 Thomas M. Morse
1*
Thomas M. Morse
1* Nicholas T. Carnevale
1
Nicholas T. Carnevale
1 Pradeep G. Mutalik
2
Pradeep G. Mutalik
2 Michele Migliore
1,3
Michele Migliore
1,3 Gordon M. Shepherd
1
Gordon M. Shepherd
1
- 1 Department of Neurobiology, Yale University School of Medicine, New Haven, CT, USA
- 2 Center for Medical Informatics, Yale University School of Medicine, New Haven, CT, USA
- 3 Institute of Biophysics, National Research Council, Palermo, Italy
The integrative properties of cortical pyramidal dendrites are essential to the neural basis of cognitive function, but the impact of amyloid beta protein (aβ) on these properties in early Alzheimer’s is poorly understood. In animal models, electrophysiological studies of proximal dendrites have shown that aβ induces hyperexcitability by blocking A-type K+ currents (IA), disrupting signal integration. The present study uses a computational approach to analyze the hyperexcitability induced in distal dendrites beyond the experimental recording sites. The results show that back-propagating action potentials in the dendrites induce hyperexcitability and excessive calcium concentrations not only in the main apical trunk of pyramidal cell dendrites, but also in their oblique dendrites. Evidence is provided that these thin branches are particularly sensitive to local reductions in IA. The results suggest the hypothesis that the oblique branches may be most vulnerable to disruptions of IA by early exposure to aβ, and point the way to further experimental analysis of these actions as factors in the neural basis of the early decline of cognitive function in Alzheimer’s.
Introduction
Interest in the pathogenesis of Alzheimer’s disease is mainly focused on molecular and biochemical changes underlying degeneration in the cell body and dendrites of cortical pyramidal cells (reviewed in Anderton et al., 1998 ; Lue et al., 1999 ). Relatively little attention has been given to the dendrites with regard to disruptions of intrinsic electrical properties related to their information processing mechanisms (reviewed in Byrne and Shepherd, 2009 ), yet these are recognized to be essential to the neural basis of cognitive function.
Critical experimental evidence for a negative impact on these mechanisms in animal models of Alzheimer’s has been obtained in dual patch recordings in rat hippocampal tissue slices, in which aβ blocked A-type K+ channels (IA) in pyramidal cell dendrites, causing an increase in dendritic membrane excitability as shown by enhanced back-propagating action potentials (bAPs) (Chen, 2005 ). This aβ block of IA has been confirmed in dissociated (Xu et al., 1998 ) and cultured (Zhang and Yang, 2006 ) hippocampal CA1 neurons and in cholinergic basal forebrain neurons (Jhamandas et al., 2001 ) and neocortical neurons (Ye et al., 2003 ). bAPs are believed to be critical for information processing and memory storage by cortical pyramidal neurons and their cortical microcircuits (Gasparini et al., 2007 ; Stuart et al., 2008 ). Disruption of bAPs can therefore be a sensitive indicator of negative effects of aβ on signal processing involved in cortical function.
These effects on bAPs are likely to occur mainly in the distal branches of the apical and possibly basal branches, where most of the integrative activity of the neuron occurs, which poses a difficult challenge to electrophysiological analysis. We have therefore utilized a realistic computational model of the hippocampal pyramidal neuron to analyze dendritic bAPs under K+ channel block in closer detail. The simulation results go beyond the experimental findings, providing evidence that the fine oblique branches of the apical dendritic tree are especially vulnerable to the increased excitability and disruption of the bAP due to the K+ channel block by aβ. This suggests that disruption of dendritic signaling in otherwise normal cells may underlie the earliest cognitive impairments in AD.
How might these changes in dendritic signaling be related to the subsequent degenerative changes in AD? The calcium hypothesis proposes that long-term changes in Ca2+ signaling lead to the degenerative changes (Khachaturian, 1987 ; see review in Stutzmann, 2007 ). In dual patch recordings from soma and apical dendrite (240–300 μm), Chen (2005) found that aβ exposure enhanced backpropagation. He suggested that the raised excitability of the cell increases Ca2+ influx which can lead to excitotoxicity and degenerative changes (Good and Murphy, 1996 ). This left unexamined the effects of aβ on obliques and more distal dendrites. To test the hypothesis that these sites might be particularly vulnerable to IA block we undertook to assess a possible link between the dendritic action potentials and their ability to stimulate Ca2+ currents at different points in the dendritic tree. These simulations provide novel insights into the relation between bAPs and Ca2+ currents at different dendritic sites, pointing especially to the sensitivity of Ca2+ currents in the oblique dendrites to the IA block. The results are of further interest in relation to the finding that oblique and basal dendrites are lost first in early Alzheimer’s before the apical tuft and shaft (Scheibel, 1979 ).
Materials and Methods
In order to reproduce and extend the results of (Chen, 2005 ) we drew on several web resources. ModelDB is a web-accessible database of curated published neuronal models. Of over 500 models now available at this site, more than 40 are of the CA1 pyramidal neuron, each simulating a representative cell under a set of physiological conditions. To evaluate the effects of aβ, we started with one of the models by Migliore et al. (2005) (ModelDB accession number 55035) that have morphologies and physiological properties similar to those of the cells illustrated in the study of Chen (2005) . The model we selected had basic membrane properties (see Table 1 ) that are robust over cells of this type. Recent experimental evidence indicates that membrane resistance in distal branches may be significantly lower than the values used in the originally published version of the model (Golding et al., 2005 ; Omori et al., 2006 , 2009 ; cf. London et al., 1999 ). Consequently we updated the model by reducing Rm to 103.5 Ω cm2) at distances greater than 350 μm from the soma. This shifted the envelope of peak bAP amplitude about 100 μm toward the soma in both the control and aβ applied cases.

Table 1. The basic membrane properties in the model. The set of constants, Kboundary, are 0 on the apical trunk, and adjusted in the obliques so that gA(d) is “continuous” from the trunk onto each oblique. The last four conductances were only present in simulations that examined calcium effects. The axon (no IA) and soma (IA with different kinetics in soma and proximal dendrites) have other simple distributions. See model code in ModelDB for details. We recommend NEURON’s ModelView tool for viewing the spatial distributions of these and other parameters (Hines et al., 2007 ).
We used an updated IA distribution that increases linearly with distance from the soma, in which the slope of the maximum conductance is three times as large in the oblique branches as along the apical trunk (consistent with experiments by Frick et al., 2003 ; Gasparini et al., 2007 ) (Table 1 ). This has the effect of making IA densities at distal ends of obliques similar to the densities at distal ends of apical tuft branches. It may be noted that none of the current pyramidal cell models includes representation of dendritic spines. For present purposes these can be considered as increasing the surface-to-volume ratios in Figure 6 (see for example, Katz et al., 2009 ), but would not affect the basic conclusions.
The effect of aβ on the model neuron was simulated by a reduction of the transient A-type potassium (K+) maximum conductances, gKA. While there are likely other properties affected by aβ, this was the only one monitored in the experiments of (Chen, 2005 ), and the only one tested in our study. The simulated aβ effect was modeled by reducing gKA by the average amounts, 47 and 32%, that (Chen, 2005 ) measured in control and 1 μM aβ applied excised patches from CA1 pyramidal neuron dendrites and somas, respectively. This caused a similar enhancement of the amplitude of the bAP at a distance on the apical dendritic trunk corresponding to the site in the labeled cells in the experiments of (Chen, 2005 ). Effects of lower aβ concentrations were simulated by proportionally smaller reductions of IA block. To determine the effects of aβ on internal calcium concentration we added calcium channels and a calcium accumulation mechanism. We used L, T, and N-type Ca2+ channels from the files associated with the publication Hemond et al. (2008) which were available in ModelDB (accession number 101629). The kinetics of these channels were derived from Jaffe et al. (1994) . Conductance density ratios were the same as those used by Song et al. (2002) who based their calculation on the measurements of Fisher et al. (1990) . More details on the anatomical distribution of these channels have been published (see review in Oh et al., 2010 ); however, electrophysiological measurements taking these new results into consideration have not been published. Conductance densities were adjusted so that a bAP produced a rise of about 200 nM [Ca2+]i in a proximal apical dendritic compartment to be consistent with experimental values (Helmchen et al., 1996 ; Maravall et al., 2000 ; Liao and Lien, 2009 ). For the purpose of these simulations of the effects of bAPs on membrane Ca2+ fluxes we omitted NMDA receptors and calcium induced calcium release from internal stores.
All simulations were performed with NEURON (Hines and Carnevale, 1997 ); model source code is available from ModelDB under accession number 87284.
Results
We first used our model to reproduce the experimental results of (Chen, 2005 ) for the normal mouse CA1 hippocampal pyramidal cell. Depolarizing current injected in a simulated axon caused the initiation of an action potential (the same protocol that Chen, 2005 used) which backpropagated along the axon, into the soma, and then further backpropagated into the dendrites. As shown in Figure 1 B, with simulated patch recordings at the soma and at a site 264 μm from the soma on the apical dendritic trunk, the responses closely resembled those shown in Figure 1 A of (Chen, 2005 ), with a large amplitude spike in the soma, and a lower amplitude spike 280 μm from the soma in the dendritic trunk. The differences between the simulations and experiment are examined further in the Discussion. Action potentials are shown in traces recorded from an oblique’s distal tip in the simulation (far right of Figure 1 B); experimental recordings of membrane potential near the distal terminations of oblique dendrites have not yet been reported in normal or Alzheimer’s animal models.
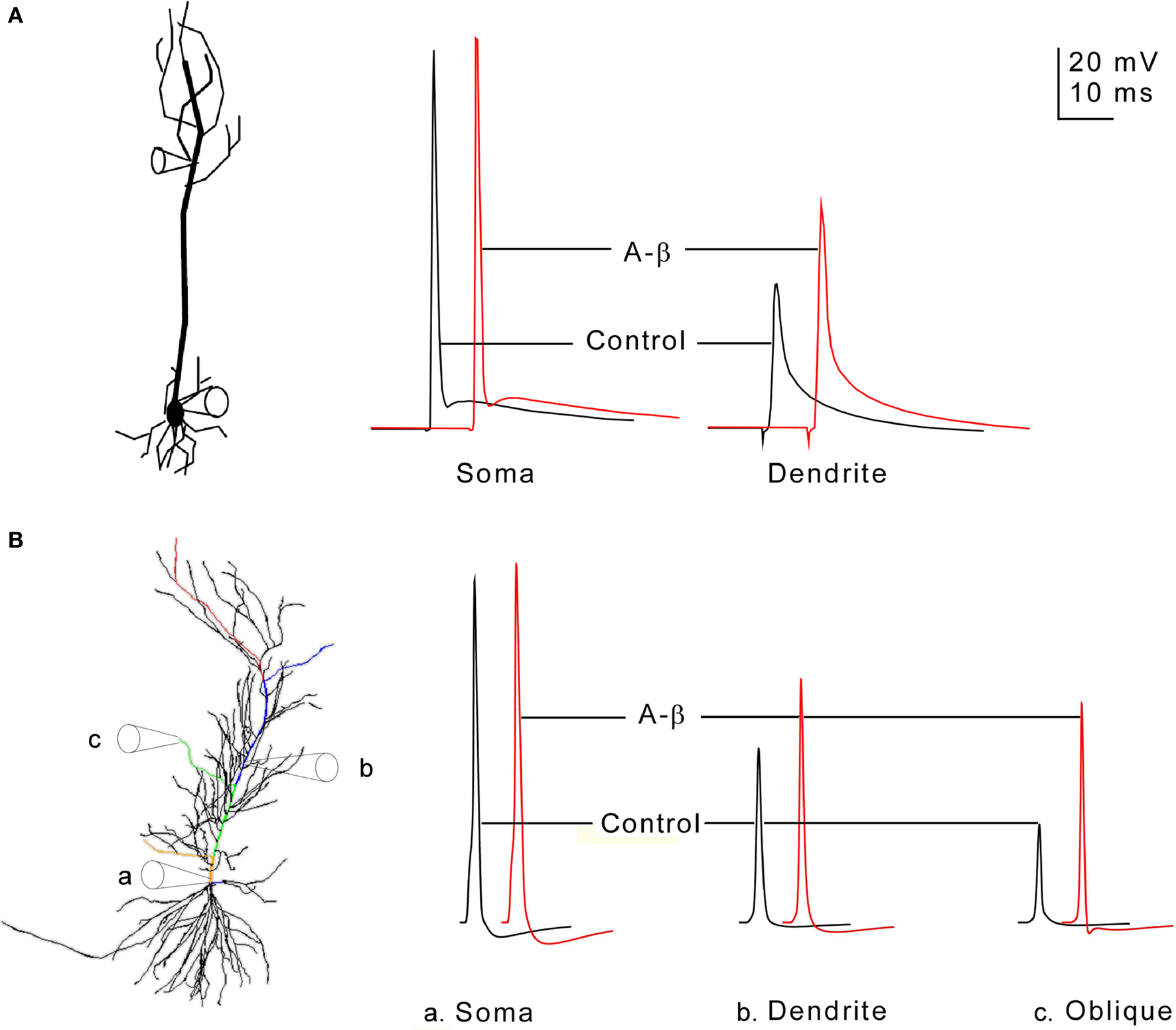
Figure 1. (A) Identified CA1 neuron with soma and primary dendritic spike recordings (modified from Chen, 2005 to transform recordings to the same scale bars); red color indicates the aβ applied condition. The voltage traces were recorded at the soma and the primary dendrite 280 μms from the soma. (B). Morphology and simulated recordings from our CA1 pyramidal neuron model. Several labeled paths through the cell have voltages displayed in Figure 3 and the movie in the Supplementary Material: from the soma to the most distal tuft (red), a distal oblique (blue), a middle oblique (green), and a proximal oblique (tan) dendrite. These paths overlap along the primary trunk. Scale for (B) traces same as indicated in (A).
Spike attenuation with increasing distance from the soma has been reported in dendrites of normal cells (see Spruston et al., 1995 ; Hoffman et al., 1997 ; Johnston et al., 1999 ; Acker and White, 2007 ; Gasparini et al., 2007 ). Golding et al. (2001) reported that back-propagating dendritic spike amplitudes in CA1 pyramidal cells in vitro usually fall into two groups, strong and weak; at 300 μm 26–42% attenuation (9/20 cells) and 71–87% attenuation (10/20 cells). Only weakly propagating bAPs (i.e., strongly attenuating) CA1 pyramidal neurons have been observed in vivo (Kamondi et al., 1998 ). Our model cell exhibited 48% attenuation bAPs at 313 μm (Golding et al., 2001 found 1/20 cells with 45% attenuation which they termed intermediate).
As noted by Hoffman et al. (1997) , IA channel density increases with distance from the soma into the apical dendritic field. The resultant shunting progressively reduces the peak amplitude of the invading action potential (Migliore et al., 1999 ; see also reviews in Gulledge et al., 2005 ; Sjostrom et al., 2008 ). This implies that the most distal apical dendritic branches are normally not subjected to a strong depolarization by the bAP, which is explored further below.
The left side of Figure 1 B uses illustrations of “electrodes” to indicate the positions in the model from which the voltage traces on the right side of Figure 1 B were recorded. The primary dendrite recording location was 264 μm from the soma.
As described in Section “Materials and Methods,” the effect of aβ was simulated by reducing IA conductance by the amounts reported by Chen (2005) . This increased somatic spike amplitude by 4.7%, and enhanced the back-propagating primary dendritic spike at a distance of 264 μ by 40%. These are near the average increases observed by Chen: 3.1 ± 1.3% in the soma and 33.9 ± 6.1% in the primary dendrite 240–300 μ from the soma. The enhancement of dendritic spike amplitude was shown experimentally to be due to the block of a transient IA current, a result in line with previous studies that examined bAP propagation with blocked transient IA current (see for example Hoffman et al., 1997 ; Migliore et al., 1999 ; Watanabe et al., 2002 ). The model supports this result by showing that the partial block of the IA current is sufficient to enhance the bAP, and that other reported excitatory effects of aβ, such as the longer time course increase in L-type Ca currents (Ueda et al., 1997 ), which likely contributes to the excitability of the dendrites, are not necessary to produce enhanced bAPs.
Back-Propagating Action Potentials in the Dendritic Tree
An advantage of the model was that it could be used to explore properties of any part of the dendritic tree that might be affected by aβ reduction of IA conductance. In particular, we were interested in thin dendritic branches which to our knowledge have not been recorded from in animal models of Alzheimer’s, but which we postulated might be especially vulnerable to aβ effects.
We first analyzed the invasion of the bAP throughout the entire extent of the modeled apical dendritic tree. Figure 2 shows the peak action potential amplitude in each compartment of the cell as a function of path distance (through the processes of the cell) from the soma. Under normal conditions (black oblique dendrites, purple trunk, green tuft) the spike is largest at the soma, but amplitude drops to ∼50% about midway into the dendrites (cf. Figure 1 ). This result is consistent with weakly propagating bAPs as seen in vivo (Kamondi et al., 1998 ). In normal pyramidal cells, this shows, surprisingly, that the bAP causes little depolarization of the most distal branches. By contrast, with reduction of IA, the amplitude of the bAP was largely maintained into the terminal oblique branches (red). There was even a 62-mV depolarization in the most proximal part of the apical tuft (tan), whereas this same region had only a small depolarization (20 mV) under normal conditions.
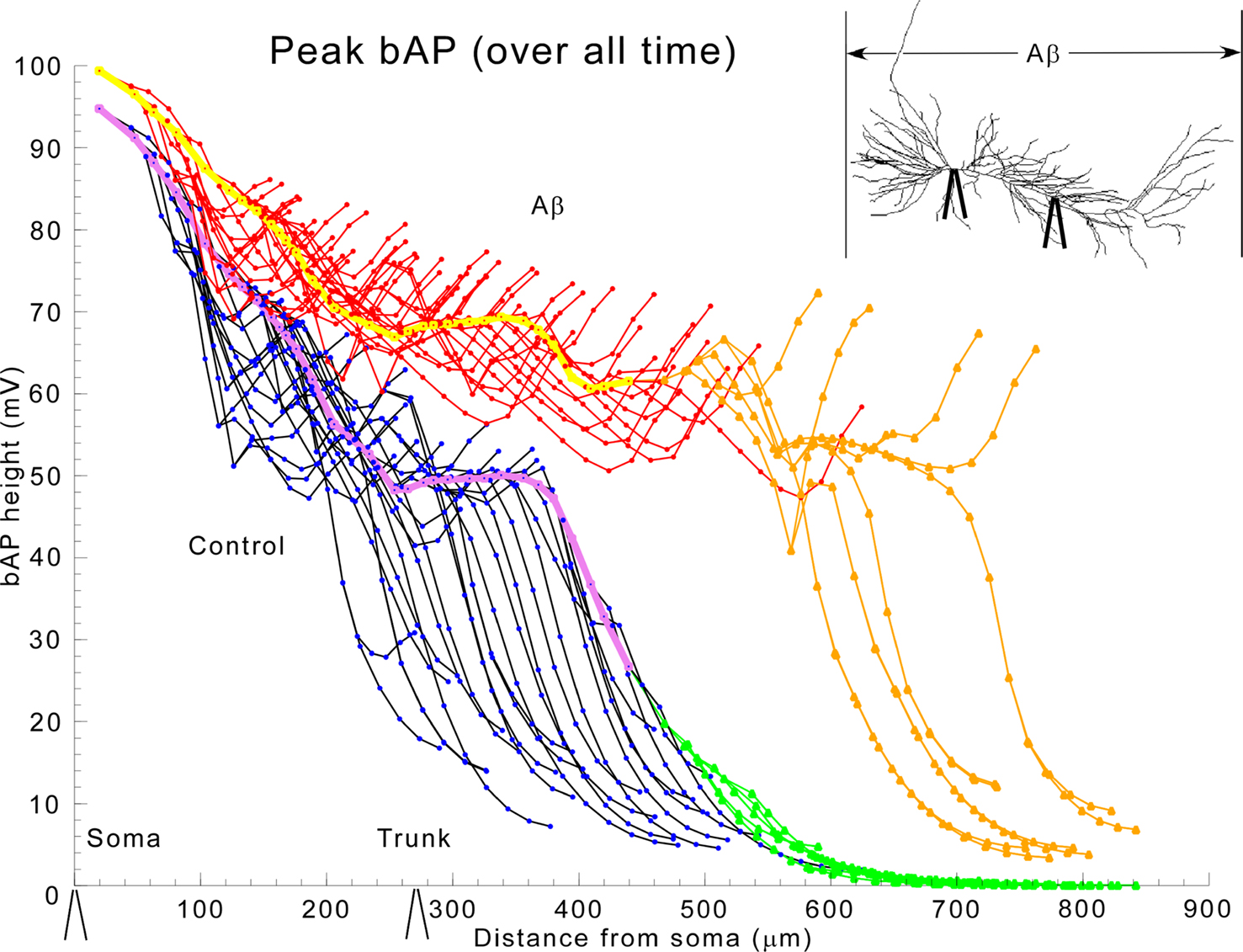
Figure 2. Global application of aβ enhances bAP invasion in the trunk, obliques, and tuft. bAP amplitudes in apical dendritic compartments of the model as a function of distance from the soma. Color key: normal conditions (black-oblique dendrites, purple-trunk, green-apical tuft), and in the presence of 1 μM aβ (red-oblique dendrites, yellow-trunk, tan-apical tuft). The inset shows that aβ was applied ubiquitously and displays equivalent locations (soma and 270 μ along primary dendrite) of the experimental recording electrodes on the model cell, which are also shown along the x-axis in this and subsequent appropriate figures to give a sense of scale.
We next analyzed the extent of bAP invasion in individual dendritic branches. A single frame from a movie, Figure 3 depicts the voltage throughout the cell at an instant of time about 2 ms after the AP has peaked at the soma. The movie (available in Supplementary Material and on the website for the CA1 pyramidal neuron in BrainPharm) shows the entire period of invasion of the bAP. The depolarizations in individual branches in the right panels are indicated in color coding and the instantaneous voltages along indicated paths (see Figure 1B left) are shown in the left panels. The bAPs in the Alzheimer model cell (bottom panels) distal dendrites have substantial amplitudes (lower left), −15 mV peak, while in the normal cell the bAP is nearly extinguished, −47 mV peak (upper left). The results in Figures 2 and 3 show that generalized block of A current by aβ allows bAPs to produce intense depolarization that extends to the most distal parts of the oblique branches.
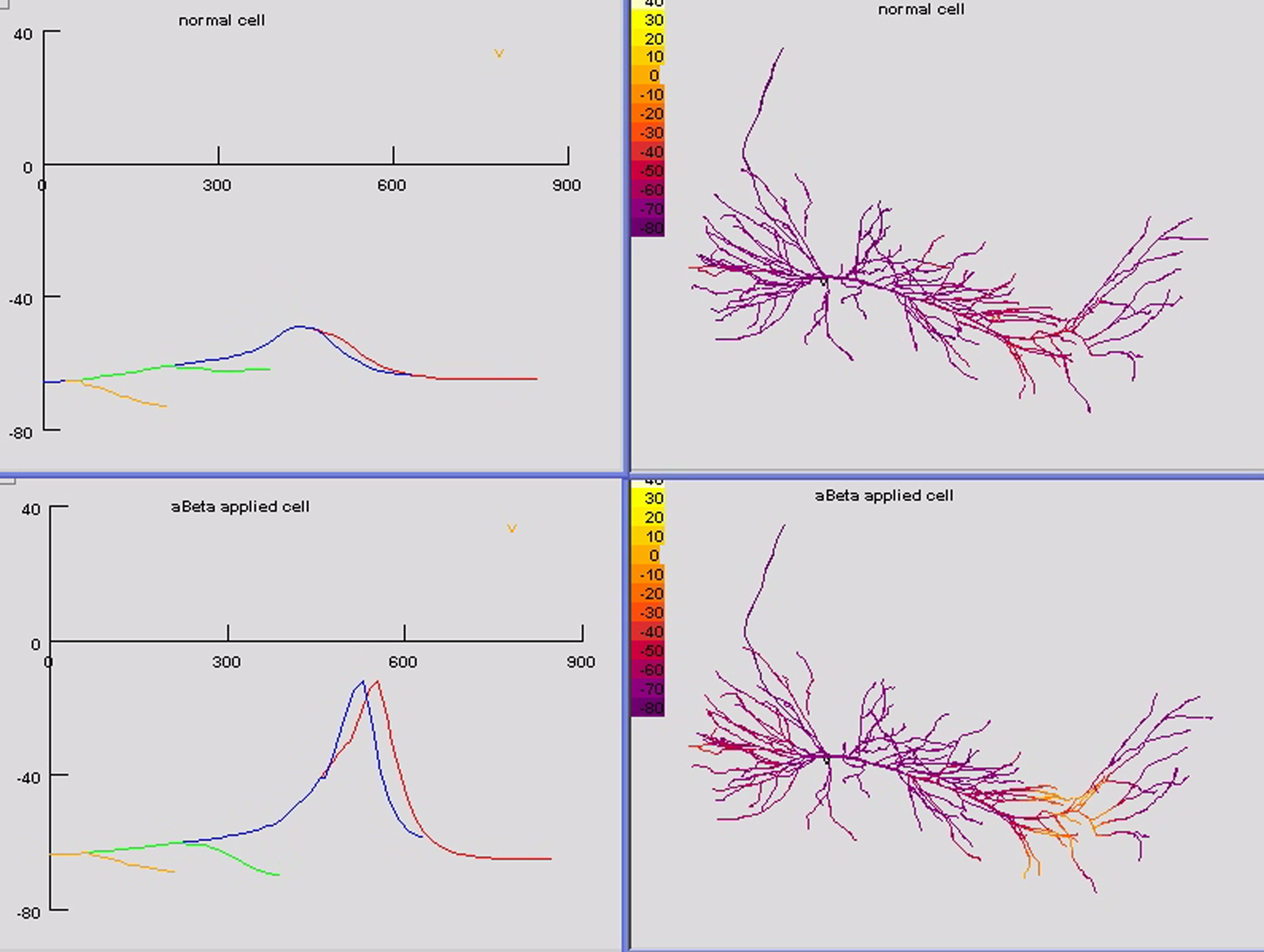
Figure 3. Movie frame of normal (top) and aβ applied (bottom) simulations 2 ms after the peak of the somatic spike. On the left are plots of instantaneous membrane potential along paths from the soma (0 μm) to a distal dendritic tuft compartment (849 μm) and oblique dendrites (x-axis in μm, y-axis in mV, paths shown in left panel of Figure 1B ). The figures on the right display membrane potential throughout the model at the corresponding time. At this instant the bAP has died out in the normal model, but it continues to propagate into the apical tree of the aβ-applied model. The movie is in the Supplementary Material.
Differential Vulnerability of Different Parts of the Dendritic Tree
The enlarged view of the entire dendritic tree afforded by the model enabled us to address the hypothesis that different parts of the dendritic tree might be differentially sensitive to aβ block of IA. To test this hypothesis we carried out two types of computational experiments. The first was to test for a possible threshold for the effect on the bAP. The model allowed us to go beyond the Chen experiments by using increasing degrees of IA block to simulate different concentrations of globally applied aβ. This revealed interesting nonlinear threshold effects. At approximately 500 μm distance on the apical dendrite there was a jump in amplitude of the bAP between 0.2 and 0.3 μM (*) and then at 600 μm in the apical tuft there was another jump (**) when aβ increased from 0.7 to 0.8 μM (Figure 4 ). These results were similar to the threshold phenomena reported by Golding et al. (2001) (their Figure 9B) when investigating effects of IA density on spike backpropagation.
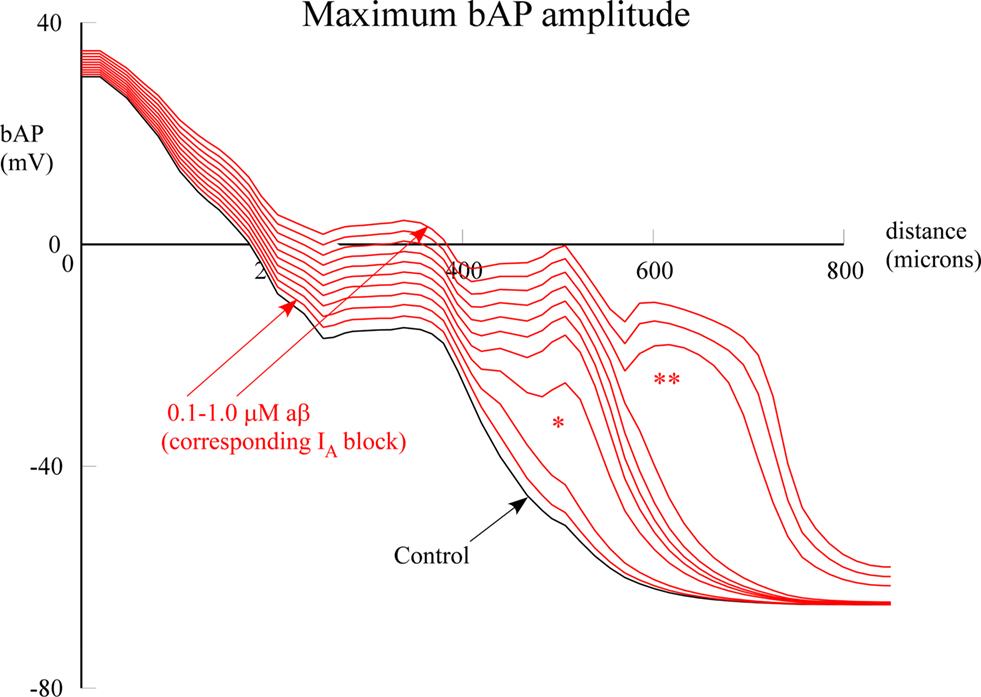
Figure 4. Effect of global (uniform) aβ concentrations on bAP amplitude. Peak bAP amplitude is plotted as a function of distance on a path from the soma along the apical trunk to the distal termination of a branch (red branch in Figure 1 B). aβ concentrations were simulated by proportional reductions of IA block, varied in 10% steps. Single asterisk indicates nonlinear amplitude jump at around 500 μm distance; double asterisk indicates jump at around 600 μm. See Section “Materials and Methods” for details. The normal (black) and 1 μM aβ (top red) correspond to the data in Figure 2 .
The second type of model experiment was to apply the IA block to specific parts of the dendritic tree. We were particularly interested in comparing the effects on the oblique branches, believed to be critically important for learning and memory (cf. Gasparini et al., 2007 ), with the effects on the trunk and distal dendritic branches.
The results showed that the oblique dendrites were the site of the most prominent effects on the bAP. Reduction of IA in the proximal compartments (<100 μ from the soma), equivalent to local application of 1 μM aβ, resulted in only a slight shift (not shown), similar to the effect of 0.2 μM uniformly applied aβ (Figure 4 ). By contrast, the same IA reduction limited to oblique dendrites caused a heightened local spread of depolarization and also spread of additional depolarization into the proximal portion of the apical tuft (Figure 5 ). In both cases, proximal (between 0 and 100 μm) bAPs and soma APs were least affected (Figure 5 left part of graph near the soma). These observations make intuitive sense considering that the larger effect of the distal vs. proximal reductions in IA was correlated with the increasing density of IA channels with distance from the soma, as well as with the decreasing diameters of the branches. Local IA reduction in only the apical dendritic tuft caused no change because the bAP died out well before it could reach the tuft, as in the control plot in Figure 5 .
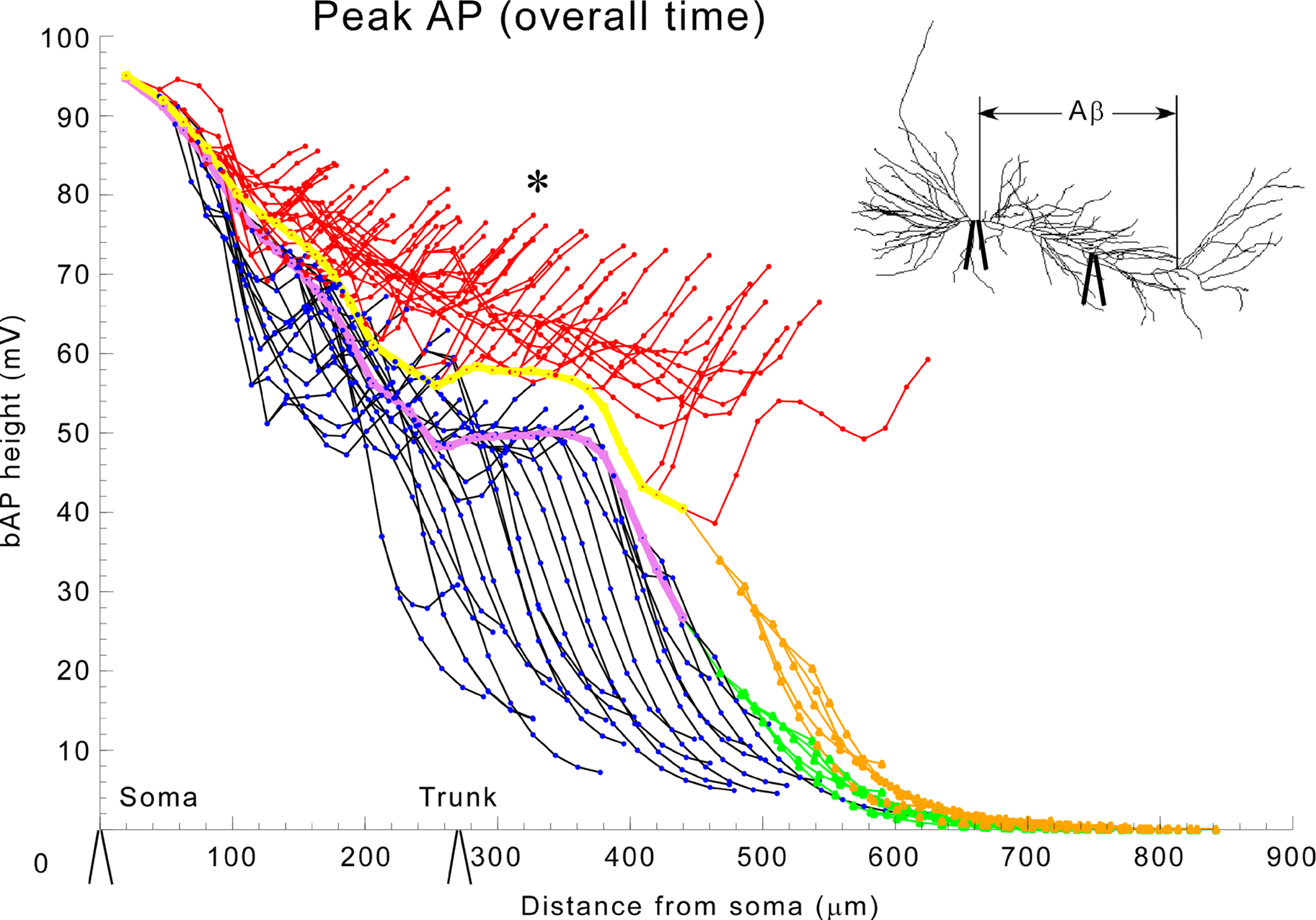
Figure 5. Local application of aβ has the greatest effects on enhancing bAP when applied to the obliques. Normal: black-oblique dendrites, purple-trunk, green-apical tuft; 1 μM applied only to oblique dendrites: red-oblique dendrites, yellow-trunk, tan-apical tuft. Asterisk indicates abnormally large amplitude in the oblique dendrites. The inset shows that aβ (IA block) was applied only to the oblique dendrites. (See Figure 2 for electrode locations).
Simulations Suggest Special Vulnerability of Obliques to Large Increases of [Ca2+]I
Given the sensitivity of bAPs in the oblique dendritic branches to IA block, we wished to test the hypothesis that Ca2+ influx in these branches would be correspondingly sensitive. To test how aβ might affect Ca2+ influx and concentration elicited by bAPs, we added calcium channels and a calcium accumulation mechanism as described in Section “Materials and Methods.” First we examined maximum [Ca2+]i elicited by a back-propagating spike as a function of location along four paths in the apical dendritic tree (Figure 6 A). One path ran from the origin of the primary apical trunk to the distal end of a dendrite in the apical tuft (thick black line), and the other three ran along oblique branches that arose from the apical trunk in the proximal, intermediate, or distal zones of stratum radiatum (thick red, blue, and violet lines).
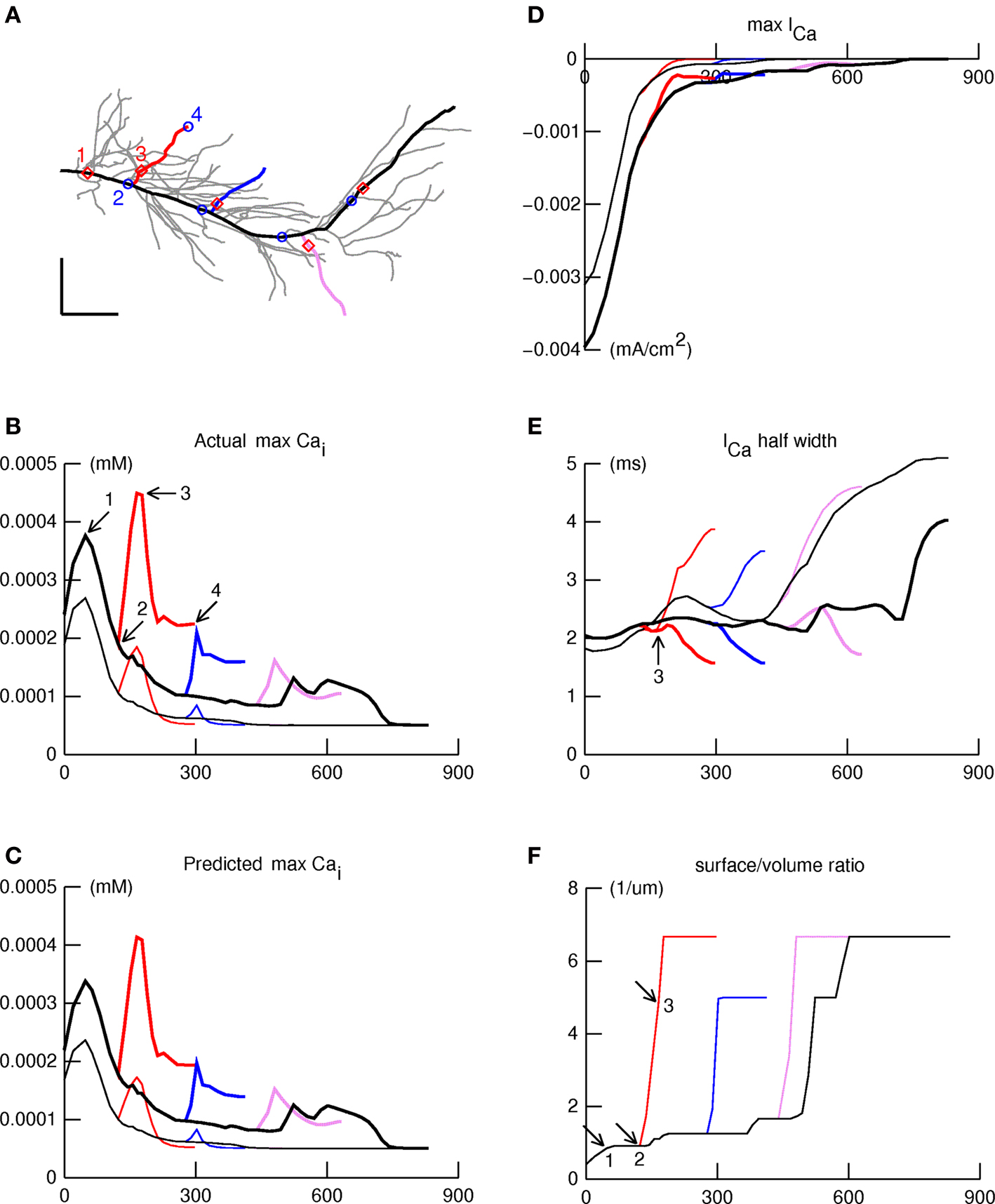
Figure 6. IA block leads to large [Ca2+]i increases in the obliques. The spatial profile of [Ca2+]i elicited by a backpropagating action potential in the apical dendritic tree was examined along four paths indicated in panel (A) by thick lines: primary apical and apical tuft (black); proximal, intermediate, and distal obliques (red, blue, violet). Panels (B–F) show simulation results plotted as functions of distance along these paths, using thin and thick lines for normal and aβ (IA blocked) responses, respectively. (B) Maximum [Ca2+]i shows a declining trend with distance which is interrupted by peaks and valleys. aβ (IA block) increases maximum [Ca2+]i but does not affect locations of the peaks and valleys. (C) The product of ICa amplitude, ICa duration, and svr, shown here, closely predicts maximum [Ca2+]i (see B). (D) ICa amplitude falls off more or less steadily with distance along the path from apical tree origin to termination in the distal tuft, similar to maximum [Ca2+]i under normal conditions. (E) Spatial variation of ICa half width. The arrow marks the location of the peak of maximum [Ca2+]i in the proximal oblique. (F). Surface/volume ratio as a function of distance along the paths. The arrows mark positions that correspond to points 1, 2, and 3 in Figures 6 A and B. The large jumps in svr happen at branch points, and encompass the locations of peaks in the spatial profile of maximum [Ca2+]i.
The plots of maximum [Ca2+]i vs. distance along these paths are shown in Figure 6 B under normal and aβ conditions. Under normal conditions (thin traces), maximum [Ca2+]i had a peak near the proximal end of the primary apical trunk (point 1 in Figure 6 B), then decayed more or less steadily with increasing distance out to the end of the apical tuft. This is similar to the spatial distribution of Ca influx associated with the bAP in the experiments of Gasparini et al. (2007) , and to the results presented in Figures 6 and 7 of Golding et al. (2001) . Smaller peaks appeared in the proximal and intermediate obliques near their attachments to the apical trunk; similar peaks were observed in Ca1 pyramidal neurons of rat by Gasparini et al. (2007) (see their Figure 1 and associated text).
The effect of aβ block of IA was to cause a large increase of maximum [Ca2+]i almost everywhere (thick traces in Figure 6 B); only the most distal part of the apical tuft (>700 μm) seemed unaffected. The increase was disproportionately large in the obliques, especially near their origins from the apical trunk. The locations of peaks, and of the valleys that separate them, are marked in Figure 6 A by red diamonds and blue circles, respectively. Note that each peak is near the proximal end of a branch, and each valley is near the distal end of that branch’s parent. It is also noteworthy that aβ block of IA does not affect the positions of these features in space.
What accounts for these features, and for the particular vulnerability of oblique dendrites, especially at their proximal ends? The model allowed us to identify the three factors that determined the maximum [Ca2+]i. The first two are maximum calcium current activated by the bAP, and the half-width of this current (Figures 6 D,E respectively); the product of these approximates the net calcium entry. The third is the surface to volume ratio (svr), which increases abruptly in the proximal ends of obliques where they attach to the apical trunk (Figure 6 F). These three factors multiplied together produced the profiles in Figure 6 C, which were almost identical to the maximum values for maximum [Ca2+]i produced by the bAP (cf. Figure 6 B).
Discussion
The simulation results reported here support and extend the experimental results (Chen, 2005 ) that a selective effect of aβ on K+ currents increases the extent of invasion of action potentials from the cell body into the apical dendritic trunk of CA1 pyramidal neurons, as has also been seen with pharmacological blockade of the A-type K+ current (Hoffman et al., 1997 ). In both the experiments and the model the effect involved a moderate increase in spike amplitude in the trunk region. The simulation results provide new evidence that aβ would have a prominent effect on the bAP in the oblique dendritic branches.
In the thin and most distal branches of the oblique dendrites the invading bAP maintained a large amplitude in the presence of aβ, whereas in the normal control the depolarization decayed rapidly. There was thus a much larger difference between the depolarizations in the aβ and normal cases in the distal oblique branches compared to the dendritic trunk. Building on previous studies of IA block (starting with Hoffman et al., 1997 , see also for example Golding et al. 2001 ), the results show for the first time that the effect of a disruption of normal dendritic electrical activity by IA blockade appears to be more severe in the small branches than other dendritic locations. This would potentially have marked effects on both dendritic information processing, as well as Ca2+ excitotoxicity induced by the excessive depolarization (Good and Murphy, 1996 ).
To analyze this further, we carried out simulations of localized A-type potassium channel block to model the effect of local applications of aβ to different parts of the tree. The local application to the proximal dendrites, dendritic trunk, or tuft had relatively little effect on the amplitude and extent of invasion of the bAP. In contrast, local application to the thin oblique dendrites caused large local bAPs and some spread to the tuft.
The prediction from these results is that the early aβ pathology in CA1 hippocampus could have its earliest and most profound effects on the thin oblique dendrites of the pyramidal cells there. This may be significant for several reasons. First, the bAP is an important property in the normal function of the pyramidal neuron. It is believed to play a key role in learning and memory, through its depolarizing actions which signal to the dendrites that a particular combination of synaptic inputs has been sufficient to generate an axonal spike output of the neuron (Sjostrom et al. 2008 ). One possible function of the bAP is to relieve the Mg2+ block of glutamatergic NMDA receptors. This depolarization is presumed to affect the readiness of NMDA receptors to be involved in activity-dependent mechanisms believed to underlie learning and memory. Experiments (Kamondi et al., 1998 ; Canepari et al., 2007 ) and our results have implications for this normal model, because they indicate that it is unlikely that normally the bAP significantly depolarizes the apical tuft. Significantly, this distal region of the pyramidal cell dendritic tree appears to be more involved in the control of NMDA receptors by local synaptic depolarizations than by the bAP (cf. Larkum et al. 2009 ). Oblique dendrites on the other hand appear to be under control of both. It is likely that when synaptic background and/or normal network activity is added to the model the distal tuft will be partially depolarized by bAPs. It may be relevant in this regard that increasing concentrations of aβ could cause the nonlinear enhancement of the bAP in the distal tuft indicated in the simulations in Figure 4 .
With regard to links between the bAPs and degenerative changes, a previous single compartment model (Good and Murphy, 1996 ) calculated changes in [Ca2+]i with respect to simulated increases in aβ. Comparisons between their work and ours are difficult because it is not clear which compartment their single compartment model should be compared to (the soma, proximal apical, oblique dendrite, etc.). Chen (2005) reported calcium dye recordings in the apical dendrite in aβ treated slices. To our knowledge, there are no publications that have measured calcium concentration changes with the application of aβ to the spatial extent of the dendritic tree of a CA1 pyramidal neuron. The model points to the need for experiments in animal models of Alzheimer’s to test these predictions and hypotheses.
Suggested Experiments
We suggest repeating the kinds of experiments that (Chen, 2005 ) performed on the primary dendrite, but targeting the oblique dendrites to examine if they are more susceptible to large bAPs with IA block by aβ. These experiments are technically very challenging due to the small diameter of the obliques. We are only aware of one report of voltage recordings in obliques; Kamondi et al. (1998) made in vivo patch electrode recordings from hippocampal CA1 pyramidal neuron dendrites and somas. They were able to record from the middle of (at least one) oblique dendrite in vivo (see their Figure 3 ). Others have recorded calcium dye activity in the obliques (complete obliques in Canepari et al., 2007 , Figures 5 F,G), and locations in the proximal half of obliques in (Gasparini et al., 2007 ; Losonczy et al., 2008 ). We suggest repeating any of these methods in animal models of Alzheimer’s (either with transgenic animals or acute application of aβ) to assess the electrical and calcium excitotoxicity vulnerability of the oblique dendrites relative to other cell areas such as the primary apical dendrite and soma.
The identification of the potassium channel subunit gene Kv4.2 underlying the A-type channel in CA1 pyramidal neurons (Kim et al., 2005 ) has allowed some details of IA activity in determining synaptic kinetics and weights to be illuminated. The Kv4.2 channels are involved in regulatory interactions with the NMDA receptor subunits (Jung et al., 2008 ). By comparing dominant negative pore mutants of Kv4.2 to uninfected or enhanced Kv4.2 with GFP infected CA1 pyramidal neurons in organotypic cultured slices, support was found for the notion that IA is a negative regulator of synaptic NMDA currents and evidence was found that the activity of IA apparently regulates the ratio of NR1 to NR2B subunits present in synaptic NMDA receptors (Jung et al., 2008 ). Furthermore, the Kv4.2 A-type K+ current has previously been shown to be necessary for normal distance-dependent scaling where the amplitude of unitary Schaffer collateral inputs increases with distance from the soma (e.g., Magee and Cook, 2000 ) and interestingly, that NMDA activity in turn regulates the expression of IA (Losonczy et al., 2008 ). It has been shown that the genetic deletion of Kv4.2 in CA1 pyramidal neurons eliminates this distant dependent scaling and increases the GABA current to these cells (Andrasfalvy et al., 2008 ). Experiments comparing normal to Alzheimer animal models could test the ratio of NR2A/NR2B and also distant dependent scaling to see if they are consistent with the changes expected from the presumed aβ block of IA (the changes should be in the direction of those associated with Kv4.2 deletion experiments). These experiments could test the hypothesis that IA block is a path of neurodegeneration in Alzheimer’s.
Experiments with uncaging glutamate or synaptic stimulation in normal and acute application of aβ slices may be able to evaluate changes in integrative properties of CA1 pyramidal cells. We plan future simulations that would predict the results of these kinds of tests by creating ePSP-spike probability graphs, (e.g., as in Figure 2 of Jarsky et al. (2005) , that could then also be tested.
A class of genetics experiments could test the hypothesis that the IA current plays a pivotal role in the progression of AD. These tests would cross any aβ overproducing mutant mouse (AD model mouse) (e.g., Wang et al., 2003 ) with a Kv4.2 subunit deleted mouse (Andrasfalvy et al., 2008 ). If deficits of the AD model mouse were rescued in these double mutant mice it would suggest that the absence of this A-type current removed a permissive factor in the progression of AD.
Potential Short-Comings and Limitations of the Model
We based our study on previous electrophysiology and modeling publications for the A-type K+ channels in the obliques (Frick et al., 2003 ; Migliore et al., 2005 ; Gasparini et al., 2007 ). There are no similar publications for CA1 pyramidal neuron basal dendrites. The computational model has a simple set of currents to model excitability (see Table 1 ) and therefore is limited by currents not represented in the model to what it can match experimentally. (See Table 1 in Demont-Guignard et al., 2009 for a summary literature review of CA1 pyramidal neuron models.) Omitted potassium channels include the KCNQ/M channels, known to limit the After Depolarization Potential (ADP). Modulation of the KCNQ/M channels can transition the neuronal firing pattern from simple to complex spiking (Yue and Yaari, 2004 ; Golomb et al., 2006 ). In addition a persistent Na+ channel (not present in the model) contributes to bursting and the ADP in these cells (Golomb et al., 2006 ). It is also known that a slowly inactivating voltage-gated Na+ channel (not represented in the model) is responsible for a declining amplitude of APs in spike trains in these cells (Colbert et al., 1997 ; Mickus et al., 1999 ). Although we found a dependence of AP half-width on IA in our model (two-fold increase in half-width when IA max conductances are set to 0) experiments with dominant negative mutant of Kv4.2 in rats showed three-fold increases in AP half-width (Kim et al., 2005 , Figure 5 B). Because of the simplified currents used in the model we only expect to capture the basic features of excitability in these cells. Detailed features such as an exact match to the change in half-width of the APs with total loss of IA, shape of ADPs (Metz et al., 2005 ) and complex bursts or AP amplitude reduction in trains of APs are not possible to reproduce in the model.
Implications for Disease Mechanisms
The large depolarizations caused by the bAP in the thin oblique branches are of particular interest. Oblique dendrites are likely more vulnerable to neurodegenerative changes and associated effects on signal processing because of their greater distance from the soma, placing them further from metabolic maintenance and repair. These effects would be magnified because of the smaller diameters, increasing the svr, with corresponding large ion fluxes (both K+ and Ca2+) into small interior volumes, and would be even greater within the small volumes of the dendritic spines arising from these branches. The results thus support the hypothesis that these sites in the dendritic tree could be differentially vulnerable to the effects of aβ.
Relatively distal locations in oblique dendrites (∼130 μm) have supra-linear boosts in the Ca2+ signal from paired bAP and synaptic stimulation, unlike more proximal locations (∼70 μm) which are linear (Gasparini et al., 2007 , Figure 9; Canepari et al., 2007 , Figure 3). Abnormally large bAPs from IA block by aβ might mimic a similar Ca2+ signal that paired stimulation produces, providing a false positive (paired signal) input to cellular calcium signaling pathways. The bAP invades locations beyond where it would have been extinguished in the normal cell. Figure 4 suggests that this may not occur in the earliest stages of aβ accumulation, but that progressive accumulation will eventually produce serious disruptions of dendritic signal processing. Background levels of network activity providing synaptic input to the cell, unpaired in the normal animal, might then be paired with these bAPs providing excess signaling to these cells. A CA1 pyramidal cell modeling study predicts that neighboring synapses to paired inputs are also potentiated (Urakubo et al., 2004 ), which would make the system even more vulnerable to excitation.
Others have recognized the importance of dendrites for neurodegenerative pathogenesis. Structural and biochemical dendritic changes that may underlie Alzheimer’s disease have been reviewed by Anderton et al. (1998) . However, there was no discussion of signaling in terms of the electrical properties analyzed here.
Beyond the early disruption of electrical signaling, it has been speculated that the heightened depolarization of the bAP causes increased Ca2+ influx and excitotoxicity leading to the degenerative changes seen in AD (Good and Murphy, 1996 ; Chen, 2005 ). Our results (Figure 6 ) suggest that these later degenerative changes would be especially severe in the oblique branches at peaks of [Ca2+]i. This would be consistent with the anatomical study of (Scheibel, 1979 ) who observed that basal and oblique dendrites were lost before the peripheral portions of the apical shafts in AD. Immunohistochemical evidence that two variants of phosphorylated A-type K+ channel subunit Kv4.2 are present in similar density in basal dendrites (stratum oriens) as in the obliques (stratum radiatum) (see Figures 6A,B in Varga et al. 2000 ) suggests that these results may also be applicable to the basal dendrites.
The present results indicate that future experiments on animal models of Alzheimer’s should focus on oblique dendritic branches and spines as critically vulnerable to the earliest effects of aβ on IA. They suggest the testable hypothesis that, in addition to degenerative effects, disruption of electrical signaling in distal dendrites by aβ may play a role in the neural basis of early cognitive decline in AD. Therapies to oppose this decline could be directed at these changes in dendritic electrical signaling.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported in part by NIH grants R01 009977-01 and R01 DC000086-42. The authors thank Michael Hines, Luis Marenco, Kei-Hoi Cheung, and Perry Miller for discussions and the reviewers for their suggestions.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/neuroscience/neuralcircuits/paper/10.3389/fncir.2010.00016/
A movie (http://senselab.med.yale.edu/BrainPharm/movies/alz_model_movie.mov ) of the entire period of invasion of the bAP in the normal cell model (top) and aβ applied model (bottom). These frames are synchronized so that a 0.2-ms 0.1 nA current injection pulse antidromically simultaneously initiates APs (and associated bAPs) in the two cells. In the left two frames (top and bottom) are the instantaneous voltages along paths from the soma to a distal point in the apical tuft and proximal, medial, and distal oblique dendrites. In the right two frames (top and bottom) are color coded voltage values throughout the cell (including the paths whose voltages are graphed on the left); the basal dendrites are on the left and the apical tuft is on the right. The time evolution shows that at the time the bAP is extinguished in the normal cell dendrites (about 2 ms after the peak of the AP in the soma which is also 4.7 ms after the antidromic current injection) the bAP in the aβ applied cell is still propagating abnormally into the apical tuft.
References
Acker, C. D., and White, J. A. (2007). Roles of IA and morphology in action potential propagation in CA1 pyramidal cell dendrites. J. Comput. Neurosci. 23, 201–216.
Anderton, B. H., Callahan, L., Coleman, P., Davies, P., Flood, D., Jicha, G. A., Ohm, T., and Weaver, C. (1998). Dendritic changes in Alzheimer’s disease and factors that may underlie these changes. Prog. Neurobiol. 55, 595–609.
Andrasfalvy, B. K., Makara, J. K., Johnston, D., and Magee, J. C. (2008). Altered synaptic and non-synaptic properties of CA1 pyramidal neurons in Kv4.2 knockout mice. J. Physiol. 586, 3881–3892.
Byrne, J. H., and Shepherd, G. M. (2009). “Complex information processing in dendrites,” in From Molecules to Networks: An Introduction to Cellular and Molecular Neuroscience, 2nd Edn, eds J. H. Byrne, and J. L. Roberts (San Diego: Elsevier), 489–512.
Canepari, M., Djurisic, M., and Zecevic, D. (2007). Dendritic signals from rat hippocampal CA1 pyramidal neurons during coincident pre- and post-synaptic activity: a combined voltage- and calcium-imaging study. J. Physiol. 580, 463–484.
Chen, C. (2005). beta-Amyloid increases dendritic Ca2+ influx by inhibiting the A-type K+ current in hippocampal CA1 pyramidal neurons. Biochem. Biophys. Res. Commun. 338, 1913–1919.
Colbert, C. M., Magee, J. C., Hoffman, D. A., and Johnston, D. (1997). Slow recovery from inactivation of Na+ channels underlies the activity-dependent attenuation of dendritic action potentials in hippocampal CA1 pyramidal neurons. J. Neurosci. 17, 6512–6521.
Demont-Guignard, S., Benquet, P., Gerber, U., and Wendling, F. (2009). Analysis of intracerebral EEG recordings of epileptic spikes: insights from a neural network model. IEEE Trans. Biomed. Eng. 56, 2782–2795.
Fisher, R. E., Gray, R., and Johnston, D. (1990). Properties and distribution of single voltage-gated calcium channels in adult hippocampal neurons. J. Neurophysiol. 64, 91–104.
Frick, A., Magee, J., Koester, H. J., Migliore, M., and Johnston, D. (2003). Normalization of Ca2+ signals by small oblique dendrites of CA1 pyramidal neurons. J. Neurosci. 23, 3243–3250.
Gasparini, S., Losonczy, A., Chen, X., Johnston, D., and Magee, J. C. (2007). Associative pairing enhances action potential back-propagation in radial oblique branches of CA1 pyramidal neurons. J. Physiol. 580, 787–800.
Golding, N. L., Kath, W. L., and Spruston, N. (2001). Dichotomy of action-potential backpropagation in CA1 pyramidal neuron dendrites. J. Neurophysiol. 86, 2998–3010.
Golding, N. L., Mickus, T. J., Katz, Y., Kath, W. L., and Spruston, N. (2005). Factors mediating powerful voltage attenuation along CA1 pyramidal neuron dendrites. J. Physiol. 568, 69–82.
Golomb, D., Yue, C., and Yaari, Y. (2006). Contribution of persistent Na+ current and M-type K+ current to somatic bursting in CA1 pyramidal cells: combined experimental and modeling study. J. Neurophysiol. 96, 1912–1926.
Good, T. A., and Murphy, R. M. (1996). Effect of beta-amyloid block of the fast-inactivating K+ channel on intracellular Ca2+ and excitability in a modeled neuron. Proc. Natl. Acad. Sci. U.S.A. 93, 15130–15135.
Gulledge, A. T., Kampa, B. M., and Stuart, G. J. (2005). Synaptic integration in dendritic trees. J. Neurobiol. 64, 75–90.
Helmchen, F., Imoto, K., and Sakmann, B. (1996). Ca2+ buffering and action-potential evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys. J. 70, 1069–1081.
Hemond, P., Epstein, D., Boley, A., Migliore, M., Ascoli, G. A., and Jaffe, D. B. (2008). Distinct classes of pyramidal cells exhibit mutually exclusive firing patterns in hippocampal area CA3b. Hippocampus 18, 411–424.
Hines, M. L., and Carnevale, N. T. (1997). The NEURON simulation environment. Neural. Comput. 9, 1179–1209.
Hines, M. L., Morse, T. M., and Carnevale, N. T. (2007). Model structure analysis in NEURON: toward interoperability among neural simulators. Methods Mol. Biol. 401, 91–102.
Hoffman, D. A., Magee, J. C., Colbert, C. M., and Johnston, D. (1997). K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387, 869–875.
Jaffe, D. B., Ross, W. N., Lisman, J. E., Lasser-Ross, N., Miyakawa, H., and Johnston, D. (1994). A model for dendritic Ca2+ accumulation in hippocampal pyramidal neurons based on fluorescence imaging measurements. J. Neurophysiol. 71, 1065–1077.
Jarsky, T., Roxin, A., Kath, W. L., and Spruston, N. (2005). Conditional dendritic spike propagation following distal synaptic activation of hipocampal CA1 pyramidal neurons. Nat. Neurosci. 8, 1667–1676.
Jhamandas, J. H., Cho, C., Jassar, B., Harris, K., MacTavish, D., and Easaw, J. (2001). Cellular mechanisms for amyloid beta-protein activation of rat cholinergic basal forebrain neurons. J. Neurophysiol. 86, 1312–1320.
Johnston, D., Hoffman, D. A., Colbert, C. M., and Magee, J. C. (1999). Regulation of back-propagating action potentials in hippocampal neurons. Curr. Opin. Neurobiol. 9, 288–292.
Jung, S. C., Kim, J., and Hoffman, D. A. (2008). Rapid, bidirectional remodeling of synaptic NMDA receptor subunit composition by A-type K+ channel activity in hippocampal CA1 pyramidal neurons. Neuron 60, 657–671.
Kamondi, A., Acsady, L., and Buzsaki, G. (1998). Dendritic spikes are enhanced by cooperative network activity in the intact hippocampus. J. Neurosci. 18, 3919–3928.
Katz, Y., Menon, V., Nicholson, D. A., Geinisman, Y., Kath, W. L., and Spruston, N. (2009). Synapse distribution suggests a two-stage model of dendritic integration in CA1 pyramidal neurons. Neuron 63, 171–177.
Khachaturian, Z. S. (1987). Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol. Aging 8, 345–346.
Kim, J., Wei, D. S., and Hoffman, D. A. (2005). Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J. Physiol. 569, 41–57.
Larkum, M. E., Nevian, T., Sandler, M., Polsky, A., and Schiller, J. (2009). Synaptic integration in tuft dendrites of layer 5 pyramidal neurons: a new unifying principle. Science 325, 756–760.
Liao, C. W., and Lien, C. C. (2009). Estimating intracellular Ca2+ concentrations and buffering in a dendritic inhibitory hippocampal interneuron. Neuroscience 164, 1701–1711.
London, M., Meunier, C., and Segev, I. (1999). Signal transfer in passive dendrites with nonuniform membrane conductance. J. Neurosci. 19, 8219–8233.
Losonczy, A., Makara, J. K., and Magee, J. C. (2008). Compartmentalized dendritic plasticity and input feature storage in neurons. Nature 452, 436–441.
Lue, L. F., Kuo, Y. M., Roher, A. E., Brachova, L., Shen, Y., Sue, L., Beach, T., Kurth, J. H., Rydel, R. E., and Rogers, J. (1999). Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 155, 853–862.
Magee, J. C., and Cook, E. P. (2000). Somatic EPSP amplitude is independent of synapse location in hippocampal pyramidal neurons. Nat. Neurosci. 3, 895–903.
Maravall, M., Mainen, Z. F., Sabatini, B. L., and Svoboda, K. (2000). Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys. J. 78, 2655–2667.
Metz, A. E., Jarsky, T., Martina, M., and Spruston, N. (2005). R-type calcium channels contribute to after depolarization and bursting in hippocampal CA1 pyramidal neurons. J. Neurosci. 25, 5763–5773.
Mickus, T., Jung, H., and Spruston, N. (1999). Properties of slow, cumulative sodium channel inactivation in rat hippocampal CA1 pyramidal neurons. Biophys. J. 76, 846–860.
Migliore, M., Ferrante, M., and Ascoli, G. A. (2005). Signal propagation in oblique dendrites of CA1 pyramidal cells. J. Neurophysiol. 94, 4145–4155.
Migliore, M., Hoffman, D. A., Magee, J. C., and Johnston, D. (1999). Role of an A-type K+ conductance in the back-propagation of action potentials in the dendrites of hippocampal pyramidal neurons. J. Comput. Neurosci. 7, 5–15.
Oh, M. M., Oliveira, F. A., and Disterhoft, J. F. (2010). Learning and aging related changes in intrinsic neuronal excitability. Front. Aging Neurosci. 2, 1–10.
Omori, T., Aonishi, T., Miyakawa, H., Inoue, M., and Okada, M. (2006). Estimated distribution of specific membrane resistance in hippocampal CA1 pyramidal neuron. Brain Res. 1125, 199–208.
Omori, T., Aonishi, T., Miyakawa, H., Inoue, M., and Okada, M. (2009). Steep decrease in the specific membrane resistance in the apical dendrites of hippocampal CA1 pyramidal neurons. Neurosci. Res. 64, 83–95.
Scheibel, A. B. (1979). The hippocampus: organizational patterns in health and senescence. Mech. Ageing Dev. 9, 89–102.
Sjostrom, P. J., Rancz, E. A., Roth, A., and Hausser, M. (2008). Dendritic excitability and synaptic plasticity. Physiol. Rev. 88, 769–840.
Song, D., Wang, Z., and Berger, T. W. (2002). Contribution of T-type VDCC to TEA-induced long-term synaptic modification in hippocampal CA1 and dentate gyrus. Hippocampus 12, 689–697.
Spruston, N., Schiller, Y., Stuart, G., and Sakmann, B. (1995). Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science 268, 297–300.
Stuart, G. J., Spruston, N., Häusser, M. (eds). (2008). Dendrites (2nd ed). New York: Oxford University Press.
Stutzmann, G. E. (2007). The pathogenesis of Alzheimers disease is it a lifelong “calciumopathy”? Neuroscientist 13, 546–559.
Ueda, K., Shinohara, S., Yagami, T., Asakura, K., and Kawasaki, K. (1997). Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J. Neurochem. 68, 265–271.
Urakubo, H., Aihara, T., Kuroda, S., Watanabe, M., and Kondo, S. (2004). Spatial localization of synapses required for supralinear summation of action potentials and EPSPs. J. Comput. Neurosci. 16, 251–265.
Varga, A. W., Anderson, A. E., Adams, J. P., Vogel, H., and Sweatt, J. D. (2000). Input-specific immunolocalization of differentially phosphorylated Kv4.2 in the mouse brain. Learn. Mem. 7, 321–332.
Wang, J., Ikonen, S., Gurevicius, K., van Groen, T., and Tanila, H. (2003). Altered auditory-evoked potentials in mice carrying mutated human amyloid precursor protein and presenilin-1 transgenes. Neuroscience 116, 511–517.
Watanabe, S., Hoffman, D. A., Migliore, M., and Johnston, D. (2002). Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc. Natl. Acad. Sci. U.S.A. 99, 8366–8371.
Xu, C., Qian, C., Zhang, Z., Wu, C., Zhou, P., and Liang, X. (1998). Effects of beta-amyloid peptide on transient outward potassium current of acutely dissociated hippocampal neurons in CA1 sector in rats. Chin. Med. J. (Engl.) 111, 492–495.
Ye, C. P., Selkoe, D. J., and Hartley, D. M. (2003). Protofibrils of amyloid beta-protein inhibit specific K+ currents in neocortical cultures. Neurobiol. Dis. 13, 177–190.
Yue, C., and Yaari, Y. (2004). KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J. Neurosci. 24, 4614–4624.
Keywords: action potential backpropagation, computer model, electrical activity
Citation: Morse TM, Carnevale NT, Mutalik PG, Migliore M and Shepherd GM (2010) Abnormal excitability of oblique dendrites implicated in early Alzheimer’s: a computational study. Front. Neural Circuits 4:16. doi: 10.3389/fncir.2010.00016
Received: 04 November 2009;
Paper pending published: 18 December 2009;
Accepted: 04 May 2010;
Published online: 31 May 2010
Edited by:
Nelson Spruston, Northwestern University, USAReviewed by:
Dalton J. Surmeier, Northwestern University, USANelson Spruston, Northwestern University, USA
Copyright: © 2010 Morse, Carnevale, Mutalik, Migliore and Shepherd. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Thomas M. Morse, Department of Neurobiology, Yale University School of Medicine, P.O. Box 208001, New Haven, CT 06520-8001, USA. e-mail:dG9tLm1vcnNlQHlhbGUuZWR1