 William M. Connelly
William M. Connelly Adam C. Errington1
Adam C. Errington1 Giuseppe Di Giovanni
Giuseppe Di Giovanni Vincenzo Crunelli
Vincenzo Crunelli- 1Neuroscience Division, Cardiff School of Biosciences, Cardiff University, Cardiff, UK
- 2Department of Physiology and Biochemistry, Faculty of Medicine, Malta University, Msida, Malta
A large body of work now shows the importance of GABAA receptor-mediated tonic inhibition in regulating CNS function. However, outside of pathological conditions, there is relatively little evidence that the magnitude of tonic inhibition is itself under regulation. Here we review the mechanisms by which tonic inhibition is known to be modulated, and outline the potential behavioral consequences of this modulation. Specifically, we address the ability of protein kinase A and C to phosphorylate the extrasynaptic receptors responsible for the tonic GABAA current, and how G-protein coupled receptors can regulate tonic inhibition through these effectors. We then speculate about the possible functional consequences of regulating the magnitude of the tonic GABAA current.
Introduction
GABA is the major inhibitory neurotransmitter in the mammalian forebrain. It is estimated that a third of synapses in the forebrain use GABA as their neurotransmitter (Bloom and Iversen, 1971). Through ionotropic GABAA receptors, GABA works to increase membrane permeability to Cl- (and to a lesser extent HCO3-) thereby reducing membrane impedance and potentially hyperpolarizing the membrane potential. The role of GABAA receptor-mediated inhibition in the control of neural function is undeniable, and can be seen in nearly every aspect of neural function (Macdonald and Olsen, 1994; Freund and Buzsáki, 1996; Farrant and Nusser, 2005).
GABAA receptors are believed to form as a pentameric assembly, out of 19 possible subunits (α1–6, β1–3, γ1–3, δ, ε, θ, π and ρ1–3), generally as a combination of α, β and γ subunits. Other combinations exist, where δ, ε, θ or π subunits replace the γ subunit. Finally, other permutations have been described, such as ρ homopentamers and receptors containing solely α and β subunits (Sieghart and Sperk, 2002). Importantly, different subunit combinations give GABAA receptors different functional properties, e.g., different activation, deactivation and desensitization rates and altering their affinity for GABA and exogenous compounds (Verdoorn et al., 1990; Sigel et al., 1991). Furthermore, specific subunit combinations have specific expression patterns, often being expressed in restricted brain nuclei or neuronal cell types (Sieghart and Sperk, 2002). Finally, even on the level of a single cell, GABAA receptors with a specific subunit make-up can be expressed in different subcellular compartments.
With this complexity in mind, a wealth of evidence has demonstrated that GABAA receptors with specific subunit compositions, which are expressed in a unique spatial distribution, mediate a persistence or “tonic” inhibitory conductance. These receptors are generally α4βδ and α6βδ (though there are also α5βγ and others). They are expressed at a high density in the extrasynaptic compartment of dentate gyrus granule cells, cerebellar granule cells and thalamocortical cells (and to a lesser extent in olfactory bulb granule cells and striatal medium spiny cells) (Brickley and Mody, 2012). Due to their high affinity for GABA, and relatively slow desensitization rates, these extrasynaptic GABAA receptors are believed to sense the activity dependent spill over of GABA from the synaptic cleft as well as the ambient concentration of GABA (and potentially they provide tonic inhibition in the absence of GABA; Wlodarczyk et al., 2013). There is a growing body of evidence showing the importance of tonic inhibition in regulating a variety of CNS functions, including sensory processing, controlling epileptiform activity and modulating anxiety states (Chadderton et al., 2004; Maguire et al., 2005; Cope et al., 2009). However, what is less clear is when and how the nature and magnitude of the tonic current are regulated. There are several studies that show that the magnitude of tonic current is altered in pathophysiological states, especially as a result of epilepsy, but it is less clear whether tonic currents are regulated during normal CNS function (Naylor et al., 2005; Payne et al., 2006; Zhang et al., 2007). Therefore, in this review, we will cover mechanisms by which tonic GABAA inhibition can be regulated, specifically focusing on metabotropic regulation. Furthermore, we highlight potential paradigms where this regulation may be used in vivo to modulate inhibitory tone.
Kinases
Phosphorylation is one of the most well understood post-translational modifications a protein can undergo. This reaction is catalyzed by kinases, and involves the transfer of a phosphate group from ATP to a serine, threonine or tyrosine residue in the target polypeptide. This phorphorylation changes the structure of the protein, and potentially its function. Due to the residues they target, kinases are generally subdivided into serine/threonine kinases such as calcium-dependent protein kinase (PKC) or cyclic AMP dependent protein kinase (PKA), tyrosine kinases such a v-Src, dual specificity kinases and histidine kinases (Edelman et al., 1987; Dhanasekaran and Premkumar Reddy, 1998; Schlessinger, 2000; West and Stock, 2001). Furthermore, while these families of kinases target a specific residue (or two, in the case of serine/threonine kinases), each individual family of kinases recognizes a general sequence of amino acid residues: a so called “consensus site.” This consensus site is in the order of 5–10 residues long, and is more or less specific depending on the family of kinases, for example, PKC is known for having a broad substrate specificity (Edelman et al., 1987). However, just because a protein contains a consensus site for a kinase, it does not guarantee that protein is a target for the kinase, for instance steric hindrance may prevent the kinase from accessing the site (Edelman et al., 1987).
PKC Mediated Regulation
One of the earliest pieces of evidence that GABAA receptors can be modulated by kinases directly was provided by Sigel et al. (1991), who demonstrated that phorbol myristate acetate (PMA) stereo-selectively reduced the amplitude of evoked GABA currents recorded in Xenopus oocytes expressing GABAA receptors with a variety of subunit compositions. Soon afterward, this effect was shown to be mediated by phosphorylation of both β and γ subunits, with serine 409 (S409) being the target on the β1 and β3 subunits and S410 being the target on β2 subunits, while S327 and S343 are the target on the γ2 subunit (Figure 1) (Kellenberger et al., 1992; Moss et al., 1992a; Krishek et al., 1994; McDonald and Moss, 1997). It is also worth noting that the alternative splicing that occurs on the γ2 subunit, which inserts 8 additional amino acids to create the γ2L subunit, adds a serine residue that satisfies the consensus site for phosphorylation by PKC and other kinases (Moss and Smart, 1996). Similarly, the β2 subunit is subjected to alternative splicing, though only in the chicken and human, and not rodent (McKinley et al., 1995). The β2L subunit is differentiated from the β2S subunit by an insertion of 17 amino acids in the chicken, and 38 amino acids in the human, both of which contain a strong consensus site for PKC (Harvey et al., 1994; McKinley et al., 1995). The α4 subunit appears to be unique amongst α subunits in that it expresses a consensus site between transmembrane domains 3 and 4 at S443 (Figure 1; Abramian et al., 2010). The recruitment of PKC to GABAA receptors (and especially their β subunits) appears to be facilitated by the receptor for activated C kinase (RACK-1; Brandon et al., 1999).
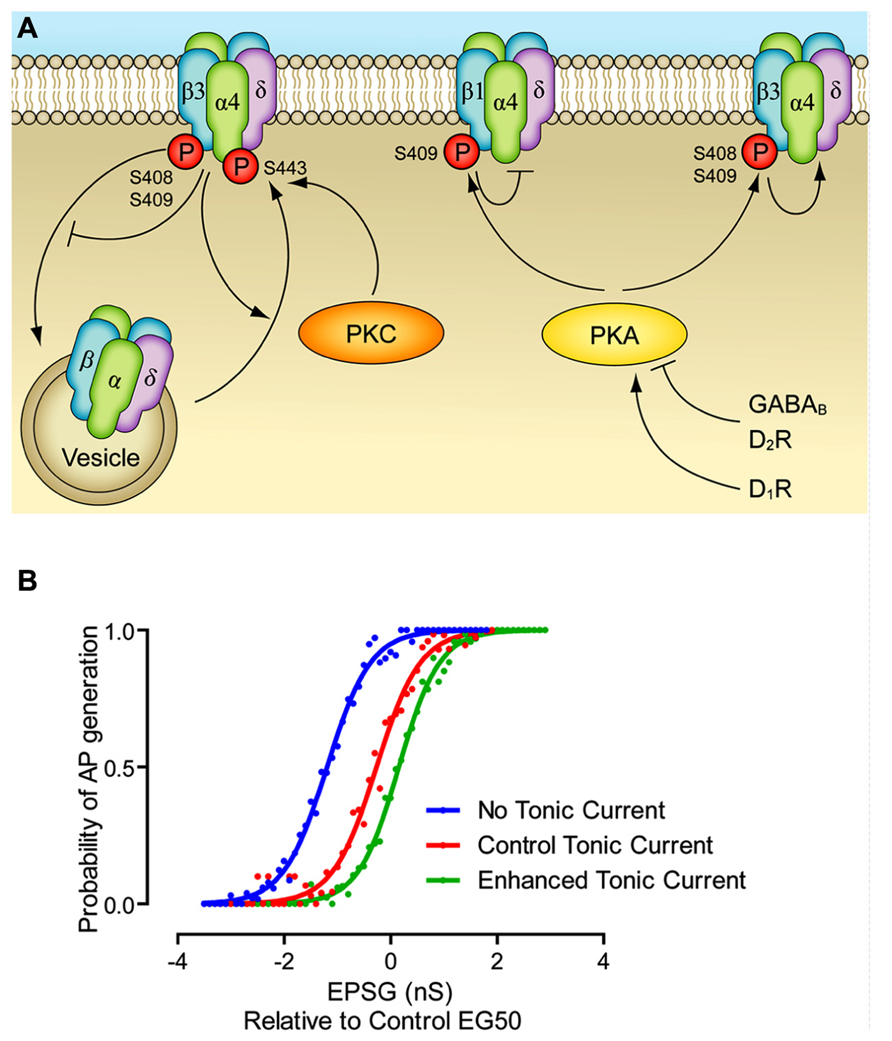
FIGURE 1. Modulation of extrasynaptic GABAA receptors. (A) PKC leads to phosphorylation of α4 and β3 subunits, which increases cell surface stability. PKA leads to phosphorylation of β1 and β3 subunits, which inhibit and enhance GABAA function, respectively. PKA mediated modulation of extrasynaptic currents has been demonstrated via dopamine D1 and D2 receptors, and via GABAB receptors. Other potential pathways have been demonstrated, but they are not included here since they have not been as fully elucidated. Enhancement of a pathway is represented by an arrow head, inhibition is represented by a bar. (B) The effect of dynamic modulation of the tonic current on the neuronal input/output (I/O) function. Dynamic clamp was used to inject excitatory postsynaptic conductances (EPSGs) into neurons in control conditions, when the tonic GABAA current was blocked by gabazine, and when the tonic current was enhanced to the level produced by GABAB receptor agonists. There was no artificial injection of excitatory noise in this experiment, though the tonic current was produced via a noisy conductance model. Note that altering the level of the tonic current changes the offset of I/O function, without affecting the gain. Modified from Connelly et al. (2013).
The effect of PKC activation on GABAA receptors is diverse, and appears to be dependent on the subunit composition in question. For instance, in hippocampal pyramidal cells, PKC appeared to have no effect on miniature inhibitory postsynaptic potentials (mIPSCs), while in dentate gyrus granule cells, PKC enhanced mIPSC amplitudes (Poisbeau et al., 1999). Furthermore, it has been shown that PKC causes an enhancement of receptor function in α1β1γ2L expressing cell lines (Lin et al., 1996) and an increase in mIPSC amplitudes mediated by αxβ3yx receptors (Jovanovic et al., 2004). Similarly, there is a large amount of evidence suggesting that PKC regulates the cell-surface expression and the stability at the membrane of GABAA receptors. In both expression systems expressing α1β2γ2 and cultured cortical neurons, where there is constitutive recycling of GABAA receptors from the cell-surface, PKC activity leads to a decrease of cell-surface GABAA receptors and associated currents (Connolly et al., 1999; Filippova et al., 2000; Balduzzi et al., 2002; Herring et al., 2005). Interestingly, this effect appears to be independent of direct phosphorylation of the GABAA receptor, and instead must involve phosphorylation of some other protein in the endocytotic cascade (Connolly et al., 1999). Thus, it is clear that synaptic GABAA receptors can be modulated by PKC. Perhaps then it is surprising that there is such a paucity of results linking kinase action to the tonic GABAA current. The following review of the available findings clearly indictes that further research into this area is warranted.
There is significant evidence that ethanol is a high affinity positive modulator of the α4/6βxδ receptors responsible for the tonic GABAA current. Furthermore, this potentiation is, at least in part, responsible for the behavioral action of ethanol (Hanchar et al., 2005; Mody et al., 2007). Curiously, it appears as if the action of ethanol at these receptors is dependent on PKC. Choi et al. (2008) demonstrated both an anatomical and biochemical linkage between the PKC isozyme PKCδ and the δ subunit of the GABAA receptor. They reported that the distribution of PKCδ protein overlapped with that of the δ subunit. They went on to show that ethanol failed to potentiate tonic currents recorded from PKCδ knockout animals. Likewise, ethanol only potentiated α4β3δ mediated currents in cell lines also expressing PKCδ. Curiously, knocking out PKCδ appeared to have no effect on the baseline magnitude of the tonic current, indicating that at least in this paradigm, PKCδ only regulated the activity of other drugs at extrasynaptic receptors, rather than the activity of the receptors themselves. These effects are relatively rapid, and likely reflect the direct interaction of ethanol with the GABAA receptor. It is worth noting that a similar effect has been observed for synaptic γ-containing GABAA receptors and neurosteroids (e.g., Fáncsik et al., 2000). However, a negative interaction between kinase activity and neurosteroid action has also been noted at extrasynaptic receptors. In rats that had kindling-induced seizures, Kia et al. (2011) demonstrated that while the tonic current in CA1 pyramidal cells (likely mediated by α5β3γx) was similar to sham-controls, the extrasynaptic receptors were completely insensitive to the neurosteroid THDOC. This neurosteroid insensitivity could be reproduced in naive animals by PKC activation, though it is worth noting that the phosphatase activator Li-palmitate could not cause the tonic current recorded in kindled rats to show its normal THDOC sensitivity.
Just as kinase activity appears to regulate cell-surface expression of synaptic GABAA receptors, there is evidence that kinases play a similar role at extrasynaptic receptors. Abramian et al. (2010) demonstrated that PKC activity in expression systems and hippocampal slices leads to an increase in α4 phosphorylation and cell surface expression, apparently at odds with what occurs at synaptic GABAA receptors (e.g., Connolly et al., 1999). This increase in cell-surface expression was mirrored by an increase in GABAA receptor mediated currents. Importantly, PKC activators could no longer enhance surface expression or currents in cell lines expressing a point mutation on the α4 subunit, whereby the phosphor-sensitive S443 residue was replaced with an alanine. These results contradict the analogous results seen at synaptic receptors, where PKC activity decreases cell surface expression, and is thought to do so independently of direct phosphorylation of GABAA receptors (Connolly et al., 1999). PKC also seems to be able to regulate cell surface expression via phosphorylation of the β subunit, though apparently in an opposite direction to α4-phosphorylation reported by Abramian et al. (2010). Application of the PKC activator PMA inhibited tonic currents in dentate gyrus granule cells and thalamocortical cells and inhibiting PKC with bisindolylmaleimide I enhanced the tonic current (Bright and Smart, 2013). This result could be replicated in HEK293 cells expressing α4β2δ receptors, where the effect was dependent on phosphorylation at the S410 residue on β2 subunits, and independent of S443 on α4 subunits. Live cell imaging revealed that PKC activity was associated with a decrease in cell surface expression of δ-subunits (Bright and Smart, 2013). On the other hand, downstream of BDNF signaling, PKC has been shown to increase the cell surface stability of δ-subunits. BDNF was demonstrated to activate TrkB receptors, which in turn activated PLCγ. Presumably this then leads to an increase in intracellular Ca2+, and PKC activity, as the increased surface expression of δ-subunits was blocked by inhibitors of PLC and PKC. Unfortunately, the exact site of PKC phosphorylation on the GABAA protein (or even whether it was on another protein altogether) was not elucidated (Joshi and Kapur, 2009).
PKA Mediated Regulation
PKA exists in many subtypes, but irrespective of the subtype, it is formed as a heterotetramer composed of two catyltic subunits held in an inactive state through an interaction with a dimer of regulatory subunits. PKA’s main regulatory mechanism is through binding of cAMP, but is also compartmentalized and regulated through an interaction with A-kinase-anchoring proteins (AKAPs; Pidoux and Taskén, 2010). PKA is a well established modulator of GABAA receptors. While other subunits may contain PKA consensus sites, so far the only subunit that appears to be phosphoylated by PKA are the β subunits (McDonald et al., 1998 and citations therein). Indeed, more selectively than that, PKA appears to act only on β1 and β3 subunits, at S409 and S408/S409 respectively (Moss and Smart, 1996; McDonald et al., 1998). In HEK cells expressing α1β1γ2, PKA activation inhibits evoked GABAA currents, while PKA enhances currents mediated by α1β3γ2 receptors (Moss et al., 1992b; McDonald et al., 1998). These results all come from synaptic subunit combinations.
However, the picture is not so clear cut for extrasynaptic isoforms. Tang et al. (2010) demonstrated that in HEK cells expressing α4β3δ receptors, PKA activation led to an increase in purely spontaneous GABA currents, that is, currents measured in the absence of GABA, while PKA had no effect on spontaneous currents measured from α4β3γ2L receptors. However, in the presence of low concentrations of GABA (1 μM), the effect was reversed, and PKA appeared to inhibit α4β3δ receptors. However, outside of expression systems, the effect of PKA becomes even more unclear. For instance, Poisbeau et al. (1999) reported that intracellular infusion of PKA suppressed mIPSCs recorded from hippocampal CA1 pyramidal cells, but had no effect on those recorded from dentate gyrus granule cells. This result cannot easily be explained in terms of differential expression of β subunits, as the both CA1 and dentate gyrus cells express all flavors of β subunit (Wisden et al., 1992). Likewise, while Nusser et al. (1999) reported that intracellular infusion of PKA enhanced mIPSC amplitude in olfactory granule cells (a cell type that only expresses the β3 subunit), Brünig et al. (1999) found that in the same cell type, dopamine D1 receptor agonists (which should stimulate adenylate cyclase and enhance PKA action) reduced evoked GABAA receptor currents, in a manner that was blocked by PKA inhibitors, suggesting that PKA inhibits GABAA receptors in these cells. It is possible to explain this result in light of the fact that Brünig et al. (1999) worked on cultured neurons, activating PKA through a more physiological G-protein coupled approach, while Nusser et al. (1999) used native tissue, but caused phosphorylation through infusing active PKA. Finally, it may also appear that the length of time PKA activity is increased for can produce different effects, as Angelotti et al. (1993) reported that expression of α1β1γ2S receptors in cell lines with higher PKA activity had higher GABAA receptor mediated currents than those with lower PKA activity, in comparison to direct application of PKA activators that usually inhibit β1 containing GABAA receptors.
Regarding PKA activity at extrasynaptic GABAA receptors, there are two papers which appear to reveal the picture. Janssen et al. (2009) demonstrated that a dopamine D1 receptor agonist enhanced a tonic current believed to be mediated by α5β3γ receptors in D1-positive striatal medium spiny neurons, while a D2 receptor agonist (which should inhibit adenylate cyclase and inhibit PKA action) reduced the tonic current (believed to be mediated by the same α5β3γ) in D2-positive neurons. Curiously, PKA infusion enhanced the tonic current in D1-positive medium spiny neurons, while it inhibited the current in D2-positive neurons. Thus, while experiments involving dopamine receptor agonists support the notion of McDonald et al. (1998) that PKA activity at β3 containing receptors enhances GABAA receptor function, the experiments involving PKA infusion paint a more complex picture. However, the results can be understood when one considers that the PKA inhibitor PKI reduced the tonic current in D2-positive cells, but had no effect in D1-positive cells, implying that β3 containing receptors are basally phosphorylated at D1-positive cells, but not at D2-positive cells. Thus application of PKA to D1-positive cells would have no action at β3 subunits, and may potentially be having its effect via a small proportion of β1 containing receptors. In a more straightforward to interpret result, Connelly et al. (2013) demonstrated that activating the GABAB receptor enhanced the α4βδ mediated tonic current in thalamocortical cells and dentate gyrus granule cells, as well as the α6βδ mediated tonic current in cerebellar granule cells, an effect mimicked by PKA inhibitors. Inversely, infusing PKA decreased the tonic current, however the β subunit involvement was not determined in this paper (also see Tao et al., 2013).
PKA is also known to regulate the cell surface stability of GABAA receptors. For instance, dopamine D3 receptor activation has been shown to increase the rate of clatherin-mediated endocytosis of synaptic GABAA receptors in a PKA-dependent fashion (Chen et al., 2006). Likewise, PKA can regulate the expression of GABAA receptors. Specifically, the expression of the δ subunit is known to be highly dynamic in cerebellar granule cells (e.g., Payne et al., 2008). Uusi-Oukari et al. (2010) reported that AMPA receptor activation led to an increase in δ-subunit mRNA in cultured cerebellar granule cells, and that this effect was dependent on PKA.
Other Kinase Mediated Regulation
There are only a small number of studies investigating the effects of non-PKA/PKC mediated modulation of the tonic GABAA current, indicating the need for more research in this area. Tyrosine kinases can phosphorylate GABAA γ2 subunits at Y365/367, which reduces the ability of the clathrin-adaptor protein, AP2, to bind, resulting in reduced internalization and the subsequent increase in membrane insertion of the channels (Moss et al., 1995; Kittler et al., 2008). It appears that this site is constitutively phorphorylated and its effect is more readily seen by blocking phosphorylation, rather than enhancing it (Brandon et al., 2001). Therefore Nani et al. (2013) used a Y365/367F mouse line, where the principle tyrosine sites were mutated to phenylalanine, blocking phosphorylation and AP2 binding. As would be predicted, spontaneous IPSC amplitude was increased, while the decay was unaffected. Curiously, the expression of α4 and δ subunits were increased, as were the tonic current recorded in dorsal lateral geniculate neurons. Furthermore, these effects seemed limited to female mice. Exactly how alterations in γ2 surface expression lead to an increase in α4 and δ subunit expression is unclear.
Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a serine/threonine that has been demonstrated to be able to modulate synaptic inhibition in a wide variety of cell types (e.g., Wang et al., 1995). Saliba et al. (2012) extended these findings by showing that CaMKII activation, subsequent to Ca2+ influx produced by Bay K 8644 application, produced a profound increase in surface insertion of α5 and β3 subunits, and an increase in a tonic current mediated by α5β3γ2 receptors. This effect was mediated by phosphorylation at S383 on the β3 subunit. While in these experiments Ca2+ influx was caused by Bay K 8644 or 4-AP application, they do suggest the possibility of activity dependent regulation of the tonic current (see Implications).
Wang et al. (2012) investigated how acute systemic inflammation leads to memory loss. Systemic interleukin-1β (IL-1β) injections caused an impairment of contextual fear memory, an effect which was absent in α5-/- animals or in animals treated with L-655, 708, an inverse agonist selective for α5-containin GABAA receptors. Acute systemic IL-1β injections or in vitro application of IL-1β both caused an increase in the tonic current measured in hippocampal CA1 cells, where there was a concurrent increase in α5 subunit surface expression. This effect was dependent on the activity of serine/threonine kinase, p38 mitogen-activated protein kinase (MAPK), though how it causes increased cell-surface expression of α5 subunit containing receptors is still unclear.
Presynaptic Regulation of Tonic Current
As the δ-containing GABAA receptors appear to sense ambient GABA and/or GABA which spills over from the synaptic cleft, it seems likely that manipulations that increase the release of GABA will increase the magnitude of the tonic current. Indeed, it appears that blocking action potential dependent release of GABA can reduce the size of the tonic current (e.g., Brickley et al., 1996; Glykys and Mody, 2007; though see Wall and Usowicz, 1997; Rossi et al., 2003). But can more subtle manipulations of GABA release alter the tonic current? Indeed, it appears that they can. Rossi et al. (2003) demonstrated that in cerebellar granule cells, acetylcholine, acting through nicotinic receptors, produces a largely vesicular, Ca2+ dependent, action potential-independent release of GABA that causes a 12 fold enhancement in the magnitude of the tonic GABAA current. The exact source of this GABA is unclear, but the authors speculate that it is caused by presynaptic nicotinic receptors on interneuron terminals, causing presynaptic depolarization, and hence vesicular GABA release. This finding is mirrored by Errington et al. (2011) who reported that group I metabotropic glutamate receptor agonists cause an increase in spontaneous IPSC frequency and tonic current in thalamocortical neurons of the dorsal lateral geniculate nucleus. While the IPSCs are clearly action potential-dependent, the increase in tonic current was independent of action potentials, again pointing to the notion that presynaptic receptors were facilitating release from interneurons (for similar findings see Krishek et al., 1994; Kullmann and Semyanov, 2002). The inverse case was demonstrated by Bright and Brickley (2008), where depolarization of ventrobasal thalamocortical cells induced a robust increase in spontaneous IPSC frequency, but failed to affect the tonic current. Thus, presynaptic modulation of GABA release can enhance the tonic current, but increasing action potential-dependent release does not necessarily enhance the tonic current.
Implications
If the tonic GABAA current simply provides a hyperpolarizing/shunting influence on the membrane, why do neurons use it, rather than classical leak potassium channels? One suggestion is that the largely shunting inhibition provided by tonic inhibition alters the input/output function of the neuron in a way that hyperpolarizing inhibition (as produced by potassium channels) cannot. That is to say, hyperpolarizing inhibition alters the offset (the excitatory input needed to bring the cell to fire) while not greatly affecting the gain, i.e., the relationship between input excitation and firing rate. Shunting inhibition is often suggested to largely have the opposite effect, reducing the gain, while not affecting the offset. However, it appears that the situation is more complex, and also depends on the nature of the excitatory drive, specifically, during tonic excitation shunting inhibition affects only the offset, while during noisy trains of excitation shunting inhibition mainly alters the gains (Figure 1B; Holt and Koch, 1997; Mitchell and Silver, 2003; Prescott and Koninck, 2003; Semyanov et al., 2004). Indeed, this is further complicated by the rectifying property of the tonic current, as seen in several cell types (Pavlov et al., 2009; Ransom et al., 2010). We suggest a reason that may also come into play is the plasticity afforded to the tonic GABAA receptor system. As described above (Table 1), there are a multitude of pathways by which the magnitude of the tonic current can be modulated, with most of them largely independent of synaptic GABA release. This means that, as opposed to regulation of the potassium channels responsible for the resting membrane potential, modulating tonic GABAA inhibition affects neuronal excitability largely independently of the resting membrane potential.

TABLE 1. Summary of the effects of kinase action on GABAA receptor mediated tonic currents.
The results cited above clearly demonstrate that the tonic GABAA system is susceptible to modulation (Figure 1). While there have been some studies showing a role of dynamic modulation of the tonic current, for instance in response to ethanol abuse or in response to epilepsy, these effects are generally seen to be due to changes in expression (Cagetti et al., 2003; Maguire et al., 2005; Payne et al., 2007). It would be fascinating to investigate whether more rapid changes in the magnitude of tonic current can occur due to kinase-dependent modulation, for instance during the switch between different levels of vigilance (in response to changing levels of brain stem neuromodulators). On a simpler, cellular level, the results summarized above show the diversity of effects caused by kinase action on extrasynaptic GABAA receptors. However, there are relatively few data demonstrating whether G-protein coupled receptors are able to induce the same effects. Similarly, while PKC has been shown to modulate extrasynaptic GABAA receptors, we are not aware of any papers that show that interventions that cause a rise in intracellular Ca2+ (and hence PKC activity) can modulate the tonic current through PKC (though see Saliba et al., 2012). Hence, the notion of activity-dependent regulation of δ-containing GABAA receptors remains attractive, yet elusive.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abramian, A. M., Comenencia-Ortiz, E., Vithlani, M., Tretter, E. V., Sieghart, W., Davies, P. A., et al. (2010). Protein kinase C phosphorylation regulates membrane insertion of GABAA receptor subtypes that mediate tonic inhibition. J. Biol. Chem. 285, 41795–41805. doi: 10.1074/jbc.M110.149229
Angelotti, T. P., Uhler, M. D., and Macdonald, R. L. (1993). Enhancement of recombinant gamma-aminobutyric acid type A receptor currents by chronic activation of cAMP-dependent protein kinase. Mol. Pharmacol. 44, 1202–1210.
Balduzzi, R., Cupello, A., and Robello, M. (2002). Modulation of the expression of GABA(A) receptors in rat cerebellar granule cells by protein tyrosine kinases and protein kinase C. Biochim. Biophys. Acta 1564, 263–270. doi: 10.1016/S0005-2736(02)00460-1
Bloom, F. E., and Iversen, L. L. (1971). Localizing 3H-GABA in nerve terminals of rat cerebral cortex by electron microscopic autoradiography. Nature 229, 628–630. doi: 10.1038/229628a0
Brandon, N. J., Delmas, P., Hill, J., Smart, T. G., and Moss, S. J. (2001). Constitutive tyrosine phosphorylation of the GABAA receptor γ2 subunit in rat brain. Neuropharmacology 41, 745–752. doi: 10.1016/S0028-3908(01)00121-6
Brandon, N. J., Uren, J. M., Kittler, J. T., Wang, H., Olsen, R., Parker, P. J., et al. (1999). Subunit-specific association of protein kinase C and the receptor for activated C kinase with GABA type A receptors. J. Neurosci. 19, 9228–9234.
Brickley, S. G., Cull-Candy, S. G., and Farrant, M. (1996). Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J. Physiol. 497, 753–759.
Brickley, S. G., and Mody, I. (2012). Extrasynaptic GABA(A) receptors: their function in the CNS and implications for disease. Neuron 73, 23–34. doi: 10.1016/j.neuron.2011.12.012
Bright, D. P., and Brickley, S. G. (2008). Acting locally but sensing globally: impact of GABAergic synaptic plasticity on phasic and tonic inhibition in the thalamus. J. Physiol. 586, 5091–5099. doi: 10.1113/jphysiol.2008.158576
Bright, D. P., and Smart, T. G. (2013). Protein kinase C regulates tonic GABAA receptor-mediated inhibition in the hippocampus and thalamus. Eur. J. Neurosci. doi: 10.1111/ejn.12352 [Epub ahead of print].
Brünig, I., Sommer, M., Hatt, H., and Bormann, J. (1999). Dopamine receptor subtypes modulate olfactory bulb gamma-aminobutyric acid type A receptors. Proc. Natl. Acad. Sci. U.S.A. 96, 2456–2460. doi: 10.1073/pnas.96.5.2456
Cagetti, E., Liang, J., Spigelman, I., and Olsen, R. W. (2003). Withdrawal from chronic intermittent ethanol treatment changes subunit composition, reduces synaptic function, and decreases behavioral responses to positive allosteric modulators of GABAA receptors. Mol. Pharmacol. 63, 53–64. doi: 10.1124/mol.63.1.53
Chadderton, P., Margrie, T. W., and Häusser, M. (2004). Integration of quanta in cerebellar granule cells during sensory processing. Nature 428, 856–860. doi: 10.1038/nature02442
Chen, G., Kittler, J. T., Moss, S. J., and Yan, Z. (2006). Dopamine D3 receptors regulate GABAA receptor function through a phospho-dependent endocytosis mechanism in nucleus accumbens. J. Neurosci. 26, 2513–2521. doi: 10.1523/JNEUROSCI.4712-05.2006
Choi, D.-S., Wei, W., Deitchman, J. K., Kharazia, V. N., Lesscher, H. M. B., McMahon, T., et al. (2008). Protein kinase Cdelta regulates ethanol intoxication and enhancement of GABA-stimulated tonic current. J. Neurosci. 28, 11890–11899. doi: 10.1523/JNEUROSCI.3156-08.2008
Connelly, W. M., Fyson, S. J., Errington, A. C., McCafferty, C. P., Cope, D. W., Di Giovanni, G., et al. (2013). GABAB receptors regulate extrasynaptic GABAA receptors. J. Neurosci. 33, 3780–3785. doi: 10.1523/JNEUROSCI.4989-12.2013
Connolly, C. N., Kittler, J. T., Thomas, P., Uren, J. M., Brandon, N. J., Smart, T. G., et al. (1999). Cell surface stability of gamma-aminobutyric acid type A receptors. Dependence on protein kinase C activity and subunit composition. J. Biol. Chem. 274, 36565–36572. doi: 10.1074/jbc.274.51.36565
Cope, D. W., Di Giovanni, G., Fyson, S. J., Orbán, G., Errington, A. C., Lorincz, M. L., et al. (2009). Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat. Med. 15, 1392–1398. doi: 10.1038/nm.2058
Dhanasekaran, N., and Premkumar Reddy, E. (1998). Signaling by dual specificity kinases. Oncogene 17, 1447–1455. doi: 10.1038/sj.onc.1202251
Edelman, A. M., Blumenthal, D. K., and Krebs, E. G. (1987). Protein serine/threonine kinases. Annu. Rev. Biochem. 56, 567–613. doi: 10.1146/annurev.bi.56.070187.003031
Errington, A. C., Di Giovanni, G., Crunelli, V., and Cope, D. W. (2011). mGluR control of interneuron output regulates feedforward tonic GABAA inhibition in the visual thalamus. J. Neurosci. 31, 8669–8680. doi: 10.1523/JNEUROSCI.0317-11.2011
Fáncsik, A., Linn, D. M., and Tasker, J. G. (2000). Neurosteroid modulation of GABA IPSCs is phosphorylation dependent. J. Neurosci. 20, 3067–3075.
Farrant, M., and Nusser, Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 6, 215–229. doi: 10.1038/nrn1625
Filippova, N., Sedelnikova, A., Zong, Y., Fortinberry, H., and Weiss, D. S. (2000). Regulation of recombinant gamma-aminobutyric acid (GABA)(A) and GABA(C) receptors by protein kinase C. Mol. Pharmacol. 57, 847–856.
Freund, T. F., and Buzsáki, G. (1996). Interneurons of the hippocampus. Hippocampus 6, 347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3. 0.CO;2-I
Glykys, J., and Mody, I. (2007). The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J. Physiol. 582, 1163–1178. doi: 10.1113/jphysiol.2007.134460
Hanchar, H. J., Dodson, P. D., Olsen, R. W., Otis, T. S., and Wallner, M. (2005). Alcohol-induced motor impairment caused by increased extrasynaptic GABA(A) receptor activity. Nat. Neurosci. 8, 339–345. doi: 10.1038/nn1398
Harvey, R. J., Chinchetru, M. A., and Darlison, M. G. (1994). Alternative splicing of a 51-nucleotide exon that encodes a putative protein kinase C phosphorylation site generates two forms of the chicken gamma-aminobutyric acidA receptor beta 2 subunit. J. Neurochem. 62, 10–16. doi: 10.1046/j.1471-4159.1994.62010010.x
Herring, D., Huang, R., Singh, M., Dillon, G. H., and Leidenheimer, N. J. (2005). PKC modulation of GABAA receptor endocytosis and function is inhibited by mutation of a dileucine motif within the receptor β2 subunit. Neuropharmacology 48, 181–194. doi: 10.1016/j.neuropharm.2004.09.015
Holt, G. R., and Koch, C. (1997). Shunting inhibition does not have a divisive effect on firing rates. Neural Comput. 9, 1001–1013. doi: 10.1162/neco.1997.9.5.1001
Janssen, M. J., Ade, K. K., Fu, Z., and Vicini, S. (2009). Dopamine modulation of GABA tonic conductance in striatal output neurons. J. Neurosci. 29, 5116–5126. doi: 10.1523/JNEUROSCI.4737-08.2009
Joshi, S., and Kapur, J. (2009). Slow intracellular accumulation of GABA(A) receptor delta subunit is modulated by brain-derived neurotrophic factor. Neuroscience 164, 507–519. doi: 10.1016/j.neuroscience.2009.08.008
Jovanovic, J. N., Thomas, P., Kittler, J. T., Smart, T. G., and Moss, S. J. (2004). Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABA(A) receptor phosphorylation, activity, and cell-surface stability. J. Neurosci. 24, 522–530. doi: 10.1523/JNEUROSCI.3606-03.2004
Kellenberger, S., Malherbe, P., and Sigel, E. (1992). Function of the alpha 1 beta 2 gamma 2S gamma-aminobutyric acid type A receptor is modulated by protein kinase C via multiple phosphorylation sites. J. Biol. Chem. 267, 25660–25663.
Kia, A., Ribeiro, F., Nelson, R., Gavrilovici, C., Ferguson, S. S. G., and Poulter, M. O. (2011). Kindling alters neurosteroid-induced modulation of phasic and tonic GABAA receptor-mediated currents: role of phosphorylation. J. Neurochem. 116, 1043–1056. doi: 10.1111/j.1471-4159.2010.07156.x
Kittler, J. T., Chen, G., Kukhtina, V., Vahedi-Faridi, A., Gu, Z., Tretter, V., et al. (2008). Regulation of synaptic inhibition by phospho-dependent binding of the AP2 complex to a YECL motif in the GABAA receptor γ2 subunit. PNAS 105, 3616–3621. doi: 10.1073/pnas.0707920105
Krishek, B. J., Xie, X., Blackstone, C., Huganir, R. L., Moss, S. J., and Smart, T. G. (1994). Regulation of GABAA receptor function by protein kinase C phosphorylation. Neuron 12, 1081–1095. doi: 10.1016/0896-6273(94)90316-6
Kullmann, D. M., and Semyanov, A. (2002). Glutamatergic modulation of GABAergic signaling among hippocampal interneurons: novel mechanisms regulating hippocampal excitability. Epilepsia 43, 174–178. doi: 10.1046/j.1528-1157.43.s.5.12.x
Lin, Y. F., Angelotti, T. P., Dudek, E. M., Browning, M. D., and Macdonald, R. L. (1996). Enhancement of recombinant alpha 1 beta 1 gamma 2L gamma-aminobutyric acidA receptor whole-cell currents by protein kinase C is mediated through phosphorylation of both beta 1 and gamma 2L subunits. Mol. Pharmacol. 50, 185–195.
Macdonald, R. L., and Olsen, R. W. (1994). GABAA receptor channels. Annu. Rev. Neurosci. 17, 569–602. doi: 10.1146/annurev.ne.17.030194.003033
Maguire, J. L., Stell, B. M., Rafizadeh, M., and Mody, I. (2005). Ovarian cycle-linked changes in GABA(A) receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat. Neurosci. 8, 797–804. doi: 10.1038/nn1469
McDonald, B. J., Amato, A., Connolly, C. N., Benke, D., Moss, S. J., and Smart, T. G. (1998). Adjacent phosphorylation sites on GABAA receptor β subunits determine regulation by cAMP-dependent protein kinase. Nat. Neurosci. 1, 23–28. doi: 10.1038/223
McDonald, B. J., and Moss, S. J. (1997). Conserved phosphorylation of the intracellular domains of GABA(A) receptor beta2 and beta3 subunits by cAMP-dependent protein kinase, cGMP-dependent protein kinase protein kinase C and Ca2+/calmodulin type II-dependent protein kinase. Neuropharmacology 36, 1377–1385. doi: 10.1016/S0028-3908(97)00111-1
McKinley, D. D., Lennon, D. J., and Carter, D. B. (1995). Cloning, sequence analysis and expression of two forms of mRNA coding for the human beta 2 subunit of the GABAA receptor. Brain Res. Mol. Brain Res. 28, 175–179. doi: 10.1016/0169-328X(94)00228-7
Mitchell, S. J., and Silver, R. A. (2003). Shunting inhibition modulates neuronal gain during synaptic excitation. Neuron 38, 433–445. doi: 10.1016/S0896-6273(03)00200-9
Mody, I., Glykys, J., and Wei, W. (2007). A new meaning for “Gin & Tonic”: tonic inhibition as the target for ethanol action in the brain. Alcohol 41, 145–153. doi: 10.1016/j.alcohol.2007.03.009
Moss, S. J., Doherty, C. A., and Huganir, R. L. (1992a). Identification of the cAMP-dependent protein kinase and protein kinase C phosphorylation sites within the major intracellular domains of the beta 1, gamma 2S, and gamma 2L subunits of the gamma-aminobutyric acid type A receptor. J. Biol. Chem. 267, 14470–14476.
Moss, S. J., Smart, T. G., Blackstone, C. D., and Huganir, R. L. (1992b). Functional modulation of GABAA receptors by cAMP-dependent protein phosphorylation. Science 257, 661–665. doi: 10.1126/science.1323140
Moss, S. J., Gorrie, G. H., Amato, A., and Smart, T. G. (1995). Modulation of GABAA receptors by tyrosine phosphorylation. Nature 377, 344–348. doi: 10.1038/377344a0
Moss, S. J., and Smart, T. G. (1996). Modulation of amino acid-gated ion channels by protein phosphorylation. Int. Rev. Neurobiol. 39, 1–52. doi: 10.1016/S0074-7742(08)60662-5
Nani, F., Bright, D. P., Revilla-Sanchez, R., Tretter, V., Moss, S. J., and Smart, T. G. (2013). Tyrosine phosphorylation of GABAA receptor γ2-subunit regulates tonic and phasic inhibition in the thalamus. J. Neurosci. 33, 12718–12727. doi: 10.1523/JNEUROSCI.0388-13.2013
Naylor, D. E., Liu, H., and Wasterlain, C. G. (2005). Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J. Neurosci. 25, 7724–7733. doi: 10.1523/JNEUROSCI.4944-04.2005
Nusser, Z., Sieghart, W., and Mody, I. (1999). Differential regulation of synaptic GABAA receptors by cAMP-dependent protein kinase in mouse cerebellar and olfactory bulb neurones. J. Physiol. (Lond.) 521(Pt 2), 421–435. doi: 10.1111/j.1469-7793.1999.00421.x
Pavlov, I., Savtchenko, L. P., Kullmann, D. M., Semyanov, A., and Walker, M. C. (2009). Outwardly rectifying tonically active GABAA receptors in pyramidal cells modulate neuronal offset, not gain. J. Neurosci. 29, 15341–15350. doi: 10.1523/JNEUROSCI.2747-09.2009
Payne, H. L., Connelly, W. M., Ives, J. H., Lehner, R., Furtmuller, B., Sieghart, W., et al. (2007). GABAA alpha6-containing receptors are selectively compromised in cerebellar granule cells of the ataxic mouse, stargazer. J. Biol. Chem. 282, 29130–29143. doi: 10.1074/jbc.M700111200
Payne, H. L., Donoghue, P. S., Connelly, W. M. K., Hinterreiter, S., Tiwari, P., Ives, J. H., et al. (2006). Aberrant GABA(A) receptor expression in the dentate gyrus of the epileptic mutant mouse stargazer. J. Neurosci. 26, 8600–8608. doi: 10.1523/JNEUROSCI.1088-06.2006
Payne, H. L., Ives, J. H., Sieghart, W., and Thompson, C. L. (2008). AMPA and kainate receptors mediate mutually exclusive effects on GABA(A) receptor expression in cultured mouse cerebellar granule neurones. J. Neurochem. 104, 173–186. doi: 10.1111/j.1471-4159
Pidoux, G., and Taskén, K. (2010). Specificity and spatial dynamics of protein kinase A signaling organized by A-kinase-anchoring proteins. J. Mol. Endocrinol. 44, 271–284. doi: 10.1677/JME-10-0010
Poisbeau, P., Cheney, M. C., Browning, M. D., and Mody, I. (1999). Modulation of synaptic GABAA receptor function by PKA and PKC in adult hippocampal neurons. J. Neurosci. 19, 674–683.
Prescott, S. A., and Koninck, Y. D. (2003). Gain control of firing rate by shunting inhibition: roles of synaptic noise and dendritic saturation. PNAS 100, 2076–2081. doi: 10.1073/pnas.0337591100
Ransom, C. B., Wu, Y., and Richerson, G. B. (2010). Postdepolarization potentiation of GABAA receptors: a novel mechanism regulating tonic conductance in hippocampal neurons. J. Neurosci. 30, 7672–7684. doi: 10.1523/JNEUROSCI.0290-10.2010
Rossi, D. J., Hamann, M., and Attwell, D. (2003). Multiple modes of GABAergic inhibition of rat cerebellar granule cells. J. Physiol. 548, 97–110. doi: 10.1113/jphysiol.2002.036459
Saliba, R. S., Kretschmannova, K., and Moss, S. J. (2012). Activity-dependent phosphorylation of GABAA receptors regulates receptor insertion and tonic current. EMBO J. 31, 2937–2951. doi: 10.1038/emboj.2012.109
Schlessinger, J. (2000). Cell signaling by receptor tyrosine kinases. Cell 103, 211–225. doi: 10.1016/S0092-8674(00)00114-8
Semyanov, A., Walker, M. C., Kullmann, D. M., and Silver, R. A. (2004). Tonically active GABAA receptors: modulating gain and maintaining the tone. Trends Neurosci. 27, 262–269. doi: 10.1016/j.tins.2004.03.005
Sieghart, W., and Sperk, G. (2002). Subunit composition, distribution and function of GABA(A) receptor subtypes. Curr. Top. Med. Chem 2, 795–816. doi: 10.2174/1568026023393507
Sigel, E., Baur, R., and Malherbe, P. (1991). Activation of protein kinase C results in down-modulation of different recombinant GABAA-channels. FEBS Lett. 291, 150–152. doi: 10.1016/0014-5793(91)81124-Q
Tang, X., Hernandez, C. C., and Macdonald, R. L. (2010). Modulation of spontaneous and GABA-evoked tonic alpha4beta3delta and alpha4beta3gamma2L GABAA receptor currents by protein kinase A. J. Neurophysiol. 103, 1007–1019. doi: 10.1152/jn.00801.2009
Tao, W., Higgs, M. H., Spain, W. J., and Ransom, C. B. (2013). Postsynaptic GABAB receptors enhance extrasynaptic GABAA receptor function in dentate gyrus granule cells. J. Neurosci. 33, 3738–3743. doi: 10.1523/JNEUROSCI.4829-12.2013
Uusi-Oukari, M., Kontturi, L.-S., Coffey, E. T., and Kallinen, S. A. (2010). AMPAR signaling mediating GABA(A)R delta subunit up-regulation in cultured mouse cerebellar granule cells. Neurochem. Int. 57, 136–142. doi: 10.1016/j.neuint.2010.05.005
Verdoorn, T. A., Draguhn, A., Ymer, S., Seeburg, P. H., and Sakmann, B. (1990). Functional properties of recombinant rat GABAA receptors depend upon subunit composition. Neuron 4, 919–928. doi: 10.1016/0896-6273(90)90145-6
Wall, M. J., and Usowicz, M. M. (1997). Development of action potential-dependent and independent spontaneous GABA A receptor-mediated currents in granule cells of postnatal rat cerebellum. Eur. J. Neurosci. 9, 533–548. doi: 10.1111/j.1460-9568.1997.tb01630.x
Wang, D.-S., Zurek, A. A., Lecker, I., Yu, J., Abramian, A. M., Avramescu, S., et al. (2012). Memory deficits induced by inflammation are regulated by α5-subunit-containing GABAA receptors. Cell Rep. 2, 488–496. doi: 10.1016/j.celrep.2012.08.022
Wang, R. A., Cheng, G., Kolaj, M., and Randiæ, M. (1995). Alpha-subunit of calcium/calmodulin-dependent protein kinase II enhances gamma-aminobutyric acid and inhibitory synaptic responses of rat neurons in vitro. J. Neurophysiol. 73, 2099–2106.
West, A. H., and Stock, A. M. (2001). Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem. Sci. 26, 369–376. doi: 10.1016/S0968-0004(01)01852-7
Wisden, W., Laurie, D. J., Monyer, H., and Seeburg, P. H. (1992). The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J. Neurosci. 12, 1040–1062.
Wlodarczyk, A. I., Sylantyev, S., Herd, M. B., Kersanté, F., Lambert, J. J., Rusakov, D. A., et al. (2013). GABA-independent GABAA receptor openings maintain tonic currents. J. Neurosci. 33, 3905–3914. doi: 10.1523/JNEUROSCI.4193-12.2013
Keywords: extrasynaptic, GABA, kinase, tonic, plasticity
Citation: Connelly WM, Errington AC, Di Giovanni G and Crunelli V (2013) Metabotropic regulation of extrasynaptic GABAA receptors. Front. Neural Circuits 7:171. doi: 10.3389/fncir.2013.00171
Received: 13 August 2013; Accepted: 03 October 2013;
Published online: 25 October 2013.
Edited by:
Matthew Walker, University College London, UKReviewed by:
Ivan Pavlov, University College London, Institute of Neurology, UKJaideep Kapur, University of Virginia, USA
Copyright © 2013 Connelly, Errington, Di Giovanni and Crunelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William M. Connelly and Vincenzo Crunelli, Neuroscience Division, Cardiff School of Biosciences, Cardiff University, The Sir Martin Evans Building, Museum Avenue, Cardiff CF10 3AX, UK e-mail:Y29ubmVsbHl3bUBjYXJkaWZmLmFjLnVr;Y3J1bmVsbGlAY2FyZGlmZi5hYy51aw==