beta frequency and blockade of their nicotinic receptor-mediated excitation (Xiang et al., 1997 ) selectively reduced beta rhythms in the local field potential in the present study. LTS cells are strongly, reciprocally connected to RS cells (Goldberg et al., 2004 ), and to IB cells in the cortex (Beierlein et al., 2003 ), and both principal cell subtypes in layer 5 were strongly activated by cholinergic receptor activation with carbachol. While the output produced by this excitation may be critical for shaping cortical interactions (see below) it may also be vital for generating the local auditory cortical beta rhythm in the first instance: RS cells in particular must maintain a high firing rate to activate LTS cells as they are preferentially responsive to sustained, rather than transient excitation (Beierlein et al., 2003 ).
Each of these phenomena were reproduced in our biologically realistic model. Manipulating model parameters clearly demonstrated that reciprocal interactions between layer 5 IB cells and LTS cells formed the core local circuit underlying auditory cholinergic beta rhythms. This is in stark contrast to the mechanism underlying the glutamatergic beta rhythm in association cortex. Here, the rhythm is resistant to blockade of fast glutamatergic synaptic excitation and almost completely inert to GABAA receptor blockade. In addition, out of all the principal neuron subtypes tested, only IB cells in deep layers were active at beta frequencies during the population beta rhythms generated by kainate.
As the rhythms have both the same frequency and both come from deep cortical layers the question needs to be asked: why have two fundamentally different generatory mechanisms centred around the same subtype of layer 5 principal cell? This paper reveals two possible advantages to having multiple, region-specific modes of beta rhythm generation: First, within the framework of communication through coherence (Fries, 2005 ) different relative amounts of cholinergic neuromodulation and cortical neuronal activity (glutamatergic drive) can result in different spatial patterns of beta rhythm generation and thus temporal interaction within this frequency band. For example, within the highly reduced cortical model used here (only association and primary auditory cortex), cholinergic or glutamatergic drive alone generated only local, within-region beta rhythms and no temporal interactions between regions at this frequency. A combination of these drives caused co-activation of the two regions and synchrony at beta frequencies occurred.
In addition, the differential involvement of chemical synaptic communication in beta generation in the two areas corresponded with analysis suggesting highly directional interareal interactions. Granger causality analysis needs to be interpreted with the caveat that it is just an estimate, and an estimate of an estimate (the AR model of the real data) at that. However, the magnitude of the directional effect was striking: Despite both areas generating the same frequency, only the associational beta rhythm has a causal relationship with the auditory beta rhythm and not vice versa. Possible mechanisms underlying this directionality include: (1) The relative insensitivity of the associational cortical beta rhythm to synaptic input. Pairing two spectrally similar rhythms, one exquisitely sensitive to glutamatergic and GABAergic synaptic inputs and one not, provides a novel means to exert the influence – via horizontal synaptic connections – of activity in one local circuit over another in a highly unidirectional fashion. (2) The existence of a small but robust phase difference between primary auditory and association cortex (with Au1 lagging behind par2) suggests a period-by-period phase reset of the synaptic auditory beta2 rhythm by the non-synaptic par2 beta2 rhythm. 3) Anatomical studies show that inputs to Au1 from other cortical areas arise predominantly from deep layers (those that generate beta2 rhythms) whereas outputs from Au1 to other cortical areas originate predominantly from superficial (non-beta2-generating) layers (Rouiller et al., 1991 ).
These observations suggest an extension to the ‘communication through coherence’ framework: Temporal interactions between cortical regions at a single modal frequency could facilitate neuronal assembly formation (the binding together of firing patterns in discrete subsets of neurons; Singer and Gray, 1995 ; Fries, 2005 ) via reciprocal interactions (Traub et al., 1996 ; Kopell et al., 2000 ). However, if the local mechanisms of generation of a single frequency in two regions are different – specifically, if they have different dependence on synaptic events associated with cortico-cortical communication – then their interactions are very much non-reciprocal. Despite the establishment of synchrony, coherence between regions is highly directed as suggested by analysis of the present data.
This region specificity and directionality of cortical interaction may be utilised by the brain in transitions from sleeping to the awake state. Levels of glutamate and acetylcholine change reciprocally in these two states, with acetylcholine highest during the awake state and glutamate highest during sleep (Miller and O’Callaghan, 2006 ; Lopez-Rodriguez, 2007 ). Extrapolating from these observations, the present data suggest that during sleep activity in deep cortical layers at beta frequency would be confined to higher order cortices, with no functional interaction between these areas and deep layers of primary sensory cortices. Only during wakefulness would temporal interactions between primary and higher order cortices be facilitated. TMS/EEG studies support this by demonstrating that cortical stimulation during quiet wakefulness induces oscillatory activity that propagates throughout the brain, whereas during sleep TMS responses are highly localised to the stimulation site (Massimini et al., 2005 ). The data presented here arise from the manipulability and accessibility afforded by in vitro and in silico models of the basic building blocks of spatiotemporal dynamics in neocortex. Further work is needed to determine how the mechanisms uncovered at this reductionist level may apply to information processing in the far richer dynamic environment of the intact brain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Medical Research Council (UK), the Wolfson Foundation, IBM, Alexander von Humboldt Stifftung and NIH/NINDS (NS044133 to R. D. Traub) for financial support, and Prof N. Kopell for valuable discussion of the manuscript. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke, or of the National Institutes of Health.
References
Baker, S. N. (2007). Oscillatory interactions between sensorimotor cortex and the periphery. Curr. Opin. Neurobiol. 17, 649–655.
Beierlein, M., Gibson, J. R., and Connors, B. W. (2000). A network of electrically coupled interneurons drives synchronised inhibition in neocortex. Nat. Neurosci. 3, 904–910.
Beierlein, M., Gibson, J. R., and Connors, B. W. (2003). Two dynamically distinct inhibitory networks in layer 4 of the neocortex. J. Neurophysiol. 90, 2987–3000.
Cui, J., Xu, L., Bressler, S. L., Ding, M., and Liang, H. (2008). BSMART: a matlab/C toolbox for analysis of multichannel neural timeseries. Neural. Netw. 21, 1094–1104.
Cunningham, M. O., Whittington, M. A., Bibbig, A., Roopun, A. K., LeBeau, F. E. N., Vogt, M., Monyer, H., Buhl, E. H., and Traub, R. D. (2004). A role for fast rhythmic bursting neurons in cortical gamma oscillations in vitro. Proc. Natl. Acad. Sci. U.S.A. 101, 7152–7157.
Donner, T. H., Siegel, M., Oostenveldt, R., Fries, P., Bauer, M., and Engel, A. K. (2007). Population activity in the human dorsal pathway predicts the accuracy of visual motion detection. J. Neurophysiol. 98, 345–359.
Ernst, M., Heishman, S. J., Spurgeon, L., and London, E. D. (2001). Smoking history and nicotine effects on cognitive performance. Neuropsychopharmacology 25, 313–319.
Fairhall, S. L., Kirk, I. J., and Hamm, J. P. (2007). Volition and the idle cortex: beta oscillatory activity preceding planned and spontaneous movement. Conscious. Cogn. 16, 221–228.
Ferger, B., and Kuschinsky, K. (1994). Activation of dopamine D1 receptors or alpha1 adrenoceptors is not involved in the effect of nicotine in the rat. Naunyn Schmiedebergs Arch. Pharmacol. 350, 346–351.
Fries, P. (2005). A mechanism for cognitive dynamics: neuronal communication through neuronal coherence. Trends Cogn. Sci. 9, 474–480.
Gibson, J. R., Beierlein, M., and Connors, B. W. (1999). Two networks of electrically coupled inhibitory neurons in neocortex. Nature 402, 75–79.
Gil, Z., Connors, B. W., and Amitai, Y. (1997). Differential regulation of neocortical synapses by neuromodulators and activity. Neuron 19, 679–686.
Goldberg, J. H., Lacefield, C. O., and Yuste, R. (2004). Global dendritic calcium spikes in mouse layer 5 low threshold spiking interneurones: implications for control of pyramidal cell bursting. J. Physiol. 558, 465–478.
Jasper, H. H., and Tessier, J. (1971). Acetylcholine liberation from cerebral cortex during paradoxical (REM) sleep. Science 172, 601–603.
Kimura, F. (2000). Cholinergic modulation of cortical function: a hypothetical role in shifting the dynamics in cortical network. Neurosci. Res. 38, 19–26.
Kopell, N., Ermentrout, G. B., Whittington, M. A., and Traub, R. D. (2000). Gamma rhythms and beta rhythms have different synchronization properties. Proc. Natl. Acad. Sci. U.S.A. 97, 1867–1872.
Langmead, C. J., Austin, N. E., Branch, C. L., Brown, J. T., Buchanan, K. A., Davies, C. H., Forbes, I. T., Fry, V. A., Hagan, J. J., Herdon, H. J., Jones, G. A., Jeggo, R., Kew, J. N., Mazzali, A., Melarange, R., Patel, N., Pardoe, J., Randall, A. D., Roberts, C., Roopun, A., Starr, K. R., Teriakidis, A., Wood, M. D., Whittington, M., Wu, Z., and Watson, J. (2008). Characterization of a CNS penetrant, selective M1 muscarinic receptor agonist, 77-LH-28-1. Br. J. Pharmacol. 154, 1104–1115.
Lopez-Rodriguez, F. (2007). Changes in extracellular glutamate levels in rat orbitofrontal cortex during sleep and wakefulness. Arch. Med. Res. 38, 52–55.
MacKay, W. A., and Mendonça, A. J. (1995). Field potential oscillatory bursts in parietal cortex before and during reach. Brain Res. 704, 67–74.
Massimini, M., Ferrarelli, F., Huber, R., Esser, S. K., Shigh, H., and Tononi, G. (2005). Breakdown of cortical effective connectivity during sleep. Science 30, 2228–2232.
McCormick, D. A., and Prince, D. A. (1985). Two types of muscarinic response to acetylcholine in mammalian cortical neurons. Proc. Natl. Acad. Sci. U.S.A. 82, 6344–6348.
Mesulam, M. M. (1995). Cholinergic pathways and the ascending reticular activating system of the human brain. Ann. N. Y. Acad. Sci. 757, 69–79.
Miller, D. B., and O’Callaghan, J. P. (2006). The pharmacology of sleep. Metab. Clin. Exp. 55, S13–S19.
Muir, J. L., Dunnett, S. B., Robbins, T. W., and Everitt, B. J. (1992). Attentional functions of the forebrain cholinergic systems: effects of intraventricular hemicholinium, physostigmine, basal forebrain lesions and intracortical grafts on a multiple-choice serial reaction time task. Exp. Brain Res. 89, 611–622.
Perry, E., Walker, M., Grace, J., and Perry, R. (1999). Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends Neurosci. 22, 273–280.
Robbins, T. W. (2005). Chemistry of the mind: neurochemical modulation of prefrontal cortical function. J. Comp. Neurol. 493, 140–146.
Rodriguez, R., Kallenbach, U., Singer, W., and Munk, M. H. (2004). Short- and long-term effects of cholinergic modulation on gamma oscillations and response synchronization in the visual cortex. J. Neurosci. 24, 10369–10378.
Roelfsema, P. R., Engel, A. K., König, P., and Singer, W. (1997). Visuomotor integration is associated with zero time-lag synchronization among cortical areas. Nature 385, 157–161.
Roopun, A. K., Kramer, M. A., Carracedo, L. M., Kaiser, M., Davies, C. H., Traub, R. D., Kopell, N. J., and Whittington, M. A. (2008). Period concatenation underlies interactions between gamma and beta rhythms in neocortex. Front. Cell. Neurosci. 2:1. doi: 10.3389/neuro.03.001.2008.
Roopun, A. K., Middleton, S. J., Cunningham, M. O., LeBeau, F. E. N., Bibbig, A., Whittington, M. A., and Traub, R. D. (2006). A beta2-frequency (20–30 Hz) oscillation in nonsynaptic networks of somatosensory cortex. Proc. Natl. Acad. Sci. U.S.A. 103, 15646–15650.
Rouiller, E. M., Simm, G. M., Villa, A. E., de Ribaupierre, Y., and de Ribaupierre F. (1991). Auditory corticocortical interconnections in the cat: evidence for parallel and hierarchical arrangement of the auditory cortical areas. Exp. Brain Res. 86, 483–505.
Sarter, M., and Bruno, J. P. (1997). Cognitive functions of cortical acetylcholine: toward a unifying hypothesis. Brain Res. Brain Res. Rev. 23, 28–46.
Silberberg, G., and Markram, H. (2007). Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron 53, 735–746.
Singer, W., and Gray, C. M. (1995). Visual feature integration and the temporal correlation hypothesis. Annu. Rev. Neurosci. 18, 555–586.
Steriade, M. (2004). Acetylcholine systems and rhythmic activities during the waking – sleep cycle. Prog. Brain Res. 145, 179–196.
Tallon-Baudry, C., Bertrand, O., Peronnet, F., and Pernier, J. (1998). Induced gamma-band activity during the delay of a visual short-term memory task in humans. J. Neurosci. 18, 4244–4254.
Tiitinen, H., Sinkonnen, J., Reinikainen, K., Alho, K., Lavikainen, J., and Naatanen, R. (1993). Selective attention enhances the auditory 40-Hz transient response in humans. Nature 364, 59–60.
Traub, R. D., Contreras, D., Cunningham, M. O., Murray, H., LeBeau, F. E. N., Roopun, A. K., Bibbig, A., Wilent, W. B., Higley, M. J., and Whittington, M. A. (2005). Single-column thalamocortical network model exhibiting gamma oscillations, sleep spindles and epileptogenic bursts. J. Neurophysiol. 93, 2194–2232.
Traub, R. D., Whittington, M. A., Stanford, I. M., and Jefferys, J. G. (1996). A mechanism for generation of long-range synchronous fast oscillations in the cortex. Nature 383, 621–624.
von Stein, A., Rappelsberger, P., Sarnthein, J., and Petsche, H. (1999). Synchronization between temporal and parietal cortex during multimodal object processing in man. Cereb. Cortex 9, 137–150.
Womelsdorf, T., Schoffelen, J. M., Oosterveld, R., Singer, W., Desimone, R., Engel, A. K., and Fries, P. (2007). Modulation of neuronal interactions through neuronal synchronization. Science 316, 1609–1612.
Keywords: EEG, oscillation, interneuron, coherence, attention, auditory
Citation: Roopun AK, LeBeau FEN, Rammell J, Cunningham MO, Traub RD and Whittington MA (2010) Cholinergic neuromodulation controls directed temporal communication in neocortex in vitro. Front. Neural Circuits 4:8. doi: 10.3389/fncir.2010.00008
Received: 07 January 2010;
Paper pending published: 12 February 2010;
Accepted: 10 March 2010;
Published online: 22 March 2010
Edited by:
Erin M. Schuman, Caltech/Howard Hughes Medical Institute, USAReviewed by:
Jozsef L. Csicsvari, University of Oxford, UKJohn A. Dani, Baylor College of Medicine, USA
Copyright: © 2010 Roopun, LeBeau, Rammell, Cunningham, Traub and Whittington. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Miles A. Whittington, Institute of Neuroscience, The Medical School, Newcastle University, Framlington Place, Newcastle NE2 4HH, UK. e-mail:bS5hLndoaXR0aW5ndG9uQG5jbC5hYy51aw==